 탄화수소의 산화반응
탄화수소의 산화반응 Ol 규완 전북대학교 화학과 졸업 연세 대 학교 대 학원 화학과 졸업 (MS) 일본 도쿄 공대 자원화학연구소 연구원 독일 Eng le r-Bunte Insti tut , Karlsruhe Univ . ( D r . Eng .) 미국 텍사스 A&M, Associa t e Researcher(Prof. D . H.R. Ba rt on) 로 연구 현재 한국화학연구소 화학기술연구단 제 1 팀장 국내외에 약 60 여편의 논문 발표
탄화수소의 산화반응
 탄화수소의 산화반응
탄화수소의 산화반응
책머리에 대우재단에서 매년 실시하고 있는 〈학술사업 논저 부문〉에 관심을 가지고 있던 중 연구 지원 과제로 〈산화반응〉이라는 제목으로 신청 한 것이 채택되었다. 그리하여 이 기회에 필자가 주로 연구해 온 산 화반응과 유리기반응에 대한 단편적인 지식을 정리하여 집대성시키 는 기회로 삼고자 본서의 집필을 시작했다. 그러나 그 동안 약 3 년간의 연구소 행정 사무를 맡아야만 할 위 치에 있어 집필에 공백 기간이 있었고, 그 후 소속된 부서에서도 책임을 맡았던 관계로 집필이 생각했던 것보다 용이하지 않았다. 심사위원들은 이렇듯 산고 끝에 나온 초고를 수정 보완할 것을 요구하였댜 필자가 보기에도 원고에 미흡함과 결점이 많아 나름대 로 수정 보완을 하지 않을 수 없었다. 그러자니 역시 시간이 필요 했다. 집필하면서부터 갖고 있던 생각은 첫 술에 배부를 수 없다는 것 이었다. 우선 책으로 발간이 되든 안 되든 이것을 기초로 계속 보 충 • 보완하면서 이 분야의 책을 쓰겠다는 것과, 좀더 나아가 국적 (國籍) 있는 책(가능하면 한국인의 논문이 많이 인용된)을 만들겠
다는 집념도 있었다. 아무튼 필자 나름대로 수소문하여 이런저런 도움도 많이 받았지만. 자료의 부족으로 미진한 점이 있을 것으로 생각된다. 그러나 이러한 작은 결과가 이 분야 연구를 수행하고 있 는 사람들에게 조그마한 보탬이 될 수 있었으면 하는 바람으로 이 책을 탈고하였다. 같은 분야의 내용이라도 최근의 연구 결과를 수록한 최신판도 있 댜 그러나 이 책을 읽는 독자의 수준(대학원생)에서 같은 진리를 취급한 결과일진대 10 년. 20 년 전의 결과를 모르고 최신 결과 를 이 해하기 어렵기 때문에 구판의 서적을 인용한 경우 인용한 서적(문 헌)의 내용을 그대로 옮겨 썼다. 예를 들면 에너지 단위의 Ca l.과 J oule 의 혼용이 그것이댜 이러한 개념을 가지고 본 원고를 탈고하 였음을 밝혀 두는 바이다 . 어려운 화학반응식을 편집하느라 애쓴 편집부 여러분과 이 책이 나오기까지 협조해 준 우리 연구실 여러분에게 마음으로부터 감사 드린다. 특히 이상봉 박사의 원고 교정 • 집팔 전기원, 김성보. 최 명재 박사들의 자기 논문 인용 및 교정에, 그리고 끝으로 대우재단 의 지원에 감사드린다. 1998 년 5 월 이규완
차례
책머리에 3제1장 서론 111.1 산화의 정의 111.2 산화제의 종류와 그 반응 161.2.1 산소분자에 의한 산화(자동 산화반응) 161.2.2 화학약품에 의한 산화 17참고문헌 27 제2장 산소분자에 의한 산화 292.1 산소의 상태와 성질 292.1.1 삼중항 산소 292.2 기질/탄화수소 342.3 산소 산화반응 기구 372.3.1 반응열 372.3.2 스핀 금제 382.3.3 기질의 활성화-래디컬 종의 생성 422.3.4 산소의 활성화-여기 삼중항 산소 442.4 속도론 452.4.1 개시반응 462.4.2 성장반응 472.4.3 정지반응 49
2.5 자동 산화반응 502.5.1 기상-액상 자동 산화반응의 차이점 502.5.2 기상 산화반응 512.5.3 액상 산화반응 53참고문헌 75제3장 금속 촉매에 의한 산화 793.1 촉매 793.2 균일 촉매와 불균일 촉매의 상이점 823.3 균일상 촉매 산화반응 833.3.1 촉매 기상 산소 산화반응 833.3.2 촉매 액상 산소 산화반응 853.4 불균일 촉매 산화반응 1133.4.1 촉매 무산소 기상 산화반응 1133.4.2 촉매 산소 기상 산화반응 129참고문헌 156제4장 과산화물에 의한 산화 1694.1 과산화수소 1714.1.1 제조 1714.1.2 물성 172
4.1.3 분해 및 반응 1744.2 알킬히드로 과산화물 1794.2.1 제조 1794.2.2 물성 1804.3 과산화지방산 1804.3.1 제조 1804.3.2 물성 1834.4 과산화 화합물의 용도 1854.4.1 올레핀과의 반응 1854.4.2 카르보닐 그룹과의 반응 1894.4.3 방향족 화합물과의 반응 1934.5 헤테로 원자 과산화물 1984.5.1 유황 과산화물 1984.5.2 인 과산화물 202참고문헌 205제5장 슈퍼옥사이드에 의한 산화 2115.1 산소분자의 환원에 의해 생기는 산소종 2115.1.1 슈퍼옥사이드 211참고문헌 222제6장 여기 상태의 산소분자에 의한 산화 225
6.1 일중항 산소 2256.1.1 일중항 산소 발생법 2276.1.2 일중항 산소반응을 지배하는 인자 2326.1.3 일중항 산소산화의 반응 예와 합성에의 응용 237참고문헌 247제7장 오존에 의한 산화 2537.1 오존 2537.2 이중결합의 오존 분해반응 2547.2.1 오존의 첫 단계 반응 2547.2.2 Crieege의 반응기구 2567.2.3 새로 제안된 반응기구 2577.3 삼중결합의 오존 분해반응 2627.4 방향족 화합물의 오존 분해반응 2647.4.1 벤젠과 그 유도체 2657.4.2 나프탈렌과 그 유도체 2657.5 헤테로 고리 화합물의 오존 분해반응 2687.5.1 피리딘 2687.6 올레핀 오존 분해반응 상대 반응속도 2707.7 산화제로서의 오존 2717.7.1 아민, 포스핀, 아르신과 황화물 271
7.7.2 카르복시산 2737.7.3 알데히드, 케톤, 알코올 및 포화탄화수소 273참고문헌 277제8장 생체계 및 생체 모방계에 의한 탄화수소의 산화 2818.1 생체계에 의한 산화반응 2818.1.1 효소 시토크롬 P-450 에 의한 탄화수소의 산화반응 2838.2 생체 모방계에 의한 산화반응 2918.2.1 포르피린계 2928.2.2 Gif 반응계 2968.2.3 Gif-Orsay 반응계 3188.2.4 Gif-KRICT 반응계 322참고문헌 345찾아보기 353제 1 장 서론 1.l 산화의정의 이 지구상에 존재하는 모든 물질은 조건이 주어지는 데 따라 어 떠한 형태든, 주어진 속도로 반응을 보이며 변화하고 있다. 이러한 반응 중에서 가장 광범위하게 일어나는 것이 산화반응일 것이다. 그 이유는 가장 자유롭게 움직이는, 죽 대기 중에 반응성이 가장 강한 산소가 약 21 vol % 포함되어 있기 때문이다.I) 우리 주위에서 보편적이고 쉽게 찾을 수 있는 산소와 결합하는 〈산화반응〉은 〈녹슨다〉라고 표현되는 빨갛게 산화된 산화철 (Fe 요)일 것이다 . 지표면에는 이산화규소 (S i 0 2 ), 산화알루미늄 (Al203) 그리고 산 화철 (Fe2 아) 등 금속 산화물의 형태로 많은 금속들이 존재하고 있 다 . 유기 화합물의 경우는 좀더 복잡하다. 탄소수가 증가할수록 이 성체가 많이 존재하게 되며(1. 2 참조) 이에 따라 1 급, 2 급 및 3 급
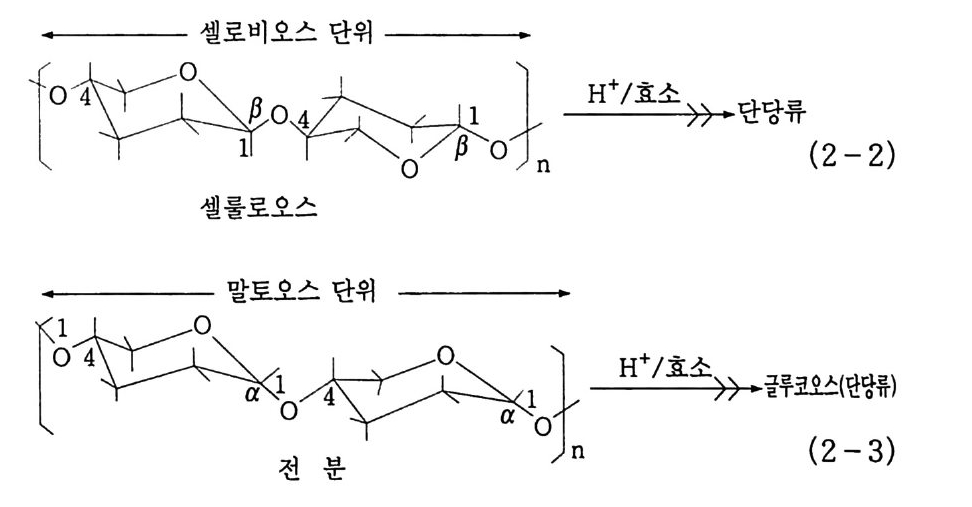
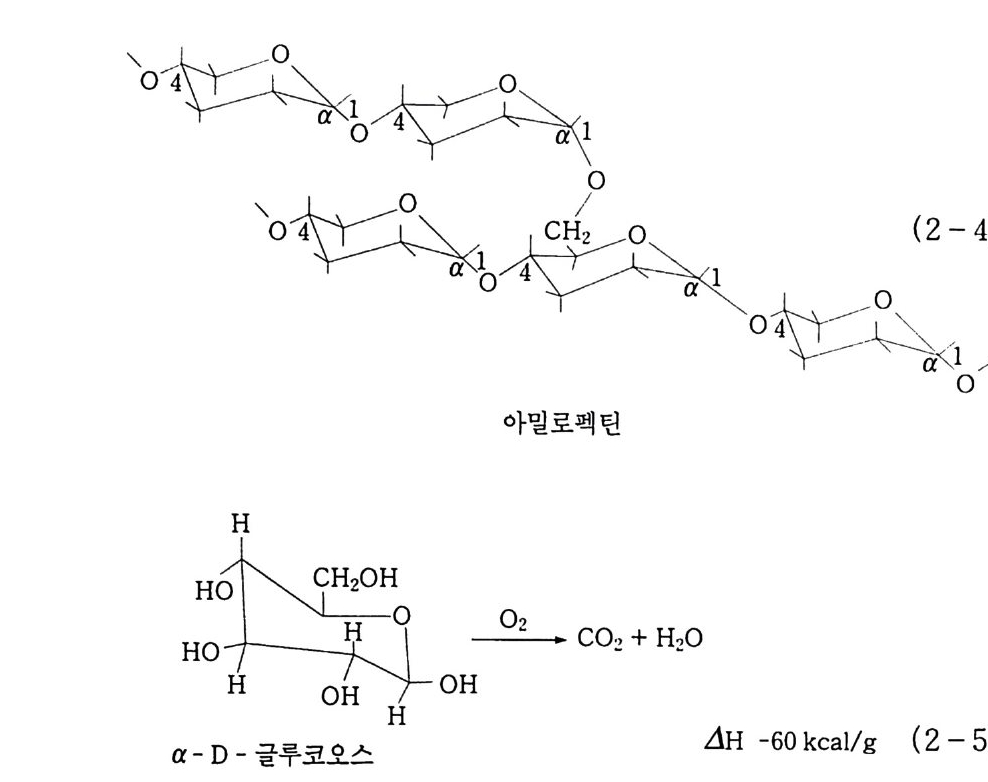
C-H 결합이 생겨서 그 반응성이 달라지기 때문이다. 유기 화합물은 여러 형태로 산화되어 인간생활에 유용하게 쓰이 지만 최종 생성물은 이산화탄소와 물이다 . 우리가 가장 혼하게 접할 수 있는 산화의 예로는 연료의 연소를 들 수 있다(식 1). 탄화수소+ 02 -CO2 + H2 0 L1H -217.8 kcal/ mo l (1) ( 1 kcal = 4.4 8 4 kJ ) 어떤 유기물을 공기 중에서 태울 때 일어나는 산화반응은 비교적 빠르지만, 체내에서의 반응은 서서히 일어난다. 즉, 동물들이 섭취 한 탄수화물은 코로 들여마신 산소와 체내에 존재하는 효소(촉매) 의 도움을 받아 다당류가 중당류를 거쳐 단당류(예를 들면 글루코 오스)로 , 다시 이것이 서서히 CO 2 와 H 2 O 로 분해되면서 우리에게 필요한 양분과 열을 공급하는 것이다. 우리 주위에서 쉽게 접할 수 있는 다당류는 셀룰로오스와 전분이 다. 이 두 다당류의 기본 골격은 포도당 단위(g lucose un it)이댜 셀룰로오스는 이당류인 셀로비오스 (Cellob i ose) 단위가 fi-1 , 4 결 합을 한 경우이고 선(線)셀룰로오스는 분자량 약 3,000 포도당 단 위로 이루어져 있다. 전분이 셀룰로오스와 다른 점은 이당류인 말 토오스 단위가 a-1, 4 결합을 하고 있다는 점이다. 전분은 1,. 4 - 결합인 아밀로오스 (am y lase , 쇄상구조)와 1, 6 - 결합인 아밀로펙틴 (am y lo p e ti n, 측쇄구조)을 가지고 있다. 또한 가 수분해에 의하여 단당류로 되지만, 1, 6- 결 합인 아밀로펙틴의 경 우는 완전히 가수분해되지 않는다. 생체 내에서나 공업적으로 효소 (enz y me) 에 의해서도 분해되는데 , a- 아밀라아제에 의하여 1, 4- 글루코오스 결합이, 1, 6- 글루코시다아제 또는 퓌아나제 (Puii ua -
nase) 에 의하여 1, 6- 결합물이 분해된다 . 분자량은 식물의 종류 , 부위에 따라 현격한 차이가 있으므로 일 률적으로 말할 수는 없다 . 아밀로오스인 경우 곡물의 것은 중합도 가 200 ~ 1,800 정 도, 뿌리 혹은 줄기 는 중합도가 1,00 0 ~ 6,200 정 도이다 . 아밀로펙틴의 분자량은 아밀로오스의 10 만 배 이상이므로 그 양이 수십만에 이른다. 이러한 전분-셀룰로오스가 우리 체내로 들어와 서서히 분해되 면서 궁극적으로는 산소와 반응하여 이산화탄소와 물(식 2-1. 2-5) 이 된다 . 이것을 소위 생분해 또는 생체 산화분해라고 하며. 온화한 조건 에서 산화반응시키는 생체 및 생체모방 산화반응 (b i o -및 bio m i m i c oxid a ti on ) 이 근래에 큰 관심을 끌고 있다. 이에 대해서 는 제 8 장에서 자세히 설명하겠다. 다당류 ―一 중당류 _ 단당류 - CO2 + H2 0 (2 -1)
 • 셀로비오스 단위 •
• 셀로비오스 단위 •
 \ o 4广 : 广\
\ o 4广 : 广\
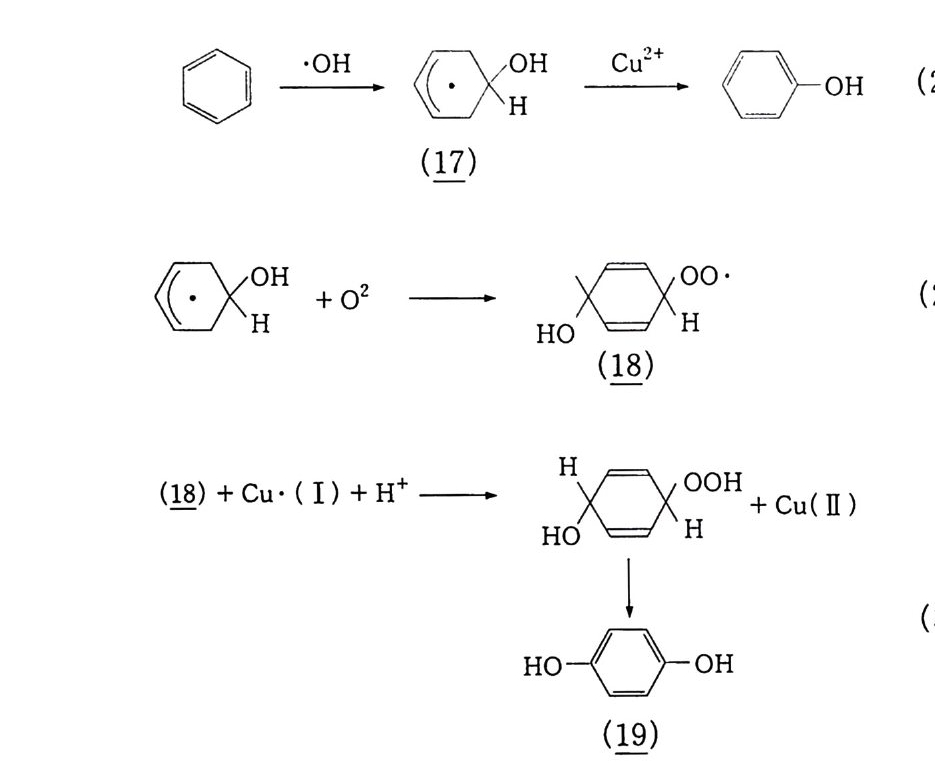
산화반응 중 가장 일반적으로 알려진 〈산소에 의한 연소〉를 예 로 들어 설명했지만, 산화는 어떤 반응물질(기질이라고 한다)에 산 소가 결합하여 산화물 상태로 있는 것이 가장 전형적이다. 예컨대 산화에틸렌(식 5) 을 들 수 있다. 물론 알코올, 케톤 등 여러 화합 물도 이에 속한다. 산화는. 반응물질이 산소와 결합하기 위해서는 대신 어떤 다른 물질이 떨어져 나와야 하고, 그러면 자연히 결합 상태가 달라지므 로 전자의 이동이 일어나야 할 것이다. 그러므로 한 화합물에서 수소원자 내지 분자가 떨어져 나오면 산 화라 하고, 전자가 없어지든지 전자 밀도가 적어지면 그 원소가 산 화되었다고 한다. 전자의 경우 크래킹이 전형적인 예이다(식 3, 4).
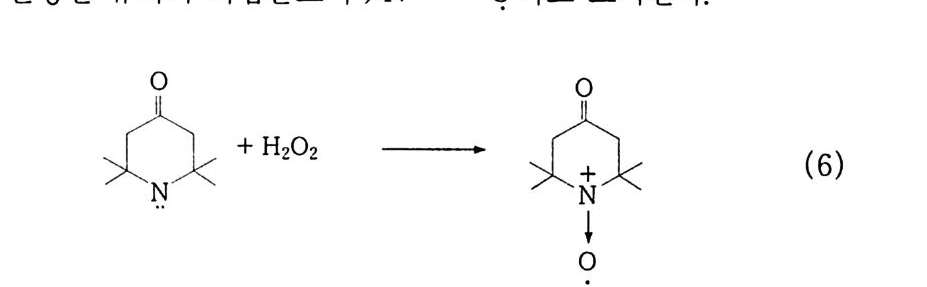
나프타 (C 5 -C 8 유분) :은七 올레핀 류 +H 2 (3) LNG (C1 -C3 유분) 브운 CH2 = CH2 + H2 (4) CH2 = CH2 으 c/H2 —°\ CH 2 (5) 후자의 경우. 트리아세톤아민 (2. 2, 6, 6-Tetr a meth y lp i pe rid - one) 을 과산화수소로 산화시키면 질소 산화물 (N-ox i de) 이 생성되 며 안정한 유리기 화합물로서 〉 N + ―-Q라고 표기한다 .2 . 3 ,4)
 二?N +H2 °2 °
二?N +H2 °2 °
지금까지 설명한 여러 가지 산화반응의 역반응은 두말할 필요도 없이 환원반응이다. 산화-환원반응은 이처럼 불가분의 관계를 가 지고 있지만 환원반응까지 취급하는 것은 너무나 방대한 작업이고 또 본 연구의 영역을 넘어서므로 본서에서는 다루지 않겠다. 그런데 왜 환원반응에 비하여 산화반응에 더 많은 관심을 가지게 되는 것일까? 그 이유는, 산화반응이 환원반응에 비하여 훨씬 완만 한 조건에서 빠르게 일어나며. 또한 우리 인간생활, 더 나아가서 우 리 인간 자체와 밀접한 관계가 있기 때문이 아닌가 생각한다. 산화반응이 산소분자에 의해서만 이루어지는 것은 아니다. 산소 를 줄 수 있는 무기. 유기 과산화물(예를 들면 과산화수소 , t-
Buty lh y d ro pe roxid e : 제 4 장 참조) 이나 전기 산화 5 1 도 빼놓을 수 없는 요소이다. 산화의 대표적인 방법으로는 열과 산화제로만 일으 키는 자동 산화 (Au t ox i da ti on) 반응과 여기에 촉매를 사용하는 촉 매반응을 들 수 있다. 각각의 반응에 대해서는 후술하는 각 장에서 좀더 상세히 설명하고자 한다. l. 2 산화제의 종류와 그 반응 산화제는 산소(자동 산화)와 화학약품을 쓰는 방법으로 대별할 수있다. 산소(공기)에 의한 자동 산화는 고전적인 방법이지만 지금도 공 업계에서 많이 이용하고 있다 . 반면 화학약품을 쓰는 방법은 제한 적으로 사용하고 있는데. 화학약품은 공기보다 비용이 많이 드는 것은 물론이고 공해문제를 야기시키기 때문이다. 공기에 의한 자동 산화는 제 2 장에서 자세히 설명할 것이다. 따라 서 여기에서는 약품에 의한 산화반응을 중심으로 간단히 기술하겠 으며, 그 중에서도 중요한 부분은 각 장에서 소개하기로 한다. 1. 2. 1 산소분자에 의 한 산화( 자동 산화반응) ―제 2 장 참조
1. 2. 2 화학약품에 의한 산화 1) 크롬산에 의한 산화 크롬산으로 이용하고 있는 것은 크롬산 무수물 (Cr03), 중크롬산 염 (M 2 Cr 요). 그리고 크롬산염 (M2Cr01) 등이다. 이것들은 산성수 용액 중에서 대부분 크롬산 (H 2 Cr01) 으로 존재하는데. 산화제로서 는 가장 오래 전부터 알려진 것이고 이미 1846 년 알코올의 산화 .6) 수산으로부터 물과 탄산가스를 생성하는 데 사용되었다.” 크롬산은 특히 황산 등 강산 존재하에서는 강한 산화력을 가지므로 초자기구 세척용액 (clean i n g solu ti on) 으로도 흔히 쓰인다. 여러 기질 중 알 코올 산화를 예로 들어 설명해 보기로 하자. (1) 알코올의 산화 1 급 알코올은 알데히드를 거쳐 카르본산 또는 그의 예스테르로, 2 급 알코올은 같은 탄소수를 갖는 케톤으로 산화되며, 3 급 알코올 은 C-H 결합이 없으므로 c-c 결합 개열이 일어난다. 이소프로 판올 (Iso p ro p anol) 을 예로 든댜 8) 3CH3CHCH3 + 2HCr04 + 8H+ 一 3CH3COCH3 + 2Cr3+ + 8H2 0 I OH (7-1) 고농도로 존재하며 산화에 관여하는 것은 HCr04 로 다음 식 (7_2) 와 같은 평형이 되며, HCr04 :;:::::::::! H+ + Cro/- (7-2)
표 1 -1 2 급 알코올의 크롬산화의 속도 상수 8 ) (물 40 C) 알코올 k 1( mo I-2 • l .2 • mm. -I ) k2( m or2 • l.3 • mi n- 1 ) 2 Prop an ol 0.053 1.94 Cy cl ooc tan ol 0.4 7 13.5 Cy cl ohep tan ol 0.61 12.4 Cy c lohexanol 0.1 6 3.2 4 Cy cl op e nta n ol 0.1 3 4.9 0 Cy cl obuta n ol 1.20 5.0 7
속도식 (7-3) 과 같이 된다. v = k[iP r OH] [HCr04- J [H +] 2 (7 -3) 산화반응 전체로는 산화제는 Cr'1-C 군로, 기질은 전자 2 개가 이용되므로 산화가 된다. 표 1-1 에 몇 가지 2 급 알코올의 속도 상 수를 나타내었다. HCr04 + H+ + R2CHOH ~k1 2R2 C HOCr03H + H2 0 ( 7 -4 ) R2CHOCr03H 一k2 R2 C O + H2Cr03 ( 7 -5 ) 2) 브산연 (TLA) 에 의한 산화 9) 4 초산연은 유기 용매에 가용하므로. 특수한 반응을 한다• 최초로 D im ro t h10) 는 균일계 반응으로 ® a- 글리콜(g l y col) 과 그 유사물 질의 개 열 ® 활성수소의 아세톡시 (aceto x y ) 치환 ® 이중결합에의
아세톡시 부가반응 등을 연구하였다. 산화제로서의 활성체는 TLA . 아세톡시 래디컬. 아세톡시 양이온 등 세 가지를 들 수 있다. Pb(OAc)1 + 2e - Pb(OAc)2 + 2Aco- (8-1) Pb(OAc)~ _-Pb (OAc)2 + 2Ac0 • (8-2) Pb(OAc) 1 - Pb(OAc)2 + Aco-+ AcO+ (8 -3) TLA 는 다른 촉매와 같이 산 촉매 작용을 받으며, 용매에 의하 여 그 생성물이 달라진다. 이것은 상술한 바와 같이 활성물이 무엇 이냐에 따라 좌우되는데. 벤젠 또는 사염화탄소 용매에 의해서는 벤젠 또는 염소가 도입된 생성물이 얻어지므로 활성체는 래디컬임 을 입증하는 예가 된댜 a- 글리콜을 예로 들어 그 반응형식을 간단히 살펴보자 .11) /\ CI -CI '/\ + Pb(OAc)4 —-I\ ' C=X + I\ ' C=Y X y H H + Pb (OAc )2 + 2AcOH (8-4) • XH. YH = OH 또는 NH2 이 식에서 보는 바와 같이 글리콜은 낮은 온도에서도 거의 정량 적, 비가역적으로 탈수소 개열반응이 일어난다. 반응속도는 용매의 영향을 크게 받아 벤젠 디옥산 둥의 비활성 용매를 사용하는 경 우 빙초산보다도 빠르다. 또한 빙초산을 이용할 경우는 물이나 알 코올을 첨가하면 속도가 증가한다 .12)
표 1 -2 Cis -시 클로핵산디올의 TLA 산화가 2 차 속도 상수에 미치는 용매 첨가량의 효과 (zo· c. 빙초산과 혼합용액) \부피(%) 에테틸이아트세 물 에탄올 아세톤 벤젠 。 4.1 4.1 4.1 4.1 4.1 0.5 5.03 10 17.l 25 90 25.2 5.6 5.4 7.0 50 850 210 12 12.6 14.0 66 2100 75 4400 1900 34 95 10000 37 670 99.3 3440
그 예를 표 1-2 에 나타내었다. 표 1-2 를 보면 알 수 있듯이 해리성 용매는 소량으로도 촉진효 과가 현저하게 나타났다. 이는 다음 식에서 보는 바와 같이 평형에 서 생기는 분해 생성물의 반응성이 크기 때문이라고 생각한다 . Pb(OAc)4 + 2H20 ~Pb(OAc)2( 0 H)2 + 2AcOH (8-5) 여기에 반하여 아세톤, 에틸아세테이트 그리고 벤젠은 첨가량 이클때효과가높다. 이것은 다음 식과 같이 중간체( I) 의 농도가 증가하기 때문이라 고본다.
Pb(OAc).1 + >C -OH-A-cO~H )\ c-o —P b(OAc)3 \IC — OH \IC —OH I I ( I) I\ C = 0 Pb(OAc)z /\ C = 0 HOAc (8-6) 여러 용매에서 반응시킬 때 속도식의 차수는 변한다 . v=k[gl y c ol] [Pb(OAc) 』 n (8-7) n 은 빙초산의 경우 l 이고 다른 용매에서는 변한다. 개열반응은 여러 설이 있으나 ( I) 이 탈식초산이 일어나며 생성 되는 Cy cl ic 중간체를 거친다고 보는 것이 일반적이다. 3) 할로겐 화합물에 의한 산화 (1) 할로겐에 의한 산화 할로겐 (Halo g en) 은 산화제로도 사용되지만 대부분 수용액 중에 서 사용된다. 알칼리 수용액에서는 다음과 같이 평형이 이루어진다. X2 + H2 0 ~ HX + HOX (9-1) 여기서 HOX 가 산화제 역할을 한다. 할로겐의 산화력은 F> Cl> Br> I 순이며 그 산화환원 전위
는 F, -2.87 : Cl. -1.36 : Br. -1.07 : I. -0.54 이다 . 불소는 너무 강력하고 요소는 너무 약해. 가격이 적당하고 취급이 쉬운 염소와 브롬이 많이 사용된다. 예를 들어 HBr 에 Cl 2 를 작용시키면 Br - 은 음전하를 잃어 Br 2 가 되고 Cl 2 는 Cl- 이 된다 . 할로겐은 구핵 시약으로 유기 화합물이 구핵 치환반응을 일으켜 가수분해에 의해 산화된다. R-CH3 + X2 -RCH2 X —-R CHX2 _-RC X3 (9-2) RCH2 X + H2 0 -RCH~ O H + HX (9-3) RCHX2 -RCHO + HX (9- 4 ) RCX3 —-R COOH + HX (9 -5) (2) 차아할로겐산염에 의한 산화 Ca-그 리고 Na-H yp ochlor it e 는 유기 화합물의 산화제 또는 표 백제인데 그 반응기구가 대단히 복잡하댜 차아염소산으로부터 생성 되어 산화제로 사용되는 것은 다음 반응식으로 나타낼 수 있다. HOX ~ HX+O (lo- 1 ) HOX + H+ ~ [H20X]+ ~ H2 0 + x+ (10-(21)0-(3_1)0 4) HOX + OH~ ~ H2o + ox- HOX :;::::::::! x+ + oH-
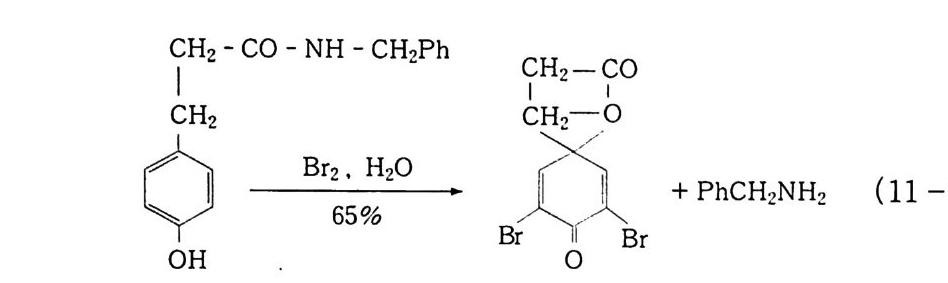
HOX .::::= HO • + •X (10 -5) 차아할로겐산염의 산화능은 할로겐과 같다. c10-) Bro- ) 10- ® 할로포름반응 CH~ CO CH3 + NaIO 一 CH3 CO OH + HCh (10 -6) CH3 CH 2 0H + NaCIO 一 CH3 CH O 一 CHCh (10 -7) C6H 5 C| H 一 CH2 CH 2 CO OH ~Br2. Na803H % . l0.C CsH s CII CH2CH2COOH OH O (10-8) ® 탈수소형 반응 13. 14) C 晶 C0II -NH-NH-C0I| -C 6 H s 쁘 c 晶0 CII -N=NC0II -C 晶 CH3 -Q -coOH ~ CH3 -o— C O OH (10-9) (10-10) ® 호프만 분해 (Hofm an De g rada ti on) 법 산 아미드에 차아염소산염을 작용시키면, 할로겐화 후 알킬기의 이동에 의해 다음과 같이 이소시안산에스테르가 생겨 가수분해가
일어나고 아민이 생성된다 . 15 ) RCONH2 쁘 RCONHBr 브 O=CNR 브으나 ~NH 2 + CO2 (11 -1) 위 반응은 산화와는 다르나 다음 반응에서는 CO-NH 간 개열 이 일어나 COOH 기가 유기 기질에 남는다.
 OCHH2 -CO -NH6 5-%C H2 P h B
OCHH2 -CO -NH6 5-%C H2 P h B
(3) 과요소산에 의한 산화 총설로 문헌 16) 을 참고하기 바란다• 4) 과망간산염 ,17) 망간산염 18) 에 의한 산화 ( 1 ) 과망간산염에 의한 산화 과망간산염은 중성一알칼리성 수용액에서 산화제로 작용하여 다 음과 같이 이산화망간으로 침전, 알칼리성이 증가하여 반응이 계속 추진되므로 선택성이 나빠진다. 2KMn04 + H20 —-2M n02 + 2KOH + 30 (12 -1 )
산성 수용액에서는 더 많은 산소를 내면서 Mn 2+ 까지 환원되어 산화력이 강하나 선택성이 나쁘다. 2KMn01 + 3H2S01 一 2MnS01 + K2 S0 1 + 3H2 + 50 (12 -2) 이 반응에 의하여 얻은 원리의 몇 가지 예를 들면 다음과 같다. ® 알코올 생성 ;C -H 一 ;C -OH (12 -3) 1, 2 급 탄화수소는 알코올을 경유하여 즉시 알데히드/케톤 (Aldehy d e/Keto n e) 으로 산화된다. ® 카르보닐기 생성 19)
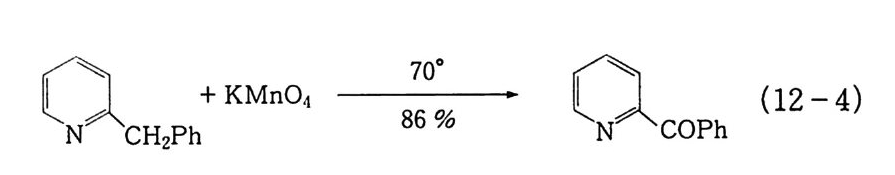 。N CH2Ph +KMn04 8760 °% 己N- COPh (12-4)
。N CH2Ph +KMn04 8760 °% 己N- COPh (12-4)
® 탄소간 결합 개열에 의한 카르본산 생성 20 , 21) p -02N-C 晶 -CH2CH3 + KMn04 ~314~~17 hr.7 p -0 2N -C sH4 -C02H + p -0 2N -C s 比 -C OCH3 (12 -5)
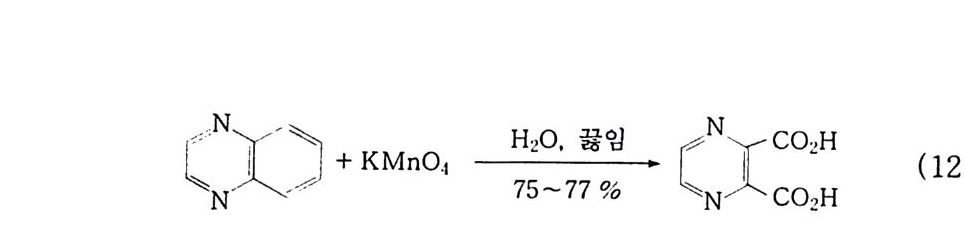 \N [>+K Mn 0.1 H752 0~, 7 7끓 %임 _f/ N xNe oHl沮c o (12 -6)
\N [>+K Mn 0.1 H752 0~, 7 7끓 %임 _f/ N xNe oHl沮c o (12 -6)
® 이중결합 탄소의 수산화와 개열 2 2 ) \CI1 / + Mn04 下=H \CI / -0 >, Mn < o - ^C H 'C\ -O O (I) ,1,I 틀'``f~무 \HH/ C CI\ -- OOHH + H20 + Mn03- (12 -7) \/ H H H 一Hoz i\CCI / \-- OOMH n02M n04~• H^\ CC I , / -- O0 MH n03- •-. \ CCI| / =-OOH +OH- + Mn02 이중결합은 알칼리성 KMn 아용액에서 히드록시화되어 ClS - g l y col 과 Ke t ol 이 생성된다. 이 반응에 대한 연구는 많이 이루어졌 다 .23~25) 이 반응은 산화제의 농도 및 PH 에 영향을 받는다. 일반적으로 산화제 농도가 높고, PH 가 낮으면 Ke t ol 이 많이 생기고 그 반대 경우는 글리콜이 많이 생성된다 .23) (2) 2 산화망간에 의한 산화 총설로 문헌을 18) 참고하기 바란다.
5) 과산화수소에 의한 산화 一 제 4 장 참조 6) 유—기제 4 과 장산 참화조물 에 의한 산화 7) 오—존제에 7 장 의 참한조 산화 8) 전—기제화 5 학 장적 참 조방 법에 의한 산화 참고문헌 1) Chem. Eng . News(March 1996). 2) a) E. G. Rozants e v,Free Nit rox y l R adic a ls, Plenum Press, New York and London( 1970) . b) A. R. Forreste r , J. M. Hay and R. H. Thoms an, Orga n ic Chemi st r y of Sta b le Free Radic a ls, 5, Academ ic Press, New York and London( 1968). 3) 이규완, 김용준, 『화학공학』, 11(1), 15(1 9 73). 4) T. Kurosak i, Ky u -Wan Lee and M. Okawara, J. Polym er Sc i., Polym er Chem. Ed. , 10, 3295 ( 1972) . 5 ) K. Yoshid a , Electr o oxid a ti on Orga n ic Chemi st r y, Joh n Wi le y & Sons, New York(1 9 84) . 6) Gmelin s Handbuch d. anorg. Chem., ill, 1, p.36 9( 1912).
7 ) E. Ludwi g, Ann. , 162, 47 (18 72 ) . 8 ) H. G. Kwi vi l a and W. J. Becker, J. Am. Chem. Soc. , 74, 5329 ( 1952 ) . 9) 총설, R. Cr iee g e, Ang ew . Chem., 70, 173(1 9 58). 10) 0. Dim roth , 0. Fri ed eman and H. Krammerer, Ber., 53, 481 ( 1920), ibi d . , 54, 3050( 1921 ) . 11) R. Cr ieg e e , Ann. , 599, 81 (19 56 ) . 12) a) R. Cr ieg ee and E. Buchner, Be,:, 73, 563(1 9 40) . b) J. M. Groshein t z , J. Ann. Chem. Soc., 61, 3379( 1939) . 13 ) E. Mohr, J. Prakt . C hem. , 70, 281 ( 1904 ) . 14) D. A. Ein h orn and R. W ill s t겼tt er, Ann., 280, 88( 1894) . 15) A. W. Hofm a n, 17, 1406, 1920( 1884), ibi d . , 14, 2725( 1881 ) . 16) E. L. Jal cson, Org. Reacti on s, 2, 341 (19 44) . 17) 총설, J. W. Ladbu ry and C. F. Cull is., Chem. Revs., 5 8, 403( 1958). 18) 총설, Qu art. Revs., 13, 61 (19 59) . 19) E. H. Huntr es s and H. C. Walte r , J. Am. Chem. Soc., 70, 3702( 1948) . 20) D. M. Kochergi n, C. A., 52, 7206( 1958) . 21 ) Org. Syn . , 30, 86( 1950) . 22) K. B. Wi be rg and K. A. Saeg eb a rth, J. Am. Chem. Soc. , 79. 23 ) J. E. Coleman, C. Ricc iu t i and D. Swem, J. Am. Chem. Soc. , 78, 5342( 1956) . 24) D. Holde and J. Marcusson, Ber., 36, 2657(1 9 03) . 25) a) R. S. Morrel and E. 0. Phil ips, J. Soc. Chem. Ind., 57, 245 (19 38). b) R. V. Lem ien x and E. Von Rudulf, Can. J. Chem., 33, 1701 ( 1955) . c) F. Oji ma and T. Osa, Bull. Chem. Soc. Jpn ., 62, 3187( 1989).
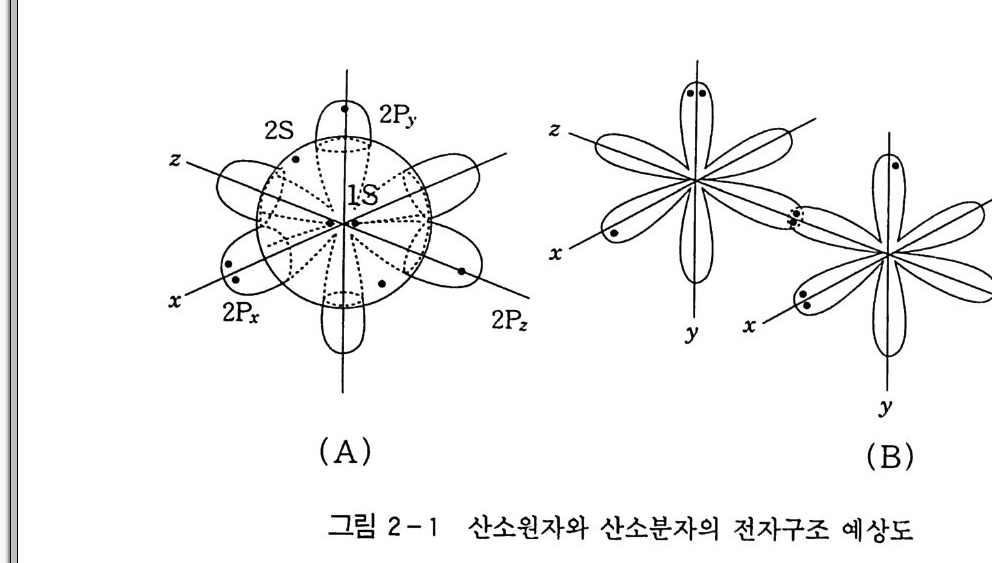
제 2 장 산소분자에 의한 산화 2. l 산소의 상태와 성질 2.1.l 삼중항 산소 산소원자 2 개가 결합해서 형성된 산소분자는 02 로 간단히 나타 내지만 그 내용을 들여다보면 매우 홍미 있다. 거의 모든 분자의 기저 상태에서 스핀 (sp in) 다중도가 1 의 일중항 상태인 데 대하여, 산소분자는 기저 상태에서 스핀 다중도가 3 의 삼중항 상태로 아주 특이하다. 그러므로 산소분자는 질소나 수소의 2 원자 분자에 비하여 높은 반응성을 가지고 있다. 기저 상태의 산소분자가 눈에 보일 정도의 속도로 유기 화합물과 반응을 하지는 않지만 어떻든 많은 유기 화 합물과 상호작용하고 또 촉매나 유리기 개시제 같은 화합물로 어떤 형태의 자극을 주면 기질(유기 화합물)과 격렬히 반응한다.
 z
z
이러한 산소분자의 반응성은 도대체 어떤 전자적 성질에서 나오 는 것일까? 1. 2. 3) 산소원자는 8 개의 전자를 가지고 있고, 다음과 같이 각 궤도에 들어 있다(식 1). 0 : (1S )2 ( 2S)2(2P)4 (1) 여기서 2P 궤도에 들어 있는 4 개의 전자가 2Px 궤도에 2 개, 그 리고 2Py 와 2Pz 궤도에 각각 하나씩 들어갔다면 식 (2-1) 과 같이 표시되고, 그 모형은 그림 2- l( A) 와 같이 나타낼 수 있다. 또 2Py 궤도에 2 개가 들어가고 나머지 궤도에 각각 하나씩 들어간다면 식 (2-2) 와 같이 나타낼 수 있을 것이다 .4) 0 : (1S )2 ( 2S) 2( 2Px)2 ( 2Py ) 1(2Pz)1 (2-1) O: (1S )2(2S)2(2Px)1 ( 2Py ) 2(2Pz)1 (2-2)
식 (2-1) 로 표시된 산소원자가 2Pz 궤도의 중복으로 (J결합을 만들든지 . 식 (2 _2) 로 표시된 산소원자가 2Py 궤도의 중복으로 1C 결합을 만들면 2Px 궤도의 고립 전자대 때문에 x 축 방향만 전자밀 도가 높게 되는 묘한 결과가 된다. 식 (2-1) 과 같이 식 (2_2) 가 동등하게 기여한다면 산소분자는 그림 2-l(B) 와 같이 나타낼 수 있다 . 이것을 루이스 (Lew i s) 식으 로 표시하면 식 (3-1) 과 같다. 1S 궤도의 4 개의 전자는 결합에 무관하므로 생략했다 . :o : :o : (3 -1) 그런데 문제는 산소분자가 자기 모멘트를 가지고 기저 상태에서 삼중항 상태임을 분광학 연구로부터 알았다는 것이다. 삼중항 상태 가 되기 위해서는 2 개의 전자가 같은 방향의 스핀으로 다른 궤도에 들어가지 않으면 안 된다. 그러므로 식 (3-2, 3-3, 3-4) 로 나타 낼 수 있다 . :.o . : :.o . : :o:o: ·O :::o• (3-2) (3-3) (3-4) 2 개의 산소원자 0A 와 OB 의 2P 원자궤도는 정상적으로 그림 2- 2 와 같은 궤도 에너지를 갖고 있고, OA 나 OB 결합에 대해서 생성 된 산소분자는 6m 元,元, 元\ 따 , 6Z · 의 6 개 분자궤도가 만들어진 다(그림 2-2). 결합성 및 반결합성의 r 궤도는 축퇴된 궤도 (Deg e nerate d orb it al) 로 순서에 따라 각 궤도에 전자를 채워 넣
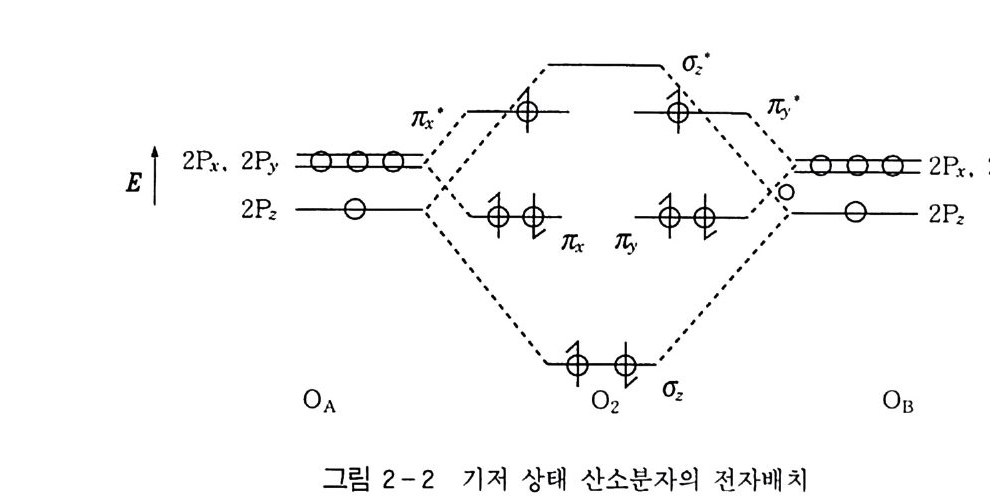 E 1 2P, ·, 22Ppz .)-=e==-,`-로\,,〈, ,군,:, 군운, , 도 :王\도 `(J: `:`\ 元홍`` · · ,三==—노 22PP ,, . 2P. I'
E 1 2P, ·, 22Ppz .)-=e==-,`-로\,,〈, ,군,:, 군운, , 도 :王\도 `(J: `:`\ 元홍`` · · ,三==—노 22PP ,, . 2P. I'
으면 반결합성의 ~·. 캄 궤도에 스핀을 같은 방향으로 전자가 하 나씩 들어간 전자배치가 가장 안정하다 (Hund 의 법칙). 이런 상태가 산소분자의 기저 상태로서 ·l z g- 라고 표시하며. 전자 배치는 식 (4) 와 같다. 0 2 (3 1:디 : ( lS1 )2( lS2)2( 2S1) 2( 2S2 ) 2 ( (Jz ) 민 ( TC,;) 2, ( n;,)민 ( 따 ) \ ( n;:) 1 ( 4) 3 I;g-에서 학는 2 원자 분자의 결합축 회전시 전자궤도 각 운동량이 O(Zero) 인 것을 나타내는 기호이며, 좌측 위의 3 은 스핀 다중도가 삼중항임을, g(g erade) 는 대칭 중심을 갖는 분자에서 대칭 중심 주위의 반전(反轉)에서 파동 함수가 부호를 변하게 하지 않는 것 (변하면 u ; un g erade) 을, 그리고 우측 상단의 -는 결합축을 포 함한 평면에 대하여 경영조작(鏡映操作, Mi rr or Op e rati on , re- fl e cti on) 했을 때 파동 함수가 부호를 바꾸는 것(바꾸지 않으면 +) 울 나타낸다. 식 (4) 의 전자배치는 언뜻 삼중결합을 갖는 식 (3-4) 와 동일하 게 보이지만, ir·궤도에 있는 2 개 전자의 반결합성에 의하여 삼중결
합의 n 결합 하나가 없어지므로 삼중결합성은 아니다. 그러므로 식 (3-2) 의 이중결합과 식 (3-3) 의 단일결합의 혼합과 같은 구조를 생각함으로써 산소분자의 성질이나 거동을 정성적으로 이해할 수 있다. 식 (3-2) 와 (3-3) 을 혼합했을 때 산소원자간의 결합은 일 종의 3 전자결합으로, 그 결합이 이중결합과 일중결합과의 중간적 성질을 가지고 있음을 나타낸댜 예를 들면 O=O 결합 해리 에너지(1 18 kca l/ mo!) 가 보통의 이 중결합 (D c;c 146kca l/ mol) 에 비해서 적다는 것, 또 원자간 거리 1.21 A 은 rc= c 1.35 A 와 rc; o 1.22 A 로부터 예상되는 ro;ol .08 A 보다 긴 것이 이해된다. 또 기저 상태의 산소분자는 삼중항 상태의 일반적 성질인 비래디 컬 (B i rad i cal) 성을 가지고 유리기에 대하여 강한 반응성을 갖는다 든지. 전자가 풍부한 물질에 대하여 구전자성을 갖고 비교적 용이 하게 전자를 취해 슈퍼옥사이드 이온 (0/) 으로 환원되는 것도 이 해가 될 것이다(제 5 장 참조).
표 2-1 기저 상태 산소분자 (3~.-) 의 여러 상수 평균핵간 거리 (ro-o ) 1.20 74 분자량 31.9 9 8 분자폭 (A) 3.18 결합 해리 에너지 118 kcal / mo! 분자길이 (A) 4.18 이온화전위 12.063 eV 비접 ('C) -182.9 7 전자친화력 0.43 土 0.02 eV 융접 ('C) -218.9 쌍국자능률 。 임계온도 ('C) -118.8 4 중국능률 -0.3 9Q xl 026 esu • cm2 동위체 분포(%) 160 (1 5.99491 )9 9.7 5 9 물에 대한 용해도 (ml) 4.9 (O 'c ) 117800 (( 11 76 ,,9999991163)) 00..02 0347 (10020gm a sl 용물)적 / 31..17 ((12000 ' C'c ) )
2. 2 기 질 1 탄화수소 5 ) 산화를 당하는 유기 물질들 중 앞 절에서 설명한 셀룰로오스 및 전분과 더불어 많이 존재하며 공업원료로 이용되는 것이 석유로부 터 얻을 수 있는 탄화수소(일명 파라핀이라고 한다)일 것이다. 그 러나 산소 존재하에서 〈태운다 〉 는 고전적인 개념으로 볼 때는 우 리 주위에서 가장 쉽게 예를 들 수 있는 화합물 LNG 를 포함한 석 유 및 석탄류 등이라고 할 수 있겠다. CnH2n+2, CnH2n+l 그리 고 CnH2 n 으로 표시 할 수 있는 화합물군으
표 2 一 2 포화 탄화수소의 이성체 수 탄소수 이성체 수 bp ('C ) mp (°C ) 5 3 36.1 129.7 6 5 68.7 -95.3 7 9 98.4 -90.6 8 18 125.7 -56.8 9 35 150.8 -53.5 10 75 174.1 -29.6 11 159 195.9 -25.6 12 355 216.3 -9.6 13 805 235.6 -5.3 14 1,858 253.6 6.2 15 4,345 270 9.9 20 366,319 345.4 36.6 25 36,797,588 53.3 30 4,111 ,84 6,763 65.8 40 62,491,1 7 8,805,831 81. 5
로는 좁은 범위로 탄소와 수소만으로 구성되어 있는 화합물을 총칭 하나. 질소. 산소 그리고 인과 같이 그 이외의 원소들이 포함되어 있는 헤테로탄화수소 (He t eroh y drocarbons) 도 존재한다. CnH2 n+ 2 로 표기하는 포화 탄화수소는 기능기가 없기 때문에 반응하기가 어 려운 화합물이다. 그러므로 단순 연료로 사용하지 않는 경우를 제 외하고는 어떠한 형태의 관능기를 형성시켜 산화반응을 시키지 않 으면 안 된다. 앞 절에서도 간단히 설명했고 또 표 2-2 에서 보는 바와 같이 포화 탄화수소는 탄소수가 증가할수록 이성체 수가 기하급수로 존
표 2-3 여러 탄화수소 C-H 결합 해리 에너지 결합 (kc에al너 I 지m o!) 결합 (kc에al너 / 지m o!) CH3 -H 105.1 (CH 깁 3C-H 85 C2 H 5 -H 98 CH2 = CH-H 104 n -C3H7-H 95 CH2 = CHCH2-H 77 i -C 3 H 7 -H 89 Ph-H 104 n -C 4H9-H 94 PhCH2-H 77.5
표 2-4 몇 가지 지방족 알칸-알켄의 산소 산화 생성물 n -도 데칸 + 02 sec-도 데칸올 n-부 탄 + 02 말레산무수물 에틸렌 + 02 산화에틸렌/아세트산 프로필렌 + 02 아세톤/아크로레인 프로필렌 + NH3 + 02 아크릴로니트릴 n -부 텐 + 02 1, 3- 부타디엔
재함을 알 수 있다. 또 표 2-3 에서 보는 바와 같이 이성체가 존재 하므로 1 급. 2 급, 3 급 탄소-수소 결합이 형성된다. 때문에 이 결합 에너지를 이용하여 반응시키든지 또는 C-H(365 kJ /m ol) 결합보 다 훨씬 낮은 C-C(245 kJ /m ol) 결합을 동시에 분해시키므로 관 능기를 도입하여 반응에 사용한다. 그 좋은 예가 나프타 크래킹 (Na p h t ha Crackin g ) 즉 에틸렌 프로필렌 등 소위 C2 - Cs 유분이다. 그외에 산소 산화반응으로 잘
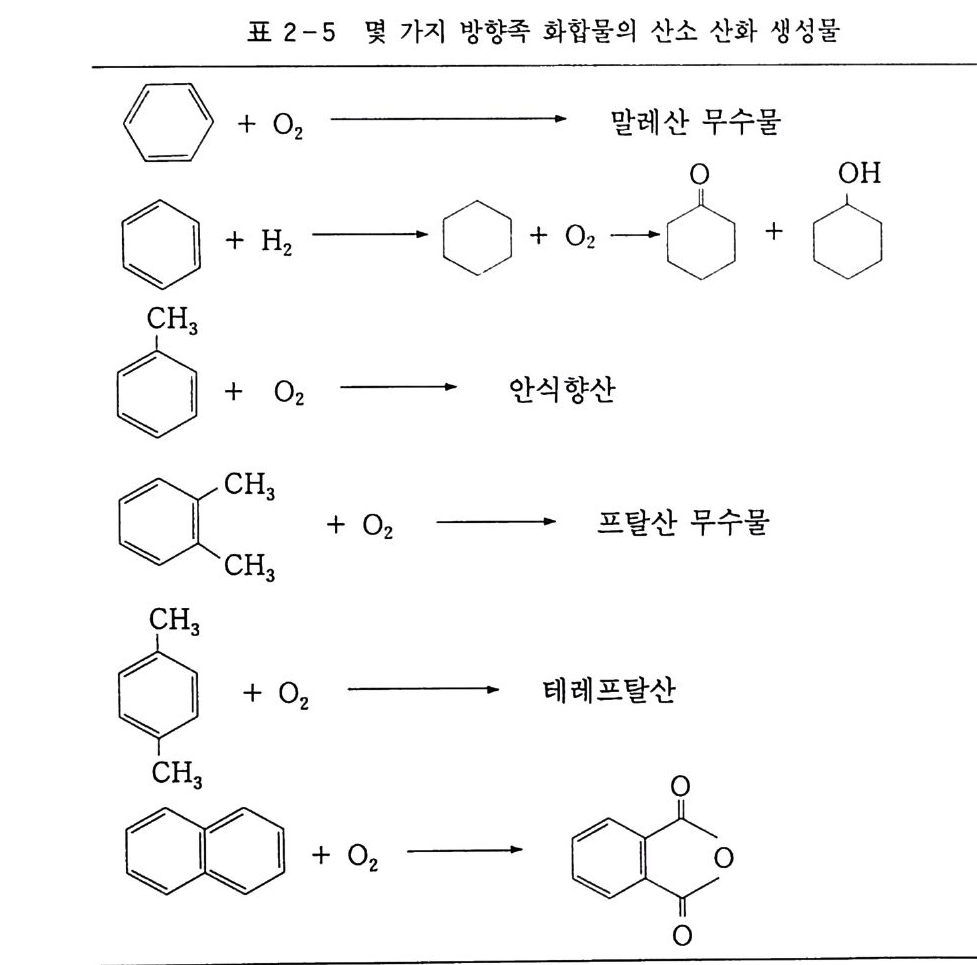 00 ++ 표 H 0,22 -一5― 몇 가―지 0 방향一 족 +화 0합2물 一말의 레산 산二소 무산수화 물 +생 성0물
00 ++ 표 H 0,22 -一5― 몇 가―지 0 방향一 족 +화 0합2물 一말의 레산 산二소 무산수화 물 +생 성0물
알려진 몇 가지 예를 들면 표 2_4 와 같다. 나프타를 촉매 리포밍시키면 많은 양의 환상 화합물을 얻을 수 있다 . 가장 기본적 화합물이 BTX. 즉 벤젠 , 톨루엔 그리고 크실렌 이댜 석유화학이 발달하기 전에는 이러한 환상 또는 방향족 화합 물들을 석탄 건유물에서부터 얻었다. 왜냐하면 석탄이 석유보다 훨 씬 낮은 C/H 비를 갖고 있기 때문이다. 몇 가지 산화반응 예를 들 면표 2-5 와같댜 산화반응의 예가 표 2_ 5 이외에 더 많이 있음은 물론이다. 이러 한 산화반응을 수행하기 위해서는 단지 공기로 반응을 일으키는 자 동 산화반응과 촉매를 사용하는 촉매반응이 있다. 후자의 경우는 더 나은 선택성과 반응조건을 최적화시키기 위하여 여러 가지 연구 가 이루어졌다 . 각 탄화수소의 산화반응은 하나씩 예를 들면서 후술하고자 한다 . 2. 3 산소 산화반응 기구 2. 3.l 반응열 유기 화합물이 산소분자와 반응하여 산소 관능기를 도입시키거 나 탈수소시키는 반응은 거의 대부분 발열반응이다. 반응의 반응열 AH 는 헤스 (Hess) 의 법칙에 의하여 다음 식 (5) 와 같이 주어진다 . AH=ZAH f° (생성물) -표 AH f°(출발 물질) (5) 죽 생성물의 생성열 총화에서 출발 물질 생성열 총화의 차이다. 한 가지 예를 들면 다음과 같다. 즉, 메탄올 산소 산화시켜 메탄
올을 생성하는 경우 출발 물질의 총생성열은 L1H=3 2 2kca l/ mo! 이 고 생성물의 총생성열은 L1H=380kca l/ mo! 이므로 생성계 쪽이 열 역학적으로 L1H=58 kcal/ mo ! 안정함을 알 수 있다. 2CH3 -H + O=O 2CH3 -0 -H D(C -H) = 102 D(O=O) =118 L'.\H = 322 L\H = 380 I D ( C -0 ) = 90 D(O -H ) = 190 2CH3 • + 2H • + 20 (6) 표 2-6 에 몇 가지 화합물의 산소 산화반응의 반응열을 나타내 었다. 이 표와 앞 절의 반응식에서 보는 바와 같이 산소를 산화제 로 쓰는 산화반응에서는 생체계이든 화학반응이든 간에 일반적으 로 발열반응이며 비가역적이라는 것이 중요한 의미를 지닌다. 또한 표 2-6 을 보면 공명 안정화가 큰 래디컬을 생성하는 경우 반응열도 낮아짐을 알 수 있다. 2. 3.2 스핀 금제 1. 1 에서도 언급하였지만, 많은 물질이 공기 중에 노출되어 있으 며 이들이 산소와 접촉함으로써 산화반응이 비가역적으로 진행되 고 있고 또한 발열반응임에도 불구하고 상온, 상압에서는 그 반응 이 빨리 이루어지지 않는다. 만일 우리 체내 물질들의 산화반응이 빨리 진행된다면 어떻게 될까? 그렇다면 인간의 수명이 무척 짧아 질 것이다.
표 2-6 산소 산화반응의 반응열 반응 [( )는 생성열 AH f o 〕〔 kca l/ m 이〕미 반응열 AH 〔 kcalIm 띠 CH~( -17.9) + ½0 2 (0 )b) 一 CH30H( _ 48.1) -30.2 C2H 6 ( -20.2) + >o2 一 C2H 5 0H ( -56.2) 一 36.0 C2H 5 0H ( -56.2) + ½0 2 一 CH3CHO( -3 9.7 ) + Hp ( -5 7.8 ) -41. 3 CH:i C HO( -3 9.7 ) + ½0 2 一 CH3COOH( -103.8 ) -64.1 C 6 H 신 19.8) + 岭 - C6H 50H( -38.9 ) -58.7 (CH3 )3 C H ( -3 2.2) + 02 一 (CH3)3COOH( -52.1 ) -19.9 2(CH 깁 3 CH( -64.4 ) + 02 一 (CH3 )3 C -0 0-C(CH 깁 3( -81. 5) + Hz0( -57.8) —74 .9 CH2 = CH-C H/4.9) + 02 一 CH2 = CH -CHzOOH( -15.1 ) _20-1-9 29 PhCH3(12) + 02 一- PhCHzOOH( -7) c I 「\ +02 一 二〉 드 (-1.7) + 02 一 b (-17.2) -15.5 ~ ( -1.7) +02 一 2CH3CHO( -7 9.4 ) -77.7 * a) AH f o 는 문헌 29)~34) 에서 인용 b) 표준 상태에서 원소의 AH f o 는 0 c) 문헌 11) 에서 인용
그러면 왜 삼중항 상태로 존재하고 있고 산소분자가 불활성인가? 그리고 그 반응을 활성화시키려면 어떻게 해야 하는가? 그 첫번째 이유는 산소가 삼중항 상태(t r ip le t s t a t e) 에 있기 때 문이다. i) 보통 일중항 상태인 기질이 삼중항 상태의 산소와 반응하면 일중 항 산화 생성물이 된다(식 7). 1A + 30 2 - 1A02 (7) 이것은 원계와 생성계 각각의 스핀 총화가 같지 않은 스핀 금제 과정으로서. 일반적으로는 용이하게 일어나지 않는다 . 그러나 광화 학 반응에서 여기 일중항 상태의 분자가 여기 삼중항 상태 또는 그 역으로 스핀 변환을 동반하는 상태 간의 전이는 잘 알려져 있다. 이러한 스핀 변환에는 1-10 -9 sec 의 시간이 필요한데, 이 시간은 보통 화학반응의 속도에 비하면 긴 편이므로 일반적인 화학반응에 서는 스핀의 금제과정이 일어나기 어렵다. 한편, 산소분자의 여기 상태인 일중항 산소분자 (10 2 ) 는 식 (8) 과 같은 스핀 허용반응으로 서 적당한 기질이라면 반응이 용이하게 일어난다. 1A + 102 - 1A02 (8) 이상의 이유 때문에 기저 상태의 산소가 일중항 상태의 기질과 스핀 금제과정을 경과하지 않고 반응을 일으키려면 식 (9— 1, 9_ 2) 와 같이 두 개의 래디컬 종이 생성되어야 좋을 것이다 . 1AH + 302 - A· + ·OOH (9-1)
1AH + 302 —-A t + 0/ (9 -2) 왜냐하면 래디컬 종은 스핀의 총화가 +1/2 또는 -1/ 2 로 되어 이중항 상태가 되므로 원계와 생성계의 각 스핀 총화가 같게 되어 스핀 허용의 반응이 되기 때문이다. 반응식 (9 기)은 산소의 래디컬에 의한 탈수소반응이다. (9-1) 의 반응열은 생성열 @H e° (02) =0, L\H c °( OOH) =4.9 kca l/ mol) 로 부터 계산해 보면 어느 반응이든 상당한 홉열반응이므로 일어나기 가어렵댜 좀더 단순하게 생각해 보면 R-H 결합 해리 에너지(대개 75~ 100 kcal/ mo l ) 와 • OOH 의 해 리 에 너 지 47 kcal/ mo l8· 9) 의 차가 반응열에 상당한 것을 알 수 있다(표 2-7 참조). (9-2) 반응은 전자 이동반응으로 50kcal/ mo l 전후의 흡열반응이어서 11) 역시 보 통의 유기 화합물에서는 상온에서 거의 일어나지 않는다. 이상의 설명을 통해 상압, 상온에서 반응이 일어나기 어렵다는 사실은 이해할 수 있을 것이다. 따라서 산소 산화반응을 일으키기 위해서는 산소와 반응하기 쉽게 기질을 활성화하든지 기질과 반응 하기 쉽게 산소를 활성화하든지. 또는 양자를 동시에 활성화시켜야 한다.
표 2 -7 RH + 02 - R, + ,OOH 의 반응열 반응 LlH ( kcal / mol) CH3CH3 + 02 - CH3CH2 • + • OOH I 6L3 CH2 = CH -C H3 + 02-C H2 =C H -C H2 • + • OOH I 35 Ph-CH3+02 - PhCH2·+·00H I 36 CH3CHO + 02 一 cHi o + • ooH I 40.3
2. 3. 3 기질의 활성화―래디컬 종의 생성 식 (9-1. 9-2) 는 앞에서 설명한 바와 같이 홉열반응이며, 바닥 상태의 산소가 비래디컬성이고 천전자반응이므로 이 산소가 반응 할 수 있는 조건을 기질에 만들어 준다면 반응이 일어날 것이다. 그 첫째 방법이 기질에 래디컬을 형성시켜 주는 일이다 . 기질을 래디컬화하기 위해서는 가열하여 산소와 직접 산화시키 는 방법과 래디컬 개시제 등에 의하여 기질을 래디컬로 만 들 어 산 화시 키 는 방법 ( radic a l chain auto x id a ti on ) 이 있 다. 그 연 쇄 반응 기구는 식 (1 0) 과 같이 표시할 수 있다. 개 시 RH + 0 2 _-R • + • OOH (10 -1. a) RH ~으므 R (10 -1. b) 생 장 R • + 30 2 쓰노 ROO • (10 -2) ROO • + RH ~노 R • + ROOH (10-3) ROO • +C=C 一 ROO-C|I -CII · (10 -4) 정지 ROO • + ·R -ROOR (lo一 5) R· +R· 一 R-R (10 -6) ROO • + ROO • 一 ROOR + 02 (10 一 7)
표 2-8 래디컬과 산소의 반응열 1 51 반응 반응열 ( kcal / mo!) CH3 • + 02 --CH3 0 0 • I -28 CH3 CH 2 • + 02 --CH3 CH zOO • I -28 .2 (CH3 )3 C • + 02 --(CH3 ) 3 C O O • I -27.7 C,;H 5C H 2 · + 02 -C6 H 5C H zOO· I -12 CH2 = CHCH2 · + 02 - CH2 = CHCHzOO·J -12 CHp • + Oz - CH: PO O • I +22
표 2 — 8 에서 보는 바와 같이 일단 생성된 알킬 래디컬과 산소의 반응은 발열반응으로서 반응이 쉽게 일어남을 알 수 있고, 또 공명 안정성이 좋은 벤질 또는 알킬 래디컬의 반응은 발열량이 적게 된 댜 10) 즉 ROO· 의 결합 해리 에너지가 R· 의 공명 안정성 때문에 적 게 되는 것이다 . CH3 0 • 은 수용성(受容性) 래디컬로 11 ) 천전자성이 므로 같은 성질을 갖는 산소와 반발하는 성질이 있다. 반대로 알킬 래디컬은 공여성의 래디컬로 전자를 내어서 카르보 늄 이온이 되려는 경향이 있어 천전자성의 산소와 쉽게 반응을 일 으킨다. 생장과정의 반응열은 .1H =DA-H + DAoo-H 로 나타내지만 둘째 항의 경우 A 에 의하여 그렇게 크게 영향을 받지 않으므로〔알킬히 드로 과산화물 ( alky l hy d rop e roxid e , HPO ) 인 경 우 보통 90 kcal/ mo l) DA-H 가 작을수록 쉽게 일어난다. 정지반응은 래디컬의 안정성, 계중의 산소 농도에 의하여 변한다 .
2. 3. 4 산소의 활성화一여기 삼중항 산소 산소분자는 여러 조건하에서 갖가지 활성 종으로 만들 수가 있 다. 여기에는 기저 상태와는 전자배치가 다른 여러 가지 산소분자 이외에 산소원자 종 0, 산소분자의 환원 종으로서 O/ . 02 -2. o; 및 프로톤화된 • OOH, -ooH, H2 0 2 , • OH 가 있는데, 이 런 것들 이 금속과 결합된 상태로 되어 여러 가지 특성을 가지고 산화반응 에 참여한다. 923 A OUD) + 0UD) 아(혹-) 1759-1950 A 아 (3Eu- ) 一 0(ID) + 0(3P ) ( 11) 2454 A 02(ZU+) 一 0(3p ) + 0(3p ) 기저 상태의 산소분자는 식 (11) 에서 보는 바와 같이 여러 파장 의 광에 의하여 각각 다른 산소 종을 생성한다. 즉, 1000 A 에 의 하여 2 개의 일중항 산소원자로 해리하고, 2000 A 전후 광에서는 단수명의 여기 삼중항 상태 3Lu- 와 32U+ 가 생성된다. 3 :E u- 는 140 kcal/ mo l, 3 착는 약 90 kca l/ mol 이 기 저 상태 보다 높다. 그리 고 3 :rg-를 여기시키는 데 110~160 kca l/ mol 의 에너지가 필요하므로 통상의 산소 산화반응에서 여기 삼중항 상태의 산소분자가 생성되 는 것은 우선 생각하기 힘들다. 물론 광산화에 의하여 생성되는 것을 생각할 수 있으나 산소 농 도보다 훨씬 높은 기질이나 용매에 의하여 광이 흡수되므로 어렵 다. 가스 상태반응에서는 가능할 것이다 . 여기 일중항 산소에 대하 여는 제 6 장을 참조하기 바란다.
2.4 속도론 래디컬 자동 산화반응에서 동력학적 거동을 아는 것은 실용적으 로 반응기구의 해명을 위해서도 중요하다. 그러나 각 소( 素 )반응에 서의 속도론 (K i ne ti cs) 적 측정은 래디컬 연쇄반응에서 특별히 어 려움과 불확실성을 내포하고 있다. 왜냐하면 부반응과 같이 병행해서 반응이 진행되며 제 2 의 반응 이 일어나고, 반응물의 농도가 아주 낮기 때문이다 . 그럼에도 불구 하고 반응의 과정들은 다음과 같은 연구로 많은 결과들을 얻을 수 있댜 즉, ® 탄화수소에서 과산화물의 분해 ® 여러 화합물의 공기산화반응 ® 기상반응에서 압력을 높이면서 ® 특별한 분석기기, 예를 들면 e. s. r, 화학형광, 래디컬 발생을 위한 u.v 흡수, 히드로 과산화물 분리를 위한 액체 크로마토 그래피, 그리고 분석을 위한 G. C 등을 이용하여 상당한 연구 가 이루어졌다. 그러나 아직도 많은 문제들이 남아 있는데 반응계측에 있는 모든 조성물들의 정량적인 측정이 그것이다 . 예를 들면 활성이 강한 중 간체는 샘플링을 한 후에도 반응이 계속 진행되므로 그 조성이 급 변한다. 반응온도가 100 ·c 이하이고 충분한 산소분압하에서 액상반응인 경우 식 (10 -1. a) 는 대단히 빠르고 R· 농도가 낮으므로 산소 홉 수 속도는 식 (12) 로 표시된다 .
-d一[d―0t2_ ]- = kpJ R OO • ][RH] (12 ) 여기서. 개시 속도가 다음과 같고. Ri = kn[ROO • ]2 + 2km[R • ][ROO • ]+ kt2 [R • ]2 Ri = (ku[ROO • ] + kt2 [R • ])2 ktI !’ . R t臼訂십 RH] 라고 한다면 [ROO • ] =(k~t1 )% (13 ) 식 (12 ) 는 식 (14 ) 로 표시 가능하다. -백d익t = -k 誌 H] (上k_tI ) ½ (14 ) 2.4 . 1 개시반응 앞의 2. 3. 3 과 래디컬 종의 생성에서 표시된 반응식 (lo) 에서 보 는 바와 같이 R 울 발생시키는 방법은. 그 반응계에 열, 광 또는 방 사선을 쪼이는 방법과, 개시제 혹은 그로부터 생성된 래디컬이나 생 성 과산화물의 분해로 생성된 래디컬에 의한 방법으로 나눌 수 있다. 작은 분자, 예를 들면 H2( 10 4.2 kcal/ mo l). CH4(C-H, 103 kca l/ mol) 과 같은 화합물은 안정한 결합을 가졌으므로 높은 해리 에너지가 필요하기 때문에. 그리고 가스 분자간의 낮은 상호반응
 b c
b c
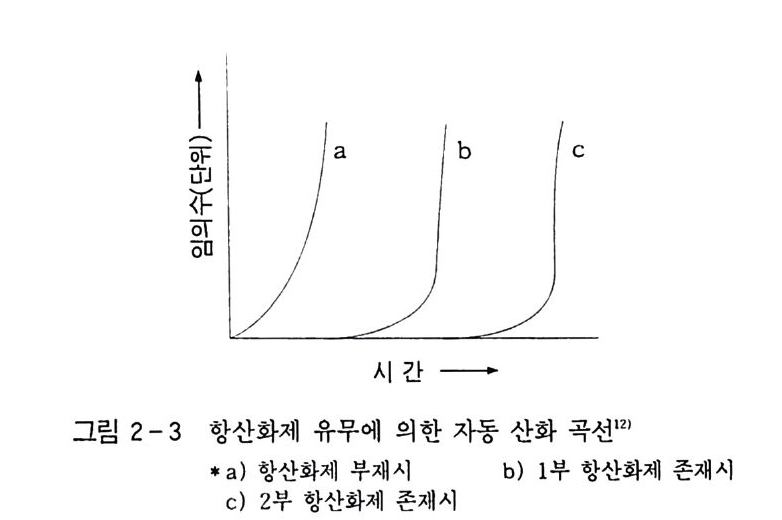
때문에 높은 온도가 필요하다. 그러나 액상 산화반응에서 고급 탄화수의 경우는 완전히 다르다. 즉, 비교적 낮은 c-c 결합 (82.6 kca l/ mol) 과 C-H 결합 (98 kcal/ mo l). 그리고 가스상보다 더 높은 상호작용 때문에 래디컬은 낮은 온도에서도 드물게, 그러나 지속적으로 일어난다(식 10-1). 비록 위에 설명한 대로 낮은 온도에서 래디컬이 지속적으로 생성 된다 하더라도 생성된 래디컬보다도 더 많이 소모된다면 유도기간 (ind uc tive pe rio d ) 이 형성된다. 이것들은 무촉매 자동 산화반응에 서 나타난댜 죽, 이 현상을 다른 말로 표현하면, 그 반응계에 항산 화제 ( anti ox id a nts ) 또는 금지 제 ( inh ib i t or ) 가 존재 한 경 우 그림 2-3 과 같다 (2.4.3 정지반응 참조). 2.4. 2 성장반응 표 2-8 에서 보는 바와 같이 생성된 R· 래디컬이 디래디컬의 산 소와 결합하여 pe roxy 래디컬을 형성하는 과정은 홉열반응으로 대 단히 빠르다( 식 10 -2).
식 (10 -3) 반응이 높은 온도에서 평형이 이루어져 R· 과 ROO· 래디컬이 존재비가 같은 온도를 천장온도 (ce ili n g t em p era t ure) 라 부른다. 예를 들면 메탈 알릴 및 벤질 래디컬의 천장온도는 0.1 산 소분압에서 각각 470 °C, 200 'C 및 100 ° C 이다 .1 3) 따라서 식 (10_3) 은 자동 산화반응에서 매우 중요하다 . 식 (10-3) 의 반응열은 L1H=DR -H -D Roo-H 로 DR oo- 11 는 많은 경우 90 kcal/ mo l 정도로 큰 차이가 없댜 그러므로 R-H 의 결합 해리 에 너지에 크게 영향을 받는다. 또한 영향을 받을 수 있는 요인으로는 그 탄화수소의 입체 효과 1 4) 와 그 용매의 극성효과 1 5) 를 들 수 있다. 자동 산화반응에 있어서 극성효과에 대해서는 많은 연구가 되었고 퍼옥시 래디컬에 의한 탈수소반응의 치환기 효과도 많이 연구되었 다 .1 6~22) 몇 가지 유기 화합물의 kp 값은 표 2-9 에 나타내었댜
표 2-9 몇 가지 유기 화합물의 래디컬 자동 산화반응에서의 kP 값 과 그에 해당하는 C-H 결합 해리 에너지 화합물 knp( l( m 6o0 l。u -)1 a) D(k(Cc -aHl)/ b ) 화합물 k(pe m( 6o0l °-1) D(k(cC a-Hl/I s·l) mol) s- 1 ) mo!) 시클로헥산 0.1 1 2 94 PhCHO 1900(5°) 74 PhCH2-H 0.9 2 85 테트랄린 27.5 CH3CH =C H2-H 1.75 80 (PhCH2)p 18 p -C H3C6H4CH2-H 2.8 74 曰o ·. H 3.4 PhC(CH3)2-H 0.72 74 Et-. ......_ / H Me/ C \ OH 2.05 * a) 문헌 23). b) 문헌 24)~30) 에서 인용
2,4,3 정지반응 반응식 (10 -5, 10-6, 10-7) 에 해당되는 반응으로 반응계 내 에 존재하는 래디컬에 의하여 상호 정지반응 (Term i na ti on) 도 일 어나겠지만. 그것보다는 반응 금지제의 역할을 빌리는 경우도 있 댜 금지제 3 ” 로는 주로 티올, 페놀류와 S-H(82 kcal/ mo l) 방향족 아민 (am i ne) 류가 사용되는데. 그 이유는 낮은 에너지에서 안정한 유리기를 형성하여 래디컬과 결합하기 때문이다. 고체 표면은 공업적으로 래디컬 촉진제로 많이 사용하는데, 예를 들면 Raschig 링이나 미세 유리관 절편 등이 있다. 식 (10 -5. 10 -6. 10 -7) 에서 산소가 충분하고 R • 이 안정하다 면(예를 들면 트리페닐 래디컬) 산소와의 반응이 줄어들므로 (10 -5. 10-6) 반응은 무시할 수 있다. 따라서 정지반응에서는 식 (1 0-7) 이 중요하다. 이 반응에서 과산화 래디컬 (ROO • )의 R 의 구조에 의하여 반응성이 현저히 달라진다. 즉, 정지반응은 ktI ){ 에 반비례 32) 하므로 3 급 수소는 1 급-, 2 급- 수소보다 102~103 정도 산화받기 쉽다 . 다시 말하면 정지반응은 3 급 <2 급 <1 급 알킬 과 산화 래디컬의 순으로 빨라진다(표 2-10 참조). 정지반응도 용매의 극성효과를 받는다. 예를 들면 테트랄린 (t e t ra li n) 에서는 극성이 높을수록 k t가 작아진다 .33)
표 2-10 연쇄 정지반응 속도 상수 화합물형 R3CH R2CH2 RCH3 C=C-CII H C=C-CIH 2- CII =C-CH3 ktI( 30°) |l~2xl03 1~10xl06 >10xl06 2~44xl04 2~6x107 ~3xl09 (e· M- 1 s- 1 )
2.5 자동 산화반응 자동 산화란 일반적으로 촉매를 사용하지 않고 기질(피산화 물질) 을 공기 또는 산소를 이용하여 산화시키는 것을 지칭한다 . 또한 자 동 산화는 산소 산화반응과 거의 동의어로서 연소나 폭발 등 격렬한 현상을 동반하지 않는 산화반응이다. 흔히 자동 산화반응이라고 하면 래디컬 자동 산화반응을 가리키는 경우가 많댜 석유화학 공업계에서는 이 반응을 이용한 많은 프 로세 스들이 실용화되어 있다. 공기는 가장 싼 산화제이므로 촉매의 유무에 관계없이 산화반응 에 가장 널리 이용된댜 유기기질(예를 들면 탄화수소)을 공기 중 에 방치하더라도 연소하는 현상이 일어나지 않는데 , 이것은 제 2 장 1 절에서 설명한 바와 같이 산소분자가 삼중항 기저 상태이고, 스핀 금제 때문에 일중항 기저 상태의 기질과 반응하지 않기 때문이다. 그러므로 기질과 산소분자를 반응시키기 위해서는 이 금제 를 풀어 주어야 하는데. 광반응에 의하여 일중항 산소(제 6 장 참조)를 만들 어 줌으로써 기질과 반응하게 하든지 유기기질을 어떤 방법으로 래 디컬 종을 반응시키면 래디컬 연쇄반응이 개시되어 자동 산화반응 이 진행된다. 2. 5. l 기상-액상 자동 산화반응의 차이점 34) 반응기구상으로 기상-액상 산화반응은 같다. 그러나 보통 공업 적 액상 산화반응은 50~200°C 사이인 반면, 기상 산화반응이 이와 비슷한 반응속도를 갖기 위해서는 100~200°C 정도 더 높아야 한 다. 왜냐하면 액상반응에서는 강한 분자간 반응이 일어나기 때문이 다. 그러나 가스 상태 반응에서 온도를 올린다면 약한 상호작용과
적은 충돌 때문에 액상반응과 다른 경로로 가게 한다. 즉, 이것이 래디컬 수명을 길게 하여 이성화반응 가능성을 더욱 높이는 것이다. 액상반응에서의 산화반응 속도는 산소 전달에 의하여 제한되지 만 가스상 반응에서는 불충분한 반응열 제거가 완전하지 못하기 때 문에 폭발을 일으킬 수 있다. 자세한 것은 문헌 35) 을 참고하기 바란다. 2. 5. 2 기상 산화반응 이제 각 기질의 실질적인 반응에 대하여 서술하겠다. 산화반응은 기질의 상태에 따라 기상 산화반응 (Gas ph ase ox i da ti on) 과 뒤에 서 설명할 액상 산화반응으로 나눌 수 있다. 다음 장에서 본격적으 로 취급하겠지만 지금부터는 촉매의 개념이 도입되어야 할 단계이 다 . 촉매 산화반응이 거의 모두를 차지하기 때문이다. 탄화수소 중 실제적으로 공업화학에 응용되는 유분은 거의 C2- C1 2 이다. 그 중에서도 C 2- C6 유분이 대부분을 차지한다고 해도 과 언이 아니댜 앞에서 설명한 바와 같이 탄화수소를 활성화시키기 위해서는 가 열해야 하므로 실온에서 액체인 저비점 액체도 기체화하지 않으면 안된다. 1) 무산소 기상 산화반응 (1) 나프타 크래킹 무산소 기상 산화반응의 가장 전형적인 예가 나프타 크래킹 반응이다. 나프타 (C5_c8. 30~12o ·c 유분)를 기질의 0.5~1 .0 무게비의 수증기와 혼합하여 격렬한 반응으로 800°C 전후에서 1 초 전후 분
표 2-11 나프타 분해방법에 의한 생성물 조성 (wt %) 16) Severity \.-六l- TO 노포으口 분해 조성물 725~800 °C 800~900 °C 1 ~1.5 sec 0.2~0.5 sec 기/- .A.l - 0.5 0.1 메탄 7.4 17.7 아세틸렌 0.1 0.5 에틸렌 17.2 32.0 에탄 3.4 4.4 메틸아세틸렌 0.4 1.0 프로펜 15.3 16.7 프로판 0.6 1.6 부타디엔 3.0 4.3 부텐 10.1 5.3 C5-204 °C 37.5 12.2
해시켜 탈수소반응으로 올레핀을 생산하는 것이다. 이 반응은 래디 컬반응으로 일어난다(표 2_11 참조). 나프타 (C5 규겁) 스一 알켄류 (15 ) - 에틸렌 -프 로필렌 -부 텐 혼합물 -펜 텐 혼합물 둥
2) 산소 기상 산화반응 여기에 속하는 반응은 완전 산화, 즉 연소반응이다. 예를 들면 빠른 반응으로는 자동차 엔진에서 가솔린 또는 디젤의 연소, 벙 커 _C 유 및 액화 천연가스 (LNG) 에 해당하는 난방용 보일러 연소 를 들 수 있으며, 느린 반응으로는 앞에서 설명한 바와 같이 우리 체내에서의 산화반응을 들 수 있다 . 따라서 이 반응은 소비되는 물질의 양으로 볼 때 가장 중요한 반 응 중의 하나이다. 2. 5. 3 액상 산화반응 1) 무촉매 액상 산소 산화반응 이 반응에 속하는 전형적인 예는 히드로 과산화물 , 알코올 , 케톤 그리고 지방산 제조를 들 수 있다. 공업적으로 사용되는 전형적인 예를 들면 다음과 같다. (1) 큐밀 및 디이소프로필 벤젠 산화 : 히드로 과산화물 ,37a,b) 방향족 알코올, 아세톤
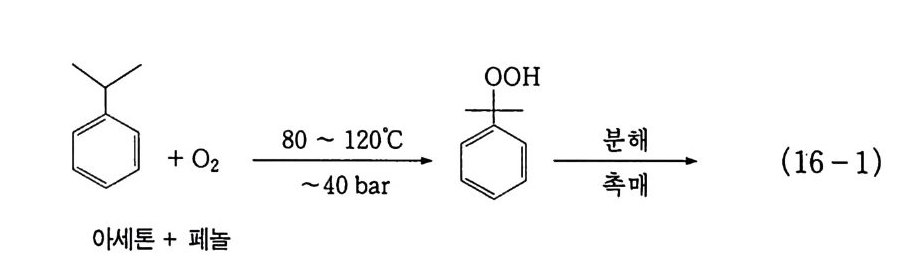 0아세톤+ + o페놀, 80~ ~4 01 b2aor·c 六OOH 분촉 매해 (16 -1)
0아세톤+ + o페놀, 80~ ~4 01 b2aor·c 六OOH 분촉 매해 (16 -1)
 \\ OOH
\\ OOH
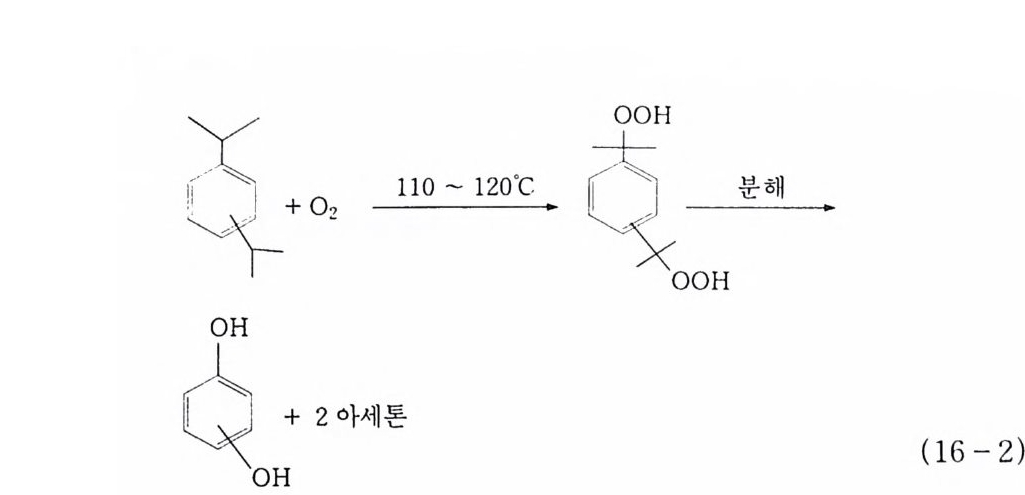
(2) t-부탄 산화 : t -부틸히드로 과산화물 . : l~l 에폭사이드. t-부탄올 十 H + 02 120~14 0‘C 一 十 OOH + (十 OH) ~40 bar 。 R-C=C ► R -C ‘/ -\C '/ + 十 OH (17 ) 촉매 에폭시 (3) 시클로헥산 산화 : 시클로헥실히드로 과산화물 , 39> KA oil, 아 디핀산.카프로락탐
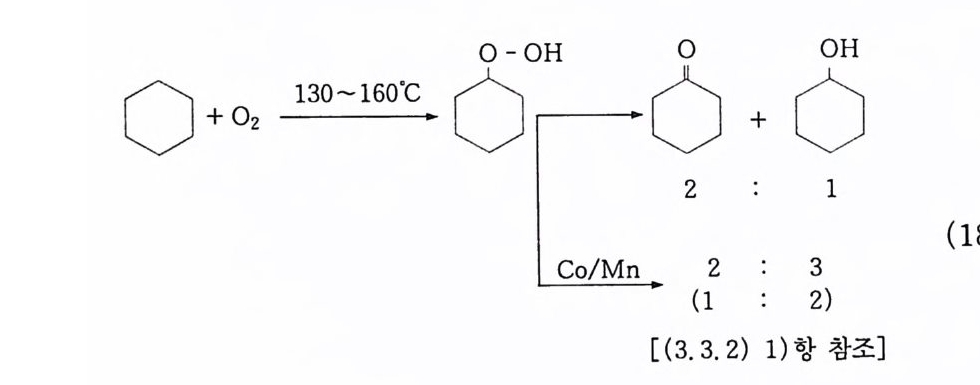 0-0H 0 oO H
0-0H 0 oO H
(4) n-파 라핀의 산화 10' (Bashk i rov 반응) : 2 급 히드로 과산화물. 2 급 지방 알코올 일명 Bashk i rov 반응이 라고 일컫는 이 산화반응은 상압에 서 과산 화 (over ox i da ti on) 를 방지하기 위해서 붕산을 사용하는 것이 특 징이댜 ® 붕산 에스테르 n -파 라핀의 액상 자동 산화반응에서는 알코올, 케톤, 산. 에스테 르 등 여러 가지 생성물이 얻어진다. 반응의 복잡성 때문에 확실하 게 밝혀진 전반적인 반응기구는 없으며. 다만 부분적인 반응기구가 여러 가지 제시되어 있다 . ' II~4 8) 특히 붕산을 첨가한 산화반응의 기 구에 대해서는 전혀 알려져 있지 않다. 일반적으로 산소에 의한 n- 파라핀 산화반응으로 이중 래디컬인 바닥 상태의 삼중항 산소분자 에 의한 래디컬 연쇄반응으로 알려져 있고, 알킬히드로 과산화물의 초기 생성물을 거쳐서 진행되는 것으로 믿어지고 있으며 n- 파라핀 산화반응의 개괄적인 반응경로는 식 (1 9) 와 같이 나타낼 수 있 댜 · 1 6) n -파 라핀 _ 알킬히드로 과산화물 _ 알코올 \ 케톤 一산 (19) Semenov49) 는 과산화물의 결합 에너지에 대하여 Q RO-OH 는 약 40 kcal 이고, Q ROO-H 는 약 90kcal 임을 구하였으며, 이 사실로부터 과산 화물이 RO· 와 ·OH 로 분해됨을 밝혔다. 그리고 일반적으로 RO· 와 ·OH 래디컬은 다른 알킬히드로 과산화물의 3 급 C-H 결합으 로부터 수소를 빼앗아 2 급 알코올과 물을 생성한다고 알려져 있다.
알코올이 생성되는 n- 파라핀의 산화반응은 래디컬반응이기 때문 에 대부분 1 급보다는 2 급 알코올이며, 반응물인 n- 파라핀과 같은 탄소수를 갖는댜 이때 붕산이 첨가되면 알코올과 붕산이 에스테르를 형성하여 과 산화를 방지할 수 있기 때문에 선택성을 높일 수 있다. 그리고 반 응 중간체로 생성되는 에스테르는 식 (20), (21) 에서 볼 수 있듯 이 m -붕 산과 알코올의 반응몰수에 따라 o -붕 산 에스테르 〔( R0)3B 〕와 m -봉산 에스테르〔( ROB0) 3 〕롤 형성할 수 있다 . 3ROH + HB02 一 (RO)3B + 2H2 0 (20) 3ROH + 3HB02 一 (ROBO)3 + 3H2 0 (21) 일반적으로 o-~ 운산 에스테르로 알려진 직쇄 알킬붕산 에스테르 는 열안정성이 매우 높으며. 특히 m- 붕산 에스테르는 47o•c 에서 도 안정하다고 보고되었다 .50) 안정성은 3 급 < 2 급 < 1 급 순으로 증 또한 알킬붕산 에스테르의
가한다 .51) 그러나 알킬붕산 에스테르는 공기 중의 수분과 같은 미 량의 물에 의해서도 쉽게 가수분해된다. 이 반응기구는 식 (22) 에 서 보는 바와 같이 친핵성 반응이므로 알킬기에 따라 가수분해 속 도가 결정됨을 알 수 있다. 그리 고 m -형 으로 알려 진 트리 알콕시 보록신 (tria l koxy b oroxin e ) RO 禪 OR [>[ OR ] \/ H20+ B 一OR 一 HO 一 B/~\ + HOR (22) OR 은 열안정성이 낮아 증류시 분해되기 쉬우며, o- 형과 마찬가지로 흡습성이 대단히 크므로 정제하여 순수한 화합물을 얻기가 매우 어 려운 반면 산소에 대한 저항성 , 죽 항산화성이 큰 것으로 알려져 있댜 본 연구에서 52) 실시한 반응온도 (150~180°C) 에서는 m- 봉산이 형성되어 알코올과 반응, 즉시 m- 붕산 에스테르가 주로 생성된다. 이 m- 붕산 에스테르는 o- 봉산 에스테르에 비하여 열안정성은 낮 으나 항산화성이 높아 알코올의 과산화를 방지하기 위한 목적에 적 당하므로 이를 가수분해함으로써 선택적으로 알코올을 얻을 수 있 는 것이다. ® 반응 속도론적 고찰 i ) 과산호頃의 생성 40 b) 반응기구의 고찰에서 언급한 바와 같이 n- 파라핀이 산화되면 우 선 알킬히드로 과산화물이 생성된다. 여기서는 n- 도데칸을 원료로 사용하여 2 급 도데실히드로 과산화물 (dodec y 1 hy dro p erox i de) 의 생성 및 분해실험으로부터 속도식을 산출하고 가정한 반응 모델에 서 유도한 속도식과 비교하여 반응 기구를 확립하였다 . n- 도데칸의 산화반응 중간체인 과산화물이 생성되는 반응차수를 구하기 위하여 일반적인 속도식 -y =dC/d t =kC 으로부터 적분법 에 의하여 얻어진 농도항에 측정값을 넣고 이를 시간에 따라 도시 하였다.
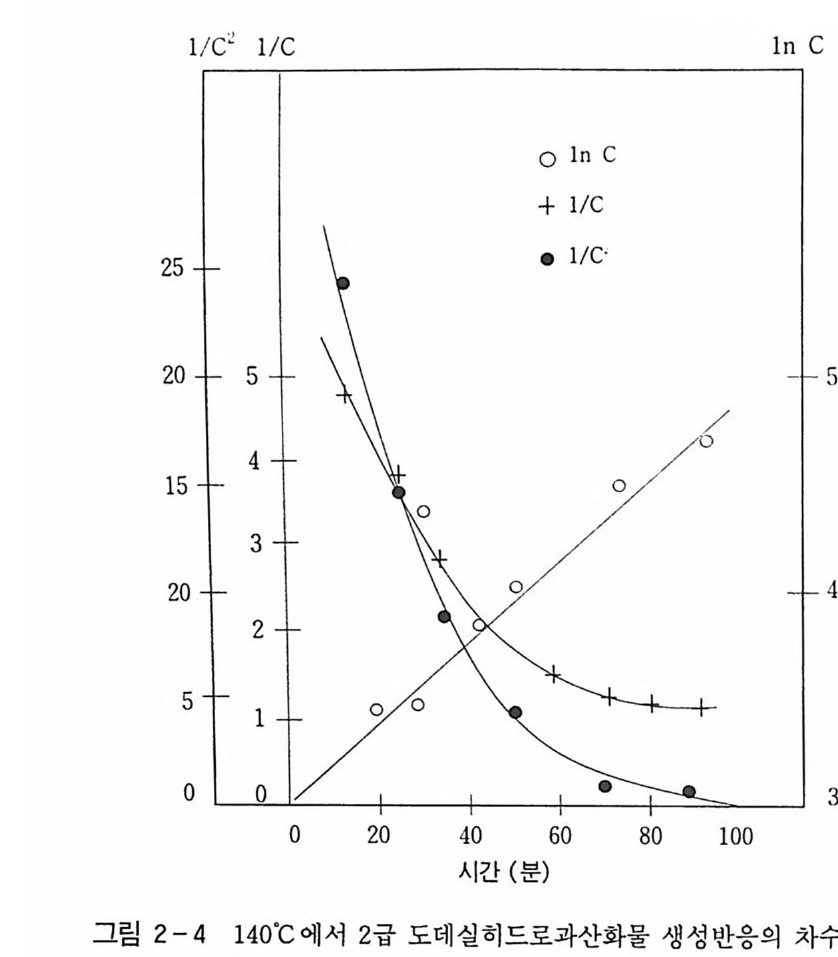 l/C2 1/C ln C
l/C2 1/C ln C
즉, 반응온도 14o·c . 공기 함량 1,1 5 0 cc/mi n/ 60g r , 봉산 5 wt % 의 조건으로부터 시간에 따라 생성되는 과산화물가를 enc, 1/C, l/C2 의 항으로 시간 t에 대해 각각 도시하였다. 이 결과 그림 2-4 와 같이 1/C, l/ C 2 에 대해서는 곡선을 나타내 었으나 enc 의 경우에는 직선을 나타냄으로써 과산화물 생성의 반 응차수가 1 차임을 알았다. 같은 방법으로 반응온도를 12o · c 에서 180 °C 까지 변화시키며 온 도가 HPO 의 생성에 미치는 영향을 조사한 결과 14o ·c 이상의 반
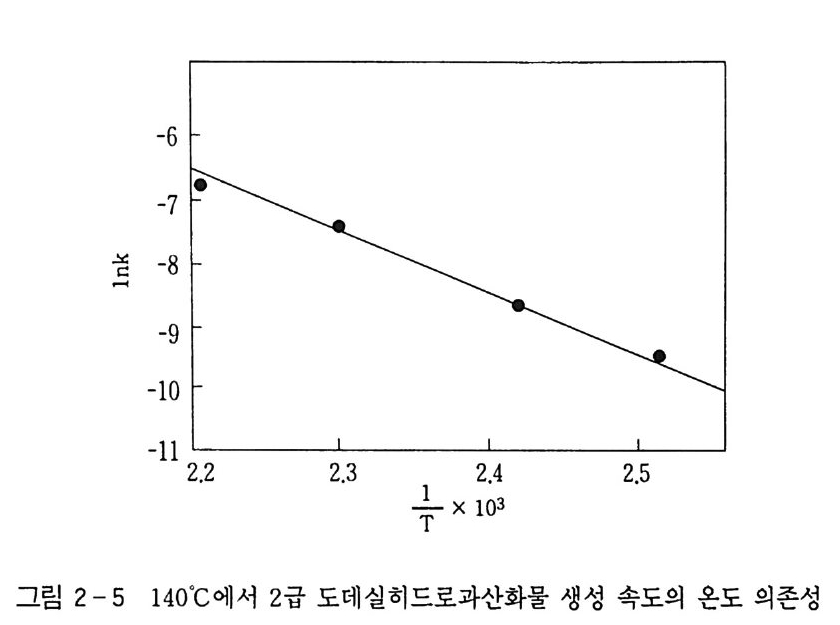 -6
-6
응온도에서는 과산화물이 생성되는 속도가 급증함을 알 수 있었다. 또한 Arrhen i us 식 k=koe-E/RT 를 이용하여 그림 2-5 와 같이 활 성 화 에 너 지 ( ac tiva ti on energy ) 를 구한 결과 약 19.2 kca l/ mol 로 서 비교적 산화반응이 용이함을 알았다. 한편 생성된 산화반응 생성물이 과산화되는 것을 막기 위하여 에 스테르화제로 사용한 붕산이 알코올의 생성에 미치는 영향을 조사 하려는 목적으로. 붕산의 양에 따라 중간체로 얻어지는 과산화물 생성반응의 속도를 추적하였다. 이 결과 그림 2-6 에서 보는 바와 같이 붕산의 양이 증가할수록 과산화물의 생성 속도가 느려짐을 알 수 있었다. 따라서 이 실험으 로 봉산은 알코올 생성을 방해하는 역할을 하고 있음을 알았고, 이 러한 현상을 정량적으로 해석하기 위하여 봉산이 작용되는 반응기 구를 조사하고자 연구하였다.
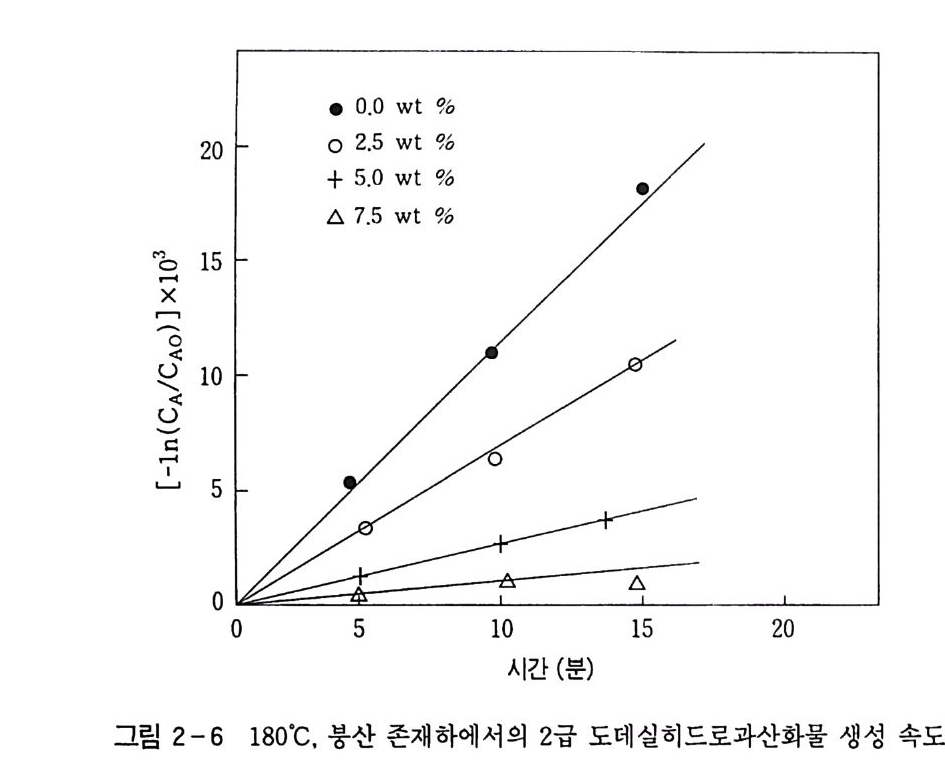 • 0.0 wt %
• 0.0 wt %
ii ) 생성반응 속도식의 고찰 2.3.3 절에서 기술한 바와 같이 n —파라핀이 산화되어 HPO 가 생 성되는 반응 모델을 다음과 같은 연쇄반응 기구로 가정하여 반응식 을 유도하고 이를 실험식과 비교하였다. 개시 RH + 02 2k1 -R • + HOO • (23 -1) R·+02 一 ROO· (23 -2) 성장 ROO • + RH 一:k2. _ ROOH + R· (23(_233) - 4 ) 정지 R·+R. 一- R-R
따라서 n -파 라핀의 감소 속도는 d 箕dt H 〕 = k! 〔 RH][O 』 + k2[ R OO· ][RH] (24) 반응조건에서 산소 농도가 일정하게 유지되고 중간체인 ROO· 에 대해서 정상 상태로 도달하게 되면 [0 2 ] 와 [ROO • ]는 일정하 다고 볼 수 있다 . d[RdtH ] = k,[RH] + k2 [RH] = k[RH] ( 단, k = k,+k2) (25) 따라서 반응속도는 n- 파라핀의 농도에 대하여 1 차가 될 것이다. 이는 앞에 나타난 실험 결과와 일치한다. 생성 속도의 고찰에서와 같이 그래프에 의한 방법으로 170 °C 의 분해반응의 시간_농도 결과를 도시하여 데이터를 처리한 결과 생 성반응과 같이 1 차반응임을 알았댜 같은 방법으로 120~180 °C 의 온도에서 분해시킨 결과 온도가 증가함에 따라 분해 속도가 증가되 었고, 특히 12o ·c 에서는 속도 상수가 0.0 0 8 m i n-1 인 데 비해서 5 wt%의 붕산을 첨가하여 같은 온도에서 분해시켰더니 0.0 4 mi n- 1 으로 5 배 증가되었다. 따라서 붕산은 과산화물의 분해반응을 촉진시킴울 알 수 있었으 며 과산화물의 분해에 따른 활성화 에너지는 약 18.2kca l/ mol 로서 생성반응 (19 . 2 kca l/ mol) 보다 더 쉽게 일어남을 알았다. iii ) 분해반응 속도식의 고찰 앞의 결과 등을 근거로 HPO 가 다음과 같이 연쇄반응 기구로 분 해된다고 가정하여 반웅 모델을 세웠다 .
ROOH 느 RO • + • OH (26) 생성된 2 급 도데실히드로 과산화물이 분해되는 단계반응에 대하 여도 앞의 생성반응에서 설명한 방법과 같이 반응차수. 반응상수 및 속도 의존성을 확인하기 위한 실험들이 수행되었다. iv ) 괴산화물의 분해 40 b) n- 파라핀이 산화되어 HPO 가 생성되고 이것이 분해되어 알코올 이 생성된댜 본 항에서는 과산화물의 분해에 관한 속도론적 고찰 을 하려 한다. 산화반응기에 n- 도데칸 300 g r 을 넣고 공기 유량을 2,000cc/ mi n/ 100 g r 으로 유지시키며 140 ° C 에서 얻은 과산화물 (P. O. V. : 44.7 mmo l/ k g)의 각 온도에서 분해 속도를 측정하였다 . 개시 RO· + ROOH 브- ·ROOH + ROH (27 -1 ) ·OH + ROOH 브 ·ROOH + R20 (27 -2) 성장 • ROOH ~노 R1COR + ·OH (27 一 3) 정지 2 ·ROOH 브 -R'-C-R+R'-CH-R +02 0II OI H (27-4) 따라서 분해 속도는 다음과 같이 쓸 수 있다. -d[ROOH]/dt = k1[ROOH] + k2[RO • ][ROOH] + k3[ ·OH][ROO 町 (28)
RO·. ·OH, ·ROOH 에 대하여 정상 상태가 이루어진다면 d[ R O• ]/dt = k1[ROOH] -k2[ R O • ] [ROOH] = 0 k1[ROOH] = k 誌 O • 〕 [ROO 町 (29) d[ ·OH]/dt = k2[ R OOH] 一 k3[ . 。띠〔 ROOH] + k1[ R OOH] = 0 k3[ • OH][ROOH] = k1[ROOH] + k4[ R 。 OH] (3 이 d[ · RO 。 H]/d t = ki( R O • ] [RO 。 H J + k3[ ·OH] [ROO 띠 -k1[ ·ROOH] -ks[ R 。。 H] 2 = 0 (31) 식 (29), (30), (31) 로부터 ks[ · ROOH]2 = 2k1[ROO 町 仁 ROOH] = (2k/k 이 %[ ROOH]½ (32) 식 (28) 의 각 항에 식 (29), (30) 및 (31) 을 대입하면 -d[ROOH]/dt = 3k1[ROOH] + k4[ · ROO 띠 = 3k1[ROOH] + k 4 (2k i/ k 깊 %[ROOH J ½ (33) 여기서 k1, ks 는 각각 개시 단계 및 종결 단계의 속도 상수이므로 일반적으로 k1~〈 k5 이고, 케톤의 생성 속도는 느리므로 k 4 ~〈 k5 이다 . 따라서 두 번째 항은 무시할 수 있다. -d[ROOH]/dt = k/ [ROOH] (단, k/ = 3k1) (34)
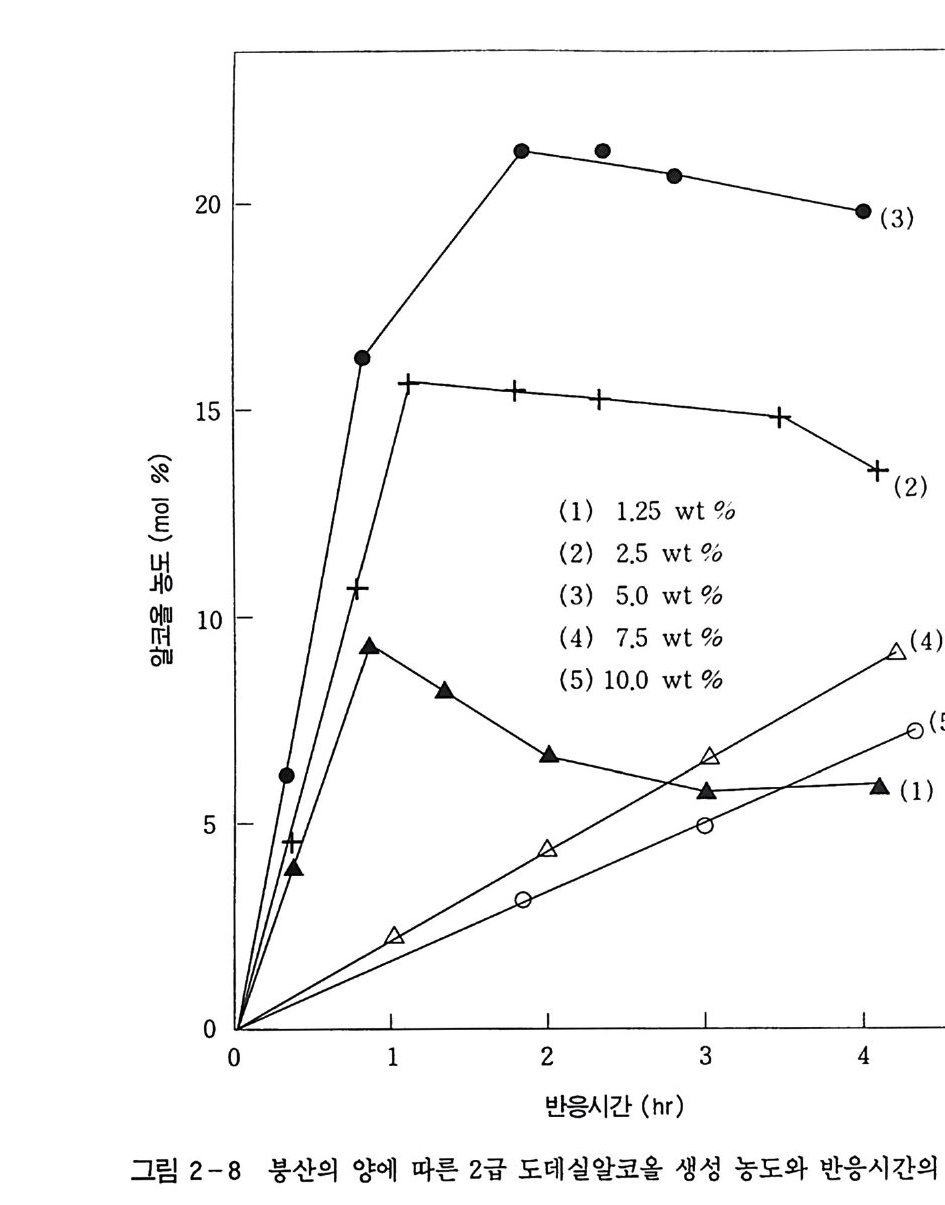
즉 HPO 의 분해 속도는 그 농도에 대하여 1 차가 된다. 이는 실 험 결과와 잘 일치한다. ®m 봉산의 영향 i ) m- 봉산 양의 영향 탈수과정에서 얻은 m- 봉산은 산화반응에서 생성되는 알코올과 반응하여 안정한 m- 붕산 에스테르를 형성함으로써 알코올의 과산 화를 방지하여 알코올 회수율을 높여 준다. 그러므로 높은 수율의 알코올 생성물을 얻기 위해서는 적정량의 붕산이 필요하나 붕산과 붕산 에스테르는 산화반응에서 반응 억제 제로도 작용하기 때문에 에스테르화제인 붕산의 첨가량이 조절되어
 6
6
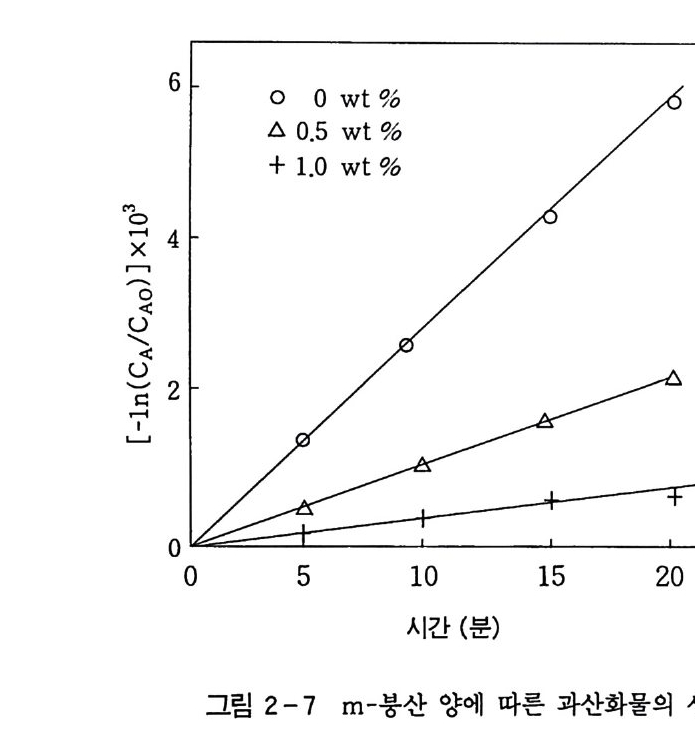
야 한다. 그러므로 m- 붕산의 양이 산화반응에 미치는 영향을 알 기 위해 실험실 규모 (Lab. scale) 의 산화반응기에 60 g의 n- 파라 핀을 넣고 m나당산의 양을 변화시키며 실험하였다. 그 결과 그림 2 一 7 에서 보는 바와 같이 붕산의 양이 증가할수록 히드로 과산화 물의 생성률이 저하되었다. 위의 결과로부터 m -붕산의 존재는 알킬히드로 과산화물의 생성 을 억제시키며. 반응의 개시는 붕산의 양에 영향받음을 알 수 있었 댜 또한 그림 2-8 에서처럼 알코올의 생성 속도도 봉산의 양이 증 가함에 따라 매우 저하되는데. 이는 과산화물의 생성과 밀접한 관 계가 있기 때문이댜 그러므로 붕산의 존재하에서 n- 파라핀 산화 반응의 래디컬반응의 메커니즘은 다음과 같이 나타낼 수 있다. RH + 02 一 R· + H00· (35) R· + 02 一 ROO· (36) ROO· + RH 一 ROOH + R· (37) ROO • + BA - ina c tive pro duc t (38) ROO • + ROO • - ina c tive pro duc t (39) 여기에서 RH 와 BA 는 n- 파라핀과 m 나당산을 의미한다. m 봉산의 농도가 임계값 (cr iti cal value) 에 도달할 때까지는 과 산화반응이 일어나지 않는다. 붕산의 양이 증가됨에 따라 임계 농 도에 가까워지면 반응단계 (38), (39) 와 같이 메타 (me t a) 봉산의 표면에서 ROO· 래디컬의 종속반웅이 일어난다. 이때 정상 상태에
 20 ,_ (3)
20 ,_ (3)
서 알킬퍼옥시 래디컬의 농도는 다음 식과 같이 나타낼 수 있다. ROO • = ½ [~ (BA) = [信 ) 2 (BA) 도t (RH) (02) 「] (40) 반응식 (40) 은 bin o mi al exp ans i on 에 의해 식 (41) 로 단순화된다. (ROO· ) = ~k1( kB 1 A) (RH)(02) (41) n- 파라핀의 산화반응 속도는 반응식 (42) 와 같이 나타낼 수 있 으며, 반응식 (42) 는 반응식 (43) 과 같이 적분된다 . -¥ = k1(RH) (02) + dfu (RH)2(02) (42) kI 122 ) + ·en· [1 + (RH)(R 。 O-O( RHO) OH) ] + en [1 一 1쓰 :노 ((R「O니O)H) 。 ] ] = t ~'t (43) k, (BA) 여기서 r 는 정상 상태의 과산화물 농도에 도달하는 시간인 유도 기간을 의미한다. 정상 상태의 과산화물 농도가 붕산입자의 분포가 일정할 때 봉산 의 농도와 비례한다고 가정하면 식 (43) 에서 t=i에서 ROOH= (ROOH)s = Q (BA) 로 나타낼 수 있으므로 r 는 식 (44) 와같이 표현될 수 있다.
r = kI(\2) [야 -? ~r + en(1-—k, _1 .Q~ )] (44) k, (BA) 반응식 (44) 는 붕산 /n? 고데칸의 농도에 대한 개시 시간을 나 타낸댜 r 값은 임계값을 나타내며 요값(표면임계상수)은 붕산의 표면 위 에서 (ROO· ) 래디컬의 종속반응에 관계하는 상수이므로 Q값은 다음 식과 같이 반응 속도이론을 적용할 수 있다. (RH) 。 요= BA Q = 요 exp ( ER /R T) (45) 여기에서 터는 붕산의 표면과 반응하는 퍼옥시 래디컬의 흡착에 기인하는 활성화 에너지이며 온도 T 는 절대온도이다. 붕산의 임계 농도는 0.2mm 의 평균입자를 가진 메타 봉산의 평
표 2-12 도데칸의 액상 산화반응에서 각 온도에 따른 붕산의 임계 농도와 표면임계 상수(요) 온(°C도 ) 초농기도 도(m데o칸l/의L ) 붕산(의m o임l /계 L )농 도 Q 150 4.4 1 0.3 0 0 14.7 0 160 4.4 1 0.723 6.1 0 170 4.4 1 1.1 4 6 3.8 5 180 4.4 1 1.81 8 2.4 3
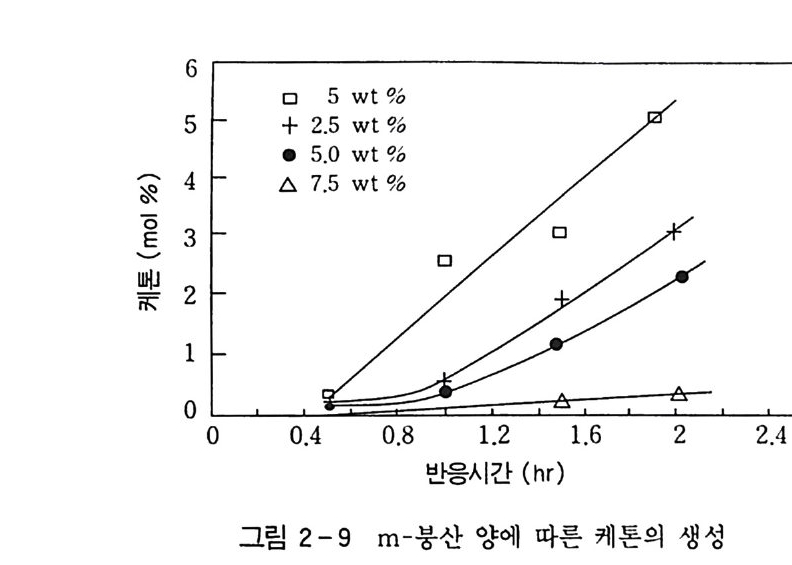
균입자에서 측정되었으며. 온도에 대한 값들이 표 2_l2 와 같이 얻 어졌고 E R 값은 T 에 대한 lo g.Q값을 도식하여 구해졌는데. 구하여진 E R 값은 19.87 kca l/ mo! 이었댜 표 2-12 에서 보는 바와 같이 n- 파라핀의 산화반응에서 반응이 거의 정지되는 붕산의 임계량은 요의 증가 및 반응온도의 감소와 함께 감소된댜 ii ) 붕산 존재하에서의 반응경로 고찰 봉산의 존 재하에서 반응경로는 다움 식과 같이 나타낼 수 있으며 n- 파라핀k〈 1 알케코톤올 느 느 지 붕방산산 에스테르 (46) k , 1 가 k1 보다 매우 빠르므로 산화반응 경로는 다음 식과 같이 단 순화할 수 있다. k1 / 붕산 에스테르 n- 파라핀 \ k2 케톤 上느 지방산 (47) 이처럼 단순화된 모델은 그림 2-9 와 같이 붕산 양의 증가와 함 께 얻어진 결과처럼 붕산의 증가와 함께 케톤의 생성이 크게 억제 되었댜 산화반응이 개시되어 정상 상태에 도달한 후 n- 파라핀의 산화반 응은 많은 연구에서 1 차반응임이 증명되었으며 산화반응 속도식은 다음과 같이 나타낼 수 있다.
 6
6
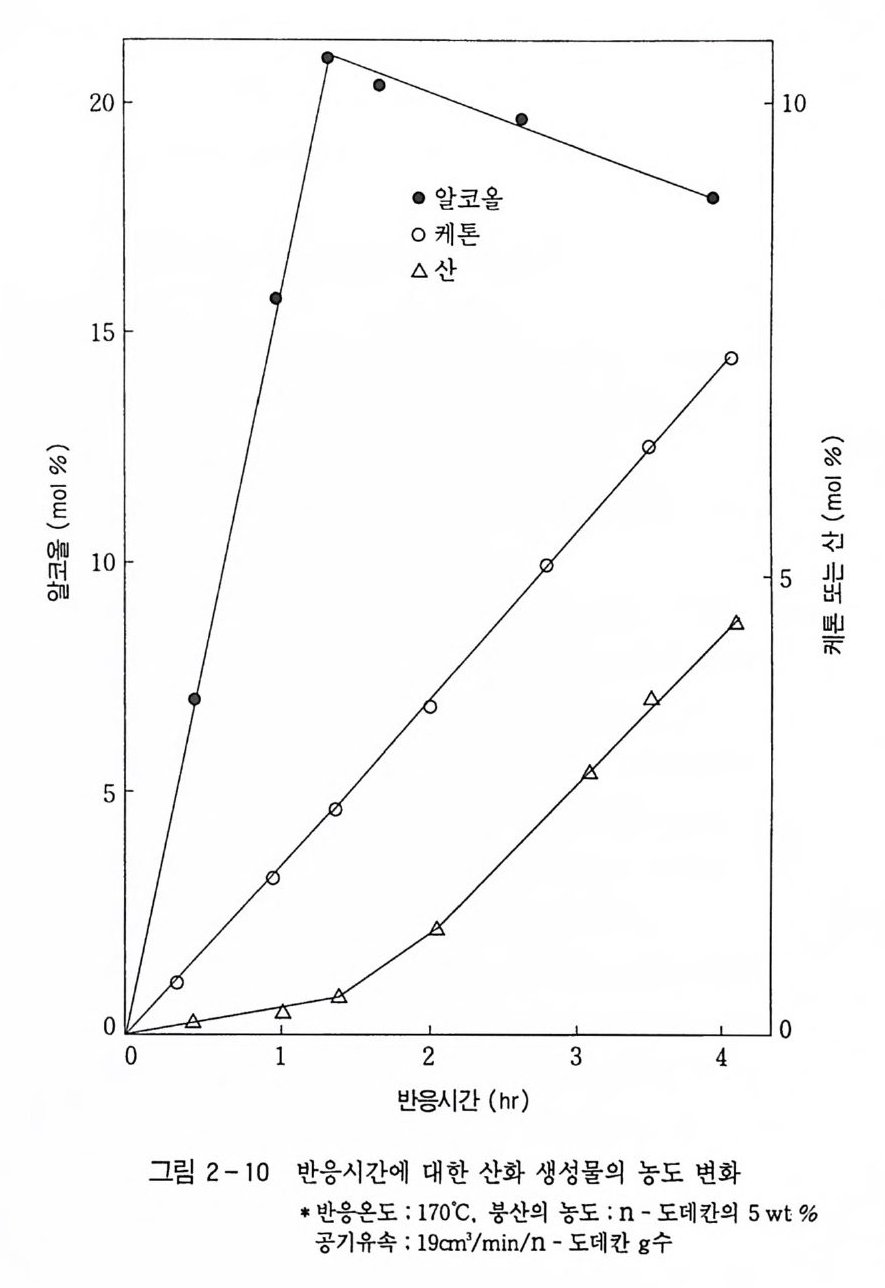
dCo/dt = -( k1 + k2) C o (48) dCB/dt = kICD (49) dCK/dt = k2C o -k 3CK (50) dCA/dt = k3CK (51) 산화반응에서. 반응시간 t에서 생성된 반응물의 각 농도는 다음 식과 같이 나타낼 수 있으며 정상 상태에서의 n- 도데칸의 산화반 응은 그림 2-10 과 같다. xd = Co/Coo = e-al (52) XB=C 십 CDo= f3 (1-e-a t) (53)
 20 10
20 10
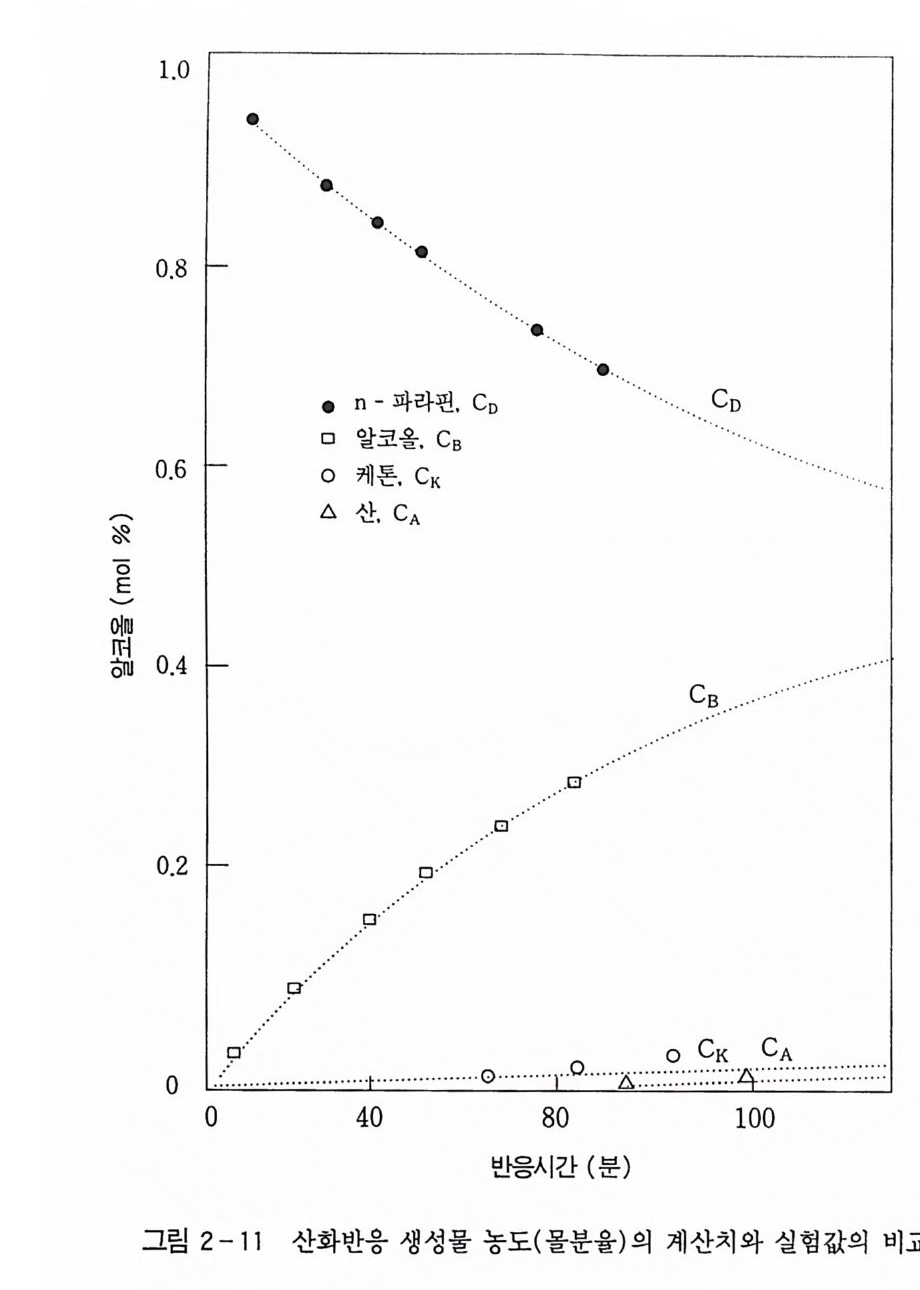 1.0
1.0
XK = CdCn o = y(e - at -e-k :tt ) (54) XA = CA /C oo = 1-e-a l -{3(1- e-•t) - y( et - e-k ) (55) 단. a=k1+k2, /3=k i/ a, y= k2/ k 3 - a n- 도데칸의 산화반응에서 반응 개시 시간은 20 분이었으며. 위의 산화반응식에서 계산된 반응속도 상수는 k1=3.63X10-3, k2 = 0.37 XlO 집 . 그리고 k3 = 4.02x10-3 m i n-1 이었다. 실험에서 얻은 결과들도 그림 2-11 과 같이 본 연구에서 제안된 반응속도 모델과 거의 일치하였다. 이외에 n- 파라핀의 산화반응에서 원료 중의 방향족 화합물 , 유 황 화합물 등과 에스테르화제로 사용되는 붕산의 양과 크기 등의 복합적인 원인에 의하여 산화반응이 억제된다. 그러므로 붕산이 과 량 존재하는 액상 산화반응에서 알킬히드로 과산화물의 생성을 촉 진시킬 수 있는 적절한 촉매의 선택이 필요하였으므로 촉매 선정을 위한 연구들이 함께 수행되었으나 산화반응에 초점을 맞추어 기술 하기 위하여 본서에서는 더 이상 다루지 않겠다. 지금까지 몇 가지 예를 들었으나 결과적으로는 래디컬과 히드로 과산화물을 형성한 다음 계속적으로 (분해)반응이 일어나 생성물 이 만들어 진다. 그러므로 제 2 장 2. 3. 3 절 반응식 (lo) 의 래 디 컬에 산소부가 ® 반응과 다른 래디컬과 재결합하는 정지반응 ®와 ® 반응이 일어난댜 공업적으로는 산소를 거의 소모시키기 때문에 반 응탑 상부에서의 산소 농도는 거의 무시할 정도이고, 반대로 탄화 수소 래디컬 농도는 증가하므로 ®와 ®과 같은 정지반응이 일어난 댜 ROO· 래디컬의 비교적 낮은 반응성 때문에 ROO· 래디컬은 감지할 수 있는 어느 농도까지 축적된다( ·OH, ·R, RO· 래디컬
은 빠르다). 실제로 큐멘퍼옥시 래디컬의 농도가 8x10-sm ol/ L( 5 x1016 분자 /cm 3 에 상당) 임을 e. s. r 로 확인했다. 그러므로 쉽게 재 결합하여 다음 식과 같이 반응한다. H R1 \ C / R\1/ c \ / H ..…..; .·.·.·.·…o 00.| /· \ R2一 RRI2 \/ ' C=O + o.` + H O~\ / C )/R. 2I (56) R2 RI RI R2 R2 RI RI R2 R\3/ C \/ 。。\ / C \/ R3 R\3/ C \ 。 。\ / C \ R3 | I I/ r _ t\ O· ·O 0-0 R2 RI RI R2 (57 -1) R\3/ C \/ 0-0\/ C \/ R 3 + 02 R\/ C \/ R R\/ C \/ R • 2 R\ / C \/ R -2 R\/ C=O + 2R· (57 -2 ) R OI OI R R + 02O · R 0-0 반응식 (56) 와 같이 1 급, 2 급 ROO· 인 경우 빠른 재결합과 분 해를 통해 케톤과 알코올이 생성된다. 그러나 a-H 이 없는 t- ROO• 의 경우(식 57-1 과 57-2) 는 식 (56) 과 같이 반응이 일어
나지 않고 분해하면서 안정한 디 -3 급-알킬 과산화물(식 57-1) 이 3 급-알콕시 래디컬로 분해(식 57_2) 되는데, 이것은 케톤, 알데히 드 등으로 다시 분해된다. 참고문헌 1) a) 松浦輝男, 『酸素酸化反應』, 제 3 장, 丸善(1 977). b) 森住洋, 井口洋夫, 『酸素의 化學』, p.2 7, 共立出版(1 967). 2) 다動起狀態의 化學강좌유기반응 기구」, 12, p.3 0, 동경화학동인 (19 67). 3 ) G. Herzberg, Sp ec tr a of Di at o m i c Molecules, p.5 60, Rein Hold, New York( 1950) . 4) W. C. Schumb, C. N. Satt er f iel d and R. L. Wentw ort h, Hy dr og en Peroxid e , Rein Hold, New York( 1955) . 5) F. Asin g er , Paraff ins , Chemi st r y and Technolog y, Perga m on Press ( 1968) . 6) D. R. Keams, J. A m. Chem. Soc., 61, 6554(1969). 7) G. A. Hami lton , Molecular Mechanis m s of Ox yge n Acti va ti on , p.40 5, 0. Hays h i ed., Academ ic P ress, New York(1 9 74) . 8) S. N. Foner and R. L. Hudson, J. Chem. Phys ., 38, 2681 ( 1962) . 9) J. H. Baxendale, M. D. Ward and P. Wardman, Trans. Faraday Soc., 67, 2532(1 9 71 ) . 10) 松浦輝男, 『酸素酸化反應』, 제 4 장 p.1 06, 丸善(1 977). 11) E. S. Huy se r, Free Radic a l Chain Reacti on s, p.7 7, W ile y - Inte r sci en ce, New York(1 9 70) . 12) Ullmann's Encyc l op e dia of In dustr i a l Chemi st r y, vol. 18, p.2 76
(19 91 ) . 13 ) S. W. Benson, J. Am. Chem. Soc. , 87, 972 ( 1965 ) . 14) J. A. Howard and K. U. Ing ol d, ibi d . , 46, 2661 ( 1968) . 15) J. A. Howard and K. U. Ing ol d, Can. J. Chem., 41, 2800( 1963). 16 ) C. Wallin g and E. A. McElhil , J . Am. Chem. Soc. , 73, 2927 ( 1951 ) . 17) G. A. Russel, ibi d . , 78, 1047(1 9 56). 18) G. A. Russel and R. C. Wi llian son, Jr., ibi d . , 86, 2357( 1964) . 19) J. A. Howard and K. U. Ing ol d, Can. J. Chem., 1 744( 1963) . 20) S. Kato and F. Mashio , Sci . Technol. K y ot o Technic a l Univ . , 12, 23(1 9 63) . 21 ) J. A. Howard, K. U. Ing ol d and M. Sy m onds, Can. J. Chem. , 4 6, 1017(1 9 68). 22) 大律陸生, 山本忠弘, 『有合化』, 23, 643(1965). 23 ) L. Saju s , Adv. Chem. Soc. , 75, 59 (19 68 ) . 24) J. B. Calvert and J. N. Pit ts, Jr. , Photo c hemi st r y, p.8 1 5, Wi le y, New York(1 9 66) . 25) 日本化學會編, 『化學 便覽 基礎編』,p .832, 丸善(1 966). 26) S. W. Benson and R. Shaw, Orga n ic Peroxid e s, vol. 1, p.10 5, D. Swem, ed., W i ley -In te r scie n ce, New York( 1970) . 27) 多羅間公雄 , 『大有機化學定數便覽』, p.5 37, 朝倉 촙 店(1 963). 28) S. W. Benson et al., Chem. Rev., 69, 279(1 9 69) . 29) J. A. Kerr, ibi d . , 66, 465(1 9 65). 30) S. W. Benson, J. Chem. Educ., 42, 502( 1965) . 31 ) G. Scott , At om osph eric Oxid a ti on and Anti ox id a nts , Elsevie r Publi shi ng Co., Amste r dam( 19 65) . 32) J. A. Howard, Advances in Free Radic a l Chemi st r y, vol. 4, p.49 , G. H. Wi lliam s, ed., Log os Press, London( 1972) .
33) J. R. Thomas, J. Am. Chem. Soc., 8 4, 2079( 1962) . 34 ) Ullmann's Encyc l op ed ia , vol. A 18, 280, VCH ( 1991 ) . 35 ) N. M. Enmanuel, G. E. Zaik o v and Z. K. Maiz u s, Oxid a ti on of Orga n ic comp o unds ( Mediu m Ef fec ts in Radic a l Reacti on s ) , p.44 2 ~495, Perga m on Press, Oxfo r d( 1984) . 36) Joh n J. McKett a ed., E ncyc l op e dia of Chemi ca l Processin g & Desig n , vol.13, 1 ~211, New York, Dekker(1 9 81 ). 37) a) Halcon, KE -OS J, 668, 200(1 9 61) . b) 이규완, 이상봉, 김우선, 장영길, 「공정부산물을 이용한 DHB 의 제조에 관한 연구」, 한국화학연구소(1 984) 및 이에 응용 된문현 38) Shell, US 2, 845, 641 (19 58) . 39) Dup on t, US3, 530, 185(1 9 70). US3, 957, 876(1 9 76). 40) a) A. N. Bashl ciro v, Khim . Nauka IPromy , 1(3), 273(1 9 56). b) 최명재, 박사학위 논문, 충남대학교(1 983) . c ) Ky u -W an Lee, My o ung -Ja e Choi, Sung -B o Kim , Cheong - Song Choi, I & ECReserch, 26, (10 ), 1951( 19 87). d) 강용, 조수행, 박소진, 최명재, 이규완, 『화학공학』, 30(2), p.2 1 2 (19 92). 41 ) 工藤土郞, 『工業化學雜誌』, 65(9), 52~58(1 9 62). 42) 倉田直次, 「日化學日報」 ,7,24~33 (1 978). 43) 高木行雄, 平野二郞, 『有機合成化學』, 18(1 9 60). 44) 小堂定次, 『油化學』, 24(7) , 427~434(1 9 75). 45) 佑久滋山, 「石油學會誌」, 19(8), 629~633(1 9 76). 46) V. V. Veselov and J. V. Sip pe va, Int. Chem. Eng. Jan ., 12~16 ( 1965) . 47 ) Bruce D. Boss and Robert N., Hazlett , Ind. Eng. Chem. Prod. Res.
Dev., 14(2) , 135 ~138( 1975) . 48 ) H. Wi liarn Preng le et a l., H. P, I 06 ~ 118 (19 70 ) . 49) N. N. Semenov(tr an slate d by J. E. S. Bradley ), Some Problems of Chemi ca l Kin e ti cs Reacti vit y , 2, 123 ~ 136 (19 59 ) . 50) S. Ma kish im a, Y. Yoneda and T. Taji ma , J. Phys . Chem., 61, 161 B( 19 57). 51) R. D. Ki rk and D. F. Ot hm er, eds., Encyc l op e dia of Chemi ca l Technology , vol. 3, 663 ~666, 2nd Edit ion , Wi le y, New York ( 1964 ) . 52) 이규완, 최명재, 김성보, 정헌창, 김영직, 「생분해성 산업용 계면 활성제의 제조 기술개발 보고서」, 한국화학연구소(1 991).
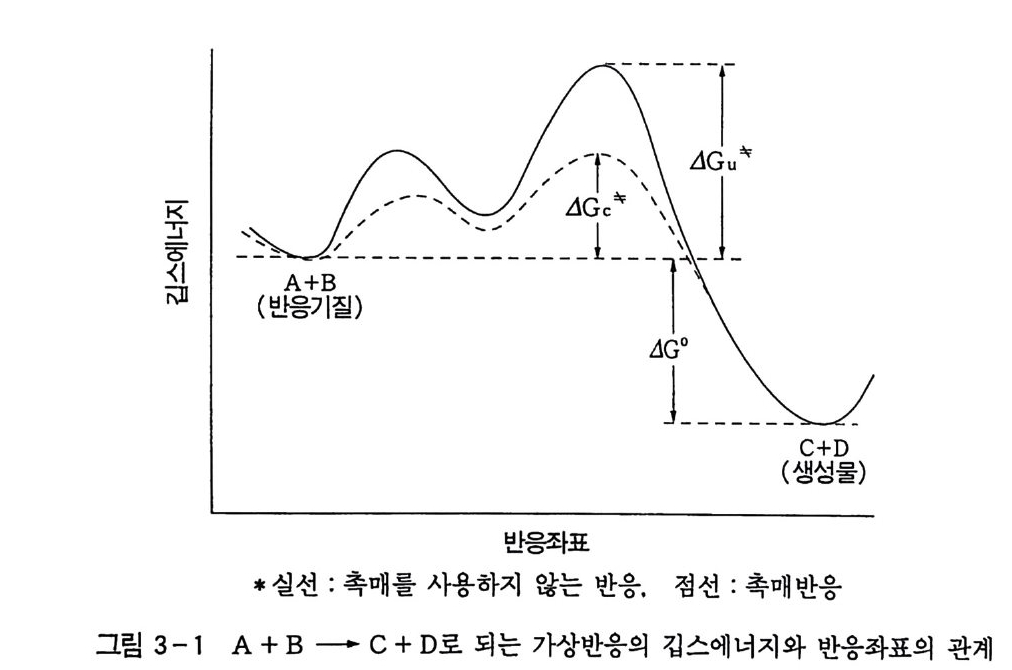
제 3 장 금속 촉매에 의한 산화 3.l 촉매 촉매란 우리들이 아는 바와 같이 반응을 원활히 하고 효율을 좋 게 하기 위해서 사용하는 물질이다. 인체 내 반응에서는 효소가 그 역할을 한다. 예를 들어 그림 3-1 에서 보는 것처럼 반응기질 (A+B) 가 반응하여 생성물 (C+D) 를 만들기 위해서는 몇 개의 장벽이 있다. 그 반응의 율속단계는 그 장벽 가운데 가장 높은 곳이다. 그 반응속도는 활성화 깁스 에너지에 의하여 결정되는데, 그림 3-1 에서 AGu .. 로 표시된다. 여기서 어떤 물질(촉매)을 넣어서 활 성화 깁스 에너지가 감소하고 (AGcT 로 표시) 속도가 빨라진다면 이를 촉매 반응이라 한다. 촉매는 반응기질과 상호작용하여 반응계 에 들어가지만 없어지지 않고 재생된다. 이와 같이 촉매는 반응속 도를 올리는 것은 가능하지만, 최종적으로 계의 화학평형 위치를
변경시키는 것은 되지 않는다. 평형은 반응기질과 생성물의 깁스 에너지차 (AG ° ) 를 이용하여 다 음 식 (1)과 같이 표시된다. L1G0 = -RTln [〔A이 ] [[BD J] (1) [A ],[ B] 와 [CJ , [D] 는 평형시의 반응기질과 생성물의 농도이 다. 어느 반응을 설계하는 경우 그 반응을 탐색하기 전에 열역학적 으로 검토하는 것이 필요하다. 즉, 가능한 반응조건하에서 평형이 어느 정도 이상 생성계로 기울지 않으면 안 된다. AG° 가 40kJ 이상일 경우 평형은 거의 원계 쪽으로 치우쳐 촉매 를 탐색할 필요가 없다. 예를 들면 다음 반응들이 그러하다.
 - i -』 -A -
- i -』 -A -
CH, + CO 一 CH3 CH O (2-1) CO + H2 一 CH2 0 (2-2) 그림 3-1 에서 보는 바와 같이 촉매는 생성물에 이르는 낮은 활 성화 깁스 에너지 (AGc 기를 갖는 반응경로를 따른다. 촉매를 사용 합으로써 AG 가 많이 낮아짐을 볼 수 있다. 즉, 반응조건을 온화하 게 하므로 에너지 절약이 되는 것이다. 또한 반응경로 AGc 적만을 선택적으로 낮춤으로써 바라는 생성물을 선택성이 높게 합성하는 것도 가능하다 .
 ( 2환중 화화,, 이躍성화화., 아크미래노킹화 등 )
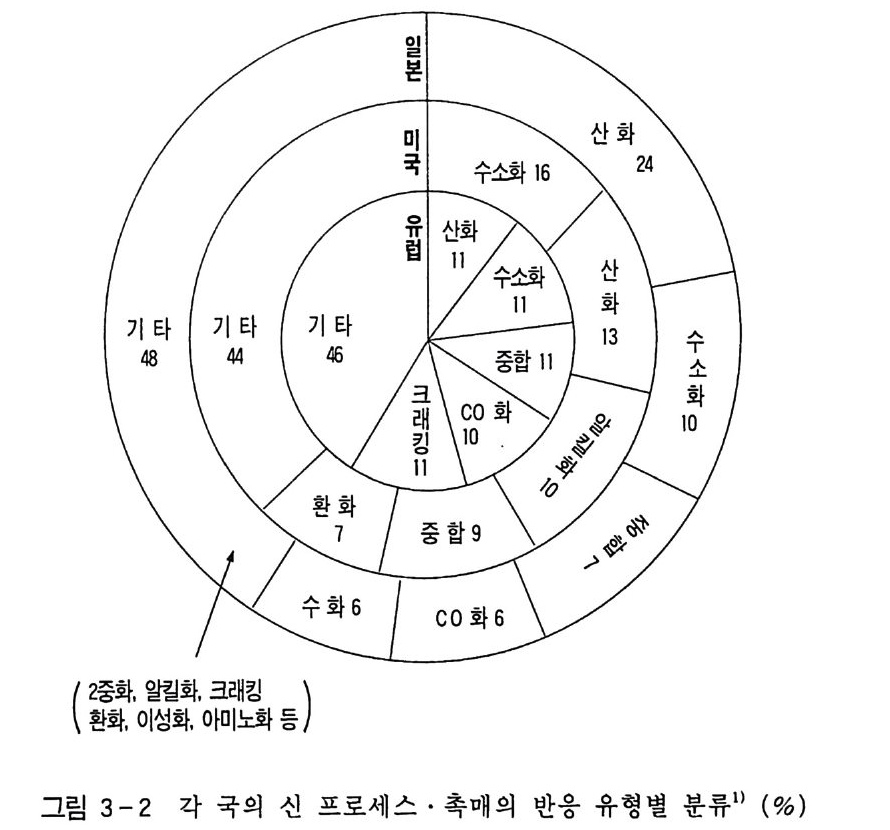
( 2환중 화화,, 이躍성화화., 아크미래노킹화 등 )
이것은 자원의 효율적인 이용인 동시에. 부산물이 생성되지 않으 므로 이러한 반응의 개발은 불가결한 것이다. 각국에서 사용중인 신 프로세스 • 촉매를 반응별로 분류한 데이 터를 그림 3 ― 2 에 나타내었다. 그림 3-2 에서 보듯이 일본의 경우 산화반응이 전체의 24 %를 차지하는 반면 미국의 경우 13 %이고, 수소화는 16 %이다. 이는 석유 유분의 수소 처리 (H y dro t rea ti n g) 및 수소 존재하 개질 (Re for mi ng ) 등에 사용되는 것으로 보인다. 3. 2 균일 촉매와 불균일 촉매의 상이점 촉매는 그 작용 상태에 의하여 균일 (homo g eneous-) 및 불균일 촉매 (he t ero g eneous ca t al y s t)로 분류할 수 있다. 그 차이점들에 대해서는 표 3-1 을 참조하기 바란다. 표 3-1 중 반응기구에 관해서 설명하겠다. 균일 촉매를 사용한 경우 일반적으로 반응한 활성종은 단일 금속 착체이고 LR 또는 N.M.R 둥의 물리화학적 수단에 의해 추적이 가능하므로 분자 상태로 이해가 된다. 또 어느 경우에는 반응 중간체를 단리하여 X 선 구조 해석 등을 행함으로써 가능하다. 한편, 불균일 촉매 반응의 경우는 여러 형태의 활성점이 표면에 존재할 수가 있댜 최근에 여러 가지의 고체 표면 분석에 유효한 기기 등이 개발되어 반응기구가 꽤 상세히 논의되게 되었다. 또 균일 촉매반응의 연구 결과가 불균일 촉매반응의 기구 해명에 큰 공헌을 하고 있다는 점도 간과할 수 없다.
표 3-1 균일 -불 균일 촉매의 비교2 1 비교항목 균일 불균일 촉매의 형태 가용 천이 금속착체 무기산화물에 담체시킨 금속 또는 금속산화물 촉매의 (염) 안정성 낮다 높다 l:I\..0.} 으 사0 액상 기상/고상 반응온도 낮다 (200 °C 이하) 높다 (250~550 °C ) 촉매 활성 낮다 높다 반응의 선택률 높다 낮다 촉매의 분리 곤란 용이 반응기구 분자규모로 꽤 분자규모의 반응기구 명확히 이해됨 해명은 꽤 어려움
3. 3 균일상 촉매 산화반응 3. 3. l 촉매 기상 산소 산화반응 촉매가 균일상이 되기 위해서는 금속이 염의 형태로, 즉 유기 금 속착체로 액상 형태로 존재하여야 하며 이것은 기질이 기상으로 통 과하여 이루어지는 반응이다. 후술하는 불균일상 촉매의 기상반응은 예를 찾을 수 있으나 이 경우는 극히 드물다. 가장 전형적인 예가 바커 공정 (Wacker p rocess)3·4) 으로 PdCl2_ CuCl2 촉매 존재하에서 반응온도 100~110 °C 그리고 공기 (10~ 15a t m) 로 에틸렌을 산화하여 아세트알데히드를 만드는 공정이다.
CH2 ---If--P dCI2 쁘 CH3 C HO + H2 0 + 2 HCI (3 -1 ) CH2 Pd + 2 CuClz 一 PdClz + 2 CuCl (3-2) 2 CuCl + 2 HCl + ½ 02 一 2 CuC12 + H2 0 (3 -3) 위 반응에서 보는 바와 같이 에틸렌이 Pd-Cb 와 ,r -com p lex 를 만들어 전자가 Pd 쪽으로 이동해 에틸렌(올레핀)에 물이 쉽게 구 핵부가 반응이 일어나게 하는 반응이다. 이런 종류의 반응은 Bay e r 법 및 ND 법으로 초산비닐 제조와. CH2 = CH2 + CH3COOH + ½ 02 llO~zoo·cP ,d 02(7at m ) CH2 = CHOAc + H20 (4) 에틸렌을 옥시염소화시켜 염화비닐을 제조하는 것을 예로 들 수 있다. CH2 = CH2 + HCl + ½02 1so ·c C. u0C2l(2 10 atm ) CH2 = CHCl + H20 (5) 이러한 종류의 산화에 대해서는 상세한 총설 5 , 6) 올 보기 바란다.
3, 3, 2 촉매 액상 산소 산화반응 촉매를 사용하여 산화반응을 수행하는 것에는 반응조건완화, 더 나은 선택도 등을 들 수 있다 . 이 범주에 속하는 것은 균일 촉매 인용이다 . 촉매를 연속적으로 사용하기 때문에 산화-환원반응이 쉽게 일어나야 하며 , 두 전자는 상태의 안정성이 비슷한 금속이 좋 다. 예 를 들면 Co2+ / Co3+ , Fe2+ / Fe3+ , cu+/Cu2+ , Mn2+ / Mn3+ 등을 둘 수 있댜 이렇게 두 전자 상태를 유지하기 위해서는 금속이 착 체로 존재하여야 하며 이의 리간드 (L ig and) 변환이 빠른 속도로 일어나야 한댜 그러므로 그 반응용매의 극성이 반응에 크게 작용 한댜 2. 3. 3 에서 설명한 히드로 과산화물울 예로 들어 설명하겠다. 제 2 장 식 (1 0-3) 에서 생성된 R-OOH 두 분자는 다음 식 (6) 과 같이 열분해된다. ROOH + ROOH 一 ROO· + RO· + H2 0 Ea=90~170 kJ/ m ol (6) 또한 금속 촉매 분해 (Meta l Cata l ys is ) 인 경우는 다음과 같다. RROOOOHH ++ MM (n++ I )-+ —R-OR • O +O M • n+( nM+lnl++ + +O Hf f+ ((78)) net, 2ROOH ~Mn+/Mn(n+l) + ROO· +H20 +RO· Ea=40~50 kJ /m ol (9)
위의 두 반응식 (6). (9) 에서 보는 바와 같이 금속 촉매를 사용 한 경우 활성화 에너지가 급감됨을 알 수 있다. 1) 시클로헥산의 산화 시클로헥산이 시클로헥산올과 시클로헥사논으로 산화되는 것을 첫번째로 택한 이유는 C-H 결합이 동등하며 c-c 결합이 잘 끊 어지지 않기 때문이다. 그러므로 겉으로 보기에는 상대적으로 단순 하며 이론적으로도 잘 정립되어 있다. 나일론 생산의 주요 중간체인 아디핀산은 시클로헥사논과 시클 로헥산올”의 산화에 의해 만들어진다. 이 중간체는 또한 시클로헥산의 공기산화나 페놀의 수소화에 의 해 생성된다(그림 3-3). 이 반응의 상업적 가치는 자체의 화학적 특성에 의해 영향을 받 는다 .7~11) 가장 단순한 예로서. 시클로헥산은 시클로헥산 히드로퍼 옥사이드로 전환되며 히드로퍼옥사이드의 일부 또는 전부는 시클 로헥산올이나 시클로헥사논과 같은 생성물로 분해된다. 시클로헥산 (l) 에 있는 C-H 결합의 산화용 공격은 느리면서 격
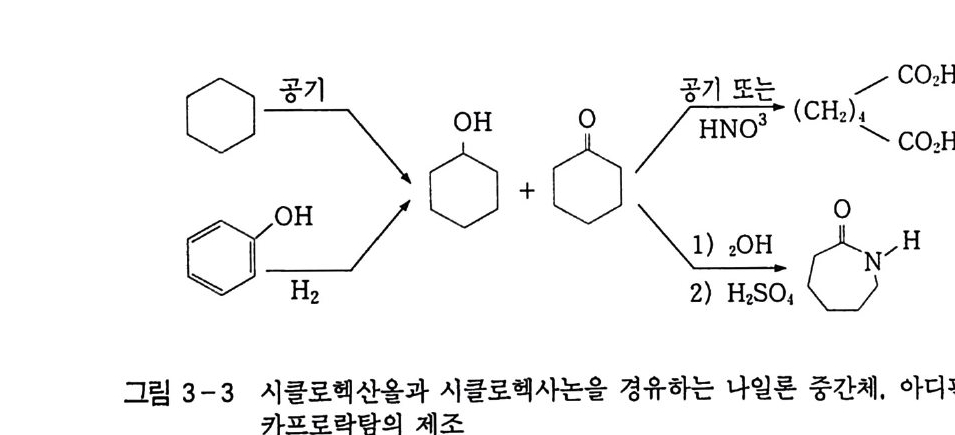 0\0 + 6( 틀 (CH: 〈:::
0\0 + 6( 틀 (CH: 〈:::
렬한 반응조건들이 요구된다 . 대조적으로 히드로퍼옥사이드(~). 알코올(한 그리고 케톤(산)은 쉽게 산화된댜 그러므로 이 반응은 원하는 생성물의 분해를 막기 위해 일반적으로 시클로핵산울 낮게 전환시킨다 .
 二 o OOH oO H
二 o OOH oO H
대표적인 산업적 산화는 연속적 공정으로서 대략 140~165°C, 10 atm 반응조건하에서 녹을 수 있는 코발트〔( Co ( II) 〕염의 시클 로핵산 용액과 공기의 반응에 의해 진행된다 . 9) 반응기에 머무는 시간은 시클로헥산의 10 % 전환이 이루어질 때 까지로 제한된다. 액체반응 혼합물은 연속적으로 배출되고 증류되 며. 미반응 시클로핵산은 산화반응기로 재투입된다. 시클로핵산올 과 시클로헥사논은 다음에서 서술될 아디핀산 또는 카프로락탐(그 림 3-3) 으로의 변환을 위해 다른 산화 공정으로 보내진다. 시클로핵산 전환율이 6~9 %인 경우 알코올과 케톤의 종합 수율 은 60~70 %이다. 또 다른 전략으로는 코발트 촉매를 전혀 사용하 지 않거나 최소화하는 첫 단계로부터. 그리고 반응기의 벽을 고정 화하는 것으로부터 퍼옥사이드의 생성을 최대화하는 것이다. 12. 13) 그때 퍼옥사이드는 시클로헥사논과 시클로헥산올이 최대 수율을 갖는 조건에서 분해된다. Halcon 에 의해 개발된 공정에서는, 형성 된 시클로핵산올 자체를 안정화시키기 위해 봉산을 첨가하면 10~12% 전환까지 수율을 상당히 높일 수 있다 .14~18) 붕산염은 히드로퍼옥사이드(~)와 반응하여 퍼옥시보레이트를 형
성한다고 알려졌으며 . 이것은 잇달아 분해되어 시클로헥산올의 보 레이트 에스테르로 변한다 .1 9 . 20 ) 시클로헥실보레이트는 연이은 산화 로부터 시클로헥산올을 보호하여 더 높은 변환율을 갖도록 한다. 개선된 수율은 붕산의 재순환을 위한 투자와 운전 비용으로부터 부 분적으로 상쇄된다. 그러나 이 기술은 몇 가지 주된 아디핀산의 제조에 이용된다 . 대 부분의 시클로헥산 산화 촉매는 나프테네이트 (Na p h t hena t e) 나 2 -에 틸헥사노네 이트 (2 -eth y lh exanonate ) 와 같은 탄화수소에 녹는 코발트 (II) 카르복실레이트이다. 생성물의 조절을 위하여 망간 (II) 이나 크롬 (II) 과 같은 다른 금속이온들이 코발트와 함께 종종 사용된다. 금속이온들은 시클로헥산울 시클로헥실 히드로퍼옥사이드로 변 환하는 데에 직접적인 역할을 하지 않는데. 그 이유는 이 반응이 단순히 래디컬 사슬 공정이기 때문이다. 그러나 이온들은 히드로퍼 옥사이드가 시클로핵산올과 시클로헥사논으로 변환되는 것을 조절 하는 역할을 한다. 덧붙여서, 금속 촉매 히드로퍼옥사이드 반응은 산화를 개시하고 유지하는 데 필요한 유리-래디컬을 공급하며. 금 속이온 농도는 전반적인 반응속도를 조절한다 . 그러나 금속이온들 은 높은 농도에서는 래디컬 사슬공정의 금지제가 되고 있기 때문에 반응 속도론적 효과는 단순하지만은 않다 .21) 코발트나 망간의 이온 촉매들은 시클로헥산올과 시클로헥사논을 생성하며 유사한 기구 22) 로 작용하는 것처럼 보인다. 크롬은 시클로헥사논 23) 의 생성을 촉진한다. 시클로헥산에 가해지는 래디컬 X· 의 공격은 시클로헥산으로부 터 수소원자의 추출에 의한 산화과정을 개시함을 의미한다(초기 화학종의 성질 X· 는 잘 알려져 있지 않으나 순수한 시클로핵산에 서 산화가 시작되기는 어렵다는 것은 잘 알려져 있다).
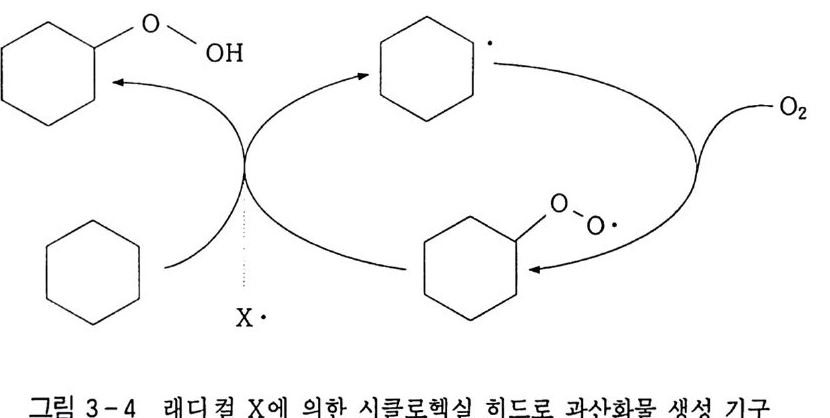 二 0 '- oH 二 02
二 0 '- oH 二 02
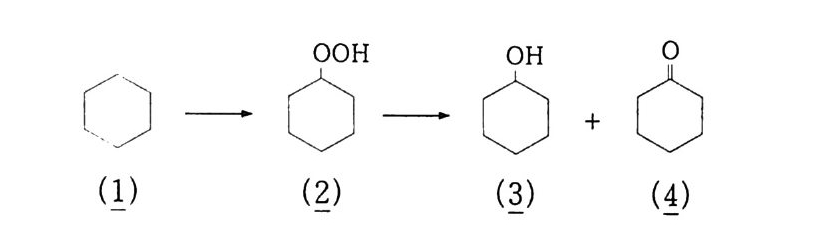
공장 규모상 시클로핵산의 흐름은 반응을 종료하는 데 도움을 주 는 시클로헥사논과 함께 오염된다. 결과적으로 시클로헥산 래디컬은 급속히 02 와 결합하여 시클로 헥실퍼옥시 래디컬을 형성한다. 후자의 경우가 시클로헥산 분자와 만날 때 C-H 결합으로부터 수소를 흡열 상태에서 빼내지만 이때 평형 유지가 중요하다. 탄소로부터 산소로의 수소 전이는 1 차 생성 물인 시클로헥실히드로 과산화물을 생성하며 시클로헥실 래디컬을 발생시킨다 . 시클로헥실 래디컬은 즉시 산소에 의해 포촉되면서 평형을 옮기 며 또 다른 반응주기를 시작한다. 한번 산화가 시작되면, 이 과정은 2. 4 절의 속도론에서 설명한 바 와 같이 개시, 생장, 정지반응들로 구성되는 대표적인 래디컬의 사 슬공정으로 이루어진다. 개시는 시클로헥실히드로 퍼옥사이드의 Haber-Weis s 경로를 거친 분해의 일차적인 결과이다. 개 시 2 Cy 0 0H 으노 Cy O • + Cy O O • + H20 (10 )
Cy O • + Cy H 一 Cy O H + Cy • (11) 높은 에너지를 가진 시클로핵실퍼옥시 래디 컬 은 과량의 시 클 로 헥산으로부터 수소원자를 빼내어 시클로헥산을을 생성한다. 긴 생명력을 가진 시클로헥실퍼옥시 래디 컬 은 반응의 홉열적 성 질에도 불구하고 시클로헥산으로부터 수소원자 를 추출할 능력 이 있다. 그 이유는 결과적으로 시클로 헥 실 래디 컬 이 급속 히 산소분자 와 결합하여 더욱더 안정한 시클로헥실퍼옥시 래디 컵을 형성합 으 로써 평형을 오른쪽으로 옮기기 때문이다. 생 장 Cy O O • + Cy H .;=! Cy O OH + Cy • (12 ) Cy · + 02 一 Cy O O· (13 ) 이 주기는 각각의 개시에 대한 몇 가지 시클로헥실히드로 과산화 물 분자를 설명하는 사슬 메커니즘을 제공한다. 낮은 전환에서 시클로핵산 산화의 주된 정지반응은 두 개의 시 클 로헥실퍼옥시 래디컬들의 2 분자 결합이다.
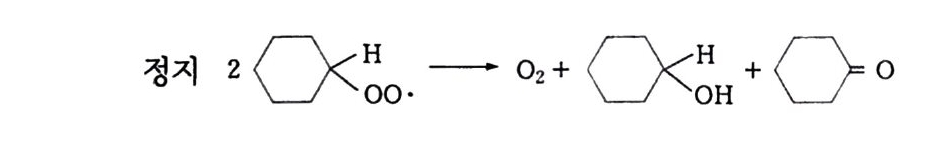 정지 2 O< :0· 一 0 2 + O< 。H H+ 0 0 (14)
정지 2 O< :0· 一 0 2 + O< 。H H+ 0 0 (14)
이 반응은 활성화 에너지가 전혀 필요없거나 아주 작은 양으로써 일어나며 본질적으로 온도와 무관하다. 이것은 분자 내 수소원자가 한 시클로헥사논과 한 시클로헥산을 로 전이되는 전이 테트록사이드 35 ) 를 통해 진행된다. 이러한 분자반 웅으로부터의 가능성은 Cy O O· 래디컬의 상대적인 안정도와 망간
이나 코발트 22) 의 배위에 의해 제고된다 . 케톤은 3 중 상태에서 형성되며 결과적으로 시클로헥산 산화나 시 클로헥실히드로 과산화물의 분해 25,26) 동안에 화학 형광체를 관측할 수 있댜 그것은 일정한 상태에서 연속적으로 같은 속도를 가진 개 시와 정지반응을 통하여 독특하게 반응이 진행되어야 하기 때문이 댜 개시는 하나의 시클로헥산을 분자를 생성하며 반면에 정지에서 는 하나의 시클로헥산을과 하나의 시클로헥사논을 생성한다. 이러 한 균형은 코발트 촉매반응에서 시클로헥사논에 대한 시클로헥산 윤의 비가 대략 0.5 로 관측되는 것으로 설명된다. 작은 반응기 물 통한 시클로헥산 산화에 대한 상당 부분의 반응 속도론 연구는 알려져 있는 래디컬 중간체로부터의 유발을 포함하 지 않고도 명백히 설명할 수 있다. 한 예로서. 관측된 생성물은 산 화된 생성물들의 잇달은 산화로부터뿐만 아니라 시클로헥산에 유 리한 래디컬 종 27) 으로부터 직접적으로 이루어진다. 그러므로 매우 낮은 변화율에서도 히드록시 카프로익산(~) 또는 아디핀산(인과 같은 생성물이 관측된다.
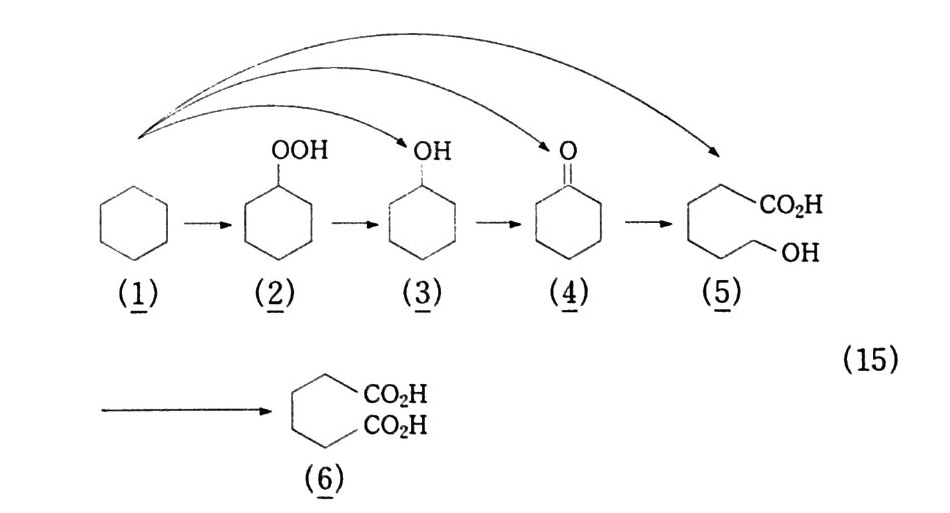 O 一二一 。야 一 : 〈二 °02HH
O 一二一 。야 一 : 〈二 °02HH
이 예는 복잡한 산화반응에 대한 대부분의 설명을 무시하고 있다. 더욱 정교한 모형은 래디컬 중간체를 포함. 150 가지의 반응이 상 업적 공정을 조율하기 위해 개발되었으며 . 새로운 반응기 형태의 효율을 예견한다. 이러한 정교한 수준에서조차도 많은 반응집단이 독립적으로 취 급되기보다는 군( 群 )으로서 이루어져야만 한다. 중간체 중 하나인 시클로헥사논의 산화에 대해 진동으로 설 명되 는 반응 속도론적 모형이 29 가지 식 때) 으로 설명된다. 산화의 초기 생성물 중 하나인 시클로헥실히드로 과산화 물 은 단 지 평범한 안정도를 갖는다. 용액들은 낮은 온도에서는 오랫동안 저장할 수 있으나 . 온도가 150 °C 가 넘으면 급속히 분해된댜 진한 히드로 과산화물의 용액은 믿을 수 없을 정도로 폭발력이 강하댜 결과적으로 히드로 과산화 물은 거의 유리되지 않으며 , 대체로 금속이온의 존재하에서 과산화 물이 생성되는 대로 분해된다. 이러한 운전방법은 통제할 수 없는 퍼옥사이드의 위험성을 제거 하며 반응경로를 유지하기 위한 래디컬의 공급원을 제공한다. 그런 곳에는 몇 가지 운전 모형이 있다. 그러나 만약 히드로퍼옥 사이드 농도가 최대치가 되고 온화한 조건 1 2 .1 3 ) 에서 분리된 반응기 속에서 분해가 수행된다면 그것은 전반적으로 아디핀산 전구체의 최대 수율을 제공한다. 높은 퍼옥사이드 농도를 얻기 위해서는 반응이 촉매 없이 진행되 고 반응기 벽 이 고정 되 어 야 한다 . 29~3 1) 다양한 금속들은 정상적인 시클로헥산 산화 온도에서 효율적이 지만 완전히 과산화물로 분해되기 위해서 더욱더 효율적인 촉매들 이 요구되는 바로 이때 촉매의 환원 온도 조건에서 과산화물의 수 율을높인다.
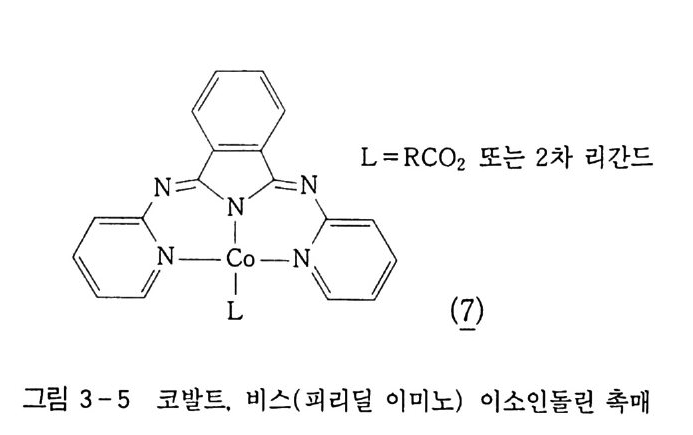 (7)
(7)
촉매 (1) 과 같은 집단의 코발트 포르피린이나 프탈로시아닌이 효율적으로 사용된다 . 32~31) 이렇게 증가된 효율은 촉매에 의해 증가된 속도라기보다는 증가 된 촉매 수명의 결과이다. Co 나 Mn 과는 대조적으로 크롬( III) 이온이 C y OOH 를 분해하여 주생성물로서 23,35,36) 시클로헥사논을 형성하는 데 촉매 역할을 한다. 이 반응은 기본적으로 히드로 과산화물의 탈수이다. 이 반응은 금속 배위 37) 를 통하여 금속이온이 0-0 결합을 끊도 록 보조하는 비래디컬 메커니즘에 의해 일어난다. 보통 관측되는 아세트산이나 포름산과 같은 부생성물은 코발트 촉매의 촉매적 활성에 영향을 준다 .38) 시클로헥실히드로 과산화물 은 강산이지만 강염기에 의해서도 역시 분해될 수 있으며, 시클로 헥사논의 비를 높인다 . 39~4 1) 비록 상업적으로 실용화되지는 않고 있으나 시클로핵실히드로 과산화물의 수소화는 시클로핵산올에 대 한 시클로헥사논의 비를 높인다. 42~45) 시클로핵산으로부터 시클로헥산올과 시클로헥사논으로 바뀌는 주 경로를 나타내는 이러한 반응들에 덧붙여서 문자 그대로 한 다 발의 또 다른 반응들이 반응 혼합물 속에서 일어난다. 대표적안 것으로 c-c 결합이 끊어져 락톤과 C4_Q 디카르복
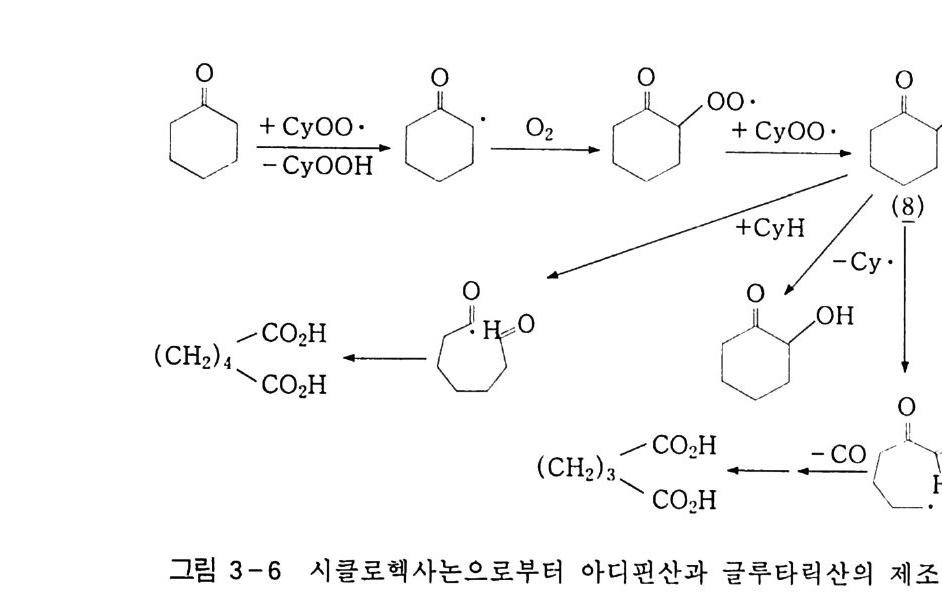 (CH〈〔。2 ) 4:::::〔〔一 —广°<> > ° 20 一 C02H 。·
(CH〈〔。2 ) 4:::::〔〔一 —广°<> > ° 20 一 C02H 。·
시산울 형성하는 반응을 지칭한다. 시클로헥사논에 있는 a-CH 2 기는 특별히 래디컬 공격에 민감하 다. 동위원소 표시 연구는 디카르복시산 2 7 , 4 6) 에 대한 전구물질로서 a- 옥시 래디컬과 밀접한 연관이 있다. 시클로헥산을 아디핀산으로 직접 산화시키는 공정은 경제적으로 매력이 있으나 수율은 낮다• 현재 상업적으로 운용되는 것은 주로 알코올-케톤 혼합물의 산화로, 아디핀산의 분리에 기초를 두고 있 다. 화학자들은 보통 화학반응에 대한 물질-전달에만 국한시키지 않고 이러한 것들이 시클로핵산 산화의 주원인이 된다고 생각한다. 때문에 반응기를 고안하고 물리적으로 그리고 화학적으로 나타나 는 효과를 연관지으며 이예 대한 반웅 생성물의 탐구에 대해 많은 노력을 기울이고 있다 .47~50)
2) 시클로헥산울과 시클로헥사논의 산화 시클로헥산 산화의 결과인 시클로핵산올_시클로헥사논 혼합물은 비록 코발트-촉매에 의한 공기 산화로 인해 아디핀산으로 전환되 지만 현재의 상업적인 운전은 보통 산화제로 질산을 사용한다 . 7.5 1 ) 그러나 질산의 사용에서도 공기는 궁극적으로 산화제인데. 이는 대 부분 의 질소산화물 부생성물이 재순환되기 때문이다. 시 클 로핵 산을과 시 클로헥사논의 혼합물은 70 ~ 80 °C S2) 에서 45 ~ 50 % 질산 속에 있는 Cu(NO) 2 와 NH 4 V0 3 의 용액으로 연속적 공 급이 이루어지며 산화는 수분 내로 완료된다 . 주로 질소산화물인 기 체 생성물은 질산 합성공정으로 재순환된다. 소량의 질산은 HN0 3 로 재산화되지 않는 N 2 나 N2 0 생성물로 전 이된다. 유기 생성물이 포함된 뜨거운 산 용액은 냉각되어 원하는 아디핀산으로 결정화된다. 대표적으로 순수한 아디핀산의 수율은 90 % 를 초과한다 . 질산 산화에 대한 화학은 매우 복잡하다. 거의 모든 시클로핵산 올이 잔존하는 HN0 253~551 에 의해 개시되는 바촉매적 반응을 통해 서 시클로헥사논으로 산화된다. 시클로헥사논은 세 가지 경로를 통 하여(그림 3-6)56, 57) 아디핀산을 만든다. 첫번째 경로는 2 초화를 포함하며 단지 고온의 조건에서 관측된다. 상업적인 조건에서 쉽게 관측되는 다른 두 가지는 케톤이 2 _ 니트로소 유도체(g)로 변환되 는 것을 의미한다. 이러한 전환 메커니즘은 잘 설명되고 있지 못하나 산 촉매반응에 있어서는 케톤 엔올화를 제안하는 또 다른 시클로헥산의 산화에 대 한유추를 할수 있다. 한 수소원자(또는 연속적인 한 전자와 한 양자)의 손실은 NO 와 결합하여 2 -니 트로소 시클로헥사논(g)을 형성하는 것과 연관될
수 있는 비편재화된 래디컬을 생성한다. 니트로소케톤은 비촉매적 경로에 의해. 또한 연속적인 바나듐-촉매 ;" 에 의해 아디핀산을 형 성한댜 HNO .i 와 함께하는 비촉매반응은 니트롤산 (n it ro li c acid ) 과 아디핀산으로 가수분해되는 니트로-니트로소 유도체(반)를 생 성한다. 오존 감소의 진범인 니트로소옥사이드는 질소가 포합되는 부생 성물이다 . 58 ) 영속적안 촉매에서 니트로소케톤(인의 상호변이(t au t omer i za ti on) 는 1, 2-시 클로헥산디온을 가수분해하는 옥심 (ox i me) (12) 울 생성한다. 이 디케톤은 두 개의 V02+ 이온을 화학양론적으로 산화합으로 써 아디핀산을 형성한다. 바나듐 (N) 공동 생성물은 질산에 의해 쉽게 재산화된다. 그러므 로 이 경로는 바나듐에 의해 촉진된다. 산화에서 Cu(NO 山의 역할은 명백하지 못하다. 구리는 부반응
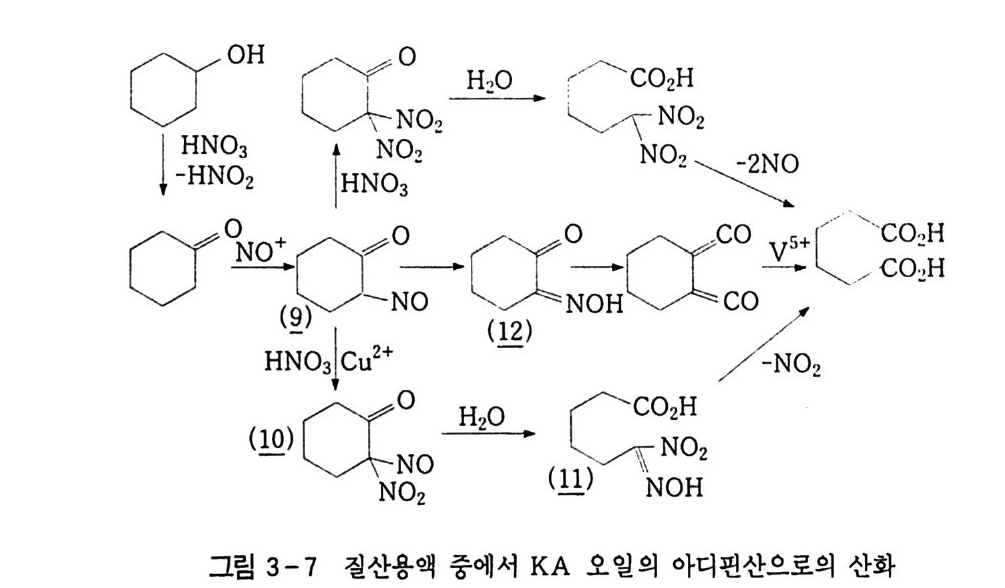 U 潭二0H 2 ::2卒 / ,\,~ co2H\
U 潭二0H 2 ::2卒 / ,\,~ co2H\
을 줄임으로써 .,” 아디핀산의 수율을 증가시키는 것처럼 보인다. 3) 부탄산화에 의한 빙초산의 합성 또 다른 예로 리간드와 관련된 용매의 영향을 기술하면 다음과 갇댜 Co2+I : n - 부탄 ―17―0 ―C 소 빙초산 (16) 선택률 40% Cc, ~ + 11+ n- 부탄 170 C, HAc • 빙초산 (17 ) 전환율 80%. 선택률 83% 위의 두 식 (16 ), (17) 에서 보는 바와 같이 식초산을 용매로 사 용하면 전환율 및 선택률이 공히 80 % 이상이 됨을 알 수 있다. 빙초산은 유기화학 산업 5'며 에서 중요한 중간체이다. 그것은 크실 렌 산화에서의 용매로서 그리고(폴리비닐 아세테이트 또는 폴리비 닐 알코올을 형성하는 데 사용되는) 비닐아세테이트를 합성하는 선구물질로서 광범위하게 사용된다. 빙초산 무수물은 셀룰로오스아세테이트 또는 아스피린의 생산을 위한 아세틸화제 시약이다. 최근 메탄올의 카르보닐화에 기초를 두어 온 빙초산의 생산을 위 한 공장이 건설된 반면 몇몇의 빙초산은 아직도 산화 공정을 통하 여 생산된다. 부탄과 다른 알칸의 산화는 최대 규모의 공장이 될 수 있댜 또 다른 주된 공정은 Wacker 공정에 의한 에틸렌으로부 터 생성된 아세트알데히드의 산화이다. 부탄과 아세트알데히드의 산화는 다음으로 분리시켜 논의하였다.
두 공정은 촉매로서 코발트 (II) 나 다른 전이 금속염을 채택하였는 데. 세밀하게 기술된 화학은 본질적으로 차이가 있댜 천연가스로부터 얻은 부탄은 값이 싸고 양이 풍부하다. 그것은 앞에서 본 식 (1 6) 과 (1 7) 에 나타난 바와 같이 산화에 의해 빙초 산 생산의 주공급원이 되고 있다. 천연가스가 풍부하지 못한 국가에서는 원유 정제에서 소량의 나 프타를 사용한다. 두 가지 공급원으로부터(비록 부탄으로부터의 수 율이 나프타로부터 얻는 것보다 높다고 할지라도) 형성되는 빙초산 의 수율은 대체적으로 적절하다 (30~60 %). 수율은 운전 조건이나 전환에 상당히 의존한다. 빙초산의 생성을 위한 알칸의 산화는 C-H 산화에 의하여 나타 나는 c-c 결합의 분열을 포함하고 있다. 지방족 탄화수소의 혼합 물이 Q 유분으로 분해되는 나프타의 산화반응 경로는 매우 복잡 하다. 이러한 산화의 성공 여부는 래디컬 공정을 위한 빙초산의 상 대적 안정도를 나타낸댜 CO2 와 달리 이것은 대단히 안정한 생성물이며 다양한 반응조건 하에서 축적되려는 경향이 있다. 산화에 대한 같은 저항성 때문에 산화과정에서 빙초산을 용매로 사용한다. 전형적으로 160~2oo ·c 와 60~80 기압하에서 촉매와 6 1) 아세트 산 용액에 있는 부탄을 통하여 그리고 간헐적으로 공기를 불어 줌 으로써 산화는 진행된다(식 17 참조). 반응 혼합물의 한 부분은 변화되지 않은 탄화수소를 회수하기 위해 추출되고 증류된다. 빙초 산. 프로피온산. 부티르산들과 2- 부탄온 같은 생성물들은 분리되 고, 잔존하는 촉매용액은 반응기로 재순환된다. 전형적으로 30 % 전환에서 45 %의 빙초산 수율을 갖는다. 이 반응은 상업적으로 메 틸에틸케톤 (2- 부탄온)의 중요한 근원이 되고 있고 MEK62) 의 수
율을 개선하기 위한 노력이 진행되고 있다. 온화한 반응조건하에서 진행되도록 하는 반응의 변경은 높은 수 율과 전환율을 준댜 통상 래디컬 사슬반응을 유지하기 위해서는 고온의 반응온도가 필요한데 그것은 부탄이 상대적으로 활성이 없 기 때문이다. 2- 부탄온 부산물의 산화반응기로의 계속적인 순환 63) 은 온화한 반응조건 (110~13o ·c) 하에서 잇따른 래디컬 사슬을 유지하기 위 해서이며, 이것은 a- 메틸렌기에 대해 래디컬의 공격이 매우 민감 하기 때문이다. 2- 부탄온 역시 빙초산 수율 64) 에 기여한다. C2H s CO CH3 + %02 一 2CH3C02H (18) 부탄과 2- 부탄온으로부터의 빙초산 수율은 85 % 부탄온 전환에 서 대략 75 %이다. 2- 부탄온의 첨가에 대한 부수적인 효과는 반응 혼합물에서 코발 트( III) 이온의 농도가 증가함에 기인한다 .6 4 ) 코발트( II) 에서 코발 트( III) 의 산화는 공정에서 느린 단계인 것처럼 보인다. 케톤의 산 화는 a-히 드로퍼옥사이드 (E) 를 경유하여 진행되는데. 이것은 Haber-Weis s C y cle65) 에서 보는 메커니즘에 의해 코발트( II) 를 산화할수 있다. CH3CII -CI HCH3 0 OOH (~) 온화한 조건에서 운전되도록 하는 또 다른 시도는 오존 66) 과 코발 트( II ) 염의 사전 산화이다. 이 처리는 부탄의 일괄 산화에 대한 유
도기간을 단축하며 110~130 · c 와 35 기압에서 연속적인 운전이 이 루어지도록 한다 . 산화는 코발트( II ) 아세테 이트롤 µ 3 -oxo -brid g e d tri m er Co 3 0(OAc) G (AcOH) 3 로 전환시키며. 이것은 알칸 산화에 대한 좋 은 개시제가 된다 . 67) 케톤, 코발트(때) 또는 브로마이드 이온과 같은 촉진제의 사용은 전반적인 산화반응 기구에서 초기 단계의 중요성을 지적한다. 부탄 의 일괄 산화나 연속적 공정의 초기 단계에서는 2 차 C-H 결합으 로부터 충분히 역동적으로 수소원자를 빼낼 수 있는 자유 래디컬 개시제의 공급이 필요하다 . RO· 와 ROO· 래디컬은 평형조건에서 적어도 이 조건을.충족시 키며 일반적으로 150 ° C 에서 산화 혼합물의 실질적인 양으로 존재 한다. 여기서 코발트 (m) 이온이 알칸이나 시클로알칸에 직접적으로 공격하는지에 대한 몇 가지 의문이 남는다 .68) 한 번 개시가 시작되면. 시클로핵산 산화에서 기술한 것과 같은 래디컬 사슬반응은 부탄을 2- 부틸히드로 과산화물 65) 로 변환시킨다. n-C 4 H1 。 + 02 一 CH3CI HC2 H s (19 ) OOH 2 차 탄소를 공격하는 선택성은 적당하고 부탄의 말단 탄소의 공 격에 의해 프로피온산과 부티르산의 실질적인 양이 된다. 초기에 생성된 2- 부틸히드로 과산화물은 다양한 경로를 통하여 최종 산화 생성물을 형성한다. 촉매가 존재하지 않는 단순한 분해 는 과산화물 이량화 2 4 ) 를 통하여 주로 Q 생성물 69) 이 생성된다고 보고되 었다( 식 20 참조) .
2C2 H s福 C H 0CHH 3 一 C2-H 5C b C H:•l• + C -晶 •• C6HHC H -3 + 0-2 + H-20 (20) 그러나 실질적으로 코발트 염에 의한 촉매 분해는 순환산화를 유 지하기 위하여 C1H g O · 과 C1H g 0 0· 래디컬 등울 형성한다. 그들은 래디컬 이량화와 불균일화 같은 사슬 정지 공정에 의해 잃어버린 래디컬을 대신한다 . 덧붙여서 C4 사슬의 분열은 늘 관측되는 C1 과 C3 부생성물로 전 환시킬 뿐만 아니라 필요한 C2 생성물로 전환시키는 반응기구를 제공한댜 2- 부독시 래디컬의 f3 - 균열은 c-c 분열에 대한 한 경로를 제 공한댜 CH 3 COHOCH 晶 브노 CH3 CtH C2 Hs 一 CH3 CH O + C2Hs • (21) 다음 절에서 논의하지만 CH3 CH O 중간체는 쉽게 빙초산으로 산 화된다. 에틸 래디컬은 에탄올로 전환되는데 이것은 반응조건 70) 하 에서 관측된다. 비록 이 자체가 부톡시 래디컬을 포함하는 분자간 반응인 것처럼 보인다 할지라도 또 다른 c-c 균열반응 기구가 히 드록시 래디컬 69) 의 분자 내 반응을 포함한다. H H H2 CH3_c|| -CH책 으 CH3-¢| 一 c| _C 比 一 CH3CHO+ CzHsO· OOH 0-0 • (22) 아마 2- 부틸 히드로퍼옥사이드의 열적이나 촉매적 산물인 2- 부
탄온을 포함하는 경로가 더 중요하댜 케톤의 a- 메틸렌 C-H 결 합은 산화에 특별히 민감하며 c-c 분열에 의한 아세트산 전구물 질을 생성하는 옥시 래디컬을 만든다. CH3CII -CI HCH3 一 CH3 CII • + CH3 CH O ( 23 ) 0 O· 0 전반적인 공정은 완만한 빙초산의 수율을 갖는다. 이러한 수율은 다음에서 논의된 바와 같이 낮은 온도의 아세트알데히드 산화에 의 해 얻어진 빙초산의 수율을 높이는 데에서 뚜렷이 대조된다. 4) 아세트알데히드의 산화 곡물에서 얻는 에탄올과 아세틸렌에 기본을 둔 아세트알데히드 는 빙초산을 생성하기 위한 주공급원이 되고 있다. 위의 두 출발 물질은 촉매로서. 코발트( II) 와 함께 공기에 의해 산화된다. 에틸렌으로부터 (3.3 참조) 아세트알데히드의 생성공정 에 대한 Wacker 의 발견은 또 다른 빙초산 제조에 대한 공정을 제 공한다. 특별히 관심을 끄는 점은 아세트알데히드의 산화 기술이 잘 개발 되었다는 것이다. 아세트알데히드는 부탄보다 산화되기 쉬우며 수 율을상당히 높인다. 부탄 산화 조건인 150~225 °C 와 60 기압과는 대조적으로 산화는 전형적으로 60~80 °C 의 온도와 3~10 기압의 압력하에서 진행된다. 대략 20 % 빙초산으로 붉혀진 아세트알데히드는 N2 로 희석한 02 와 함께 반응기로 공급되며, 코발트( II) 나 망간( II) 아세데이트 가 촉매로 사용된다. 종종 코발트 71). 니켈 72) 또는 구리 73) 의 염들을
가하여 생성물의 분포를 조절한다. 니켈 첨가로 빙초산의 수율은 아세트알데히드의 전환율 92~97 %에서 90 %를 넘는다. 최종 생성물의 순도는 99 %를 넘는다. 빙초산 무수물은 코발트와 구리염들의 혼합물 촉매 74) 를 사용하여 생성할 수 있댜 순간적인 증류에 의한 생성물의 급속한 분리는 부 수물의 가수분해를 피할 수 있다. 아세트알데히드로부터 과빙초산으로의 초기 산화는 알칸산화와 동류인 래디컬 사슬 메커니즘에 의해 일어난다. 알데히드 C-H 는 RO· ROO· 또는 Co(m) 에 의해 쉽게 공격 되어 순환산화 과정을 진행하는 아세틸 래디컬을 만든다. 코발트 포르피린 착물의 두 산소 아닥트는 10•c7 s ) 에서조차도 알 데히드 수소를 빼낼 수 있다. 아세틸 래디컬은 0 2 와 결합하여 아세 틸퍼옥시 래디컬을 만든다 . 이것은 선택적으로 알데히드 수소를 빼 내어 과빙초산(브)과 아세틸 래디컬을 만들면서 반응경로를 반복 순환한다(그림 3_8 참조). 과빙초산의 수율은 낮은 온도와 낮은 전환에서 거의 정량적이다. 주목되는 바와 같이 과빙초산은 주로 아세트알데히드 아닥트 그 자체로 존재한다. 산화는 주생성물로서 과빙초산을 합성하는 방식으로 진행된다 .7 4 ) 주된 사슬一정지 반응은 메탄올, 이산화탄소、 포름알데히드 그리고 포름산과 같은 부생성물로 분해될 수 있는 테트록사이드 76) 를 형성하 기 위한 아세틸퍼옥시 래디컬의 2 분자 결합이다. CH3-C \~ oH +02 一- CH3-(C14 \, p)- 0O OH (24)
CH3 CH O + X • -CH3 -c f +。 HX
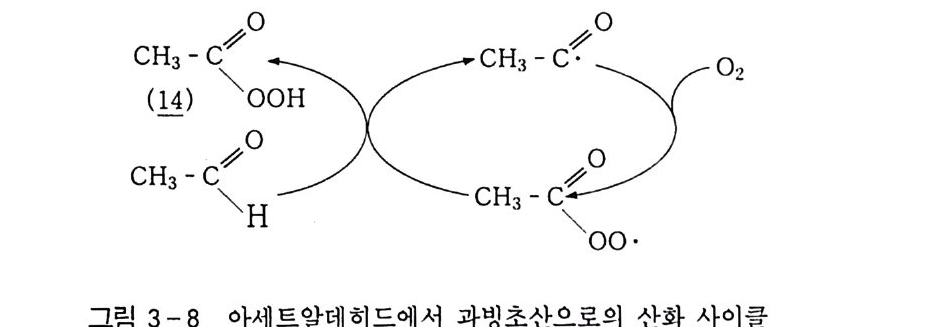 c\0 \\\\\0\ °2
c\0 \\\\\0\ °2
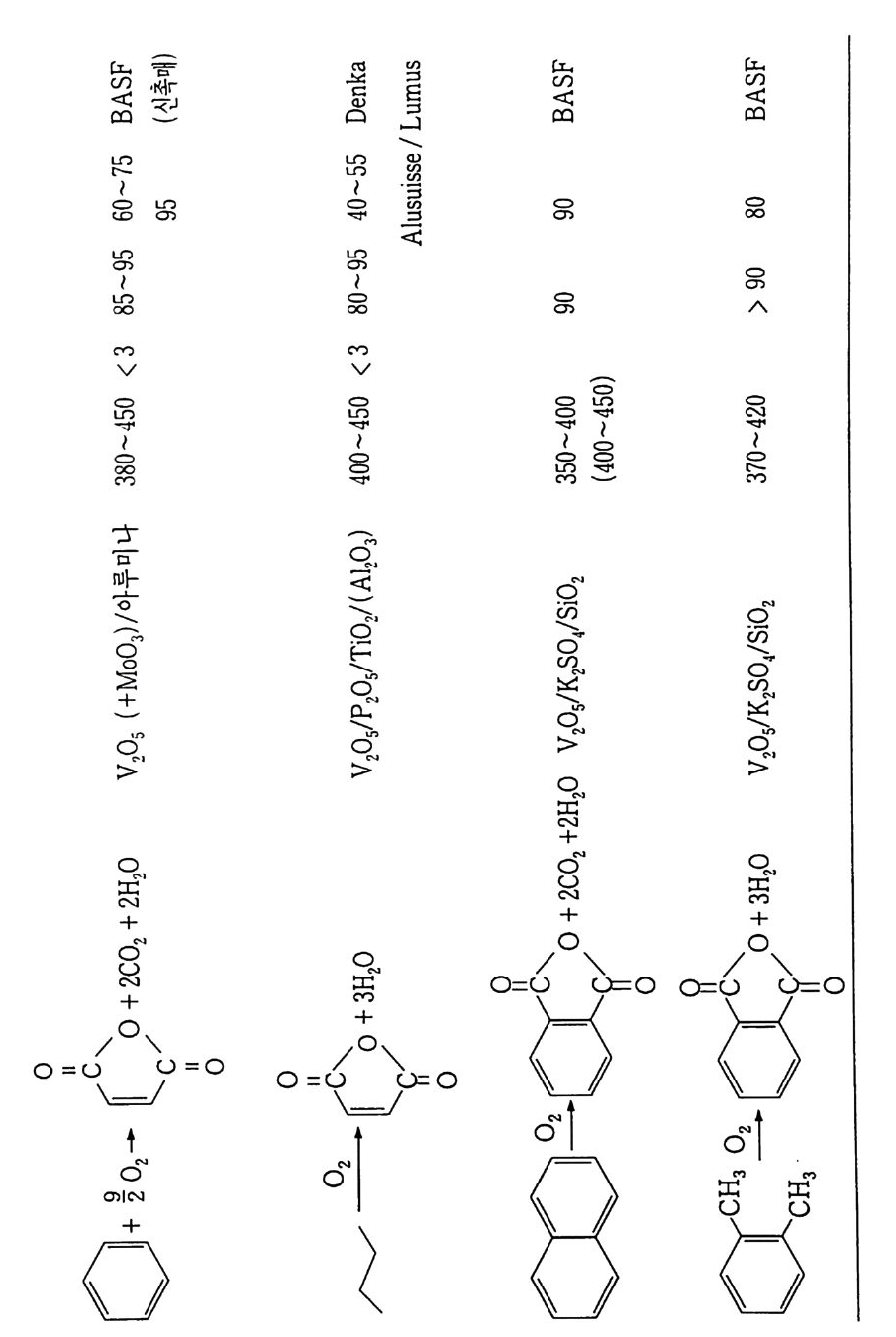
과빙초산은 낮은 온도에서 부가적으로 아세트알데히드와 함께 반응하여 아세트알데히드 모노퍼아세테이트(~)9} 같은 부가 생성 물을 합성한다. CH3 -C\/ O。O H+ 0H `/ C -C H3 ~ CH3 -C/ \ 00 -H0O/ \ C -CH3 (25) (15) 이것은 열에 의해 분해되어 30 % 과빙초산 용액을 만드는데, 이 용액은 의약품 합성에서 에폭시화제로 많이 사용된다. 5) 방향족 화합물의 수산화 (1) 벤젠 수산화 벤젠을 수산화하여 페놀을 만드는 공정은 Cumene 법으로 이미 널리 알려져 있다. 이 공정은 CD Cumene 제조 ® 액상 산화에 의
한 Hy d ro-p e roxid e 제조 ® HP 의 분해 등 다단계로 구성되어 있 다. 그러므로 많은 장치비가 들어가는데. 이때 페놀뿐만 아니라 아세 톤도 부생되어 두 화합물의 소모에 의하여 생산이 좌우된다. 그러 나 최근 들어 벤젠을 직접 수산화하여 페놀울 제조 처리하는 연구 가 활발히 진행되었다. 가장 성공적인 예가 H202 를 이용한 펜톤 (Fento n ) 형 시 약을 이 용하는 방법 이 다.77) 벤젠을 직접 수산화시켜 페놀울 산화하고자 하는 노력은 모노옥 시게나아제 효소의 발견으로 시작되었다 .78,79) 그리고 수산화의 활 성종은 • OH 래디컬이나 8 0) Oxeno i d 종 81) 이라고 생각된다.
 /N\
/N\
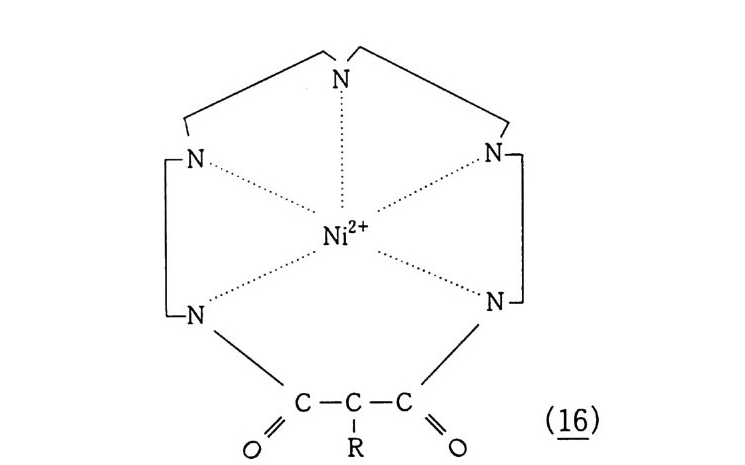
환상펜타아민의 Ni ( II) 착제(표)는 붕산 완충액 중에서 그리고 실온에서 벤젠의 페놀화반응을 촉진시킨다 .82) 동위원소를 이용한 연구에서 페놀의 산소원자는 전부 공기의 산 소분자로부터 왔음을 알았다. N i 3+-02- 형의 Sup e roxo 착제가 형 성됨은 E.S.R 측정과 자장감응으로부터 확인된다. 0.05 황산 수용액 중 CuCl 은 산소 존재하에서 벤젠을 페놀과 HQ ( Hy d ro Q u i none) 로 산화시킨다 .83)
그 농도는 각각 24 %. 8 %이며. 반응은 다음과 같이 진행되어 ·OH 를 생성한다. 02 + 2Cu+ + 2H+ 一 H 오 + 2Cu2+ (26) H2 02 + Cu+ + H+ 一 ·OH + H2 0 + Cu2+ (27) 이 ·OH 래디컬이 벤젠을 공격하여 히드로시클로헥사디에닐 (Hy d roxy c y c lohexadie n y 1 ) 래디컬(끄)을 만들고 이것은 Cu2+ 존 재하에서 페놀로 쉽게 산화된다.
 0 그 广『 Cu2 + • 0-oH (28)
0 그 广『 Cu2 + • 0-oH (28)
H Q(브)의 생성은 (끄)가 생성되어 퍼옥시 래디컬(년)을 만들
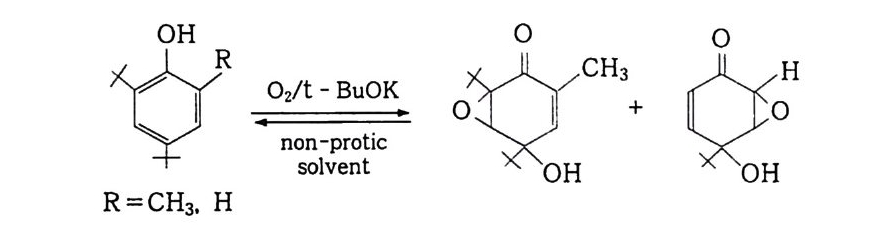
고 이것이 C 마 에 의하여 환원되어 생성된다. 조금 더 진보된 방법은 메탄올 용매에서 동, 아스코르브산 (V it a mi n C) 존재하에서 산소로 산화시키는 것이다 .84) ® 불균일 촉매 Pd-Cu/Sil ic a 존재하에서 0 2 와 H 2 를 흘려보내 벤젠을 페놀과 H Q로 전환시키는 연구 결과도 있다 . 85) P t/ V 2 0 s /S i 0 2 를 촉매로 하고 60 °C, 50 기압에서. 벤젠, 산소, 수 소로부터의 페놀 생성 속도가 1230 g /k g -ca t. hr 라는 보고도 있 다 . 86 ) 또한 환원제 라고 말할 수는 없지 만 일산화탄소 존재 아래 Pd - Rh -V /Sil ica 촉매 에 서 130 °C , 30 기 압에 서 1750kg -c at. hr 의 높 은 활성을 보고한 예 87) 도 있다. 이 방법에서 앞으로 해결해야 할 점 은 폭발 회피, 반응기당 생산성의 확보(페놀 STY) 200g I elhr), 촉매의 수명(1년) 확보. 또 다른 환원제를 사용할 경우 환원제의 가격과 환원제가 산화되어 생산되는 부산물의 처리 등을 충분히 고 려해야 한다 . 다음으로 톨루엔을 산화시켜 안식향산-안식향산울 산화분해시 켜 두 단계로 만드는 방법이다. 현재 기상 산화분해에 의한 수율 향상을 검토중에 있다 . 88) (2) 치환 페놀의 산화 벤젠환에 전자 공여기를 가지고 있으면 redox p o t en ti al 을 내려줌 으로써 쉽게 산화가 일어나지만 전자 흡인기를 가지면 그 반대이다. N i sh i na g a89) 는 2. 6-과 2, 4- d i- t-B uty lp h enols 산화에서 다 음과 같은 reg io 선택성을 발견했다 . 즉, 비양자성 용매 (non p ro tic solven t)에서 2, 6- 과 2, 4- 유도체는 산소와 반응하여 P- 위치에
서만 반응하여.
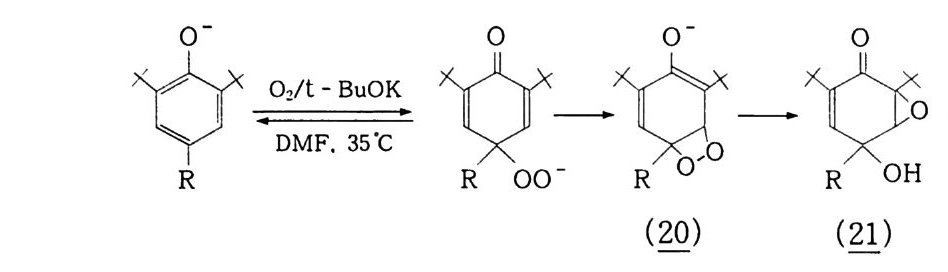 :Ro - 틀 〉一 广 (20) 一 *(21) (31)
:Ro - 틀 〉一 广 (20) 一 *(21) (31)
p-히드로퍼옥시페놀레이트 음이온 (찬)을 거쳐 에폭시 -p-퀴 놀(낌)로 전환된다. 이와 같이 2.4- 유도체는 양자성 용매에서는
 ~R 言 (32)
~R 言 (32)
다른 reg io 선택성을 보인다 .
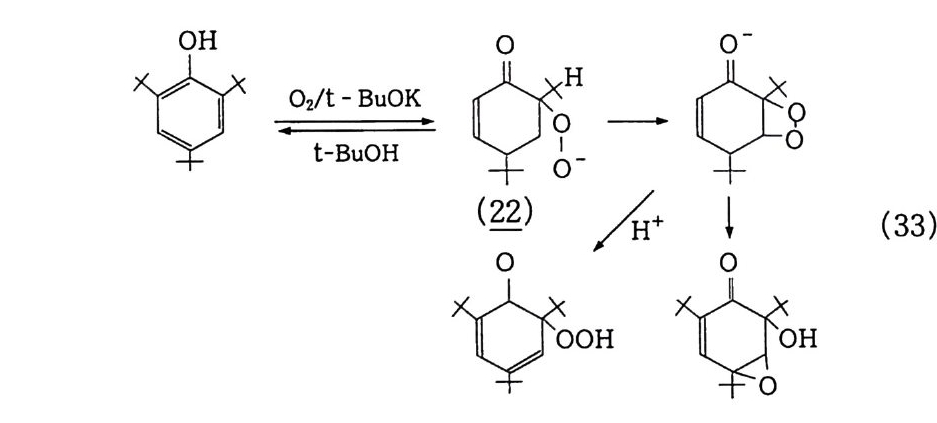 OH 02 /t - Bu OK :。H ~
OH 02 /t - Bu OK :。H ~
Tr i-t-부틸페놀의 경우 t-부탄올 용매에서는 주로 o- 퍼옥시 페놀레이트 중간체(꾸 ) 를 거쳐 에폭시 -o- 퀴놀로 전환된다. (3) 코발트( II ) Schif f 염착체 촉매 Co(salen) 과 Co(salpr ) 그리고 이들의 제 5 배위자 유도체 LCo(salen) 과 LCo(salpr ) 은 치환페놀울 산소로 산화시키는 촉매 작용을 한다 . 9 0) 이 것은 양자성 용매 ( ap ro ti c solvent) 에 서 가역 적 으로 산소착체 를 형성하기 때문이라 생각한다. 한 가지 재미있는 사실은 Co (salen) 착체는 Su p eroxo 와 Peroxo 종을 형성하는 반면. Co(sal- pr) 는 1 : 1 Su p eroxo 종만 형 성 한다는 점 이 다 .91 ) LCo(salen) + 02 ~ LCo(salen)02 (34) LCo(salen) + LCo(salen) ~ L (salen)Co02C o(salen)L (35) LCo(salpr ) + 02 ~ LCo(salpr )02 (36) 단,
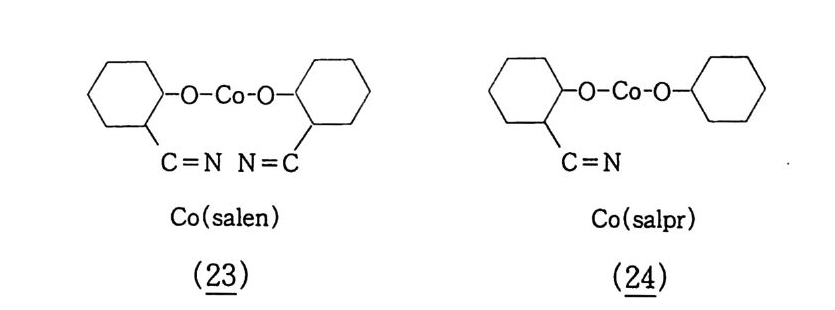 Q-0 -Co~O- O Qo -co-0- 0
Q-0 -Co~O- O Qo -co-0- 0
Co(salen) 및 그 유도체에 의하여 2 ,6 -d i-t-부틸페놀 산화시 그 반응의 선택성은 수직에 붙은 리간드와 용매에 의존한다. Ko t har i와 Tazuma 가 연구한 결과를 표 3 -2 에 나타냈다 . 92) 표 3-2 에서 보듯이 그들은 DMF 가 아주 좋 은 용매임을 발견했 댜 즉 Co(salen) py를 사용하는 경우 DMF 용매에서 B Q에 대한 선택성이 100 % 에 도달할 수 있댜 Mn(salen) 인 경우 DMF 에서 DP Q에 대한 선택성을 99 .9 % 까 지 올릴 수 있다.
표 3-2 Co(salen)p y 촉매에 의한 페놀유 도 체 산 화 (0 2 pre s s ure : 3.5 bar : 1 mo! % cat a l ys t : ph enol conve rs io n : 95-1 00 % ) Phenol Solvent Temp (° C ) BQ SelecDt ivPi Qt y ( % )O t he r 2,6-DTBP DMF 20~4 6 98 .8 1.2 2,6- D TBP DMSO 15~4 0 84 1.5 14.5 2,6-DTBP 1-M e-2-p yrr o 20~40 95.2 4.8 -lid i n o ne 2,6-DTBP DMFa) 20~45 99.5 0.5 2,6-DMP b) DMF 25~45 98 2.0 2,6- D MPb) DMFa) 20~45 100 2,6-DTBP DMFc) 22~30 <0.l 99.9 2-TBP b) DMFd) 20~35 96e) 2-M e th yl p h e n ol DMF 22~40 92f) Phenol DMF 68~73 100g) a) Co(salen) as cata l ys t ; d) 2-t- b y tylp h enol ; b) 2,6-dim eth y lp h enol : e) 3 % Co(salen) as cata l ys t : c) Mn(salen) as cata l ys t : f) at ca. 50 % ph enol conversio n :
만약 4 위치에서 알킬 (Alk y l) 이나 아릴 (Ar y l) 기가 있다면 다른 생성물로 나타난다. 예를 들면 Co(sal p r) 은 상온, 메탄올(염기성 알코올 )4 -알 킬 -2 . 6 -di- t- 부 틸페놀울 산화시켜 p-퀴놀울 거 쳐 옆 탄소로 R -기 의 전위 (NIH -shif t) 와 함께 같은 구조의 H Q를 생성한댜 93)
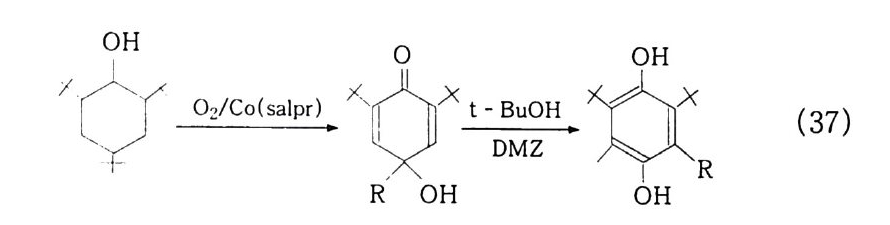 二OH 三 ;,(` \R o o : :OXH R (37)
二OH 三 ;,(` \R o o : :OXH R (37)
만약 페놀의 Para 치환체가 방향족이라면 오직 orth o - pe roxy co balt 착체만 형성되며. 반응기구는 식 (37) 의 경우와 같다.
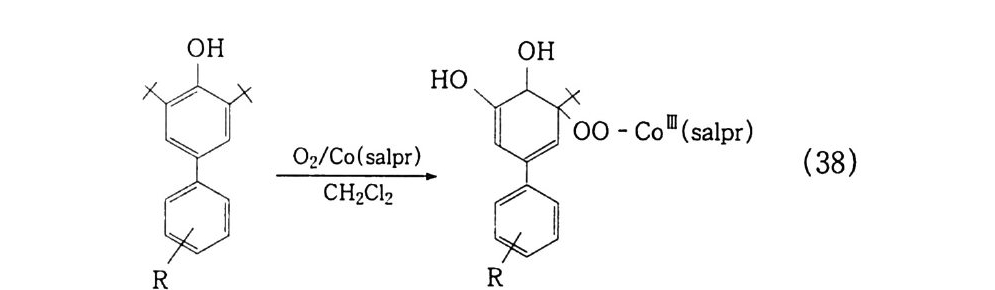 OH H。 :OH :
OH H。 :OH :
6) 데레프탈산 폴리에스테르의 한 성분으로써 대부분 사용되는 TPA( 혹은 그 의 Es t er 인 DMP) 는 공업적으로는 P- 크실렌을 액상과 공기로 1 단계 또는 2 단계 산화시키는 것이다.
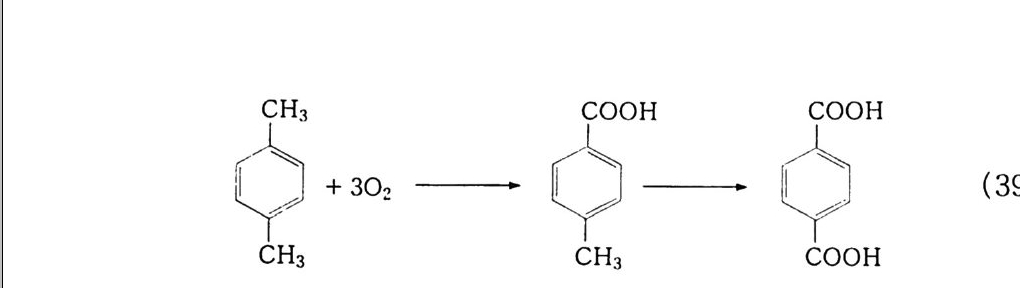 _r” \〉멕叫 + 3 5』 一 ¢CCHO3 O H- : CC OOOOHH (39)
_r” \〉멕叫 + 3 5』 一 ¢CCHO3 O H- : CC OOOOHH (39)
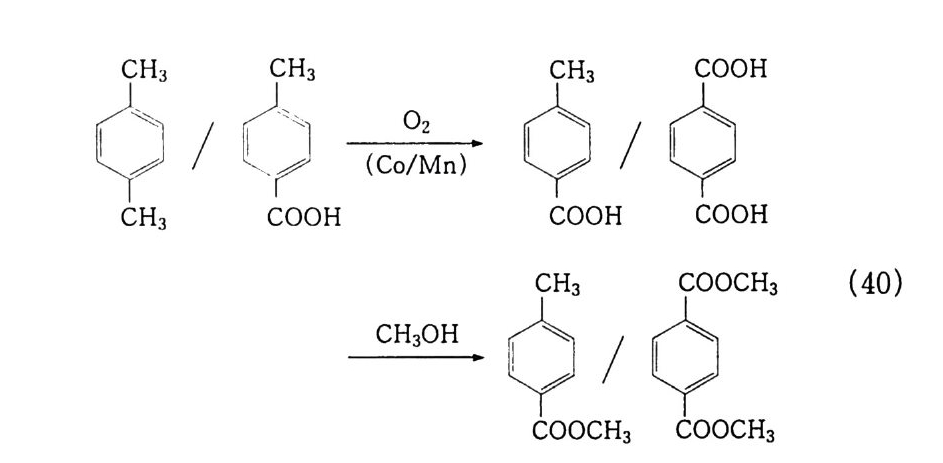
빙초산 용액에서 Co/Mn- 아세테이트를 주촉매로. NaBr. NH1 Br. 테트라브로모에탄 등을 조촉매로 사용하여 일단으로 산화시 켜 테레프탈산 (Tere ph t hal i c acid ) 을 만든다. 부식성의 브롬을 사용하므로 공정의 재질은 티타늄을 이용한다. 이때 반응조건은 195~225 ·c. 15~ 3 0 a t m 으로 0.5~ 2 시간 동안 접촉시킨다. 그 후 반응 혼합물을 냉각시켜 조생성물을 얻는다. 생 성된 조테레프탈산을 에스테르화하든지. 고온 (225~275 · c) 에서 압력을 가해 물에 녹여 Pd 촉매하에서 부산물로 존재하는 4- 카르 복시벤젠 알데히드를 P- 톨룰산으로 수화시킨다 . 전환율은 95% 이 상이 고 수율은 90 mol % 이 다 . 9l 95) Amoco 의 주부산물은 약 25 pp m 의 4- 호밀안식향산이다. 이를 물과 같이 현탁액 (slurr y) 형태로 반응기에 수송하고 25o ·c 이상 의 귀금속/탄소 촉매로 수침시켜 4- 호밀안식향산을 P- 톨루엔산 으로 환원하고 착색물질도 무색으로 바꾼다 . 9G) 테레프탈산의 메틸에스테르화는 극소량의 브롬 유도체 등 불순 물을 포함하고 있고 에스테르화함으로써 비점이 낮아져 증류로 쉽 게 고순도화할 수 있으므로 DMT 로 만들어 반응에 참여시킨다. 촉 매 없이도 에스테르화는 잘 일어나나 보통 250~3oo ·c 에서 Zn, Mo, Sb 등을 사용한댜 전환율은 평형까지 일어나며 보통 96 %이 다 .97, 98) 독일의 W itten9 9> 및 Imhausen 에서는 P- 크실렌과 Mono- 메틸 테레프탈산을 동시에 산화시키는 법이 있다. 죽, 140~160°C 에서
유기산의 Co/Mn 염 존재하에 공기로 산화시켜 P- 톨룰산울 중간 체로 만든다. 카르복실기의 과잉산화를 막기 위하여 240 ·c. 30~40a t m 에서 메탄올과 무촉매로 에스테르화시킨다. 그리고 둘 째 메틸 그룹은 역시 Co- 촉매하에 180~210·c . 1~5a t m 에서 산 화시킨다 . 에스테르화 후 미반응의 P- 크실렌과 P- 톨룰에스테르 는 재순환시키고. 생성물은 메탄올이나 크실렌으로 재결정시켜 순 도를높인댜
수득률은 90 %(P - 크실렌대) 및 80 %(MeOH 대)이다. 3. 4 불균일 촉매 산화반응 3. 4. 1 촉매 무산소 기상 산화반응 1) 나프타 크래킹 2. 5. 2 기상 산화반응의 1) 무산소 기상 산화반응, 죽 나프타 크
래킹에서 촉매를 사용한 경우이다. 2. 5. 2 에서 서술한 열분해 반응의 높은 반응온도 를 내리고. 생성 물 조성을 변화시키기 위하여 이 방법을 사용한다. 즉, 원료 가스 와 분말 상태의 촉매를 반응기(유동층)에서 탈수소 분해반응을 시 킨다. 반응온도는 500 °C 전후. 촉매로는 제올라이트. 실리카. 알루 미나가 사용되고 있댜 촉매를 사용하므로 카르보늄이 형성되어 {3- 분열이 일어나고. 그렇기 때문에 에틸렌보다는 프로필렌, 부탄류
표 3-3 C.I - 유분의 조성 (vol %) 조성 b.p (°C) 증기 크래킹 촉 매 크래킹 프로필렌 -47.7 0.2 3.1 프로판 -42.1 프로파디엔 -34.5 0.1 프로판 一 23 .2 0.3 0l 소부탄 -1 1. 7 1.0 24.4 이소부텐 -6.9 23.5 20.2 n -부 텐 -1 -6.3 12.6 13.4 부타디엔 -I. 3 -4.4 44.6 0.4 n- 부탄 -0.5 4.7 15.6 t-부 텐 -2 0.9 6.4 13.3 cis - 부 텐 -2 3.7 5.4 9.0 비닐아세틸렌 5.1 0.8 부틴 -1 8.1 0.2 디아세틸렌 10.3 흔적 부타디엔 -1, 2 10.8 0.2 부c틴; -탄2 화수소 26.9 혼혼적적
등이 더 많이 생산된다(표 3-3 참조) . 2) co 계 의한 메탄리포밍 산업의 발달과 더불어 많은 화석 연료를 사용하게 됨으로써 배기 가스에 들어 있는 탄산가스 때문에 대기 중의 이산화탄소 농도가 280 pp m 이던 것이 1950 년대 말에는 315 pp m 이었으며. 1990 년대 에는 353 pp m 으로 계속 증가되고 있다. 이 산화탄소는 화학적으로 볼 때 AG f 가 94.26 kca l/ mol 로 열 역 학 상 대단히 안정한 화합물이다. 그러므로 현재까지 주로 냉매나 탄 산음료 제조에 사용되고. 약 10 % 정도만 화학제품 제조에 이용되 고 있다. 예를 들면 아스피린 원료인 살리실릭산을 제조하는 Kolbe- Schmi dt 공정과 산화에틸렌으로부터 제조되는 유기 카르보네이트 정도이다. 이처럼 이산화탄소의 공업적 이용은 극히 제한되어 있으며, 특히 환경문제와 관련하여 이산화탄소의 이용을 생각할 때는 CO2 활성 화에 관한 근본적인 화학 발전과 함께 대규모의 수요처 발굴이 필 수적이라 하겠다. 그런 면에서 C 아와 암모니아의 반응을 통한 요소의 제조와 메탄 올 합성공정에의 수요를 생각할 수 있다. 앞에서 설명한 바와 같이 CO2 는 안정하므로 그 활성화는 높은 에너지의 환원제나 외부에서 에너지를 가해야 하는 고난도의 기술이 요구된다. 접촉 수소화법은 촉매를 이용하여 이산화탄소와 수소를 접촉시 켜 메탄이나 메탄올 등으로 환원하는 기술이다. 촉매로서는 Cu— Zn 계를 주로 이용하며, 온도와 압력을 각각 250 °C, 10 MPa 로 반 응시켜 생성물을 얻는다. 이와 같은 반응은 천연가스를 이용한 메
탄올 제조공정으로 활용되고 있어 앞에서 언급한 다른 고정화법보 다는 기술적으로 쉽게 상용화에 접근할 수 있는 방법이다. 그러나 이 방법의 최대 난제는 염가의 수소를 다량으로 공급할 수 있는 방 법을 동시에 개발해야 한다는 점이다. 앞 절에서 설명한 바와 같이 나프타를 수증기 접촉 분해시키면 올레핀과 수소가 생성된다. 같은 원리로 천연가스 중에 풍부히 존 재하는 메탄을 산화제로 사용하여 이산화탄소를 환원시켜 화학제 품으로 전환할 수 있는 촉매의 개발 및 방법에 대한 연구는. 이산 화탄소의 재활용 측면뿐 아니라 4 개의 1 급 C-H 결합을 가지고 있기 때문에 가장 안정한 탄화수소 가운데 하나인 메탄의 활성화 연구 측면에서도 의미가 있다. 이러한 관점에서 천연가스의 주성분안 메탄에 의한 이산화탄소 의 개질을 통해 합성가스를 제조하여 기존 공정 접목 가능성에 대 해 논하겠다. 합성가스라 함은 수소와 일산화탄소의 혼합물로, 기존 석유화학 의 기초 원료인 에틸렌처럼 F i scher-Tro p sch 반응에 의한 탄화수 소 및 알코올류의 제조. 암모니아 합성. 메탄올 합성. 옥소반응에 의한 알코올류 제조. 디메틸에테르, 아세트산의 합성에 사용되는 기초 출발 물질이다. 그 가운데 메탄을 반응물로 한 촉매에 의한 수증기 개질 (S t eam Re fo rm i n g)은 이미 상업화되어 있으며. 산소 개질 (Ox y re fo rm i n g)과 이산화탄소 개질 (Carbon Dio x id e Re- fo rm i n g)반응이 촉매반응 관점에서 최근 큰 관심이 되고 있다 . 이 들 각각의 반응식과 반응 엔탈피 및 양론적 H2/CO 비는 표 3-4 와같다. 표 3-4 에서 보는 바와 같이 기존에는 공업적으로 수증기 개질이 주로 활용되 어 왔으나 그 용도에 따른 H2/CO 비 의 요구에 따라 방법을 달리할 수 있다. 그리고 이산화탄소와 함께 강한 홉열반응
표 3-4 메탄의 촉매 개질 촉매 개질 반응식 L1H02 98( kJ/ m ol) H/CO 증기 개질 CH ., + H20 - CO + 3H2 206 3 옥시 개질 CH., + 21 0 2 一 CO + 2H2 -36 2 이산화탄소 개질 CH1 + CO2 - 2CO + 2H2 247 1
인 점과 수증기의 잠열에 의한 에너지 흡수 등이 문제됨에 따라 산 소 개질이 의미가 있댜 또한 이산화탄소 개질은 이산화탄소가 지 구 온난화의 주범이며, 잠열에 의한 에너지 손실을 극복할 수 있다 는 장점 때문에 더욱더 관심이 집중되고 있다. 현재 이산화탄소 개질반응을 공정에 적용하는 것은 확실하지 않 지만 기존 개질공정의 보유 회사들로부터 몇 가지 가능성 있는 자 료들이 제시되고 있다. 특히 합성가스를 제조하는 경제적인 측면에 서 볼 때 메탄의 이산화탄소 개질공정은 메탄의 수증기 개질공정과 대등하다고 평가되고 있다 . Haldor-To p soe 에서는 메탄 개질반응 의 산화제로서 수증기와 이산화탄소를 함께 사용하는 SPARG ( Surfu r Passiv a te d Refo r mi n g ) 공정 을 제 시 한 바 있 다 .100) SPARG 공정이란 개질공정의 운전시 니켈 촉매에서 코크 형성을 유발하는 활성점을 황에 의해 봉쇄하고 개질반응의 활성점은 그대 로 유지 시 키 는, 이른바
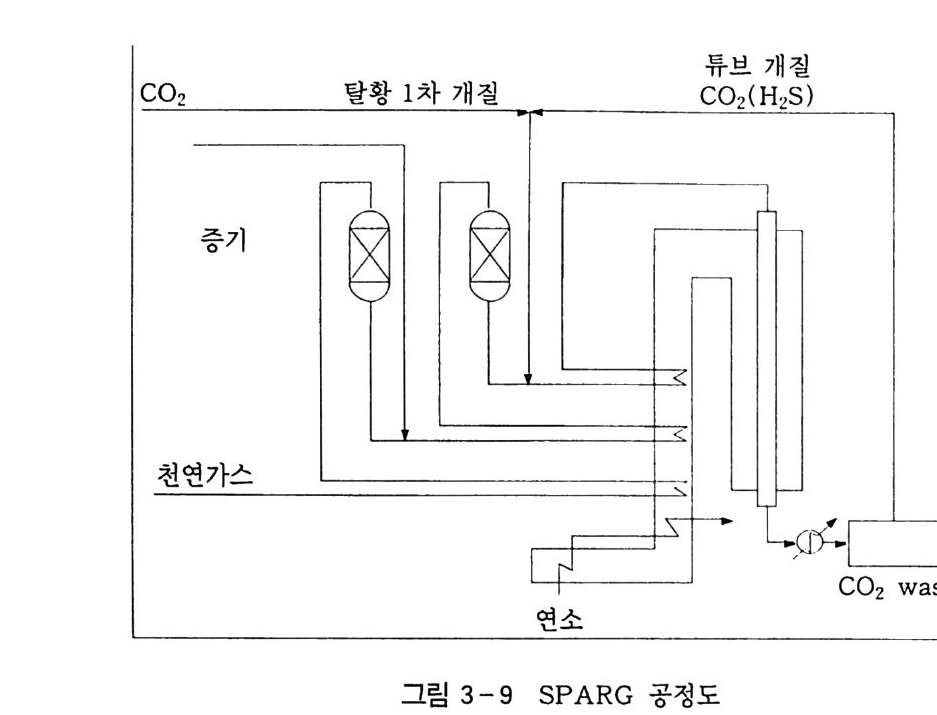 튜브개질
튜브개질
정에 이용할 수 있는 가능성이 있었다. 때문에 이러한 SPARG 공정은 값싼 이산화탄소를 공급할 수 있을 경우 경제성이 있다는 분석이 나왔다. 한편, 독일 Caloric GmbH 사의 Teuner 는 합성가스를 제조할 수 있는 새로운 공정으로 LPG 의 이산화탄소 개질공정을 제시한 바 있다 .IOI) 이 공정은 천연가스 또는 LPG 가스와 이산화탄소의 다 단계 촉매반응에 의해 일산화탄소로 전환시키며, 기존의 수증기 개 질공정에 비해 경제적일 뿐 아니라 고순도의 일산화탄소를 얻을 수 있다는 장점이 있다(그림 3-10). 이산화탄소 개질반응의 용도로서 가장 중요한 것은 무엇보다도 생성되는 합성가스의 화학원료로서의 응용이다. 수증기 개질로부터 얻어지는 합성가스가 메탄올 합성의 원료로 널리 이용되고 있으며, 암모니아 합성, 수소 제조, 탄화수소 합성, 함산소화합물 합성 둥에
도 이용되고 있음은 주지의 사실이다. 수증기 개질로부터 얻어지는 합성가스 (H 2 /C0=3) 는 수소 함량이 높으므로 수소 제조에 이용되 기도 하고, 여러 단계 공정을 거쳐 H2/N2 = 3 의 몰비를 필요로 하 는 암모니아 합성에도 이용되고 있다. 특히 가장 중요한 용도는 메탄올 합성인데, 메탄올 합성에 필요
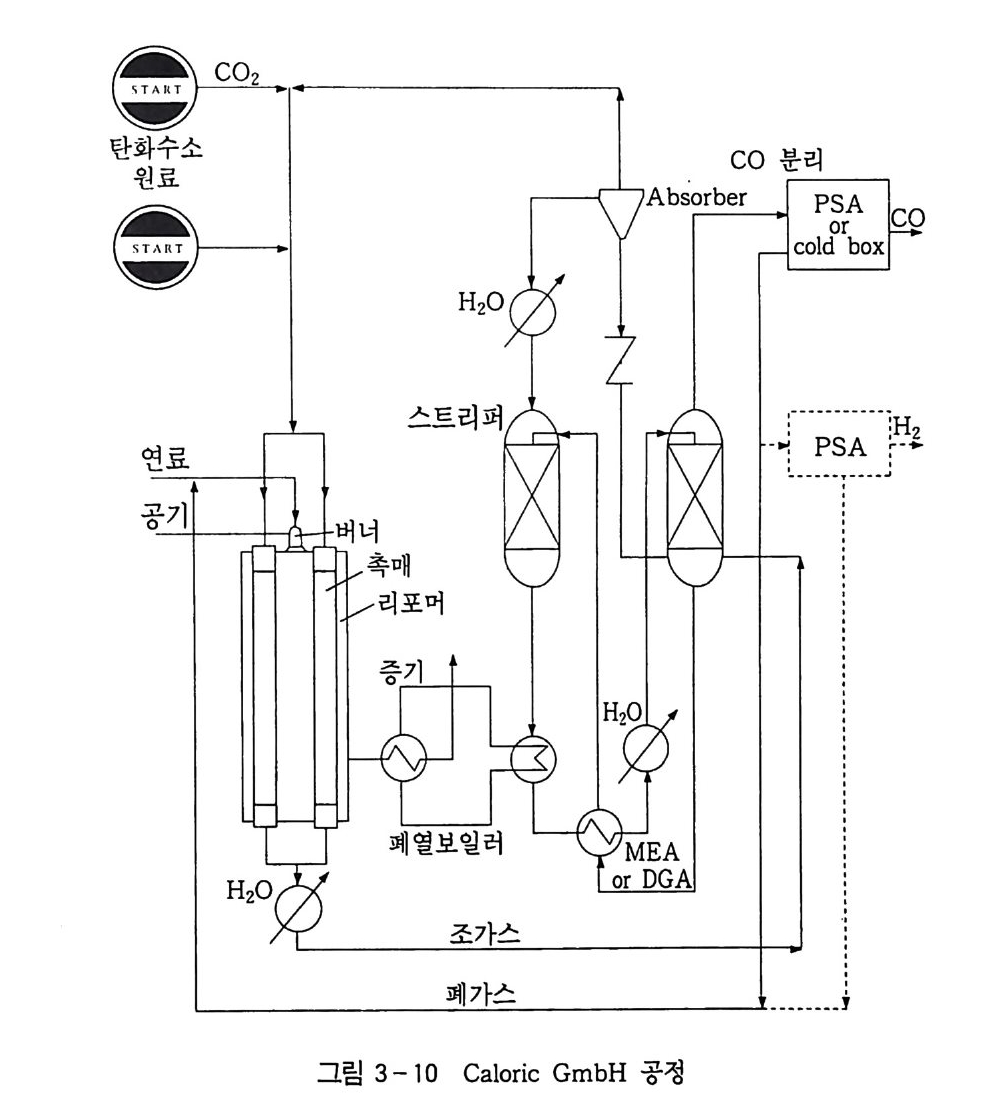 -탄화수소 CO 분리
-탄화수소 CO 분리
한 합성 가스비는 H i C0=2 이므로 수증기 개질에 의해 얻어지는 H2 /C O 비를 낮추기 위해, 수성가스화 반응에 연결하여 여분의 수 소를 전환시키거나 자동열 개질과 같은 이차 개질반응으로 낮은 H2 /C O 비를 공급함으로써 조절한다. 이산화탄소 개질로부터 얻어 지는 합성가스도 이와 유사한 용도로 이용될 수 있으나 합성가스의 CO 함량이 매우 높기 때문에 H 2 /CO=l 의 몰비를 필요로 하는 옥 소공정 이 나 Fis c her -Trop s ch 합성, CO 및 수소 제조 등에 이 용 할수 있다 . 그리고 다른 개질반응과 연결하여 합성가스비를 조절할 경우 메 탄올 합성이나 암모니아 합성 등에 이용할 수 있다. 특히 옥소에서 는 코발트 . 루테늄 둥의 균일계 촉매상에서 합성가스와 올레핀과의 반응에 의해 알데 히드를 합성 하고 , Fis c her -Trop s ch 합성 에 서 는 철 , 코발트, 루테늄 등의 불균일계 촉매상에서 합성가스를 탄화수 소로 전환시킨다. 그 다음으로 가능성 있는 분야는 이산화탄소 개질반응을 화학 에 너지 전송 시스템 (Chemi ca l Energy Transmi ss io n Sy s te m , CETS) 에 적용하는 것이다. 그림 3-11 과 같이 CETS 는 가역반응 이면서 홉열반응인 이산화탄소 개질반응에서 태양 에너지, 핵 에너 지 혹은 화석 에너지를 반응열로 저장하여 파이프라인을 통해 원하 는 위치까지 전송시키고, 이산화탄소 개질반응의 역반응으로 발열인 메탄화반응에 의해 에너지를 발산하는 시스템을 예시하고 있다. 메탄의 수증기 개질반응에서는 이러한 CETS 가 이미 적용되고 있기 때문에 보다 심한 홉열반응인 이산화탄소 개질반응에서도 CETS 로서의 이용 가능성이 충분하다고 여겨지며, Ric h ardson 등 을 비롯한 몇몇 그룹에서 시도하고 있는 것으로 보고되고 있다• Ric h ardson 등은 나트륨 열파이프를 반응기로 한 태양열 이용 CETS 를 제시한 바 있다 •102)
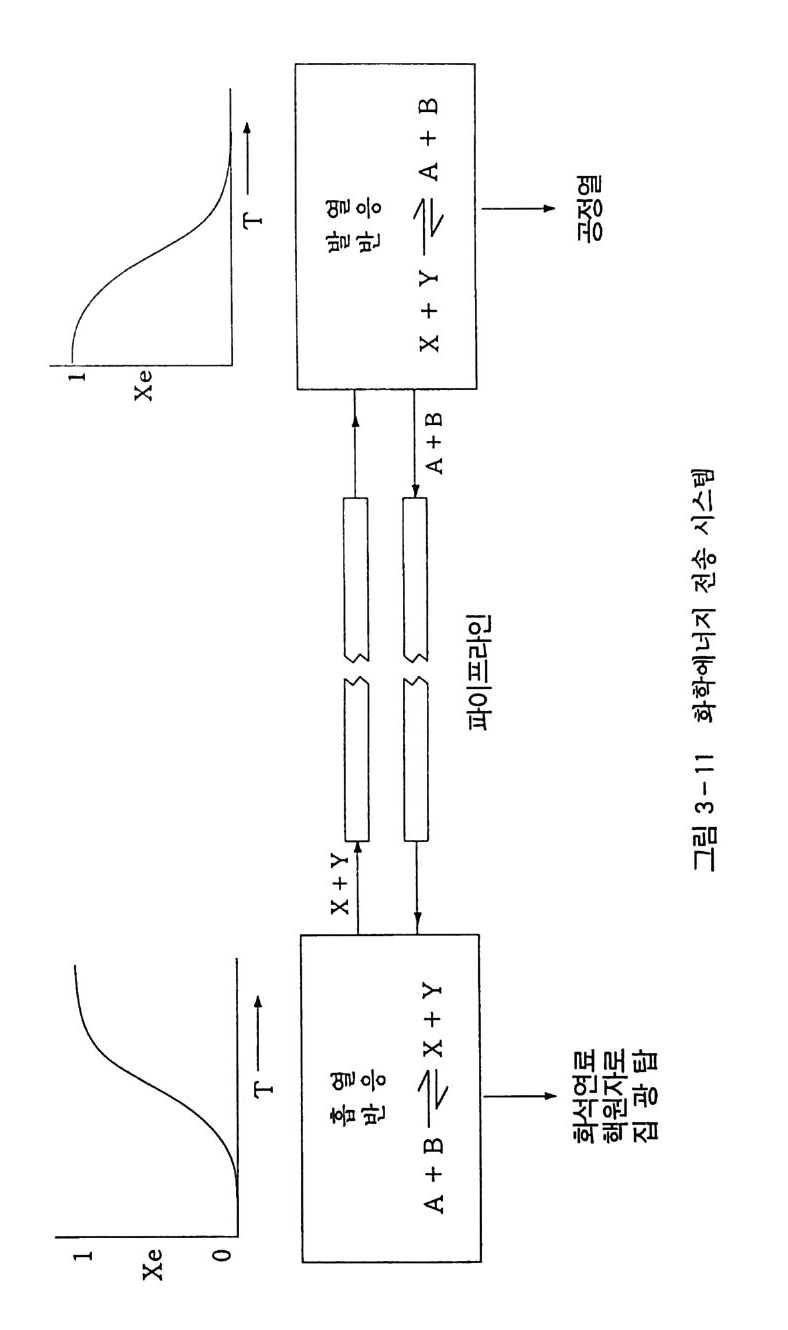 띠
띠
이와 유사한 시스템을 Levy 등도 제시하였는데 . 수직형 열파이 프 반응기에 로듐 촉매를 충진시켜 이용하였다 .1 03) 이들의 열파이 프 반응기는 태양열을 6.3kW 까지 홉수하였으며, 반응기체의 최대 유속은 11,000 L/h, 메탄의 전환율은 85 %에 이르렀다 . 또한 이보다 훨씬 전에 Chubb 등 10 ~ ) 과 McCar y 1 05) 도 CETS 의 일종인 Solchem 공정, 즉 태양 에너지를 화학열 에너지로 저장할 수 있는 시스템을 제안한 바 있댜 이산화탄소 개질반응에 관한 연구는 현재까지 주로 촉매 및 반응 조건의 선별에 초점이 맞춰져 있으며. 반응 메커니즘 규명에 관한 연구는 거의 없는 실정이다. 이산화탄소 개질반응의 촉매는 처음에 수증기 개질 촉매를 그대 로 적용하면서 연구되기 시작하였다. 표 3-5 에는 현재 메탄의 이 산화탄소 개질반응을 연구하고 있는 세계 주요 그룹들의 촉매계와 그 특징을 요약하여 나타내었다. 이 표에 제시된 바와 같이 현재까 지의 연구 흐름을 살펴볼 때 이산화탄소 개질 촉매는 크게 두 종류 로제시되고 있다 . 하나는 이리듐, 로듐, 팔라듐, 백금 둥의 귀금속 담지 촉매이며. 다른 하나는 개질반응의 촉매로서 널리 사용되고 있는 니켈 담지 촉매이댜 제시된 촉매는 거의 대부분 Vlll 족 전이금속들로, 주어진 반응조건하에서 평형 전환율에 근접하는 높은 활성들을 나타낸다. 이러한 고활성을 얼마나 유지할 수 있는지가 촉매 개발의 관건이 다 . N i수 el증se기n 이 개보질고 한촉 바매1 로06) 서에 따널르리면 알N려i >진 P t니 >켈 R u촉 >매 는Ir 의 R순os서tr 로u p 메- 탄의 탄소화 반응에 활성을 나타내며, 니켈 촉매상의 코크 형성에 의한 촉매 비활성화를 막기 위해 메탄에 비해 수증기를 이용하고자 하는 것이 가장 먼저 시도되었으나 반응물 중에 수분이 없을 경우
심한 코크 형성으로 촉매 활성이 저하되는 문제점을 안고 있다. 한편 박상언 등은 최근 제올라이트 담체에 K-Ni - CaOx 산화 물을 담지하여 이산화탄소 개질반응에 이용하였는데, 그 결과는 다 음과 같다 .10 7) 표 3-5 에는 또한 여러 종류의 니켈 촉매상에서 메탄의 CO2 개 질반응에 대한 활성을 비교하여 나타내었다 . 촉매 활성을 명확히 비교하기 위해 반응성이 못 미치는 조건하에서 실험하였다. 그런데 700 °C, 2.2 X 10·1 h -1 이하의 공간 속도로는 고활성을 갖
표 3-5 메탄에 의한 CO2 개질 연구현황 연구그룹 촉매 비고 귀금속 B. L. Gusta f s o n ( 미 국 ) Pd. Pt / Alz03 특허 출원중 (Eastm an Kodak) J. T . Ric h ardson( 미국) Rh / Alz03 CETS(Chem. Energy Trans. Sy st e m ) A. K. Cheeth a m ( 영 국) Ir / Alz03 고활성 및 안정성 Solym osi ( 헝 가리 ) Pt / Alz03 CO2 분해에 대한 반응차수 Lev y(이스라엘) Rh/ Alz03 열파이프 (CETS) N i -촉매 A. M. Gadalla( 미국) Ni I r-Alz03, Sp ine ! 지지체 효과와상변화 I. Mats u ura( 일본) Ni /u se Mg O 초미세 Mg O 단결정 K. Fujim o to ( 일본) Ni /M g O CO2 개질 및 F-T 합성 KRICT( 한국) K-Ni- C a-Ox/ Zeoli te 용융열 기술 Mamedov ( 러 시 아) Ni- c ata l ys t s C/C2 탄화수소로 CO2 개 질 Kryl o v ( 러 시 아) K-Cr-MnO / Si0 2 CO2 에 의 한 Cl-( 겁 알칸 산화
는 니켈 담지 촉매에서 반응 활성의 차이를 비교하기가 곤란하였 다. 그러나 N i O/a-Ab 아 촉매의 경우 반응온도 7oo·c. 6x1oh-1 의 매우 큰 공간 속도에서 50 % 이하의 비교적 낮은 메탄 및 CO2 전환율과 합성가스 수율밖에 얻지 못하였다. 한편. 펜타실형 세공 구조를 갖는 제올라이트 담체상에 니켈을 담지한 경우 a-Al203 담체에 비해 약 20 % 이상의 전환율 및 수 율 향상이 관찰되었다. NiO /ZSM 촉매의 경우 함침법과 용융법에 의해 제조된 촉매의 활성이 큰 차이를 보이지 않았지만. Ni O /ZSM 에 알칼리 및 알칼리 토금속이 조촉매로 사용된 K-Ni -C aOx/ ZSM 에서는 용융법에 의해 제조된 촉매가 함침법에 의해 제조된 경우에 비해 활성이 약 20% 이상 높게 나타났다. 용융법에 의해 제조된 K-N i -CaOx/ZSM 은 700 °C 에서 0.05 sec 의 짧은 접촉시 간 (WHSV=6X104h-l) 에서도 평형에 가까운 활성을 보였으며, 800 ·c 에서는 열역학적인 평형치와 일치하는 활성을 얻을 수 있었다. 이러한 결과로부터 용융법에 의해 제조된 니켈 촉매상에서 K 과 Ca 이 메탄의 CO2 개질반응에 활성을 증진시키는 역할을 한다는 것 을 알 수 있다. 용융법에 의해 제조된 K-Ni -C aOx/ZSM 촉매상 에서 반응온도에 따른 메탄의 CO2 개질반응의 영향을 그림 3-12 에 제시하였다. C02/CH4 =1 인 경우 메탄의 CO2 개질반응은 반응 식 (41-1) 과 같이 진행되는데, 공간 속도 4.4X104h-1, 반응온도 700 °C 이상에서는 물이 거의 생성되지 않았으며, 메탄과 CO2 전환 율, 합성가스 수율이 모두 평형에 근접하는 결과를 얻었다. CH4 + CO2 一 2CO + 2H2 (41 一 1) CO2 + H2 一 CO + H20 (41 —2)
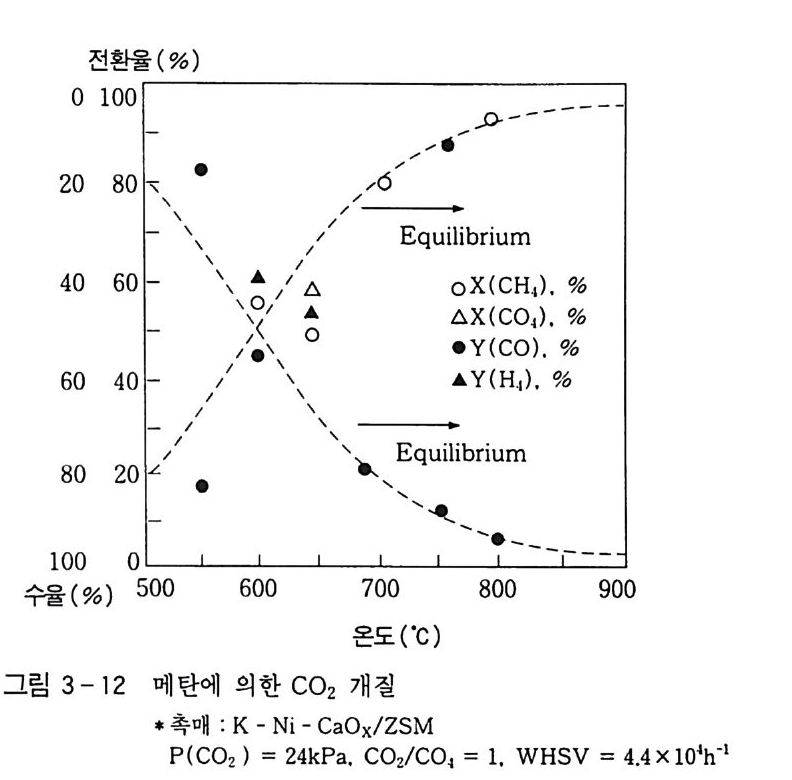 전환율(%)
전환율(%)
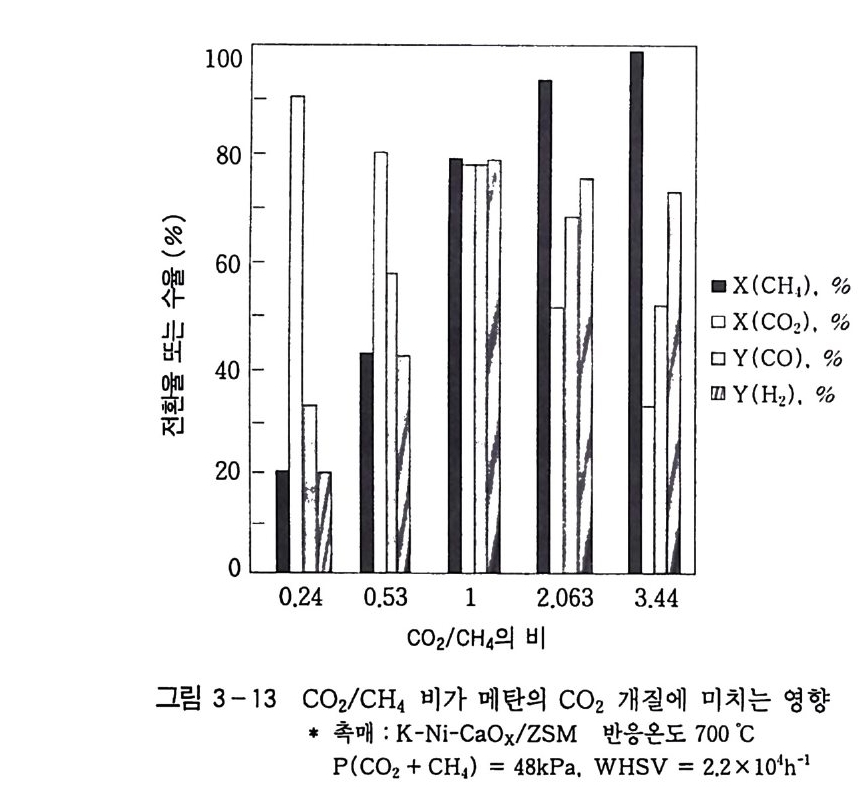
히 706000° C~ 6이50하 °에C 에서서는는 소 H량z의 /C O 물 <이 1 합로성 메가탄스에와서 함의께 수 생소성원되자었가다. 수 특소 분자 외에 물로 손실됨을 보여 준다. 이러한 물은 반응식 (41-2) 의 수성가스 전환반응으로부터 CO2 와 수소와의 반응에 의해 생성 되는 것으로 보인다. 그림 3-13 에 700 °C 에서 C02/CH4 비율의 변화에 따른 반응의 영향을 나타내었다. 메 탄의 전환율은 COz /CH 4 비 가 증가할수록 증가하며, COz /C H4> 3비 일 가 때감 는소 거할 수의 록10 0증 가%하에며 ,가 까C웠O다z ./C H 반4 대<로o . 3C 일O2 때전 환90율 %은 이CO상z이 /C H 얻4 어졌다. CO 및 H2 수율은 C02/CH4=l 일 때 가장 높게 얻어졌으 며, 생성된 H2/CO 비는 반응물의 COz /CH 4 비가 0.24 에서 3.44 로
증가함에 따라 0.62 에서 1. 50 으로 점진적으로 증가하였다 . 따라서 최고의 CO 및 H2 수율을 얻기 위한 최적의 조성은 COJ C H,1 = 1 이며 . 이때 생성되는 합성가스비도 CO/H 2 =l 임을 알 수 있다 . 니켈이 실리카에 담지된 Ni /Si0 2 촉매의 경우 니켈 산화물과 담 체와의 반응에 의해 니켈 h y dros i li ca t e 가 생성됨이 보고된 바 1 08) 있고. Ni /Al 20 3 촉매에서는 N i Al 2 0 4 와 같은 스피넬 산화물이 관찰 되 기도 하였다 .1 09) 본 연구에서 합성된 니켈 촉매의 XRD 분석 결과로부터 니켈과 담체와의 반응에 의한 화합물은 관찰되지 않았으며 . 담체상의 니켈 은 Ni O 산화물로서 유일하게 관찰되었댜 특히 ZSM 담체상에서 는 XRD 패턴으로 겨우 관찰될 정도의 아주 낮은 세기의 NiO 회
 100
100
표 3-6 메탄에 의한 CO2 개질 반응에서 각 촉매 활성 : 촉매 Ni / 지지체. 반응온도 : 1oo ·c 촉매 CH전4 환 율 %C O2 co 수율. % H2 Ni O /a -Alz03 45 47 46 46 NiO / T/ -Alz03 78 78 72 78 NiO / Si0 2 (l ) 75 77 76 76 K-Ni -C a-Ox I Si0 2 ( I ) 13 21 17 18 NiO / ZSM( l ) 78 78 77 77 NiO / ZSM( II ) 77 78 76 77 K-Ni -C a-Ox / ZSM ( I) 79 78 79 78 K-Ni -C a-Ox I ZSM ( Il ) 58 59 59 59 * 반응조 건 : P(CH4) = 24 k Pa. CO2 /C H•I = 1. WHSV= 6.0 x lO4'h. -I co 수율 = P(CO) 。 ul / [P( C H4),n + P(C0 2 ) 』 . H 2 수 율 = P(H2 )o u l / ZP(CH4),. N i량 : 6.6 w t %, K-Ni -C a- O x ; K 0_08 N i1. 0 C 또 Ox
절 피크가 얻어져 담체상에 매우 잘 분포되어 있다고 여겨졌다. K -N i -CaOx/ZSM 에서는 Ni O 외에 CaAl2 아가 역시 관찰되었으며, 소량 존재하는 K 와 관련된 상은 관찰되지 않았다 . BET 흡착에 의해 측정한 K-N i -CaOx/ZSM 의 표면적은 137 m2/ g으로 니켈울 포함한 금속 성분이 약 20 % 정도 담지되었음에 도 불구하고 상당히 높은 표면적을 나타냈다. 이것으로 볼 때 용융 법에 의해 K 一 N i -CaOx 가 제올라이트 담체에 잘 분포되어 있음을 알 수 있다(표 3-6). 메탄의 수증기 개질 촉매로 널리 사용되고 있는 니켈 담지 촉매. 그 가운데서도 대표적인 Ni/ Al 203 촉매의 가장 큰 문제점 중 하나
는 코크 형성에 의한 촉매의 바활성화로 지적되고 있다. 이러한 단 점은 메탄의 CO2 개질반응에서도 제시된 바 있다. Gus t a fs on 은 메탄의 C 아 개질반응의 촉매로 P t /Al 2 0 3 와 Pd/Al 오 촉매를 제시하면서. 비교 예로. N i /Alz0 3 의 경우 800°C 에서 반응시 초기 활성은 매우 높지만 반응 2 시간 뒤 코크 형성에 의해 반응이 멈춘다는 사실을 들어 보고한 바 있으며 .11 0> Fuji m oto 등은 9 % N i/ Alz 이 촉매가 800 °C 이상의 반응조건에서 코크 침적 으로 인해 반응시간에 따라 급격한 반응성의 저하를 일으킨다고 보 고한 바 있다.Iii) 본 연구에서는 니켈 담지 촉매의 이러한 단점을 극복하기 위해 열안정성이 높은 펜타실형 ZSM 제올라이트를 담체로 . K 과 Ca 을 조촉매로 사용하여 활성 향상과 함께 안정성을 높일 수 있었다. K 은 CO2 에 의한 탄소의 기화반응시 CO 로의 전환 촉매로서 작용한 다는 보고가 있으며. 수증기 개질반응에 메탄으로부터 촉매상의 코 크 형성을 막기 위해 알칼리 조촉매의 첨가가 효과적이라고 알려져 있다 .11 2) 그리고 염기성 산화물인 K-CaOx 화합물은 안정한 탄소 의 수증기 기화반응에 촉매작용을 한다는 보고 11 2) 가 있어 메탄의 CO2 개질반응에서 생성되는 코크의 누적을 억제하는 데 유리하게 작용할수 있다. 여기에서 제올라이트에 니켈만을 담지한 경우 반웅 수시간이 지 난 후 심한 코크 침적이 일어났으나 용융법에 의해 제올라이트에 니켈과 함께 K 과 Ca 울 첨가함으로써 높은 활성을 유지할 수 있었 고, 800 °C 에서 140 시간 이상 반응시킨 경우에도 코크 생성 없이 반응활성이 그대로 유지되는 촉매의 안정성도 크게 향상되는 결과 를얻었다• 따라서 메탄을 환원제로 혹은 이산화탄소를 산화제로 해서 가장 안정한 두 화합물을 활성화하여 기초 출발 물질인 합성가스를 평형
전환율과 수율에 이르도록 하는 안정된 촉매 시스템이 확인된 것이 다. 그러므로 상황에 따른 이의 적용. 즉 수증기 개질의 부분적 대 체. 화학 에너지 운송 시스템에 의한 태양 혹은 원자력 에너지의 활용 등에 적용될 수 있는 가능성이 높다고 하겠다. 3. 4, 2 촉매 산소 기상 산화반응 촉매 산화반응의 거의 대부분이 이 범주에 속한다. 너무나 광범 위하여 공업적으로 잘 알려진 예를 표 3-7 에 나타내었다. 후술할 액상 산화반응과 같이 이 경우에 가장 큰 문제는 촉매의 피독, 열 화 등에 의한 비활성, 즉 촉매 표면에서 일어나는 반응의 해석이다. 이 경우 촉매 표면에서 일어나는 반응을 직접 관찰할 수 없기 때문 에 전부 간접적인 방법, 즉 분광학적 방법들을 쓸 수밖에 없다. 그 러므로 그 반응기구도 가정적이다. 불균일 촉매 존재하에서 일어나는 반응은 촉매의 물리적인 면에 서 보면 다음 과정을 따라서 일어난다. ® 반응물이 촉매 표면에 확산 ® 촉매 표면에 흡착 ® 촉매 표면에서 반응 ® 촉매 표면에서 반응물의 탈착 ® 반응물이 촉매 표면에서 떨어져나와 확산 촉매반응의 속도와 생성된 생성물의 성질은 물질 전달 속도에 크 게 영향을 받는다고 할 수 있다. 즉, 얼마나 빨리 반응물 분리가 촉매 표면에서 전달되고, 얼마나 빨리 생성물 분리가 제거되는가에 따라 영향을 받는다.
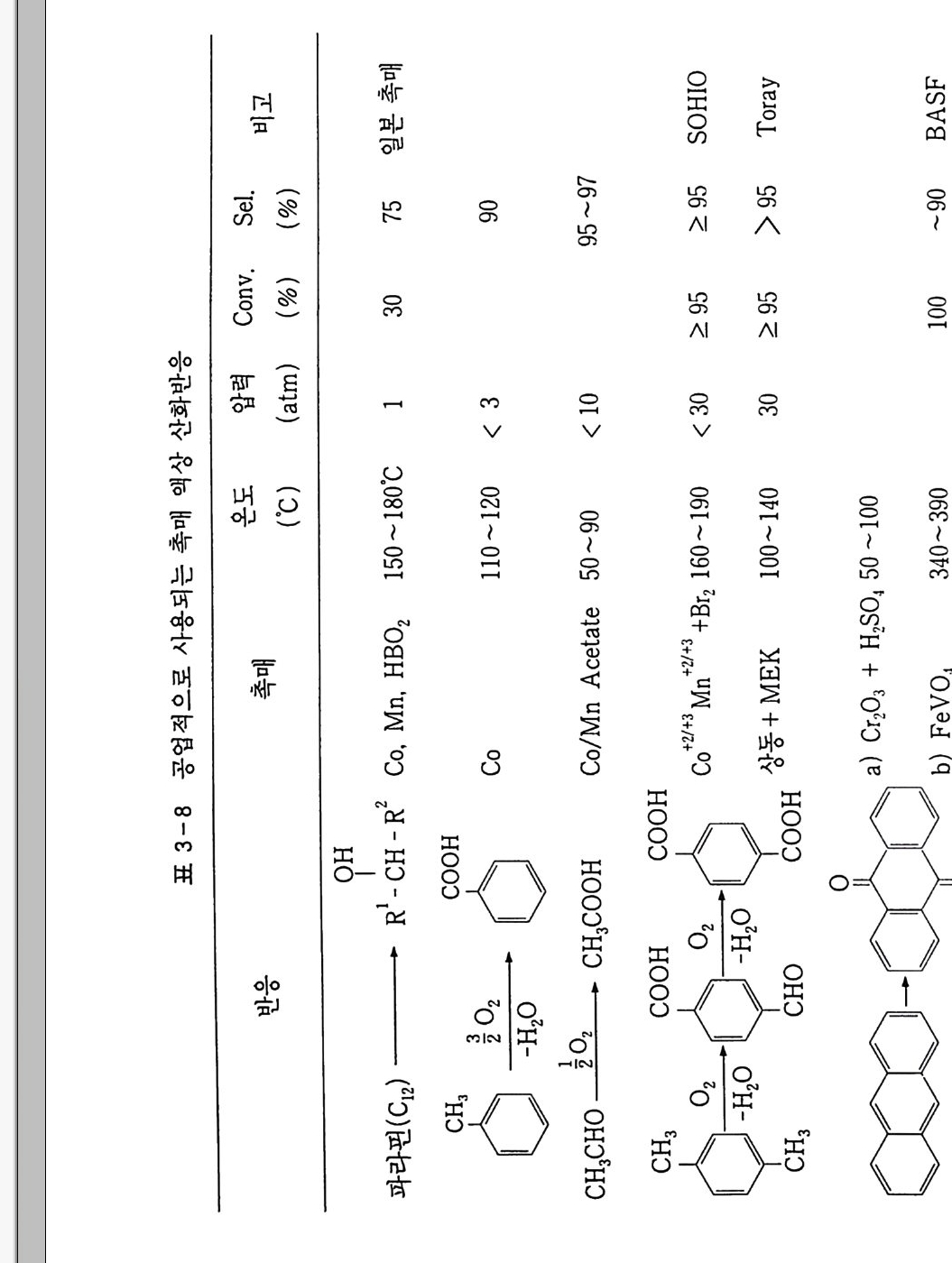
뎌띠 i。ui』〕([iO ]늦。( [UQ] {}。<。m ”<。。} { 역。디l。 럭띠mι 。ιEgx S훤,(-뼈 ·ωm니다>.][ (§))(ξ( at∞m∞ ‘。(。[l)。 1 (l!aP∞a.vf N'F)) 。∞M。l.f ' i) lζ。l∞。∞.fn' ) }어|∞l∞∞。빙∞ σl∞<@.f ' 〉 ) ∞m*tl{∞{Y} {{}} 혜칭{파빼*깐뎌메매해이P핸{꾀〈Z에*J양때버|배이Fe - K 어πM아{버 」에큼효。 l 」 。 μ()5움) :낙。‘좋u,。표z국z’iQ그>+1.。얘 3디‘、 N‘‘ -ι:u:d:。‘om+~νN연그i;i-*【υU훈umw:g‘NN。tN1: +1 I I디디 r I NrNr NN); : ‘’ - - u {{u r。\/ ν@.、-*럭←‘N영!이Nm|4+앞“”Q써|。Q£NN * $ Q 。llN~”>Q며엽’δN어+N퍼NφZι며U。Q잊”Q””Nl영a~+-。。E응。N J } I { QQz “여 ‘。i@i띠Y*i’띠ιu。”}’”N4}{Q。l”}{。NNu}}니+{ Z · { { }Q Q {|Q 。~팩o ~ ;、Q쉴며。여N헌t。。르。S>、 ( J) ∞*(∞l〕(。~。N+}、{、/、벼응.김-응]+다앓N”。N。+\N 4에 > | |/=∞μ。ω므X。응。\ <。Ni!}。Q{}{~(nN。써]b ( 예 r }O: { } -。띠어!않‘ι〔。。응〔。N~P
 회‘μ깅 ‘ E,픔써=「 (]∞ν{ωS ω【능그~그i -럭ι띠ω ιm,∞럭
회‘μ깅 ‘ E,픔써=「 (]∞ν{ωS ω【능그~그i -럭ι띠ω ιm,∞럭
 뎌 흐디。i}{ {mi」 ιm
뎌 흐디。i}{ {mi」 ιm
일반적으로 ®. ®. ©단계는 동시에 일어나는 것으로 생각되며. 확산과정보다 더 천천히 일어나서 화학적인 속도를 측정할 수 있다. 한편. 촉매의 화학적인 면에서 살펴보면 위에서 기술한 물질 전 달 또는 확산 문제를 제거시키든지. 무시한다 하더라도 불균일 촉 매 산화반응을 적절하게 기술한다는 것은 어려운 문제이다. 그 문 제 들을 Vo g e 는 다음과 같이 지 적 하고 있다 .1 6 4 ) ® 활성화 상태에서 고체 표면의 특성 ® 고체 표면 위의 산소 및 다른 종들의 확인과 각각 이루어진 반응 확인 ® 반응경로에서의 각 단계 ® 중간체의 구조와 에너지 등 이러한 문제들이 여러 학자들에 의하여 많이 규명되고 있음에도 불구하고 아직도 규명중이다. 지금까지의 연구 결과에 대해서는 문 헌과 책을 참고하기 바란다 .111 , 11 2, 1 64 ~1 6 8) 표 3-7. 3-8 을 보면 공업적으로 사용되는 촉매들을 귀금속 촉 매와 Cu-Fe 계 촉메 V 2 아 Mo03 촉매계, 3 종류로 나눌 수 있음 을 알 수 있다. 1) 산화에틸렌 산화에 틸 렌 ( Et hy le noxid e , Oxir a ne) 은 가장 간단한 환화 에 테 르 (e t her) 로서 환의 높은 응력 때문에 반응력이 강하다. 산화에틸 렌은 1859 년 War t z113) 가 에피클로히드린 (E pi chloroh y dr i ne) 에서 KOH 로 염화수소를 떼어냄으로써 얻었고 1914 년부터 공업화되었다. 1931 년 Le fo r t 114) 가 에틸렌을 공기/산소로 직접 산화시켜 제조
하는 방법을 발견한 이래 클로로히드린 (Chloroh y dr i ne) 공정은 점 점 사라져가고 있다. (1) 촉매 은 {A g)보다 더 좋은 촉매는 아직 발견하지 못했다. 은은 지지체 에다 7~20 % 함입시켜 사용한다. 지지체로서는 고온에서 처리한 고순도 (99 % 이상) Alum i n 으로 공정(p ore s i ze) 이 0.5~50µm 이 고 표면적은 <2m2/ g이 좋다 .115, 116) 표면적이 넓으면 반응성은 좋 지만 선택성이 낮아진다. 그리고 세슘 (Ces i um) 같은 알칼리, 알칼 리성 금속 같은 프로모터(p romo t er) 를 100~500m g /k g를 넣으면 더욱 효과적 이 다 .117) 선택성을 증가시키기 위해서는 반응가스에 염소를 첨가하는 것 이 좋다. 소위 금지제 (1, 2-D i chloroe t hane) 는 에틸렌을 탄산가스 와 물로 연소되는 것을 막는다. 이는 염소가스가 촉매 표면의 은 촉매를 균일하게 덮기 때문인 것으로 알려졌다 .118) (2) 촉매의 비활성화 근래의 촉매는 79~81 %까지, 최고로는 83 %까지 선택성을 나 타낸다. 이렇게 높은 선택도를 나타내는 촉매는 쉽게 열화되는데, 그 이유는 다음과 같이 추정한다 .119~12 1) ® 마모에 의한 먼지가 기공을 막음 ® 해로운 불순물의 축적으로 반응가스에 혼입 (예:반응가스 중의 유황) ® 응집현상에 의한 은 입자의 대형화로 은 입자의 불균일화 비활성화 촉매의 재생에 대하여는 알려져 있지 않다. 세슘의 메
탄올 용액으로 처리하는 방법 1 22) 이 있지만 재생이 불가능할 경우 교환해야 한댜 일반적으로 촉매의 수명은 2~5 년이댜 (3) 반응기구 촉매 표면에서는 다음과 같은 두 반응이 동시에 일어난다 (250 °C, 1.5 MPa) . CH2 = CH2 + ½ 02 ---ClH o2 - \ C H2 L1H = -106.7 kJ /m ol (42) CH2 = CH2 + ½ 02 一 2C02 + 2H2 0 .1H = -1323 kJ /m ol ( 43 ) 은(銀) 반응기구의 해명은 어렵지만 각 주장의 요건은 다음과 같 이 나눌 수 있댜 ® 산소원자의 반응 참여 ® 산소분자의 반응 참여 ® 아표면 (subsur fa ce) 산소 참여 이 중 산소분자의 반응 참여가 여러 면에서 설득력을 지닌다. 6Ag + 602 -― ► 6Ag • 02ads (44)
6Ag • 02a d s + 6H2 C = CH2 —- ( 45 ) 6H2 C -CH2 + 6Ag • Oad s \o/ 6Ag • Oa ds + H2 C = CH2 -6Ag + 2C02 + 2H2 0 (46) 반응식 (44) ~(46) 에서 보듯이 한 분자의 산소는 한 분자의 에 틸렌을 산화하여 한 분자 산화에틸렌을 형성한다. 한편. 산소원자 는 CO2 와 물을 형성시키며. 염소(금지제로부터)는 은 촉매 표면에 산소원자 흡착을 막는다. 그러므로 적당히 금지제로 도포된 은 촉 매 면이 분자산소를 흡착하여(식 44) 에틸렌과 반응, 산화에틸렌 을 제조한다(식 45). 또한 식 (45) 와 같이 탈착된 한 산소원자는 에틸렌과 반응하여 완전연소를 일으켜서 CO 2 와 물을 생성한다(식 46). 따라서 하나의 에틸렌 분자를 완전히 산화시키기 위해서는 6 산 소원자가 필요하며, 6E.O 분자가 생성되어야 한다. 2) 아크로레인 (1) 프로필렌 산화에 의한 아크로레인 제조 이중결합과 카르보닐기를 가지고 있는 아크로레인은 반응성이 강하여 중요한 중간체로 이용된다. 상업적으로는 1942 년 Deg u ssa 에 의하여 아세트알데히드와 포름알데히드를 축합시켜 얻었으나 최근 들어서는 프로필렌을 불균일 촉매상에서 기체반응으로 산화 시켜 얻는다 . 촉매는 처음에는 산화동으로 반응시켰는데 전환율이 20 % 정도 밖에 되지 않았다• 그 후 1957 년 SOHIO 에 의하여 Mo -Bi 산화
ωQ φU다·i } 어[N {Niy 띠N[ ∞[N ‘[nN N[∞ m[N 。m[ {여{ 여{N 여{어 v이{i φ ¢s‘ {‘E。」 。α.* [-@. N.∞∞ .α*여 ∞••. ∞[∞ mN@. α[{ } ∞.。• Nα。 ←∞。 αN.。 。i 때=빠뼈칭힘p{ m-‘-l띠φ임C-m〕{(F〉gi 」5。u3i럭』U므SuQmi .αh[.a{* 。m∞∞.。 aN∞。. @ m.∞m∞.∞ αω‘k .{∞{ .{∞a•{. 띠∞[*.• ∞.m∞{∞. 。.N∞am. ∞{.m.∞Nh .α•N∞.• ∞m∞영*. 비 럭 W빼{숨아써{ φ。。다다·u>a•i ‘§ .a∞∞ .aα∞ aα← 。.∞∞ ∞∞m .•aα ∞‘nm •α.m 。。α {@ @ α。.* .*@*v 킹
며때숨。{|∞ m。*u다ν;U φigωEmi·E3 。U (]여。 Sm이 。N여 。N여 ∞N이 。mm m여m 여。Ng 띠m어 mm。 [이m 이。N 벼이 >]m;다Q。i。eig。gm m Q다()벼ω〉다ω잉{。〕다어u야.여 응ωω£;.iι。i띠。:zN 。ι。입N띠;-물로 약 60 % 전환율과 72 %의 선택률을 얻었댜 최근 각 회사별로 Mo-B i를 주촉매로 하여 3~5 개의 금속을 첨 가시킨 다성분 촉매를 개발하였다. 이 다성분 구성은 촉매환원 c y cle( 선택적인 제품 형성)과 촉매재산화 c y cle( 라티스 산소 재생) 을 얻기 위함이다. 아크로레인의 수율은 화학적 조성뿐만 아니라 촉매의 모양 다공 성, 다공성 분포, 그리고 반응조건에 의존한다. 현재의 촉매는 5~8 vol % 프로필렌을 반응온도 300~400 °C, 접촉시간 1.5 ~3.5 s 조 건 에서 전환율 90~95 %, 수율 80 % 정도이다. 그리고 부산물로 5~10 %의 아크릴산을 포함한다. 촉매의 수명은 2~4 년이고 그 후 에는 교환한다. 표 3-9 에 지금까지 문헌상에 알려진 촉매와 그 반 응결과를 나타내었다. (2) 프로판 산화에 의한 아크로레인 제조 프로필렌은 이중결합의 관능기를 가지고 있기 때문에 반응에 쉽 게 참여한다. 그러나 프로판은 포화 탄화수소이기 때문에 반응성이 대단히 낮다. 반면 프로필렌에 비해 자원적으로 풍부하며, NCC 에서도 많은 양이 부생되므로 이의 용도 개발은 당연한데, 문제는 촉매 사용온 도에서의 프로판에서 프로필렌으로의 탈수소 반응 전환율이 낮다 는점이다. Y. S. Yoon 과 Y. Morooka13s. 1 36) 둥은 Co -M o 촉매 를 사용하며 프로판 탈수소반응을 많이 연구하였다. 그들의 결과에 의하면 소성 온도 55o •c 에 영향을 받고, 반응온도 45o ·c 에서 19 %의 전환율 과 60% 의 선택률을 나타내었다.
3) 아크릴산 지금까지 아크릴산은 아크릴로니트릴을 가수분해 13 7) 시켜서, 그리 고 Rep pe1 38 ) 반응을 통하여 만들어 왔으나, 최근 들어서는 프로필 렌을 불균일 촉매 존재하에서 앞에서 설명한 아크로레인을 경과하 여 제조하는 방법이 주류를 이루고 있다. 그리고 아크릴산과 이의 메틸-, 에틸-, n- 부틸-, 2- 에틸헥산 에스테르는 폴리머 제조에 사용되고 있다. 아크릴산은 무색 액체로서 물리 상수비점 141 .0 ·c. 융점 13.5 ·c, 그리고 밀도는 1.06 0( 10 ·c). 1.04 0(30 ·c). 1.01 s(so ° C)g /cm 2 이댜 그림 3-14 에 아크릴산 수용액의 비중. 표 3-10 에 각 용매에서 의 빙점을 나타내었다.
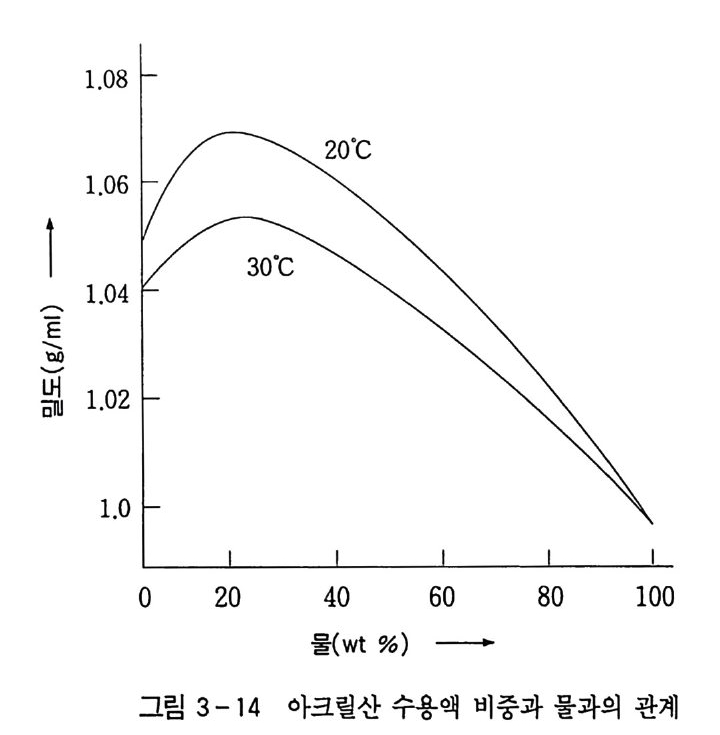 1.08
1.08
표 3-10 아크릴산 혼합물의 어는접 (A : 빙초산. 8 : 물) 시스wt템 % A. 어는·c 점 시스wt템 % B. 어는·c 점 아세트산 물 。 13.5 。 13.5 10 7.5 5 5.5 20 0.7 10 1.0 40 -14.1 20 -5.5 50 -23.5 30 -10.3 50.2 -23.8 37 -12.5 60 -13.4 40 -12.0 80 3.7 60 -8.0 100 16.6 80 -4.0 100 。
(1) 단계별 반응 프로필렌을 산화하여 아크릴산을 제조하는 것은 1950 년대부터 시작되었는데, 이 공정은 1 단계 반응과 2 단계 반응으로 나누어졌다. 1 단계 반응 CH2 = CHCH3 + 3h02 一 CH = CH2 CO OH L1H = -59 4.9 kJ /m ol ( 47 ) 2 단계 반응 CH2 = CHCH3 + 02 ― ► CH = CHCHO AH = 一 340.8 kJ /m ol (48)
CH2 = CHCHO + ½ 02 一 CH2 = CHCOOH L1H = -254.1 kJ /m ol (49) 1 단계 반응에서 최고의 수율은 50~60 %137 , 1 39 ~1 4 이며. 이때의 촉매는 Mo 산화물을 주금속으로 한 텔루륨 (Tellur i um) 산화물을 소량 넣은 것이고. 그 수명은 길지 않다. 그 이유는 텔루륨 산화물 의 승화성 때문이다. 2 단계 반응은 물론 다른 촉매와 반응조건을 필요로 한다. 프로필 렌을 산화한 경우 첫 단계 반응은 전술한 아크로레인 제조 촉매이 다 . 아크로레인과 아크릴산의 수율은 85 % 이상이다. 아크로레인에서 아크릴산으로의 제 2 단계의 초기 촉매는 Co- Mo 산화물 M 2) 이었으나 높은 반응온도에서 70mol% 을 넘지 못했 댜 근래에는 Mo:V=l:1 원자비를 주로 한 다성분 촉매를 사용 하였다. 다성분 촉매의 조성은 표 3-11 에 나타내었다 . 4) 말레산 무수물 제조 말레산 무수물은 이중결합을 가진 이염기성 산이므로 축합 및 부 가반응이 사용된다. 불포화 수지, 폴리에스테르. 래커 , 가소제, 공중합체, 그리고 윤 활유에 사용되며, 최근에는 수소화에 의해 1,4- 부탄디올과 THF 제조의 중간체로 쓰이고 있다 . 폴리에스테르와 알킬 수지는 특히 섬유유리 보강 플라스틱에 사 용된다. 이러한 말레산 무수물은 다음과 같은 여러 가지 제조방법 이 알려져 있다.
<」Q¢』ωφ}ωω 여*{v [**Y 띠*{ i{{v} i‘n{v 영[∞ {αiv μ
(1) 벤젠 산화에 의한 말레산 무수물 제조
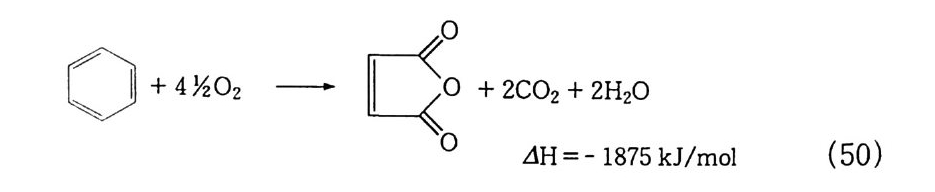 。
。
식 (50) 에서 보는 바와 같이 높은 발열반응이므로 열 제거가 가 장 큰 문제이다 . 그러므로 수천 수만 개의 직경 25mmx 길이 3.6 m 인 관에 촉매를 넣어 염융으로 열을 처리하고 , 이것을 열교환기 에 보내 고압증기를 생산한다. 고온이므로 반응온도 조절이 중요하 댜 촉매는 \O s -Mo0 3 / 담체를 주로 사용하며, 수명은 약 4 년이 다. 100 kg 벤젠을 사용하며 120 kg MA 를 생산한댜 (2) C, 1 탄화수소 산화에 의한 MA 제조 벤젠을 원료로 사용할 경우 6 개의 탄소에서 2 개의 탄소를 잃게 된다 . 그러므로 같은 탄소를 갖는 c 4 - 유분으로부터 얻고자 하는 노력이 많이 이루어졌다. C~H1 。 + 3 ½02 一 MA + 4H2 0 Aft= -1260 kJ/ m ol (51) 부탄법은 1974 년 Monsanto 사에 의하여 세계 최초로 공업화되 었는데, 이때는 고정상을 사용했다. 부탄의 유동상 반응기술 개발 154a) 은 촉매의 강도 요구와 scale u p의 곤란 등으로 10 년 후인 1980 년대 후반에야 공업화되었다. 표 3-12 에 현재 가동되고 있는 중요한 부탄법 공정을 실었다 .154b}
표 3-12 상업화된 공정(부탄으로부터 MAN 제조) Industr i a l tec hnolog ies for th e sy n th esis of maleic anhy d rid e Process, lice nsor co. Ty pe of reac tor Produc t rec o very ALMA ( Alusuis s e-Lummus) Fluid - bed Anhy d rous Alusuis s e Itali a Fix e d-bed Anhy d rous or aq u eous Mi tsu bis h i Chem. Fluid - bed Aq u eous Monsant o Fix e d-bed Anhy d rous Seie n ti fic Desig n Fix e d-bed Aq u eous DuPont Ci rc ulati ng Aq u eous flui d bed- ri s e r reac tor Sohio - UCB Fluid - bed Aq u eous
®촉매 현재 세계에서 사용하고 있는 부탄 산화반응은 기본적으로 Di- vanady l pyro nic acid , (VO) 2 P 오를 활성성분으로 하고 있다. (VO)2P201 의 구조는 단결정 X 선 회절에 의하여 그림 3-15 와 같 이 2 개의 V 원자가 능을 공유하는 8 면체로 되어 있다. (V0)2P 2 01 의 반응성 활성은 조제방법에 따라 다른데, 일반적으 로 유기용매법이 수용매법보다 좋다고 한댜 선택성은 유기용매법이 낫다 155) 는 설과 수용매법이 낫다 1 56, 1 5 ” 는 설 두 가지가 있댜 국내에서도 C 4 로부터 MA를 제조하려는 연구는 오래 전부터 연 구되었다. 김재승 158) 등은 V-P/Ti0 2 촉매로 1 _부텐 산화시켰다. 이 결과를 표 3 가 3 과 그림 3-16 에 나타내었댜 MA 에 대한 선 택도는 부타디엔울 중간체로 하고 반응온도 270~31o ·c 범위에서 가가장좋았다.
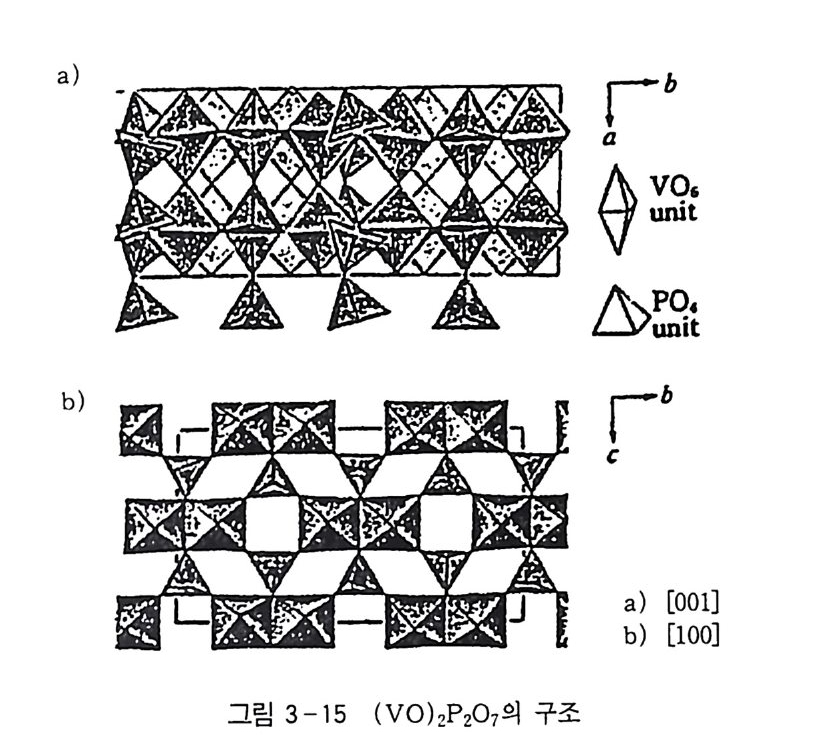 a) 广 b
a) 广 b
표 3-13 부텐산화에 의한 제품 분포 Reac tion tem p . 『 C J 270 310 Conversio n [ % J 50 81 Selec tivi t y [ % ] 57 63 Produc t [%] buta d ie n e 14 7 maleic anhy d rid e 27 51 carbon oxid e s 7 21 fur an tra ce tra ce aceta l dehy d e tra ce trac e aceti c aci d 1 1
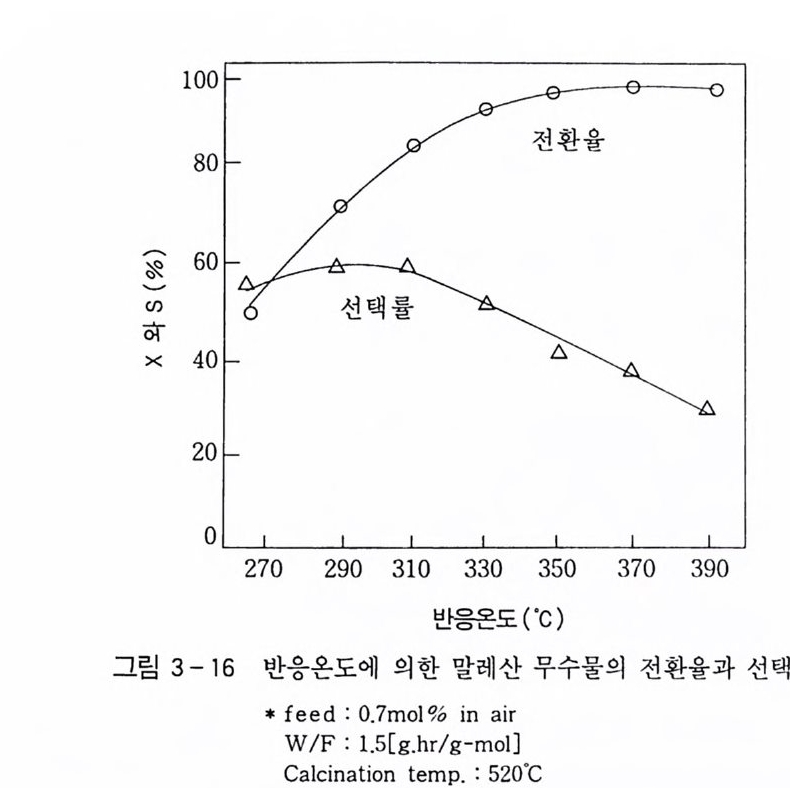 100
100
홍석인 등 159) 은 바나듐계 촉매를 제조하여(표 3-14 참조) 부탄 과 부텐으로부터 말레산 무수물을 제조하는 선택 산화반응에서 촉 매 제조시 사용되는 환원제와 첨가제인 비스무스의 영향을 살펴본 결과, 그림 3-18 과 3-19 에서 보는 바와 같이 환원제로 이소프로 판올과 벤질 알코올 혼합물이 사용된 촉매의 선택도가 가장 높았다 고보고하였다. 촉매에 비스무스를 첨가한 경우, 그림 3-19 에서처럼 부탄의 산 화반응에서는 VPO 촉매에 비하여 전환율과 말레산 무수물에 대 한 선택도가 감소되었으나, 부텐의 반응에서는 말레산 무수물에 대 한 선택도가 증가되었다(그림 3-20 참조). UV 확산 반사분광 분 석 결과, 부탄의 경우는 vs+ 의 양에 관계없이 vs+ 에서 02- 로의 전 자 전이 에너지가 상대적으로 높은 비스무스를 첨가하지 않은 촉매
표 3-14 각각 다른 환원제와 금속 조성에 의해 제조된 촉매의 비표면적 Cata l ys ts Reducin g Comp o sit ion s Sup po rts Sp e cif ic ag e nts surfa c e area V P Bi (m2/g) C41 HCI 1 1.1 6.4 3 C42 이소프로파놀 1 1.1 14.8 + 벤질 알코올 C43 이소부탄올 1 1.1 25.1 + 벤질 알코올 C44 옥살산 1 1.1 Ti 02 5.72 C45 이소프로파놀 1 1.1 0.2 9,98 + 벤질 알코올 C46 이소부탄올 1 1.1 0.2 9.95 +벤질 알코올 C47 이소부탄올 1 1.1 0.2 Ti 02 4.32 + 벤질 알코올
상에서 말레산 무수물의 선택도가 높게 나타났으며, 부텐의 반응시 에는 전자 전이 에너지의 크기보다는 활성점인 V5+ 의 양이 많은 비스무스를 첨가한 촉매를 사용한 경우에 선택도가 더욱 높게 나타 났다. x- 선 회절 분석 결과 비스무스가 첨가된 촉매에서 p-V OP04 결정이 크게 성장되었으며, p-V OP04 결정이 클수록 부텐에서 말 레산 무수물에로의 선택도가 높음을 알 수 있었다. 또한 (V0)2P201 결정에서 [02 0] 평면의 [20 사 평면에 대한 상 대적인 비율인 morph olog ica l fact or 와 말레산 무수물의 최대 선 택도도 서로 비례함을 알 수 있었다. 바나듐의 평균 원자가는 비스
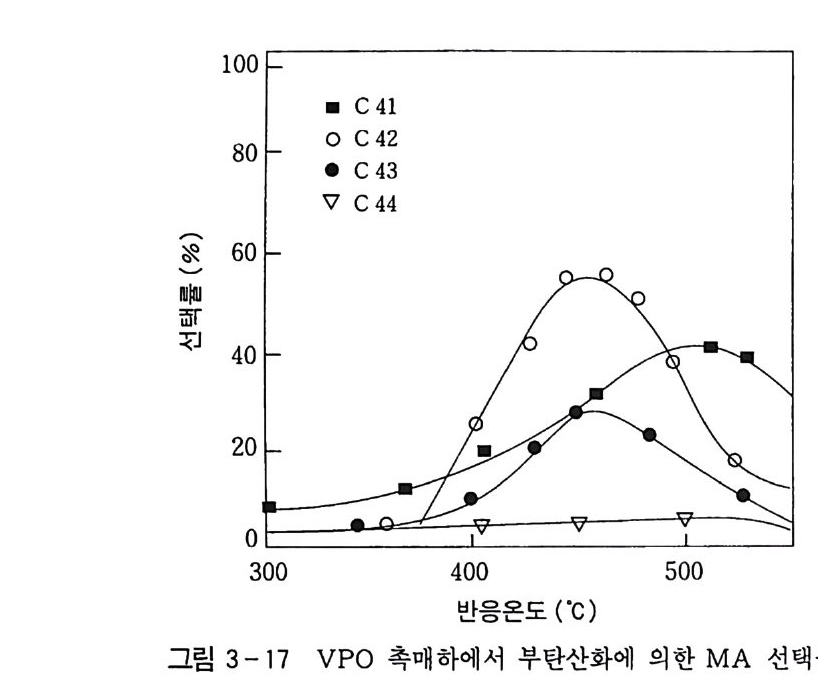 100
100
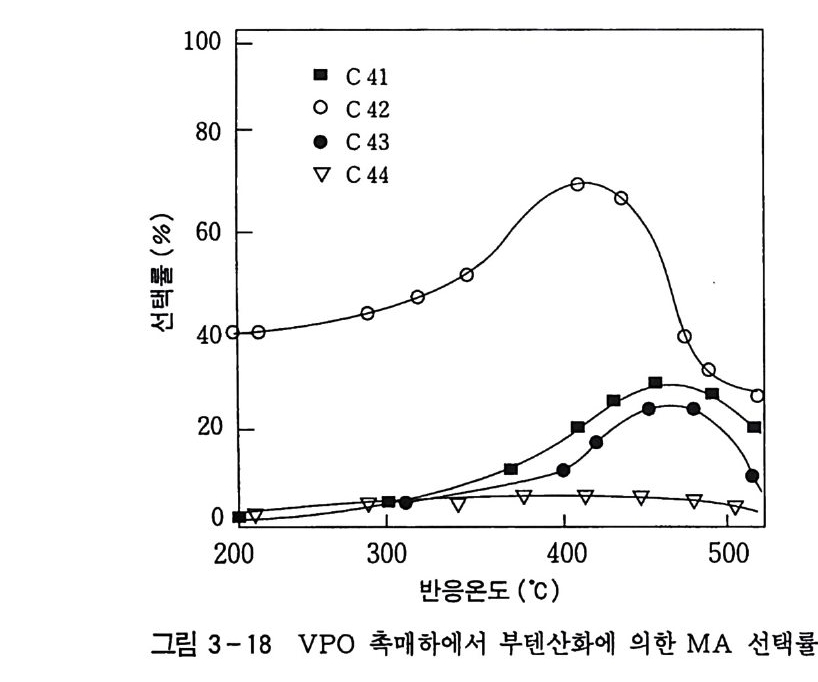 100
100
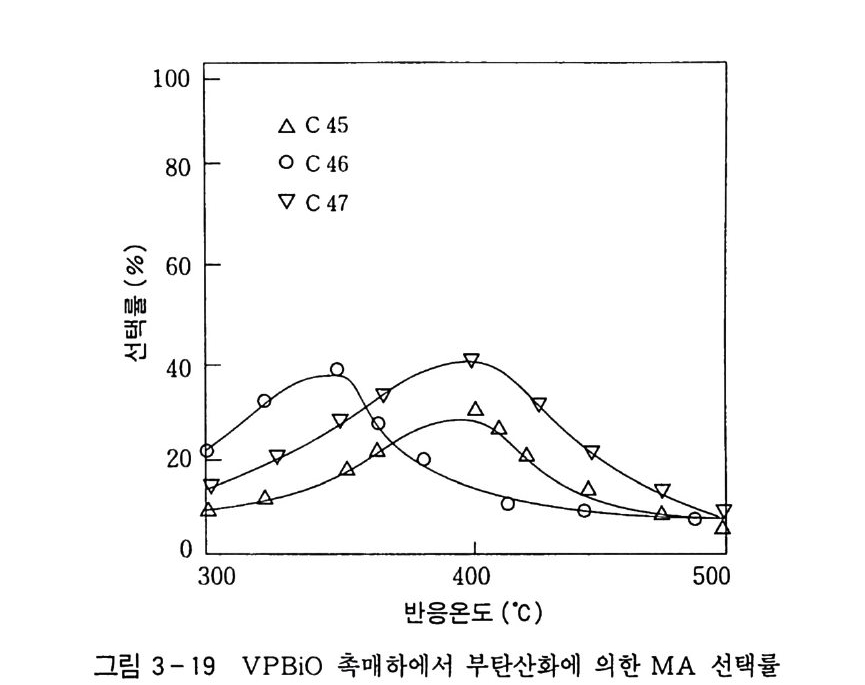 18000 [| 'v C 47
18000 [| 'v C 47
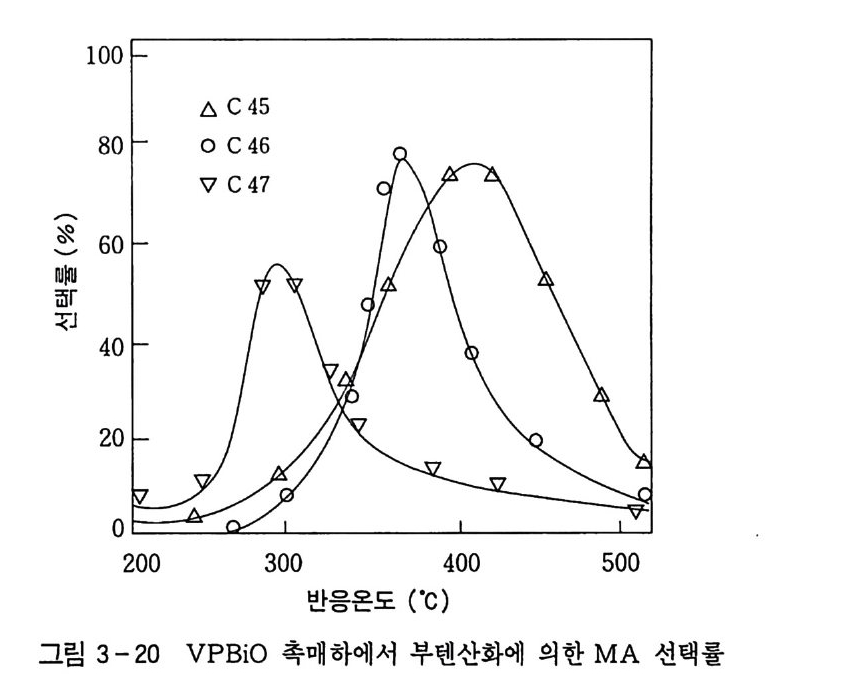 100
100
무스가 첨가된 촉매에서 상대적으로 높았는데, 이는 평균 원자가가 활성점인 /3- VOP01 결정의 크기와 서로 비례하기 때문이댜 따라 서 평균원자가가 큰 비스무스 촉매가 말레산 무수물의 선택도도 높 게 나타남을 알 수 있었다. (3) C5 유분으로부터 말레산 무수물 제조 앞에서 벤젠과 c4 유분으로부터의 말레산 무수물 제조에 대하여 기술했댜 벤젠은 유용한 화학원료이며 또한 탄소 2 개가 CO 2 로 소 실된다. 한편 c4 유분의 각 성분도 분리과정을 통해 여러 가지 원료로 사 용되고 있다. 그러나 c4 유분은 나프타 크래커에서 생산되는 양이 한정되어 있음을 감안할 때 G와 C6 의 중간 부분인 C5 유분의 용도 개발은 중요하다. C5 유분에서 말레산 무수물을 제조하고자 하는 연구가 많이 수행 되었다. Hon i cke 과 Gr i esbaum160) 은 V-Mo-P 촉매를 사용하여 시클로펜타디엔과 여러 펜텐울 산화시켜 말레산 무수물이 주산물 로, 그리고 프탈산 무수물이 부산물로 생성됨을 발표했으며, 그 생 성 반응기구도 제안하였다. 한편 전기원, 이규완 161) 등은 V:Mo:P 의 여러 가지 촉매에 프 로모터로서 Ag을 극소량 (V20s:A g =1:0.0003) 넣은 촉매를 제조 하여 그 성능을 조사해 보았다. 그 결과 지지체로 ana t ase 형 T i 02 를 사용하는 경우 알루미늄보 다 A g의 분산도가 좋으며 MA/MA+PA 선택도도 좋은 것으로 나타났다. A g의 양을 변화시키면서 얻은 반응성 MA+PA 수율을 그림 3-21 과 3-22 에 나타내었다. 한편, 국내에서는 다른 학과에 의해서도 연구되었다 .16 2. 163) 홍석인 162) 등은 시클로펜탄과 시클로펜타디엔을 원료로 사용하여
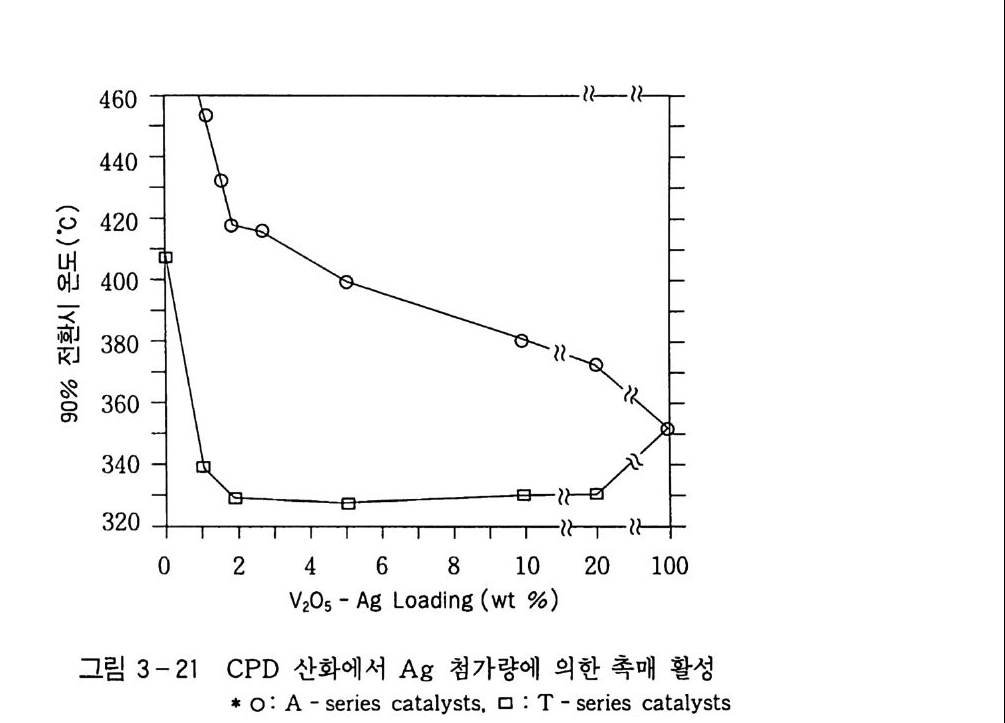 460 II— II- ,-
460 II— II- ,-
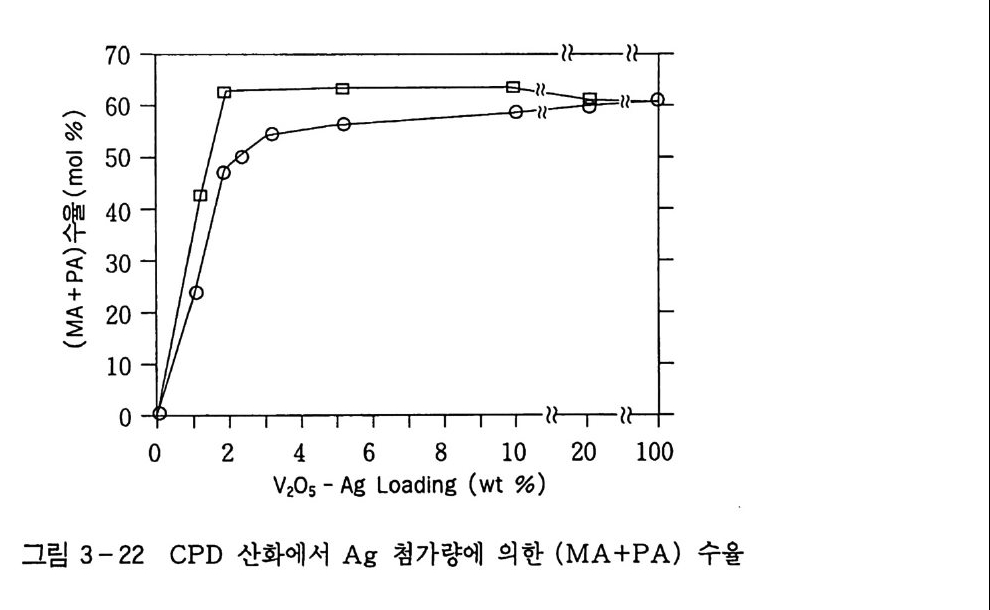 ..-.. 7600 도II II Il
..-.. 7600 도II II Il
표 3-15 제조한 유리 촉매의 조성과 물성 Cata l ys t Batc h comp o sit ion ( mo! % ) tAem nnp eea rlaltiun rg e Surfa c e area v2°5 P2° 5 B2°5 (°C ) (mz /g ) Pl0-450 90 10 450 1.11 P20-450 80 20 450 1.24 P30-450 70 30 450 P40-450 60 40 450 1.46 P13B3-410 84 13 3 410 9.4 Pl3B3-450 84 13 3 450 10.2 P13B3-450(0.5) 84 13 3 450(0.5 ) * 5.6 * 온도승은 : O.5 ° C/ 분
표 3-15 에서 보는 바와 같이 바나듐계 이성분계 결정질 유리를 제조, 촉매로 사용하여 다음과 같은 결론을 얻었다. [1] 급랭된 모유리는 무정형이며 200 °C 로 1 차 재가열한 후에는 결정성장이 관찰되지 않았다. 2 차 재가열을 45o ·c 에서 시행한 후 에 뚜렷한 결정이 성장되었고, 결정은 분상을 이루어 1 % HCl 로 에칭하였을 때 화학적 저항이 약한 상이 쉽게 용출되어 다공성이 되었다(표 3-16 참조). [2] V20s 一 P 요 이성분계 촉매의 경우 P205 의 첨가량이 증가할 수록 V”의 비율이 증가하였으며 이에 따라 전환율은 감소한 반면 선택률은 그림 3 국 ~5 와 3 -26 에서 보는 바와 같이 증가하였다. [3] V20s-P 요 -B 요 삼성분계 촉매에서 Cs 유분 부분 산화반
표 3-16 이원 유리 촉매로 MCP, CP, CPD 의 산화반응 온도에 의한 결과 Pl0-450 P20-450 P40-450 반응온도 MCP CP CPD MCP CP CPD MCP CP (°C ) Cv. Se. Cv. Se. Cv. Se. Cv. Se. Cv. Se. Cv. Se. Cv. Se. Cv. Se. 250 5912 51l51l1810 2598 122352513752 2l51232 511 5 1155 200513 081830227810 3720 246 46 5 300 18 020 252 55852480 0 0545 350 1012802222 2 0 058 0 55 。 400 88 65 450 92 78
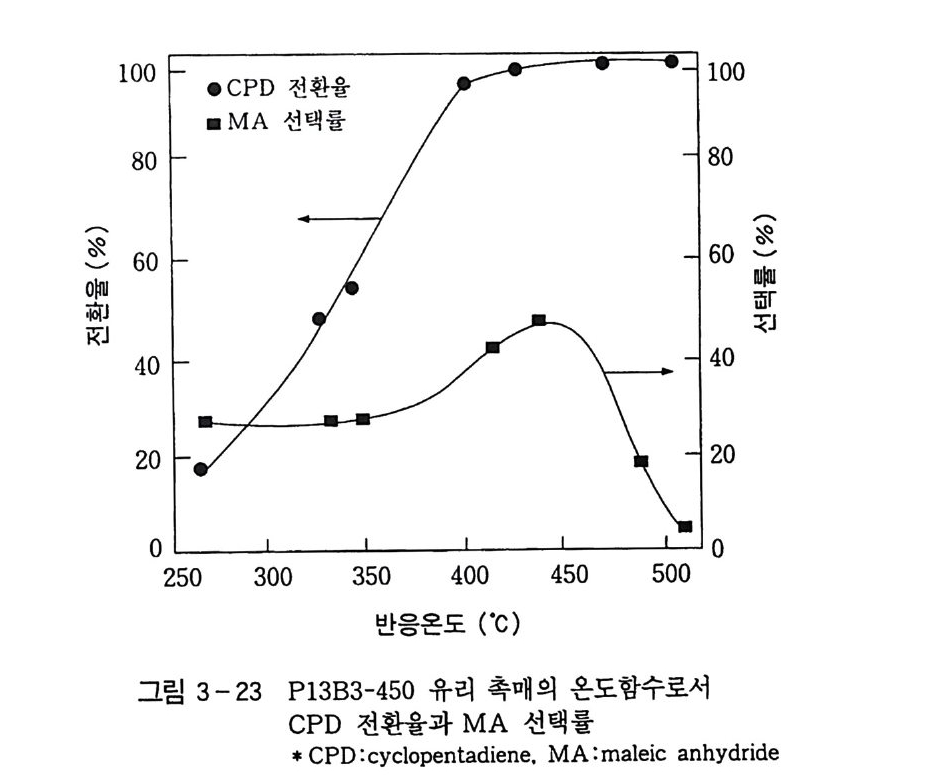 100 •CPD 전 환율 100
100 •CPD 전 환율 100
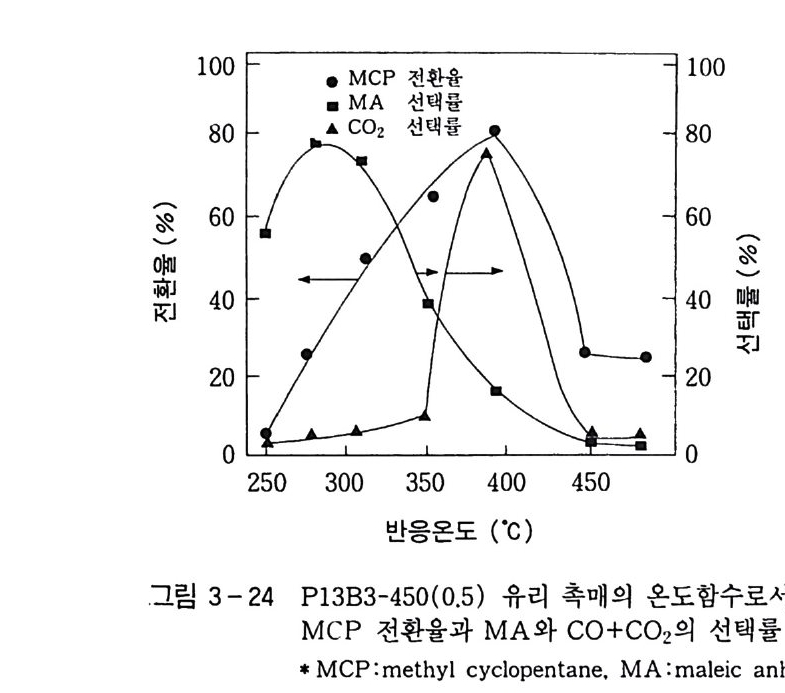 100 100
100 100
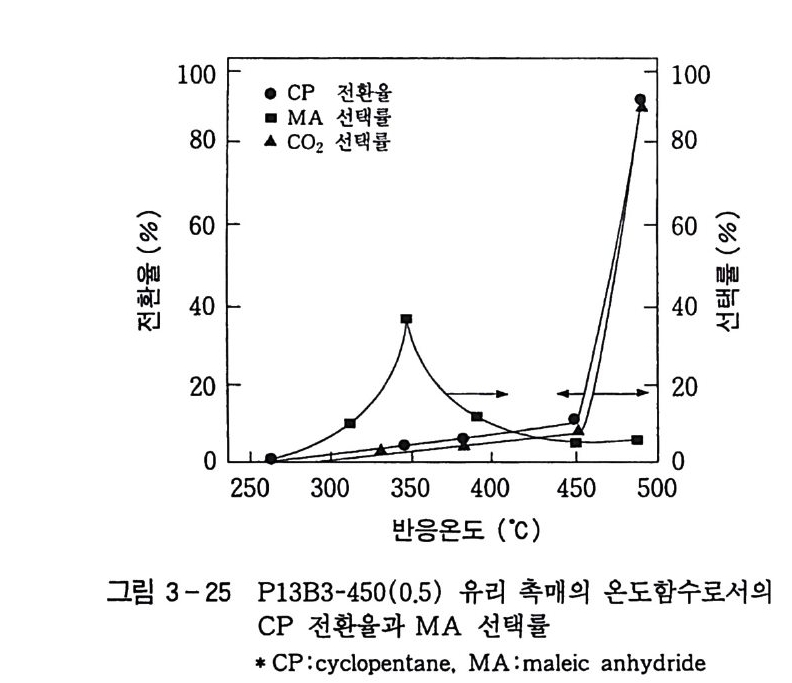 100 • CP 전환율 100
100 • CP 전환율 100
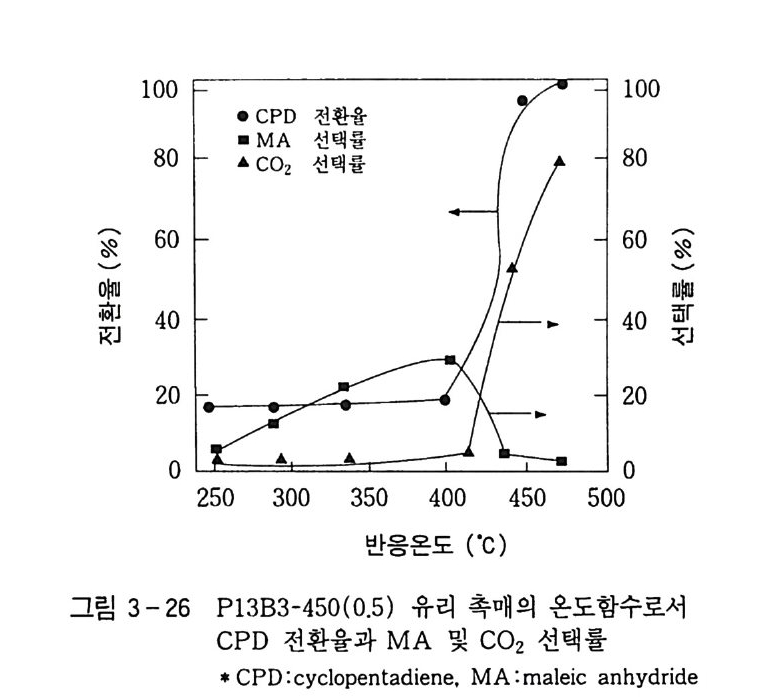 100 100
100 100
응에 대한 촉매활성이 우수하였고, 선택률이 높아 MCP 의 경우 18 %(409 °C), CP 의 경우 1o %(320 ·c). CPD 의 경우 48.3 %(453 ·c) 의 말레산 무수물의 수득률을 보였다. [4] V20s-P 요 -B 요 결정질 유리의 경우 400~450 °C 사이에 서 전환율이 급격히 상승하고 선택률이 저하되는 전이현상을 보였 다. 이는 직류 전기전도도 측정 결과에 비추어 보면 400~45o·c 부 근에서 가역적인 상변화가 일어나며 전도도가 급격히 감소하는데, 이는 V 오의 결정상의 불안정성에 기인하는 결과로 볼 수 있다.
참고문헌 1) N. Noji ri and M. M iso no, Ap pl. Cat. A. ge nenral, 93, 103~ 122(1 9 93) . 2) 千魚眞信, 市川勝, 『均一觸媒와 不均一觸媒入門』, 4, 丸善(1 983) . 3 ) a) F. C. Phil ips, J. Am. Chem. Soc. , 16, 255 ( 1894) . b) J. Sm idt et al., Ang ew . Chem., 71, 176(1 9 59) . 4) J. Sm idt et al., ibi d ., 74, 93 (19 62) . 5 ) 橋本春吉, 『化學과 工業』 , 20, 1077 (19 67 ) . 6) K. Tatu s umi and M. Tsuts u i, in M. Tsuts u i ed., Funda-menta l Research in Homog en eous Cata l ys i s , 53~72, Plenum Publish in g Co., New York(1 9 79). 7) V. D. Luedecke, Adip ic Acid , in J. J. Mckett a, W. A. Cunnin g ha m, Eds., Encyc l op e dia of Chemi ca l Processin g & Desin , 2, 128~146, Marcel Dekker, New York( 1977) . 8) I. V. Berezin , E. T. Den iso v and N. M. Emanuel, The Oxid a ti on of Cy cl ohexane, Perga m on, New York ( 1966 ) . 9) H. J. Boonsta and P. Zwi et e r in g, Chem. Ind., 2309( 1966) . 10) S. A. M 沮 er, Chem. Proc. Eng ., 50, 63(Ju n e, 1969). 11 ) M. T. Musser, Cy cl ohexanol and Cy cl ohexanone, in W. Gerha rtz, Eds., Ullmann's Ency lo p e dia of Industr i a l Chemi st r y, vol. 48, 217~226, VCH Verlags g e s ellscha ft( 1987) . 12) M. Costa nti, N. Crenne, M. Jou ff et and J. Nouvel, U. S. Pate n t 3, 923, 895(1 9 75) 13) J. C. Brun ie, N. Crenne, U. S. Pate n t 3, 925, 316:U . S. Pate n t 3, 927, 105(1 9 75). 14) J. L. Russell, U. S. Pate n t 3, 665028(1 9 72) .
15) J. L. Russell, ibi d . , 9 32, 513(1 9 76). 16) Eur. Chem. News, 15, 22(March 29, 1969) . 17) J. Alagy , L. Asselin ea u, C. Busson and B. Cha, French Pat e nt 1, 573, 834(1 9 69). 18) R. L. Marcell, U. S. Pate n t 3, 317, 614(1 9 67) . 19) A. N. Bashk irov , Dok/. Akad. Nauk. SSSR., 1 18, 149(1 9 58) . 20) P. L . Won, V. A. Its k ovic h , V. M. Pote k h in, Neft ekh i- m i ya, 20( 4) , 568~572(1 9 80) . 21 ) A. V. Ni ki t in, V. V. Kaspa r ov, V. L. Rubail o and A. B. Gag arina , Izv . Akad. Nauk SSSR, Ser. Khim . , 547 ( 1975 ) . 22) N. M. Emanuel, Z. K. Maiz u s and I. P. Sk ibi d a , Ang ew . Chem. Int. Ed., 8, 97( 1969) . 23 ) G. F. Pusta r nakova, V. M. Soli an ik o v and E. T. Den iso v, Izv. A kad. Nauk SSSR. Ser. Khim . , 547 (19 75 ) . 24) G. A. Russell, J. Am. Chem. Soc., 79, 3871 (19 57) . 25 ) R. E. Kellog g, J. Am. Chem. Soc. , 91, 5433 ( 1969 ) . 26 ) R. F. Vassile v , Prag. React. Kin e t. , 4, 305 ( 1967 ) . 27) a) C. A. Tolman, J. D. Druli ne r, M. J. Napp a and N. Herron, Alkane Oxid a ti on Stu d ie s , in Du Pont' s Centr a l Research Depa rtm ent, in C. b) L. Hi ll, Ed., Acti va ti on and Funti on ali za ti on of A lkanes, Wi le y -Inte r scie n ce, 303~360, New York(1 9 89) . 28) J. D. Drulin e r, L. D. Greller and E. Wasserman, J. Phys . Chem., 95( 4) , 1519~1521 (19 91 ) . 29) J. P. M. Bonna rt, U. S. Pate n t 3 , 510, 526(1 9 70). 30) N. Ri eb er, R. Platz , W. Fuchs, J. Sta b enow, G. Herrmann, H. J. Wi lfi n g er and H. Hellbach, Europ ea n Pat e nt 31, 113(1 9 81 ) .
31 ) H. A. Sorge n ti and S. N. Rudn ick , U. S. Pate n t 3 , 949, 004( 1976). 32) C. A. Tolman, J. D. Drulin e r, P. J. Krusic , M. J. Napp a , W. C. Seid e l, I. D. Wi lliam s and S. D. Itte l , J. Mo!. Cata ! . 48, 129( 1988) . 33 ) A. Yash im a, T. Mats u da, T. Sato , K. Sakai and M. Takahashi, Eur. Pate n t 164, 463 ( 1985 ) . 34) P. E. Ellis , J. E. Ly on s and H. K. Meye r s, U. S. Pate n ts 4, 895, 682 & 4, 895680( 1990) . 35 ) T. Adams, U. S. Pate n t 3, 404, 185 ( 1968 ) . 36) W. J. Barnett , D. L. Schm itt and J. 0. Whit e, U. S. Pate n t 3, 987, 100(1 9 76) . 37 ) A. M. Sy ro ezhko, V. A. Proskurya kov, Mechanis m of Cata l yt ic Decomp o sit ion of Cy c lohexy l Hy d rop e roxid e , in A. I. Rakhim ov, Ed., Org. Peroksid y Homoli ticl1 eskie Reakts . lkh. Uchasti em , p.5 1 ( 1989) . 38) R. V. Kucher, A. P. Pokuts a , V. I. Tim okhin and T. B. Cherny ak , Dolk. Akad. Nauk SSSR. , 313 ( 2) , 373~376 ( 1990 ) . 39) M. S. Furman and I. L. Arest -Y akubovic h , V. V. Lipe s, F. A. Geberge r , L. E. Mi tau er and G. Z. Lip k in a , ZH. Khim . , 15B, 1175 (19 75) . 40) W. 0. Bry an , U. S. Pate n t 4, 238, 415 (19 80) . 41 ) W. W. Croch and J. C. Hilly e r , U. S. Pate n t 2, 931, 834 ( 1960) . 42) J. P. M. Bonna rt, Y. Bonnet and P. P. M. Rey, U. S. Pate n t 3, 557, 215(1 9 71 ). 43 ) C. G. M. van der Moesdij k and A. M. J. Thomas, U. S. Pate n t 3, 927, 108 (19 75 ) . 44) A. M. J. Thomas and C. G. M. Vande Moesdij k, U. S. Pate n t 3, 927, 108(1 9 75) .
45 ) J. Nouvel, U. S. Pate n t 3, 694, 511 (19 72 ) . 46) J. D. Drulin e r, J. Org. Chem., 43, 2069(1 9 78). 47) K. Tanaka, Chem. Tech ., 555(1 9 74). 48) T. Alagy , P. Trambouze and H. van Landeg h em, ind . Eng. Chem. Proc. Des. Dev. , 13, 317 (19 74) . 49) A. K. Suresh, T. Sri dh ar and 0. E. Pott er , AIChE Jo urnal, 34( 1) , 55, 69, 81 (19 89) . 50) A. K. Suresh, T. Srid h ar and 0. E. Pott er , A/ChE Jo urnal, 36n( 1) , 137~140(1 9 90) . 51 ) D. D. Davis , Adip ic Acid in Ullmann's Ency c lop e dia of Industr ia l Chemi str y , 5th Ed., vol. Al, VCH Verlag s- ge s ellsha ft, 269~278(1 9 85) . 52) A. F. Li nd say, Chem. Eng . Sc i., 3(Sp e cia l Sup ple ment) , 78~93 (19 54) . 53) W. J. van Asselt a nd D. W. van Krevelen, Chem. Eng. Sc i., 18, 471~483 (19 63) . 54) W. J. van Asselt a nd D. W. van Krevelen, Reel. Trav. Chim . , 82, 51~56, 429~437, 438~449(1 9 63) . 55) H. Godt and J. Qu in n , J. Am. Chem. Soc., 78, 1461~1464 (19 56) . 56 ) I. Y. Luby an it sk i i, R. Mina ti and M. Furman, Russ. J. Phys . Chem. , 32, 294~297(1 9 62). 57) Ya. I. Luby an it sk i i, Zh. Prikl . Khim . , (Lenin g ra d) 36, 819~823 (19 63) . 58) M. H. Thie m ens and W. C. Trog le r, Sc ien ce(4996), 251, 932~934 (19 91 ) . 59 ) K. S. McMahon, Acetic A cid , in J. J. McKetta , W. A.Cunningh am, Eds., Ency c lope dia of C hemi ca l Processin g andDesig n , vol, 1, 216,
240, 258, New York( 1976) . 60 ) R. P. Low ry and A. Ag ui lo , Hy dr ocarbon Proc. , 103 ( Nov. 1974 ) . 61) F. Broic h , H. Hofe r mann, W. Hunsmann and H. Sim mrock, Erdol Kohle -Erdga s Petr o chemi e, 16, 284 ( 1963 ) . 62) N. Cox, U. S. Pate n t 3, 196, 182( 1965) to Unio n Carbid e . 63 ) J. G. D. Schulz and R. Seeki rch er, U. S. Pate n t 4, 032, 570 ( 1977 ) . 64) A. Onop ch enko and J. G. D. Schulz, J. 01:, Chem. , 3 8, 909( 1973) . 65) a) F. Broic h , Chem. Ing. Tech., 36, 417( 1964) . b) F. Harber and J. Weis s , Natu n - viss enschaft en, 20, 948 ( 1932) 66) J. S. Ba rthle tt , B. Hudson and J. Pennin g ton , U.S . Pate n t 4, 086, 269(1 9 78). 67) T. Szy m anska-Buzar and J. J. Zi o lkowski, Koord. Khi m ., 2, 1172(1 9 76) . 68) R. A. Sheldon and J. K. Koehl. Adv. Cata / ., 25, 272( 1976) . 69 ) N. M. Emanuel, Z. K. Ma izu s and I. P. Sk ibi d a , A ng ew . Chem. Int. Ed., 8, 97(1 9 69) . 70) C. C. Hobbs, T. Horlenko, H. R. Gerberi ch and F. G. Mesic h , Ind. Eng. Chem. Proc. Des. D ev., 11, 59( 1972) . 71 ) D. C. Hull, U. S. Pate n t 2, 578, 306 (19 51 ) . 72) G. Roscher, H. Schaum and H. Schmi tz, Briti sh Pate n t 1, 483, 724( 1977) to Hoechst. 73) G. C. Allen and A. Ag u il o , Meta l -Ion Cata l yz e d Oxid a ti on of Aceta l dehy de , in F. R. May o, Ed., Oxid ati on of Orga nic Comp o unds vol. 2, Advances in Chemi st r y Serie s , 76, 363~381, Ameri ca n Chem ica l Socie ty , Washin g ton , DC ( 1968 ) . 74) G. H. Twig g, Chem. Ind., 476( 1966) . 75 ) M. Tezuka, 0. Seki guc hi , Y. Ohkats u and T. Osa, Bull. C hem. Soc.
Jpn ., 49, 2765(1 9 76). 76) N. A. Clin t o n , R. A. Kenley and T. G. Tray lo r, J. A m. Chem. Soc., 97, 3746, 3752, 3757 (19 75 ) . 77) C. Wallin g , Acc. Chem. Res., 8 , 125(1 9 75). 78) 0. Hay as hi, 0. Kata g iri and S. Roth b erg, J. Am. Chem. Soc., 77, 5450( l95 5). 79) H. S. Mason, W. L. Fowlks and E. Pete r son, ibi d . , 77, 2914(1 9 55) . 80) R. Breslow and L. N. Lukens., J. Bio l . Chem., 235, 292( 1960) . 81 ) a) G. A. Hami lton , J. P. Frie d man and P. M. Camp be ll, J. Am. Chem. Soc., 8 8, 5266( 1966) . b) K. B. Sha rple ss and T. C. Flood, ibi d . , 93, 2316(1 9 71 ) . 82) E. Komura and R. Machid a , J. Chem. Soc. Chem. Commun., 499(1 9 84) 83) S. Ito , A. Kunai, H. Okada and K. Sasaki, J. Org. Chem., 53, 296( 1988) . 84) H. Orit a, T. Hay ak awa, M. Shim i zu and K. Takehir a , J. Mol. Cata ! ., 42, 99( 1987). 85) K. Sasaki, S. Ito and A. Kunai, in New Develop m ents in Selecti ve Oxid a ti on edit ed by G. Centi and F. Tri firo, Stu d ie s in Surfa c e Sc ien ce and Cata l ys i s , vol. 55, Elsevie r Scie n ce Publis h ers, Amste r dam, p.12 5 (19 90) . 86) 日特 平 5-4935. 87) 森憲二 등, 『觸~. 37(2), 178(1 9 95). 88) J. Mi ki, M. Asanuma, Y. Tach ikan a and T. Sh ikad a, Ap pl. Cata / ., 123, 111 (19 95) . 89 ) A. Ni sh i na ga , T. Itah a ra, T. Sh imizu and T. Mats u ur a, J. Am. Chem. Soc., 100, 1820(1 9 78).
90) L. H. Vog t, Jr. , M. Faig e nbaum and S. E. Wi be rley, Chem. Rev., 63, 269 ( 1963 ) . 91 ) A. Ni sh in a g a and H. Tom ita, J. Mo/. Cata / ., 7, 179( 1980) . 92) V. M. Koth ari and J. J. Tazuma, J. Cata ! ., 41, 180( 1976) . 93) A. Nis h in a ga , T. lta h ara, T. Mats u ura, S. Berge r , G. Henes and A. Riek er, Chem. Be,:, 1 09, 1530(1 9 76) . 94) U. S. Pat, 3, 092, 658 (Jun e 4, 1963, to Sta n dard Oi l, I ndia n a) . 95) P. H. Towle and R. H. Baldwi n, Hy d rocarbon Process, 43( 11) , 149(1 9 64) . 96) Brit. Pat, 994, 769(Ju n e 10, 1965, to Sta n dard Oi l, India n a) . 97 ) U. S. Pat, 2, 976, 030 ( Mar 21, 1961, to Sta n dard Oi l I ndia n a ) . 98) Brit. P at, 1, 202, 560(Ang . 19, 1970, to Toxay ) . 99) Ency c lop e dia of Chemi ca l Processin g and Desig n vol. 16, 38~41 ( 1982) edit ed by Joh n. J. McKett a. 100) J. R. R. Ni el sen, Meth a ne Conversio n , Stu d . Surf . Sci. Cata / ., 3 6, 73(1 9 88). 101 ) S. Teuner, Hy dr ocarbon. Process., 64(5) , 106(1 9 85) . 102) J. T. Ri ch ardson and S. A. Parip a ti ya dar, Ap pl. Cata ! . , 61, 293(1 9 90). 103) M. Levy, R. Levit an , E. Meir ov it ch , A. Seg a l, H. Rosin and R. Rubin , Sol. Energy , 48(6), 395(1 9 92). 104) T. A. Chubb. Sol. Energy , 24, 341 ( 1980) . 105 ) J. H. McCa ry, T. A. McCa ry, T. A. Chubb, J. J. Nemecek and D. E. Sim mo ns, Sol. Energy , 29, 141 ( 1982) . 106) J. R. R. Ni el sen, Cata l ys is Scie n ce & Technolog y, vol. 5, 1~118( 1984) Sp ringe r-V erlag. 107) S. E. Park and K. W. Lee, Proc. t he 1st C O2 Workshop at KRICT,
67~85 (19 93 ) . 108) A. S. Al -U b 과 /nd. Eng. C hem. Res., 27, 790( 1988) . 109) A. M. Gadalla and B. Bower, Chem. Eng . Sci ., 42( 11) , 3049 (19 88) . 110) B. L. Gusta f s o n and J. T. Waldon, US Pat, 5, 068, 057 ( 1991 ) . 111 ) K. Fuji mo to , K. Omata , T. Nozaki , 0. Yamazki and Y. Han, Energy Convers. Mg m t. , 3 3(5~8), 529(1 9 92). I I2) F. Kap tei jn , R. Meij er , B. Van Eck and J. A. Moulijn , NATO ASI. Ser. E., 192, 221 (19 91 ) . 113) A. Wa rtz, Jus tu s Lieb in g s A nn. Chem., 110, 125~128 (18 59). 114) Sp co ete Framcais e de Cata l ys e Generalise e, FR 729952, 739, 562(1 9 31 ). 115 ) S. A. Mi ller, Eth y l e ne and its I ndustr i a l Deriv a ti ve s, Ernest B enn Lim ited , London, p.54 6~548 (19 69 ) . 116) R. Wolf and H. Gotz e , Chem. Tech ., (Leip zi g ) 14(no.10) , 600~ 606(1 9 62). 117) R. P. Nie ls en and J. H. La Rochelle, Shell US, 4012452 (19 72) , M. M. Bashin and G. W. Warner, UCC . EP 75938( 1982) 118) G. L. Montr a si and G. C. Batt es to n , Oxid . Commun., 3(3~4) , 259~267 (19 83 ) . 119) G. Leofo n ta n i, G. R. Tauszik and M. Padovan, J. Thenn. Anal. , 30(No. 6) , 1267~1272( 1985) . 120) G. L. Montr a si, G. R. Transzik , M. Solari a nd G. Leofo n ta n i, Anal. Cata ! . , 5, 359 ( 1983 ) . 121 ) W. D. Mross, Ber. B useng es . Phy s. Chem. , 8 8, 1042~ 1053 (19 84). 122) S. Rebsta t , S. Maye r and J. Alfr an seder, Chem. Ing. Tech., 5 3
( no. 11 ) , 850~854 (19 81 ) . 123) G. Yamag u chi and S. Takenaka, Nip po n Kay ak u, US 3454630 (I96 9) . 124) S. Takenaka, Y. Ki do , T. Shim abara and M. Og a wa, Nip po n Kaya k u, US 3778386( 1973) . 125) T. Ohara, M. Ueshim a and I. Yanag isa wa, Ni ppo n Shokubai Kaga k u, US 3825600 ( 1974 ) . 126) T. Ohara et al., Nip po n Shokubai Kaga ku, JP 42242(1 9 72), US 3833649. 127) H. Ito , S. Nakamura and T. Nakano, Toa Gosei Chemi ca l Co., FR 2028164. 128) T. Kita and H. Ishii , Mi tsu bis h i Rayo n , JP 32048(1 9 72) . Chem. Abstr. , 83, 27611 a(1 9 74) . 129) Sumi tom o Chemi ca l Co., BE 769508( 1972) . 130) Y. Wata na be et al., Mi tub is lz i Petr o chemi ca l Co. , JP 41329 , DE 2165335, us 4008280(1 9 72) . 131 ) G. Duembg en et al., BASF, DE 2338111 ( 1973) . 132) Y. Wata na be et a l., Mi tsu bis h i Petr o chemi ca l Co., JP 34111 ( 1973) . 133 ) D. Arntz , G. Prescher, W. Burkhardt and J. Heil o s, Deg u ssa, DE 3125062 and 3125061 ( 1981 ) . 134) R. K. Grasselli , Sohio , EP 663( 1977) . 135) Y. S. Yoon, N. Fuji saw a, W. Ueda, Y. Morooka and K. W. Lee, Cata l ys i s T oday, 24, 327~333 (19 95 ) . 136) Y. S. Yoon, N. Fuji wa ra, W. Ueda and Y. Morooka, Chem. Lett ., 1635(1 9 94). 137) F. T. Maler and W. Baye r , Incyc l . Chem. Process Des., 1 , 401 (19 76).
138) L.S .Luski n, H ig h Polym . ,2 4, I05(1970). 139) B. F. Goodric h Co., US 3392196(1 9 64) , Nip po n Shokubai, US 3475488(1 9 69) . 140) S. Sakuy am a, T. Ohara, N. Shim i zu and K. Kubota , Chemt ec h, 3(6), 350(1 9 73). 141 ) D. J. Hucknall, Selecti ve Oxid a ti on s of Hy d rocarbons, Academ ic Press, London. p.5 2( 1974) . 142) Dis t i ller s, JP 20838, GB 915799(1 9 63) . 143) M. Yanag ita and M. Ki tah ara., Ri ka g a ku Kenky u sho, JP 26287( 1969) , us 3567772( 1971 ) . 144) S. Takenaka, Nip po n Kaya k u, BE 698273 (19 61) . 145) G. C. Allen, Celanese, G B 1267189, 1972, US 3644509(1 9 72). 146) R. Krabetz and H. Eng el back, BASF, FR 2032915, 1970, US 3845120( 1974). 147) J. Hensel, Deg u ssa, JP -Kokai 8360(1 9 72), DE-OS 2055155 ( 1972) . 148 ) M. Wada, I. Yanag isa wa, M. Ni m orn iya and T. Ohara, Nip po n Shokubai, JP 11371 (19 14) , DE 2152037(1 9 72) . 149) T. Shir a is h i, S. Kish i wa da, S. Shim izu , S. Honmaru, Y. Naga oka and K. Jinp o , Sumi tom o Chemi ca l, JP -Kokai 31923 ( 1972) , Chem. Abstr. , 78, 30464g . 150) M. Wada, I. Yanag isa wa, N. Ni no mi ya and T. Ohara, Nip po n Shokubai, JP -Kokai 1 17419, DE2413206(1 9 14). 151 ) Y. Kadowa ki and T. Koshi lcaw a, Mi tsu bis h i Petr o chem. , J P 169 ( 1974) , DE 2164905( 1972) . 152) S. R. Dolhy i and E. C. Milbe rge r , Sohio , JP -Kokai 8 3280, 1975, DE 2448804(1 9 15) .
153) N. Ferlazzo, Soc iet a Ital ia n a : JP 28889, 1978, DE 2456100 (19 75) . 154) a) G. Em ig et al., Cata ! . Today 1, 477~498( 1987) . b) F. Tri frro et al., Chemt ec h. , Ap ril, 18( 1994). 155) G. Busca et al., J. Phy s. Chem. , 9 0, 1337( 1986) . 156) G. Centi , Stu r d. Surf . Sc i. C ata ! ., 55, 563( 1996) . 157) T. Sh im oda et al., Bull. Chem. Soc. Jpn ., 58, 2163(1 9 85) . 158) 김재승, 박남국, 김영철 『화학공학』. 27( 1), 42~49(1 9 89). 159) 정지훈, 허성 기, 홍석 인 , 『화학공학.!l. 33(3) , 273~281 (19 95) . 160) D.Hon ick eandK .G r ies baum,Ap pl. Cata ! .,2, 177(1 9 82). 161 ) a) Ki-W on Jun and Ky u- Wan Lee et al., Korean J. of C hem. Eng ., 3(2) , 135~139( 1986) . b) Ki -Won Ju n, Ky u -Wan Lee and Hakze Chon, Ap pl. Cata l . , 63, 267~278(1 9 90). c) 이규완, 전기원, 「 C5 유분 산화에 대한 연구 보고서」 , 과학 기술처(1 987). 162) 홍석인, 정지훈, 이인화, 『화학공학』 ,31(6),683~691 (1 993). 163) 김재승, 박남국, 김영철, 『화학공학』, 26(2), 149~157(1 9 88). 164) H. H. Vog e, Oxid a ti on of O rga n ic Comp u nds. p.2 42, ACS vol. II, Washi ng ton ( 1968) . 165 ) K. Weis e mel and H. J. Arpe , Industr i e l le Orga n is c he Chemi e, Verlag Chem ie, Wein he im ( 1988) . 166) D. J. Hucknall, Selecti ve Oxid a ti on of Hy d rocarbons, Academ ic Press, London( 1974) . 167) R. Higgins and P. Hayd e n, Cata l ys i s vol. 1, Chem. Soc., London (19 77) . 168) Cata l yt ic A ssoc iat e s , Selecti ve Cata l yt ic Oxid a ti on of H y d rocar-
bons,, A Criti ca l Analys i s , Stu d y no. I077, Santa Clara, Calif (Oct. 1977) .
제 4 장 과산화물에 의한 산화 퍼옥시 (pe roxy ) 화합물은 -0-O -결 합을 갖는 화합물로서 유기 과산화물인 경우 최소한 하나 또는 두 개 등의 탄소결합을 갖는 화 합물을 말한다 .1~ 8) 퍼옥시 화합물 중 가장 잘 알려진 것은 벤조일 퍼옥사이드 9) 로, 이미 1858 년에 합성되었으며, 표백제로 많이 이용 하였다. 이 과산화물은 또 산화제보다는 고무, 지방, 기름, 페인트 등의 노화제로 사용하였다. 1970 년대 중반에는 자연에서 존재하는 퍼옥소(p eroxo) 화합물 들이 단리 및 확인되었다. 1983 년 미국에서 사용된 과산화물을 종 류별로 보면, 디알킬 과산화물 (1,995 톤), 디아실 과산화물 (4,855 톤), MEK- 과산화물 (3,855 톤), 퍼옥시에스테르 (4 , 035 톤) 그리 고 기타 (1,225 톤)이댜 과산화물은 비닐 단량체의 래디컬 중합 개시제로 쓰여지면서 중 요하게 되었댜 그 밖에는 폴리올레핀의 가교제, 탄성체의 경화제, 그리고 이 책에서 취급하는 산화제로서 극히 소량이 사용되고 있
다. 일본에서는 크게 나누어 중합 개시제로 약 50 %, 가교제 25 % 그리고 경화제로 약 25 %가 각각 쓰이고 있다 .10 ) 사실 유기 화합물들은 과산화수소의 수소를 여 러 유기 그룹으로 치환한 것이다. 이들울 분류하면 표 4-1 과 같다. 비교적 약한 -0 -O- 결합은 광이나 열에 의해서 균일 개열하는 것 이 특징이다. 그 반응성은 과산화물의 구조에 의하여 크게 좌우되 며 불균일 개열 및 가수분해성 개열로 알려져 있다. 산화 퍼텐셜은 다음 순서로 감소한다. 퍼옥시 지방산> 히드로 과산화물> 디아실 과산화물> 퍼옥시 지방산 에스테르> 디알킬 과산화물 주어진 동종 과산화물에서의 안정성은 탄소수가 많을수록 높다. 품열 화안된정 과성산은화 반물감의기 반또감는기 는10 시래간디 컬반을감 기형의성 하온면도서로 t1나12 타<내 1는0-데 3 ,se c 상로
표 4-1 과산화물의 종류 과산화수소 H-0-0-H 알킬히드로 과산화물 R-0-0-H 디알킬 과산화물 R I -0-0-R2 。 II 퍼옥시 지방산 R-C-0-0-H 。 。 디아실 과산화물 R1 - CI..I -0-0-CI.I. -R 2 퍼옥시 지방산 에스테르 I R -C。 -0-0-R I .II. 2 。 퍼옥시 카보네이트 RI -O-C.I.I -0-0-R2
대단히 짧다. 그리고 반감기 온도는 25~170 °C 이고, 이 분해는 전 이금속에 의 해 촉진(p romo t er) 된다 .II) 4. l 과산화수소 12 ) 과산화수소는 기초 과산화물로서 매우 중요하며, 과산화수소 자 체나 유기 화합물을 과산화물로 만드는 데 공기와 같이 긴요하게 쓰이고 있다. 4. 1. 1 제조 1) 안트라퀴논 공정 13) 과산화수소 제조의 총 85 %가 안트라퀴논 (an t ra q u i none) 공정 을 채택하고 있댜 반응식으로 간단히 표시하면 다음과 같다•
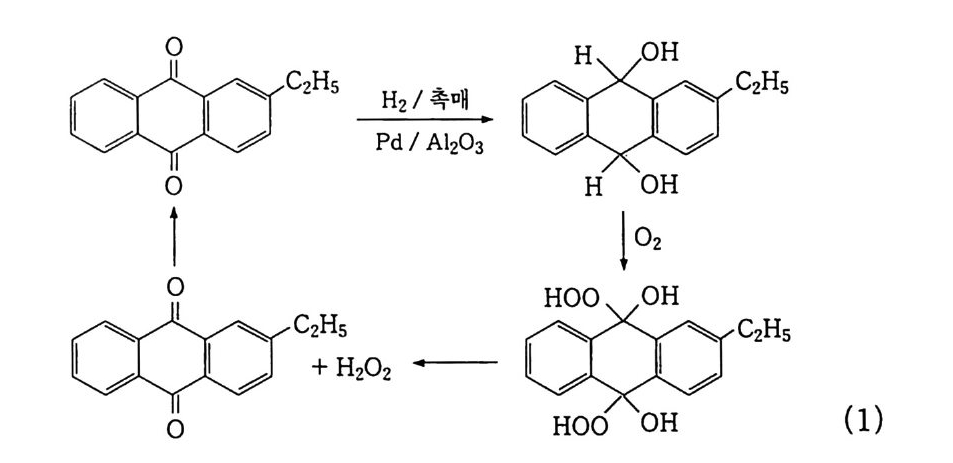 :三?二 따+ H;릅2°2- :二CH 2OHH C52H5 (1)
:三?二 따+ H;릅2°2- :二CH 2OHH C52H5 (1)
각 공정에서 퀴논 및 히드로퀴논 용해도가 다르므로 선택적으로
녹이 는 용매 ( workin g solvent ) 가 사용되 는데 . 마지 막 단계 에 서 물 로 과산화수소를 추출, 45~50 %로 농축시킨 다음 원하는 농도의 과산화수소 수용액으로 만들어 시판한다. 2) 이소프로판올 공정 액상반응으로 유도 기간을 줄이기 위하여 0.5~1 .5% 과산화수소 를 2- 프로판올에 넣어 공기로 산화시킨다. HO-OH-2HO· cg/ C3\ /0 H •OH CH/\ 3C / . OH 02 C/\H ,c/\ 3 ' 0 0· 2- 프로판을 CH3H CH3 CH30H CCH_/H \3C3 \/ O0H-0 H + CCH\/H 3C3 \ . OH CC/H\H C33 =O + H2 02 (2) 4. 1.2 물성 고농도 (>65w t%)의 과산화수소는 많은 유기용매에 녹는다. 물과 공비점을 형성하지 않으며, 물과 과산화수소와의 물리적 성질 비교는 표 4_2 에, 그리고 여러 수용액 등의 물성은 표 4-3 에 나 타내었다• H2 야는 수용액에서 약산성이며 해리상수는 1.78 x10-12 (pk 11.75 at 20 ·c) 이다.
표 4-2 과산화수소와 물의 물리적 성질 성질 값 H 2° 2 H 2° mp °C -0.4 3 。 bp (l01 .3k p a ). °C 150.2 100 융해열 Jig 368 334 기화열 Jg-I K-1 at 25 'c 1519 2443 at bp 1387 2258 비열 Jg- I K -1 용액 (25 °C ) 2.6 2 9 4.1 8 2 기체 (25 ·c ) 1.35 2 1,86 5 상대밀도, g/ cm 3 o·c 1.47 00 0.9 9 98 2o· c 1.45 00 0.9980 25° C 1.44 25 0.9971 점도. mPa·s o·c 1.81 9 1.79 2 20 ·c 1.24 9 1.00 2 임계온도, ·c 457 374.2 임계압력 MPa 20.99 21 .44 굴절률 nD2 0 1.40 84 1.33 30
표 4-3 과산화수소 수용액의 물리적 성질 성질 H 오 농도. wt % 35 50 70 90 상대밀도. g/c m 3 o·c 1.14 41 1.21 10 1.30 71 1.41 36 2o·c 1.13 12 1.1 9 53 1.28 86 1.39 20 25°C 1.12 82 1.19 14 1.28 39 1.3867 점도. mPa·s o·c 1.82 1.87 1.93 1.88 2o·c 1.11 1.17 1.23 1.26 굴절률, nD20 1.35 63 1.36 72 1.3 8 27 1.39 95 mp , ·c -33 -52.2 -40.3 -11 .9 bp ( lOl .3 kPa), ·c 107.9 113.8 125.5 141 .3 H202 분압 (30 °C )kPa 0.05 0.1 1 0.1 7 0.29
4. 1.3 분해 및 반응 과산화수소의 분해는 불균일반응으로 일어나므로 보관시 주의하 여야한다. H202(e) 一 H20(e) + ½02 -98kJ/ m ol (3) 식 (3) 에서 보는 바와 같이 이 분해반응은 극열 발열반응이므로 극소량의 촉매 존재하에서도 분해된다. 균일계 촉매로는 녹아 있는 중금속인 철, 동. 망간 등이며, 불균 일 촉매로서는 이들 금속 외에 팔라듐, 수은의 산화물 또는 수산화
물 등이고 백금 , 오스뮴 그리고 은 금속에 의하여 분해가 촉진된다. 1) 촉매 존재하에서 H 2 0 2 의 반응 (1) 금속이온에 의한 반응 중성 또는 산성 매체에서 H 2 0 2 는 낮은 산화 상태의 금속이온 존 재하에서 전형적인 유리기 반응이다. 예를 들면 기질이 없는 상태 에서 Cu + 와 Fe ” 는 H 2 0 2 를 자동 분해시켜 식 (4) 에서처럼 물과 산소로 분해된댜 이것을 Harber-Weis s c y cle1 4) 이라 한댜 H-0-0-H + Fe2+ 一 Fe3+ + OH- + ·OH (4-1) H-0-0-H + Fe3+ 一 Fe2+ + H+ + ·OOH (4-2) ·OOH 一 0 2 + ·H (4-3) 2H2 0 2 ~ 02 + 2H2 0 (4) 그러나 기질 (RH) 존재하에서는 이 기질이 ·OH 래디컬과 반응 하여 치환반응이 일어난다. 전형적인 반응은 펜톤 시약을 15) 이용한 방향족 화합물의 수산화 반응이다. H- 0 -0-H + Fe2+ 一 Fe3+ + OH-+ ·OH (4 -1) RH + ·OH 一 R· + H20 (5-1) R • + Fe3+ 一 R+ + Fe2+ (5-2) R++oH-一 R 一 OH (5-3) H 요 +RH ~ R-OH + H2 0 (5)
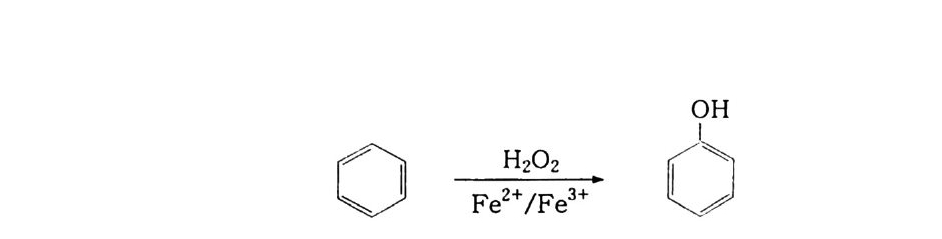
(2) 금속 또는 금속 산화물 존재에서 H20 2 반응 전이금속 산화물을 H 2 0 2 와 반응시키면 여러 종류의 무기 과산화 물이 형성되는데, 이는 후술할 유기 산화물 형성 1 6 ) 기구와 비슷하다. _O H -M=O+H20 2 - -M (7) OOH 이러한 금속 산화물은 계속적으로 올레핀 존재하에서 에폭시화 가 일어난다. \/ C = C\/ + H2 0 2 ---M=-O::::x.. ::.. /\ C\ -/ C \/_ + H2 0 (8) 。 2) 알칼리성 용액에서의 H 2 0 2 의 반응 알칼리 수용액에서 H2 야는 OH- 와 반응하여 퍼옥시수산기 이온 을만든다. H202 + OH- ---Ho o-+ H20 (9) HOO- 은 OH - 이온보다 200 배 이상의 반응성을 가진 〈슈퍼 구핵 제〉이다 .17) 기질이 없는 경우 OOH- 는 제 2 의 H202 와 반응하여 다 음 식 (10) 과 같이 분해된다.
\\ :/- H 一 [<:/ H ] 一 。 2 + H20 + 0H- (10 ) 만약 구전자 센터를 갖는 올레핀 기질과 반응하면 에폭사이드를 생성한다. CC/\HH C33 = CH -C0|l -C H3 + OOH- 一 CC `/H H`J`3 CO I O=H C _H -0CIl -C H3 一 C/`H ``.C 3 / 0- C\ H-C0l|- CH3 + OH- (11) CH3
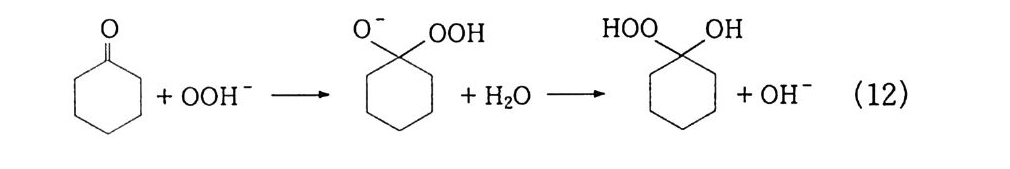 () + OOH 一 \° °+\。 一H OOH+ ow ( 12)
() + OOH 一 \° °+\。 一H OOH+ ow ( 12)
3) 산성 용액에서의 H20 2 반응 산성 용액에서 H202 는 안정하다• /H H202 + H30+ 一 Hoo+ + H20 (13 ) \H 알칼리성 수용액에서는 반대로 H202 가 구전자 반응 (ele ct ro p h ili c) 성이 되어 더 안정적이 된다. 그 반응계 내에 기질, 예를 들
어 지방산. 알코올 또는 케톤이 있으면 이를 프로톤화시켜 새로운 구전자 중간체를 만들고. 과산화수소와 반응한다. 이는. 후술하겠지만 (4. 3.1 참조). 강산의 촉매 작용에 의하여 과 산화지방산(식 14)18) 과 히드로퍼옥사이드(식 15)1 9) 를 만든다. (11) / OHo __ ,,. H ,, OH CH3COH + H+ 一 CH3-c \+ + oI 一 CH3 - c \+ + H2 0 :O H \ H .O. H 一 CH3-C『 -O OH + H+ (14) CH3 CH3 『比 CH3-CI -O H + H+ -CH3-cI + + H2 0 -CH3-c + I I I CH3 CH3 CH3 / H CH3 0 | + oI \ -H CH3-CC| H -3O OH + H+ (15 ) 방향족 탄화수소와 같은 약한 염기성 기질이 존재하면 H2 아의 옥소늄형이 주를 이루고, 이것으로부터 OH+ 양이온이 생성되어 페 놀이 생성 20) 된다고 제안했다.
H2 ° 2 + H+ 一 [H0-0< H l + 一 0H+ +H2 ° H
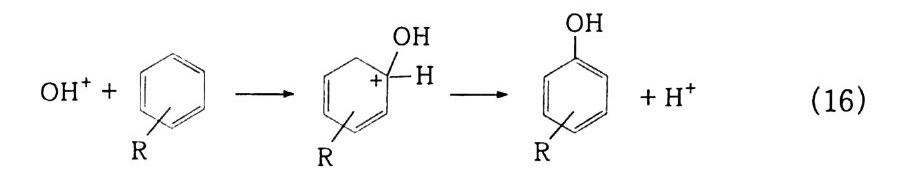 OH OH
OH OH
4. 2 알킬히드로 과산화물 4. 2. l 제조 여러 가지 방법이 알려졌지만 원칙적으로 퍼옥시 그룹의 근원에 의하여 다음과 같은 세 가지 방법이 있다. ® 과산화수소로부터 제조 ® 산소를 이용하여 제조 ® 다른 퍼옥시 화합물로부터 제조 과산화수소로부터 제조하는 것에 대하여는 4.1 의 설명을 참조하 기 바란다. 산소를 사용하여 제조하는 것에 대하여는 제 2 장의 반응 기구 (2.3) 에서, 그리고 자동 산화반응에서 부분적으로 언급하였다. 반응기구에서도 본 바와 같이 반응은 높은 온도에서 비교적 천천 히 지속적으로 일어나기 때문에 합성에는, 특히 공업적으로는 제 한적으로사용된다. 2.5.3 절 액상 산화반응에서 설명한 큐밀-과 t-부틸 과산화물 및 에틸벤젠히드로퍼옥사이드. 1,4- 디이소프로필 벤젠 과산화물.
피낸(pi nane) 히드로퍼옥사이드가 공업적으로 응용되고 있다. 4. 2. 2 물성 알킬 수산화 과산화물 등은 대개 안정적이어서 80°C 이하에서 결정 또는 증류할 수 있다. 열 안정성은 -0-0-결합 해리 에너지로 결정되며 균일 분해인 경 우 180~184k J /mol 이다 .2 1. 22) 온도가 중가함에 따라 분해가 빨라 지는데, 폭발이 일어날 수도 있다. 몇 가지 알킬 과산화물의 물성을 표 4-4 에 나타내었다. 4.3 과산화지방산 과산화지방산은 높은 산화 퍼텐셜로 인하여 제조 및 공업적으로 광범위하게 응용되고 있다. 하지만 불안정성 때문에 몇 가지 화합 물들을 동시에 제조하여 사용하든지. 비활성화시켜 사용해야 한다. 4. 3. l 제조 여러 방법으로 제조할 수 있는데, 그 중 몇 가지만 소개한다. 1) 지방산과 과산화수소로부터 제조 4.1 .3 과산화수소의 분해반응 3) ‘산성용액에서의 반응’란에서 설명한 바와 같이 지방산울 산성용액에서 H2 아와 반응시켜 제조하 는 방법이 가장 잘 알려져 있다.
때갖。 u 칭써 )샘(ξ 어여.어 a[N. {Na. {.h• m。{. )g건(μ 륭 m‘F。。op ‘ 11 ~.0ζζ*。o:) i‘> ~u ζN0[o ' 。 〉' ‘l ∞∞.。…a} [{。.{{}} ζ。 )(μg .Y{[이{* ~.0。r0o... η0. 1.. ~u‘0~0.. . 。。 껴0... .이{{*。 .NN[∞*v 띠•[N*Nv } 뻐F <ζa μ)( .*。 Nζ i l‘ma(]* 하
찌{디칭패빼{「빙K { K Qja· . ιs)(이 )g(여∞.∞Pl y *v(*)l여* * 어(띠.N• ) ∞g‘nl g ({{)(] N.어()m이 {•(@。)•m 。여[。).여(m 耶-- 응 *∞.*。Yy ∞。∞ 。h∞.∞ •[aN@ [•…{[@ } {@•N[m Nam.p∞ -‘--버 gE{바에써 。。 칭디뼈줌{”빠{γ서。[。U}l} { { 피숭피〔N‘피lQQ U ”비m때wl다}{{lUN、UR}UN영 -。m때w비서l[‘} }{{Q ) N( Q ]|〕μ@빼마{Q)이(lU‘}η 탱합-빼때<{잉l【‘【}Q 힘T”QQll(jιRN})‘{ 갱다h‘I’n }{ g Q 〕이』〕며(μm‘{}.(”N(.•μ ummp m N 배표 4-5 평형 조성과 H 2 0 흔 의 전환 (AA-H p 2 -PAA-H p, at 2 5· c) 2J> (H 오) 몰비 조성. wt %(mo! %) H.2 ° 2 수용액 AA:H 오 AA PAA H.2° H2 0 .2 전환(%) 50 1:1 22.0(9.7 ) 29.1 (16.2) 31. 5( 58.4 ) 16.0(1 5 .7 ) 37 2:1 22.8(1 1,6) 45.2(29.2) 23.6(50.9) 7.2 ( 8.2) 70 4:1 18.6(11 .1) 62.4 ( 47.2 ) 15.3 ( 38.5 ) 2.4 ( 3.2 ) 76 6,66:1 13.8(9.0) 73.3(60.8) 10.4 ( 28.8 ) 9.2 ( 1 .3) 86 10:1 10.1 (7.0) 81. 0( 70.8) 7.4 ( 21 .6) 0.5(0.7 ) 90 90 1.5 : 1 45.0(27.8 ) 35.0(27.4 ) 14.0 ( 36.5) 6.0(8.3) 77
탄소수가 적은 지방산은 산 촉매(강산)하에서 과산화수소와 혼 합함으로써 반응이 일어난다. 평형은 출발 물질들의 몰비나 과산화수소의 농도에 의해서 좌우 된다. 빙초산의 경우 AA-H202 - PAA-H20 평형 조성을 표 4-5 에 실었다. 반응 후 생성물의 정제는 진공 증류에 의하여 얻어진다 . 2 4) 표 4-6 에 몇 가지 지방산 과산화물과 물의 공비 혼합물을 실었다. 물론 유기 과산화물은 유기 용매 25,26) 와 다른 산 촉매 27) 중에서도 합성할 수 있다.
표 4-6 과산화지방산과 공비 혼합물 화합물 과산화 지방산 wt % 물 wt% 과산화식초산 61 .0 39.0 과산화프로피온산 51 .5 48.5 과산화부탄산 42.0 58.0 과산화 이소부탄산 55.0 44.5
2) 지방산 크로리드와 H 2 0 2 로부터 제조 염기 촉매에서 지방산 크로이드와 과산화수를 반응시켜 방향족 과 산화 지방산을 만드는 것은 중요한 방법이며 , 또 가장 잘 알려 져 있기도 하다. 3- 염소과산화 안식향산 28 ) 이 대표적인 화합물이다 . 용매는 물 2q) 또는 물-유기 용매 30,3 1 ) 혼합물에서 여러 가지를 만들 수 있다. 4. 3. 2 물성 단쇄 과산화물은 물에 녹으나 Q 이상은 물에 잘 녹지 않는다 . 고체인 화합물은 (2) 와 같이 2 량체의 형태로 존재하고 배위하지 않은 용매에서는 (l) 과 같이 존재한다 .
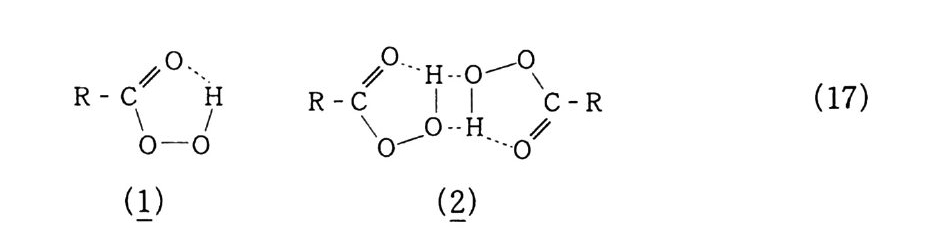 R-C \ o/ - 0` o` `H I R-C\, yoo .- __ -0HI -· -·oH- I - . -~ O0 /\ C -R (17 )
R-C \ o/ - 0` o` `H I R-C\, yoo .- __ -0HI -· -·oH- I - . -~ O0 /\ C -R (17 )
과산화지방산의 PKa 는 지 방산과 같이 공명 안정화하지 못하므 로 지방산보다 3~4 단위가 낮다. 어떤 환경하에서는 열이나 충격에 의하여 폭발적으로 분해할 수 있다. -0-0- 결합 해리 에너지는 80~90k J /mol 이므로 히드로 과산화물이나 디알킬 과산화물보다 대단히 낮다 .32~3 4 ) 몇 개의 물성은 표 4-7 에 정리하였다.
-m{mNmiiviiyis . ω{{K vaζigh선@. SF。.-{〔 --*i.。【gv - Is。z ;<- - 빼m{체-·킹K이파훤rM파칭한뼈마W-{|날배버FK {”w*?r경석되g·μ 강써마응mmι -μ”ν{α a r(im)ι { -챔W* z i 。{。}{.。Q}”m{m ∞n‘.N.∞au--애.。. N. . 여{().어 (。Q므얘·)E@gi。건 ζ[l。[}QQ}.띠)∞N{(•.{∞.〕NQN〉〕[m잉∞.{{υ@어강{{ 〉 ;건u어』u강다@mφ)( QQ맞픽':-∞{{l.(hm)l.애.∞∞{.u•--{a.:. N JN N r{ } :-F% s며며 N∞l∞{N*.。{{{-。:Q--.}im{쩌며A;v NM N : . l :. s 며rF U나i•,rN[띠어(.){∞vi ∞N.;띠l·J∞Q이.j: F QM , . l : 9m며. :Pr나 δ},a며a∞*i.{N∞여n:iL v.j u•{,o r ’U m : 、_벌〔: U0:。(。{la∞ {.@{∞ ∞m.{m(]쇠{〕강_•·「;.디mt∞) :U Nm M :: ’ ' Q _ 〔r‘ $6피i;∞{Q。{:‘n∞∞{z• @’{ :7- 「T - N M: ::Q ’ : 피 u m」편필/rcs∞∞밍N).:(∞N띠{띠. ’U이김〕 @띠.t←.Q여∞、 :얻) , 、 ' . .디.. {〕애·1i〕Q강。 。엉띠c{NN}{〔•(。。N며QQ)’Q”lN•a}{。..∞ > m 1 Q Qmu-김。므 。。N{R。l(}∞NQ)’QR}Q。N.여*{•여..’ω!Cmu{kp〕∞.mg∞。{{{ m 3 u 1iQ이。i〕l잉 rζ@N(.Egω1)g(<〕.N여}{〔。(Ql)。‘lQ”I。{.NN‘Nn } { -QU-uimg ∞•Nl[i∞어}[Q。’ω{.}{-{υ.{∞{v- *{ y Q므m( h여{h0。•{.∞N}’{l잉〔니EQ}{u}〔l。,’~{X여 0。 므Q∞uSNω겁(} ih∞(.{∞gy)∞임[.피〔。.’k‘?}∞{z잉Qlu’QN。a g p{〕강여QSN다U(} {]∞’∞QN.(<∞}〔OQl,}〕·}{l{{N{。잉υla{{.∞Ql즐。』@gg。응∞.∞ {1애〕UQ-£-£ma
4. 4 과산화 화합물의 용도 4. 4. l 올레핀과의 반응 1) 올레핀의 에폭시화 니트릴과 같은 염기성 용매에서 H 2 0 2 는 다음과 같이 반응한다 . 0 0 \C = CI + H2 0 2 + R -CN 一 \CI - \CI + RCII -NH2 (18 ) I \ I \ 산성하에서는 과지방산으로 반응하여 공업적인 규모로 사용된다. 불포화 지방산을 저급 지방산과 과산화물로 에폭시화시켜 PVC 의 가소제로서 널리 사용하고 있다(콩기름 에폭사이드). 이 경우 산 촉매에 의하여 개환반응이 일어나 디올 (d i ol) 과 에스테르를 형성할 가능성이 크다. R\I C =C(\IC HIC 2O)n+O H R0CI| OOH 一H+ R \Ic /-O \ c(/\C HIC 2O)n+O H R?CO H (19 ) 。 \ / \ / H+ \ / C-C+H 2 0 一 C-C I \ / OI H OI H (20)
0 0 \ / \ / || H+ \ / C -C + RCOH 一 C -C / I 1 \ OH OCOR (21) 그러므로 분자량이 큰 m- 염소과산화 안식향산이 많이 사용된댜
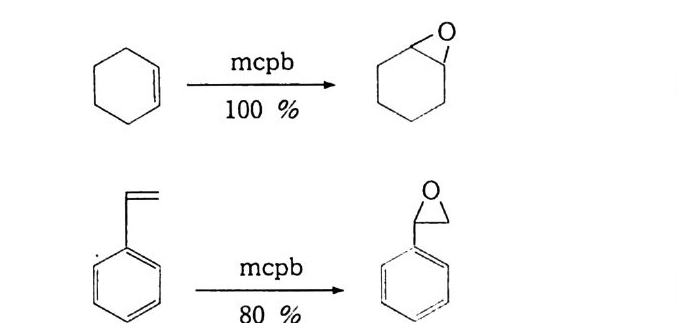 。 1m00c p %b < (22)
。 1m00c p %b < (22)
비교적 불안정한 디할로겐 프로판 에폭사이드도 합성되었다 . 35 , 36) RI R3 R1 0 R3 R\/2 C=XC| -CX| \/ R4 mcp b • R/\2 c /-\cX| -cX1 /\ R4 (24) H202 및 수산화 과산화물은 관능기가 없어 스스로 분해하기 어 려우므로 금속 촉매 존재하에서 반응시킨다. 특히 H202 인 경우 존 재하는 물 때문에 촉매의 비활성화가 일어나 전환율과 선택률에 많 은 영향을준다. 가장 오래 전부터 알려진 것이 텅스텐산 (H2W04) 이고, 알릴 (allyl ) 알코올을 기질로 하는 연구도 많이 이루어졌다 .37~39)
CH2 = CH -C H2 0 H H2 040 2 ~- 5H02 W% 04 C/H 。2 -\ C H -C H2 -O H (25) CH3 C H = CHCH2 0 H H20 2 8 -0H% 2 W 04 CH3C/H0 -'- --C- H -C H20H (26) 최근에 H 2 0 2 의 높은 전환율과 에폭사이드의 높은 선택률의 반응 결과가 알려졌다. 즉, 폴리에테르를 용매로 물량을 적당히 조절하 는 방법 이 다 .40 ~ 42 )
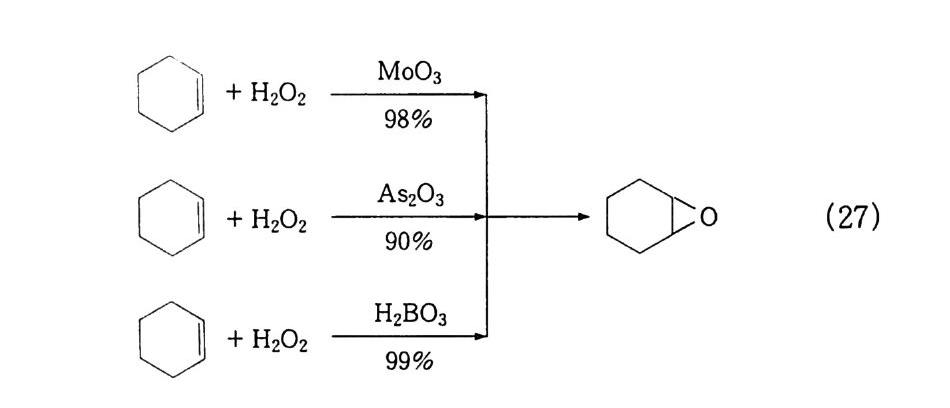 。 + H2 0 2 M98o%0 3
。 + H2 0 2 M98o%0 3
CH 3 CH=CH 군 %02 ~一 CH3-C/ H。 _ \ CH2 (28) 공업적으로는 t-부틸히드로 과산화물 (TBHP) 을 사용하여 프로 필렌옥사이드를 제조한다 . 4 3) TBHP 를 사용할 경우의 이점은 다음과 같다. ® 높은 열 안정성 ® H202 나 과지방산보다 높은 안정성 ®무부식성 ® 비극성 용매에 높은 용해도 ®중성 pH ® 선택적인 산화제
® 부산물인 t -부탄올은 증류로 쉽게 분리 가능 十 OOH + CH3CH=CH2 ——-C H3-CH-CH 군 十 OH (~ 8 0%) ,,,,,0 (29)
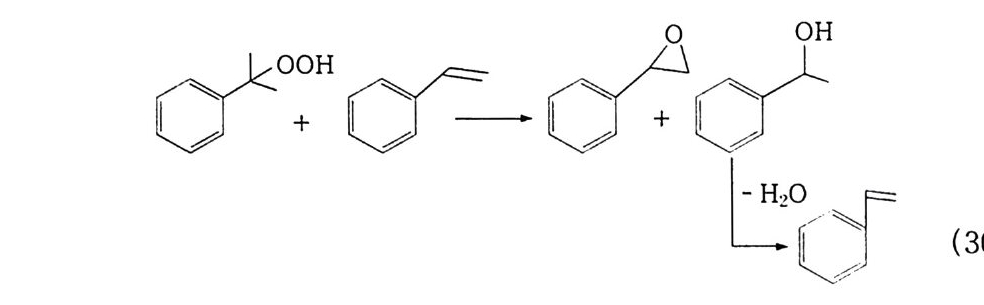 : 0+0H : 二 : \0 〉OH
: 0+0H : 二 : \0 〉OH
위의 여러 반응기구에 대하여는 참고문헌 41 . 15) 올 보기 바란다. 2) 올레핀의 수산화 이것은 과산화지방산으로 올레핀을 에폭시화시킨 후 산 촉매하 에서 가수분해시켜 얻는다. 0 0 \ / II \/ov |I / C=C\+ R -C -OOH - / H~+- lCH .\ 2 0+ CH3COH \/ c I -c1·/ +\ H+ (31) OH OH 글리세린을 공업적으로 제조할 목적으로 다음과 같은 방법이 연 구되 었다 •46~48)
CH2 = CHCH2 0H + H2 0 2 뽑우 CH2 -CH2 -CH2 (32) I I I OH OH OH 다른 촉매의 경우도 올레핀 수산화에 대하여 많은 연구가 이루어 졌으며, 그 촉매들은 주로 0s04, Fe(N03) 2 , Hg S 01, P 2 아, Fe(S01) , Fe(S0 1 ) 3 이다. 특히 0s0 1 가 많이 연구되었는데, 이것은 올레핀에 cis -부가를 일으키며, Mi la s rea g en t라 불린댜 그러나 고가인데다가 독성 때 문에 공업화되지는 못하고 있다 . 49)
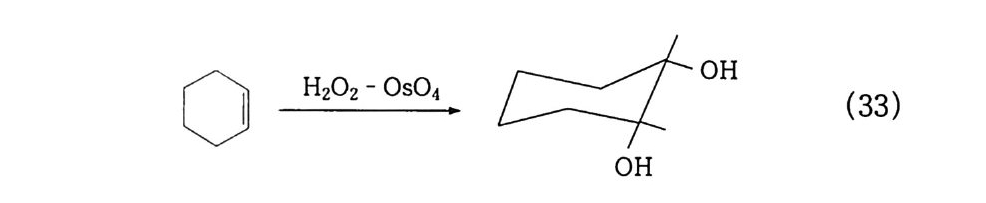 。 H20 2 -0 s04 二 OH (33)
。 H20 2 -0 s04 二 OH (33)
4. 4. 2 카르보닐 그룹과의 반응 H2 0 2 -c 알데히드, 케톤과 쉽게 반응하며 과산화물 혼합물을 형 성한다. 이 과산화물 혼합물은 산화력이 낮으며 대단히 불안정하 므로 주의해야 한다. 과산화지방산으로 카르보닐 화합물을 산화시킬 때에는 지방산이 나 에스테르를 형 성 한다 . 이 반응은 Baey e r -Vil lige r 반응으로 알 려져 있다. 1) 과산화물의 형성 산성하에서 H202 가 다량으로 존재하는 경우 g em- 디히드로 과
산화물이 생성된다. R OH R 0-0H I\c< + H202 一H+ /\ C,\' + H2 0 (34) R' 0-0H R' 0-0H 반대로 과량의 알데히드 및 케톤이 존재하면 a- 히드로 과산화물 과 혼합하여 a, a'- 디히드로 과산화물이 형성된다. RR/\' c < O0 H-0 H + O=CI\R R ' 一 RR/\ ' C.\ ‘ O0H-0O\.H 'C R ' R ' (35) 표 4-8 에 알데히드와 각 케톤으로부터 얻을 수 있는 a- 디히드 로 과산화물 및 에스테르를 정리하였다. 2) B aey e r -Vi llige r 반응 1899 년 Bae y er 와 V illig er 는 과산화지방산이 케톤을 에스테르로 변환시키는 것을 발견했다 .53) 이 반응은 케톤과 과산화지방산에 대 하여 각각 1 차 반응이고 일반적으로 산 촉매하에서 일어난다. R\ C=O 과산화지방산 R -?C - OR1 (36 -1 ) R<
표 4-8 a-히 드로 과산화물의 물리 적 성질 ?2 R 느 c_ 。― O ― R 3 晶 RI R2 R3 b.p( tor r) 참고문헌 H H H oil 5 CH3 CH3 H oil 5 CH3 CH3 H n.m.r 로 확인 50 Et Et H 반응으로 확인 51 D H 129~130 52 。 H 155~156 52 H H CH3 45(1 7 ) 5 H H Et 46~48(1 3 ) 5 H H n -C 4H 9 52~53(8) 5 CH3 H CH3 25~27(1 7 ) 5 CH3 H c2H 5 48~50(65) 5 Et H Et 50~52(50) 5 n -C 3H7 H n -C 4H9 34~37(1) 5
Cr i e g ee 는 다음과 같이 반응기구를 제안했댜 R~ R~ ~ C=O+H+ - c+-oH+R3C-OOH - / / R2 R2 R1 R:x\H o \ \ 0 0 2/ > > cIIO한 브 노 R1 -CI I -O R2 + R3 -CI I -O H (36 -2) 가장 공업적으로 응용되고 있는 것이 Unio n Carb i de 51) 가 개발 한 카프로락톤 제조를 위한 락톤 제조이다.
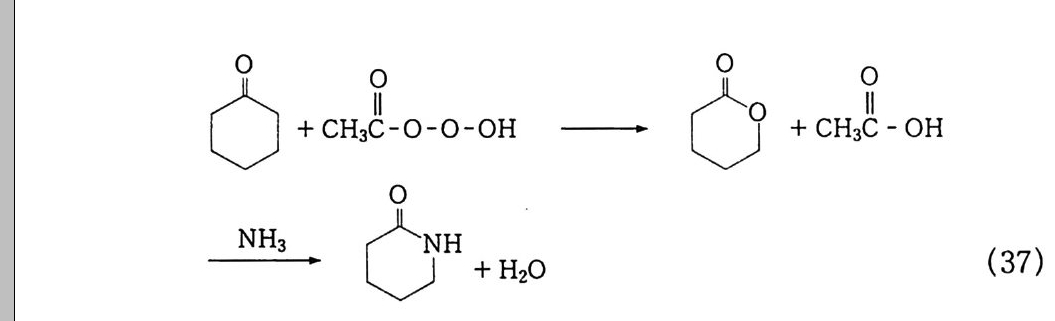 두\』 :H -。 0+-0H-02H ° 一 〔。〈 + CH3?C - OH (37)
두\』 :H -。 0+-0H-02H ° 一 〔。〈 + CH3?C - OH (37)
m-CPBA 를 사용하여 락톤도 합성하였다 .55) 3) 케타진 형성 56a) 케톤과 과산화수소 혼합물에 암모니아 가스를 통과시키면 반웅 식 (38) 처럼 케타진이 형성된다. 이것을 물로 가수분해시키면 (l)
과 같이 히드라진과 케톤이 되고, 청산을 부가시키면 히드라진 니 트릴 (~)를 거쳐 산화에 의해 아조니트릴(예를 들어 AIBN) 을 제 조한댜 이것은 과산화물과 더불어 중합 개시제 및 발포제로 널리 이용된댜 RI RI RI 2 \ C=O + H2 02 + 2NH3 ..N..a:2.H.:P.O::4. :..\ C=N -N =C/ + H20 R: /R2/ lHCN \R 2 RI RI RI NH2 ·NH2(1 +)R ; C=O R /\2 ICC _ NN( H _N) H02 ICC2N / - 판\ 아조니트릴 56b) (Azonit ril e ) (38) 4. 4. 3 방향족 화합물과의 반응 1) 래디컬 반응_펜톤 시약 4 . 1. 3 의 1) 에서 설명한 펜톤 시약과의 반응이 전형적인 예다. 한 전자를 주고받음으로써 산화환원 (Redox) 반응을 일으키는 금속은 과산화수소의 분해를 촉진한다. 앞에서 본 바와 같이 이 반
응은 ·O H 래디컬이 중간체로 생기고. 이것이 구전자 반응으로 방 향족 화합물과 반응하여 페놀을 생성한다. e . s . r 측정으로 히드록시 시클로헥사디에닐 {h y drox y cy c lohexadie n y l) 래디컬이 공명으로 안정하게 존재함을 증명하였다 . 57 , 58)
 >O/\ 二O H 二〔 \O H (39)
>O/\ 二O H 二〔 \O H (39)
·OH 래디컬이 구전자 성질을 가졌다는 증거는 전자를 주는 치 환기를 가진 방향족 화합물이 미치환 방향족 화합물보다 더 빨리 반응한다는 점과 o- 및 p-이성체를 얻는다는 데서부터 증명되었다. 이 반응에는 용액의 p H 가 큰 영향을 미친다. 톨루엔의 경우 pH 를 낮추면 디벤질 (d i benzy l) 양이 증가하며 , 벤젠의 경우에는 반대 로 페놀-디페닐 비가 증가한다 .
 0 一 0언 + 0-0
0 一 0언 + 0-0
 |o덱 ¢CH3 + 6CH3 °: 6CH3 0 : ¢CH3 亡 CH3
|o덱 ¢CH3 + 6CH3 °: 6CH3 0 : ¢CH3 亡 CH3
이 현상은 중성-약산성 용매 중에는 히드록시 시클로헥사디에 닐 래디컬이 크레졸로 쉽게 산화되고. 강산 용매에는 H20 가 빠짐 으로써 벤질 래디컬이 형성되기 때문이라고 설명할 수 있다 . 59~6 1)
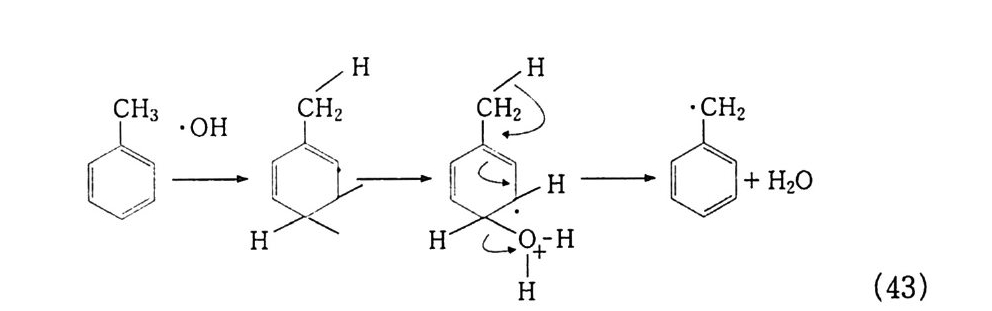 H (43)
H (43)
2) 구전자 히드록시화 H-과0-산\ ? 화저수나소 를히 드산로 촉옥매소하늄에 이서온 ,사 O용H할+ 이때 에생성는된 다히.드 로방퍼향옥족소환늄에 이대온한 H 구전자 반응성은 방향족환의 수산화에 많이 응용하였다. H\ 0-0\ H 느H+ H-H-/ -o ~ + ·- o \H . OH+ . OH+ (44)
이 경우 단일화만 일어나지 않고 계속해서 여러 수산화가 일어나 기 때문에 메시틸렌 62) 같이 OH 가 들어갈 곳이 막혀 있지 않으면 폴 리 수산화 화합물이 생성된다. 그러므로 펜톤 시약의 경우 벤젠에 서 페놀을 선택적으로 제조하기는 대단히 어렵다.
 C: CH 3 二 C: CH3 (4 5 )
C: CH 3 二 C: CH3 (4 5 )
그러나 펜톤 시약과는 달리 이 경우 첫째 ` OH 그룹에 대하여 메 타 위치의 치환은 관찰되지 않았고 63) 둘째, or t ho/ p ara- 치환제의 비는 사용하는 산 촉매의 성질에 의하여 좌우된다 回 그러므로 페 놀로부터 카테콜과 히드로퀴논을 선택적으로 제조하는 것이 가능 하다. 실질적으로 H 2 0 2 -HF 와 H2 0 2 -H Cl04 (C F3S 03H ) 이 공업적으 로쓰인다.
 。
。
알킬 벤젠인 경우 프리델 -크라프트 알킬반응의 촉매가 쓰이는
데. 알킬 벤젠의 수산화에는 알킬 그룹의 이동 또는 탈리반응이 동 반된다.
다음과 같은 경우 벤젠화에 수산화가 일어난다.
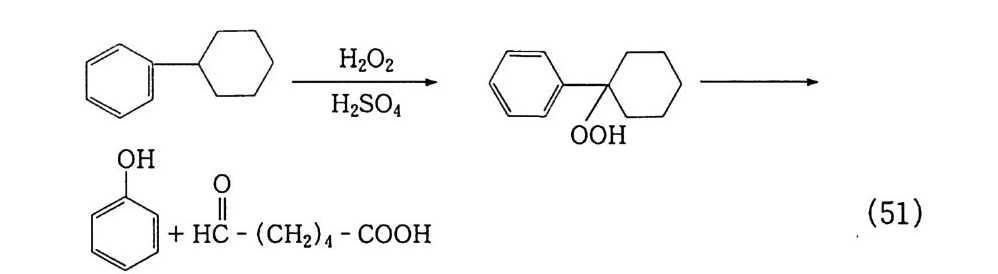 :6O
:6O
공업적으로 히드로퀴논을 제조하는 공정은 식 (47) 에 따라 제조 한다. 즉,
® 프랑스 Rhone-Poulenc 은 이가페놀을 85~90 %로 얻는다. 1;5. GG) ® Univ e rsal O il은 HF 를 사용하여 H Q와 카테콜을 각각 52 % 와 35 %로 제조한댜 6 - 1. 67) ® 이탈리아의 Bric h im a S. p. A 는 무기. 유기산 코발트 염을 사 용하여 이가페놀울 90% 로 얻는다 .6 8) 이 이외에 다음 방법이 사용되고 있다.
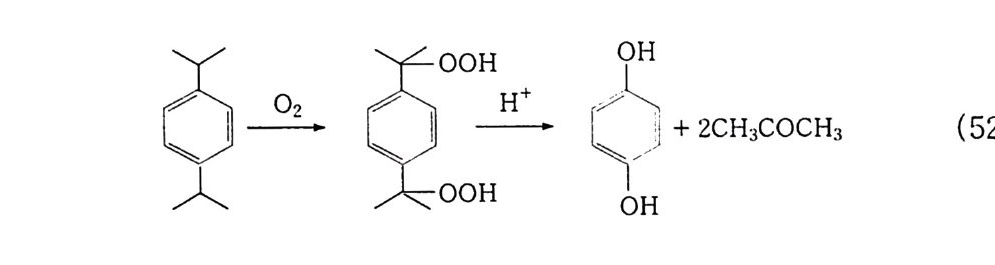 二《 一02 00 00 HTH 一 \OO\HH + 2CH3COCH3 (52)
二《 一02 00 00 HTH 一 \OO\HH + 2CH3COCH3 (52)
4. 5 헤데로 원자 과산화물 69 ) 지금까지는 탄소를 포함하는 과산화물에 대하여 기술하였다. 이제 유황과 인을 포함하는 과산화물에 대하여 간단히 서술하고자 한다. 4. 5. l 유황 과산화물 유황 과산화물의 종류를 그림 4-1 에 나타내었다. (l) 으(퍼옥시모노황산)는 Caros 산이라고 하며 H202, K2S 2 아이나 암모늄, 유황 염으로부터 직접 만든다 .70) (!)!?_(퍼옥시모노 포타슘유산염)는 Oxone 이란 상표로 팔리고 있 는데 유기용매에 잘 녹지 않아 용도에 제한이 있지만 알코올을 케 톤과 에스테르로, 아민을 니트로소 화합물로, 그리고 술파이드를
 OIISIIO( 0 0
OIISIIO( 0 0
술폭사이드로 산화시킨다四 유기용매에 잘 녹지 않으므로 상이동 촉매를 사용한다(식 53). OIIs OIISIIO R-S-R KHCSH025C, l2B - uH4 N2 0 +. B 2r5-. c( c a t) R R + R R (53)
디메틸 아니솔의 메톡시 그룹에 대해 o-또 는 p-위 메틸기를 선 택적으로 레지오 산화반응하여. 메톡시 메틸 안식향산 알데히드의 제 조는 K2S 2 아 -C uS04 계 에 서 잘 일 어 난다 . 72)
 CH30- Q-CHC3 H3 KC2 HS2 3 0 C 8 N , C-HuS2 00 4 CH3 0 - Q-CHC3 H O (54)
CH30- Q-CHC3 H3 KC2 HS2 3 0 C 8 N , C-HuS2 00 4 CH3 0 - Q-CHC3 H O (54)
화합물(한은 다음과 같이 합성한다 .7 3) 0 0 2R -S 02C l + H2 0 2 ~H2 0 --2E0t. C - C HCl3 R-S0IIII -0-0-S0IIII -R (55) 이 종류는 몇 가지밖에 알려져 있지 않다. 죽 , 디메틸 비스술포 닐 과산화물 (m, p 79 ·c) 은 실온에서 1 주일 간 안전하나 74) 대부분 은 상온에서 불안정하다. 1 급 및 2 급 아민은 4- 니트로벤젠 술포닐 과산화물과 -78 ° C 에 서 이민을 생성하고 이것은 좋은 수율 (66~91 %)로 카르보닐 화 합물로 전환한다( 식 56) _75 ) R1CH2NHR2 + (R S020 ) 2 EtO A-7c8.. C N aOH [R1CH2 -N (R2)0S02R] 브-- R1CH=NR2 + RS03- H20 ► R1CHO + R2NH2 (56) 비대칭 술포닐 과산화물 (산)와 (~)는 일반적으로 대칭물보다 더 불안정하다. 그러므로 이 비대칭 성질이 대칭물과는 아주 다른 반
응으로 유도된다. 몇 개의 아실 술포닐 과산화물(산)는 과산화 술포 닐 크로리드와 염기 존재하에서 반응시켜 얻는다 . 76) 벤조일 4- 톨루엔 술포닐 과산화물은 트리페닐 포스핀과 반응하 여 트리페닐 포스핀 산화물을 생성한다(식 57).
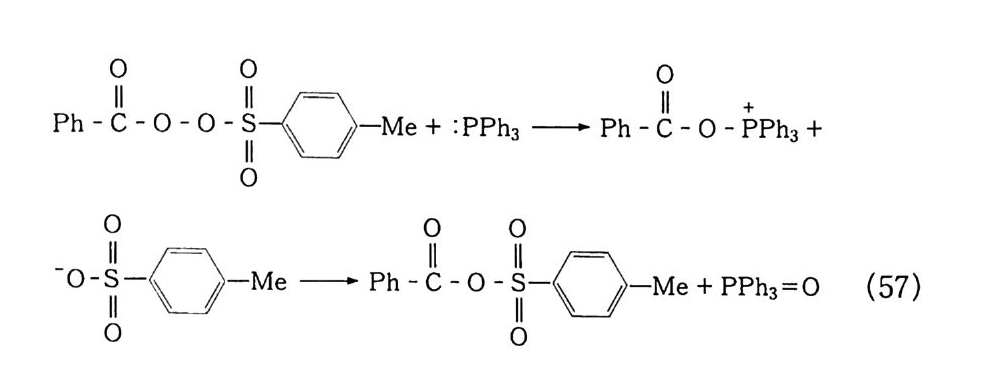 Ph-t o-o -[- O-M e+: PP ha 一 PhJ _0- PPh,+
Ph-t o-o -[- O-M e+: PP ha 一 PhJ _0- PPh,+
슈퍼옥사이드 래디컬 음이온 (0/) 은 4 가지 중요한 역할을 한 다. 77) 즉, 염기로서 탈프로톤, 래디컬로서 H- 원자 탈리, 음이온으로서 구핵반응 그리고 전자 전달체로서 작용하는 것이다. 또한 슈퍼옥사 이드는 적당한 환원제로서뿐만 아니라 약한 산화제로도 작용하는 것으로 알려 졌다 .78) 디술파이드, 티오술피네이트 또는 티오술포네이트가 슈퍼옥사이 드 음이온과 반응하여 생성된 퍼옥시 유황 중간체들에는 산화 기능 이 있다는 보고가 있다 .79) 2- 니트로, 4- 니트로벤젠 술포닐 크로리드는 쉽게 아세토니트릴 용매 중에서 02 효과 반응하여 4- 니트로 벤젠 술포닐 퍼옥시 중간체 를 생성한다.
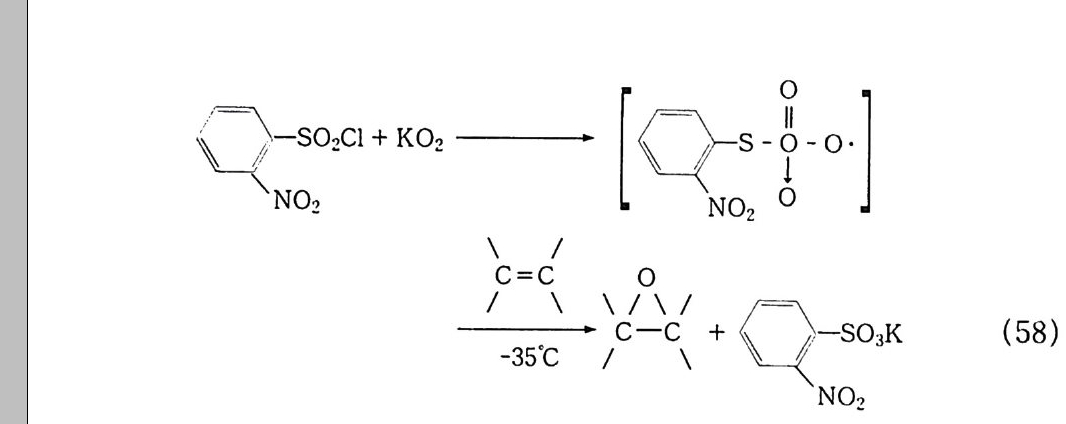 \: ,0 2Cl+KO2 一 [\\ \ -o l
\: ,0 2Cl+KO2 一 [\\ \ -o l
이것들은 한 개 또는 두 개의 이중결합을 가지고 있는 알켄에 아 주 높은 수율과 레지오 선택성으로 에폭시화한다 .MO, Kl ) 이 결과를 표 4-9 에 나타내었다. 4.5.2 인 과산화물 인 과산화물의 구조를 그림 4-2 에 나타내었다. 유기 인 과산화물은 일반적으로 불안정하다. 몇 가지 안정한 유 기 인 화합물이 보고되었지만 대부분은 분리하지 않고 가능한 중간 체로 연구되었다. 안정한 비스디페닐 포스피닐 과산화물은 식 (59) 와 같이 합성되 었다 .82)
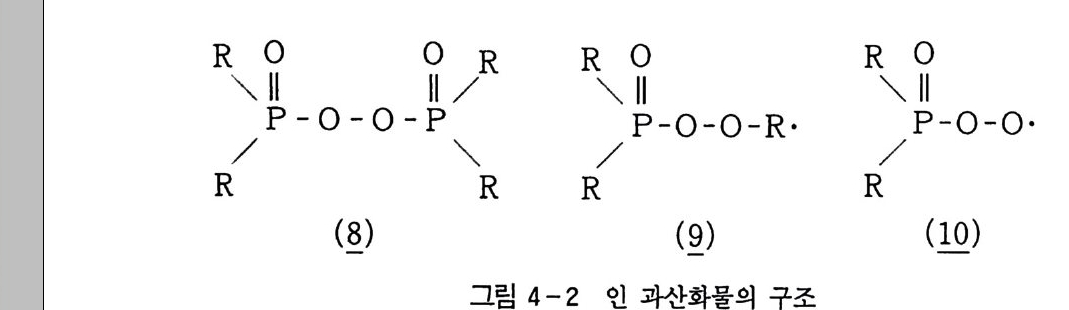 RR/\ OP |I -0-0-POII \/ RR RR/\ 0P || -0-0-R· RR/\ P0 II -0-0·
RR/\ OP |I -0-0-POII \/ RR RR/\ 0P || -0-0-R· RR/\ P0 II -0-0·
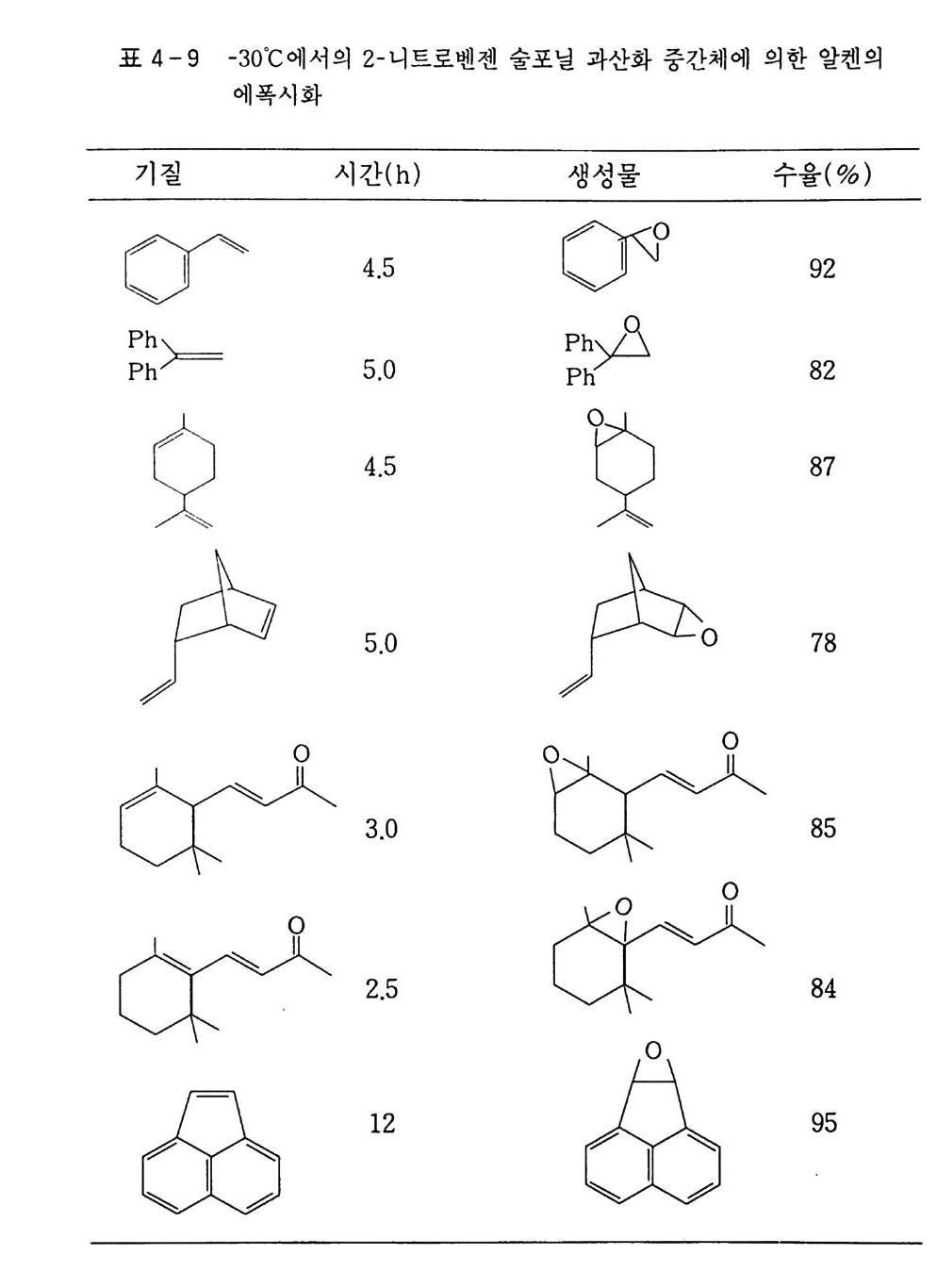 표 4-9 -30 ° C 에서의 2 - 니트로벤젠 술포닐 과산화 중간체에 의한 알켄의
표 4-9 -30 ° C 에서의 2 - 니트로벤젠 술포닐 과산화 중간체에 의한 알켄의
PPhh\/ ’ P°II - C l + M 20 2 PP► hh \/ °P|| -0-0-P°II \, / PPhh (59) 화합물델)은 여러 술파이드와 반응하여 2 차 산화 없이 좋은 수 율로 온화한 조건에서 술폰을 생성한다(식 60).8 3) R -S -R' P+h \P 『 -0 -0 -P? / -P h R\ S+ -^0 -~P + -o -~P, ,P h Ph / \ Ph R/' \ Ph \ Ph M20e.Cc N ’ RR \/' S=O +PP hh \/ ?P -0-P? \/ PPhh (60) R, R' = alky l, ary l 비대칭 인 과산화물류들은 약간 알려져 있다. 과산화 에스테르 也) 제법은 참고문헌 8 4) 을 보기 바란다. 이 화합물은 에틸에테르 중 5 °C 에서 며칠간 안정하다. RO\ OP| I -C l + Na 아 C/ -RR11 드R O \OPI I -OOC-RR1 1 (61) RO/ \ R2 RO/ \R2 (브) 디페닐 포스포닉 크로리드를 아세토니트릴 용매와 온화한 조건
에서 슈퍼옥사이드 음이온 래디컬 (0 2 ;) 과 반응하여 디페닐 포스포 닉 퍼옥시 래디컬 중간체 (旦)를 만드는데, 이것은 올레핀의 에폭 시화, 술파이드를 술폭사이드로, 티오아미드를 탈류 반응을 통하여 아미드로 그리고 트리알릴 포스핀을 포스핀으로 강력하게 산화시 킨다. 또한 중간체(프)는 디메틸 피롤리딘 N- 옥사이드 (DMPO) 와 래디컬 아닥트 (브)을 형성한다 .
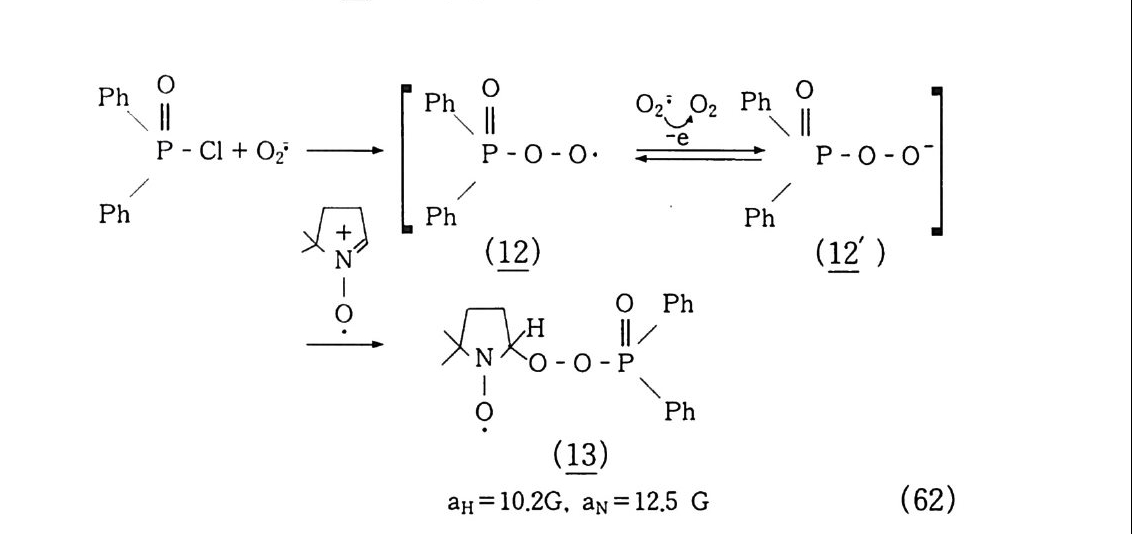 Ph> Lei + o ,一 [ 三 。 。 >-OP II- 02(。, • •
Ph> Lei + o ,一 [ 三 。 。 >-OP II- 02(。, • •
일반적으로 인 과산화 래디컬은 저온에서 인 염화물보다 슈퍼옥 사이드와 쉽게 반응하고 또 비교적 안정하므로 앞으로 많이 사용되 리라고 생각한다. 참고문헌 1 ) D. Swem, Orga nic Peroxid e s, Wi le y -Inte r scie n ce, New York, vol. I a) 1970, b) vol. 11:1971, c) vol. ID 1972. 2) Ullmann's Encyc l op ed ia , vol. 19, p.1 98~233 (19 91 ) .
3) Kir t-O t/ zm er Ency cl op e dia of Chemi ca l T/ze chnolog y, vol. 17, p.2 7~89, Joh n Wi le y & Sons, New York ( 1982 ) . 4) S. Pata i , The Chemi st ' )' o f Peroxid e s, Joh n Wi le y & Sons, New York, Chic h este r (19 83 ) . 5) A. G. Davis , Orga nic Peroxid e s, Butt er worth , London( 1961 ) . 6) E. G. E. Hawkin s , Orga nic Peroxid e s, E and F. F. Sp on , London( 1961 ) . 7) J. C. Mi tch ell, C hem. Soc. Rev., 14, 339(1 9 85) . 8) D. Swem, D. H. R. Barto n and D. W. Ollis eds., Comp re hensiv e Org, Chem. , vol. I, p.9 0 9, Perga m on Press, Oxfo rd ( 1979 ) . 9 ) B . C. Brodie , Jus tu s Lie b ig s Ann. Chem. , 108. 79 ( 1858 ) . 10) 「 日 本工業新聞」( 1983. 1. 21 .) 11) 日本油脂(株) , 「有機過酸化物」, 카탈로그,p .5. 12) Jea n-Pie r re Schin na nn and Serge Yvon Delavarenne, Hy dr og en Peroxid e in Orga nic Chemi st r y , edit ion et document at i on ind ustr iell e. 13) Ulhnann's Ency cl op ed ia, v ol.13, p.4 43~466(1 9 89) . 14) J. W eis s , Advances in Cata l ys i s , vol. 4, p.3 43, Academi c Press (19 52). 15) 문헌 1) b) 의 p26 9. 16) J. Rouchaud, Ind Chim . Belg., 37, 741 ( 1972) . 17) J. E. Mclssac et al., J . Org. Chem., 37, 1037(1 9 72) . 18) 문헌 1) a) 의 p.3 13. 19) 문헌 1 ) a) 의 p.2 5. 20) J. A. Vesely and L. Scherin g, J. Org. Chem., 3535, 4028 ( 1970) 21 ) K. S. Pit zer . J. Am. Chem. Soc., 7 0, 2140(1 9 48) . 22) S. W. Benson, J. Chem. Phys . , 40, 1007 (19 64) .
23) Kirk -Ot hm er, 1st S up pl. vol. , p.6 6 2, 1957. 24 ) Deg us sa, DE 1, 170, 960 (19 62 ) . 25 ) Prop ylo x, EP 4407 ( 1978 ) . 26) Produit s C him i qu es Ug ine Kuhlmann, EP 22396( 1979) . 27) Degu s sa, DE 2, 018, 713(1 9 70). 28) R. N. McDonald, R. N. Ste p p e l and J. E. Dorsey, Org. S yn t h . , 50, 15( 1970) 29) P. R. H. Sp ea kman, Chem. Ind., 579, London( 1978) . 30 ) Y. Og at a and Y. Sawa ki, Tetr a hedron, 23, 3327 ( 1967 ) . 31 ) Food Machin e ry , US 3, 485, 869 ( 1967 ) . 32) S. W. Benson, J. Chem. Phy s., 40, 1007(1 9 64). 33) K. S. Pit zo r, J. Am. Chem. Soc., 7 0, 2140(1 9 48). 34) A. H. Sheon et a l., Can. J. Chem., 41, 1819(1 9 63) . 35 ) K. W. Lee and K. Gr ies baum, Chem, Ber. , 114, 3273 ( 1981 ) . 36) K. W. Lee and K. Gr ies baum, J. Org. Chem., 49(4) , 679(1984). 37) Z. Raciz e wski , J. A . Chem. Soc., 82, 1267(1 9 60). 38) Shell, C. A. 52, 16367f. 39) M. Iga r ashi and H. Mi do ri kaw a, J. Org. Chem., 32, 3399( 1967) . 40) C. A. 89, 59832 P, Ug ine Kuhlmann. 41 ) C. A. 86, 29606 P, Un ion Carbid e . 42) French Pate n t, 2378773, Ug ine Kuhlmanm( 1977) . 43) R. Landau, G. A. Sulliv a n and D. Brown, Chem. Technol., 602~607 (19 78) . 44) H. Kwa rt and D. M. Hoff ma n, J. Org. Chem., 31, 419(1 9 66) . 45) K. D. Bin gh am, G. D. Mea kins and G. H. Whitam , Chem. Comm., 445(1 9 66) . 46) K. A. Saege b a rth, J. Org. Chem., 24, 1212(1 9 59).
47) US. Pate l lf , 2773909, Shell, C. A. 52, 10153b. 48) French Pate n t, 1298983, Ug ine Kuhlmamn, C. A. 57, 15868b. 49) C. J. Nort on and R. E. Whit e, Advan. Chem. Se,:, n o. 51(1 9 65) . 50) W. Di et h e y, M.lnckel and H. Ste p ha n, J. Prakt. Chem., 154, 219 (19 20). 51 ) N. A. Mi las and A. Golubovic , J. Am. Chem. Soc., 81, 3381, 5824, 6461 ( 1959) . 52) N. A. Mi la s, S. A. Ma rris and P.C . Panag iot a k os, ibi d . , 61, 2430 (19 39). 53) A. Baeye r and V. Vil liger , Ber. Deut. Chem. Ges., 32, 3625 ( 1899) . 54) Unio n Carbid e , US 3064008( 1960) . 55 ) G. Mag nu sson, Tetr a hedron Lett er , 2713 (19 77 ) . 56) a) 이규완, 김성보, 장우상, 마 l y draz i ne hy dr ate 제조 연구보고 서」, 한국화학연구소 및 인용문헌(1 986. 6) . b) 이규완 , 한국특허 제 10094 호(1 98 1. 7. 29) . 57) W. T. Di xon and R. 0. C. Norman, Proc. Chem., 97( 1963) . 58) W. T. Di xo n and R. 0. C. Norman, J. Chem. Soc., 4857( 1964). 59) J. R. Rind say -S m ith and R. 0. C. Norman, J. Chem. Soc., 2897 (19 63). 60) C. R. E. Jef c o ate , J. R. Rind say -S mi th and R. 0. C. Norman, J. Chem. Soc. , par t B, 1013 (19 69 ) . 61) S. L. Cosgr ov e and W. A. Wate r s, J. Chem. Soc., 1726(1 9 51 ). 62) D. H. Derby sh i re and W. A. Wate r s, Natu r e, 165, 401 ( 1960) . 63) J. D. McClure and P. H. Wi llam s, J. Org. Chem., 27, 24(1 9 62) . 64) J. A. Versely and L. Schmerlin g , J. Org. Chem., 35, 4028 ( 1970) . 65) J. Varagn a t, Ind. Eng. Chem. Prod. R es. Dev., 15, 212 (1976). 66) Ger. Pate n t, 2658545 Rhone-Poulenc, C. A. 87, 184204v.
67) US. Pate n t, 3461170, Univ . O il . Prod. C. A. 68, 68700h. 68) French Pate n t, 2 218312, Br ich im a, S. p. A, C. A. 81, 151787k. 69) Y. H. Ki m , Orga nic Peroxid e s, W. Ando ( Eds ) Chap ter 8, p.38 7~423, Joh n Wi le y & Sons( 1992). 70) G. B. Payn e , Org. Sy n. , Coll. vol. 5, 805(1 9 73) . 71 ) I. A. Perl , ibi d . , Coll. vol. 4, 972 ( 1963 ) . 72) F. M. Hauser and S. R. Ellenberge r , Syn t h esis , 723 (19 87) . 73) R. L. Dannley, J . E. Gag en and 0. J. S te w a rt, J. Org. Chem., 35, 3076(1 9 70) . 74) C. J. My al l and D. Pletc h er, J . Chem. Soc., Perkin Trans, 1, 953(1975) . 75) R. V. Hoff m an, J. Am. Chem. Soc., 9 8, 6902(1 9 76) . 76) R. Hi sa da, M. Kobay as hi and H. Mi na to , Bull. Chem. Soc. Jpn ., 45, 2035 ( 1972) . 77) D. T. Sawy er and J. S . Valenti ne , A cc. Chem. Res., 14, 393 (19 81 ) 78 ) D. T. Sawy er , M. J. Gi bi a n , M. M. Mori so n and E. T. Seo, J. Am. Chem, Soc., 1 00, 627( 1978) . 79) S. Oae, T. Tanaka and Y. H. Kim, Terrahedron, 37, 37(1 9 81 ). 80) Y. H. Kim and B. C. Jun g , J. Org. C hem., 48, 1562(1 9 83) . 81 ) Y. H. Kim and K. S. Kim and H. K. Lee, Terrahedron Lett. , 30, 6357(1 9 89). 82) R. L. Dannley and K. Kahre, J. Am. Chem. Soc. , 87, 4085 ( 1965 ) . 83) S. C. Lim and Y. H. Kim , Hete r oato m , Chem. , 1 , 261 ( 1990) . 84) G. Sosnovsky and H. E. Zaret, J. Org. Chem., 3 4, 968( 1969).
제 5 장 슈퍼옥사이드에 의한 산화 5. l 산소분자의 환원에 의해 생기는 산소종 생체 내에서 산소는 산화제 또는 탈수제로서 작용하여 효소제인 다이옥시게나아제 (D i ox yg enase) 와 같이 산소분자를 그대로 반응 시키는 경우를 제외하고는, 전자 수용체 역할을 하여 최종적으로 4 개의 전자를 받아 환원됨으로써 물이 된다. 이러한 환원의 대부분 의 경우에는 단계적으로 1 전자 환원을 일으켜 산소, 물의 중간 단 계에 있는 몇몇 종류의 환원종. 예를 들면 슈퍼옥사이드 (Su p er oxid e ) 이온 (0 간 이나 과산화 이온 (02-2) 등이 산소 산화반응에서 중요한 역할을 하고 있음이 실증되었다. 5.1.1 슈퍼옥사이드 1~3) 표 5 一 1 에서 보는 바와 같이 반응계의 산도에 따라 여러 가지의
표 5 -1 산소분자의 환원종 산성 Eo(V) 알칼리성 Eo(V) 탈프로톤종 02 + e -+H + -0.1 3 02 +e - - 0' -0.563 0 2召 슈퍼옥사이드 -·OOH ·OOH+e -+H + + 1.49 5 O· +e+Hp +0.4 1 3 살: 퍼옥사이드 -H2° 2 --OOH+-OH Hp 2+ e -+ H + +0.71 OOH+e-+Hp -0.262 O· : 산소원자 - ·OH+Hp - ·OH+2- 0 H 음이온 래디컬 ·OH+e -+H + +2.85 ·OH+e---O H +2.02 02- : 옥사이드 -H2°
산소종이 생성된다. 프로톤 존재하에서는 히드로퍼옥시 래디컬( ·OOH). H2 0 2 , 수산 래디컬( ·OH) 그리고 물이 생성된다. 알칼리성 수용액에서는 이 것으로부터 하나의 프로톤이 해리한 슈퍼옥사이드, 히드로퍼옥시 음이온 (OOH 기 수산 래디컬( ·OH) 과 수산 음이온 (OH - ) 이 생성 된다. 이 환원종의 전 프로톤이 해리된 형태를 표 5-1 의 우측란에 나 타내었다. 슈퍼옥사이드 이온 02 먹는 기저 상태의 산소분자보다 0.43eV 낮은 에너지를 갖는 상자성 산소분자종이다. 슈퍼옥사이드 이온을 확인하는 방법은 U. V. 스펙트럼 5) 으로, 산 소를 포함한 물을 펄스 전자선 조사로 생성된 0 2 먹는 l=240nm, ·OOH 는 l=230nm 에서 측정되었다. 이 방법보다는 e. s. r 스펙트럼 6, 7) 이 용이해 , 촉매에 흡착시킨 상태나 전이 금속과 착체화한 상태로 많이 측정되었다 . 그러나 그 상태나 매체에 의하여 스펙트럼 형태나 G- 값이 변하므로 간단하 게 측정할 수 있다고는 말할 수 없다.
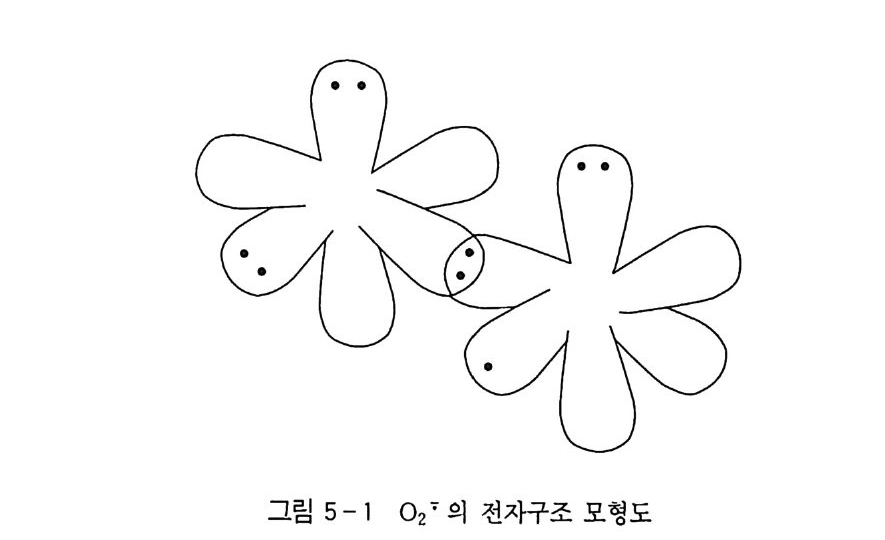 그림 5-1 02-;-의 전자구조 모형도
그림 5-1 02-;-의 전자구조 모형도
비프로톤성 용매 중 전해환원으로 생성된 0/ 는 실온에서 수분 에서 수시간의 반감기를 가지고 있다산 예를 들면 디메틸포름 아미 드는 o ·c 에서는 76 분의 반감기를꼬 그리고 77 °K 에서는 수주간의 수명을 가지고 있댜 8) 1) 제조 슈퍼옥사이드 발생은 앞에서 설명한 바와 같이 산소분자의 1 전 자 환원에 의한다. 환원제로는 다음을 들 수 있다. ® 알칼리 금속이나 알칼리 토금속이 있다. M+02-M02 (1) 이것들을 산소와 가열하든지 또는 액체 암모니아 중에서 만든 다 .2, 10) 나트륨을 사용하는 경우 -78 ·c 에서 반응시키 면 Na20 와 Na202 혼합물이 생성된다.
® 전자를 내어서 케톤이 되기 쉬운 성질을 가진 케틸 (Ke ty l) 래디컬 (R 2 C-O · )을 생성하는 계에서는 이것이 산소를 슈퍼옥사 이드로 환원한다. ¢ ¢ ¢ ONa \ C=O 一Na \ C• -ONa 一o 2 \ c / / / / \ ¢ ¢ ¢ 00· ¢\ 一 C=O + Na02 (2) / ¢ ® 전이금속 이온에 의한 산소의 환원 8 족에 속하는 금속 이온은 산소와 여러 가지의 착체를 형성한다. oo 0-; / / 。 。 | Con + 02 一 Con 一 com (3) (1) (2) 식 (3) 의 Co 와 같이 1 : 1 착체는 특히 안정하다. e. s. r 로부터 이 착체는 (l) 와 (~) 공명 혼합체 임을 알 수 있다 .6) ® 그리고 마지막으로 산소의 전해 환원이 있다. 이에 관해서는 참고문헌 6, 11, 12) 올 보기 바란다. 슈퍼옥사이드 이온의 물리화학적 성질은 표 5-2 에 실었다.
표 5-2 슈퍼옥사이드 이온의 물리화학적 성질 항목 수치 결합 거리 1.3 3A 결합차수 1.5 결합 해리 에너지 88.8 , 69.92 kcal • mor1 AHf ° -24 . 6 士1. 8 kJ • mor1 ASf° 79 J • mor1 • K- 1 신축 진동 1,14 5 cm- 1 최대흡수파장 255nm(e=1 .46 0, 아세토니트릴 중) 462 nm( 피 리 딘 중) ESR g값 g.J..= 2.008, g // =2 . 083( 아세토니트릴 중) 염기도 K =2.5 X 108 수화열 -418kJ • mor1
슈퍼옥사이드 염도 약간 알려져 있지만 K20 가 가장 안정하다. 이 문헌은 Ga y -Lussac(1810 년)시대부터 이미 알려졌었으나 최종적 으로 X 선 결정해석 (K20, K202 에 대해서)에서 0 2 ; 의 존재가 확인 된 것은 1937 년이다 .13> K 2 0 는 물에 의해 다음 식과 같이 분해한다. 2 K20 + 2 H20 一 2 MOH + 2 • OOH 2 ·OOH 一 H 요 +02 (4) CO2 와 반응하여 K02 1 g당 0.34 g의 산소를 발생 한다. 이 성 질 울 이용하여 우주선, 잠수함. 고산등산의 산소 보급에 사용된다. 동 중량의 산소통보다 훨씬 다량의 산소를 내고. 호흡에 의하여 배출 되는 CO2 를 효율 좋게 흡수하는 이점이 있기 때문이다 .2)
2 K20 + 2 CO2 !.:_모l.. K2 C 03 + 02 (5) 슈퍼옥사이드는 전자구조로부터 알 수 있듯이 음이온과 래디컬 성질을 반영해서 구핵성과 래디컬 반응성을 가지고 있다. 또 환원 성과 산화성이라는 아주 다른 역의 성질도 겸비하고 있다. 산화 0/ + X —一 X++022- 환원 0/ + Y —-y - +02 (6) 식 (3) 에서 보듯이 H202 를 생성하면 산화제가 산소를 생성하는 환원제가 된다 . 2) 산화반응 2 급 알코올은 THF 중 Na02 로 산화시키면 대응하는 케톤이 된 다. 반응기구는 다음 식 (7) 에서 보는 바와 같이 히드로 과산화물 래디컬에 의하여 개시하는 자동 산화반응이다 .14) R2CHOH + Na02 一 R2CHONa + • OOH 2 ·OOH 一 02+H 요 RCHONa + • OOH 一 R2CONa + H202 R2CONa + 02 - R2C = O (7) 비프로톤 용매 중에서 아에 의한 산화반응은 다음과 같은 세 가 지 형태를 들 수 있다. ® 전자이동 반응으로 개시되는 산화반응
® 수소원자 Abs t ra ct로 개시되는 산화반응 ® overall 반응은 산화반응이지만 02 가 구핵 시약, 염기 또는 _전자 환원제로서 작용하여 개시되는 산화반응 이 중 ® 반응은 비프로톤성 용매에서는 02 가 안정하므로 반응이 일어나기 어렵다. ®반응은 다음과 같다. 9, 10- 디히드로 안트라센을. 피리딘 중에서 Et 4N ·Cl04 를 지지 전해질로서 산소 존재하에 -0.8 V(sec) 정전위 전해하면 안트라 퀴논과 안트라센이 생성된다 .15)
 CXX)+0z 전해+ o二co (8)
CXX)+0z 전해+ o二co (8)
이외에 N, N' -Dihy d rop h enazin e , N, N' -Dihy d rofl ua vin , N- Benzy l - 4, 4 -d i hy dron i co ti nam i d 도 이 방법으로 산화가 가능하다. 02; 는 N it ro 기에 의해 활성화된 벤질 위의 수소를 빼어 반응을 계속함으로써 대응 니트로 안식향산을 생성한다 .16)
 〔人x N 02+ 。2 -· •
〔人x N 02+ 。2 -· •
®반응은 카테콜 a- 토코페놀. 아스코르브산 둥 산성을 나타내 는 화합물과 반응하여 1 7 ) a- 치환 케톤, c-c 결합 절단 반응 1 8 ) 이 일어난다.
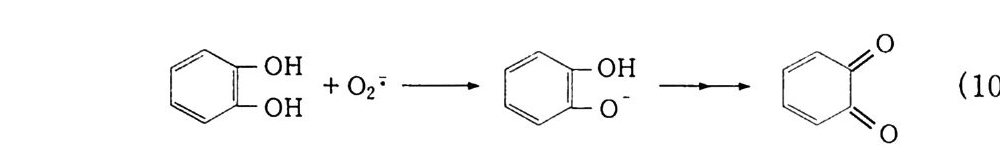 二 :[ +02· 一 二 °0H --二 ° (10 )
二 :[ +02· 一 二 °0H --二 ° (10 )
R-C0II -00II - R 0/ . H2 0 . aR-\C -0- H (11 ) 악에 의한 올레핀의 에폭시화 반응은 홍미가 있댜 Baiz e r19 > 등 은 Meth y l -D iet h y l -m alonate ( PKa= 15~16), 또는 디페닐아세 토나이트릴 (D ip hen y lace t on it r il e) (PKa = 17.5 ) 의 존재하에서 0.2 M(Bu)4NBr/DMF 용액에 산소를 불어 넣으면서 수은 음극에 대하 여 -0.1 V vs SCE 정전위 전해를 행하면 에폭시화가 일어난다.
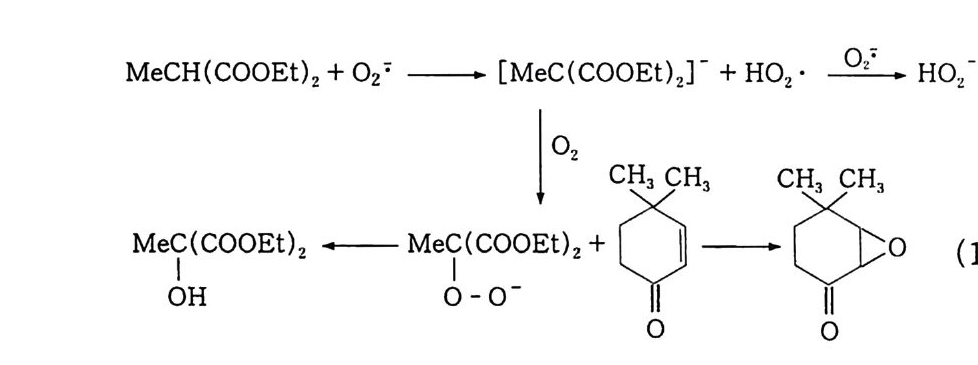 MeCH(COOEt )2 + 02 ~ 一 [MeC(COOE t ) 2 「 + H02 • 으 HOZ -
MeCH(COOEt )2 + 02 ~ 一 [MeC(COOE t ) 2 「 + H02 • 으 HOZ -
3) 구핵반응 02 은 구핵 시 약으로서 Alky lh alid e , 카르보닐 화합물과 반응한 다. Alk y lhal i de 와의 반응은 다음과 같다.
RX + 0/ -一 R02 • + x- 1,, ')) R02 • + 0/ 一 R02- + 02 1, Ro2- + RX - ROOR + x- (15 ) 상대적 반응속도는, 알킬기에 대해서는 벤질 >1 급 >2 급 >3 급> 방향족 순이고 할로겐에 대하여는 Iodid e> Bromi d e> Chlorid e 순이댜 20) DMF 중에서 할로겐화니트로벤젠과 02 가 반응하여 0-, p-이 성체에서 반응이 일어나 대응하는 페놀이 생성된다 .21)
 x上〔〉 N +002 2 N 02 (16 )
x上〔〉 N +002 2 N 02 (16 )
meta 이성체는 반응을 하지 않거나, 하더라도 반응속도가 대단 히 느리다 . 최근 말론산디에스테르, 빙초산에스테르, 아세틸아세톤 의 카르본산과 탄소수 2~6 개의 a, (J)-디브로모 알칸 공존하에 DMF 중 산소를 넣으면서 전해 환원하면 a, p-Di bromoalkane 보 다 탄소수가 하나 더 큰 지방족환화합물이 고수율로 생성되는 것을 발견했다 . 22)
 EEtt O0 0O CC /\ CH2 + BrRnBr ----o-- :-:= --EE tt O0 0O CC \/ C/\ \Rn
EEtt O0 0O CC /\ CH2 + BrRnBr ----o-- :-:= --EE tt O0 0O CC \/ C/\ \Rn
이 반응기구는 다음 Scheme 과 같이 추측된다 .
RR丈< CH 2 + Br(CH2 \ B r 一 RR\/12 c /\ (HC H 라 Br+ Br- RR\1/ 2 C \( CH2 )n Br 一RR \/12 \ C/ \ ' 一\ /( . / C H 라 + Br- R1. R2 : 전자구인기 (EWG) Oi; 에 의한 카르본산으로부터 Carban i on 의 생성반응 (0 / 는 EGB 로 작용) 과 a. (J)- 의 브롬알칸과의 구핵치환반응 (02 는 구핵 시약으로 작용)의 쌍방의 결과로서 치환화합물이 생성된다. 그러나 전극반응을 이용한 양반응 속도의 시뮬레이션 결과는 Carbanio n 생성 속도 정수 1 x 102e m o1- 1 s -1 . 구핵치환 속도 정수 5~6 X 102e mol-1 s - 1 이므로 후자의 반응기대가 더 크다고 할 수 있댜 4) 양극 산화반응 벤젠류의 전해 산화에 의한 페놀류 , 퀴논류의 합성은 공업상 중 요한 과정으로서 오래 전부터 잘 알려져 있다. 그 후에도 황산수용 액 중 Pb02 양극을 이용해서 에멀션 (emuls i on) 계에서 벤젠으로부 터의 P-BQ 산화가 밝혀졌다 . 2 3 )
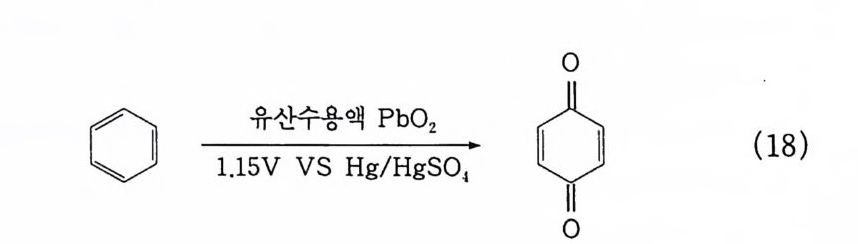 。 1.15유 V산 수VS용 액H g P/H bg0 S2 0 4
。 1.15유 V산 수VS용 액H g P/H bg0 S2 0 4
이 반응기구는 산성 조건에서 양극상의 Pb0 2 가 래디컬 종의 공 급원이 되고, 그 래디컬 종이 벤젠을 산화시켰다. Pb0 2 는 OH - 의 전해 산화에서 재생된다고 생각된다. 참고문헌 1) 松浦輝男 「化學」, 28, 1106(1 9 73). 2) I . I . Volnov, Peroxid e s, Sup er oxid e s and Ozonid e s of Alkali a nd Alkali Earth Meta l s, Plenum, New York( 1966) . 3 ) I . Fri do vic h , Acc. Chem. Res. , 5, 321 ( 1972 ) . 4) G. Charlot et al., Selecte d Consta n ts of Oxid o -Reducti on Pote n ti al , Perga m on Press ( 1958 ) . 5 ) G. Czaps k i and L. M. Dorf ma n, J. Phy s. Chem. , 68, 1169 ( 1964 ) . 6) M. E. Peover and B. S. Whit e, Electr o chim , Acta . , 1 1, 1061 ( 1960) . 7) L. Greenlee, I. Fri do vic h and P. Handler, Bio c hemi st ry , 1, 779( 1962). 8 ) E. Seyb . Jr. and J. Klein b erg, Anal. Chem. , 23, 115 (19 51 ) . 9) R. Die t z . A. E. J. Forno, B. E. Lacombe and M. E. Poever, J. Chem. Soc., pa rt B, 816(1 9 70) . 10) A. W. Petr oc ell i and D. L. Klaus, J. Chem. Ed., 40, 146( 1963) . 11 ) D. L. Ma ricle and W. G. Hodg so n, Anal. Chem. , 37, 1562 (19 65 ) . 12) J. E. McCord and I. Fri do vic h , J. Bio l . Chem., 244, 6049( 1963) . 13) H. Helms and W. Klemm, z. Anorg. Allg. Chem., 241, 97( 1939) : 242, 201 (19 39) . 14) A. Le Berre and Y. Bergn e r, Bull. Soc. Chem Fr. , 2363, 2368( 1966) . 15) a) T. Osa, Y. Ohkats u and M. Tezuka, Chem, Lett ., 99(1 9 73) . b) T. Osa and Y. Ohlcats u , Bull. Chem. Soc, Jpn . , 48, 1471 (19 75 ) .
16) H. Sag ae , M. Fuji hi r a , T. Osa and H. Lund, Chem. , Lett ., 793(1 9 77). 17) E. J. Nanni Jr. , M. D. Sta l l ing and D. T. Sawy er , J. Am. Chem. Soc., 1 02, 4481 ( 1980) 및 이에 인용된 문헌 18) a) J. San Fil ip o , C. I. Cheru and J. S. Valenti ne , J. Org. Chem., 41, 1077(1 9 70) . b) K. Boujl el and J. Sim onet, Tetr a hedron Lett ., 1 063(1 9 79) . 19) a) M. Sug aw ara and M. M. Baiz e r, Teta h edron Lett ., 24, 2223 (19 83) . b) M. Sug aw ara and M. M. Ba ize r, J. Org. C hem., 48, 4931 (19 83) . 20) H. Yamane, N. Yada, E. Kato r i, T. Masahin o , T. Naga n o and M. Hir o be, Bio B io p h ys . R es. Commun., 142, 1104(1 9 87). 21 ) H. Saga e , M. Fuji hir a, K. Komazawa, H. Lund and T. Osa, Bull. Chem. Soc. Jpn ., 53, 2188( 1980) . 22) a) F. Oji ma , T. Mats u e and T. Oae, Chem. Lett ., 2235(1 9 87) . b) F. Oji ma and T. Osa, Bull. Chem. Soc. Jpn ., 62, 3187(1 9 89) . 23) J. S. Clarke, R. E. Ehig a musoe and A. Kuhn, J. Electr o canal. Chem., 70, 333 ( 1976) .
제 6 장 여 기 상태 의 산소분자에 의 한 산화 1~5) 6. l 일중항 산소I) 삼중항 상태에 있는 기저 상태의 산소분자 (02) 16 전자의 배치를 정성적으로 정리하면 그림 6-1 과 같다. 같은 방향의 스핀울 갖는 전자는 같은 궤도에 들어가지 않으므로 축퇴한 두 가지의 반결합 궤도 짜와 ry·가 존재하지만, 이 두 개의 전자스핀이 역평행한 경우 축 일중항 상태를 생각해 보면 r· 궤도 한쪽에 편재한 경우( II) 와, 양쪽의 亢·궤도에 들어간 (m) 의 전자배 치를 가진 두 종의 일중항 상태의 산소분자 존재가 가능하다. ( II) 는 I L:::,_g라고, (m) 은 1E g+의 일중항 산소 (S i n gi e t Ox yg en) 라고 부 르며, 기저 상태( I), (3P, 3 :Eg-)보다 각각 22.5kcal(7,882cm-1) 와 37.5 kcal (1 3 ,1 21cm 기 더 높은 에너지를 갖고 있다. 후술하겠지만 여러 가지 방법으로 일중항 산소를 발생시킬 때 많 은 경우 이 양자가 존재한다는 것이 확인되었으나 여러 유기분자와
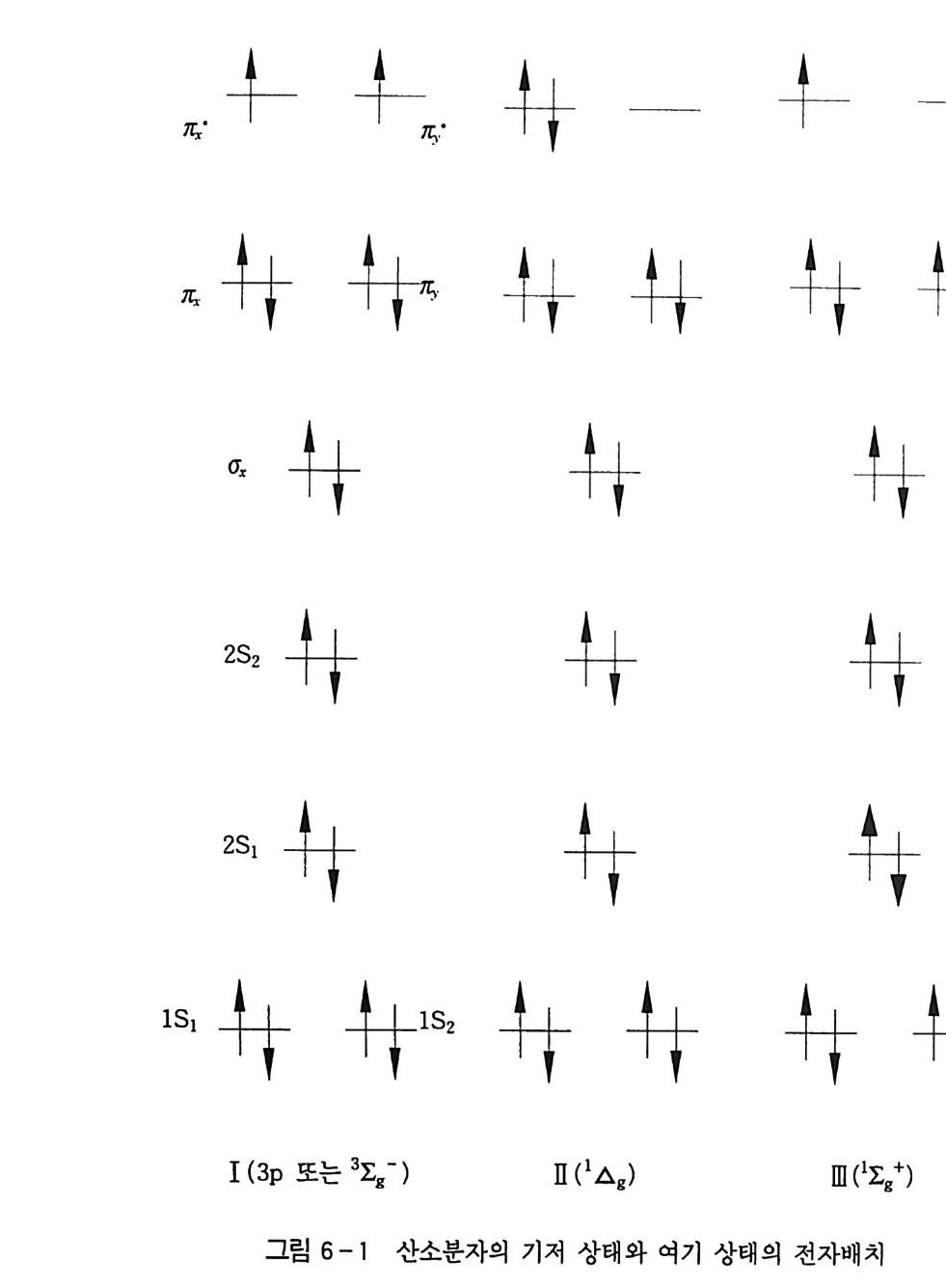 0�N\. GrL t
0�N\. GrL t
반응할 때 !스g 상태의 산소는 기저 상태의 산소보다도 구전자성이 풍부하므로 올레핀, 디엔, 다환성 방향족환 등에 쉽게 부가해서 과 산화물 내지 그 분해물이 생성되며. 또 아민류와 같이 이온화 전위 가 낮은 화합물과 용이하게 상호작용을 일으킨다 . I/:::,_g 산소분자는 자신이 일중항임에도 불구하고 궤도 각 운동량 (m) 때문에 상자성이며 e. s. r 스펙트럼을 나타낸다 . 6,7 ) 6.1.1 일중항 산소의 발생법 1 ) 광증감 산소 산화반응 광증감 산소 산화반응 (Pho t osens iti zed ox yg ena ti on) 은 오래 전 부터 연구되어 온 분야이댜 A+02 으므~ A02 (1)
표 6-1 광증감 산소 산화반응에 이용되는 광증감제와 그 특성 • - l 0) 광증감제 Et (k cal/ mo l)
식 (1)에서 보는 바와 같이 산소 존재하 광증감제 (Pho t osens i tize r : sens. 으로 약함) 가 흡수하는 광을 조사할 때. 기 질 A에 산 소를 부가하여 과산화물 A02 또는 그 분해 생성물이 생기는 반응 이다 . 몇 개의 광중감제를 표 6-1 에 정리했다. sens. + h v -1s ens' ―― ► 3se ns' (2) 3sens· + 302 一 sens + lo 2 心) (3) 3sens· + 302 一 sens + '0 2 ( 나 (4) 3sens· + 302 一 sens + 30 2 ( 작 ) (5) 식 (2)~(5) 에서 보는 바와 같이 광증감 산소 산화반응은 모든 경우, 증감제의 여기 일중항 상태 (1sens·) 로부터 계간 교차 ( int e r sy s te m crossin g : ISC 로 약함) 에 의 해 생 긴 여 기 삼중항 상 태 (3sens•) 가 관여한다. 따라서 증감제로서는 효율 좋게 여기 삼중 항 상태를 생성하는. 죽 계간 교차의 양자 효율이 높은 반면 삼중 항 상태의 수명이 긴 것일수록 좋다. 광원으로서는 텅스텐-할로겐 램프 등이 좋다. 2) 기저 상태의 산소분자로부터 발생 기저 상태의 산소분자는 진공 자치 영역으로부터 자치 영역에 겹 쳐서 많은 흡수대를 가지고 있다. 이 광을 흡수하면 분자가 두 개 의 산소원자로 해리한다• 이 산소원자가 자신의 재결합에서 산소분 자를 재생하든지 산소분자와 반응해서 오존을 생성하는 것이다.
02 ~ hv0 +0 O+O 一 0 2 (6) 0+02 一 03 이 반응은 성층권이나 오존 형성의 기본이 되는 반응이다. 이 성 층권에서의 형성은 로켓을 이용하여 일중항 산소가 생성됨이 확인 되었댜 o~+ hv( (3103 A) ― 一 Oz(I t_니 + o O + O 一 02 (I f::: ..g ) (7) 0 + 03 一 02 (I f::: ..g ) + 02 기저 상태의 산소분자가 광을 흡수하여 일중항 산소로 직접 천이 遷 移 )하는 과정은 스핀에 대하여는 금제천이이므로 상압에서는 기 상이나 액상에서 거의 관측되지 않는다. 그러나 상술한 고공 오존 충 즉 고농도 산소가 존재하는 곳에서는 가능한데, 실제 고압산소 가스. 액체산소 11 ) 또는 CFC 같은 난연성 용매에 고압에서 녹인 산소 1 2) 에서는 직접 스펙트럼이 측정된다. 또한 산소분자에 초단파 방전(주로 2,540MHz) 을 시켜 얻는 방 법도 있다. 이때 생성된 산소원자가 재결합에 의하여 일중항 산소 로 된댜 이 경우 일중항 산소의 실활(失活)을 막기 위해 5mmHg 이하의 감압(식 7) 에서 행한다. 3) 화학적 방법 일반적으로 기저 상태가 일중항인 과산화물의 분해로 산소분자 가 생성되는데, 이것이 일중항인 경우에는 반응이 단계적인 래디컬
 iy in{ NN
iy in{ NN
개열 반응이 일어나고 그렇지 않으면 생성된 산소분자가 스핀 허용 의 반응에 의해서 일중항 상태로 될 것이다(식 8). 1A02 —-1A + 102 (8) 원칙적으로 이러한 반응으로 일중항 산소를 생성하는 계는 어느 정도 알려져 있으므로 이를 표 6-2 에 나타내었다 . 표 6-2 의 반응들은 일중항 산소만 반응에 참여하는 이점이 있 지만. 기질의 성질에 의하여 일중항 산소가 발생하기 전에 반응이 완료되거나. 부반응이 일어난다. 하나의 예를 들면 다음과 같다. 라이모넨(Li monene) 을 표 6-2[1] 의 방법, 즉 H202 -N aOCI 로 산화시키면 광증감 산소 산화반응 (RB 로 사용)과 아주 다른 생 성물 분포를 보인다 . 8) 그러나 여기에 래디컬 저해제인 t-부틸페놀 을 조금 가해 산화를 행하면 광증감 산소산화의 경우와 일치한 다 . 25)
 〉《:二 。 〈 。〉 。:。p (9)
〉《:二 。 〈 。〉 。:。p (9)
(9) 식 반응에서 [a] 소값을 보면 광중감제 산화반응과 H202 -NaOCI 반응이 같은 일중항 산소에 의하여 일어남을 알 수 있다.
6. l. 2 일중항 산소반응을 지배하는 인자 1) 구전자 반응성 일중항 산소 (IA g)의 전자배치는 그림 6-1 에서도 볼 수 있는 것 처럼, 축퇴한 반결합성 궤도의 한쪽에 두 개의 전자가 역평행으로 들어가 있는 것과 같댜 그러므로 나머지 하나의 빈 궤도에 의해서 구전자성이 생긴다고 생각하면 일중항 산소가 비교적 전자가 풍부 한 계에 반응성을 가진다는 것이 정성적으로 이해된다 . 일중항 산 소의 반응에 대해서는 많은 이론적 고찰이 이루어져 있다 . 3. 26~30) 여러 결과에 따르면 일중항 산소분자와 올레핀류와의 상호작용에 는 전자이동 (Char g e Transfe r : CT 로 약함) 상호작용 에너지가 안정 화에 가장 중요한 기 여를 한다 . 26 ~ 30) 일중항 산소를 수용체 (acce pt or) 로 . 올레핀류를 공여체 (donor) 로 하는 CT 상호작용이 중요한 인자가 된다면 공여체의 전자 풍 부성의 척도가 되는 이온화 전위가 낮을수록 일중항 산소반응이 일 어나기 쉽다. Kop e cky 31 , 등이 여러 가지 올레핀을 메틸렌 불루 증감광산소 산화시킨 결과 알킬기 치환이 많을수록 반응성이 높다 는 발표를 한 후, 많은 올레핀의 상대 반응성이 측정되어 이 결과 가 더욱 적중되었다 . s. 3 2. 33 ) Foote 3 2) 등의 연구 결과를 표 6-3 에 정리하였다 . 한편, 같은 수의 치환기가 있다 하더라도 알킬기의 입체 효과에 의하여 반응성이 다름을 식 (1 0) 에서 알 수 있다 . 34)
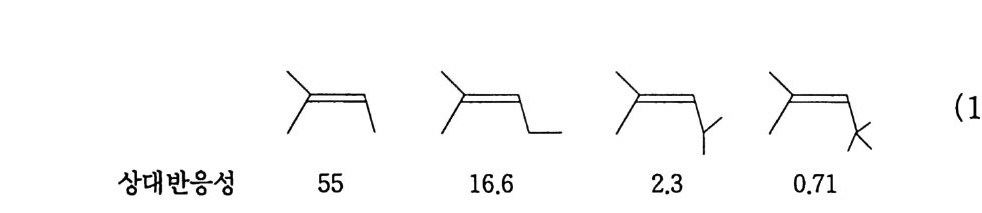 \ \ 三 三 (10 )
\ \ 三 三 (10 )
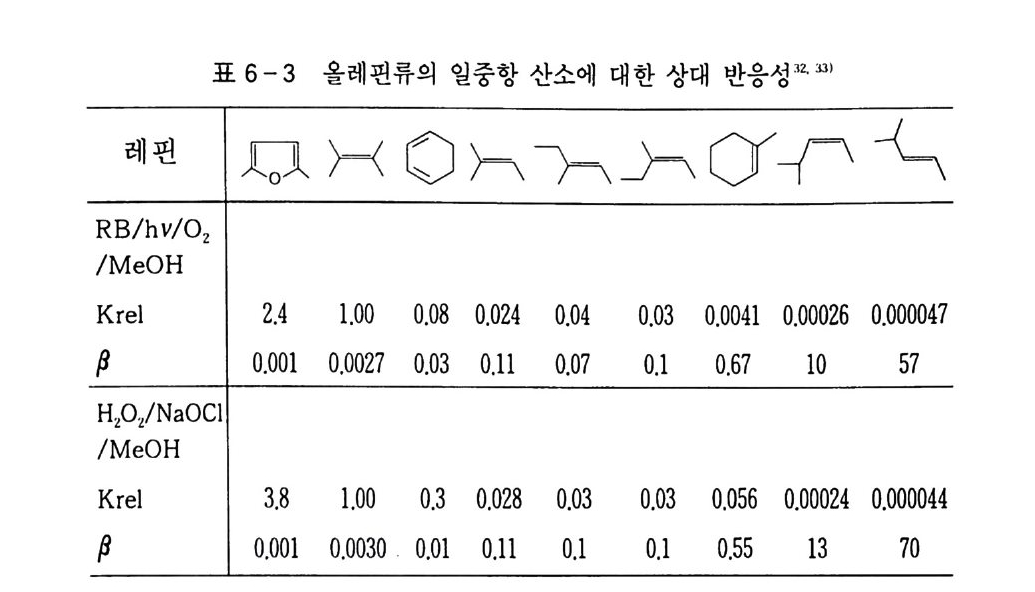 표 6 一 3 올레 핀류의 일중항 산소에 대 한 상대 반응성 12, 9)
표 6 一 3 올레 핀류의 일중항 산소에 대 한 상대 반응성 12, 9)
2) 입체 선택성 보통의 화학 시약에 비해서 분자의 크기가 대단히 작음에도 불구 하고 일중항 산소분자는 일반적으로 입체 장애가 적은 측으로부터 공격한다. 이 특징은 합성 화학적으로 매우 중요한 점이다. 또한 알릴 위의 수소를 가지고 있는 엔 (ene) 반응에도 현저한 입체 선택성이 보인 다. 이 경우 알릴 위의 수소가 C=C-이 중결합 면에 대하여 수직 축 (ax i al) 상의 입체 배치가 가장 안정한 전이 상태를 만든다는 반 응기구를 식 (11) 과 같이 제안하였다 . 8,35 . 36)
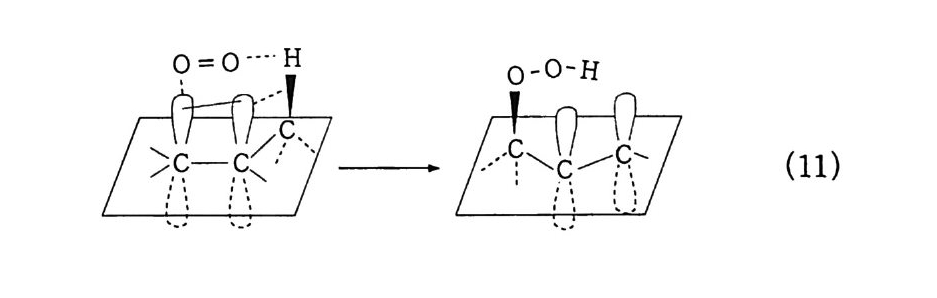 ¢〈 H
¢〈 H
이 기구에 의하면 전이 상태는 생성물에 의한 것이라기보다는 출 발 물질의 입체 배치에 의하여 정해진다 . 37) 이러한 예를 두 가지 들어 보겠다.
 HO W,(溫 ~o 二H (D) (12 )
HO W,(溫 ~o 二H (D) (12 )
식 (12) 에서 7 위의 수소를 중수소화한 콜레스테롤의 광증감 산 소산화시. 7a-D 화합물에서 생성 히드로 과산화물을 환원해서 얻 는 알릴 알코올에는 8.5 %밖에 중수소가 남지 않으나 7/3 _ D 화합 물로부터의 알릴 알코올에는 95 %의 중수소가 남아 있다 . 35) 이것 은 입체 장애가 적은 a 측, 죽 수직축에 수소가 있는 측에서부터 일중항 산소의 공격이 우선적으로 일어나고 있음을 뜻한다. 스테로이드와 같이 입체 배치가 고정되어 있는 것 외에는 입체 배치의 우위에 의하여 알릴 위 수소간의 입체관계가 중요해진다. 즉, 2. 4- 디메틸 -2- 부텐의 경우 2 급 히드로 과산화물이 우선적으 로 생성된다. 일반적으로 생각하면 3 급 히드로 과산화물이 우선적으로 생성될 것 같으나 식 (1 3) 에서 보는 바와 같이 3 급 수소가 이중결합에 대 하여 수평축 (e qua t or i al) 으로 놓여 있어 3 급 히드로 과산화물은 아 주 적게 (5 %) 생성된다 .8)
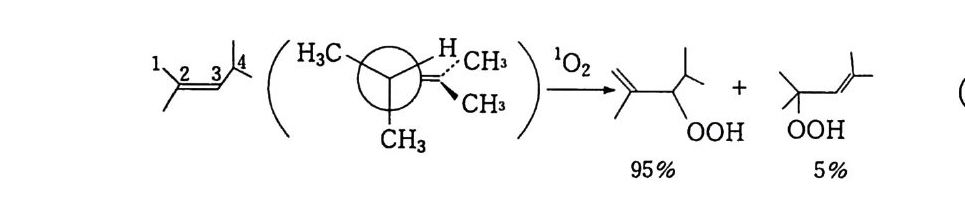 l 一 (H3C?H、C CHH: )느三 + 尸)- (13)
l 一 (H3C?H、C CHH: )느三 + 尸)- (13)
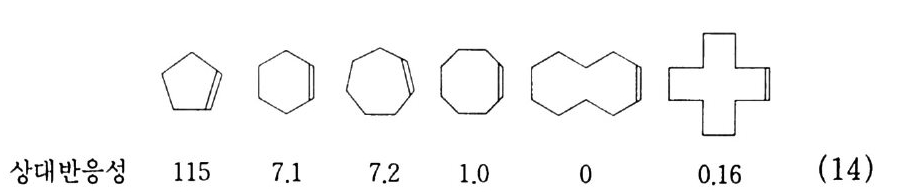 0000 二 〈
0000 二 〈
또 다른 예로 식 (1 4) 에서 보는 바와 같이 환상 알켄의 경우를 들 수 있다. 5, 6, 7 환의 경우, 알릴 위 수소가 수직축으로 놓였을 때는 우선적이나 그 이상을 넘으면 이중결합이 폴리메틸렌기에 의 해 싸여져서 입체 장해를 받기 쉬운 위치만 얻을 수 있다 .38) 3) 용매 효과 유기반응에서 보듯이 일중항 산소의 반응은 여러 가지의 입체 효 과를 받는댜 그 중 두 가지를 살펴보자. 첫째 일중항 산소의 수명에 대한 용매 효과이다. Kearns 등은 메 틸 렌 블루 -증 감광산소 산화반응을 Laser-Pulse 조사법으로 조사해서 여러 종류 중 일중항 산소의 수명 r=(1/kd) 를 구해 표 6-4 에 나타내었다.
표 6-4 각 용매 중에서 일중항 산소의 수명(-rµs )39) 용매 | 물 메탄올 벤젠 아세톤 CS2 물-메탄올 중수-메탄올 중수-중메탄올 'l' I 2 7 24 26 200 3.5 11 ~35
표 6-4 에서 보는 바와 같이 CS2 는 수명이 상당히 긴데, 중수 중메탄올 중에서는 물-메탄올보다 약 10 배 정도 길다. 다른 예는 문헌 40) 을 참고하기 바란다. 둘째, 한 기질로부터 2 개 또는 그 이상의 반응이 경쟁적으로 일 어날 경우 반응 선택성에 대한 용매 효과이다.
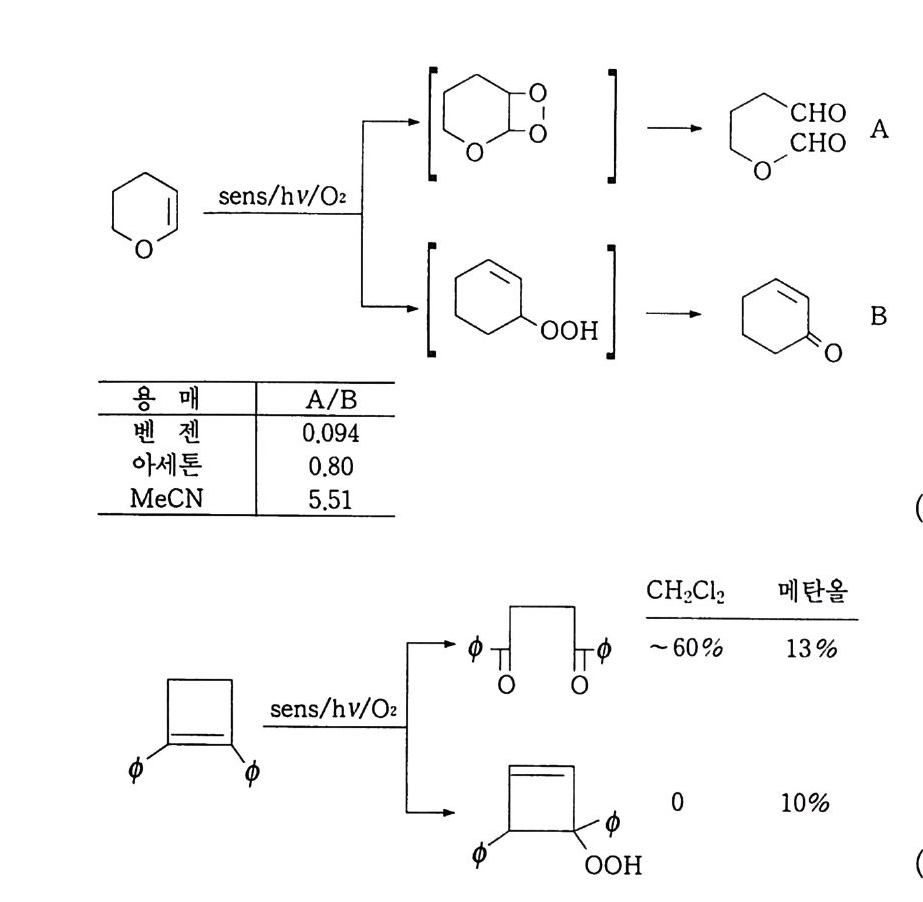 一 (。 /‘ 잡 g A
一 (。 /‘ 잡 g A
식 (1 5) 와 (16) 에서 기질에 대한 일중항 산소의 반응은 >C =C < 에 1, 2- 부가반응을 나타내며 C=C 결합의 개열반응과 ene- 반응 에 유래하는 생성물을 주지만 , 식 (1 5) 의 경우 용매의 극성이 높을 수록 1, 2 개열이 우선적으로 일어나는 데 반해서 식 (1 6) 의 경우 는 극성이 높아지면 1, 2 개열이 어렵게 된댜 41) 이 두 경우를 비교해 보면 식 (15) 의 비대칭 올레핀과 식 (16 ) 의 대칭 올레핀의 일중항 산소와의 반응에서, 천이 상태에 대한 극 성의 기여가 다르기 때문에 역의 결과를 나타낸다고 추론할 수 있 다.'l:l . 28 )
6. 1. 3 일중항 산소산화의 반응 예와 합성에의 응용 광층감 산소산화에 의해 발생시킨 일중항 산소를 이용한 반응은, 반응조건이 비교적 온화하므로 여러 형태의 화합물에 산소관능기 를 도입하는데 합성화학(合成化學)도 대단히 적합하며 반응 예도 여럿 있다. 특히 이 반응은 꽤 낮은 온도에서도 행해지므로 종래의 산소산화 법에서는 반응조건에서 분해하는 불안정한 과산화물을 분리하든지, 저온에서 만든 과산화물을 선택적으로 분해해서 여러 종류의 화합 물로 유도하는 일들이 가능하다. 일중항 산소에 의한 유기 화합물 의 산화반응에 대 하여 는 다수의 총설이 있다 .42, 43) 1) 공역 디 엔계에의 1. 4- 부가 공역 디엔 (d i ene) 계에의 1, 4- 부가는 환상 디엔계의 예가 압도 적으로 많다. 쇄상 디엔도 s-cis 형태를 취하면 1, 4- 부가가 일어 난다. 일반적으로 6 환 디엔계로부터 생성하는 과산화물은 상온에서 안 정하다. 그러나 5 환 또는 쇄상 디엔계로부터 생성되는 과산화물은 불안정한 것이 많으며 반응중에 2 차 생성물로 분해하는 경우가 자 주 나타난다. (1) 6 환 및 중급환 디엔계 전술한 바와 같이 6 환 이상의 지환식 공역 디엔으로부터 생기는 디엔 과산화물은 일반적으로 안정하게 실온 부근으로 쉽게 단리된 다. 가장 간단한 예는 식 (17).44) (18).45) (19 ).46) (20)47) 그리고 (21)47) 에 나타내었다.
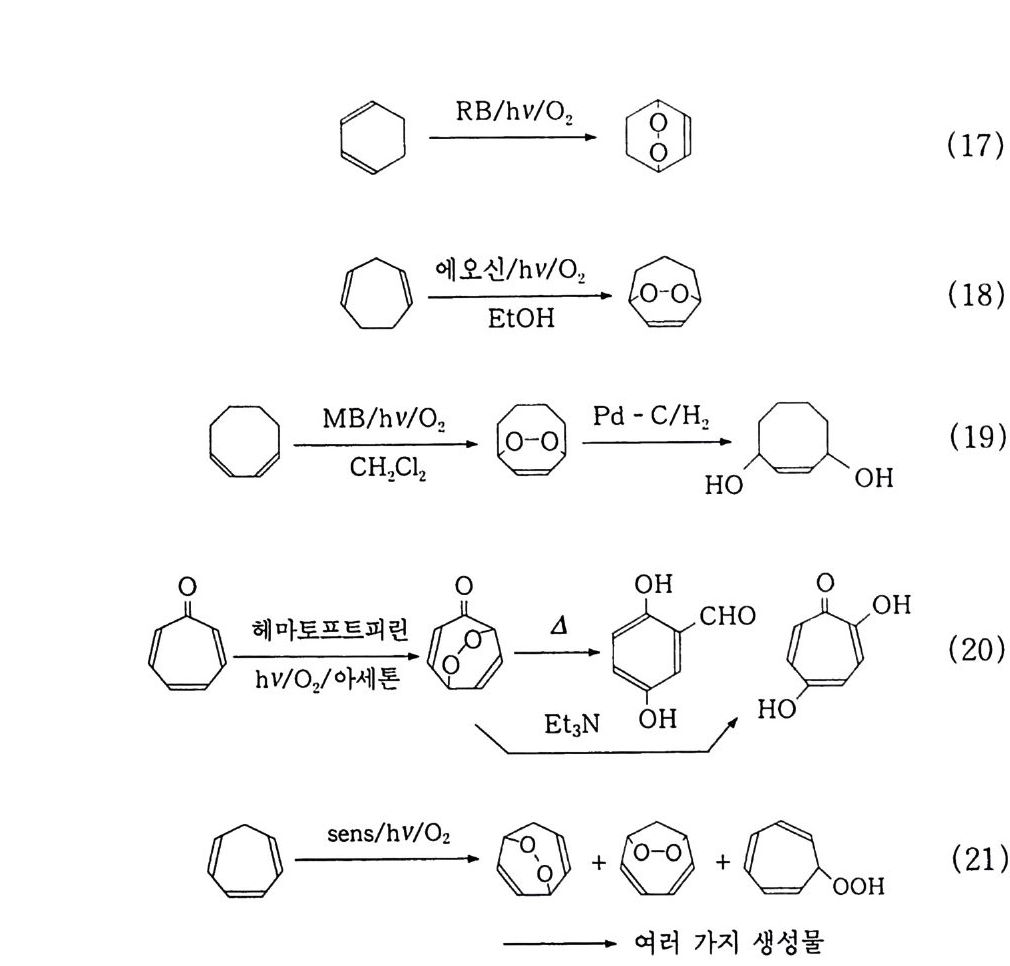 。 RB/hv/02 @ (17 )
。 RB/hv/02 @ (17 )
벤젠, 나프탈렌, 페난트렌 자신은 일중항 산소와 반응하지 않지 만 적절한 위치에 약간의 전자 공여기를 집어넣으면 반응한다. 반 웅 (22) 와 (23) 에 나프탈렌 치환체의 예를 나타내었다 .20 , 49,50)
 MeOOM•eM B/hAv /02 • 二MeM (22)
MeOOM•eM B/hAv /02 • 二MeM (22)
〔。 一 :MeO] 广O (23) e 환반이응것들도 을일 으가킨열다하.면 일중항 산소를 방출하나 식 (M23) 과 같이 변 아자스연카 리식돌물은에 서그 림쉽 게6 -얻2 와을 같수이 있 는여 러a - 가테지르 피형엔태으의로 e 부산터소 생관성능하기는를 가진 화합물로 유도된다 .42, 43 ) 치환 페놀류는 일중항 산소의 소광제로 되지만 반응할 때는 식 (26)5 1.5 2) 과 식 (27)52 . 5 3) 에서 보는 바와 같이 페놀 수산기의 파라 위가 산화된 히드로 과산화물 케톤 또는 그 분해물이 얻어진다. 케
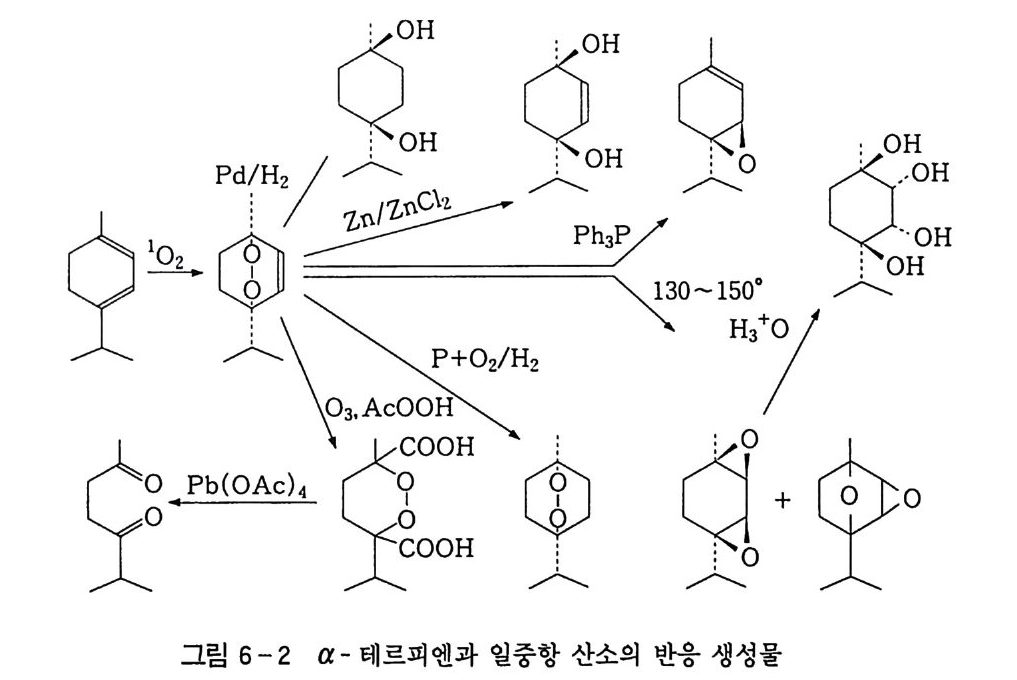 OH:
OH:
톤 히드로 과산화물은 1,4- 엔도 과산화물을 통해 생성되지만 〈엔 (ene) 〉형의 반응에서 생성되는지는 잘 알려지지 않았다. (2) 5 환 디엔계 5 환 디엔계의 대다수 화합물에 대하여 일중항 산소를 1, 4- 부가 하지만 생성된 엔도 (endo) 과산화물은 일반적으로 불안정하며 꽤 낮은 온도에서도 분해해서 2 차 생성물을 낸다 . 분해 양식도 다양해 서 출발 물질의 구조. 특히 치환기의 종류나 반응조건에 의해서 생 성물이 다른 경우가 많다. 전형적인 화합물의 광증감 산소산화 생성물을 표 6-5 에 나타내 었다. 이 생성물에 대하여는 그림 6-3 의 반응기구를 참고하면 대개 해석할 수 있을 것이다.
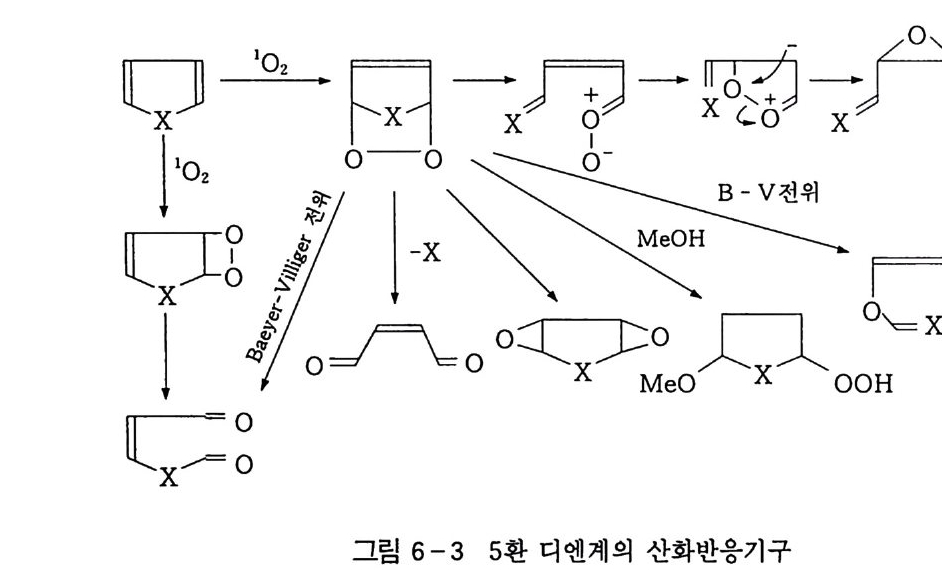 。
。
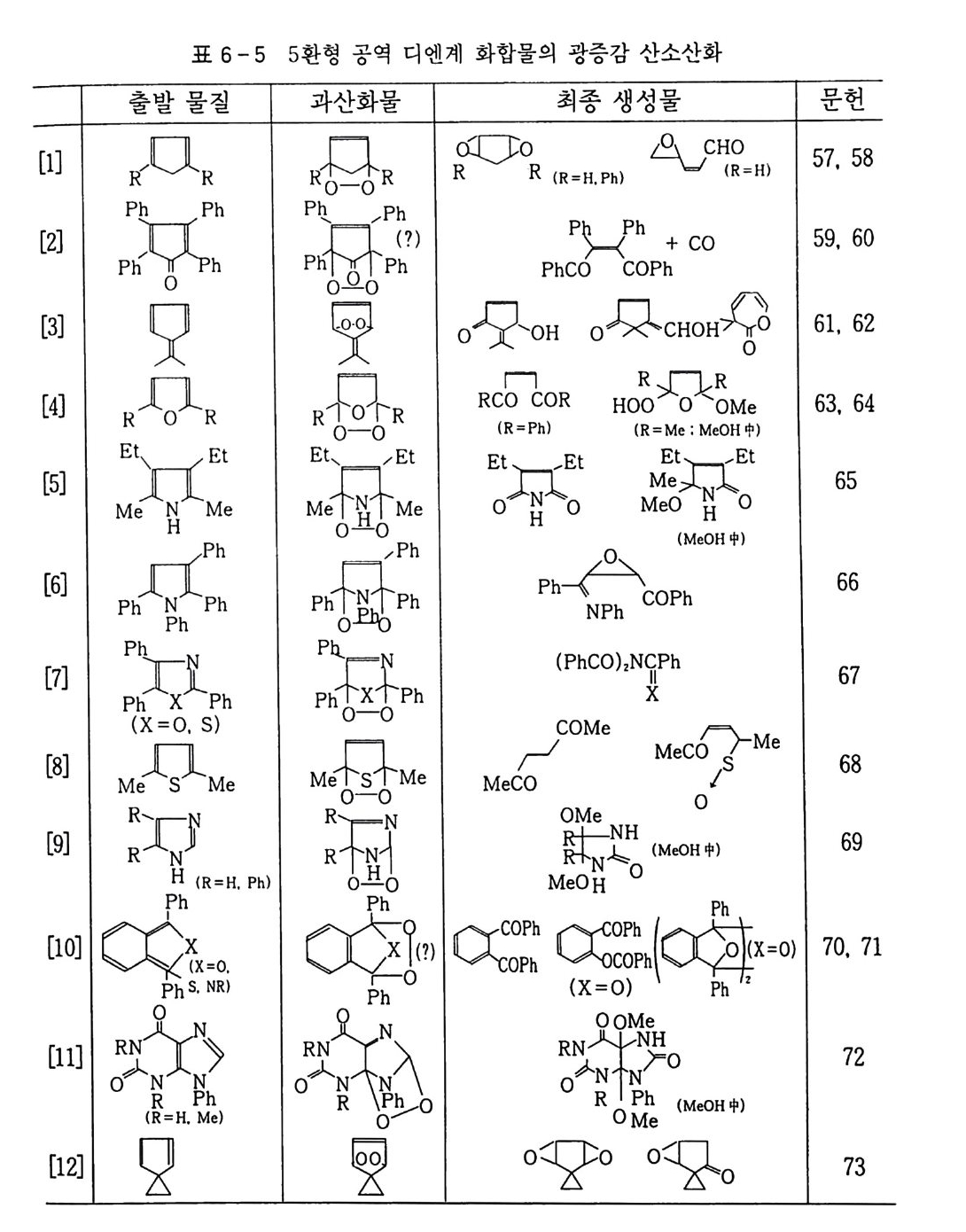 표 6-5 5 환형 공역 디엔계 화합물의 광증감 산소산화
표 6-5 5 환형 공역 디엔계 화합물의 광증감 산소산화
(3) 쇄상 이중결합을 포함한 공역 디엔계 쇄상 이중결합을 포함한 디엔계나 입체 배치가 고정되어 있지 않 은 공역 디엔계에 대해서 일중항 산소가 부가하는 예는 그렇게 많 지 않았지만(식 24)42 ) 근래 들어 이 방면의 연구가 많이 진행되었 다. 즉 , 치환 부타디엔의 어떤 것들은 1, 2- 부가를 해서 6 환 과산 화물을 만든다(식 25).
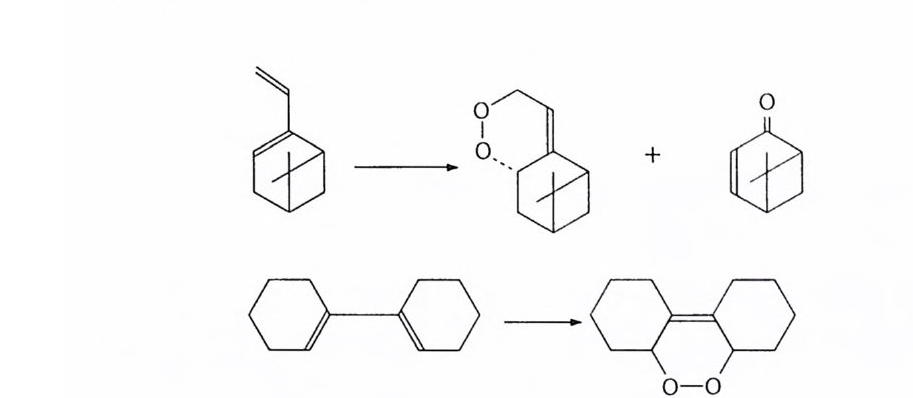 \ :1+ <0 >
\ :1+ <0 >
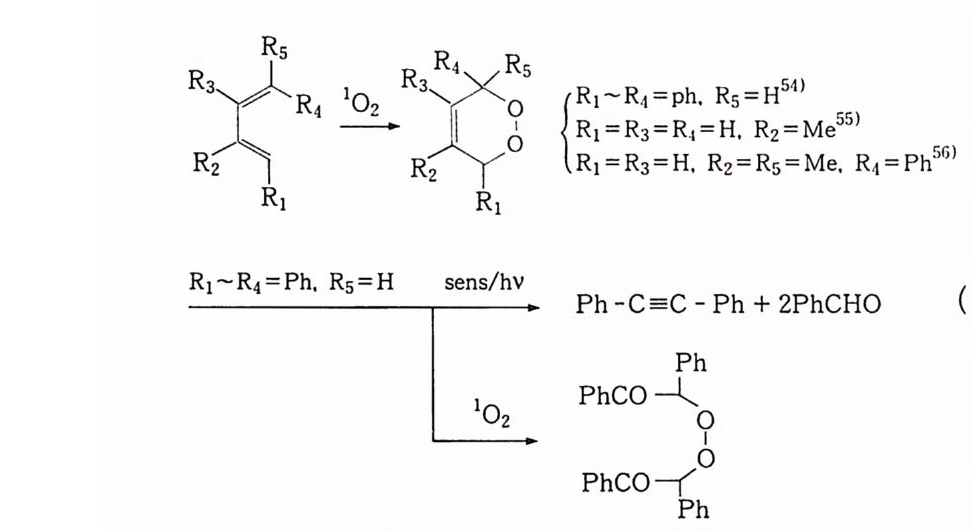 氏/Rz /RR sl R 4 021一 RR3 z :广 lR OIR s O{ :R :1 := R::3 := H[\. '=RR 2H =5' =RRs\ 5:=) MM ee 5.5 )R ~ =P hsr ,)
氏/Rz /RR sl R 4 021一 RR3 z :广 lR OIR s O{ :R :1 := R::3 := H[\. '=RR 2H =5' =RRs\ 5:=) MM ee 5.5 )R ~ =P hsr ,)
그 밖에 배위가 고정된 환외 이중결합을 가진 공역 디엔에서 식 (26) 과 같은 예가 알려져 있고 1, 4- 부가물은 -70 °C 에서 확인되
었다.
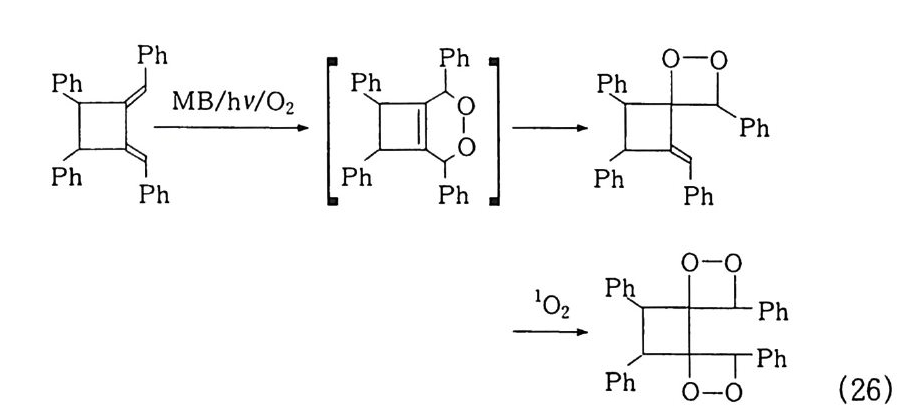 :〈三 [\\ l 一 :\\Ph
:〈三 [\\ l 一 :\\Ph
2) 전자가 풍부한 올레핀에의 1, 2- 부가 산소산화에 대하여 올레핀 이중결합이 개열할 경우 중간체로서 디옥세탄 (D i oxe t ane) 이 오래 전부터 생각되었지만 Ko p eck y 74) 가 처음으로 디옥세탄을 합성하여 단리하기까지는 아주 불안정한 화 합물이라고 믿었다. 디옥세탄은 열분해에 의하여 식 (27) 과 같이 두 개의 카르보닐 화합물이 되며, 이때 전자 여기한 카르보닐 화합 물이 생성된다 . 75 )
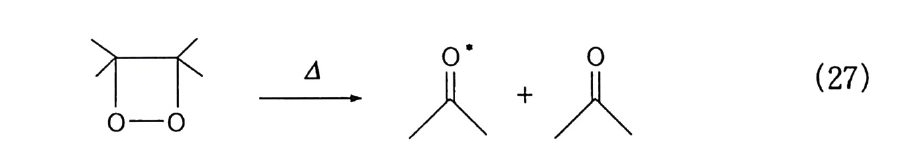 二 + Ol (27)
二 + Ol (27)
이 반응은 화학 발광의 하나의 기구로서 중요하며, 또 적당한 디 옥세탄에 의하여 광을 사용하지 않고 광화학 반응을 일으키므로 최 근 특히 주목되고 있다. 일중항 산소의 1, 2- 부가에 의한 디옥세탄 생성반응의 특징으로 는 첫째 반응이 입체 선택적으로 일어난다는 점이다 .76)
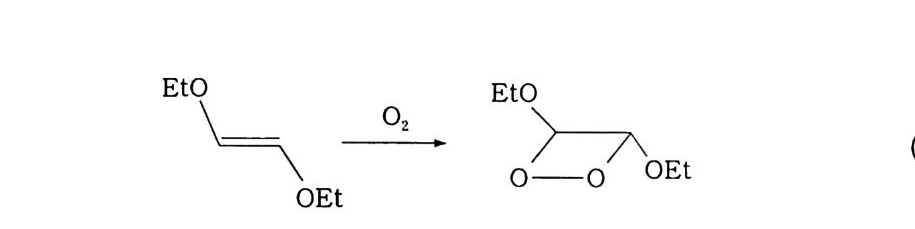 Et ~ 02 • E0t \OEt (28)
Et ~ 02 • E0t \OEt (28)
둘째로는 디옥세탄 생성이 [2s+2a] 의 반응이지만 퍼에폭사이드 와 같은 양성 이온을 경유하는 이단계 반응인지에 대해서는 아직 확실치 않다. 그러나 디아다만탄 (D i adaman t ane) 이 피나콜론 존재 하에서 광중감 산화반응시키면 에폭사이드가 얻어지는 것으로부터 식 (29) 의 반응기구가 제안되는데.77) 어떤 종류의 디옥세탄은 트 리페닐포스핀에 의하여 식 (30)78) 에폭사이드로 환원되는 것이 알 려져서 꼭 퍼에폭사이드를 중간체로 생각하지 않더라도 설명이 가 능하다.
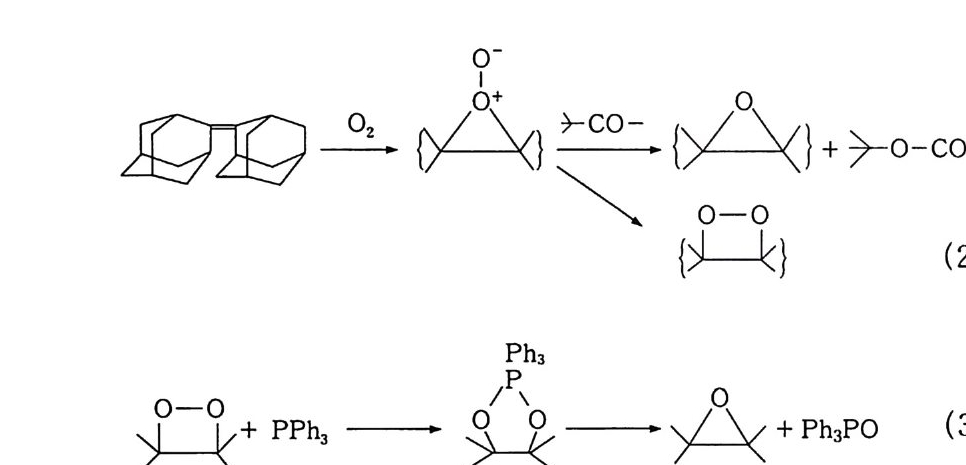 二느 二〈o-} \二{二二〈}} +戶 0-C(O2-9 )
二느 二〈o-} \二{二二〈}} +戶 0-C(O2-9 )
그러나 Dewar 의 계산 결과는 퍼옥사이드 경로를 지지한다 .28)
3) 알릴 수소를 가지고 있는 올레핀의 〈엔〉 반응 알릴 수소를 갖는 올레핀은 일중항 산소와 반응해서 식 (31) 과 같이 이중결합 전이를 수반. 알릴히드로 과산화물을 생성한다. /\I\C +I /I0 C --0_ C.IH _c I _c_H - = C (31) I I I -C-C=C- boH 이 생성물은 보통 원래의 올레핀보다도 현저하게 반응성이 저하 되므로 이 이상의 산화는 일어나지 않는다고 할 수 있다. 여러 종류의 환원제를 사용하여 히드로 과산화물기를 환원해서 대응하여 알릴 알코올을 용이하게 얻을 수 있으므로 합성적으로 유 용하다. (1) 올레핀류 일반적으로 알킬 치환기가 많을수록, 죽 전자가 풍부할수록 반응 이 쉽게 이루어진다 (6. 1. 2 의 1) 참조) . 표 6-6 에 여러 가지 올레핀에 대하여 전형적인 예를 모아 놓았 다. 모노 알킬 올레핀은 반응이 높고, 한 생성물만 생성하는데, 치환 체가 둘 이상의 비대칭 구조를 가지면 위치 선택성이 문제가 된다.
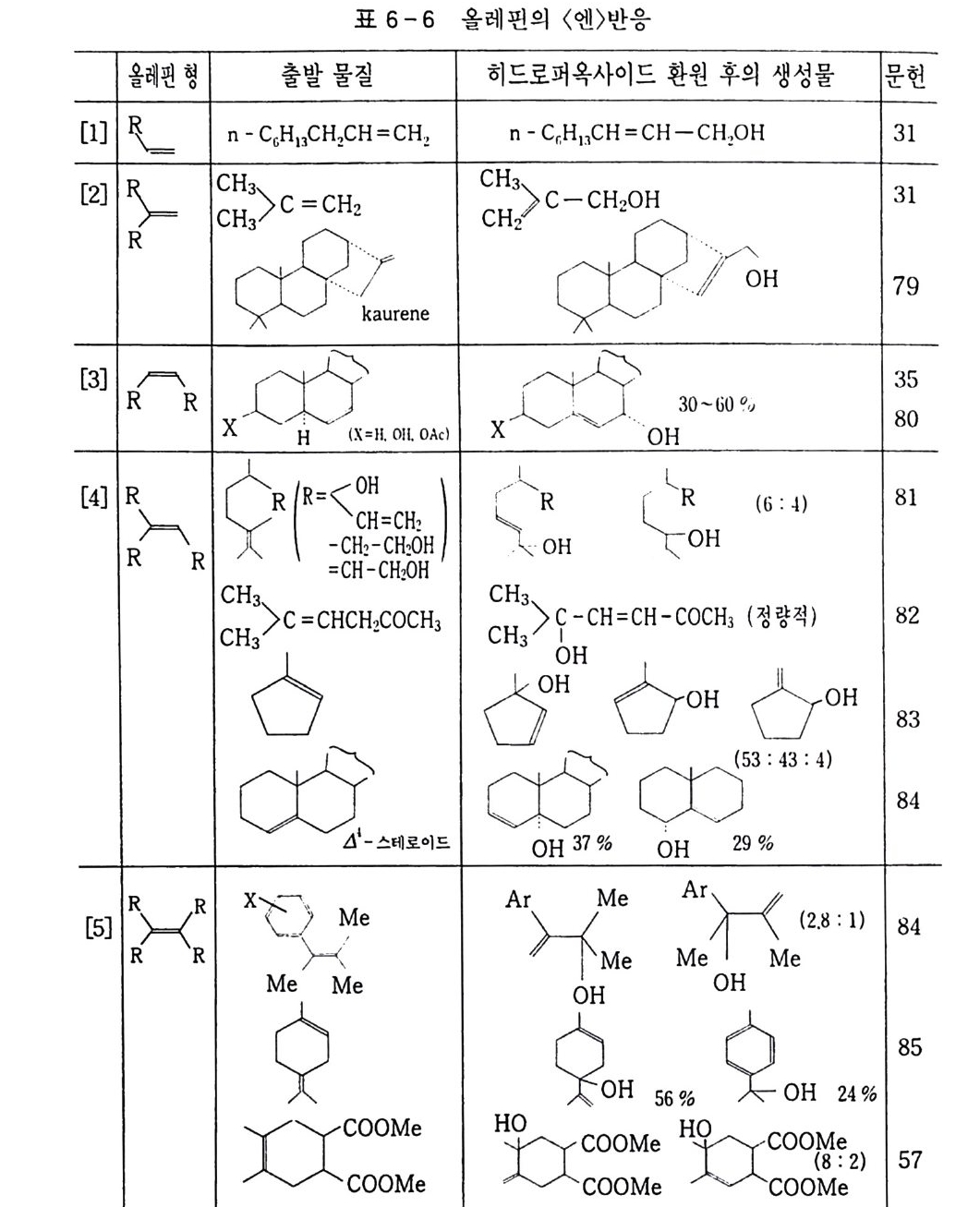 표 6-6 올레핀의 〈 엔 〉 반응
표 6-6 올레핀의 〈 엔 〉 반응
참고문헌 1) R. P. Way ne ,Adv. Photo c hem, 1, 311( 19 69). 2) 森佳伴 井口 1 羊夫, 「酸素化學 共立化學」, Lib r ar y- 4, f惡丸克己編 (19 73). 3) D. R. Kearns, Chem, Rev., 71, 395(1 9 71). 4) M. Kasha and A. U. Khan, Ann. N. Y. Acad. Sc i., 171, Artic l e l(1970) 에 일중항 산소분자 특집이 수록되어 있음. 5) C. S. Foote , Acc. Chem. Res., 1, 104(1 9 68). 6) E. Wasserman, R. W. Murray, M. L. Kap la n and W. A. Yag er , J. Am. Chem. Soc., 90, 4160( 1968). 7) A. M. Falic k and B. H. Mahan, J. Chem. Phys ., 47, 4778( 1967) . 8 ) K. Gollnic k and G. 0. Schenck, Pure Ap pl. Chem. , 9, 506 ( 1964) . 9) K. Gollnic k , Adv. Photo c hem. , 6 , 1( 19 68) . 10) Y. Usui, Chem. Lett ., 7 43(1 9 73). 11 ) A. U. Kahn and M. Kasha, J. Am. Chem. Soc., 92, 3293( 1970). 12) D. F. Evance, Chem. Commun., 367(1 9 69). 13) C. S. Foote and S. Wexler, J. Am. Chem. Soc., 86, 3881 ( 1964) . 14) E. McKeon and W. A. Wate r s, J. Chem. Soc., B, 1040 ( 1966) . 15) R. P. Ste e r, K. R. Darnall and J. N. Pit ts, Jr., Terrahedron Lett ., 3765 (19 69 ) . 16) H. H. Wasserman and J. R. Sheff er, J. Am. Chem. Soc., 89, 3073 (19 68). 17) H. H. Wasserman, J. R. Sheff er and J. L. Coop er , ibi d ., 94, 4991 (19 72). 18) S. R. Abbott , S. Ness and 0. M. Hercules, ibi d . , 92, 1128Ll970). 19) A. M. Trozzolo and S. R. Fahrenholtz. Ann. N. Y. Acad. S ci ., 171,
61 (19 70) . 20) H. H. Wasserman and D. L. Larsen, Chem. Commun., 2 53( 1972) . 21 ) M. M. Murray and M. L. Kap la n, J. Am. Chem. Soc., 91 , 5358 (19 69). 22) L. M. Ste p h e nson and D. E. McClure, ibi d . , 95, 3074( 1973) . 23 ) R. M. Murray, W. C. Luman, Jr., I. Rosenth a l and H. Fuhr, ibi d . , 92, 4348( 1972) . 24) J. W. Pete r s, J. N. Pit ts, Jr. , I. Rosenth a l and H. Fuhr, ibi d . , 92, 3205(1 9 70). 25) C. S. Foote , S. WexlerandW. Ando, Tetr a hedron Lett ., 4 111( 1965). 26) D. R. Kearns, J. Am. Chem. Soc., 91, 6554( 1969) . 27) 山口非, 『化學總說』, 1, 292(1973). 28) M. J. S. Dewar and W. Thie l, J. Am. Chem. Soc., 97, 4439(1 9 75) . 29) S. Inag a ki, S. Yamabe, H. Fuji mo to and K. Fukui, Bull. Chem. Soc. Jpn ., 45, 3510(1 9 72). 30) S. Inag aki and K. Fukui, ibi d . , 46, 2240( 1973) . 31 ) K. R. Kop ec ky and H. J. Reic h , Can. J. Chem. , 43, 2265 ( 1965 ) . 32) R. Hi ggins , C. S. Foote and H. Cheng , Adv. Chem. Ser. , 77, 102(1 9 68). 33 ) B. Ste v ens, Acc. Chem. Res. , 6, 90 (19 73 ) . 34) K. Goll nick , Adv. Chem. Ser., 77, 118 (19 68) . 35) A. Ni ck on and J. F. Bag li, J. Am. Chem. Soc., 83, 1498( 1961 ) . 36) F. A. Lit t and A. Ni ck son, Adv. Chem. Ser. , 77, 118(1 9 68) . 37) A. Ni ck on et al., J. Am. Chem. Soc., 94, 5517( 1972) . 38) T. Mats n ura, A. Hor ina ka and R. Nakashim a, Chem. Lett . , 887(1 9 73). 39) P. B. Merckel and D. R. Kearns, J. Am. Chem. Soc., 94, 1029, 1030,
7244 (19 72 ) . 40) R. H. Young , K. Wehrly and R. L. Ma rtin, Adv. Photo c hem., 93, 5774 (19 71 ) . 41 ) A. G. Schultz and R. H. Schlessin g e r , Tetr a hedron Lett ., 2731 (19 70). 42) K. Gollnic k and G. 0. Schenck, J, 4 -Addit ion Reacti on , p. 225, J. Hamer, ed., Academ ic P ress, New York( 1964) . 43 ) R. W. Denny and A. Nic k son, Org. React, 20, 133 ( 1973 ) . 44) G. 0. Schenck and D. E. Dunlup , Ang ew . Chem., 68, 248 ( 1956) . 45) A. C. Cop e, T. A. Lis s and G. W. Wood, J. Am. Chem. Soc., 79, 6287 (19 57 ) . 46) A. Hori na ka, R. Nakashi m a, M. Yoshik aw a and T. Mats u ura, Bull. Chem. Soc. Jpn . , 48, 2095 ( 1975 ) . 47 ) M. Oda and Y. Kitah ara, Tetr a hedron Lett. , 3295 (19 69 ) . 48) A. S. Kende and J. Y -C . Chu, Tetr a hedron Lett. , 4837( 1970) . 49) J. Rig a udy, C. Deleta n g and J. J. Basseli er, Comp t. Rend., 2 68, 334( 1969) . 50) P. S. Song and T. A. Moore, Photo c hem. Photo b io l ., 1, 113(1 9 68) . 51 ) I. Sait o, N. Yoshim ura, T. Arai , K. Omura, A. Nis h in a ga and T. Mats u ura, Tetr a hedron, 28, 5131 (19 72) . 52) T. Mats u ura, K. Omura and R. Nakashi ma , Bull Chem. Soc. Jpn ., 38, 1358(1 9 65) . 53 ) P. L . Julian , W. Cole and E. W. Mey er , J. Am. Chem. Soc. , 67, 1724(1 9 45) . 54) G. Rio and J. Bert he lot, Bull. Soc. Chim . Fr. , 1664(1 9 69), 2938 (19 71 ) . 55) N. Hastry , P. B. Merkel, P. Racl lick and D. R. Keams, Tetr a hedron
Lett ., 49( 1972) . 56) K. Kondo and M. Mats u moto , Chem. Commun., 1332( 1972) . 57) K. H. Schulte -Elte , B. W iel halm and G. Ohloff , Ang re w. Chem., 81, 1045( 1969) . 58) W. R. Adams and D. J. Trecker, Tetr a hedron, 21, 2631 ( 1971 ) . 59) N. M. Bik a les and E. J. Becker, J. Org. Chem., 21, 1405(1 9 56) . 60) J. H. Beyo n , R. F. Curt is a nd A. F. W illiam s, Chem. Commun., 237 ( 1966) . 61 ) W. Skoria n etz , K. H. Schulte -Elt e and G. Ohloff , Helv. Chim . Acta . , 54, 1913(1 9 71 ). 62) N. Harada, S. Suzuk i, H. Uda and H. Ueno, J. Am. Chem. Soc., 94, 1777( 1972), Chem. Lett ., 8 03, 807( 1972) . 63) C. S. Foote , M. T. Wuesth o ff , S. Wexler, I. G. Burnste i n , R. Denny, G. 0. Schenck and K. H. Schult e -Elite , Tetr a hedron, 23, 2583(1 9 67). 64) A. M. Trozzolo and S. R. Farenholtz , Ann. N. Y. Acad. Sc i., 171, 61 ( 1970) . 65) D. A. Ligh tn e r and G. B. Qu in s ta d ,Ang ew . Chem., 84, 216( 1972). 66) H. H. Wasserman and A. Mille r, Chem. Commun., 199( 1969) . 67 ) T. Mats u ura and I. Sa ito, Bull. Chem. Soc. Jpn . , 42, 2973 ( 1969 ) . 68) C. N. Skold and R. H. Schlessin g er , Tetr a hedorn Lett ., 791 ( 1970) . 69) H. H. Wasserman,Ann. N. Y. Acad. Sc i., 171, 168(1 9 70). 70) F. Naharandi, F. Razmara and M. P. Ste v ers, Tetr a hedron Lett . , 301( 19 73). 71) P. B. Merkel and D. R. Kearns, J. Am. Chem. Soc., 97, 462( 1975) . 72) T. Mats u ura and I. Sa ito, Tetr a hedron, 25, 541, 549 ( 1973 ) . 73) H. Takesh ita, H. Kanamori and T. Hats u i, Tetr a hedron Lett ., 3139
(19 73) . 74) K. R. Kop ec ky, J. H. Van de Sende and C. Mumf old , Can. J. Chem. , 46, 25 ( 1968 ) . 75) N. J. Turro and P. Lechtk e n et al., Acc. Chem. Res., 7, 97( 1974) . 76) A. P. S chaap and N. Tonta p a nisc h, Chem. Commun., 490( 1972). 77) A. P. Schaap and G. R. Faler, J. Am. Chem. Soc., 95, 3381 ( 1973) . 78) P. D. Bartl e tt , A. L. Baumsta r k and M. Landis , ib id . , 95, 6486(1 9 73) . 79) R. A. Bell, R. E. Ireland and L. N. Mander, J. Org. Chem., 31, 2536(1 9 66) . 80) A. Ni ck on, N. Schwarz, J. B. Di gior gi o and D. A. Wi dd owson, ibi d . , 30, 1711 (19 65) . 81 ) E. Klein and W. Roja h n, Chem. Ber., 97, 2700(1 9 64). 82) N. Furuta c hi, Y. Nakadair a and K. Nakanis h i, Chem. Commun., 1625(1 9 68) . 83 ) A. Nic k on and W. L. Mendelson, Can. J. Chem., 43, 1419( 1965) . 84) C. S. Foote and R. W. Denny, J. Am. Chem. Soc., 93, 5162( 1971 ) . 85) E. Klein and W. Ra jah n, Tetr a hedron, 21, 2173(1 9 65) .
제 7 장 오존에 의한 산화 7.1 오존 최근 오존층의 파괴로 인하여 지구 환경변화에 큰 물의를 일으키 면서 우리에게 갑자기 심각한 문제로 대두된I) 오존은, 1840 년 Schonbein 2 ) 이 전기스파크 실험에서 알지 못하는 어 떤 화합물의 냄새라고 정의를 내렸다. 그 후 번개가 친 후에 나는 냄새와 같다 는 관찰로부터 〈오존〉이라 명명했는데. 이는 희랍어 〈 Oze i n 〉에서 유래한 것으로, 〈냄새난다〉는 뜻이다. 302 一 203 .1H 0t = 144.8 kJ/ m ol (1) 식 (1) 에서 보는 바와 같이 오존생성은 홉열반응이며, 열역학적 으로 불안정하여 즉시 산소분자로 분해된다. 오존의 구조는 식 (2) 의 (!) 과 (~) 구조로 공명 함을 알 수 있다 .3)
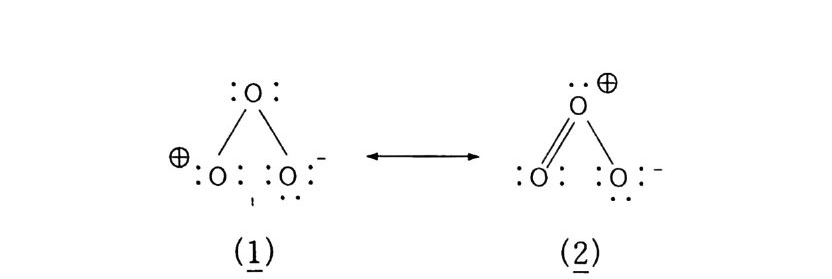 락!\I :• -• • • ..。 /: 0二 :\® 。 · · (2)
락!\I :• -• • • ..。 /: 0二 :\® 。 · · (2)
위 구조에서 보듯이 오존은 구전자 성질이고 전자쌍을 가지고 있 으며, 산소와 같이 유리전자를 지니고 있지 않기 때문에 상자성을 띠지 않는다. 오존은 상온에서 연청색 가스로 폭발성이 있고, -119 ·c 에서 액화되는 암자색 액체이며. -192.7 · c 에서 고체화된다. 오존 은 또한 강한 산화력이 있어 무기. 유기 화합물과 반응한다. 유기 물과의 반응은, 1885 년에 역시 Schonbe i n 이 올레핀과 반응하여 탄 산포름 알데히드, 포름산이 생성됨을 보고한 이래.1) 많은 연구가 진 행되었으며 여러 총설들이 s .6 ) 보고되었다. 7. 2 이중결합의 오존 분해반응 7. 2. l 오존의 첫 단계 반응 올레핀의 오존 분해반응이 래디컬 반응인가 이온성인가에 대해 서는 많은 연구가 이루어져 있다. W i bau t와 공동 연구자들”은 오 존의 이중결합과 반응이 천전자성 첨가임을 보고한 바 있다. Huis g e n8) 은 오존이 이중결합에 첨가될 때는 1, 3-쌍 극자 첨가임 을 제안하였다. 여하튼 첫 단계 반응 생성물은 네 고리 구조를 가 진 (한 이거나. Cr i e g ee 와 Greenwood 가 제안한 다섯 고리 구조인 년)일 것이다.
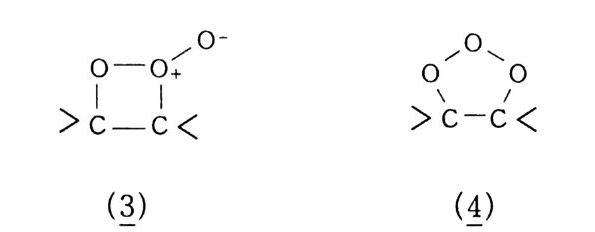 >cOI —— 0cI+ /< o - >0 \c /-。 c`I0<
>cOI —— 0cI+ /< o - >0 \c /-。 c`I0<
Baile y 9> 등은 트랜스-디 -t-부틸에틸렌의 nmr 스펙트럼에서 메 틴 양성자의 단일선 신호를 얻었는데, 이 경우에는 적어도 1, 2, 3- 트리옥솔란인(산)의 구조가 옳다. Duhran 과 Greenwood10) 는 몇 가 지 다른 트랜스-올레핀에 대해서도 비슷한 결과를 얻었다. 이들은 얻기 어려운 트랜스-올레핀에 대한 첨가 생성물의 nmr 을 얻었는 데. 이 경우에는 더 낮은 온도 (-130 · c) 가 필요하다. 또한 첫 단 계 생성물의 구조(산)가 아닌 경우가 있다. 1- 올레핀과 같이 이중결합의 주 오존 분해 생성물은 에폭사이드 이다 . 에폭사이드 생성물은 다섯 고리 구조(산)로부터 얻어지기도 하지만 대부분은 이와 다른 경로를 통해 얻어진다. (§)와 같은 7T:- 복합체 또는 (~)과 같은 c- 복합체로부터 산소분자가 제거되며 에 폭사이드가 생성된다. 또한 이 중간체로서 열려진 6- 복합체 (7) 가 제안되었다.
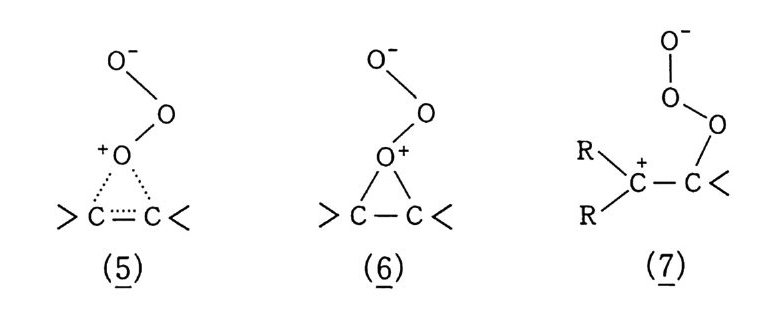 >c+..°::.o.:. . /.:\. . 。: c< >c/°—o\+ /\ c 。 < RR\/+c -o。Ic -><°
>c+..°::.o.:. . /.:\. . 。: c< >c/°—o\+ /\ c 。 < RR\/+c -o。Ic -><°
7, 2. 2 Cr i e g ee 의 반응기구 올레핀(이중결합 및 삼중결합 화합물)과의 반응은 Crie g e e 반응 기구에 의하여 잘 설명되었다 .1 1. 1 2) Cr i e g ee 에 따르면 올레핀 (8) 은 오존과 같이 1. 3- 쌍극환상 부가 로 1, 2, 3_ 트리옥솔란인(산)을 형성한다. 이것은 약간의 예를 제외 하고는 거의 -78°C 에서 불안정하여 카르보닐 화합물 (g)가 양성 자이온 (브)으로 분리되는데. (브)은 카르보닐 옥사이드라고도 부 른다. 이상의 반응경로는 출발 물질의 구조와 반응조건에 달렸다 . 펜탄 또는 클로로포름 같은 불활성이고 반양성자 용매에서 분해 생성물인 (g)와 (브)은 醫+끽 부가로, 역시 오조니드인 1, 2, 4- 트리옥솔란 (브)로 된다 . 또한 (g)가 (브)과 반응하는 것이 어렵 기는 하지만 (g)와 (拉)이 각각 더 반응하여 2 량체 (~)와 다량체 의 산화물 (브)이 생성된다.
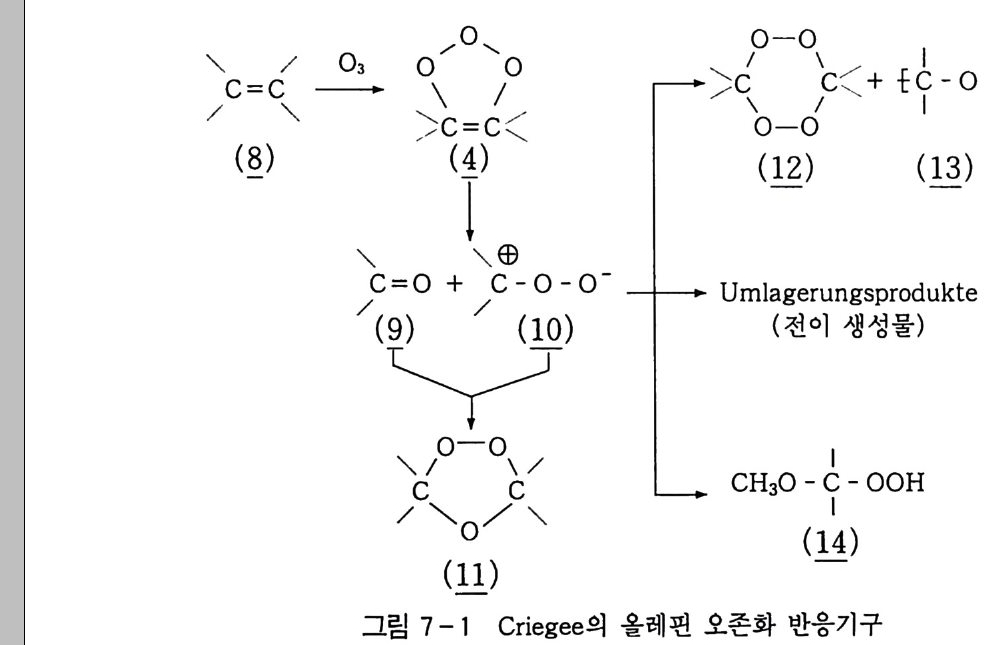 \/ C(=8C)/\ 一03 〉0\ /? 。- '「 -/ 0 > C/\0 0 (1 一— 2 0o) / \ C < + t c(1I1 -3 o) - O 뇨
\/ C(=8C)/\ 一03 〉0\ /? 。- '「 -/ 0 > C/\0 0 (1 一— 2 0o) / \ C < + t c(1I1 -3 o) - O 뇨
양성이온 (브)에서 치환체의 성질에 따라 이것은 전이반응이 일 어날 수 있고. 열이 c-c 단일결합을 분해시킨다. 메탄올과 같은 반양성자 용매에서는 양성이온 (!Q)에서 a- 메톡시 알킬히드로 과 산화물 (브)가 형성된다 . 7. 2. 3 새로 제안된 반응기구 Cr i e g ee 가 제안한 반응 메커니즘은 많은 실험 결과를 잘 연결시 켜 설명해 주고 있댜 그러나 이는 입체적 효과에 대한 언급이 없 어, 점차 수정되거나 경쟁적인 반응경로를 통하여 새로운 메커니즘 이 제안되게 되었다 . 이 새로운 메커니즘은 올레핀의 기하학적 배 치에 대한 오존 화합물의 시스-트랜스 비의 의존성을 나타내는 실 험 결과로부터 이루어졌다 . Sto r y 13· 14) 등은 Cr i e g ee 가 제 안한 메 커 니 즘을 기 본으로 해 서 어 떤 상태에서는 적어도 또 다른 과정을 거치는 새로운 메커니즘을 제안하였다. 이 과정이란 초기 오존 첨가물과 알데히드 사이에서 일어나는 반 응이다. 알데히드는 Crie g e e 반응과정에서 생성된다. 시스-올레핀 에 대한 이 반응 메커니즘을 그림 7-2 에 나타내었다 . 올레핀에 오 존이 첨가되면 다섯 고리 첨가물인 (산)나 6- 복합체 (인이 되고 특 수한 상태에서 (인은 년)로 변환된다 . 구조 (산)의 산소-산소 결 합이 끊어지면 양성이온 (臣)가 되며, 양성이온 (旦)가 분해되면 알데히드와 Cri eg e e 양성이온 (브)으로 된다. 양성이온 (旦)은 2- 메틸프로판알과 결합하여 시스-트랜스 혼합 물인 오존 화합물 (끄으:p)로 된다. 양성이온 (끄)은 알데히드와 반응하여 새로운 중간체 혹은 전이 상태인 (登으~만)가 된다. 구조 (旦)은 알데히드 교환이 일어나서 오존 화합물 (끄으 :E) 와 알데히
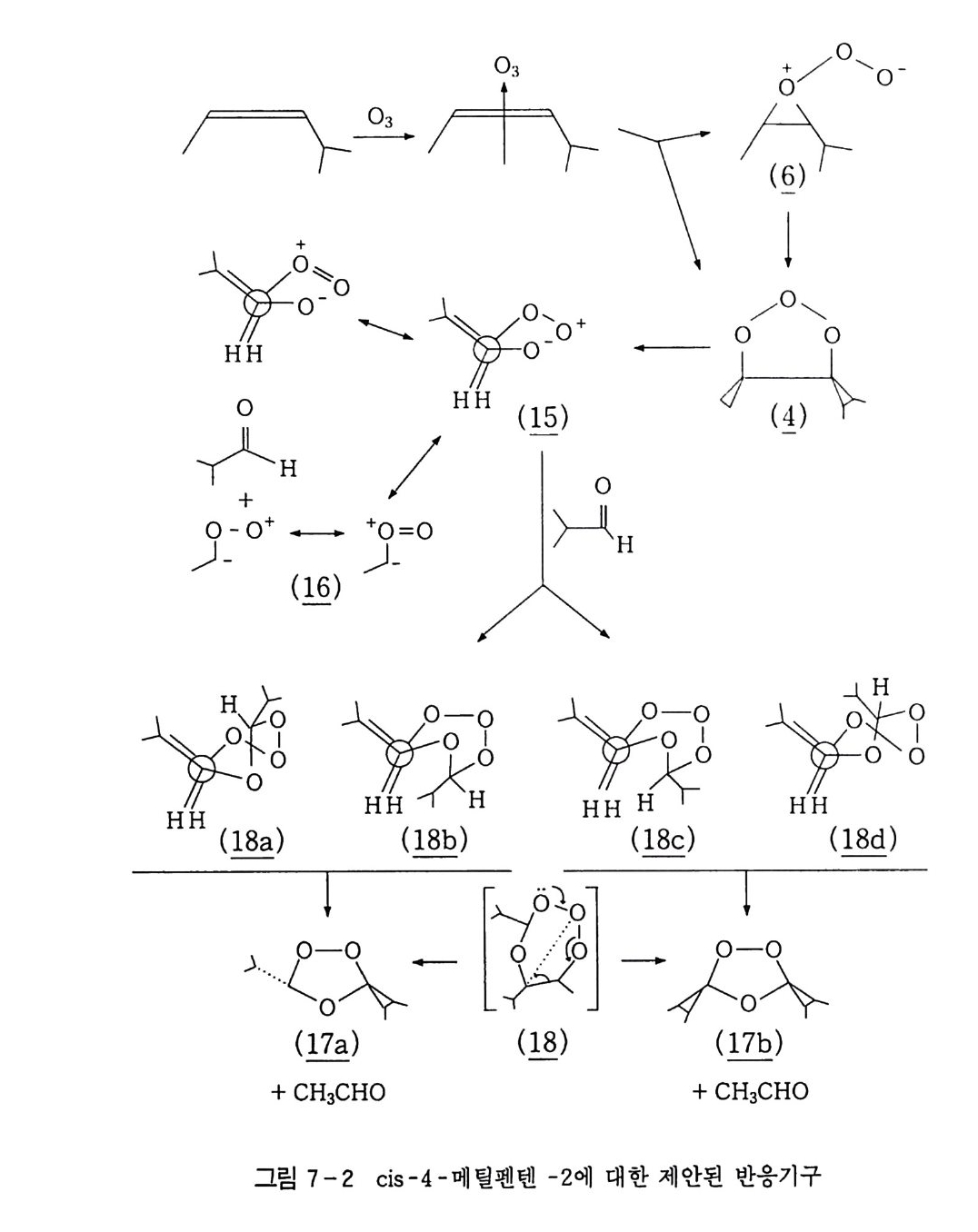 三느三O:i \ :0 \ o -
三느三O:i \ :0 \ o -
드가 된다 . 이 모델을 살펴보면 올레핀의 입체적 효과가 오존 화합 물의 시스-트랜스 분배비에 영향을 미치고 있음을 나타낸다. 이 경우 (년오와 같은 이형태체 (con fo rmer) 는 수소-수소의 적 은 입체 장해 때문에 유리하게 진행된다. 이형태체 (년으)는 시스 오존화물을 생성하므로 이 상호변환 반응에 의해 시스-오존화물이 트랜스-오존화물보다 더 많이 생길 것으로 기대된다. 트랜스-올 레핀에 대해서도 같은 고려를 적용할 수 있으나 이 반응에서는 시 스一트랜스 오존화물의 어느 것에 대해서도 큰 이점을 주지 않고 있댜 적어도 다소의 선택성은 있다 하더라도 시스의 경우에서처럼 현저한 이점을 나타내 주지 않는다는 결론을 얻게 되는 것이다. 아무튼 올레핀의 입체화학과 입체적 요인이 오존화물의 시스 트랜스 생성비에 영향을 주는 것은 분명하다. 생성되는 오존화물이 Crie g e e 통로를 거치느냐에 따라 알데히드의 산소가 결합되는 위 치가 달라진댜 이를 알아보기 위해 01 8 로 표식된 알데히드를 써서 오존화반응을 시켰다 .1 5) 그림 7 -2 를 보면 알데히드 교환 메커니즘은 018 이 오존화물의 과산화결합다리에 들어가나. Crie g e e 통로에 따르면 에테르다리에 들어간다. 오존화물을 환원하고 생성하는 알코올의 분배로부터 오 존화물에 있는 018 의 위치를 결정할 수 있다. 오존화물의 산소 중 수소화 리튬 알루미늄으로 환원할 때 어느 산소를 잃게 되는지 결정하는 연구가 보고되었다. 표식된 아세트알 데히드 존재하에서 트랜스-디 -이소프로필에틸렌을 오존화하였다. 이렇게 해서 생성된 메틸 이소프로필 오존화물에 대해 018 분배 를 결정하였다. 환원은 오존화물의 입체적 효과에 영향을 미치게 되므로 수소화 리튬 알루미늄, 메틸 리튬으로 이루어졌다. 환원제 로 수소화 리튬 알루미늄을 사용했을 때 오존화물의 31 %가 에테 르 산소다리에 018 로 된 오존화물이. 68 %가 과산화다리에 018 이
결합된 오존화물이 얻어졌댜 환원제로 메틸 리튬을 사용하면 오존 화물의 23 %가 에테르다리에, 77 %가 과산화다리에 01 8 이 결합되 었다. 이 두 환원 결과의 차이는 환원제에 의한 입체적인 효과 때 문이라고 보여진다. 그림 7-3 에 이 반응 메커니즘을 나타내었다. 그림 7-3 에 의하면 이 물질에 대해서는 Sto r y 통로가 Crie g e e 통로보다 더 유리하게 진행된다. ol8 로 표식된 알데히드를 사용한 유사한 연구 결과가 Flisz ar Iii> 등에 의해 이루어졌다. ol 8 의 위치를 결정하기 위해 질량분광법을 도입하였는데, 이들의 실험 결과에 의하면 대단히 낮은 온도를 제외 하고는 대부분의 018 은 에테르 다리에 결합되어 있다. 낮은 온도에 서는 10 % 정도만이 018 의 과산화물다리에 결합되어 있을 뿐이다. 그림 7-4 는 이 반응의 메커니즘을 나타내었다. 이 메커니즘에서 볼 수 있는 바와 같이 Fli sz ar 등은. Crie g e e 메 커 니 즘과 알데 히 드 교환이 일어나는 Sto r y 메커니즘이 서로 경쟁적으로 일어난다는 결론을 내렸다. 또 올레핀의 입체화학을 고려한 새로운 메커니즘이 Ba i le y 17l 에 의해 제안된 메커니즘의 보완이라고 볼 수 있다. 즉, Cr i e g ee 의 양성이온이 신 (s y n) -안티 (an ti) 이성질체로 존재한다 고 가정한 . 것이다. Bail e y 제안에 의하면 반응 상태가 달라짐에 따라 생성되는 신-안티 양성이온의 바가 달라지고, 신-안티 양성이온은 알데히 드와 반응하여 시스-트랜스 비가 다른 오존화물을 생성한다. 낮은 온도의 오존 분해반응에서는 신-안티 양성이온이 따로따로 존재 할 수 있다. 이 제 안은 첨 가물의 동시 분해 ( concerte d decomp o si- ti on) 를 가정하고 있으며, 최종 오존화물이 시스-트랜스 비롤 결 정하는 데는 세 가지 규칙이 적용된다. 초기 첨가물의 수평치환체 는 안티-양성이온으로, 수직치환체는 신-양성이온으로 분해된다. 수평치환체는 수직치환체보다 양성이온으로 더 잘 생성된다. 알데
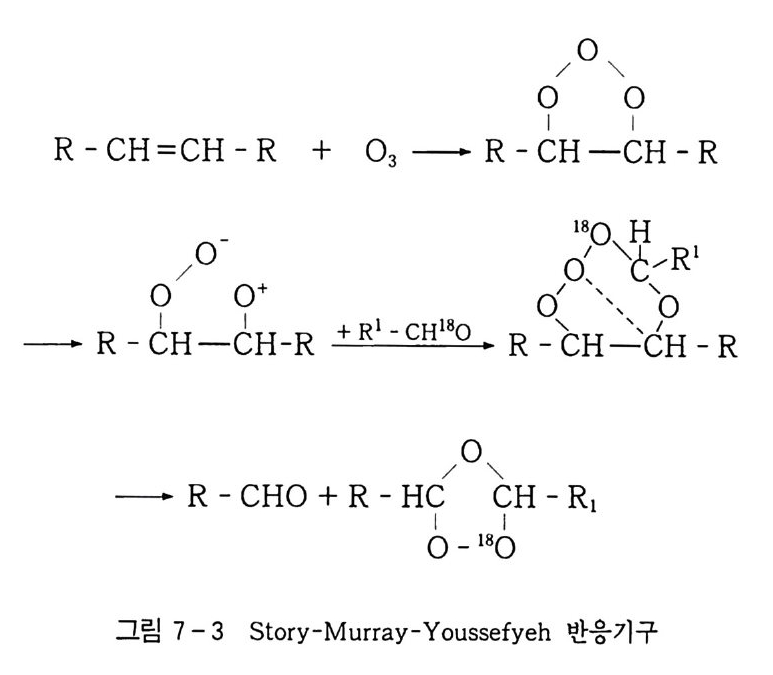 / 。 \
/ 。 \
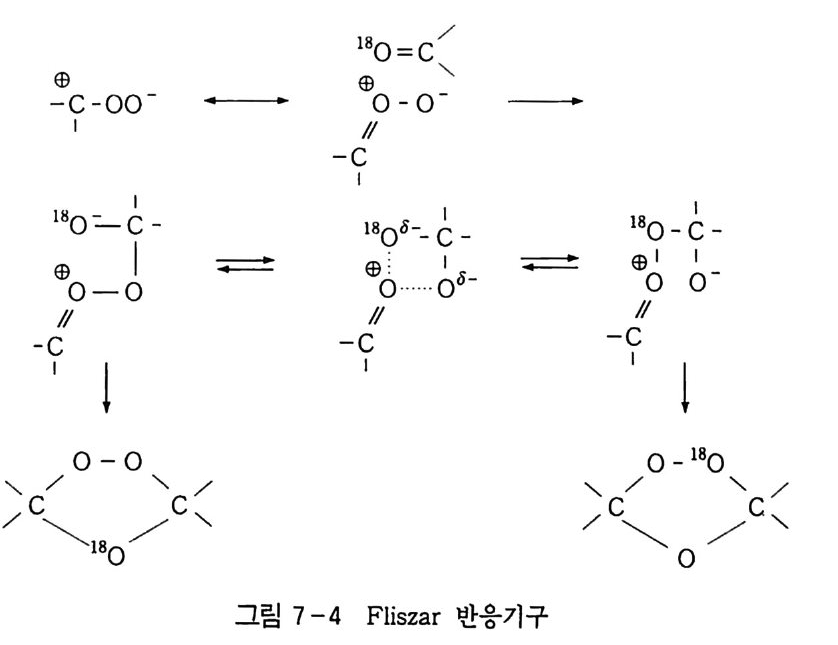 180 = C /
180 = C /
히드와의 반응에서 안티-양성이온은 시스-오존화물을. 신 - 양성 이온은 트랜스-오존화물을 생성한다. 큰 치환기를 가진 시스-올레핀은 시스-트랜스 비가 1 보다 큰 오존화물을 생성하며 . 트랜스-올레핀은 초기 첨가물의 형태에 의 존한댜 비교적 작은 치환기를 가진 올레핀은 신-양성이온을 형성 하고 트랜스-오존화물이 예상된다. 더 큰 치환기를 가진 트랜스 올레핀은 더 많은 안티-양성이온을 형성하고 따라서 더 많은 시 스-오존화물이 생성된다. 이 반응 메커니즘은 홍미의 대상이 되고 있는데. 특 히 신-안티 이성질체의 평형에 관한 연구에 관심이 집중되고 있다. 오 존 화물의 시스-트랜스 분배에 대한 온도 효과에 관한 연구는 아직도 거의 이루어지지 않고 있다. 어떤 온도에 도달하면 동일한 시스-트랜스 이성질체로부터 얻 어지는 신-안티 분배가 같아지게 될 것으로 예상된다. 그러나 현 재로서는 완전한 오존 분해반응의 메커니즘을 확정하기 어렵다 . 이 문제를 해결하기 위해서는 간단한 알켄에 대한 더 많은 속도론적인 연구가 필요하며 동위원소로 표식된 알데히드를 사용한 연구가 활 발하게 이루어져야 할 것이다. 7. 3 삼중결합의 오존 분해반응 아세틸렌 화합물의 오존 분해반응은 올레핀 화합물의 그것보다 많이 연구되어 있지 않다. 두 가지 형태의 비과산화물이 얻어졌는 데 카르복시산(삼중결합의 완전 분해로부터) 6- 카르보닐 화합물 은 항상 주생성물이고 카르복시산 형성의 중간체로 고려된다.
CH3 (C H2 ); C =C(CH2 )1 C OOH HOOC(CHz ); C O OH (18 ) (19 ) CH3 ( C H2 ) ; CO CO ( CH2 ) 1 CO OH (20) 전형적인 예는 스테어롤산 (s t earol i c acid ) (旦)으로, 69~80 % 수득률로 아젤라산 (azela i c ac i d)(l2) 과 4% 의 9, 10- 디케토스테 아린산(쁘)을 형성한다. 예외적으로 아세틸렌으로부터 81 %의 수 득률로 글리옥살을 얻을 수 있다. Pa ill ard 와 W i eland18 ) 는 그림 7 -5 에서 보는 바와 같이 카르복시산의 생성에서 무수 중간체를 제안하였댜 1, 4- 디벤족시 -2- 부틴의 오존 분해반응에 대한 연구로 올레핀 에서와 동일하게 아세틸렌 화합물의 오존 분해반응에 대한 메커니 즘이 제안되었다. 불활성 용매로부터의 생성물은 중간체인 케토 과산화물이 되고. 사염화탄소_아세트산 혼합용매로부터는 아세톡시히드로 과산화물 이 생성된다 .1 9 ) (盤)의 환원과 재배치로 디케톤(쁘)과 카르복시산(완!)이 각각 높은 수득률로 생성된다. 또한 (쁘)의 열분해로 무수물 (~)o] 된 댜 디케톤으로의 환원과 산으로의 재배치는 아세독시히드로 퍼옥 사이드(챔)를 분리하지 않고 이루어진다. 같은 결과가 1.4- 디아세톡시 -2- 부틴에서도 얻어졌다. 중간체 (쁘)의 구조는 확실치 않으나 r - 복합체이거나 첨가 화합물일 것 으로 고려된다. 양성이온 중간체 (亞)는 위에 나타낸 네 가지 방법 으로 반응한다. 또한 삼중결합과 이중결합을 동시에 함유한 탄화 수소는 이중결합에서 반응이 더 잘 일어난다 .
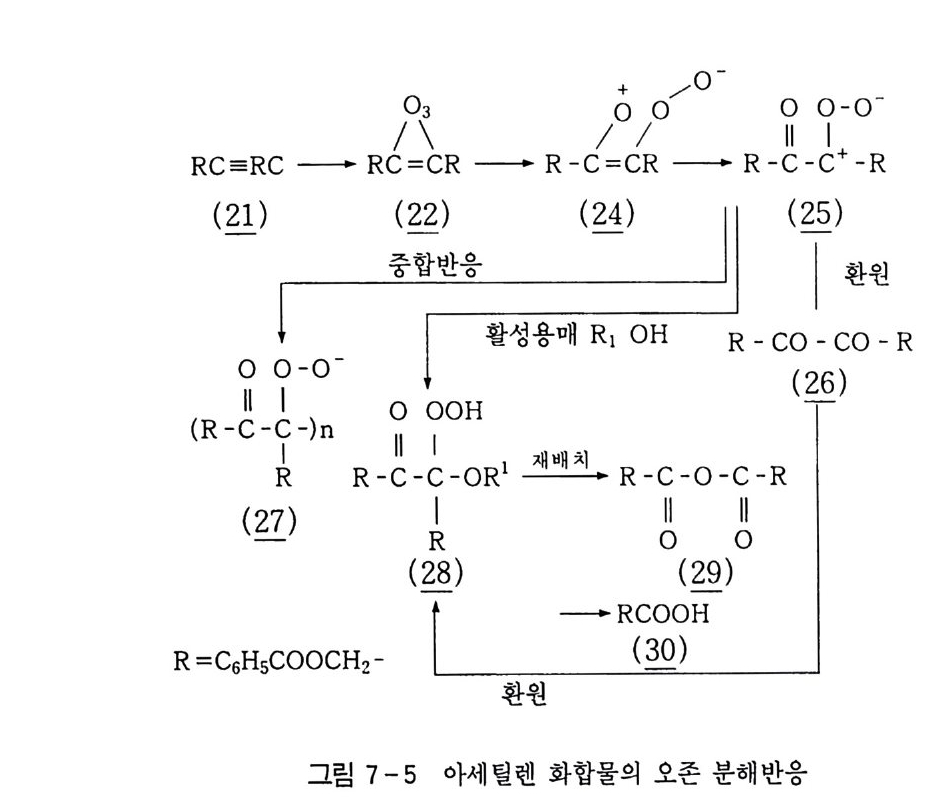 + ,,,,.o -
+ ,,,,.o -
7.4 방향족 화합물의 오존 분해반응 방향족 화합물에 대한 오존 분해반응은 1940 년부터 널리 연구되 어 왔다. 일반적으로 이들 화합물은 올레핀 화합물의 오존화반응보 다는 서서히 일어난다. 여러 가지 방향계는 또한 서로 다른 반응성 울 나타낸다. 간단한 계의 정상적인 오존 분해반응 용이도 순서는 다음과같다. 간단한 알켄>안트라센>페난트렌>나프탈렌>벤젠
7.4.l 벤젠과 그 유도체 W i bau t와 공동 연구자들 20) 은 벤젠, 나프탈렌과 그 유도체의 오 존화에 대한 속도론적 연구 결과를 보고하였고, 이 반응들로부터 비과산화물을 분리하였다. 할로겐, 니트로기와 같은 전자끌기는 방 향족 고리를 비활성화하고 알킬기, 히드록실기, 메톡시기와 같은 전 자 주기는 활성화한다. 메틸기로 치환된 벤젠의 동족열 화합물에 대한 오존화반응의 속도는 치환된 메틸기의 수가 증가함에 따라 빨 라진댜 올레핀계 화합물과 삼중결합의 탄소에 결합된 기에 대해서 도 동일한 결과가 얻어졌다. 이 두 경우에서 오존은 천전자계로 작 용함을 나타내 준댜 아무튼 대부분의 올레핀에 대한 천전자성 끝에서 더 강한 결합형 성을 나타내는 전위 상태를 거쳐 1, 3 -쌍극자 고리화 반응으로 진 행된다. 오존 분해반응이 일어나는 방향계에 있어서도 같은 메커니 즘임을 가정할 수 있다. o- 크실렌의 오존 분해반응은 20~26 %의 수득률로 글리옥살, 메틸글리옥살, 바이아세틸이 각각 3:2:1 의 물 비를 갖는 혼합물로 얻어졌다. 케쿨레 공명구조 (완)과 (~)71- 동 일하게 반응한다는 가정하에서 기대되는 비(比)다. 7. 4. 2 나프탈렌과 그 유도체 나프탈렌은 쉽게 오존 2 몰과 반응하고, 그 후 계속해서 5 몰의 오 존이 반응할 때까지 서서히 오존을 흡수한다. 메틸화된 고리에서는 반응이 더 잘 일어난다. 식 (3) 에서 보는 바와 같이 2, 3- 디메틸 나프탈렌으로부터는 바이아세틸(~). 글리옥살(~). 메틸글리옥살 (완) 및 4, 5- 디메틸 프탈산이 낮은 수득률로 얻어지고 프탈산 (무)과 프탈알데히드 유도체(쓰브)가 90~95 % 수독률로 생성
된다. 이것은 틀립없이 양성이온(브)과 메탄올의 상호작용으로 첨 가 생성물(선)의 고리화가 일어난 결과이댜 양성이온 (션)은 나 프탈렌의 1. 2 와 3, 4 결합에 오존이 공격해서 r- 복합체 (전)이 형성되고. 좀더 반응하여 (쁘)과 (쁘)를 거쳐 생성된다.
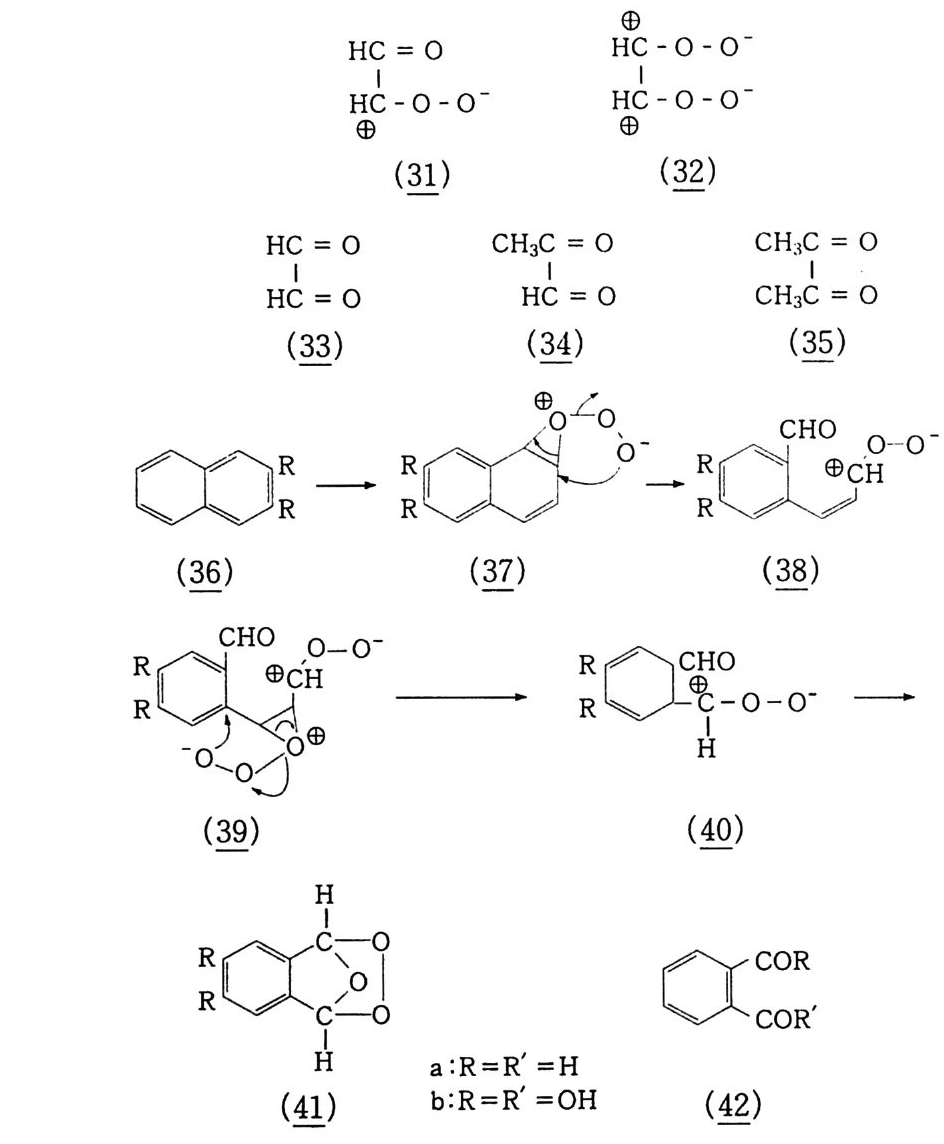 HC = 0 H®C -0 -0 ~
HC = 0 H®C -0 -0 ~
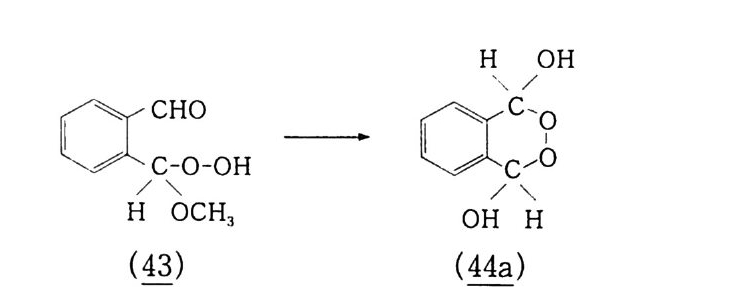 ~ H/ C\O-0 C-H03H oOHH/ c\\HOg H
~ H/ C\O-0 C-H03H oOHH/ c\\HOg H
W i bau t와 Kam p schm i d t 2 는 불활성 용매 내에서는 디오존화 물이 생성된다고 가정하였다. 이 생성물은 모노오존화물이나 (션~) 의 중합체인 것으로 기대된다. 분리된 물질은 대단히 불안정하여 따)의 구조를 가질 것으로 여겨진다. 마찬가지로 2, 3- 디메틸나 프탈렌의 주생성물은 (선으)이고 부생성물은 (산上)이다. 바이아세 틸(亞)과 글리옥살(盤)은 아마도 (브으)와 (산上)를 통해서 만들 어지는 양성이온 (완)과 그 유도체의 환원으로부터 생성되는 것 같다. 메틸글리옥살(선)은 오존화물(선上)이 더 생성되는데 , 메틸 글리옥살(센)은 오존화물(신上)이 더 오존 분해되어 생성되는 것 으로 예상된댜 히드록시, 메톡시, 할로겐, 니트로, 카르보닐 및 페닐기로 치환된 나프탈렌 유도체들의 오존화반응의 결과도 보고되었다. 메틸, 히드 록시, 메톡시기가 치환된 고리가 먼저 공격을 받는다. 1- 페닐 나 프탈렌에서는 두 고리가 동등하게 공격을 받고 전자끌기로 치환된 나프탈렌은 치환되지 않은 고리가 먼저 공격을 받는다.
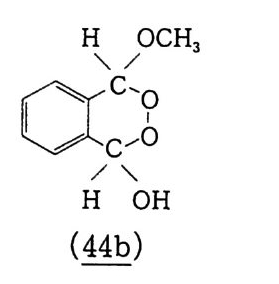 OH\ c/ \Og CH 3
OH\ c/ \Og CH 3
메탄올 속에서 나프탈렌은 (브上)와 같은 고리화 과산화물로 오 존화한다. 이 과산화물은 쉽게 메탄올울 잃고 프탈알데히딕산이 되 거나 산화하면 프탈산이 된다 . 22) 수용액 속에서도 유사한 오존화물이 생성되고 용이하게 프탈알 데히드로 환원한다 .23) 7.5 헤데로 고리 화합물의 오존 분해반응 7. 5.l 피리딘 피리딘 고리는 벤젠 고리보다 오존에 대한 반응성이 적다. 그러 므로 피리딘 유도체와는 반응하지 않고 피리딘 유도체보다 오존에 대한 반응성이 약한 용매를 발견하기 어렵다. 비대칭으로 치환된 피리딘 동족체의 오존화반응으로 케쿨레형의 구조에서 기대되는 생성물을 얻었다. 그리고 포름산. 아세트산, 피브르산, 수산, 글리 옥살산과 암모니아 등이 분리되고 확인되었다. 비록 수득률은 낮지 만 오존화반응은 피리딘 화합물의 구조를 확인하는 데 이용할 수 있다. 오존은 피리딘 핵의 탄소-탄소 결합을 공격하여 (산)과 같 은 물질이 (座)로부터 생성되는데 곧 아미드로 가수분해되고. 결국 암모니아가 된다.
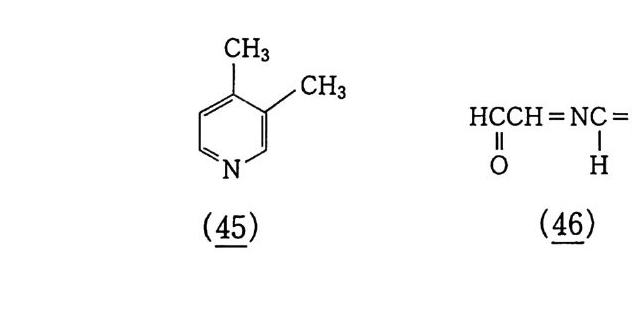 〈멕멕HCCH=NC=O
〈멕멕HCCH=NC=O
생성되는 암모니아가 오존을 흡착하기 때문에 흡착비가 2 라는 것이 S i xma 24) 에 의해 발견됨으로써 이 사실이 뒷받침되었댜 두 경우에서 아미드가 분리되었는데 (47) 로부터 (48), (49) 로부터 (완)이 분리되었댜
 二CH3 ― CCHH33 二N뻔 CH 3 二멕N H2
二CH3 ― CCHH33 二N뻔 CH 3 二멕N H2
이 결과는 2, 3-혹 은 5, 6- 결합에 오존의 초기 공격으로 이루 어진다 . 피리딘의 결합은 전부 이중결합을 띠며, 그 이상의 오존의 공격으로 곧 (완)와 (완)이 생성되고 분해 성성물이 얻어진다. 또 가수분해하면 벤젠에서와 같은 생성물이 얻어진다.
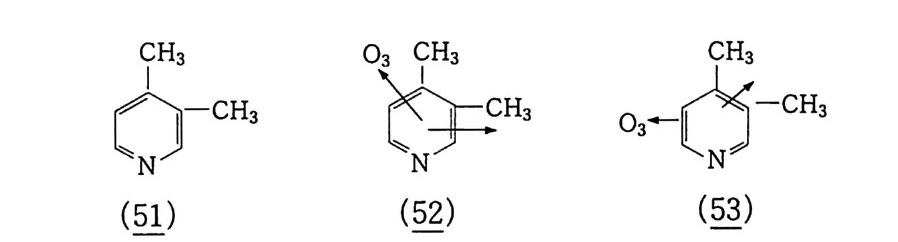 0\3 CH3뻔 0:—CH3
0\3 CH3뻔 0:—CH3
구조 (산)과 (速)에서 탄소-질소 결합이 피리딘 핵 자체보다
오존에 대한 저항을 더욱 나타낸다. 그리고 오존이 피리딘의 질소 원자에서 복합체를 만들어 피리딘 산화물이 형성될 가능성이 있다. 7. 6 올레핀 오존 분해반응 상대 반응속도 여러 올레핀 오존 분해반응의 속도론적 연구 결과 치환체에 의하 여 반응속도가 다름을 알았다. 이중결합에의 탄소원자에 올레핀으로부터 전자를 잡아당기면 속 도가 저하된다. 알킬 25 ,26) 과 알콕시 27 ) 그룹은 이중결합을 활성화시키 는 반면에 할로겐 ,28,29) 니트로뽀 시안 3 1 ) 또는 카르보닐 치환체 3 1, 32) 는 오존의 이중결합 공격성을 약화시킨다. 표 7-1 에 사염화탄소에서 몇 가지 올레핀 오존 분해반응의 상 대 속도 함수를 실었다 .25) 이 표에서 보듯 수소 대신 메틸기가 늘어 나면 속도는 8 배까지 증가한다. t-부틸 그룹이 치환될 경우 그 자
표 7-1 몇 가지 올레핀 오존 분해반응의 상대 속도 함수 올레핀 K,.1(e /m ol sec) in CCl,1 CH2=CH2 I 1 CH3CH = CH2 I 3.2 (CH3)2C=CH2 I 3.9 cis - C H3CH = CHCH3 6.5 (CH3)2C = CHCH3 6.7 (CH3)2C = C(CH3)2 8.0 C6H5-CH = CH2 4.1 cis - C IHC = CHCI 0.00144 tra ns -C IHC = CHCI 0.02364
신이 강력한 전자주기 영향을 가지고 있지만 입체 장애 때문에 오 존은 거의 반응하지 않는댜 이 이유로 인해 1, 2_ 이치환에틸렌 경우 시스-가 트랜스 이성 체보다 천천히 반응하고 33) 한 페닐 치환체는 한 메틸 치환보다 에 틸렌 이중결합을 활성화시키며. 한 이중결합이 바편재화에 의하여 역시 이중결합의 활성을 높인다. 즉, 부타디엔. 은 에틸렌보다 3 배 더 활성 화시 킨다 . 34. 35) 7. 7 산화제로서의 오존 오존이 포화 탄화수소 , 알코올, 알데히드, 에테르, 아민, 포스핀 유기 황화물과 같은 물질과의 반응을 설명하는 데 상당한 진전이 이루어졌다. 이 반응들은 분명히 적어도 두 가지 형태로 이루어진 댜 하나는 오존에 의한 천전자성 반응을 의미하며 다른 하나는 산 소가 주반응물인 오존에 의해 유도되는 산화반응을 의미한다 . 아직 확실치는 않으나 오존은 또한 찬핵체로서 반응할 가능성도 있다. 7. 7. 1 아민, 포스핀, 아르신과 황화물 36 , 37) 오존과 삼차아민, 포스핀, 아르신 (ars i ne), 황화물 및 술폭사이드 와의 반응은 오존에 의한 천전자성 반응의 범주에 속한다. 일차, 이차 지방족 아민은 오존에 의해 분해되고 삼차아민은 대응하는 아 민 산화물로 변환된다. 불포화 디아조 화합물에 대한 산화물 생성이 보고되었다.
CH3 BrCl 6 H 1N = NC = CHCOOC2 H s 一。 3 0 H BrC6H4 ( N/ = N ) COCH3 + 0 = C| COOC2 H s ( 4 ) 삼차아민에 의한 오존 흡수는 대단히 빠르다 . 대략 1 몰의 오존이 반응하여 높은 수득률로 아민 산화물을 생성한다. 트리페닐포스핀 과 트리페닐아르신에 대해서도 같은 결과를 얻었다. 이 메커니즘은 다음과 같다.
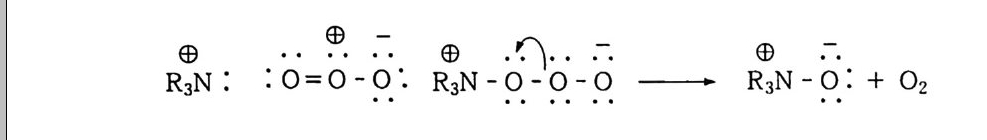 Re3N : : •0 •= E0•B • - 0•- ·•. R®3 N.-?0-·. 0· -·0-. 一 R®3N -0•-. : + 02 (5)
Re3N : : •0 •= E0•B • - 0•- ·•. R®3 N.-?0-·. 0· -·0-. 一 R®3N -0•-. : + 02 (5)
유기 황화물도 이와 비슷하게 반응한다. 그 생성물은 술폰이고 때때로 술폭사이드가 중간체로 분리된다.
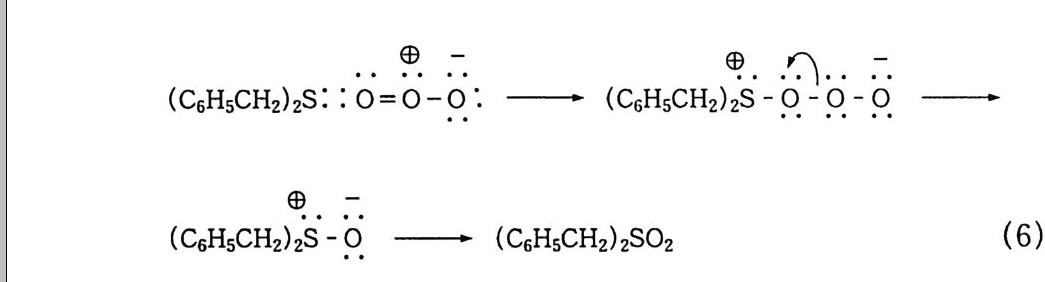 (C6HsCH 사 2S : : .0. = (0B -0- •. (CGHsCH 사® 2S• -.•O?. - ..O. .- ..O_ . •
(C6HsCH 사 2S : : .0. = (0B -0- •. (CGHsCH 사® 2S• -.•O?. - ..O. .- ..O_ . •
이 황화물 티오술핀에이트 다 황화물 (R-S-S-S-S-R) 은 오존에 의해 술폰산 무수물로 변화된다.
7. 7.2 카르복시산 카르복시산은 정상 상태에서 극히 소량만이 오존과 반응한다. Paill ar d 등은 극히 적 은 양의 빙초산만이 과초산으로 변환됨을 발 견하였댜 Taube 381 는 UV 광을 조사하면서 오존으로 아세트산을 산화하여 상당량의 과초산을 얻을 수 있었다. 이 반응에서 친전자 체는 오존이 아니라 여기된 산소원자이다. RC8 - 0 -H + 03 R-C0Il -0e0e|. ) -; - H 一 R8C- O OH (7) 7. 7. 3 알데히드, 케톤, 알코올 및 포화탄화수소 산화제로서 오존의 나머지 반응들은 오존이 반응의 개시제로 사 용되며. 실제 반응에서는 산소가 관여하는 형태의 반응이다 . 오 존-산소 혼합물이 벤즈알데히드와 다른 알데히드를 대응하는 산 소 과산으로 산화시킨다. 오존 흡수는 대단히 느리고, 산화하는 정 도는 홉수된 오존의 양으로 기대할 수 있는 정도보다 크다. 그러나 IR 데이터는 오존이 존재함으로써 산화반응 속도가 급격 히 증가함을 나타내준다. 벤즈알데히드에 0.76 %의 오존을 10 분간 처리해 주었더니 과벤조산의 특성 피크가 산소만을 3 시간 처리했 을 때보다 더 강하게 나타났다. 오존의 농도를 3.4 %로 증가시켜도 과벤조산 생성 속도를 촉진시키지는 못한다. 오존의 가속화 속도는 낮은 오존 농도, 즉 0.01 % 이하에서도 같은 효과를 나타낸다. 주 산화제가 오존이 아니라 산소라는 증거로는 오존의 양을 일정하게 유지하고 산소의 양을 감소시키면 산화 생성물이 감소한다는 사실
을 들 수 있다. 1 % 이하의 산소를 함유하는 오존-질소 혼합기체를 사용하면 과벤조산이 아니라 벤조산이 얻어진다 . 이것은 아마도 오존이 다음 반응과 같이 친핵체로서 작용하기 때문인 것으로 보인다.
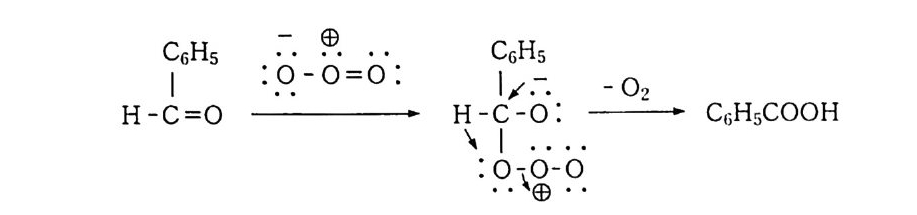 eX3
eX3
올레핀은 다른 산화제보다 오존과 더 쉽게 반응하기 때문에 오존 으로 개시되는 자동 산화반응을 일으키지 않는다. 오존-산소 혼합기체를 써서 케톤을 산화하면 좋은 수득률로 카 르복시산이 얻어진댜 이 반응은 알데히드의 반응보다 천천히 일 어나며 중간체나 부산물이 분리되지 않는다. 예를 들면 시클로펜 타데카논으로부터 펜타데카노산 , 디에틸케톤으로부터 아세트산과 프로피온산 등이 생성되는 반응이다. 이 반응은 오존의 천핵성 공 격에 의해 일어나며 에스테르가 생겼다가 더 산화되어 최종 생성 물이 된다.
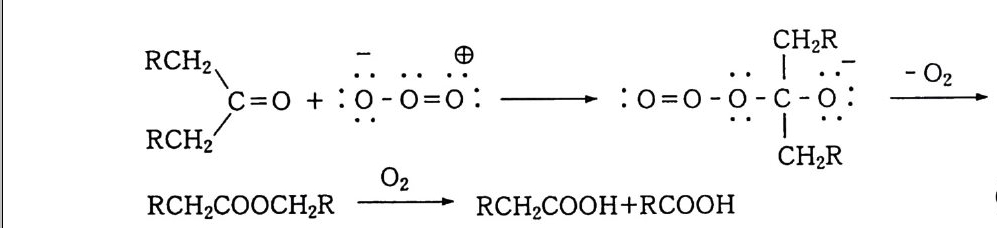 RCH2 \ c=o + :.o-. -o..=.o® .: - :o=o-..o .. -CcIIH -2 o..R.. -: -02 .
RCH2 \ c=o + :.o-. -o..=.o® .: - :o=o-..o .. -CcIIH -2 o..R.. -: -02 .
물론 산소가 없을 때 오존의 친핵성 공격이 중요하기는 하나 이 반응도 역시 오존 개시 산화반응이다.
알코올과 에테르는 오존과 반응하여 알데히드, 카르복시산, 에스 테르, 과산화물을 생성한다. 이소아밀에테르로부터 이소아밀벨러레 이트가 70~80 %의 수득률로 얻어진다. 오존에 의한 알코올의 산 화는 탄수화물 분야에서 이용된다. 수용액에서 당을 오존화하면 페 닐히드라존 오사존 등으로 확인할 수 있는 알도스가 생긴다. 이차 알코올은 오존으로 산화하면 높은 수득률로 케톤이 생성된다. 오존은 활성인 메탈 메틸렌 메틸렌기와 반응한다. 최근에 알려 진 몇 가지 예를 들면 데카린을 9_ 데카롤, 크산덴을 크산톤, 플루 오렌을 플루오레논, 안트론을 안트라퀴논, 디페닐 메탄을 벤조페논 등으로 변환시킬 수 있댜 메탄을 오존-산소 혼합물로 산화하면 메탄올과 포름 알데히드 의 혼합물이 생성된댜 높은 온도에서는 오존이 분해하여 생긴 산 소원자에 의한 반응이 일어난다. 오존_산소 혼합기체에서 프로판, 부탄 헥산. 옥탄 및 기타 알칸류의 산화는 높은 온도가 필요하다 는 것을 제외하면 알데히드의 산화 결과와 유사하다. 여러 가지 실 험 사실을 근거로 해서 Schuber t와 Pease39) 는 알칸의 산화에 대한 다음과 같은 메커니즘을 제안하였다(식 10). RH+03 一 RO· +HOO· (10 -1) RO • + RH -ROH + R • (10-2) RO ·一 R' = 0+R· (10 -3) R·+ 02 一 ROO· (lo- 4) ROO • + RH -ROOH+R • (10-5)
낮은 온도에서 곧은 사슬과 가지 달린 사슬 탄화수소가 반응의 차이점을 보이는 것은 반응 (l 0-3a) 가 일어날 수 있기 때문이다. (CH3}3CO • 一 CH3COCH3 + CH3 • (lo - 3 a) 높은 온도에서의 반응과 낮은 온도에서의 반응의 차이점은 낮은 온도에서는 반응 (1 0-5) 가 일어나지 않는다는 점이다. 그 대신 H02 • 와 R02 • 래디컬의 연쇄종결이 일어난다. 여하튼 높은 온도 에서는 히드로퍼옥사이드가 생겨 연쇄반응이 계속된다. CH3· 으노 CH 요· (1o(_16o)_ 7 ) CH302 • 一 HCHO + • OH (CH3}3COOH + HCHO 一 (CH3}3C OOCH20H (10 -8) 산소에 의한 산화반응은 항상 적은 양이 존재하는 오존에 의해서 개시되며, 3oo ·c 근처에서 다음과 같은 반응으로 오존의 양이 지 속된다 . 스모그와 관련된 최근 연구에 따르면 대기 중에서 탄화수 소가 산화될 때 오존이 생성된다. 이와 유사한 산화반응 메커니즘 을 알데히드, 케톤, 알코올, 에테르 등에도 적용시킬 수 있다. R02·+02 一 RO·+03 (lo- 9) Lon g에 의하면 아미노산은 오존과 반응하지 않는다. 그러나 Berge l 등은 이 반응은 일어나며 그 주생성물은 알데히드, 암모니 아, 과산화수소임을 밝혔지만, 이 반응 메커니즘은 아직 잘 알려져
있지 않댜
참고문헌 1) Pamela S. Zurer, C & EN p.8 ~18(May 24, 1993) . 2) C. F. Schonbein and C. R. Hebd, Seances Acad. Sci ., 10, 706( 1840) . 3) R. Trarnborlalo, S. Ghosh, C. Burrus and W. Gordy, J. Chem. Phys . , 21, 851~855(1953). 4) C. F. Schonbein and J. Prakt, Chem., 66, 282( 1855) . 5) J. S. Murp h y and Jan et, R. Orr, Ozone Chemi str y and Technolog y, The Tranklin Instit ute Press, Ph ilad elph i a ( 1977) . 6 ) C. Bis c hoff and A. Riec he, Z. Chem. , 5, 9 7 (19 65 ) . 7) J. P. W i ba ut and F. L. J. Six r na, Rec. Trav. Chim ., 71, 761 ( 1952) . 8) R. Huis g e n ,A ng ew . Chem. Inte m . Ed. Eng l., 2, 565, 633( 19 63). 9) P. S. Baile y, J . A. Thomp so n and B. A. Shoulders, J. Am. Chem. Soc., 8 8, 4098( 1966) . 10) L. J. Durham and F. L. Greenwood, Chem. Commun., 2 4(1 9 68) . 11 ) R. Cri eg e e , Ang ew . Chem. , 21, 765 (19 75 ) . 12 ) R. Cri eg e e , Ang ew . Chem. Int. Ed. Eng l. , 14, 745 (19 75 ) . 13 ) R. W. Murray, R. D. Youssefy eh and P. R. Sto r y , J. Am. Chem. Soc., 8 9, 2429( 1967) . 14) R. W. Murray and N. Y. Trans, Acad. Sc i., 2 9, 854(1 9 67) . 15) P. R. Sto r y , C. E. Bis h op, J. R. Burge s s, R. W. Murray and R. D. Youssefy eh , J. Am. Chem. Soc., 90, 1907(1 9 68) . 16) S. Fli sz ar, J . Carles and J. Renard, J. Am. Chem. Soc., 6 0, 1364( 1968) .
17) N. L. Bauld, J. A. Thomp so n, C. E. Hudson and P. S. Bail e y, J. Am. Chem. Soc. , 90, 1822 (19 68 ) . 18) H. Pa illa rd and C. Wi el and, He/v. Chim ., Acta 21, 1356(1 9 38) . 19) R. Cr ieg e e and M. Lender, Ann., 583, 6( 1953). 20) J. P. Wi ba ut, J. Chim . Phy s. , 53, 111 (19 56) . 21 ) L. W. F. Kamp sc hm idt and J. P. Wi ba ut, Rec. Trm1. Chim ., 73, 431 (19 54). 22) P. S. Ba ile y et al., J. Org. Chem. , 2 9( 6) , 1400( 1964) . 23) M. C. Stu rr ock, B. J. Cravy and V. A. Wi ng , Canadia n J. Chem., 49( 18) , 3047( 1971 ) . 24) F.L.J.S ix m a,Rec. Trav. Chim .,71, 1124(1952). 25) D. G. Wi lliam son and R. J. Cveta n ovic , J. A m. Chem. Soc., 90, 3668, 4248( 1968) . 26) J. Carles and S. Flis z ar, Adv. Chem., 311, 689( 1969) . 27) U. Schm idt and P. Grafe n , Ju stu s Lie b ig s Ann. Chem., 565, 97 (19 62) . 28) G. Nag e ndrapp a , K. Gr ies baum and J. Ag ric, Food Chem. , 26, 581 (19 78) . 29) a) K. Gr ies baum and J. Brug ge mann, Adv. Chem. Ser. , 112, 50 (19 72). b) T. S. Huh, J. Neumeis t e r and K. Gr ies baum, Can J. Chem., 59, 22, 3188(1 9 81). 30) W. Pri tzk ow and G. Schop pe , J. Prakt . Chem., 311, 689( 1968) . 31) a) S. D. Razumovsk ii and G. E. Za iko v, J. Org. Chem. SSSR., 8, 468(1 9 72). b) K. Gries baum, T. S. Huh and S. M. Guts c he, Tetr a hedron Lett ., 31, 23, 3299(1 9 90).
32) P. L. Sto t le r and J. B. Ep pn er, Tetr a hedron Lett ., 26, 2417(1 9 73) . 33) E. R. Altw ic k er and J. Basila , Tetr a hedron, 29, 1669( 1973) . 34) S. A. Adenij i, J. A. Kerr and M. R. Wi lliam s, Int. J. Chem. Kin . , 8, 209 ( 1981 ) . 35) K. Grie s baum and G. Zwi ck , Chem. Ber., 119, 229( 1986) . 36) L. Homer, H. Schaefe r and W. Ludwig , Chem. Rev., 7 1, 75(1 9 58) . 37) A. Mag giclo and S. J. Nie g o wski, Advances in Chem. Ser., 21, 202(1 9 58) . 38) H. Taube, Trans. Faraday Soc., 53, 656(1 9 57) . 39) C. C. Schubert and R. N. Pease, J. Am. Chem. Soc., 78, 5553( 1956).
제 8 장 생체계 및 생체 모방계에 의한 탄화수소의 산화 8. l 생체계에 의한 산화반응 상온, 상압의 온화한 조건에서 탄화수소의 선택적 산화반응은 생 체계에서 산화반응 효소인 모노옥시게나아제 (Monoox yg ena g e) 가 발견된 이래 생, 유기화학상 홍미 있는 연구 분야가 되었다.I. 2) 시토크롬 (C yt ochrome) P-450 은 헴 (heme) 단백질의 일종으로 모노옥시게나아제에 속하는 효소 무리에 대한 명칭이며, 희랍어 〈 Cell( 세포) + pig men t(안료)〉라는 복합어에서 나왔다. 시토크롬 P-450 은 일산화탄소와 큰 안정도 상수를 가진 안정된 착화합물을 만든다. 이렇게 형성된 일산화탄소와의 착체 흡수 스펙트럼이 450 nm 부근에서 극대 흡수를 가짐으로써 450nm 에서 흡수 극대 (p eak) 를 갖는 시토크롬이란 뜻으로 명명된 것이다. 시토크롬 P- 450 은 생소한 이물질, 죽 식품을 통해 섭취된 각양각색의 식물 성 분 또는 미생물의 2 차 대사산물 (secondar y meta b oli tes ), 식품 첨
가재 색소. 조미 성분은 물론 의약품, 농약. 공해물질. 여러 독성 물질 등 비정상적인 물질이 체내에 출현할 때 대사를 위해서 산화 반응을 통해 발동되는 효소이다. 이 시토크롬 P-450 은 한 개의 효소가 아니라 동위효소(i so z y me) 로 구성되어 있다는 사실이 밝혀졌으며. 약 10 종에 가까운 동위효소들이 분리되고 있다. 예를 들면 시토크롬 P-450 이외에 a(580~590 nm), b(556~558 nm), c(549~551 nm) 그리고 d(600~620 nm) 등이 있으며 앞으로 더욱 많은 동위효소가 알려 질 가능성이 있다. 그러나 비록 그렇다 하더라도 수백 개의 동위효 소가 존재한다고 보기는 어려우며, 수많은 기질에 대해서 이 효소 가 다양하게 대처할 수 있는 능력을 나타내는 것은 마치 면역반응 의 항체 항원간의 다채롭고 특이한 반응에 비유되기도 한다. 이와 같은 시토크롬 P-450 은 그림 8-1 과 같이 헴 구조를 포함하고 있 는데. 이의 활성점은 포르피린(p or p h y r i n) 구조인 F 설 옥세노이드
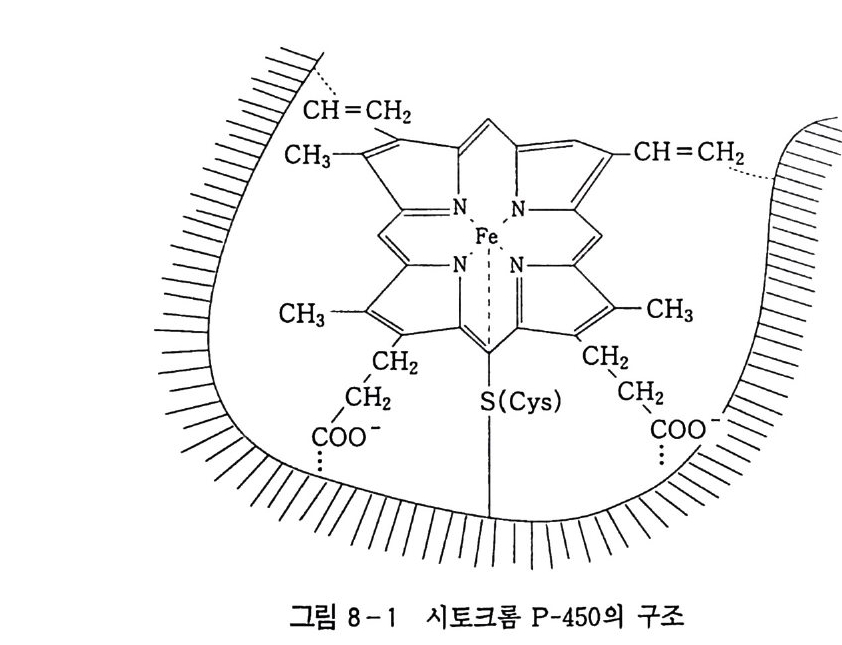 .,' ,
.,' ,
(oxeno i d) 로 이루어져 있다 . 3 ) 시토크롬 P-450 효소를 유기 화합물의 산화반응에 이용하고자 하는 연구가 활발히 시도되었다. 시토크롬 P-450 의 연구는 크게 세 가지로 대별된다. 첫째 생체에서 얻어지는 효소를 직접 반응에 이용하는 경우, 둘 째 효소의 활성 부위인 포르피린을 합성하여 반응에 이용하는 경 우. 셋째 포르피린을 화학물질로 모방하는 경우이다. 이제 각각의 방법에 대해 언급하고자 한다. 8. l. l 효소 시토크롬 P-450 에 의한 탄화수소의 산화반응 1) 반응기구 효소 시토크롬 P-450 은 어려운 반응현상 및 홍미 있는 분광학 적인 성질로 과거 오랫동안 화학자와 생화학자에게 매우 큰 호기심 을 일으켰다. 이 효소는 전형적인 모노옥시게나아제이며 식 (1) 과 같이 산소의 한 원자가 기질과 반응한다. RH +02 ~2Ce,p 4z 5H0 + ROH+H20 (1) 즉, 이 효소는 분자산소 하나를 반응물에 결합시키면, 한 산소원 자는 반응물에 그리고 나머지 다른 산소원자는 물로 환원되며, 비 활성인 알킬 알랄 아민은 수산화반응시키고 알켄은 에폭시화시킨 다. 그림 8-2 와 같이 3 가의 철착체가 한 전자와 반응하여 2 가 상 태의 철착체로 변화한 후 산소와 반응하여 강한 산화제인 옥소-메 탈을 생성한다. 생성된 옥소-메탈은 탄화수소의 탄소-수소 결합을 활성화하여
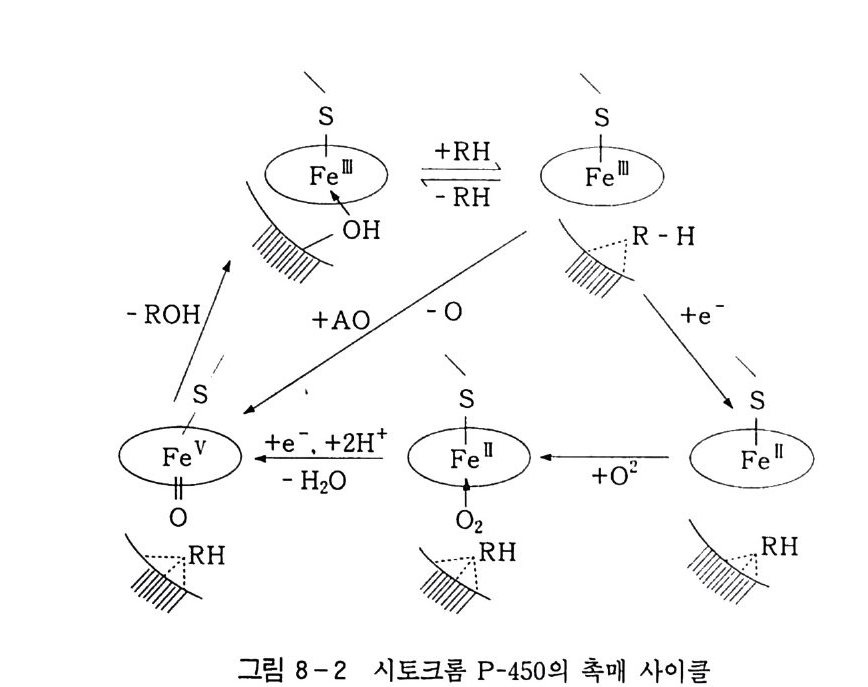 \ \
\ \
탄화수소를 산화한다. 또한 이 효소는 C-H . C-0 와 c-c 결합 에 대한 산화적 절단반응을 촉진한다 . 4) 그 동안 시토크롬 P-450 과 유사한 합성 헴 구조를 가진 계 (he me s y s t em) 에 대한 연구로부터 많은 메커니즘의 정보를 얻었다. 이들은 근본적으로 헴에서 산소를 활성화하며 시토크롬 P-450 에 의해 촉진되는 반응은 부분적으로 헴에 있는 철에 결합된, 특수한 5 번째 배위자에 의해 일어난다. 죽, 그것은 시토크롬 P-450 의 헴 의 철에 결합되어 있는 유황 배위인데, 이것의 첫번째 증명은 EXAFS 분광학 (s p e ct rosco py)을 사용하여 이루어졌으며 5) 화학 적 변형, 효소의 분광학적 연구》 합성된 모델착체 7) 에 의해 더욱 확인되었다. 결정적인 증거는 기질과 결합된 박테리아 (bac- ter ia l ) 시토크롬 P-450 의 결정 구조인데 8) 이것은 헴의 철에 결합된 시스테인 티오 데 이 트 배 위 체 ( cys te i n thi o d ate liga ti on ) 로 되 어 있으며 , 안정 한
촉매 중간체임이 증명되었다 . 9) 시토크롬 P-450 에 대한 많은 연구로부터 그림 8-2 와 같은 반 응기구가 제안되었는데, 이 연구에 사용된 효소는 시토크롬 P-450 cam 이라 불리는 캄퍼 (cam p hor) 를 히드록시화하는 박테리아 효소 였다 .10 ) 헴은 다른 효소에서 저스핀(l ow spi n, s=1/2) 철 (m) 이온울 가 지는 6 번째 배위자를 지니며, 이 배위자는 NMR11>, EWDOR12>, UV vis i b l e, MCD, CD 분광학적 연구 13) 로부터 물이 나 수산기 그 룹임이 확인되었다. 기질과의 결합은 6 번째 배위자를 해리함으로써 스핀 상태 (s pi n s t a t e) 를 고스핀 상태 (h ig h spi n, s=5 /2 )14) 로 변 화시키면서 환원 퍼텐셜이 증가된다. 곧 , 시토크롬 P-450cam 은 캄퍼와 결합하여 헴의 환원 퍼텐셜 울 -300mV 에서 -170mV 로 증가시키는데 1 5 ) 이것은 사이클의 다음 과정. 즉 환원과정을 확실히 해준다. 한 예를 들면 생리학적 환원제인 p u ti dare-dox i n( 철 -유황 단백질)은 시토크롬 P-450 cam 에 결합되어 있을 때 -196mV 의 환원 퍼텐셜을 가지며 ,15) 다 른 기질과 결합되었을 때 퍼텐셜의 증가는 다른 시토크롬 P-450 에서도 관찰된댜 16) 여러 연구들을 통해 비활성 탄화수소의 탄소-수소 결합의 절단 은 많은 경우에 H· 탈리를 통해 진행한다는 사실을 알았다. 이 결 과의 일차적인 증거는 kH/kn> 9.5 정도의 큰 분자 속도론적 동위 원소 효과(i so t o p e e ff e ct)로부터 얻어졌다 D 이렇게 큰 동위원소 효과는 동위원소 감응 단계가 기본적으로 둥에너지이므로 가역반 응임을 의미한다 .18) 현재의 의구심은 Fe(N) =O 종이 비활성화되 어 있는 메틸이나 에틸렌에서 수소원자를 떼어내 Fe(ID)-OH· 롤 생성하며. 이것은 래디컬 재결합에 의해 기질을 수산화하는 것에 있다(식 2 참조) . 래디컬 재결합 반응은 상당한 발열이며 기본적
으로 비가역 반응이기 떄문이다. RH ·R ·R HOR — F°sIe 2 4-- :!: ..느, 一―H FOsIe - 드 一 —HFs?I e 3.. :t. .._ - —FsIe 3 ..: t. .._ ( 2) II II '/ / /' / 그렇게 반응성이 강한 중간체의 가정은 시토크롬 P-450cam 에 의한 H 제거에서의 특성보다는 오히려 입체 (s t ereo) 와 레지오 선 택성 (re gi o sele cti v ity)의 관측으로 지지된다 .1 9) 이것은 철종에 붙 어 있는 기질에 하나의 면보다 여러 면에 접근할 수 있음을 의미한 다. 시토크롬 P-450 효소가 입체 선택성을 수행하는 대부분의 경 우. 그 특성은 기질이 활성점의 정확한 배열로 결합되는 것에 의해 결정된다고 할수 있다. 기발한 실험의 결과들은 래디컬 재결합 메커니즘을 좀더 확실히 해준다. 그러한 메커니즘은 알킬시클로프로판의 수산화 반응에서 시클로프로필 카르비놀 (c y clo p ro py l carbin o l) 래디컬이 부테닐 (bute n y l) 래디컬로 부분적으로 전위되어 개환 생성물이 생김을 설명한다. 히드록시 생성물과 부테닐 생성물의 상대적인 양은 래디 컬 재결합과 중간체 개환반응의 경쟁 속도에 의존한다 . 메틸시클로 프로판의 시클로프로필 카르비놀 래디컬은 K=l08s-1 로 개환되며 메탈시클로펜탄의 개환 생성물은 감은지 되k지 > 않 l0는9 다s-.1 로 개환되며 그 바이시클로 (2.1. 이펜타닐 래디컬
것은 수산화 생성물과 개환 생성물을 생성케 한다. 이 결과는 래디 컬 중간체의 증거뿐 아니라 래디컬 재결합반응의 속도가 109s-1 이 상임을 알려준다.2) 에폭시화 반응 시토크롬 P-450 은 알켄울 에폭사이드로 산화하는 촉매 역할을 한다. 이 반응은 알켄의 입체 화학을 유지하며 탄소-탄소 결합에 동시 산소를 삽입시킨다的 여러 증거로부터 비동시 삽입 (noncon cert ed ) 반응경로가 가능한 것으로 제안되었다 . 첫째 증거로 , 시토크롬 P-450 에 의한 말단 올레핀의 헴-질소의 알킬화 반응에서 가끔 에폭사이드가 먼저 생성되지 않는다는 점을 들 수 있다 . 2 1. 22) 그 새로운 결합은 비대칭 중간체를 나타내면서 모 든 경우 기질의 마지막 탄소에 결합을 생성한다 . 2 3 ) 두 번째로는, 어떤 경우에는 상당량의 알데히드가 수소의 1, 2 이동에 의해 생성된다는 점이다 언 이것으로 예정된 산소 삽입이 아니라는 것이 증명된다. 이는 또 다시 그림 8-3 과 같은 네 가지 메커니즘으로 설명할 수 있다 . 25)
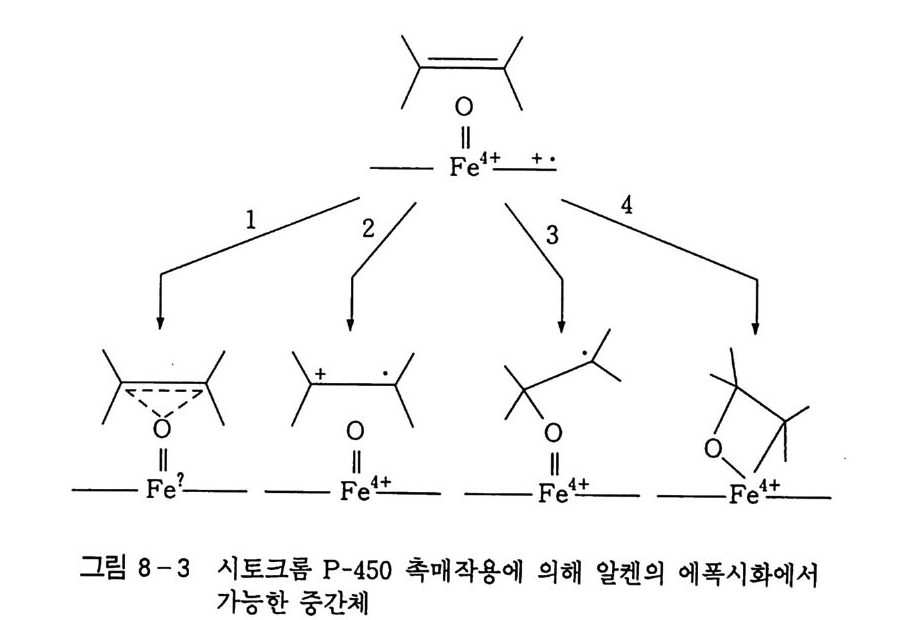 ―广 Fe 4 느
―广 Fe 4 느
® 동시 산소원자의 c_c 결합에 삽입 : 이것은 헴 알킬화나 알 데히드 생성과 일치하지 않으나 반응은 개별 경로에 의해 생 성된다. ® 기질에서 효소로 전자이동 : Fe(N) =O 와 기질의 래디컬 양 이온간의 래디컬 재결합은 환화되어 에폭사이드를 형성하거 나. 전이되어 알데히드를 생성할 수도 있다. 아세틸렌 산화의 경우 래디컬 양이온은 중간체로서 제외시킬 수 있을 것이 다 .26) ® 알켄의 한 탄소로 산소 전달 : 생성되는 래디컬 중간체는 환 화, 전이, 헴을 알킬화한다. ® 금속 옥세탄 (Me t aloxen t ane) 형성 : 합성된 헴의 착체 (heme com p lex) 를 설명하기 위해 제안된 이 메커니즘은 금속환상 중간체를 포함한다 .27) 환 개환반응성에 의존하면 에폭사이드, 알데히드 생성 및 헴의 알킬화 반응은 그와 같은 중간체로 설 명된다. 관찰된 각 효소반응은 각각 뚜렷한 반응경로가 있다 고 말할수 있다. 3) 수산화 반응 마지막으로 시토크롬 P -450 은 NIH(Nati on al Insti tut e of Health ) sh ift라 불리는 1, 2- 수소원자의 이동에 의해 방향족 화 합물을 수산화시킨다 .20) 이는 NIH 에 근무하던 W it ko p와 Uden- fri end 에 의하여 발견되었으므로 NIH-Sh ift라고 명명한 것이며, 그에 대한 총설을 참고하기 바란다 .20-b,c) 이 반응은 수산화 효소가 작용하는 과정에서 수산화되는 위치의 수소(다른 원자 또는 원자 단의 경우도 있다)가 인접 위치에 전위하는 1, 2- 전위를 말한다. 반응식 (3) 에서 보듯이 4-3H- 페닐알라닌 기질 떄 간장(肝藏) 페
닐알라닌 수산화 효소를 이용하여 수산화하면 90 % 이상의 3 중수 소를 3 위에 갖는 티로신을 얻는다.
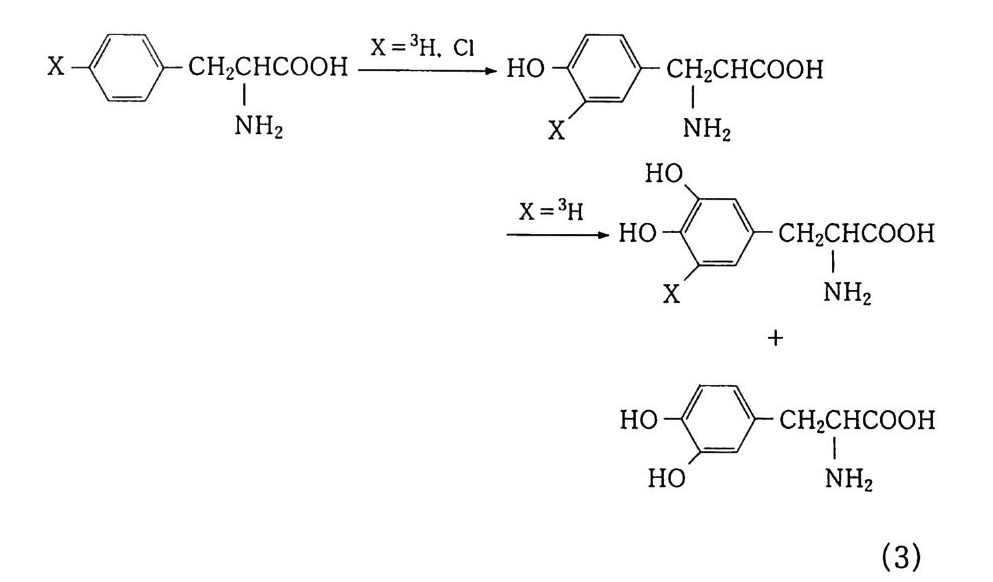 x -0- CH 『: 00H 드
x -0- CH 『: 00H 드
이것을 다시 티로신수소화 효소로 수산화하면 생성된 3, 4- 디히 드록시페닐알라닌 (DOPA) 의 5 위에 의해 3 중수소가 약 반 정도 남 는다. 또한 4- 염소페닐알라닌으로부터 출발하면 85% 의 3- 염소 티로신이 얻어진댜 이 반응은 1, 2- 수소의 이동과 에폭사이드 개 환이 페놀 생성물의 케토 토토머 (ke t o- t au t omer) 를 만드는 알켄 에폭시화의 특수한 경우이다. 최근에 히드록시디에닐 (h y drox y -d i en y l) 래디컬 중간체의 일전 자 산화반응은(그림 8-3 의 3 경로 생성) 그림 8-4 와 같이 수소 이동을 이끈다. 어떤 경우에는 메타 할로겐 치환체의 반응물에 대해 NIH-shif t 메커니즘이 관찰되지 않으며, 반응은 알칸의 수산화와 비슷하게 C-H 결합에 산소를 삽입한다 .28.29)
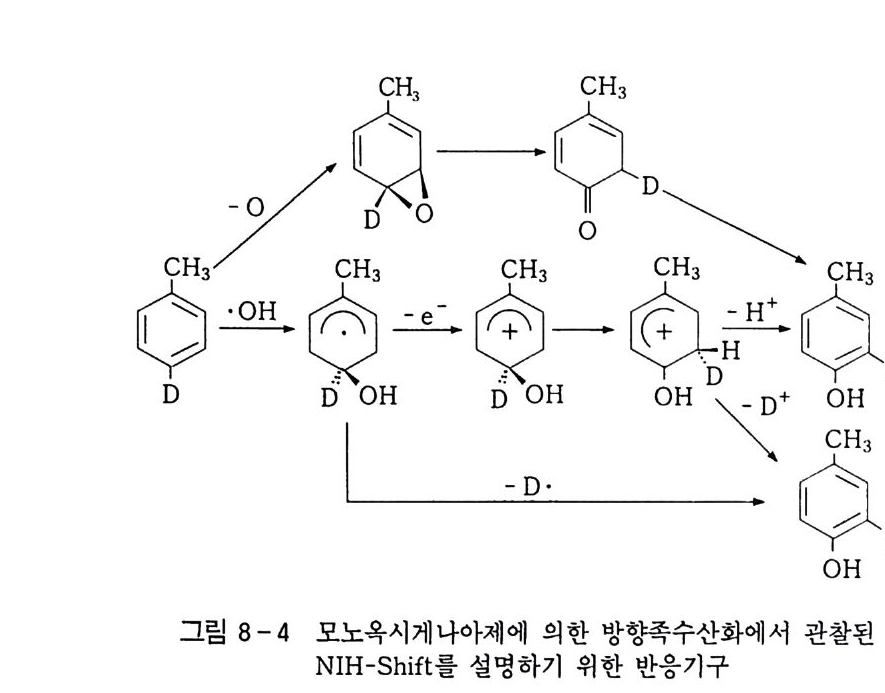 I -D. -` : H
I -D. -` : H
지금까지 살펴본 것을 정리하면 시토크롬 P-450 은 산소 전달반 응을 수행하기 위해 산소와 인접한 C_H 결합이나 비활성화되어 있는 C-H 결합(에테르나 에스테르)의 균일화 전달반응이 포함되 며, 그 과정에는 고가의 철_옥소 종을 사용한다. 전자이동이 쉬운 아민이나 티오에테르의 경우, 이 반응은 일전자 산화반응 대신 탈 프로톤에 의해 이루어진다. 위에 설명한 두 경우는 래디컬 재결합 에 의해 히드록시화 반응을 이룬다. 철-옥소 종에 의한 亢결합의 산화반응은 여러 종류의 메커니즘 이 가능하며 활성화된 중간체에 균일. 불균일 1, 2- 수소 이동반웅 을 포함한다. 표 8-1 에 시토크롬 P-450 이 일으킬 수 있는 촉매 작용 반응을 제시하였다.
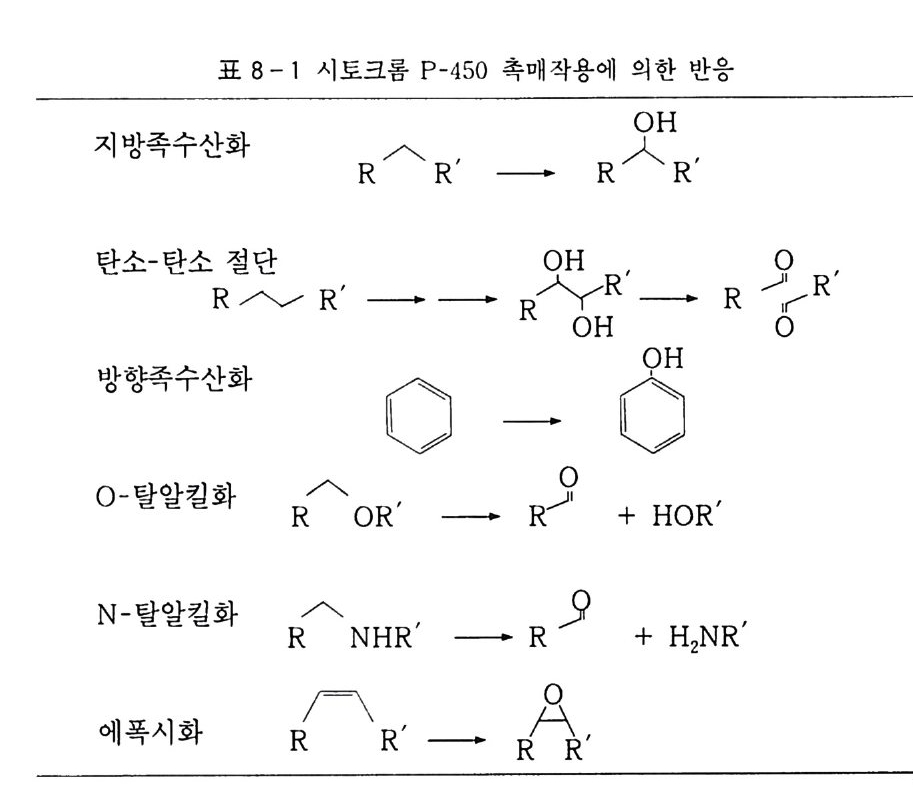 표 8-1 시토크롬 P-45 0 촉매작용에 의한 반응
표 8-1 시토크롬 P-45 0 촉매작용에 의한 반응
8. 2 생체 모방계에 의한 산화반응 앞에서 언급한 효소를 직접 산화반응에 이용하기에는 효소를 생 체계에서 분리 보관하여야 하는 어려운 문제와 반응 후 처리 등의 문제가 있으므로, 자연에서 시토크롬 P-450 에 기초를 둔 철포르피 린이나 헴울 갖지 않는 단백질인 모노히드록실라제에 의한 탄화수 소 선택적 수산화반응을 비교적 간단한 화학약품을 사용하여 효소 의 활성을 모방하는 연구가 시도되었다. 지금까지 발표된 Cp - 450 모델계를 정리하면 다음과 같다.
® 포르피린계 ® Gi f 반응계 @ Gi f Orsay 반응계 @ Gi f -K RICT 반응계 8. 2. l 포르피린계 생체계 중 모노옥시게나아제. 즉 시토크롬 P-450 은 헴을 포함하 고 있으며 이의 활성점은 포르피린 구조에 철옥세노이드 (Fev oxeno i d) 로 이루어져 있다 .3) 앞에서 설명한 바와 같이 생체에서
 z
z
분리한 시토크롬 P-450 은 여러 가지 문제점(분리. 보관. 가격 및 재사용 등)이 있으므로 활성점인 포르피린 구조 대부분의 모델계 (Porph y r in s y s t em) 는 금속一포르피린 구조를 모방하고 있으며. 또한 금속 이온 이외에도 반응식 (1)에서 보는 바와 같이 환원제 (전자원). 양성자 (H+원). 산소와 용매가 필요하다. 시토크롬 P-450 과 모델계들은 일반적으로 탄화수소의 수산화에 서 3 급> 2 급> 1 급 순의 래디컬 선택성을 보이며 30) 활성점인 포르 피린의 구조는 그림 8-5 와 같다. 그림 8 ― 5 에서 F 설 옥세노이드를 좀더 간단히 표기하면 식 (4) 와같댜 [ Fevl = O ] 二 [ FeIV -OH + ·R] 一 Fem+ HO-R [ Fe IV -0 · ] ( 4 ) 이 메커니즘을 적용하는 데 있어서 문제점 중의 하나는 어떤 알 칸을 수산화시킬 때 그 입체 (s t reochem i s tr y)의 부분적 변화, 죽 e pi mer i za ti on 이 일어난다는 점이다 .31) 또 다른 문제는 반응식 (5) 와 같이 수산화 과정에서 전이반응 생성물이 생긴다는 점이다 .32)
 DD二 :三
DD二 :三
식 (5) 에서 보는 바와 같이 바이시클로 [2, 1, O] 펜탄 (b icy-
c 叫 2, 1, O] p en t ane) 을 산화시키면 전이된 화합 물 3- 시클로펜텐- 1- 올이 생성된다. 이러한 여러 문제점들을 해결하기 위하여. 즉 좀더 선택적인 생산물을 얻기 위하여 다양한 방법이 시도되었다. 동물(생물체)로부터 얻어지는 효소인 각종 시토크롬과 그 금속 종류에 의하여 반응성이 각각 다르므로 계통적으로 정리하기에는 무리다. 예를 들면 토끼 간으로부터 얻은 효소인 시토크롬 P - 4 50LM 2 는 페닐 에탄을 수산화시킬 때 안정한 벤질 래디컬과 효소의 강한 반 응성 때문에 25~40 %의 입체 반전이 일어난다 .33) 이상 서술한 효소 이용시의 문제점 때문에 Groves 등은 생체 모 방계를 이용한 촉매를 개 발하여 요도소벤젠 (Iodosobenzene) 류를 산 화제로 사용한 반응계를 개발하였다 안 그 후, Fe(TPP)Cl. (iro n- tet r a ph eny l po rph y ri n ) Cl, Fe ( TDCPP ) Cl, ( iro n -tet r a -2. 6 -dic h loro ph eny l po rph y ri n ) Cl 또는 Mn(TTPPP)OAc, Mn( III ) [ tet r a kis ( 2, 4, 6 -trip h eny l) ph eny l po rph y ri n ] ( 그 림 8 一 6 참 조) 등이 개발되었으며 Mn(TT -P PP) OAc 로 n- 헵탄을 산화시 킬 때 그림 8-7 과 같은 결과를 얻었다 . 35)
 cl >5 NI\ vb l \_ N汀〉C/D /~ccP PF 1, Ic e lc l 타 \닙 Cl 广타N ~N 〈 Br
cl >5 NI\ vb l \_ N汀〉C/D /~ccP PF 1, Ic e lc l 타 \닙 Cl 广타N ~N 〈 Br
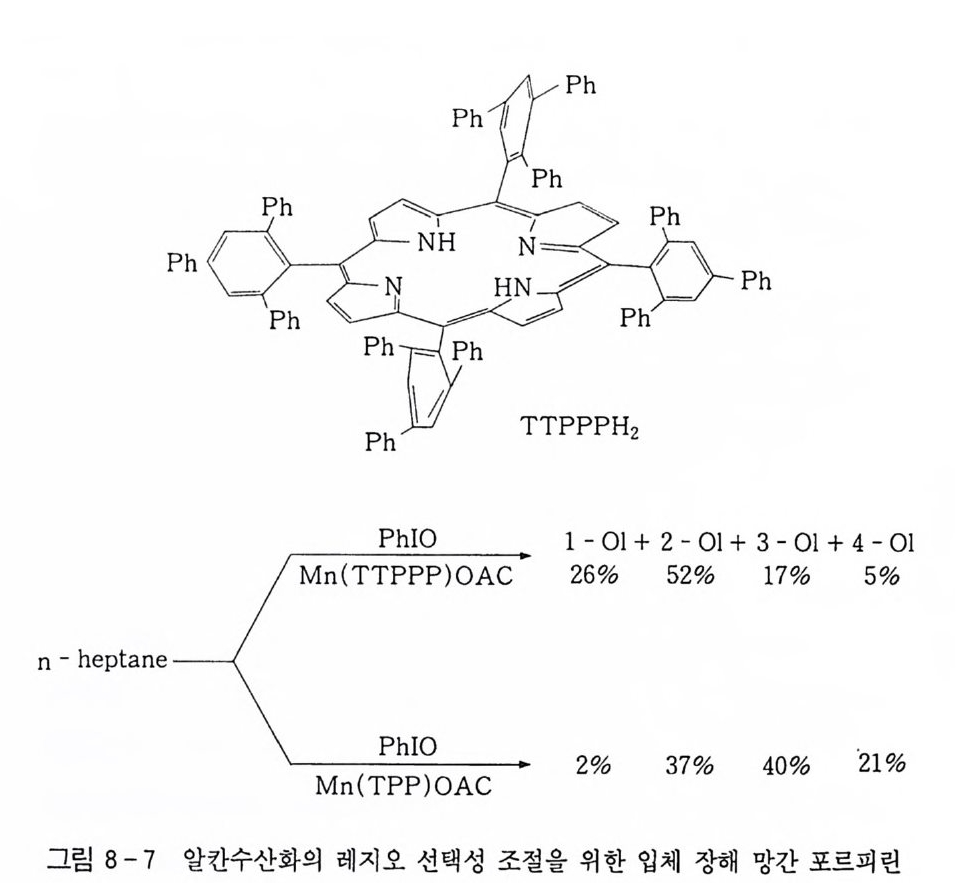 Ph
Ph
한편, Fe(TPP)Cl 과 큐멘히드로 과산화물을 산화제로 사용하여 시클로핵산을 산화시킨 결과는 식 (6) 과 같다 .3 6)
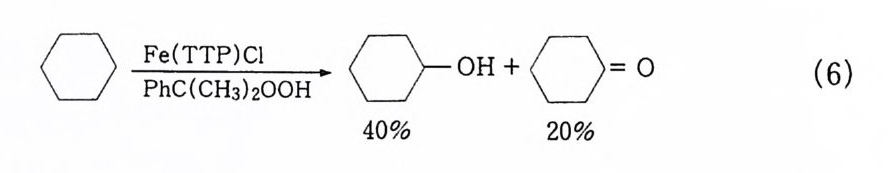 0 릅 0- 0H+ 0 =O (6)
0 릅 0- 0H+ 0 =O (6)
다음으로 공업적으로 싼 과산화수소를 사용하는 경우 이미다졸 같이 반응시킬 때 그림 8-8 과 같은 좋은 결과를 얻었다. 여기서 아다만탄의 경우 거의 전부인 95 %가 전환되었다. Mn (TDCPP)Cl- 과산화수소-이미다졸 계에서 이미다졸은 다음과 같
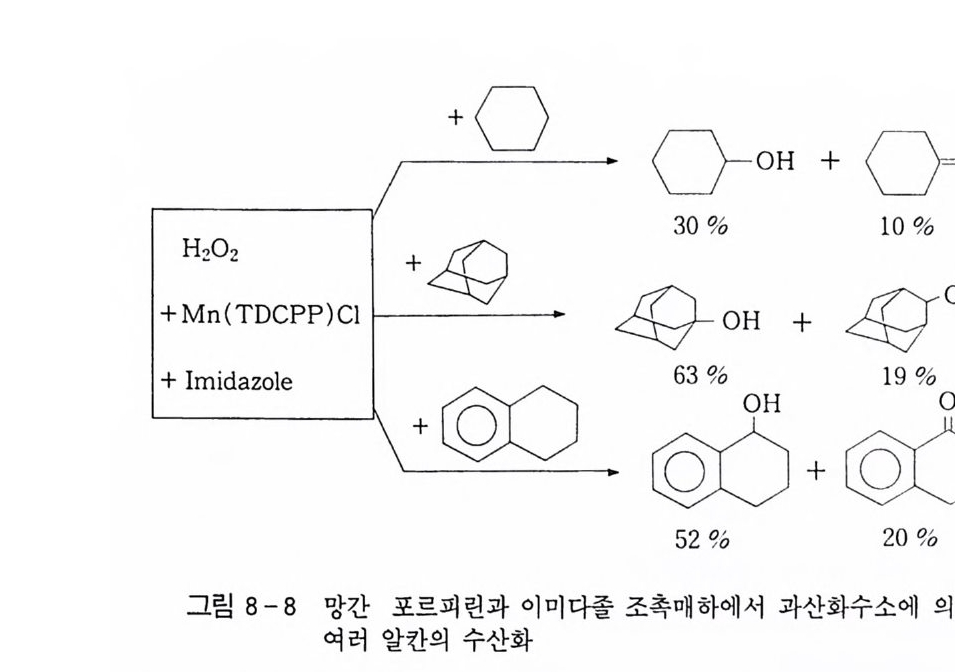 +0 0-0H + <>o
+0 0-0H + <>o
이 주요한 역할을 하는 것으로 생각된댜 ® 망간(lil)포르피린은 과산화수소의 -0-0-결 합을 비대칭적 으로 절단시켜 이미다졸 -Mn(V) =O 중간체로 되고 망간의 배위자 및 산-염기 촉매로 이중 역할을 한다. ® 이 미 다졸 존재 하에 서는 과산화수소 분해 ( dim uta t i on ) 의 상대 적 중요성이 감소되었다. 이상에서 시토크롬 P-450 과 포르피린을 이용한, 잘 알려진 예들 울 설명하였다. 이 경우 주로 수산화가 일어난다. 또한 올레핀은 에폭시화 37) 시키고 술파이드는 술폭사이드로 38) 변화시킨다. 8. 2. 2 Gi f 반응계 Gi f 반응계는 Bar t on 이 1980 년 초에 개발한 방법이며 첫번째 연
구가 프랑스의 G if -Sur-Yve tt e 에 있는 CNRS 에서 수행되어 Gi f 라는 명칭을 사용하게 되었댜 이 Gi f 반응계는 10 년간의 연구로부 터 표 8-2 와 같이 여러 종류가 개발되었으며 . 39 ~4u Fe=O(V) 옥 세노이드를 제공하기 위해 Fe(Il) 와 과산화물과의 반응, 혹은 Fe( ill ) 와 과산화수소와의 반응에 기초를 두고 있다. Fe=O(V) 종들은 시토크롬 P- 4 50 모델과 같으나 포르피린과 같 이 전자이동에 의해 환원되지 않는 매우 다른 화학 반응성을 가 지며. 효소계와 다른 반응성을 가지므로 관심을 끌고 있다. 1) Gi f 반응의 선택성 기하학적으로 대칭인 포화 탄화수소 아다만탄(_!_)은 4 개의 3 급과 12 개의 2 급 탄소-수소 결합을 가지고 있으므로 이 위치에 대한 반응성이 연구되었다 . c 2 /C 3 의 비율이 검토되었는데 C2 는 2 급 탄 소 위치에 산화된 생성물이며. c 3 는 생성된 3 급 알코올의 총량이고 이로부터 반응의 선택도를 측정할 수 있다. 아다만탄의 경우 c 2 /C 3 의 비율은 12/4 로 3 인 데 비해 래디컬
표 8-2 10 년간 개발된 여러 종류의 Gi f 반응계들 Gi f I Gi fII Gi f III Gi fN Gif -O rsay GoAg g 산용전반양매자화응성 제주자물 게주 게 N피초공탄a기리산화2S 딘 수 소 H-一一--2> S F-一一e Z一-一->n 처'-一一一-.-:>::-: 1 I. HH一一22 °° 2 2 촉매 Fe0 Fe2+ --> Fe3+
메커니즘에 의해 생성되는 반응의 C 2 /C 3 의 비율은 약 0 .1 5 이다. 이 결과는 포르피린을 기초로 한 계의 결과와 비슷하다. 실제로 G if (ID) 를 사용하는 경우 C 2 /C 3 는 3.7 이며 이는 이 반응이 래디컬 메커니즘이 아니라는 증거이다 .1 2) 아다만탄(.!.)의 경우에는 3 급 위치에 반응하였지만 이소펜탄(~) 과 메틸시클로핵산(합)의 산화반응에서는 3 급 알코올이 생성되지 않았다. 그것은 상응하는 3 급 알코올이 반응조건하에서 안정함을 보여준다.
 二 >
二 >
또한 표 8_3 에서 보는 바와 같이 Gi f 반응계의 결과들은 시토 크롬 P-450 효소계와 다르다. 즉, Gi f 반응계에 의한 주생성물질은 케톤인 반면 Cp -450 계는 알코올이다. 2) 촉매 이 Gi f 반응계에서는 철 클러스터 (Fe clus t er) 가 촉매로 작용하 며 3 핵 유기철 카르복시레이트 클러스터(t r i nuclear orga noir o n carboxy la te cluste r ) 인 Fe II Fe2 ill O ( OAc ) 6 PY3 . s 의 결 정 으로 분 리되어 43) 자기, 분광학적 방법, x- 선 분석에 의해 구조가 그림 8-9 와 같이 확인되었다 . 이 클러스터는 수용성 착화합물 (a q uo com p lex) 로부터 리간드 교환에 의해 제조할 수 있다. 이러한 방법에 의해 여러 가지 클러
298
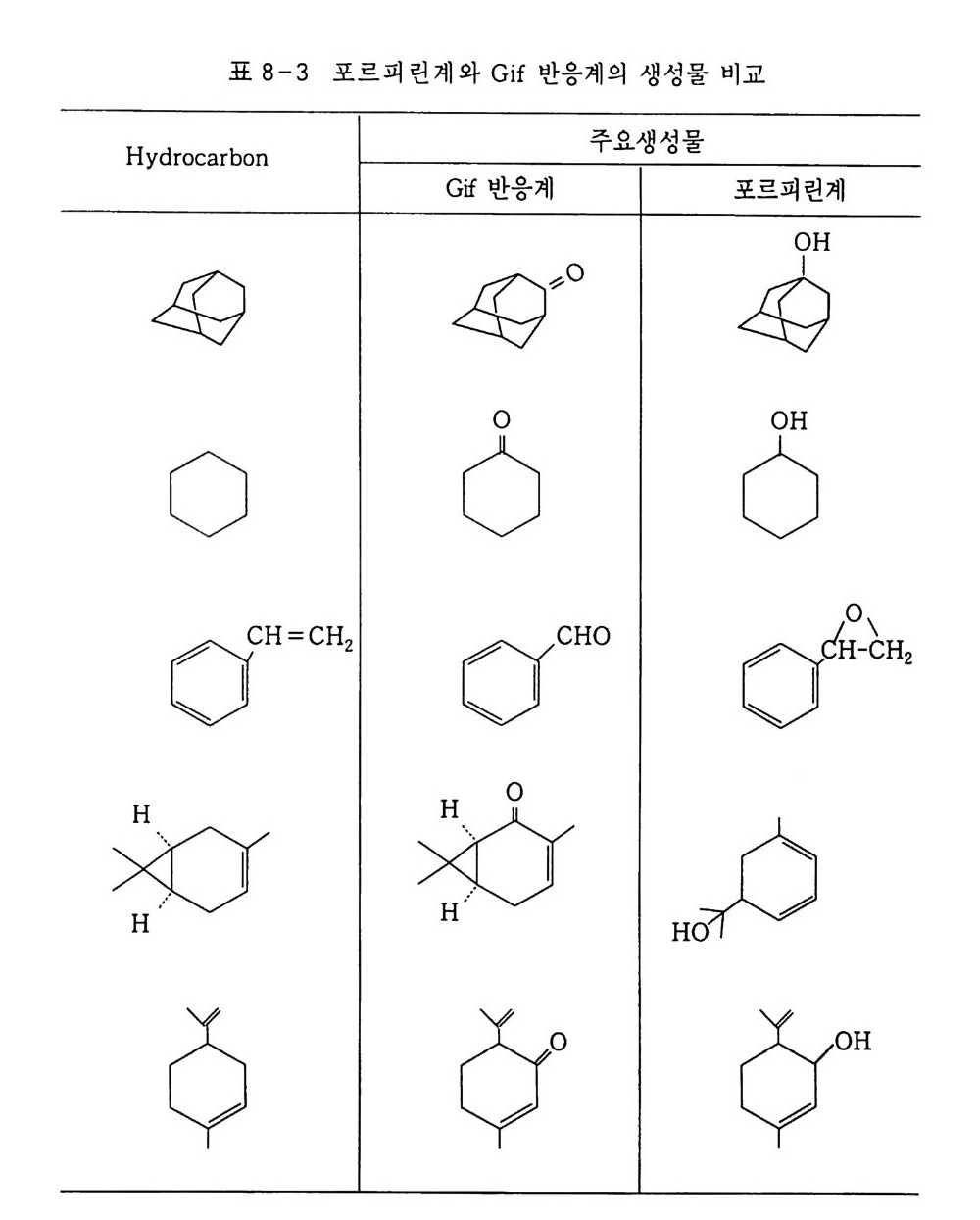 표 8-3 포르피린계와 Gi f 반응계의 생성물 비교
표 8-3 포르피린계와 Gi f 반응계의 생성물 비교
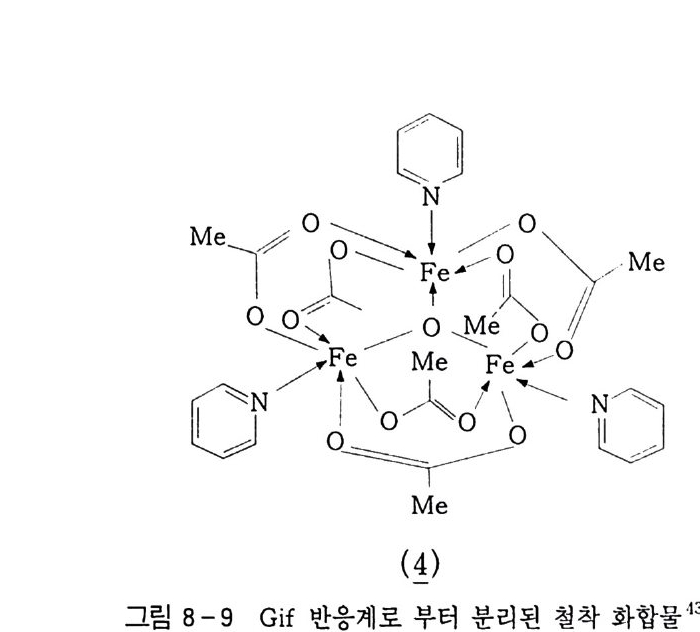 ON
ON
스터를 제조하여 실험한 결과는 표 8-4 와 같다 . 표 8-4 의 결과와 같이 이러한 클러스터 화합물은 효과적인 촉매이며 포르피린이나 salen, [N ,N -eth y le nebis ( salic y li d e ne ami na to ) ] 모델보다 우수 한 결과를 보였다. 또한 주기율표에 있는 거의 모든 금속의 활성이 검토되었는데 루 테늄염 (ru t hen i um sal t)만 촉매적으로 활성이 있었고, 특히 코발 트, 니켈 망간염들은 완전히 불활성이었다. Gi f 반응계는 반응물 에 대해 화학 선택성 (chemosele cti v ity)을 가지고 있으며 초기 Gi f 반웅계에서는 유화수소를 사용하여 선택적으로 포화 탄화수소 를 산화하였으며 이때 사용된 유화수소는 래디컬을 매우 효과적으 로 억제한다고 알려졌다. 그러나 디페닐술파이드는 시클로헥산을 거의 산화하지 못하는 것이 발견되었다. 그리고 시클로헥산과 시클로헥센의 반응성 차이 는 거의 없었다. 또한 시클로헥센은 알릴 위치에 산화되었고, 에폭 시화반응은 일어나지 않았다• Gif 산화반응에서 여러 가지 산화종이 사용될 수 있으나 아연은
표 8-5 여러 촉매 농도의 아다만탄 Gif (N) 산화반응 촉매의 농도 생성물(%) 수율 촉매의 사용 횟수 (mmol X lO- 3 ) l- 올 2- 올 3- 올 (%) (tur novre No. ) 0.8 0.7 3.4 5.6 9.7 263 0.5 1.6 4.6 7.7 13.8 584 0.2 0.7 6.3 4.4 11. 4 1232 0.1 0.8 5.8 3.4 9.9 2087 *촉매의 사용 횟수=생성물의 몰농도/촉매의 몰농도
산소의 한 전자에 의한 환원 수단으로 효과적이다. 용매가 피리딘인 경우 아연으로부터 전자를 쉽게 받아 래디컬 종이 생성되며 합체반 응에 의해 바이피리딘을 생성한다 .44 ) 또한 환원제로 아연을 사용하 여 피리단 초산의 Gi f(N ) 반웅계에서 아다만탄을 반응물로 일련의 산화반응을 통해 실험한 결과, 표 8-5 와 같이 촉매 사용 횟수가 2,000 이상임 이 확인되 었다. 이 효과적인 촉매계의 발견 후 레지오 선택성과 화학 선택성이 개선된 연구들이 이루어졌다. 1 급, 2 급 및 3 급 위치를 갖는 대칭의 포화탄소인 트랜스 -1, 4- 디메틸 시클로헥산의 산화반응은 소량의 알데히드 (1) 를 포함한 주요 케톤(이을 생성하였으며. 다른 가능한 산화반응 생성물은 GLC 에 의해 검출되지 않았다 45)( 식 7 참조).
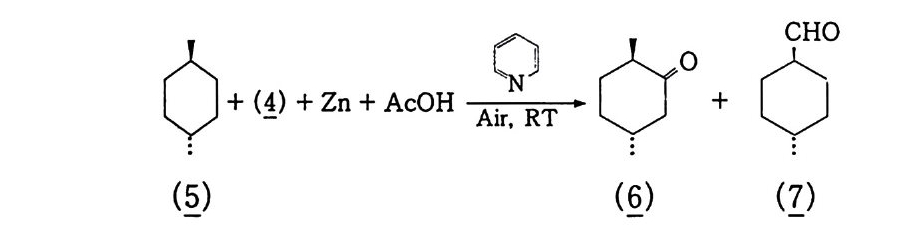 亡맷 )+Zn+AcOH½ 亡° + 广〕O (7)
亡맷 )+Zn+AcOH½ 亡° + 广〕O (7)
3) Gi f 산화반응기구 Gi f 반응계는 앞에서 언급한 바와 같이 시토크롬 P-450 의 생체 모방 모델계에서 관찰되는 결과들과 매우 다른 양상을 보인다. 예 를 들면 포르피린계는 3 급 탄소와 반응하는 데 비해 Gi f 반응계는 2 급 탄소를 공격하며 올레핀 화합물을 에폭시화반응시키지 않고 다른 화학적 선택성의 결과를 준다. 산화반응은 철 촉매에 의해 촉진되며 2 급 탄소의 공격은 펜턴 메 커니즘이나 단순한 자동 산화반응과 다르다. 반응물의 활성화는 중
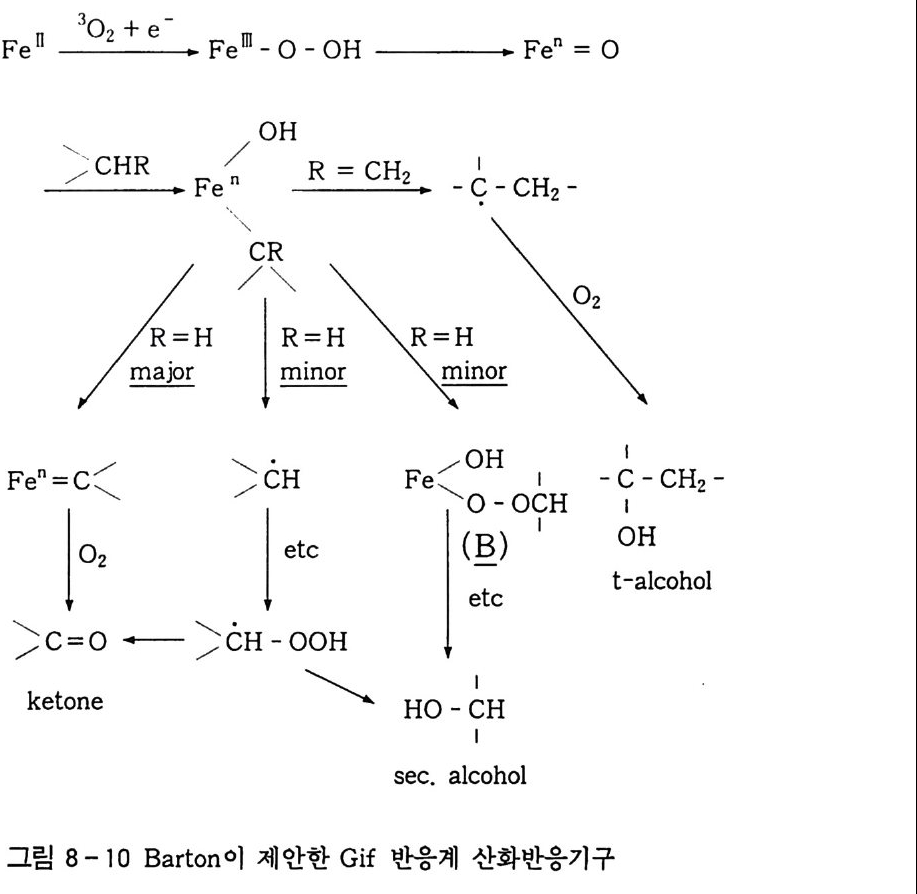 3 야 + e-
3 야 + e-
수소화 실험에서 증명된 것처럼 비가역적 반응이며 Barto n 등은 그림 8-10 과 같은 반응 메커니즘을 제안하였다. 초기에 촉매는 슈퍼옥사이드 래디컬 철( II )종과 반응하여 히드 로 과산화물을 만든다 . 이 중간체는 옥세노이드종 n 이 5 가 아니라 4 나 심지어 3 이나 2 로 바꾼다. 시토크롬 P-45 0 반응계와 다르게 n=5 는 그들이 올레핀을 에폭시화하거나 3 급 탄소를 선택적으로 공격하는 것과는 다른 면을 보인다. 철카벤 반응기구는 케 톤 이 주 요한 생성물질인 이유를 설명해 준다. 과산화물 중간체 (B) 는 산소가 crFe-C 결합으로의 삽입에 의해 형성되며 균일 절단에 의해 2 급 알코올을. 불균일 절 단에 의해 케 톤이나 물. 옥세노이드를 형성한다 . 래디컬 반응의 아다만탄 실험 에서 C 2 /C3 의 비율은 0.0 3 ~ 0 .1 5 이다. Gi f 반응의 이러한 새 결과 들은 3 급의 아다만타닐 래디컬이 형성되지 않고 직접 6- 철결합을 통하여 생성됨을 강하게 지지한다. 또한 일반적 산화조건에서 시클로핵산은 단지 14 %의 케톤을 생 성했으며 많은 양의 저급 알코올(메탄올, 이소프로판올, 3 급 부탄 올)이 산화되지 않았댜 이러한 결과는 케톤으로 진행하는 주요 경 로는 중간체로 알코올울 포함하지 않음을 말해 준댜 일반 조건하 에서의 아다만탄 산화반응에서는 14.1 %의 수율로 디케톤인 (~). @)가 생성되었다. Gi f 반응계의 근본적으로 중요한 양상은 산소 의 부분압에 대한 선택도의 비율 C 2 /C3 의 의존성이댜 16)
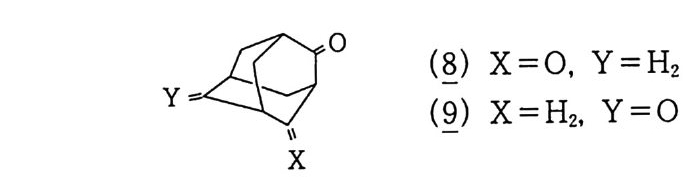 y4?0 ((~9)) XX==OH,2 , Y Y==HO2
y4?0 ((~9)) XX==OH,2 , Y Y==HO2
Gi f 반응에서 순수한 산소 대신 공기를 사용한 경우 아다만탄의
산화반응에서 더 높은 C 2 /C 3 의 비율을 보였으며. 반응 혼합물의 분 석으로부터 바이피리딘 이성체(i somer i c b ipy r i d i ne) 와 피리딘-탄 화수소 결합 생성물 (lQ). (브)과 (프)의 혼합물이 얻어졌댜 1 2) 이 생성물은 피리딘 탄화수소의 3 급 위치를 공격하여 얻어진 것이다.
 (10 ) (11 ) (12)
(10 ) (11 ) (12)
더 많은 양의 피리딘-탄화수소 결합 생성물이 높은 C 2 /C3 반응 에서 얻어진 것은 3 급 아다만틸 래디컬에 대한 산소와 피리디늄 이 온(혹은 피리디닐) 사이의 경쟁반응에 대한 확실한 증거이다. 또 한 산소와 두 개의 다른 탄소 래디컬의 이분자반응 속도 상수의 비 로부터 래디컬 반응은 3 급 위치에 포함되며. 2 급 위치에는 비슷한 래디컬 반응이 있을 수 없음을 보여준다. 피리딘-탄화수소의 결합 생성물은 다른 많은 반응물에서도 단지 1~2 % 정도 검출되었으며 주로 3 급의 알킬피리딘이었다. 최근 연구에서는 제한된 산소의 양에서 아다만탄의 산화반응이 재 현되 었다. 아르곤에 10 % 산소를 사용할 때 100 이상의 C2/C3 의 값이 관찰되었으며, 탄화수소의 3 급 위치에서의 반응은 거의 피 리딘에 결합되었다. Gi f 반응계에 의한 선택도 (2 급> 3 급> 1 급) 는 C-H 결합력 (1 급 >2 급 >3 급)과 입체 장애 (3 급 >2 급 >1 급)에 의해 설명될 수 있으며, 일반적으로 2 급 위치가 산화된다. 3 급 위치에서의 반응은 C-H 결합이 특별히 노출되었을 때 볼 수 있다. 또한 다중수소화
된 시클로핵산을 사용한 일련의 산화반응에서는 탄소가 활성화 과 정에 관계되어 있음을 보여주며, 활성화 과정에서 기질과 생성물 간의 수소 교환이 없음을 나타내준다. 이러한 실험들과 함께 알코올은 케톤의 환원반응으로부터 생성 되지 않음이 명백하댜 2.5 라는 속도론적 동위원소 효과는 시클로 헥산과 그것의 다중수소 치환체의 경쟁적인 산화반응으로부터 얻 어진다민 이 값은 시토크롬 P-450 이나 그것의 Fe=O(V) 모델 에서 얻은 결과 49) 와는 다른 것이다. 그리고 그 결과는 비포르피린계와 비슷하였으나 :, 0) 알콕시 혹은 과산화 래디컬 반응계와는 달랐다. 가능한 활성산소 가운데 슈퍼옥 사이드는 Gi f 반응계에서 가장 바람직한 산소활성종이다. 그 이유 는 K02 에 의해 산소분자를 쉽게 교환할 수 있으며, 퀴논이 있을 때 산화반응 수율도 증가시킬 수 있기 때문이다. 퀴논은 전자전달 시약으로 작용하며 슈퍼옥사이드 농도를 크게 한다언 과산화수소는 피리딘-초산 혼합물에서 아연에 의한 환원 으로 생성되는 중간체이지만 , 5 2) 탄화수소를 산화하지 못하고 철 이 온에 의해 분해된다. 이 실험은 G if와 Gi f-O rsay 반응계에서 수 차례 시도되었다. ·OH 에 의한 펜톤류의 반응기구는 Gi f 반응계에 서 레지오 선택성의 결과와 다르며, 더구나 ·OH 래디컬은 피리딘 과 쉽 게 반응한다 .53) 4) 용매의 영향 Gi f 반응계에서 피리딘의 역할에 관한 정보는 이론적으로나 실 용적으로 매우 중요하다. 피리딘은 Gi f 반응에서 가장 좋은 용매이 며. 염기나 철에 대한 배위자로서 작용할 수 있다. 양성자로 치환된 피리딘은 철종을 쉽게 주기 위해 아연으로부터 전자를 받아 환원된
상태가 되며 산소에 대해 환원제로 사용된다 . 5 , 1) 이 가정은 아연 존재하 FeCb 촉매에서 피리딘의 래디컬 결합에 의해 바이피리딘을 제조한다는 사실로써 확인된다. 그럼에도 불구 하고 환원된 형태의 피리딘과 바이피리딘 유도체는 Gi f 반응계에 서 산소분자를 대신한다. 즉, 산소 하나의 전자가 환원된 상태인 0 2 ( 혹은 H0 2 ) 를 주기 위해 삼중항 산소에 환원제로 사용될 수 있 다. 이처럼 퀴논은 피리딘이나 바이피리딘과 같이 삼중항 산소와 아연 사이의 전자이동에 간여하는 역할로 사용할 수 있으며. 환원 제는 아연 환원된 피리딘, 바이피리딘과 슈퍼옥사이드 래디컬일 수 있다. 낮은 수율로 얻어지는 이와 같은 반응은 식 (8) 과 같이 일련의 산화. 환원반응으로 낮은 전기적 수율이 설명된다.
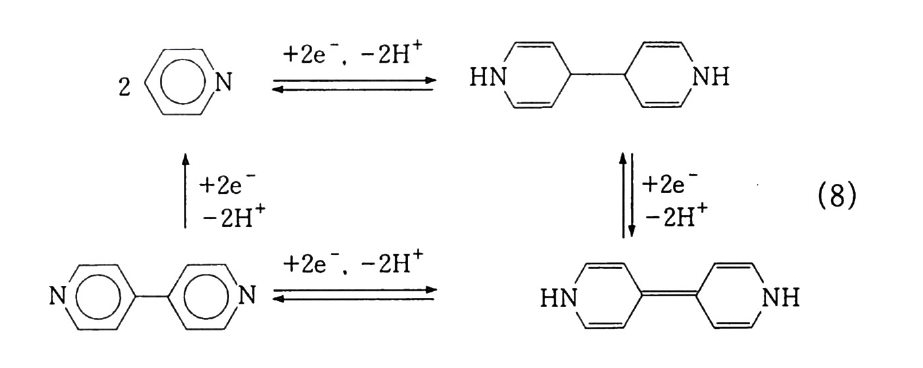 2
2
대부분의 환원제 아연과 산화제 산소는 아연아세테이트와 물을 생성하는, 필요 없는 반응에 소비될 수 있으며, 이 연구로부터 바이 피리딘 생성이 가역반응임을 증명했다. 염기도의 중요성을 측정하기 위해 피리딘이 트리에틸아민, 피페 리딘, N-N- 디메틸아닐린으로 대치되었다. 또한 디메틸포름아미드, 아세토니트릴과 디메틸술폭사이드가 용매로 사용되었다. 모든 경우, 낮은 수율의 산화반응물(약 0.5 %)과 낮은 선택도를 얻었다. 더욱 홍미로운 사실은 피리딘과 구조적으로나 화학적으로 비
표 8- 6 용매의 영향 용매(1 5ml) 아다만탄+아연+초산+촉매(산) 산소. 24 시간. 4o·c 용매 t-아다 산화생성물(%) 만탄온 (~) 아다만탄온 수율 c2 / C3 피리딘 3.1 5 1.80 12.1 0 17.0 5 4.4 3- 메틸피리딘 0.50 0.70 4.3 0 5.50 10.0 2. 6- 디메틸피리딘 0.35 0.1 0 0.05 0.5 0 0.4 2- 메틸피리딘 0.1 0 0.0 1 0.05 0.1 6 0.6 4- 메틸피리딘 1.40 0.4 0 1.30 3.1 0 1.2 2, 4. 6 - 트리 메틸피리딘 0,50 0,05 0.95 1.50 2.0 4- t-부틸피리딘 0.55 0.0 5 0.25 0.85 0.5 2, 6- 디-t-부틸 -4- 메틸피리딘 0.75 0.1 0 0,05 0.9 0 0.2 2- 시아노피리딘 0.1 5 0.60 0.4 5 1.20 7.0 2- 플루오르피리딘 0.1 0 0.1 0 0.02 0.22 1.2 퀴놀린 0.05 1.35 0.1 0 1.50 29 이소퀴놀린 0,05 1.05 2.7 5 3.8 5 76 퀴녹살린 0.3 0 0.25 0.1 0 0.05 1.2 피리다진 0.1 0 1.25 1.00 2.3 5 23 피라진 0.25 0,35 1.00 1.60 5.4 4 - 페닐피리딘 2.2 5 1.30 3.20 6.75 2.0
 L{Jx
L{Jx
슷한 2- 메틸피리딘, 2, 4, 6- 트리메틸피리딘, 2- 풀루오르피리딘, 4- 페닐피리닌 퀴놀린, 이소퀴놀린, 피리다진 등 많은 용매들을 사 용한 경우는 비효과적이었고 피리딘이 가장 바람직했다는 점이다. 피리딘은 단순히 용매로만 아니라 염기. 철의 리간드로도 작용할 수 있다. 피리딘을 아세토니트랄 N,N- 디메틸아닐린으로 바꾼 결 과 표 8-6 에서 보는 바와 같이 낮은 수율과 선택도를 보였다• 즉 피리딘과 유사한 용매들은 피리딘에 비해 비효과적이었으며 3- 메 틸피리딘이 그 중 좋은 결과를 나타내었다. 5) 산화반응에의 응용 (1 ) n- 도데칸의 반응 유기물의 많은 부류 가운데 알칸은 화학 에너지로서 또한 유기 화합물의 원료로서 가장 큰 자원 가운데 하나이다. 알칸의 풍부함 과 상당히 낮은 반응성은 과학자들에게 큰 홍미를 불러일으켜 알칸 을 기능화하거나 활성화하는 방법이 많이 개발되었으며, 지난 많은 연구자들은 수년간 저온에서 여러 가지 반응계를 사용하여 알칸의 활성 화나 기 능화에 대 한 연구를 수행 하였다 .55~57) 여기에서는 Gi f 산화방법을 이용한 n- 도데칸의 산화반응에 관 해 언급하고자 한다. n- 도데칸을 산화반응하여 얻어지는 알코올은 연성세제의 원료로 많은 양이 사용되고 있는데, 현재 개발되어 있 는 공정은 다음과 같이 복잡한 과정을 거쳐야 한다한 I 봉산의 탈수공정 十―_〔 5 군:군〕 ―一 〔군군군군〕 _ | 7 뮤분해공정 | ―一 〔군굳굳: -—_ 〔군군군〕
표 8-7 Gi f 반응계를 이용한 n-도 데칸의 산화반응 ' Gi f 반응형태 도데칸 도데칸올 전환율 케톤의 (%) (%) (%) 선택도(%) Gi f III 5.7 0.3 6 95 Gi f IV 6.8 0.2 7 96.5 GoAg gil 6.0 0.5 6.5 93
표 8-8 Gif 반응계와 공업화 기존 공정과의 비교 Item 반응온도 반응압력 전환율 케톤의 (°C) (atm ) (%) 선택도(%) Gi f 반응계 25 1 7 100 공업화공정 170 1 20 75
그러나 Gi f 반응계를 이용하면 직접 간단히 제조할 수 있다. Gi f 산화방법을 이용하여 얻은 결과는 표 8-7 과 같댜 이 표에서 보는 바와 같이 전환율은 7% 였으며 선택률은 다른 부산물이 생성 되지 않아서 거의 100 %를 나타내었다. Gi f 반응계에서 얻은 결과를 기존 공정과 비교해 보면. 표 8 -8 에서 보는 바와 같이 기존 공정에 비해 전환율은 다소 적으며. 부 산물이 거의 생성되지 않아 높은 선택성을 보인다. (2) 시클로 화합물의 반응 필자의 연구팀에서 Gi f 반응을 시클로 화합물에 이용해 실험해 보았다. 현재 공업적으로 산화반응 공정에 이용되고 있는 시클로 화합물은 시클로헥산과 시클로도데칸이다. 시클로헥산으로부터 얻 어지는 시클로헥사논은 나일론의 원료로 사용되는 카프로락탐과 아디픽산의 중간체로 사용되며 시클로도데칸으로부터 얻어지는 시
클로도데칸온은 가소제 제조의 중간체로 많은 양이 사용되고 있다. 현재 시클로헥사논의 제조는 코발트―나프테네이트의 촉매로 150 °C 전후의 반응온도와 5 k g/c남의 반응압력 에서 운전되고 있 댜 이러한 반응조건을 Gi f 산화방법에 의해 저온, 저압의 공정으 로 변환하기 위한 연구가 수행되었다. Gi f 산화방법에 의해 시클로 헥산 및 시클로도데칸을 산화반응한 결과 다음과 같은 생성물의 분 포를 보였으며 주로 케톤이 생성되었다.
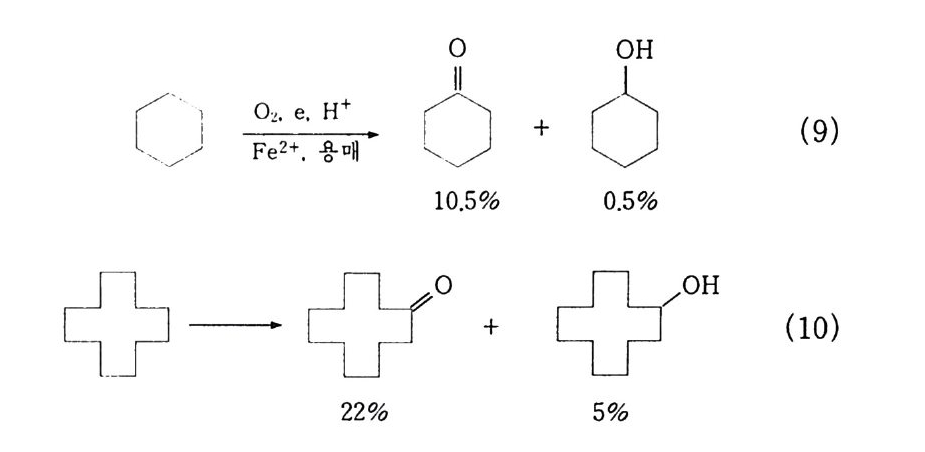 。 OH
。 OH
또한 시클로헥산의 반응에서 각 Gi f 산화방법에 의해 얻어진 결 과는 표 8-9 와 같다.
표 8-9 여러 가지 Gif 반응계를 이용한 시클로헥산의 산화 결과 반응형태 시클로헥사논 시클로헥산올 전환율 케톤의 (°C) (atm ) (%) 선택도(%) Gif ID 9.0 1.0 10 90 Gi fN 10.5 0.5 11 95 GoAg g II 10 2 12 83
(3) 테르펜류의 반응 ® 라이모넨의 반응 59) 테르펜 (Ter p ene) 류는 식물을 증류함으로써 얻어지며 이소프로 필렌 골격이 연결된 구조를 취하였음이 밝혀졌다. 자연에서 얻는 테르펜류 중 공업상 제조하는 대표적안 예는. 테르펜류인 라이모넨 (L i monene) 을 식 (11) 과 같은 방법으로 제조하는 카르본 (Car vone) 이며. 카르본은 국내에 연간 10 톤씩 전량 수입되는 향료이다.
 NOC! •
NOC! •
Gi f 반응은 (+) -라이모넨으로부터 한 단계로 쉽게 제조할 수 있으나. 식 (1 2) 와 같이 반응혼합물이 얻어진다 .
 ',',, 1', 이르 모본:Y +4페
',',, 1', 이르 모본:Y +4페
312
표 8-10 여러 조건하에서 Gi f 반응계에 의한 R-(+)- 라이모넨의 산화반응 결과 생성물 회수 수득률 (%) (%) 반응조건 (15 ) (16) (끄) (14 ) 물질수지 Gi fN , 상온 0.1 0 mmol FeCl2 7.7 3.7 0.9 88 100 Gi fN . 상온 0.0 5 mmol FeCl2 9.7 4.9 1.3 83 99 Gi fN . 상온 0.01 mmol FeCl2 10 5.3 1.5 80 97 Gi fN . O. 0.05 mmol FeCl2 12 5.4 n.d 68 ~86 Gi f N. -25° 0.05 mmol FeCl2 13 5.2 n.d 63 ~82 GoAg g II 0.5ml 과산화수소 2.1 4.3 n.d 88 ~ 95 GoAg g II 2.0ml 과산화수소 6.0 6.1 n.d 61 ~ 73 GoAg g II 1.0 ml 과산화수소 2.5 4.5 n.d 69 ~ 76
또한 Gi f 방법에 의해 여러 조건을 변경하여 표 8_l0 과 같은 결과를 얻었다. Gi f 방법으로 얻은 반응 혼합물을 칼럼크로마토그 래피 방법에 의해 분리하여 얻은 각각의 산화물의 광학활성은 표 8-11 과 같다. 생성된 케톤(프)인 4- 아세틸 -1- 메틸 시클로헥센 은 광학활성이 있었으며 카르본은 라세미체로 얻어졌다. 이와 같이 카르본의 생성에서 광학활성이 없어지는 이유를 알기 위해 여러 가
표 8-11 반웅으로부터 얻은 생성물의 광학활성 생수[a율]성D(물 % ) (9+1—.105 16) 6 .3° =g8。 (0+1—.177 12) 9 °
표 8-12 여러 온도에서 얻어진 카르본刊( 브})의 [a ] Dl l 값 반응온도 카르본 [a]D1 3 순도 (GC%) 반응계 25 °C -2.1 ( C = 0.1 8 CHCI 깁 91 Gi f IV 5°C -0.8 ( C = 0.1 7 7 CHCl3) 93 Gi f IV -10 ·c -1.33 (C = 0.1 8 6 CHCl3) 91 Gi fN
지 변수를 변화시키며 그 영향을 검토하였다. 즉 반응온도가 광학 활성에 미치는 영향을 검토하였는데. 그 결과는 표 8_12 와 같다. 표 8-12 에서 보는 바와 같이 실험으로부터 얻어진 카르본은 반 응온도에 관계없이 라세미화되었다 . 또한 라이모넨이 GoAg g 반응 에 의해 산화한 결과들은 표 8-13 에서 보는 바와 같이 촉매와 관 계없이 라세미화되었다. 표 8-12 와 8-13 에서 보는 것처럼 Gi f 반응에서 얻은 카르본은 라세미체로 얻어졌으며, 시광성이 거의 나타나지 않는 이유를 설명 하기 위해 그림 8_11 과 같은 대칭 亢_알릴 중간체 메커니즘이 제 안되었다.
표 8-13 여러 촉매에서 얻어진 카르본의 [a]D13 값 촉매 카르본 [a 〕 013 순도 (GC%) 반응계 Fe( III ) +0.53(C=0.1 8 7 CHCI 깁 95 GoAg gI I Cu( II ) +l.4( C = 0.1 1 4 CHC13) 89 GoAg gI I
 〈〔R畑 〔〔3 〕 Fe\\\。
〈〔R畑 〔〔3 〕 Fe\\\。
® ( + ) -car -3 -ene 의 반응 60) 시클로프로판 환을 함유하는 car-3-ene( 브)을 원료로 하여 시 클로프로판 환에 대한 Gi f 반응성을 알기 위해 연구가 수행되었다. (+)-car-3-ene 의 산화반응 연구는 오래 전에 여러 종류의 산
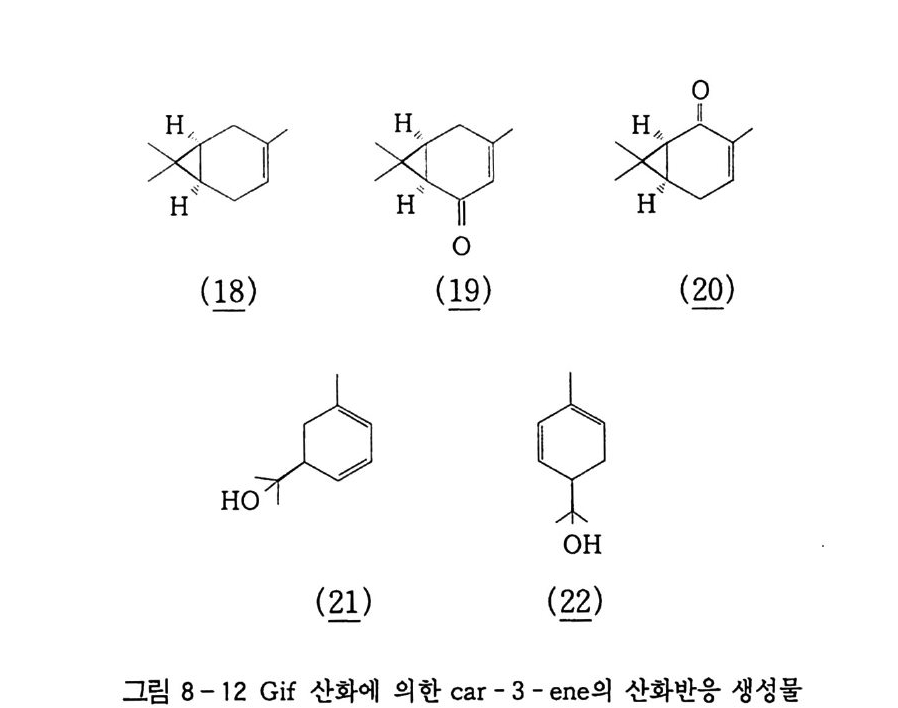 > H <>IO>
> H <>IO>
화제와 반응계를 이용하여 이루어졌다. r. 1, 62) 반응에 사용된 산화제 는 반응의 선택성이 없어서 여러 가지 생성물이 생성되었다. 즉, 바이시클로 [4. 2. OJ 헵탄은 방향족환이나 a. {3, y. 6- 불포화 시클 로헵타디에논으로 반응되었는데, 이는 ( -) -car -3-en -5-one (브)이나 (+)-car-3-en-2-one( 깐)이 산이나 염기 조건에서 불안정하기 때문이다. Gi f 반응을 이용하여 car-3-ene 을 반응시킨 후 칼 럼크로마토그 래피 방법으로 분리한 결과 그림 8-12 와 같은 생성물을 얻었다. 그립 8-12 에서 보는 바와 같이 케톤 (브)와 (四)이 각각 9 % 와 7 %로 생성되었으며 부반응으로 알코올인 극성 화합물이 7 % 생 성되었다. 또한 시클로프로판 환의 개환으로 생성되는 (-) -메타 멘타 -4. 6- 디엔 -8- 올(갇)과 (+) -파라-멘타-1. 5 디엔 -8- 올 (空)의 두 화합물은 1 % 이내로 거의 생성되지 않았다. 이러한 결 과들은 앞에서 언급한 Gi f 반응의 선택성과 온화성을 보여준다. (4) 콜레스탄 유도체의 산화반응 이 반응은 포화 탄화수소의 2 급 위치의 쉬운 산화와 산소를 포함 한 분자에서 비활성화되어 있는 C-H 결합이 우선적으로 산화되 는 Gi f 반응계의 두 가지 특수한 특징을 조합하여 자연으로부터 얻어지는 기질 (na t ural p rodu ct)을 산화시킬 때 레지오 선택적 반 응에 이용될 수 있는 편리한 방법이다. 이러한 목적에 이용될 수 있는 화합물로는 스테로이드가 있다 .63 > Gi f(N ) 반응계에 의한 콜 레스탄 (choles t an) 유도체의 산화반응은 곁가지가 분해, 생성되어 얻어지는 공업적으로 중요한 반응물인 20 위 -케톤을 얻을 수 있다. 예를 들어 프로제스트론(p ro g es t rone) (왼)은 콜레스테논 (chole ste n one) (堅) 에서 한 과정에 의해 얻을 수 있다. 곁가지 결합 분 해에 관한 주요 메커니즘은 그림 8-13 의 중간체들을 포함한다고
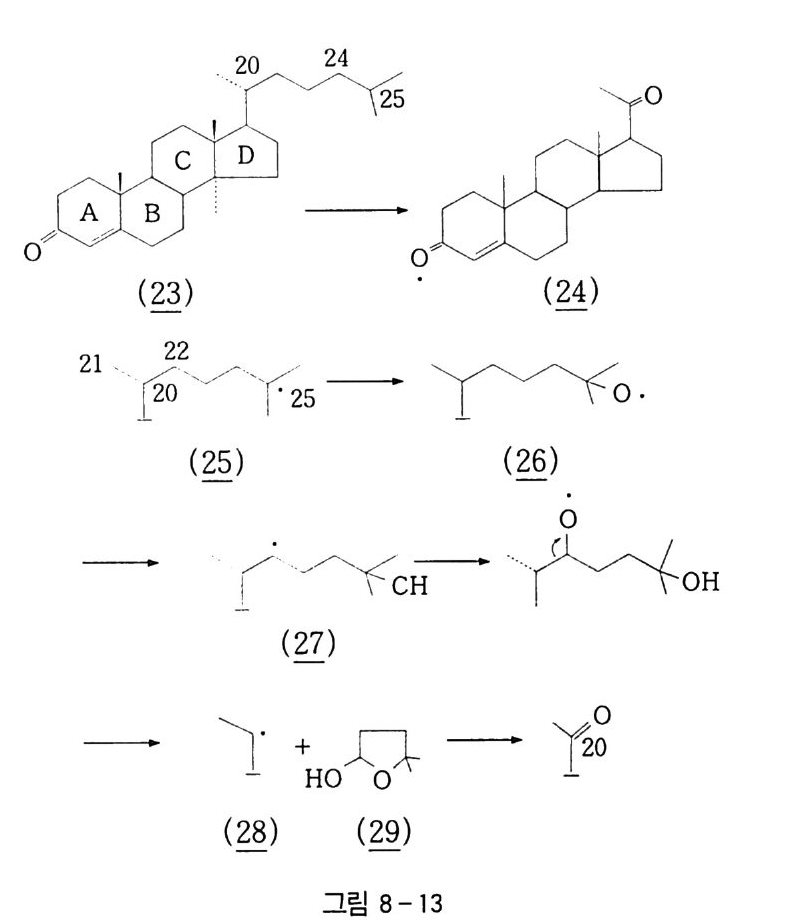 24
24
알려져 있다. C 2s 탄소의 말단수소는 입체 장애를 받지 않고 , 철-탄소 결합은 탄소 래디컬(亞)로 전개된다. 슈퍼옥사이드(혹은 산소)와의 반응 과 산소-산소 결합의 펜톤 형태의 분해는 알콕시 래디컬(~)을 생성한다. 잘 알려진 분자 내 1-5 수소이동은 탄소 래디컬(끄)을 생성한 다. 래디컬 반응이 더 진행되어 탄소 래디컬(쁘)이 된 후 케톤으로 더 산화되거나 C-6 분해물 (쁘)가 되며 분해물 (空)는 분리하여 확인되었다. 적당한 25 - 메틸 콜레스테롤 유도체의 Gi f 산화반응은
20 위-케톤을 만드는 다른 경로가 있음을 확실히 해준다 . 만일 제 안된 메커니즘이 적용되면. 곁가지 분해에 필요한 25 수소가 없을 때에는 20 위-케톤이 생성되지 않고 단지 출발 물질의 옥소-유도 체가 생긴다 .63) 8. 2. 3 Gi f -O rsay 반응계 64) 지금까지 Gi f 반응계에 대해 설명하였다. 이 반응계는 기잘 용 매(피리딘). 전자원(철 또는 아연 분말), 양자원(지방산). 촉매(철 이온) 그리고 산화제로 산소 또는 과산화수소 (GoA gg계)가 필요 하였다. 이 반응에서의 문제점은 공업화하는 데 많은 제약이 따른 다는 점이다. 즉, 균일계 촉매반응이므로 생성물의 분리. 반응수의 제거. 촉매의 재활성화, 촉매계의 재순환 그리고 용매의 독성. 용매 와 기질과의 반응 등이다. 그러므로 반응계의 단순화를 위한 연구 가 많이 이루어졌는데. 그 중 하나가 전자원으로서 전기화학을 이 용하자는제안이다. 아연이나 철 분말의 환원에 의한 Gi f 반응은 전기화학적으로 재 현될 수 있으며. 이 전기화학적인 방법은 Gi f 반응계를 산업적으로 이용할 수 있는 방법이다. 실험들에 의하면 -0.6 에서 -0 .7 V( 표 준전극에 대해)의 수은전극에서 산소분자를 피리디늄 이온의 직접 적 환원 없이 환원할 수 있었으며. Gi f 반응계와 거의 같은 수율로 아다만탄올 산화하였다 . 6 4) 이 단계에서 음극 전해질은 아다만탄, 철 촉메 피리딘에 산소가 포함된 혼합물이었고, 전도성 전해질로 서 테트라에틸암모늄염이었다• 3 불화빙초산은 피리딘을 프로톤화 하여 화학반응에서 산화반응을 지연시키는 테트라암모늄염을 제거 하는 데 유익하게 사용된다. 이러한 전기화학적인 방법을 아다만탄, 시클로헥산, 트랜스-데
표 8-14 G if -Orsa y계에 의한 탄화수소의 산화반응 반응물 전환율(%) C2/ C 3 피리딘-탄화수소의 쿨롱 결합생성물 수율(%) 아다만탄 18 8.5 10.5 4.2 시클로도데칸 21. 1 1.0 5.2 데칼린 22.2 36 1.4 5.4
칼린의 산화반응에 응용한 결과 화학계에서 사용된 아연과 동량인 전기량 (3,000C) 을 사용하여 표 8-14 와 같은 결과를 얻었다. 확산과 저항의 칸막이인 세라믹 벽에서 야기되는 문제는 전기분 해의 증가와 함께 전해 음극질의 염기성 증가인데. 3 불화빙초산의 연속적인 첨가에 의해서 해결된다. 더구나 반응의 수율은 전기분해 초기 단계(1 ,000~1,500C) 에서 급격히 감소한다. 이 문제에 대한 간단한 해결은 단간셀 (un i cellular) 계의 도입이며 이 계는 Gi f- Orsa y계라 불린다. 이 계에서 산소분자는 가장 쉽게 환원될 수 있 는 종이며. 산성매체에서 산소의 효과적인 환원은 산화반응에 충분 하다. 이는 곧 환원종을 간단하게 균일 전류로 대치함으로써 가능 하다는 점을 보여준다. 시클로헥산 (48 mmol) 의 산화에서 45 %의 쿨롱 수율은 초기 2,000 쿨롱에서 얻을 수 있으며 8,700 C 이후에는 36 %로 감소되 었 다. 쿨롱 수율의 증가는 p ara q ua t나 4, 4- 디피리딜 (3 불화빙초산으 로 프로톤화)과 같은 전자전달 시약의 이용에 의해 이룰 수 있으 며, 이러한 시약 존재하에 9,400 C 에서 49 %만큼 높은 전기적인 수 율이 얻어진다. 시토크롬 P-450 식에 의해 계산된 탄소-수소의 결합을 산화시 키는 데 사용하는 아연의 효율은 2.0 mmol 규모 반응에서 10 % 이하이다.
전기화학적 산화에서 쿨롱 수율의 증가는 더 많은 탄화수소를 사용해 단순히 긍정적으로 증가시킬 수 있다. 이 때문에 아다만탄 . 시클로핵산. 메틸시클로펜탄. 3- 에틸펜탄. ci s- 와 t rans- 데칼린 등 일련의 포화 탄화수소들은 피리딘의 포화용액에 가까운 농도에 서 G if (N) 와 G if -Orsa y계에 의해 산화되었으며 65) 다음 결론들은실험으로부터 유추되었다 .
표 8-15 Gi f-O rsay 반응계에 의한 tra ns -. cis -데 칼린의 산화반응 트랜스데칼린 산화반응 생성물 Q10(0쿨0롱 ) 0(3.024) (33) 0(.3=0 4=4 )7 0(.3355)7 0합.7계64 쿨롱수2율9 ( % ) C2z1/ .C8 3 2000 0.05 0.8 6 3 0.828 1.74 33 34 .5 3000 0.035 0.053 1.46 7 1.72 2 3.265 42 23.1 Q(쿨롱) 멸\l8(완7) 시8스( 데브)칼 린 (산쁘)화 반(응꼰) 생성물합 계 쿨롱수율(%) C2/ C 3 21000000 xc邸臨邸oH00]..22 02 00..36 39 00..24 49 01..H50c86 t21..r8652 q b54 39..12 y23..3855 3000 0.245 0.96 0.7 8 1.50 3.8 6 45.7 5.22 x (30)X=H (브 )X=H (~)X=O. Y=H2 (쁘 )X=O. Y=H2 (꼰 )X=OH (33)X=OH (~)X=H2. Y =O (프 )X=H 2 ,Y=O
320
® 전기화학적인 계에서 쿨롱 수율은 화학적인 계에서보다 높다. ® 주생성물은 항상 케톤이다. 케톤/알코올 비에 의한 레지오 선 택성과 C2/ C 3 비도 거의 같았다. ® 아다만탄은 두 산화반응계에서 가장 많은 피리딘-탄화수소 결합 생성물질울 얻었다. 최근에 t rans- 와 c i s- 의 산화반응은 전기화학적으로나 화학적으 로 매우 상세하게 연구되었는데 , 65 ) 전기화학적인 계에서의 결과들 울 표 8-15 에 나타내었댜 t rans- 데칼린은 3 급 위치에서 거의 산화되지 않고 대개 케톤이 생겼다. c i s- 데칼린은 다르게 나타났는데, C 2 /C 3 는 8 이하였으며 연 구된 어느 다른 탄화수소의 결과보다 3 급 알코올이 주요 생성물이 었댜 쿨롱 수율은 양탄화수소에 대해 만족할 만하였다. 양쪽 경우에 t rans-3 급-알코올에 대한 c i s 의 비율은 거의 같은데, 이것은 철 탄소 결합의 래디컬로의 분해로 설명할 수 있다. t rans- 와 ci s- 데 칼린의 화학산화에 대한 결과들은 전기화학적 결과와 같은 경향을 보였다(표 8-16 참조) . tra ns -와 cis - 데 칼린의 자동 산화반응은 1oo ·c 로 가열하여 줌
표 8 -16 Gif 반응게 에 의 한 tra ns -. cis - 데 칼린의 산화반응 기질 산화생성물 전체 전환율 f c2/ C 3 (迎) (33) (34) (완) (36) (37) (%) 38 0.055 0.042 0.7 7 5 0.808 1.68 16.4 16.3 39 0.54 0.3 5 1.29 0.3 2 1.39 3.89 34.6 3.3 7 * f = 전기적 수율 : 1.31 g의 아연은 3865 쿨롱에 해당한다.
으로써 두 가지의 t-산화물이 생성되는데. 이것들은 트리메틸포스 파이트에 의해 알코올로 환원된다. tra ns -데 칼린의 tra ns -와 cis - 의 비 율은 1.43 이 고 cis -데 칼린은 1. 89 였다. 또한 ci s- 데칼린은 3.5 4 mmol 의 3 급 알코올이 얻어졌으 며 t rans- 데칼린은 겨우 0.309 였다. 그러므로 ci s- 데칼린의 자동 산화가 트랜스보다 10 배 빠르며 약 간의 제 2 의 산화반응이 있었다 . 8. 2. 4 Gi f-K RICT 반응계 1) Gi f-K RICT I 반응계 Gi f 산화반응에는 금속착물 촉매 및 전자공급원(예 :Zn) 이 반응 용액에 녹아버리는 것과 반응물에 비해 과다한 양성자원 및 용매를 사용하는 것 등의 제약이 따른다. 이규완 등 66 ) 은 이러한 문제점들 을 해결하기 위하여 우선 촉매의 담지체 위에의 고정. 즉 불균일화
 철착제(촉매) lg r
철착제(촉매) lg r
를 기하였으며 동시에. 촉매 외에도 전자 및 양성자 공급원과 용매 등이 필요한 복잡한 계를 단순화한. Gi f-K RICT 반응계를 개발하 였다 . 이규완 등은 Barto n 등 67) 이 Gi f 반응에서 분리한 철착화합물인 [Fe II Fe ill 0(CH 3 COO) r, (C s H s N) 』 (C 晶 N) 。 568, 69) 을 합성하여 촉 매의 불균일화를 시도하였다 . ( 1) Fe II Fe ID 20(C H3COOMCsHs N) 3] (잉 HsN) 。 s 의 헤테로화 앞에서 제조된 철 촉매인 [FeIIFe ill 2 0(CH 3 COO) 6 (C s H s N) 』 (C H N) u . 5 를 담체인 폴 리 -4- 비닐 피리딘, 앰버라이트. a- 알루미 나와 실리카에 그림 8-14 와 같은 방법으로 불균일화하였다. 그림 8-1 4 와 같은 방법에 의해 불균일화한 촉매를 이용. 반응하 여 표 8-17 과 같은 결과를 얻었다. 표에서 보는 바와 같이 담체로 실리카를 사용한 경우 반응성이 가장 우수하였다. 이 불균일화한 촉매의 불균일 정도를 알기 위해 표 8-18 과 같
표 8-17 불균일화한 철 촉매를 이용하여 얻은 반응 수율 지지체 케톤수율 (mo! %) 5.3 폴리비닐피리딘 3.1 앰버라이트 2.1 실리카 5.5 알루미나 3.5 * 반응조건 : 시클로헥산 ( 4 6mmol) , 철 화합물 (0.005mmol) . 아연 (40mmol), 초산 (35 mmol) , 반응시간(1 6 hr), 반응온도(상온)
표 8-18 실리카 지지 촉매를 추출한 후 반응 결과 촉매 시클로헥사논 수율 (mo!%) 추출 후 용액 a) 5·22·4 추출후고체촉매 * a) 추출은 피리딘 42ml 와 초산 2ml 용액에서 12 시간 교반하였으며 그 후 여과하 여 용액과 고체 촉매 를 분리하였다 . 반응조건은 표 8-7 과 같 았다 .
은 실험을 하였으며. 그 결과 상당한 양의 금속이 용매로 용출되어 나왔다. 그러므로 그림 8-15 와 같은 새로운 헤테로화 방법을 사용하였다. 이 방법은 철 화합물을 담체에 입힌 후 공기에 의해 철을 산화하 는 방법이다. 이 방법에 의해 헤테로화한 촉매를 이용하여 Gi f 방 법을 사용, 표 8 ― 19 와 같은 결과를 얻었다. 표에서 보는 바와 같 이 이 반응에서는 철 촉매가 거의 용출되지 않았으며 추출 후 고체
 물 :20ml 시릴카겔
물 :20ml 시릴카겔
표 8-19 철/실리카 촉매 추출 후의 반응결과 촉매 케톤수율 (mo! %) 지지 촉매 5·50 52 추출 후 용액 a) 추출후고체촉매 · * a) 추출 은 피리딘 42 ml 와 초산 2 ml 용액에서 12 시간 동안 교반하였으며 그 후 여과하여 용액과 고체 촉매 를 분리하였다. 반응조건은 표 8-17 과 같았다 .
촉매는 추출 전 촉매를 사용한 경우와 거의 같은 수율로 생성물을 얻었다. 이 헤테로화된 촉매계를 Gi f-K RICTI 반응계라 명명하였댜 불 균일화된 촉매는 여러 가지 기기 분석을 통하여 분석되었으며, 그 결과는 표 8-20 과 같다. 촉매의 XRF 결과를 보면 Cl/Fe 의 값이 염화 제 1 철의 2 보다 훨 씬 적게 나타났댜 이는 촉매의 제조 과정에서 염소이온의 약 절반 가량이 손실되었음을 반영해 준다. 또한 XPS 의 결과를 보면 금속 상태 철의 707 eV 보다는 산화철 의 711 eV 에 훨씬 가깝다. 이러한 결과는 표면의 철종이 대기중에 방출되었을 때 산소에 의하여 재산화되었기 때문일 것이다. 이와 같은 불균일화된 철 촉매를 이용하여 Gi f(N ) 반응에서 철
표 8-20 철/실리카 촉매의 분석 방법 결과 XRF Cl / Fe 원자비 = 0.95 XRD 무정형 XPS Fe2 p 312 의 결합 에너지 = 711 .3 eV
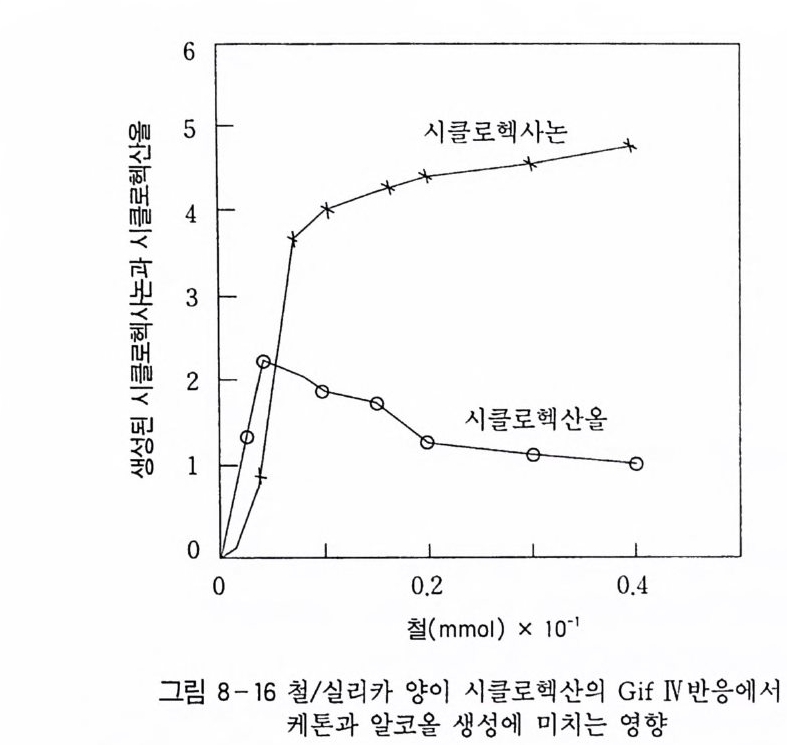 6
6
촉매의 양에 따른 영향을 검토하여 그림 8-16 과 같은 결과를 얻 었댜 그림에서 보는 바와 같이 시클로헥사논과 시클로헥산올의 생 성은 촉매의 양에 따라 큰 영향을 보였다. 특히 알코올의 생성은 그립 8-17 과 같이 촉매의 저농도에서 높 은 값을 보였으며, 케톤의 생성은 일정하게 증가하였다. 철의 저농 도에서 알코올의 생성이 최대점을 보이는 것과 촉매의 산화 상태에 밀접한 관계가 있음이 예상되어 촉매의 산화 상태에 따른 영향이 고찰되었다. 반응에서 사용되는 촉매의 산화 상태를 변경하기 위해 불균일화 한 촉매를 수소 기류하에서 환원하여 산화 상태의 변화를 시도, 표 8 -21 과 같은 결과를 보였다. 표 8-21 에서 보는 바와 같이 환원조건이 심할수록 반응물의 전
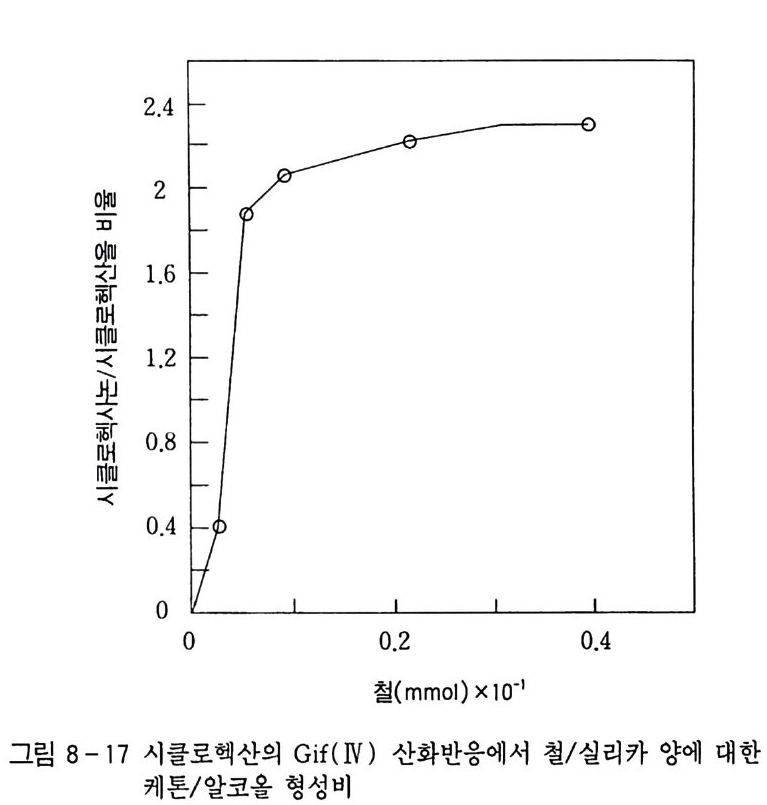 2.4
2.4
환율과 케톤/알코올의 비율은 환원 전보다 낮은 값을 보였다. 이는 촉매의 산화 상태가 반응의 전환율과 선택성에 영향을 줌을 암시해 준다 . 그러므로 Fe, FeO, Fe203 등 여러 종류의 산화가를 가지는 철 산화물을 사용하여 반응성 및 알코올, 케톤으로의 선택성이 검토되 었다. 그 결과 표 8-22 와 같이 철의 산화가에 따라 전환율과 알코올, 케톤의 비율에는 매우 큰 차이가 있었다 .
표 8-21 시클로핵산의 Gif (N) 산화반응에서 환원된 철 촉매의 영향 철 화합물 전환율 케톤/알코올 비 알코올 케톤 (wt %) 철분말 0.72 0.99 1.71 1.36 철/실리카 1.1 1 4.9 5.8 8 4.24 환원된 a) 철/실리카 I 1.84 2.71 4.57 1.47 (10 cc /분) 환원된 b) 철/실리카 0.85 1.37 2.7 2.2 (60 cc / 분) * 반응조건 : 시클로헥산 (46 mmol). 철 화합물 (0.005 mmol). 아연 (40 mmol). 초산 (35 mmol) , 반응시 간(1 6 시 간) . a) 촉매는 5 % 수소와 95 % 질소의 혼합가스를 10cc/ 분의 유속에서 400 ' C 의 온도에서 환원하였다. b) 촉매는 5% 수소와 95% 질소의 혼합가스를 60CC/ 분의 유속에서 4oo · c 의 온도에서 환원하였다 . 표 8-22 시클로핵산의 G if (IV) 반응에서 철 화합물의 산화 상태의 영향 철화합물 전환율 ON/OL 비 OL ON (wt %) 철분말 0.72 0.99 1.71 1.36 FeO 2.58 2.4 7 5.05 0.96 Fe2°3 0.36 2.52 2.88 7 * 반응조건 : 시클로헥산 (46 mmol), 철 화합물 (0.005 mmol) , 아연 (40 mmol), 초산 (35 mmol), 반응시간(1 6 시간)
2) Gi f-K RICT II 반응계 이규완 등이 개발한 Gi f-K RICTI 반응계는 산화제로 사용하는 과산화수소를 계속 부가하여야 한다는 단점을 가지고 있었다. 이 단점을 개선하기 위해 개발한 것이 과산화수소를 팔라듐 촉매에 의 해 직접 생성시키는 새로운 Gi f-K RICTII 반응계이다 . 7 0~ 72) 이 실험에서 사용된 촉매는 철의 원료로서 FeC12 • 4H20 이며, 건 조 및 소성은 환원분위기 즉 , 4 % 수소가 함유된 질소 분위기에서 행한 촉매를 사용하였댜 이 촉매에서의 철과 팔라듐의 함량은 각 각 3.0w t%, 0.6 wt%였으며 사용된 실리카 촉매의 특성 분석 결 과는 표 8-23 과 같댜 팔라듐 금속과 염화 팔라듐에서의 Pd 3ds12 전자들의 결합 에너 지는 각각 335.1 eV 와 338 eV 이댜 촉매에서의 Pd 3ds12 결합 에 너지 결과는 팔라듐종이 수소/질소 혼합기체하의 소성 중에 금속 상태로 거의 환원되었음을 알려준다. 이 연구에서 실험시 부산물의 생성 가능성에 대해 조사해 본 결 과 아무런 부산물이 검출되지 않았으며 기체 유출물 중에도 완전산 화 생성물인 일산화탄소나 이산화탄소는 검출되지 않았다. 이는 기 존 시클로헥산의 액상산화공정에 비하여 고선택성의 측면에서 매
표 8-23 불균일화된 철-팔라듐/실리카 촉매의 특성 분석 방법 결과 XRF Cl /Fe 원자비 = 0.95 XRD 무정형 XPS Fe2p 3/ 2 결합 에너지 = 711.3 eV Pd3d5/2 결합 에너지 = 335.9 e V
표 8-24 시클로헥산의 산화반응 촉매 수율 al( mol % ) 아;...l-디오근 b ) 케톤 c) 전환율 철-팔라듐/실리카 2.8 7 0.4 2 3.29 철/실리카 팔라듐/실리카 철/실리카 1 g + 팔라듐/실리카 3.75 0.7 8 4.53 철-팔라듐/실리카 추출액 d ) 0.09 0.0 1 0.1 0 * 반응조건 : 촉매(lg), 시클로핵산 (S g), 아세톤 ( 20 ml). 수소 ( 20ml/ 분). 산소 (20m l / 분). 반응온도 (30°c). 반응시간 (3 시간) a) 시클로헥산 기준 수율 b) 시틀로헥산 을 c) 시클로헥사논 d) 추출은 철-팔라듐/실리카 톡 매 를 아세톤 용매하에 서 3 시 간 환류 후 얻어짐
우 획기적인 결과라 할 수 있다 . 철-팔라듐/실리카, 철/실리카, 팔라듐/실리카 그리고 그들의 혼 합물, 촉매의 유출물을 촉매로 사용하여 얻어진 실험 결과는 표 8-24 와 같다. 철-팔라듐/실리카는 시클로핵산의 산화반응에 활성을 보이지만 철/실리카나 팔라듐/실리카 단독 촉매만으로는 활성을 보이지 않 는다. 철/실리카와 팔라듐/실리카를 혼합 사용할 때는 철-팔라듐/ 실리카 촉매보다 더 나은 수율을 낸다. 이러한 결과는 촉매의 능력 이 두 가지 금속종의 기능을 결합시킨 데 기인한다는 점을 뒷받침 해 준다. 죽, 팔라듐 금속은 수소와 산소를 과산화수소로 전환하여 주고, 철 이온은 생성된 과산화수소를 탄화수소의 수산화에 활용시 킬 수 있다는 것이다.
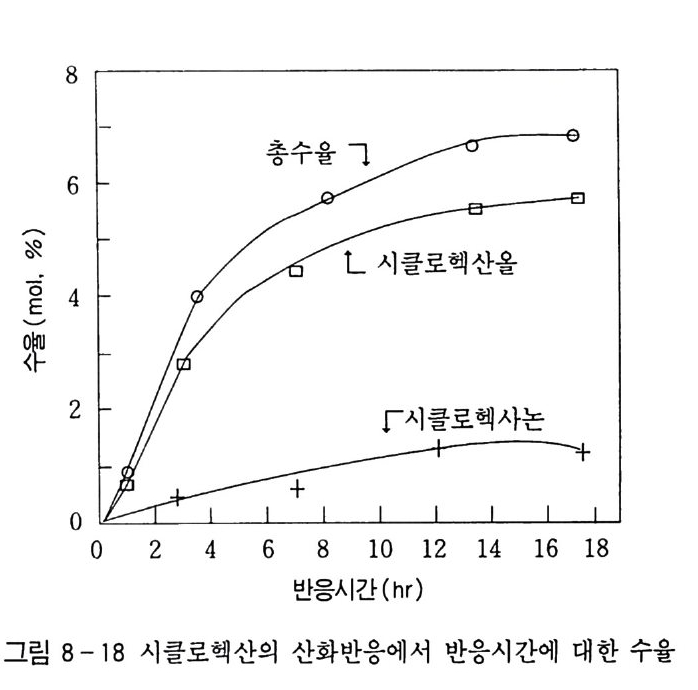 8
8
철-팔라듐/실리카의 아세톤 추출 용액을 이용한 반응이 철一팔 라듐/실리카 촉매 자체보다 훨씬 낮은 수율을 보였다는 것은 본 촉 매반응이 주로 표면에 의한 불균일 촉매 작용에 의해서 일어남을 알려주고 있다. 그리고 반응중에 유출된 철과 팔라듐의 분석 결과, 촉매 속에 함유된 각 금속에 대하여 철은 2.7 %, 팔라듐은 0.7 % 에 불과하였댜 반응시간에 따른 수율의 변화는 그림 8-18 과 같다. 반응시간이 증가함에 따라 반응은 약 7 %의 수율까지 진행되었다. 이는 촉매 중 철 원자수에 대하여는 약 8 배의 반응물 분자가, 팔라듐 원자수 에 대하여는 약 69 배의 반응물 분자가 전환된 것이다. 즉, 촉매 사 이클이 이루어지고 있음을 뜻한다. 그 이후에는 반응이 더 이상 진 행되지 않는데, 반웅중에 부산물로 생성된 물이 촉매를 불활성화하 기 때문인 것 같다. 반응온도. 아세톤 양, 기체유량 등의 반응조건을 변화시킬 때의
결과는 표 8-25 와 같다. 반응온도를 올려주면 40°C 에서는 수율이 증가하지만 50 ° C 에서 는 급격히 감소하는데. 그 이유는 높은 온도에서 과산화수소의 열 분해가 크기 때문이다. 아세톤 양의 변화 실험에서 적절한 아세톤 양은 30ml 인 것으로 나타났으며. 기체의 유량을 변화시켜 본 결과 반응속도에는 별 영향이 없는 것으로 나타났다. 반응계를 변화시키면서 비교 실험한 결과는 표 8-26 과 같다. 수 소를 흘려주지 않았을 때 알코올과 케톤 생성물은 만들어지지 않았 다. 이러한 사실은 본 반응이 산소에 의한 산화가 아니고 과산화수 소에 의한 수산화반응이라는 사실을 알려주고 있다. 또 수소와 산
표 8-25 Gif -K RICTII 반응계에서 반응조건에 따른 실험 결과 반응조건 수율a )(mol %) OEL ...l-더오근 b) 케톤c ) 초o 人T 멸0 반응온도= 4o·c 3.94 0.53 4.4 7 반응온도= 50°C 0.7 6 0.09 0,85 반응온도 = 40 °C, 아세톤의 양 = 10 ml 2.21 0.29 2.50 반응온도 = 4o ·c. 아세톤의 양 = 30 ml 2.6 5 0.4 3 3.08 반응은도= 40°C, 아세톤의 양= 40ml 2.68 0.4 4 3.09 반응온도 = 4o ·c. 수소 = lO ml /mi n 3.91 0.52 4.4 3 산소 = lO ml /mi n 반응온도 = 4o ·c. 수소 = 30 ml /mi n 3.99 0,56 4.55 산소 = 30 ml /mi n * 반응조건 : 촉매(철-팔라듐/실리카). 다른 조건은 표 8-24 와 같다. a) 시클로헥산 기준 수율 b) 시클로헥산을 c) 시클로헥사는
표 8-26 Gif -K RICT II 반응계에서 반응 변수의 비교 결과 a) 반응조건 수율 b l( mol %) 알코올c ) 케톤 d ) 초o 人T 걷0 수소 기류 없이 물 5g 부가 0.8 2 0.84 1.66 59 % 과산화수소 3 g (수소 산소 없이) 0.7 6 0.7 6 1.52 28 % 과산화수소 5 g (수소. 산소 없이) 0.7 0 0.7 2 1.42 아세톤 없이
소의 혼합 기체를 흘려주지 않고 과산화수소 수용액을 넣었을 때 반응 수율은 감소하였다. 이것은 과산화수소와 함께 들어간 물이 수산화를 방해하는 효과 롤 내기 때문이다• 아세톤 없이는 반응이 거의 일어나지 않는다. 아세톤과 과산화수 소로부터 아세톤히드로 과산화물 (Ace t one hy d rop e roxid e ) 이 생 성되고 이것이 반응중에 산화제 역할을 할 수도 있을 것이다. 그러 나 이러한 가능성은 아세톤 과산화물을 직접 제조하여 넣고 반응시 킨 결과를 보아 거의 배제될 수 있었다 . 그리고 시클로핵산을 빼고 반응 실험한 후 아세톤히드로 퍼옥사이드를 추출한 결과 그 양은
표 8-27 Gif -K RICT II 반응계에서 용매 효과 a) 용매 수율 b l (mol %) 알코올 c) 케톤 d) 초o 人T 멸0 에탄올 0.52 0.1 2 0.6 4 이소프로필알코올 0.5 6 0.1 0 0.6 6 메틸에틸케톤 1.18 0.25 1.43 메틸이소부틸케톤 0.1 3 0.1 9 0.32 에틸아세테이트 0.4 3 0.23 0.6 6 에틸에테르 0.03 <0. 01 0.03 * a) 아세톤은 표 8-24 에서와 같이 다른 용매로 교환되었다. b) 시클로헥산 기준 수율 c) 시클로헥산을 d) 시클로헥사논
약 0.04g 정도로 극히 적었다. 표 8-27 은 아세톤 대신 다른 용 매를 넣고 반응 실험한 결과이다. 아세톤 용매를 사용할 때보다 다른 용매들을 사용했을 떄 수율이 감소함을 알 수 있었다. 용매가 아세톤이 아닌 경우에도 극성 용매 에틸알코올이나 비극성 용매 에틸에테르보다는 아세톤과 극성 정 도가 가장 유사한 메틸에틸케톤이 가장 높은 수율을 나타냄으로써 용매의 극성과 수율이 관계가 있음을 알았다. 즉, 용매의 극성이 중간 정도일 때 좀더 효율적이라는 결론이다. 이는 용매가 모든 종류의 반응물, 죽 수소, 산소, 시클로헥산, 과산 화수소 등을 잘 녹일 수 있어 촉매 표면에 반응종들을 잘 전달시켜 야 하기 때문일 것이다. 이러한 요구는 아세톤에 대하여 가장 잘 일치하는 것으로 보인다.
3) 촉매 제조법의 영향 (1) 팔라듐 촉매 팔라듐을 실리카에 지지시킨 촉매의 제조를 위해 PdClz 0.1 g을 20ml 의 증류수에 녹인 후 실리카겔 9 g에 침윤시켰다. 여분의 물 은 증발시키고 공기 기류하에 150 °C 에서 2 시간 동안 건조, 4oo ·c 에서 3 시간 동안 소성했다(팔라듐/실리카). 위의 방법에서 염화팔라듐울 증류수에 녹일 때 경우에 따라 lN- 염산을 8.3ml 가하거나(팔라듐/실리카 -H), 30% 암모니아 수 1. 7 g을 가한 후(팔라듐/실리카 -N) 침윤시켜 촉매 제조에 변 화를주었댜 이렇게 제조된 팔라듐 촉매를 염화철과 혼합 사용하여 촉매능을 비교, 그 결과를 표 8-28 에 나타내었다. 표 8 -28 에 나타난 결과에서 보듯이 촉매 제조법에 따라 수율은 약간 차이가 있었으며 그 중 염산이나 암모니아수를 가하지 않고 제조한 팔라듐/실리카가 가장 좋은 결과를 보였다.
표 8-28 팔라듐 촉매 제조 방법에 의한 결과 a) 촉매 수율 bl(mol %) 알코올c ) 케톤 d) 총수율 FeCl2 • 4Hp 0.1 g + 팔라듐/실리카 1g 3.1 7 0.52 3.69 FeCl2 • 4HP 0.1 g + 팔라듐/실리카 1 g 2.8 5 0.4 5 3.26 FeCl2 • 4H20 0.1 g + 팔라듐/실리카 -N 1 g 2.66 0.4 1 3.07 • a) 다른 조건은 표 8-24 와 같았다. b) 시클로헥산 기준 수율 c) 시클로헥산을 d) 시클로헥사논
(2) 철 촉매 철을 실리카에 지지시키는 촉매 방법을 여러 가지로 변화하였다. 철 원료로서 먼저 FeCl2 • 4H2 0 1 g을 20 ml 의 증류수에 녹인 후 실리카겔 9 g에 침윤시켰다. 여분의 물은 증발시키고 공기 기류하 에 150°C 에서 2 시간 동안 건조한 후 400 ° C 에서 3 시간 동안 소성 시켜 완성하였다(철/실리카 -C2). 위의 방법에서 경우에 따라 철 원료로 FeCb • 6H20 1.45 g(철/실리카 _C3) 나. Fe(N0:1 L i • 9H2 0 2.04 g(철/실리카 -N). FeS04 • 7H2 0 1.40 g ( 철/실리카_ S) 를 사용하여 제조법을 변화시켜 보았다. 다음은 철/실리카 -C3 제조법을 변경하여 침윤 후의 촉매에 과
표 8-29 철 촉매 제조법에 의한 반응 결과 a ) 촉매 수율 bl(mol%) 알코올 c ) 케톤 d) 총수율 FeCl2 • 4Hz0 0.1 g + 팔라듐/실리카 1 g 3.1 7 0.52 3.69 FeCl3 • 6Hz0 0.1 4 g + 팔라듐/실리카 1 g 2.4 9 0.4 7 2.96 Fe(NO 갑 3·9H p 0.21 g + 팔라듐/실리카 l g - FeS04 • 7Hz0 0.1 4 g + 팔라듐/실 리 카 l g 3.62 0.4 0 4.0 2 Fe/ 실리카 -C2 l g + 팔라듐/실리카 l g 1.87 0.09 1.96 Fe/ 실리카 -C3 1 g + 팔라듐/실리카 l g 2.83 0.20 3.0 3 Fe/ 실리카 -N l g + 팔라듐/실리카 l g 1.58 0.22 1.80 Fe/ 실리카- S 1 g + 팔라듐/실리카 l g 2.2 0 0.32 2.52 Fe/ 실리카 - OH 1 g + 팔라듐/실리카 l g 1.93 0.36 2.29 * a) 다른 조건은 표 8-24 와 갇았다. b) 시클로헥산 기준 수율 c) 시클로헥산을 d) 시클로헥사논
량의 암모니아수를 가하여 수산화철로 전환시킨 다음. 고형물을 여 과한 후 증류수로 세척하고 건조 및 소성을 행하여 촉매들을 제조 하였다(철/실리카 -OH). 이렇게 제조된 촉매들과 각 철염들에 대 하여 비교 반응하여 그 결과를 표 8-29 에 나타내었다. 위 결과에서 보는 바와 같이 철염을 촉매로 사용할 때는 철염의 종류에 따라서 수율에 상당한 차이를 나타냈는데, 이는 철의 산화 가 상태와 음이온의 영향을 받은 것으로 보인다 . 2 가 상태의 철이 3 가 상태의 철 보다 활성이 좋아 보이며 음이온으로는 황산염이 가 장 높은 수율을 나타냈다. 특이한 점은 질산염은 전혀 활성을 나타 내지 않았다는 것이다. 반면 질산철을 곧바로 촉매로 사용할 때와는 달리 이를 원료로 하여 소성한 촉매에서 활성을 보이는 것은 산화철도 활성을 나타냄 을 의미한다 . 철 고체 촉매들도 제조시의 철염 원료에 따라 활성에 많은 차이 를 보이는데. 이들 중에서는 철/실리카 -C3 가 가장 높은 수율을 나타냈다. 이는 제조 후 산화철로 상당 부분 바뀌는데 잔존하는 음 이온과 철염에 따른 소성시의 분해 정도 그리고 철산화물의 실리카 표면에서의 분산도 등에 영향을 받기 때문일 것이다. 4) 철 촉매 활성에 미치는 영향 분석 철 촉매 제조시 철염의 종류에 따라 촉매 활성이 차이가 나고 이 러한 활성 차이는 여러 가지 복합적인 요인에 기인할 것이라는 점 은 이미 기술한 바 있다. 촉매 활성에의 영향 요인을 분석하여 바 람직한 촉매 제조법을 확립하기 위해서는 먼저 그 요인을 단순화하 고 철산화물의 특성 분석을 통하여 촉매 활성과의 관계를 파악하는 일이 필요하다.
이 목적을 달성하기 위하여. 대표적인 실리카에 지지된 철 촉매 제조법에 대하여 제조시 철의 함량을 변화시켜 가면서 촉매의 특성 과 반응시의 활성에 대한 영향을 살펴보았다. 철/실리카 -C3 와 철/실리카 -N3 촉매들의 철의 함량을 0.1 5 ~ 8.3 wt%로 변화시켰다. 철/실리카 -OH 촉매들도 철염의 투입량 을 변화시켜 가면서 제조하였는데 . 제조 과정에서 증류수의 세척시 철의 손실이 있기 때문에 제조 후의 촉매들에 대해서 철의 함량을 알기 위해 XRF 측정을 행하였다. (1) 촉매 특성 분석 철 촉매 중 철/실리카 -C3 계열과 철/실리카 -OH 계열에 대하 여 XRF 분석방법으로 염소 분석을 하였다. 철/실리카 -C3 촉매 들에 대한 결과는 표 8-30 과 같으며. 철/실리카 -OH 촉매들에서 염소는 거의 남아 있지 않았다 . 3 가 염화철 (FeCh) 을 출발 물질로 한 철 촉매에서의 Cl /Fe 값 은 0.6~0.8 로 원래 출발 물질의 3 에 비하여 상당히 감소되었다. 이는 소성 중에 염화철이 산화철로 바뀌면서 염소의 많은 부분을
표 8-30 각 촉매 XRF 분석으로부터 얻은 염소/철의 함량 촉매 염소/철 비율 0.7 7 % . 철/실리카 -C3 0.67 1.5 % 철/실리 카 -C3 0.7 5 3.0 % 철/실리카 -C3 0.60 4.4 % 철/실리카 -C3 0.68 5.7 % 철/실 리 카 -C3 0.61 8.3 % 철/실리카 -C3 0.75
잃어버렸기 때문일 것이다. 다음은 철/실리카 -C3, 철/실리카 _OH, 철/실리카 -N 각 계열 촉매들에 대한 XRD 구조 분석이다. 그 결과들은 그림 8-19, 8- 20, 8-21 와 같다. 철/실리카一 C3 와 철/실리카 -N 촉매들에서는 Fe20 3 결정 구조 들이 나타났으나 철/실리카 -OH 촉매들은 결정 구조를 전혀 보이 지 않았다. 특히 철/실리카 _C3 촉매들은 철/실리카 _N 촉매들에 비해 산화철의 구조가 잘 발달되어 있음을 알 수 있었다. 촉매에서의 산화철 결정 크기를 측정하기 위하여 X 선 광폭화 (X-ray line broadening analysis) (XLBA) 분석을 행하였는데 철/실리카 -N 촉매들은 X- 선 광폭화가 너무 심하여 결정 크기를 구할 수 없었댜 철/실리카 _C3 촉매들에 대해서는 표 8-31 에 나 타내었다. 표 8_31 결과를 보면 촉매에서의 철의 함량이 증가할수록 산화 철의 결정 크기가 점차 커짐을 알 수 있다. 그러나 철 함량의 증가 에 비해서 결정 크기의 증가폭이 적음을 볼 때 미세 결정의 수가 증가하는 것으로 보인다. 철/실리카 _C3, 철/실리카 -N , 철/실리카 -OH 촉매들에서 XPS 로부터 구한 철의 바인딩 에너지를 표 8-32 에, 철의 함량에 따른 철/규소 표면 비율을 그림 8-22 에 나타내었다. 각 촉매들에 서의 바인딩 에너지로 보아 표면이 거의 산화철 상태인 것으로 보 인다 . 그림 8_22 의 철 함량 증가에 따른 철/규소 비의 증가를 보면 표면에서의 철의 분산도 (d i s p ers ity)를 비교할 수 있다. 철 함량이 증가할 때 철/실리카 -OH 촉매들에서 철/규소 비의 증가가 가장 크므로 철의 확산이 가장 잘되어 있음을 알 수 있다. 그 외에 철/ 실리카 _C3 촉매가 철/실리카 _N 촉매보다는 철의 확산도가 더
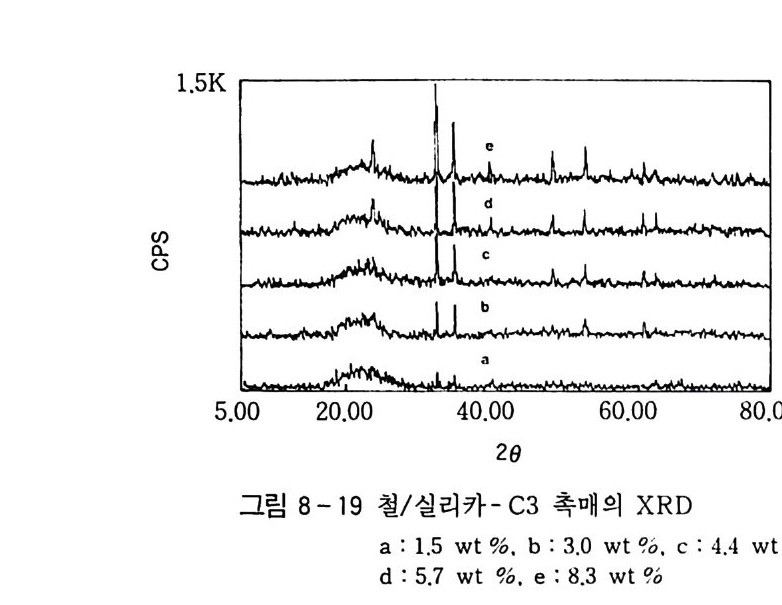 1.5K
1.5K
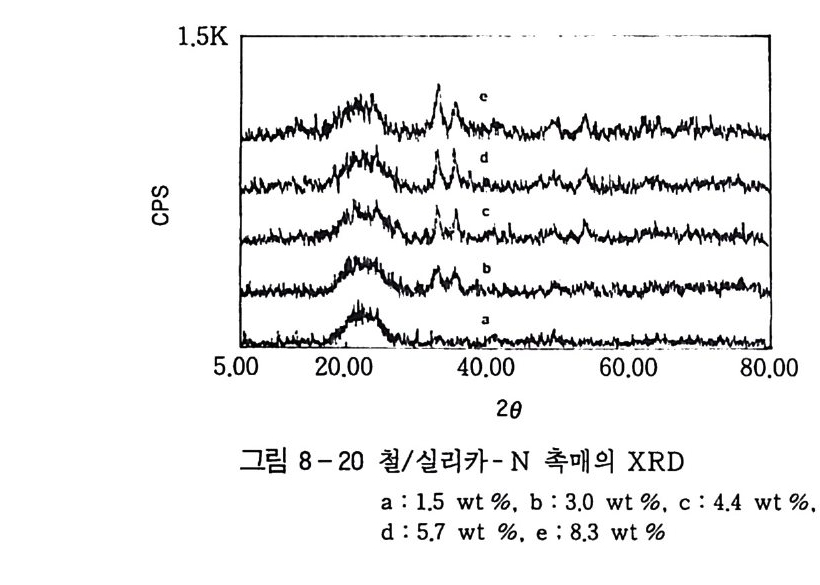 1.5 K
1.5 K
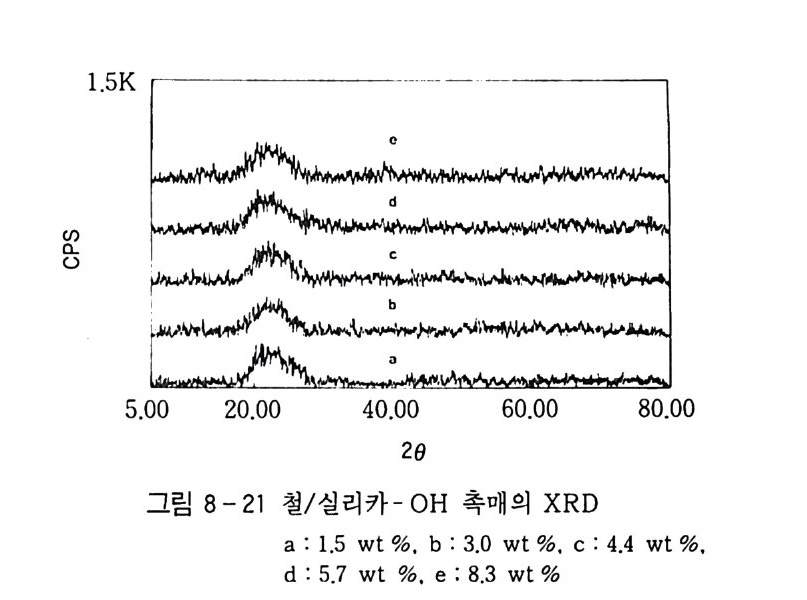 1.5K
1.5K
표 8-31 XLBA 로 측정한 산화철 결정 크기 결정 크기 (nm) 촉 매 L(l04)a) L(llO) L(024) L( l16 ) 3.0 % 철/실리카 -C3 69 34 49 55 4.4 % 철/실리카- C3 72 62 74 76 5.7 % 철/실리카- C3 73 73 77 65 8.3 % 철/실리카 -C3 75 75 88 63 * a) l04 면에 수직인 방향으로 결정의 평균 길이
표 8-32 철 촉매의 Fe 2p3 1 2 전자의 결합 에너지 촉매의 종류 B. E. (eV) 철/실리카 -C3 계열 711 .0 土 0.5 철/실리카 -N 계열 711 .5 士 0.4 철/실리카 -OH 계열 711 .5 土 0.2
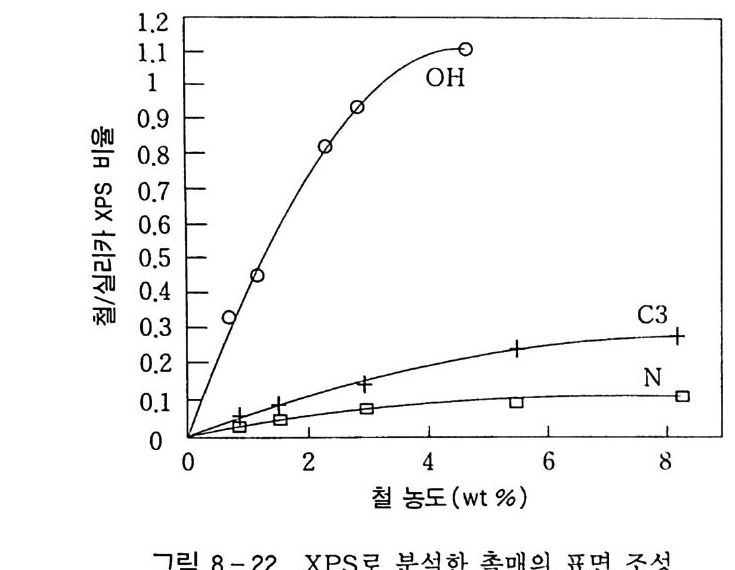 1.2
1.2
높음을 알수 있다. (2) 반응 결과 철 원료와 함량을 변화시키면서 제조한 철 촉매 l g씩과 팔라듐/ 실리카 l g을 혼합하여 반응시킨 후 그 결과를 촉매 중의 철 함량 에 대한 수율의 함수로서 그림 8-23 에 나타내었다. 촉매의 활성은 철/실리카 -C3> 철/실리카 -OH> 철/실리카 N 순으로 나타났다. 철/실리카 -C3 에서는 철의 함량이 1.5 w t% 일 때, 철/실리카 -OH 에서는 철의 함량이 0.72 wt%일 때. 철/실 리카 -N 에서는 철의 함량이 2.5 wt%일 때 각각 최고 수율을 나타 냈다. 이중 철/실리카 -OH 촉매는 상대적으로 적은 철의 함량에 서 최고 수율을 나타냈기 때문에 단위 철 원자당 생성물의 분자 몰 수가 14 에 이르렀다• 또한 촉매 활성에의 요인이 될 수 있는 것으로 철 산화물의 구조 상태, 철의 분산도, 철 산화물의 결정성 등을 들 수 있다.
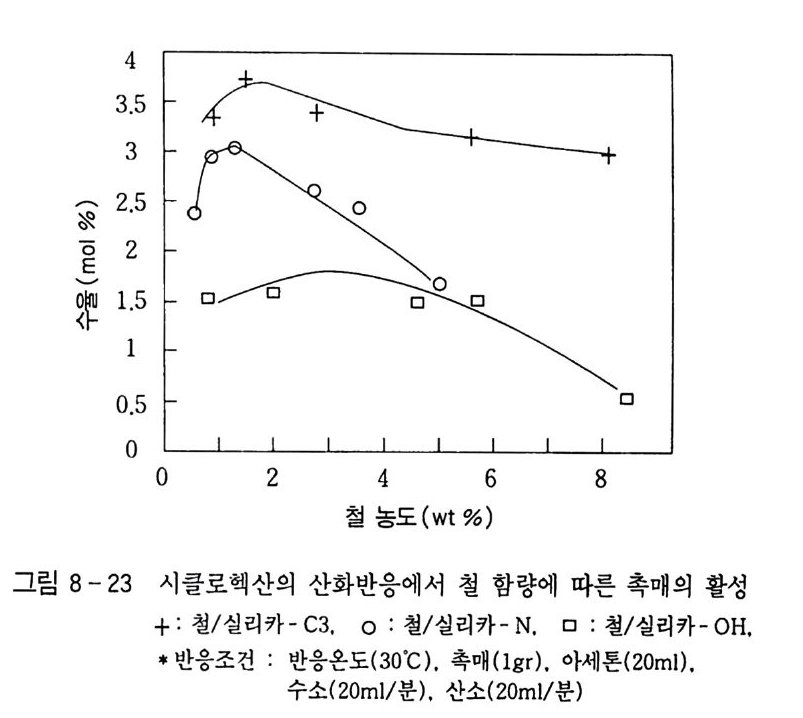 4
4
먼저 철 산화물의 상태는 철의 원료에 관계없이 XPS 바인딩 에 너지로 보아 3 가의 산화 상태인 것으로 여겨진다. 그리고 철/실리 카 _C3 와 철/실리카 -N 은 XRD 결과로 보아 거의 대부분이 Fe20 3 구조 상태로 존재하나 철/실리카 -OH 는 무정형의 철 산화 물인 것으로 보인다. 그러나 철/실리카 -C3 는 철 원료의 염소 음 이 온이 약 23 % 잔존해 있어 평 균화학식 FeCl 。 , oL15 로 볼 수 있다 . 철의 분산도를 보면 철/실리카 _OH 가 분산이 가장 잘되어 있으며. 다음은 철 산화물의 결정성이 더 큼에도 불구하고 오히려 철/실리 카 -C3 가 철/실리카 _N 보다 분산도가 좋다. 위의 결과를 보면 철의 결정성과 반응에서의 활성을 관련하여 결 과를 분석하기는 어렵다. 또한 철의 분산도 한 가지만으로는 촉매 의 활성 순서를 획일적으로 설명하기가 곤란하다. 본 촉매 작용에 서의 활성점을 제공하는 철이 표면에 많이 드러나 있을수록 촉매의
활성이 높아질 것이다. 따라서 철의 분산도가 가장 낮은 철/실리 카 -N 촉매들은 활성도 낮지만 철/실리카 -C3 와 철/실리카 -OH 에 있어서는 반대의 현상을 보인다. 이는 철/실리카 _C3 에 염소 음이온이 잔존해 있어 이의 영향으로 촉매의 활성이 높아지기 때문 인 것으로 여겨진다. 철의 함량이 증가할 때 촉매의 활성이 촉매 종류에 따라 각각의 최고점을 나타내고, 그 이후에는 촉매의 활성이 감소한다. 이는 철 의 함량 증가시 실리카와 결합해 있는 표면 산화 철종 위에 bulk 산화철종이 쌓이면서 촉매의 활성이 감소하는 것으로 보인다. 또 실리카와 결합되어 있는 표면 산화철종이 이온 상태의 철과 유사하 기 때문에 bulk 산화철보다 활성이 높을 것으로 생각된다. 위의 대표적인 촉매에 대하여 반응중 철의 유출량을 AA 분석을 통하여 측정한 결과를 표 8_33 에 나타내었다. 촉매 제조법에 따른 철의 유출량을 비교해 보니 철/실리카 -C3> 철/실리카 _OH> 철/실리카 -N 순이었다.
표 8-33 각 촉매의 반응 중에 용출된 철의 양 촉매 용출량(%) 1.5 % 철/실리카 _C3 7.2 3.0 % 철/실리카 -C3 7.0 4.4 % 철/실리카 _C3 4.1 1.5 % 철/실리카 _N 0.3 3.0 % 철/실리카 -N <0 .1 4.4 % 철/실리카 -N <0 .1 1.3 % 철/실리카 -OH 0.3 2.5 % 철/실리카 -OH 0.9 2.7 % 철/실리카 -OH 1.1
반응시 철의 유출량이 적을수록 촉매의 재사용이 유리해지고 반 응 후의 분리가 수월할 것이다. 이러한 관점에서 보면 철/실리카 N 이 좋으나 촉매 활성이 낮기 때문에 바람직한 촉매는 못 된다 . 철/실리카 -OH 촉매는 철의 유출량도 적고 수율도 높은 편이어서 좋은 촉매 제조법이라고 할 수 있다. 참고문헌 I ) 0. Haya s hi, 0. Kata g i ri and S. Roth b erg, J. Am. Chem. Soc., 7 7, 5450 (19 55 ) . 2) H. S. Mason, W. L. Fowlks and E. Peetr so n, J. Am. Chem. Soc., 77, 2914(1 9 55). 3 ) J. T. Groves, R. C. Haushalter , M. Nakamura, T. E. Nemo and B. J. Evans, J. Am. Chem. Soc., 103, 2884(1 9 81 ). 4) a) P. R. Ortiz de Monte l lano, In Cy toc hrom P -4 50 Str uc tu r e, Mechanis m and Bi oc hemi st r y Ed., Plenum Press, New York( 1986) . b) T. C. Bruic e, In Mecha nisti c Princ ip le s of Enzym e Acti vi ty , J. F. Lie b man and A. Greenberg, Eds, : Molecular Str uc tu re and Energe t ic s , vol. 9 : VCH : New York, pp.2 27~228(1 9 88). 5) S. P. Cramer, J. H. Dawson and L. P. Hag er , J. Am. Chem. Soc., 100, 7282~7290(1 9 78) . 6) a) H. S. Mason, J. C. Nort h and M. Vanneste , Fed. Proc., 24, 1164 (19 65). b) J. H. Dawson, J. R. Trudell, R. E. Lin d er, G. Barth and E. Bunnenberg, C. Dje r assi, Bio c hemi st ry 17, 33~42(1 9 78) .
7 ) a ) J. 0. Ste m and J. Peis a ch, J. Bio l . C hem. , 249, 7495~7498 ( 1974 ) . b) S. Koch, S. C. Tang , R. H. Holm, R. B. Frankel and J. A.lb ers, J. Am. Chem. Soc. , 97, 917~991 (19 75 ) . c) J. P. Collman, T. N. Sorrel and B. M. Hoff ma n, J. Am. Chem. Soc., 97, 913~914(1 9 75) . d) S. C. Tang, S. Koch, G. C. Papa e ft hy m iou , S. Foner, R. B. Frankel, J. A. lbers and R. H. Holm, J. Am. Chem. Soc. , 98, 2414~2434( 1976) . 8) T. L. Poulos, B. C. Fin z el and A. J. Howard, J. Mo/. Bio l ., 195, 687~700( 1987) . 9) J. H. Dawson and M. Sono, Chem. Rev., 87, 1255~1276( 1987) . 10) S. G. Sli ge r and R. I. Murray, In Cy toc hrom P -450 Str u ctu re, Mecha nism and Bio c hem istr y : P. R. Or tiz de Monte l lano, Ed, : Plenum Press : New York ; pp. 429~503 (19 86 ) . 11) a) B. W. Gr iffin and I. A. Pete r son, J. Bil o . Chem., 250, 6445~ 6451 (19 75) . b) S. B. Phil so n, P. G. Debrunner, P. G. Schmi dt and I. C. Gunsalus, J. Bil o. Chem., 254, 10173~10179(1 9 79) . 12) R. Lobrutt o, C. P. Scholes, G. C. Wang er , I. C. Gunsaulus and P. G. Debrunner,Am. Chem. Soc., 102, 1167~1170(1 9 80). 13) J. H. Dawson, L. A. Andersson and M. Sono, J. Bil o. Chem., 257, 3606~3617(1 9 82) . 14) a) R. Tsai , C. A. Yu, I. C. Gunsalus, J. Peis a ch, W. Blumberg, W. H. Orme-J oh nson and H. Bein er t , Proc. Natl. Acad. S c i., USA 66, 1157~1163 (19 70) . b) S. G. Sli ger , Bio c hemi str y , 15, 5399~5406( 1976) . 15) S. G. Slig e r and I. C. Gunsalus, Proc. Natl . Acad. Sc i., USA 73,
1078~ 1082(1 9 76) . 16) D. R. Ligh and N. R. Orme -J oh nson, J. Bio l . Chem., 256, 343~ 350(1 9 81 ) . 17) J. T. Groves, G. A. McClusky, R. E. Whit e and M. J. Coon, J. Bio c hem. Bio p hy s . R es. Commun., 8 1, 154~160(1 9 78). 18) R. P. Bell, The Proto n in Chemi st r y 2nd Ed. ; Cornell Univ e rsit y Press : Ita c ha, N ew York( 1973) . 19) a) M. H. Gelb, D. C. Heim brook, P. Mallconen and S. G. Sli ga r, Bio c hemi str y 21, 370~377(1 9 82) . b) K. S. Eble and J. M . Dawson, J. Bio l . c hem., 259, 14389~ 14393 (19 84) . 20) a) P. R . Or tiz de Monte ll ano and N. 0. Reic h , In Cytoc hrome P- 450 Str uc tu re , Mechanis m , and Bio c hem ist ry : P. R. Or tiz de Monte l lano, Ed. : Plenum Press : New York : pp. 217~ 271( 19 86). b) B. W itk o p(金剛佑一譯), 『化學의 鈴城』, 23, 949(1 9 68). c) D. M. Jer i na , Chem. Technol. , 4, 120(1 9 73). 21 ) P. R. Orti z de Mote l lano and N. 0. Reic h , ib id . , pp.2 73~314 (19 86) . 22) P. R. Or tiz de Monte l lano and B. A. Mi co , Mol. Pharmacol, 18, 128~135 (19 80) . 23) a) P. R. Or tiz de Monte l lano, K. L. Kunze, H. S. Beil a n and C. Wheeler, Bio c hemi st r y 21, 1331~1339( 1982) . b ) P. R. Or tiz de Monte l lano, R. A. Ste a ms and K. C. Lang ry, Mol. Pharmocol, 25, 310~317(1 9 84). 24) a) R. E. Miller and F. P. Gueng er i ch , Bio c hemi str y 21, 1090~1097 (19 82).
b) D. C. Lie b ler and F. P. Gueng er ic h , Bio c hemi st , )' 2 2, 5482~ 5489 (l98 3 ) . 25) J. M. Garris o n and T. C. Bruic e , J. Am. Chem. Soc., 111, 191~ 198(1989). 26 ) E. A. Kom ive s and P. R. Or tiz de Monte l lano, J. Bio l. Chem. , 262, 9793~9802( 1987). 27 ) J. P. Collman, J. I. Brauman, B. Meunie r , T. Hay as hi, T. Kododek and S. A. Rayb u ck, J. Am. Chem. Soc., 1 07, 2000~2005( l98 5) . 28) J. E. Tomaszewski , D. M. Jer i na and J. W. Daly, Bio c hemi str y • 14, 2024~2030 ( 1975 ) . 29) D. B. Presto n , J. A. Mi ller and E. C. Mi ller, J. Bio l . Chem., 258, 8304~8311 (19 83 ) . 30) a) J. T. Groves, G. A. McClusky, R. E. Whit e and M. Coon, J. Bio c hem. Bio p hy s . Res. Commun., 76, 541~549(1 9 77). b) J. P. Jon es, K. R. Korzekwa, A. B. Rett ie and W. F. Trage r , J. Am. Chem. Soc., 108, 7074~7078(1 9 86). 31 ) R. E. Whit e, J. P. Mi ller, L. V. Favreau and A. Bhatt ac hary ya, J. Am. Chem. Soc., 108, 6024( 1986) . 32) P. R. Or tiz de Monte l lano and R. A. Ste a rns, J. Am. Chem. Soc., 109, 3415~3420 ( 1987 ) . 33 ) T. J. McMu rry and J. T. Groves, In Cy toc hrome P -450 Str uc tu re , Mechanis m and Bi oc hemi st r y , P. Orti z de Mante l lando, Ed., Plenum Press, New York( 1985) . 34) J. T. Groves, T. E. Nemo and R. S. My er s, J. Am. Chem. Soc., 101, 1032( 1979) . 35) B. R. Cook, T. J. Rein e rt and K. S. Suslic k , J. Am. Chem. Soc., 108, 7281 (19 86) .
36) D. Mansuy , P. Batt ion i and J. P. Renaud, J. Chem. Soc. Chem. Commun., 1 255(1 9 84). 37 ) D. H. R. Ba rton , N. Ozba lik, In Acti va ti on and Functi on a liza ti on of Alkanes, C. L. Hi ll, ed., Joh n Wi lly & Sons, New York, p.2 81 (19 89). 38) D. H. R. Ba rton , K. W. Lee, W. N. Nehl, N. Ozbalik and Li Zhang , Tetr a hedron, 46 ( 11 ) , 3753~3768 (19 90) . 39) D. H. R. Ba rton , M. J. Gastig e r and W. B. Mort he r-well, J. Chem. Soc. Chem. Commun. , 41 (19 83 ) . 40) D. H. R. Ba rton , J. Boiv i n , W. B. Mort he rwell, N. Ozbalik , K. M. Schwartz e ntr u ber and K. Jan kowski, Nouv J. Chim . , 10, 387(1 9 86) . 41 ) D. H. R. Ba rton , J. Boiv i n , N. Ozbalik and K. M. Schwa rtze ntr ub er, Tetr a hedron Lett. , 25, 4219 ( 1984 ) . 42) D. H. R. Ba rton , J. Boiv i n , N. Ozba lik and K. M. Schwa rtzen tr ub er, Tetr a hedron Lett . , 26, 447 ( 1985 ) . 43 ) D. H. R. Ba rton , R. K. Hy ne s, G. Leclerc, P. D . Magn u s and I. D. Menzie s , J. Chem. Soc., Perki n 1, 2055(1 9 75). 44) A. T. Ni el son, D. W. Moore, G. M. Muka and K. M. Berr y, J. Org. Chem. , 2 9, 2175(1 9 64). 45) D. H. R. Ba rton , J. Boiv i n , W. B. Mort he rwell, N. Ozba lik and K. M. Schwartz e ntr u ber, N ew Jo urnal of Chemi st r y, 10( 17 ) , 387 (19 86). 46) D. H. R. Ba rton , J. Boiv i n , N. Ozba lik and K. M. Schwa rtze ntr ub er, Tetr a hedron Lett. , 25, 4219( 1984) . 47) F. Mi ni s c i, A. Ci tter io and F. Valeri a, J. Org. Chem., 45, 4752 (19 80).
48 ) D. H. R. Ba rton , J. Boiv i n , N. Ozbalik and K. M. Schwa rtze ntr ub er, Tetr a hedron Lett . , 26, 447 ( 1985 ) . 49) R. E. W血 e and M . J. C oon, Annu. Rev. B io c hem., 19, 315 (19 80) . 50) D. H. R. Ba rton , J. Boiv i n , W. B. Mort he rwell, N. Ozbalik and K. M. Jan kowski , Nouv. J. Chim ( 1986 ) . 51 ) D. T. Saw ye r and M. J. Gi bi a n , Tetr a hedron Lett . , 35, 1471 (19 79 ) . 52) P. Cop re and D. T. Saw yer , Anal. C hem. , 5 8, 1057(1 9 86) . 53 ) J. L. Jr. Robert s, M. M. Mori so n and D. T. Sawy er , J Am. Chem. Soc., 100, 329( 1978) . 54) D. H. R. Barto n , S. D. Bevie r e, W. C. Chavasir i , E. Csuhai, D. Doller and W. G. Liu , J. Am. Chem. Soc., 114, 2147~2156( 1992) . 55 ) D. Mansuy, J. F. Barto l i a nd M. Momente a u, Tetr a hedron. Lett . , 23, 2781~2784( 1982) . 56) B. R. Cook, J. J. Rein e rt and K. S. Suslic k , J. Am. Chem. Soc., 108, 7281~7286( 1986) . 57) D. Mansuy , J. F. Ba rtol i, J. C. Chott ar ol and M. Lang e, Ang ew . Chem. Int. Ed. Eng l., 19, 909~910(1 9 80) . 58) K. W. Lee, M. J. Choi, S. B. Kim and C. S. Choi, Ind. Eng . Chem. Res., 26, 1951~1955(1 9 87). 59) 이규완, 김성보, 전기원, 특정연구과제 보고서 「저온에서 선택적 산화반응」 pp.34--38 , 화학연구소( 1992) . 60) K. W. Lee, S. B. Kim, D. H. R. Ba rton and D. Doller, Bull. Kor. Chem. Soc. , 12, 459 ( 1991 ) . 61 ) P. M. Boy le , W. Cocker, D. M. Gray so n and P. V. R. Schannon, J. Chem. Soc., (C), 1073(1 9 71). 62) M . S. Carson, W. Cocker, D. M. Gray so n and P. V. R. Schannon, ibi d . , 2220(1 9 68) .
63) D. H. R. Barto n , A. K. Goktu rk, J. W. Morzy ck i and W. B. Mort he rwell, J. Chem. Soc. , Perk.i n I 1985b, 583 ( 1983 ) . 64) G. Balavoin e , D. H. R. Ba rton , J. Boiv i n , A. Gref., P. Le Coup an ec, N. Ozbalik , J. A. X. Paste n a and H. Ri vi e r e, Tetr a hedron 44, 1091 (19 87) . 65) B. K. Bower and H. G. Tennett , J. Am. Chem. Soc., 94, 2512 (19 72) . 66) K. W. Lee, S. B. Kim , K. W. Jun and E. K. Sh im , New J. Chem., 1 7, 409(1 9 93). 67) D. H. R. Barto n , M. J. Gasti ge r and W. B. Moth e rwell, J. Chem. Soc. Chem. Commun. , 731 (19 83 ) . 68) A. Chereti n and E. Lous, B ull. S oc. Chim . Fr. , 11, 446 ( 1944) . 69) D. Lup u and R. Ripa n, Rev. Roum. Chim ., 16, 43(1 9 71 ). 70) K. W. Lee, K. W. Jun , E. K. Shim and N. S. Cho, Ap pl. Cat. , 9 6, 269(1 9 93) . 71 ) K. W. Lee and K. W. Jun , Chemi e Jng e nie u r Technik , 64, 637 (19 92). 72) K. W. Lee, K. W. Jun and K. Y. Choi, Stu d . Surf Sc i . Cata l ., 66, 55 (19 91 ). 73) N. A. Mi las 印 d A. Golubovic , J. A m. Chem. Soc., 81, 6461 ( 1959) .
찾 0~ 보?|
7 가교제 169 가소제 185 가수분해 170, 268 가역반응 120 개질 (Re fo rm i n g) 82 개환반응 185 결합 해리 에너지 180, 183 경화제 169 계간 교차 228 고공 오존층 229 GoAg g 반응 314 공명 혼합체 214 공비 혼합물 182 과빙초산 103 과산화물 53, 170 과산화수소 171 과산화 에스테르 204 과산화 이온 211 과산화지 방산 178, 180 광증감 산소 산화반웅 227, 231 광증감제 (Photo s ensit ize r) 228 Gusta f s o n, B. L. 128 구전자 반응 177, 232 구전자 센터 177 구전자 중간체 178 구핵시약 220 균일 촉매 82, 174
글루타릭산 94 Gri es baum, K. 150 금속이온 175 금속 촉매 분해 (Me t al Cata l ys is ) 85 금지제 (inh ib i to r ) 47 L 나프타 98, 116 나프타 크래 킹 (Naph t h a Cracki ng ) 36, 51, 113 나프테네이트 (Na p h th ena t e) 88 냉매 115 노말 (n) -도데칸의 반응 309 노말 (n) - 파라핀 산화반응 55, 65, 69, ·73 노화제 169 Nie ls en, J. R. R. 122 Nis h i na ga , A. 107 니켈 102 니켈 hyd r osil ica te 126 니트로-니트로소 유도체 96 니트로벤젠 술포닐 과산화물 200 니트로벤젠 술포닐 크로리드 201 니트로소옥사이드 96 니트로소케톤 96 니트롤산 96
c: 다량체 256 다이옥시게나아제 211 단쇄 과산화물 183 데칼린의 산화반응 320 도데실히드로 과산화물 (dodec y l hy dr o- per oxid e ) 57 도데칸의 액상 산화반응 68 동시분해 (concerte d decomp o sit ion ) 260 동-아연 (Cu-Zn) 115 동위원소 94, 105 동위원소 효과 285 디메틸 비스술포닐 과산화물 200 디메틸에테르 116 디메 틸 피롤리딘 N -옥 사이드 205 디바나딜 피론산 (D i vanad y l pyro nic aci d) 144 디술파이드 201 디 아다만탄 (D i adaman tan e) 244 디아실 과산화물 169, 170 DMF 110 DMT 112 디오존화물 267 디옥세탄(Di oxe tan e) 243 디이소프로필 벤젠 과산화물 179 디카르복시산 94 디페닐 포스포닉 크로리드 204
DPQ 110 근 라이모넨(Li monene) 231 라이모넨의 반응 312 락톤 93 래디컬 반응 88, 193, 254 래디컬 아닥트 205 래디컬 연쇄 산화반응 (rad i cal chain auto x id a ti on ) 42 래디컬 재결합 285 래디컬 종 91 래디컬 중합 개시제 169 래커 141 Levy , M. 122 레지오 산화반응 200 레지오 선택성 (reg io selecti vi t y) 202, 286,316 레지오 선택성 조절 295 Repp e 139 루테늄 120 리 간드 (L ig and) 85 Rich ardson, J. T. 120
□ 말레산 무수물 141 망간 88 Mn(salen) 110 메시틸렌 196 메타 (m) - 봉산 에스테르 56, 64 메타 (m)- CPBA 192 매탄리포밍 115 메탄올 97 메 틸에 틸케 톤 (2 -부 탄온) 98 메틸 콜레스테롤 유도체 317 모노메틸테레프탈산 112 모노오존화물 267 모노옥시 게 나아제 (Monooxy ge nag e) 105, 281 Monsanto 143 Mo-Bi 136 Morooka, Y. 138 Mi las reag en t 189 l::t 바나듐 (N) 96 바나듐 V-Mo -P 150 바나듐 V -Pff i02 144 바나듐 V20s -MoO/ 담체 143 바나듐 V20s -P20s 152
바나듐 V20s -P20s -B 2 야 152 바나듐 (VOhP207 144, 145 바나듐-촉매 96 Wa rtz, A. 133 바이시클로 (2. 1. 0] 펜타닐 래디컬 286 바인딩 에너지 339 바커 공정 (Wacker p rocess) 83 Ba rton , D. H. R. 96 반감기 170, 213 반응기구 216, 256 반응속도 모델 73 반응 속도이론 68, 79 반응 억제제 64 발열반응 174 발포제 193 Baeye r -Vil lige r 반응 190 Ba y er 법 84 백금 Pd-Rh-V/Sil ica 107 백 금 PdCl2 -CuCl2 83 베타(~) - 균열 101 베타 (~)-VOP04 147 벤젠 수산화 104 벤조일 4 -톨 루엔 술포닐 과산화물 201 벤질 래디컬 195 벤질 알코올 146 부반응 96 부탄산화 97 부테닐 (bu t en y l) 래디컬 286 부록시 래디컬 101
부티르산 98 분해반응 174 분해반응 속도식 61 불균일반응 174 불균일 촉매 82, 107 불균일화 철/실리카 촉매 324 불포화 디아조 화합물 271 붕산 87 Br ich i m a 198 VPO 146 비과산화물 262 비대칭 성질 200 비대 칭 술포닐 과산화물 200 비래디컬 (B i ra di cal) 33 비스디페닐 포스피닐 과산화물 202 비스무스 146 BQ 110 BTX 37 비편재화 271 비프로톤성 용매 213 빙초산 97 人 사슬 메커니즘 90 사슬-정지 반응 103 4- 카르복시벤젠 알데히드 112 4-호 밀안식향산 112
산소 개 질 (Oxy re fo rming ) 116 산소 전달반응 290 산처니켈 (N i O/ZSM) 124 산화 11 산화반응 83, 216, 221 산화성 216 산화에틸렌 133 산화제 128, 169 산화 지방산 183 산화철 결정 크기 339 산화 퍼텐셜 170, 180 산화-환원반응 85, 193 산화환원 퍼텐셜 (redox pot e n ti al ) 107 살리실릭산 115 삼중결합 262, 263 삼중항 산소 29 삼중항 상태 29,40 상대 속도 함수 270 상이동 촉매 199 상자성 254 상호변이 (Tauto m eri za ti on ) 96 생성물 80 생성반응 속도식 60 생체 모방계 291 선택률 97 선택성 81 세슘 (Ces i um) 134 셀룰로오스아세테 이트 97 소광제 239
SOHIO 136 속도 정수 221 Solchem 공정 122 쇄상 디엔 237 수산 래디컬 212 수산 음이온 212 수산화 104, 188, 330 수산화 반응 175, 288 수산화 효소 288 수성가스 전환반응 125 수소화 82,86 수용매법 144 수용성( 受容 性) 래디컬 43 수율 87 수증기 개질 (S t eamRe formi n g) 116 수직치환체 260 수평치환체 260 슈퍼 구핵제 176 슈퍼옥사이드 (Su p er-ox i de) 211 슈퍼옥사이드 래디컬 음이온 201, 211, 307 Sup er oxo 109 Sup er oxo 착제 105 스테어롤산 263 Sto r y- M urray -Y oussefy e h 반응기 구 261 SPARG (Su rfur Passiv a te d Refo rming ) 공정 117, 118 스피넬 126
스핀 금제 38 시그마((j) - 복합체 255 시그마 (cr) -카 르보닐 화합물 262 시스_오존화물 262 시스-트랜스 비 257 시스-트랜스 이성질체 262 시클로펜타디엔 150 시클로프로필 카르바놀 (c y clo p ro py l carbin o i) 286 시클로헥사논 86, 95 시 클 로 헥 사 디 에 닐 (hy dr oxy cy cl o- hexadie n y l) 래디컬 194 시클로핵산 86 시클로헥산 산화 54, 311, 331 시클로핵산을 86, 95 시클로핵실보레이트 88 시클로헥실퍼옥시 래디컬 89, 90 시클로 화합물의 반응 310 시토크롬 P-450 281 식초산 97 신一안티 양성이온 260 실리카 114 。 anata s e 150 아다만탄 298 아디핀산 86, 87, 91
Arrhen i us 식 59 Amoco 112 아세독시히드로 퍼옥사이드 263, 334 아세트산 93 아세트알데히드 83, 97, 102, 136 아스카리돌 239 아스코르브산 107, 218 아스피린 97, 115 아실 술포닐 과산화물 201 아젤라산 263 아조니트릴 192 아크로레 인 136, 138 아크릴산 139 아표면 (subsu rf ace) 135 안식향산 107 안식향산 알데히드 200 안정성 187 안트라센 217 안트라퀴논 (an tr a quin one) 217 안트라퀴논공정 171 알데히드 교환 257 알릴 수소 245 알켄 에폭사이드 반응 287 알코올-케톤 94 알킬 수산화 과산화물 180 알킬붕산 에스테르 56 알킬히드로 과산화물 57, 65, 73, 179 알파(
알파(a.) - 메 틸렌 102 알파 (a) - 치환 케톤 218 알파 (a) - 테르피엔 239 알파 (a) - 토코페놀 218 알파 (a) - 히드로 과산화물 190 알파 (a) - 히드로퍼옥사이드 99 알파 (a) -A l203 124 Ensemble contr o l 117 액상 산화반응 53 양성이온 257 양성이온 중간체 263 양자성 용매 (ap ro ti c solvent ) 109 에스테르화 ll2 에틸렌 83, 116 에틸벤젠히드로퍼옥사이드 180 에폭사이드 255 에폭시 -o-퀴 놀 109 에폭시화 176, 185, 218, 287 에피클로히드린 133 EX 사 :.s 분광학 284 X 선 결정해석 215 X 선 구조 해석 82 ND법 84 NIH -shif t 111 MEK 98 MEK-과 산화물 169 여기 삼중항 상태 44 연쇄반응 276 연쇄종결 276
열분해 85 열 안정성 170, 180 열화 129 염화비닐 84 오르소 (o) - 봉산 에스테르 56 오르토 / 파라 (o rt ho/ p ara) - 치 환제 196 오조니드 256 오존 253 오존 개시 산화반응 274 오존 분해 반응 254, 262, 264, 268 오존층 253 5 환 디엔계 240 Oxeno i d 종 105 옥소늄형 178 옥 소 다 리 삼 량 체 ( 3 -oxo -b rid g ed trim er) 100 Oxone 198 옥시 지방산 에스테르 170 올레핀 수산화 188 올레핀 오존 분해반응 270 완전 산화 53 요도소벤젠 294 요소 115 용매 효과 235 유기 금속착체 83 유기용매법 144 Univ e rsal Oi l 198 유도기간 47,67 유리전자 254
UV 확산 반사분광 146 유황 과산화물 198 Yoon, Y. S. 138 은 (A g) 134 이가페놀 198 이규완 150 2 -니 트로소 시클로헥사논 95 2 량체 256 2-부 록시 래디컬 101 2 -부 틸히드로 과산화물 100 2 -부 틸 히드로퍼옥사이드 102 이산화탄소 115 이 산화탄소 개 질 (Carbon Di ox id e Re- fonning ) 116 이소프로판을 146 E. S. R 105 2-에 틸헥사노네이트 88 이온성 254 2, 6-d i-t-부 틸페놀 110 이 형 태 체 (con form er) 259 인 과산화물 202 l-부 텐 144 일산화탄소 116 1, 2 -시 클로헥산디온 96 일중항 산소 225, 235 일중항 산소의 발생법 227 1 차반응 69 임계값 68 입체 선택성 233, 286
입체 장해 259 입 체 효과 235, 257, 259 츠 자동 분해 175 자동 산화반응 16, 37, 179, 274 자장감응 105 전기원 150 전가화학적 방법 318 전기화학적 생성염기 220 전이금속 171, 176, 212, 214 전이반응 257 전이 상태 257 전이 테트록사이드 90 전자끌기 265 전자쌍 254 전자주기 265 전자 흡인기 107 전환율 97 접촉 수소화법 115 정전위 전해 218 제올라이트 114 gem -디 히드로 과산화물 189 중수-중메탄올 235 중합 개시제 169, 193 지방산 크로리드 183 Gif반웅계 297,312
Gi f 산화반응기 구 303 Gi f-Or say 반응계 306, 318 Gi f-K RICT II 반응계 329 Gif -K RICT I 반응계 322 지환식 공역 디엔 237 직쇄 알킬봉산 에스테르 56 진동 92 질량분광법 260 질산 95 질소산화물 95 * 차아할로겐산염 22 천연가스 98 천이(遷移) 229 철옥세노이드 293 철-옥소 종 290 철의 분산도(di s p ers ity) 339 철의 유출량 345 철 촉매 336 철 클러스터 298 철-폴리할로겐 페닐 포르피린 294 철-팔라듐/실리카 촉매 329 초산비닐 84 촉매 79 촉매반응 37 촉매 제조법 345
취화나트륨 (NaBr) 112 취 화암모늄 (N fti Br) 112 치환반응 107, 175 치환 페놀류 239 천전자성 반응 271 찬전자성 첨가 254 천전자체 273 친핵성 274 친핵성 공격 274 천핵체 274 =, car -3 -en e 의 반응 3l5 Calori c G mbH 118, ll9 Caros 산 198 Carba nion 221 카르보닐화 97 카르본 (Carvone) 312 카르본의 광학활성 314 카테콜 196 카프로락톤 l92 캄퍼 285 케쿨레 공명구조 265, 268 케타진 192 케토 과산화물 263 케틸 (Ke ty l) 래디컬 2l4 Koth ari, V. K. llO
Co/M n -아 세테이트 I 12 코발트, 비스(피리딜 이미노) 이소인돌 린 93 Co(salen) 109 Co(salen)py 110 Co(salpr ) 109 코발트카르복실레이트 88 코발트 포르피린 93 콜레스탄 유도체의 산화반웅 316 콩기름 에폭사이드 185 퀴논 171 Cumene 법 104 큐밀 및 디이소프로필 벤젠 산화 53 Chubb 122 크레졸 195 크롬 88 Cri eg e e 반응기구 256 Cri eg e e 양성이온 257 드 Tazuma, J. J. 110 탄산음료 115 탄산포름 알데히드 254 탄소-탄소 (C-C) 결합 절단 반응 218 탈리반응 197 탈수소 분해반응 114 탈프로톤 201
터셔리(t) - 부탄 산화 54 터셔리(t) - 부틸 과산화물 179 터셔리(t) - 부틸히드로 과산화물 187 텅스텐산 186 테레프탈산 11l 테르펜류의 반응 312 테트라브로모에탄 112 텔루륨 (Tellu ri um) 141 트리 알콕시 보록신 (tri a l koxy b oroxin e ) 56 트리옥솔란 256 Tr i-t-부 틸페놀 109 트리페닐포스핀 산화물 201, 244 티로신수소화 효소 289 티오술피네이트 201 TPA 111 고: 파라(p) -c omp le x 84 파라(p) - 퀴놀 111 파라(p) - 크실렌 113 파라(p) - 톨룰산 113 파라(p) - 톨룰에스테르 113 팔라듐촉매 335 퍼에폭사이드 244 Peroxo 종 109 pero x y 래 디 컬 47
퍼옥시보레이트 87 퍼옥시수산기 이온 176 퍼옥시에스테르 169 퍼옥시지방산 170 퍼옥시페놀레이트 109 페놀 86 펜톤 (Fen t on) 시약 105, 175, 193 평형 80 평형 조성 182 포르피린계 292 포르피린 구조 282 포름산 93, 103 포름알데히드 103, 136 포타시움 -니 켈 K -Ni -C aOx/ ZSM 124, 127 폴리비닐 아세테이트 97 폴리비닐 알코올 97 풀리 수산화 화합물 196 풀리에스테르 ll1, 141 표면 산화 철종 344 표면임계상수 68 표면적 134 프로모터 (Promote r ) 134 프로톤화 178 프로피온산 98 프리델-크라프트 알킬반응 196 프탈로시아닌 93 Flis z ar 반응기구 261 피나콜 244
피독 129 피리딘 핵 268 Fil s cher-Trops c h 116 s Harber -Weis s cyc l e 99. 175 함산소화합물 119 합성가스 119 합성 화학( 合 成化學) 237 항산화제 (anti ox id a nts ) 47 해리상수 172 헤스 (Hess) 의 법칙 37 해테로 원자 과산화물 198 헤 테 로탄화수소 (Hete r ohy dr ocarbons ) 35 헴 (heme) 단백질 281 현탁액 (slurr y) 112 Hei n ic k e, D. 150 호프만 분해 23 홍석인 146 화학 선택성 300 화학 에너지 전송 시스템 120 화학 형광체 91 환상 알켄 235 환상펜타아민 105 환원성 216 환원 온도 92
환원제 107, 128, 213, 216 활성점 344 활성화 깁스 에너지 79 활성화 에너지 90 Fuji mo to , K. 128 홉열반응 116, 120, 253 히드라진 니트릴 193 히드로 과산화물 85, 170 히 드로시 클로헥 사디 에 닐 (Hy d roxy - cyc l ohexadie n y l) 106 히드로옥소늄 이온 195 히드로퀴논 171, 196 히드로퍼옥사이드 86, 178, 276 히드로퍼옥시 음이온 212 히드록시 래디컬 101 히드록시 카프로익산 91 히드록시화 195
이규완 전북대학교 화학과 졸업 연세대학교 대학원 화학과 졸업 (MS) 일본 도쿄 공대 자원화학연구소 연구원 독일 Eng le r-Bunte Insti tut , Karlsruhe Univ . (D r. E ng .) 미국 텍사스 A&M, Associa t e Researcher(Prof. D. H.R. Bar t on) 로 연구 현재 한국화학연구소 화학기술연구단 제 1 팀장 국내외에 약 60 여편의 논문 발표 탄화수소의 산화반응 대우학술총서 자연과학 123 1 판 1 쇄 찍음 1998 년 7 월 20 일 1 판 1 쇄 펴냄 1998 년 7 월 30 일 지은이 이규완 펴낸이 朴孟浩 펴낸곳 (주)민음사 출판등록 1966. 5. 19. 제 16-490 호 서 울시 강남구 신사동 506 대표전화 515-2000 팩시밀리 515-2007 © 이규완, 1998. Print e d in Seoul, Korea. 유기화학 산화 및 환원 탄화수소 KDC I 437.3 4 4 값 22,000 원 ISBN 89-374-3623-X (94430) 89-374-2000-2 세 트
대우학술총서 사인과야 1 1 1 소립자와 게이지 상호작용 김진의 43 후리에 해석과 의미분 작용소 86 등각장론 임채호 2 동력학특론 이병호 김도한 87 방사선생물학 남상열 3 질소고정 송승달 44 한국의 고생물 이하영 88 석유지질학 이용일 4 상전이와 임계현상 김두철 45 질량분석학 김명수 89 베르누이 시행의 통계적 분석 5 촉매작용 진종식 46 급변론 박대현 배도선 김성인 6 뫼스바우어 분광학 욱항남 47 생체에너지 주충노 90 신경세포생리학 강만식 7 극미량원소의 영양 승정자 48 리이만 기하학 박을룡 91 생리활성을 가진 C ― P 화합물의 8 수소화붕소와 유기봉소 49 군표현론 박승안 화학 김용준 강익중 화합물 윤능민 50 비선형 편미분 방정식론 하기식 92 생물유기화학 서정헌 9 항생물질의 전합성 강석구 51 생체막 김형만 93 조직배양 김승업 10 국소적 형태의 At1 y ah - si n g e r 52 수리분류학 고철환 94 유기전이금속화합물 조남숙 외 지표이론 지동표 53 찰스 다윈 정용재 95 실내한경 과학 김윤신 11 Mucop ol y sacchar i des 의 54 금속부식 박용수 96 유한요소법 정상권 생화학 및 생물리학 박준우 55 양자광학 이상수 97 대수적 위상수학 우무하 김재룡 12 천체물리학 홍승수 56 효소반응 속도론 서정현 98 파인만 적분론 장건수 13 프로스타글라딘 합성 김성각 57 화성암 성인론 아민성 99 응용 미생물학 박무영 14 천연물화학연구법 우원식 58 확률론 구자홍 100 리보플라빈 이상선 15 지방영양 김숙희 59 분자분광학 소현수 101 노화 김숙희 김화영 16 결정화유리 김병호 60 벡터속 이론 양재현 102 매트릭스 격리분광학 정기호 17 고분자에 의한 화학반응 조의환 61 곤충신 경 생리학 부경생 103 신경계 조직배양 김승업 18 과학혁명 김영식 62 에너지띠 이론 모혜정 104 지구화학 김규한 19 한국지질론 장기홍 63 수학기초론 김상문 105 은하계의 형성과 화학적 진화
