 Sol id Prope llant
Sol id Prope llant 노만균 고려대학교 화학공학과를 졸업하고 동대학원에서 공학박사 학위를 받았다. 국방과학연구소 추진제 실장 , 추진기 관 부장 및 로켓 추진제 공장장을 역임하였다 미국(미해군 지상무기연구소, Aeroje t Co., Univ . o f Mass.) 과 프랑스 (S . N.P.E. Co.) 에서 추진제에 관한 연구를 하였다. 현재 국방과학연구소 책임연구원으로 재직중이다.
고체 추전제
Sol id Prope llant
 고체 추전제
고체 추전제
책머리에 주로 군사용으로 세계 각국에서 널리 사용되고 있는 추진제는 최근들어 우주개발을 위한 우주발사체 로켓의 연료로 사용되면서 민수산업에서의 중요성이 새롭게 부각되는 추세에 있다. 또한 추 진제의 일종인 자동차용 안전백 (에어백) 가스발생기를 비롯한 각 종 민수용 추진제의 종류도 다양화되고 있고 수요도 확장되고 있 다. 이와 같이 추전제가 특수장치들의 부품으로 사용될 수 있는 것은 추전제가 짧은 시간 내에 가스 상태로서 큰 힘을 발휘할 수 있는 특이한 성질 때문이다. 미국을 위시한 선진국들에서는 추진제 기술에 관련된 연구 인 력이 대학교, 기업체, 연구소 등에 폭넓게 분포되어 있다. 우리 나라의 경우는 선전국들에 비하면 연구 인력도 일부 연구소에 편 중되어 있고, 기술수준도 매우 뒤떨어전 상태에 있다. 그러나 우 리나라에서도 1970 년대를 전후로 니트로셀룰로오스 계열 추전제 기술이 도입되어 활용되고 있고, 혼합형 추진제를 포함한 각종 추진제 연구도 상당한 수준으로 진척되고 있다. 1994 년도에는 추 전제를 포함한 추전기관 기술에 관한 추진기관 공학회가 창설되 어 추전제 기술 발전을 위한 산· 학· 연 간의 공개적인 기술교류 가 활발해침에 따라 연구내용의 깊이도 더 충실해지고 있다.
현재 우리나라는 국가 경제력이 크게 성장하고 있어 선전국으 로의 진입을 목전에 두고 있다. 따라서 선진국들이 주도하고 있 는 우주개발과 관련된 우주산업 분야, 죽 과학 통신위성 및 위성 발사체 기술의 개발에도 국가에서 많은 관심을 기울여야 마땅하 며, 실제로 단계적인 두자가 진행하고 있는 것으로 알고 있다. 이와 관련하여 발사체 개발과 관련된 주요 핵심 기술 중의 하나 인 추전제 기술에 대해서도 많은 연구와 두자가 필요하다고 생각 된다. 선전국과는 달리 국내에는 추전제 기술 분야에 종사하는 인력 도 많지 않고, 추전제 기술과 관련되어 발간된 책자도 거의 없는 형편이다. 그러나 추전제 기술 분야의 범위는 매우 넓어 추진제 의 종류만 해도 고체 추진제와 액체 추전제로 대별되며, 본서에 서 주로 다룰 고체 추전제와 직접 관련된 기술 분야만도 니트로 셀룰로오스 계열 추전제, 혼합형 추진제, 내탄도(그레인) 설계, 점화 및 착화 현상, 연소 메커니즘, 배출가스(p lume) 분석, 연소 관 내열재 기술, 물리적 및 기계적 성질, 고에너지 물질 등이 있 다. 이 모든 기술 분야를 한 권의 책에 다룰 수는 없으므로 본서에 서는 우선 고체 추진제 중 총 • 화포용으로 광범위하게 사용되는 니트로셀룰로오스 계열 추전제(제 2 장), 로켓용 추전제로 주로 사 용되는 혼합형 추전제(제 3 장)와 점차 추진제 원료로서 사용 범위 가 넓어지고 있는 RDX 와 HMX 를 중심으로 니트라민계 원료(제 4 장) 및 최근 활발히 연구가 전행중인 차세대 추전제 기술(제 5 장) 을 포함시켰다. 특히 개발 역사가 오래되고 자료가 방대한 니트 로셀룰로오스 계열 추진제 기술과 니트라민계 원료에 관해서는 Urbansk i가 저 술한 Chemi st r y and Technolog y of Ex p los i ve” 의 영 문 판을 많이 참조하였다.
부족한 점은 많이 있지만 고체 추전제 분야의 전문 서적을 국 내에서 처음으로 집필했다는 점이 매우 기쁘다. 이 책의 원고 작 성시 많은 도움을 준 홍명표 박사, 임유전 박사, 이범재 박사를 위시한 동료 연구원들과 권은경 양에게 감사를 표하고 싶다. 또 한 원고 작성 전후 항상 도움말을 주신 김용준 선생님과 이 책의 집필을 배려해 주신 대우재단에 깊이 감사드린다. 1998 년 2 월 노만균
차례
책머리에•5제1장 서론---------------11참고문헌•17제2장 니트로셀룰로오스 계열 추진제-------------192.1 NC 계열 추진제의 개발•212.2 물리적 성질•262.3 연소, 폭발 특성•282.4 섬광 발생 및 섬광 억제•442.5 스모크•512.6 침식•512.7 추진제의 안정성•542.8 로켓용 복기 추진제의 물리적 및 서베일런스 특성•792.9 NC 계열 추진제의 제작•92참고문헌•116제3장 혼합형 추전제-----------------123
3.1 추진제 그레인•1263.2 추진제의 에너지 및 연소 속도•1293.3 혼합형 추진제의 성분과 역할•1313.4 추진제 제조•1783.5 추진제의 기계적 성질•1883.6 추진제의 연소 및 내탄도 특성•193참고문헌•208제4장 니트라민계 원료--------------2154.1 니트라민의 화학적 성질•2164.2 니트라민의 합성•2264.3 RDX•2344.4 HMX•292참고문헌•296제5장 차세대 추진제와 고에너지 물질 3035.1 무연 및 스카벤저 추진제•3055.2 둔감 추진제•3125.3 고에너지 고밀도 물질•317참고문헌•331찾아보기/335제 1 장 서론 통상 넓은 의미에서 폭발물이라고 부르는 물질은 폭발할 수 있 는 물질, 죽 짧은 시간 안에 수백, 수천 배 이상으로 급격한 부 피 팽창을 일으킬 수 있는 물질들을 말하며 그 속에는 추전제, 화약류, 폭발물(엄격한 의미에서의 폭발물) 및 이들을 원료나 원 재료로 하여 제조된 무기, 물건 등이 모두 포함된다. 그 중 화약 은 오래 전부터 사용되어 온 단어로서 흑색화약과 같이 불을 내 며 빠른 속도로 연소하는 물질들을 지칭했으나, 현재는 화약류에 속하는 물질들을 제조하는 목적에 따라 추진제로서 혹은 폭발물 로서도 사용될 수 있으므로 추진제와 폭발물을 모두 포함하여 넓 은 의미로 화약이라고 부르기도 한다. 추전제와 폭발물은 적당한 조건하에 점화(또는 폭굉, 기폭)되면 매우 짧은 시간 안에 많은 양의 뜨거운 가스를 발생시킬 수 있는 고에너지 물질들이다. 추진제와 폭발물의 차이점은 그 작동시간 의 길이에 따라 구분될 수 있는데 추전제는 초당 연소되는 속도 가 최고 10 여 cm 이하의 연소 속도 범위에 있는 반면, 폭발물은
초당 수 km 에 이르는 폭굉속도를 갖고 있다. 다시 말하면 추전 제는 연소됨으로써, 폭발물은 폭굉됨으로써 그 성능이 발휘된다 고 볼 수 있다. 추진제나 폭발물이 발생하는 에너지는 그 반응 물질의 열화학적 특성에 따라 차이가 많겠지만 일반적으로는 그 램당 1000cc 정도의 가스와 1000 kcal 수준의 높은 에너지를 발 생시킨다. 이러한 고에너지 물질들은 의부로부터 산소의 공급 없 이 반응이 진행되므로 의부와 완전히 고립된 밀폐용기 속에서 혹 은 물 속에서도 반응이 일어날 수 있다. 이 물질들은 비교적 많 은 양의 에너지를 좁은 용기 속에 쉽게 저장하여 운반할 수 있으 므로 군사적으로 또는 일반 산업적으로 여러 분야에서 이용될 수 있다. 그 중 추진제는 로켓이나 유도탄을 발사시킬 때, 총이나 화포로부터 탄환이나 포탄을 발사시킬 때 또는 필요한 가스(유 체)를 공급해 주는 장치에 흔히 사용되고 있다. 추전제가 적용되 는 또 다른 분야는 터빈을 작동시킬 때, 피스톤을 움직일 때, 항 공기로부터 긴급사태 발생시 조종사를 탈출(방출)시킬 때, 볼트 나 철사 등을 절단할 때, 로켓의 방향조종 날개를 절단할 때, 로 켓의 방향조종 날개를 작동시킬 때, 항공기 엔진을 시동할 때 등 이 있다. 추전제는 상기와 같이 비교적 짧은 시간 내에 큰 힘을 필요로 하는 장치들에 사용된다. 따라서 추전제의 가장 큰 적용 분야는 군사적인 분야이지만 추진제의 안정성과 적용시의 간편함 으로 인해 일반 산업계에도 그 수요가 점차 증가하는 추세에 있 다 .1,2) 추진제는 오래 전부터 총, 화포, 곡사포, 박격포 및 각종 재래 식 무기에 적용되고 있는데, 이와 같은 무기에 적용되는 추전제 들은 흔히 주요 성분(원료)으로 가소화된 니트로셀룰로오스 (NC) 를 포함하고 있다. 추진제에는 NC 이의에도 사용하는 목적에 따라 여러 가지 다른 종류의 화학물질들을 첨가한다. 죽, 성능을
증가시키기 위해서는 니트로글리세린 (NG) 과 같은 고에너지가소 제를 첨가하며, 추진제의 탄도적 특성을 조절하기 위해서는 니트 로구아니던과 같은 유기물질을 첨가하기도 하고, 추진제의 물리 적 특성이나 제조상의 특성을 개선하기 위하여 프탈산디부틸과 같은 가소제를 첨가하며, 추진제의 화학적 안정성을 증가시키기 위하여는 디페닐아민과 같은 질소 화합물을 안정제로 첨가시키 며, 점화성질이나 취급시의 안정성을 개선하기 위해서는 흑연, 황산칼리, 질산칼리 등의 분말을 첨가하기도 한다. 이와 같은 NC 를 주성분으로 하는 추진제 중 산화제로서 NC 만을 포함하는 추전제를 단기 (sin g le base) 추진제라고 부르며, NC 와 NG 를 포 함하는 추진제롤 복기 (double base) 추전제, 니트로구아니딘과 같은 니 트로 유기 화합물이 더 첨 가되 면 삼중기 (trip le base) 추전 제라고 부른다. 이러한 단기, 복기, 삼중기 추진제는 매우 소량 의 고체 상태 첨가제가 특수 목적으로 첨가되기도 하지만 대부분 그 성분이 균일한 혼합물 상태이므로 균일 또는 균질 추전제 (homog e neous pro p e llant) 라고도 분류한다. 또한 그 주요 성 분 이 NC 이므로 NC 계열 추진제, 그 적용 분야가 주로 총 • 화포 의 장약이므로 총 • 포 추진제 또는 연소시 연소가스가 무연 상태 에 가까우므로 무연화약 (smokeless p owder) 으로 분류되기도 한 다 .1,3,4) 이상과 같은 NC 계열 추전제가 실제로 적용되는 화포의 구성이 그림 1. 1 에 포신 (barrel), 포탄(p ro j ec ti le), 기폭제 (pr im er), 추전제 장약(p ro p ellan t bed) 과 함께 나타나 있다. 또 한 대표적인 총·화포의 튜브 설계도와 추전제가 연소하는 chamber 의 상세 설계가 그림 1. 2 에 있다. 5) 한편 로켓 추진기관용 고체 추진제로서는 복기 추진제와 혼합 형 추진제가 사용되고 있다. 혼합형 추전제가 개발되기 이전부터 로켓에 사용된 복기 추전제는 혼합형 추진제에 비해 각종 성능이
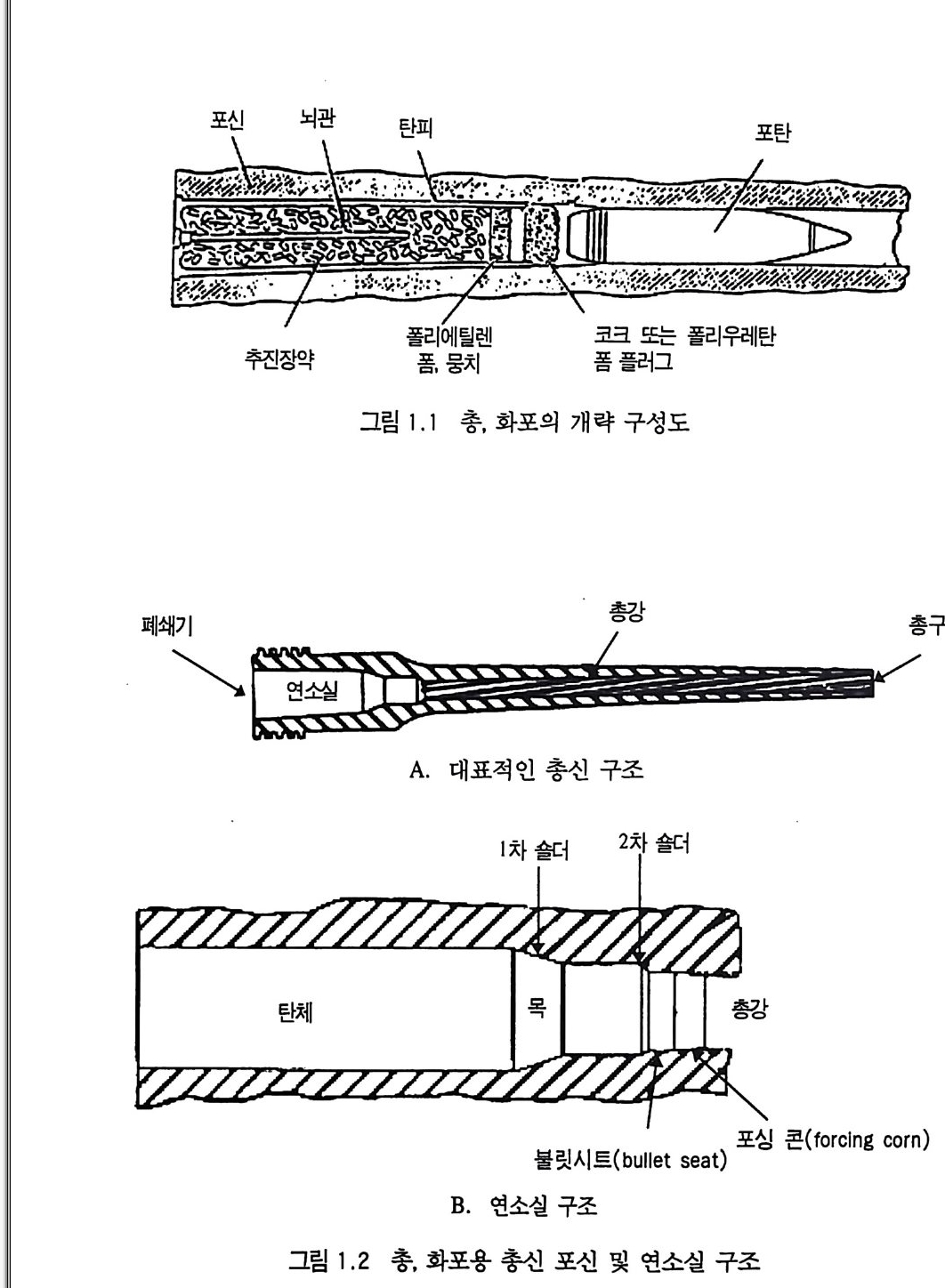 포신 뇌관 탄피 포탄
포신 뇌관 탄피 포탄
떨어지므로 점차 로켓 추진제로서의 사용량이 감소되는 추세에 있다. 그러나 최근에는 알루미늄, 과염소산암모늄 등의 고체 연 료 및 고체 산화제를 복기 추진제에 첨가시켜 각종 성능을 크게 향상시킨 추진제를 개발하여 적용하고 있는데, 이러한 추전제를 혼합형 개 선 복기 추전제 라고 부른다. 2,3,4 ) 혼합형 추진제는 고분자를 바인더 메트릭스 및 연료로 사용하고 고체 산화제와 금속 연료들을 혼합하여 제작한다. 이와 같이 산 화제와 금속 분말을 충전시킨 바인더 메트릭스상, 즉 고무상의 구조를 지닌 불균일 추전제인 혼합형 추진제는 1950 년대 이후 로 켓 추진제로 사용할 목적으로 개발되기 시작했으며, 초기의 PS (p ol y sul fi de) 계, PVC, 폴리우레탄계 등의 바인더를 거쳐 최근 에는 폴리부타디엔계 추진제가 개발되어 로켓 추진제 그레인 설 계시 필요로 하는 각종 성능이 크게 향상되고 있다 .5,6,7) 유도탄, 무유도 로켓, 위성 발사체용 추전기관에 적용되는 추 전제가 그레인 형상으로 추진기관 모터에 충전된 대표적인 로켓
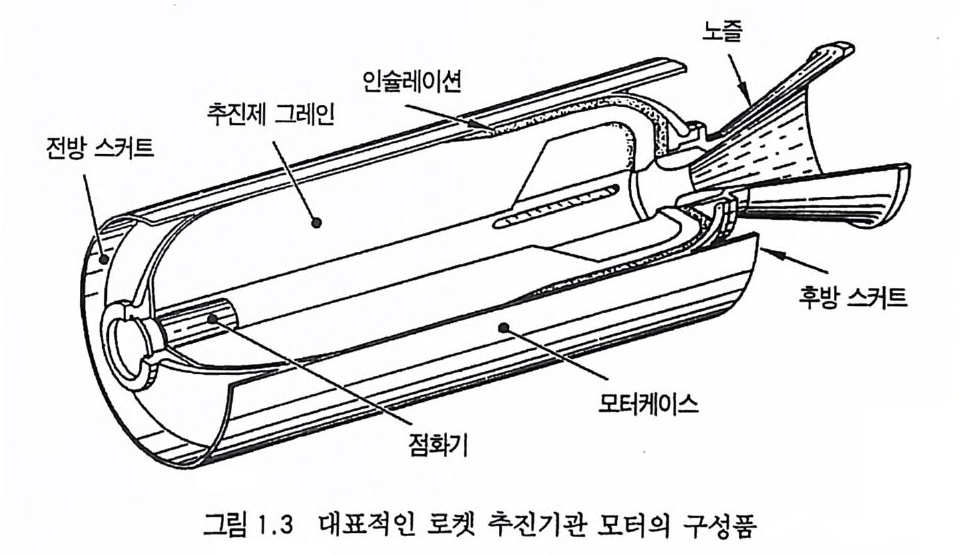 인
인
구성품이 그림 1. 3 과 같이 인슐레이션, 점화기, 모터케이스 및 노줄 등과 함께 나타나 있다. 8) 최근 급격히 발전한 고분자 및 정밀 화학 기술에 힘입어 혼합 형 추진제는 그 물리적 성질, 기계적 성질, 내탄도적 성질 등 각 종 성능 및 제작 공정성이 개선되었다. 이와 같이 각종 성능 및 제작성이 우수해짐에 따라 대형 추진기관용 추전제 제작이 가능 해지고 대륙간 탄도탄과 같은 군사용 추전제로서, 위성 발사체 추진제와 갇은 민수용 추전제로서 혼합형 추진제의 수요는 점차 증가하게 되었다. 그러나 추전제의 성능, 제작성 등의 기술이 크 게 발전하였음에도 1) 추전제 연소시 배출되는 공해성 연소가스 로 인한 심각한 환경오염, 2) 추진제 연소시 배출되는 유연가스
 그림 1.4 미국의 챌린저호 야간발사 장면 그림 1.5 소련의 A2 발사체 발사 장면
그림 1.4 미국의 챌린저호 야간발사 장면 그림 1.5 소련의 A2 발사체 발사 장면
로 인한 송수신 교란 및 위치 노출, 3) 고의 또는 우연한 충격이 나 화재로 인한 대형사고, 4) 더 높은 비추력을 갖는 고성능 추 진제의 필요성 등과 같은 문제점들이 남아 있다 .9,10) 그립 1. 4 는 미국의 챌린저호 발사 장면이며, 그림 1. 5 는 소련의 A2 발사체 발사 장면으로서 발사시 엄청난 양의 유연 배기가스를 배출하고 있음을 알 수 있다. 11) 앞에서 언급한 여러 가지 문제점들이 해결된 추전제는 연소시 환경을 오염시키지 않고, 깨끗한 무연의 가스를 배출하며, 비추 력은 기존의 혼합형 추전제와 동등 또는 그 이상으로 높아야 하 며, 그 추전제가 충전된 로켓이 우연한 사고(충격, 화재 등)를 당 했을 때 폭발하지 않고 안전하게 소화시킬 수 있을 정도로 안전 (둔감)한 추진제여야 한다. 이와 같은 추진제를 제작한다는 것은 현재로서는 불가능한 것같이 생각되지만 새로운 종류의 고밀도 고에너지 물질들이 개발되고 있고 추진기관 모터 설계, 제작, 연 소이론 등이 발전하고 있으므로 가까운 시일 내에 이상형에 가까 운 추전제가 등장할 수 있으리라 추측된다 .3,12) [참고문헌〕 1) T. Urbanski, Chemi stry and Technolog y of Ex plo siv e s, Vol. m, Eng lish Ed., Perga mon Press, New York, 1985. 2) V. Lind ner, Expl o siv e s-P r op el lants , Theory and Practi ce, U.S. Arma-ment Command, Dover, N.J., 1978. 3) A. Davenas, Solid Rocket Prop ul sio n Technology , Perga mon Press, New York, 1993. 4) Y. M. Ti m nat, Advanced Chemi ca l Rocket Prop ul s ion , Academi c Press Inc., 1987. 5) W. H. Drys d ale and B. P. Burns, Str u ctu r al D 函':g n of Proje c ti les ,
Prog res s in Astr o nauti cs and Aeronauti cs, Vol. 109, Chap e r 6, 1988. 6) R. F. Gould, Advances in Chemi st r y Serie s , Prop e llants Manufa c - tur e, Hazards, and Testi ng , Amer. Chem. Soc., 1969. 7) A. E. Oberth , CPIA Publica ti on No. 469, Pr inc ip le s of Solid Prop el lant Develop m ent, 1987. 8) MATRA Defe n se DAST/PC 28/no 1917, Rocket Moto r Techno- log y, MATRA Defe n se Comp a ny, 1993. 9) A. Yamamot o, ]. Industr i a l Exp lo siv e s Soc., Jap an , 1 (1) , 47 (1991) . 10) H. J. Pasman, ]. Industr ial Ex plo siv e s Soc., jap a n , 2 (2) , 94 (1992) . 11) D. Hobbs, Spa c e War fare (Chape r 111) , In Advanced Technology War fare , Harmony Books Inc., New York, 1985. 12) D. H. Lie b enberg, R. W. Armstr o ng , and J.J. Gi lm an, Str u ctu r e and Prop e rt ies of Energe t i c Mate r i als , MRL Sy mpo siu m Proceed- ing s, Vol, 296. 1992.
제 2 장 니트로셀룰로오스 계열 추전제 19 세기 중반 새로이 개발된 니트로 및 질산 유도체의 고에너지 물질들은 그 이전에 사용되던 흑색화약울 대신하여 총 • 화포용 추진제로 광범위하게 사용되기 시작하였다. 이때 등장한 물질 중 가장 중요한 물질이 니트로셀룰로오스 (NC) 이다. 그러나 NC 를 추진제로 사용하려고 시도했던 초창기에는 많은 기술적인 문제점 이 나타났으며, 그 중 특히 심각한 문제점은 NC 의 불안정성이 었다. 초기에는 NC 를 제조하는 공정 중 니트로화 공정이 끝난 후 셀룰로오스 내에 잔류하는 미량의 질산과 황산, 황산셀룰로오 스 및 분해 생성물들에 의해 NC 의 불안정성이 나타나는 것으로 해석되었다. 그러나 NC 의 정제공정을 크게 개선하여 불순물들 울 대폭 감소시킨 후에도 NC 는 보관 도중 느린 속도로 계속 분 해되어 질소산화물을 방출하고, 이 질소산화물들은 NC 의 분해 반응을 촉진시켜 결과적으로는 NC 의 분해속도가 크게 증가되는 현상이 나타났다. 이러한 현상을 충분히 이해하지 못했던 1900~1910 년 사이에
NC 를 저장하거나 사용중이던 세계 각국의 군용 선박, 화약 저 장고 등에서 많은 대형의 폭발사고가 발생하여 수많은 재산과 인 명피해가 있었다. 이때 Nobel 은 NC 추전제에 디페닐아민 (dip h eny la mi ne ) 과 같은 약알칼리 성 유기 화합물을 첨 가하여 추진 제의 저장 도중 발생한 질소 화합물과 결합 반응시켜 추진제를 안정화시키는 방법을 제시하였다. 이 방법이 추전제, 화학류에 안정제를 사용하게 된 효시가 되었으며, 그후 디페닐아민 이의의 많은 화합물들이 추진제, 화약을 안정화시킬 목적으로 검토되고 실험적으로 확인되었다. 1912 년에는 에틸센트럴라이트 (e t h y l centr a li te) , 1950 년대에는 2- 니트로디페닐아민이 안정제로 새로 이 개발되었다. 이와 같이 안정제를 사용함으로써 NC 추진제의 불안정성 문제는 해결되어 NC 추진제의 수명은 대략 25 년 이상 으로 증가하게 되었다. NC 는 그 섬유 성분으로 인해 다공성이고 밀도가 낮은 결점이 있는데, 이를 개선하여 기공을 없애고 밀도를 증가시키는 방법이 19 세기 말 Nobel, Vi ei l le, Abel 등에 의해 개발되었다. 즉, NC 롤 가소화시키거나 젤라틴화하여 그 섬유 성분을 제거시킨 후 사 출하거나 롤링 공정을 거치게 하면 밀도가 증가된 얇은 판이나 국수가락 형상으로 만들 수 있다. 이와 같이 NC를 용매로 처리 하여 가소화시킨 후 롤링, 사출하는 공정은 총 • 포 및 로켓용 추 전제를 제작하는 현재의 NC 추진제 제작공정에도 이용되고 있 다. NC 계 추전제에 대한 또 다른 획기적인 기술 발전은 볼 파 우더 (Ball po wder) 의 개 발이 었다. Olsen, Tib b ets , Kerone 등에 의해 개발된 볼 파우더 제작공정은 NC 를 물-초산에틸 관계처럼 서로 섞이지 않는 혼합 용매에 용해시켜 섬유구조를 완전히 제거 한 후 생 성 된 래 커 (lac qu er) 를 증류 및 그레 이 닝 (grai n i n g ) 공정 에 의해 구형 내지는 타원형의 작은 알맹이로 만드는 것으로서
소구경 탄환용 추진제 제작에 사용되고 있다. 일반적으로 사용되는 NC 추진제는 그 사용 목적에 따라 조성 이 크게 달라질 수 있으며 NC 이의에 에너지성 가소제, 안정 제, 섬광 감소제 동 여러 종류의 첨가제들이 사용되고 있다. 한편 제 2 차 세계대전 이후 크게 발달한 각종 전술 • 전략 유도 탄, 대륙간 탄도탄 및 위성 발사체 등에 요구되는 추진기관의 성 능이 매우 대형화되고 각종 성질이 크게 개선된 고성능의 추진제 를 요구하게 됨에 따라 추전제 그레인의 직경과 길이도 크게 증 가되고 있고, 그 형상도 복잡한 모양으로 설계되고 있다. 따라서 1940~1955 년대 이전에 주종을 이루던 NC 계 추전제 성능으로는 상기와 같은 추진기관에서 요구하는 성능 및 제작 능력에 어려운 문제점들이 발생하여 불균일계 혼합형 추진제의 개발이 새로운 고분자 재료의 등장과 함께 신속히 이루어지게 되었다. 그러나 현재 혼합형 추진제 산화제로 주로 사용되는 과염소산암모늄은 연소될 때 유색의 유독성 가스를 방출하여 전술적 운용과 대기오 염에 문제점이 있으므로 무색의 배기가스를 요구하는 유도탄이나 로켓 및 혼합형 추진제로 적용이 불가능한 총 • 포용 화약, 장약 등에는 균일계 추진제인 NC 추전제가 현재에도 범용적으로 사 용되고 있다. 최근에는 NC 추전제의 특성 (장점)들을 이용하고 내탄도 성질을 크게 증가시키려는 노력도 활발히 진행되어 CMDB 와 감은 새로운 추전제가 개발되어 적용중인 것으로 알려 져 있다 .1) 2.1 NC 계열 추진제의 개발 셀룰로오스를 질산으로 처리하면 연소성 물질로 변한다는 사실
울 발견한 후 이 물질을 흑색화약 대신 추진제로 사용하려는 시 도들이 있었다. 그 중 Shonbe i n2) 과 Pelouze3) 는 1846 년 니트로 셀룰로오스 (NC) 의 에너지를 측정하여 탄 효과를 실험한 결과 흑색화약보다 3 배 정도로 강력한 성능을 나타내는 현상을 발견하 였다. 그러나 NC 의 이러한 우수한 성능을 알면서도 NC 를 화 약·추진제로 적용하기까지에는 많은 시일이 소요되었다. 1896 년 Lenk4) 에 의해 NC 의 대형 제작 공정아 처음으로 개발되어 오스 트리아 군대에서 NC 를 화포용 추전제(장약)로 사용하려고 시도 하였으나 이때 제작된 NC 추진제의 품질균일성 문제 및 발사시 의 과도한 압력 발생으로 인하여 포가 크게 손상되는 문제점들이 나타났다. 그후 밀폐된 공간에서는 NC 가 흑색화약보다 훨씬 빨리 연소 된다는 사실이 발견되었다. 또한 NC 를 압축시켜 밀도를 증가시 킴으로써 연소속도를 떨어뜨리기 위한 물리적인 방법만으로는 연 소속도 감소 효과가 별로 나타나지 않으므로 왁스나 지방 같은 연소 방해제를 NC 에 가하는 방법들이 시도되었다. 이러한 일련 의 실험 중 Schul t z 는 부분적으로 매우 성공적인 화약을 얻었다 .5) 죽, 목재를 1~2mm 크기로 잘게 부순 그레인으로 만들어 가성 소다 용액과 함께 끓인 후 차아염소산칼슘으로 표백시키고, 혼산 (질산+황산)으로 니트로화시켰다. 이 니트로화물을 탄산나트륨 용액과 함께 가열하여 안정화시킨 다음 건조시킨 그레인울 질산 칼륨 및 질산바륨 용액으로 처리한 후 다시 건조시켜서 파라핀 왁스로 처리하여 다음과 같은 조성의 화약을 얻었다. 니트로셀룰로오스 (NC) 성분 50 % 목재 (펄프) 성분 13 % 질산칼륨 및 질산바륨 33 %
파라핀 4% 그러나 이 화약은 군용 소총 화약으로 사용하기에는 너무 빨리 연소하여 군용으로는 사용이 불가능했으나, 단구경 (shot gun ) 소 총용 화약으로는 적합했으므로 스포츠 경기용 화약으로 영국 등 에서는 Shul t z 형 화약이라는 이름으로 많이 이용되었다. 그후 NC 를 아세톤, 초산에틸, 알코올 또는 에테르 등의 유기 용매에 용해시킨 후 용매를 건조시키고 남는 두명한 고밀도 필름 형태인 NC 는 원래의 NC 보다 훨씬 천천히 연소된다는 성질이 발견되었다 .6) 용매에 대한 수많은 연구가 NC 연소특성 개선을 위해 수행되어 많은 특허들이 얻어졌지만 실제로 추전제 화약에 적용하기에는 대부분 쓸모없는 결과들이었다. 단 Dutt en hofe r 7) 에 의해 개발된 , 약간 카본화된 셀룰로오스를 니트로화시키고 안 정화시킨 다음 젤라틴화될 때까지 초산에틸로 처리하고 건조시켜 얻은 NC 는 RCP(Rott we il C ellulose Powder) 라는 이름으로 한동 안 사용되었다. 그러나 RCP 는 그레인 모양이 불균일하여 연소 상태가 균일하지 못한 단점이 있었다• 1879 년 V i e ill e 는 8) 봄베 (manometr i c bomb) 속에 서 화약류의 연소에 대한 조직적인 연구를 시작하였으며, 흑색화약의 비중이 1.80 이상이 되면 화약이 평행충에 따라 연소한다는 사실을 발견 하였다. V i e ill e 는 NC 밀도를 변화시키면서 같은 종류의 실험을 여러 번 하였으나 NC 는 아무리 압력을 가하여도 그 비중이 증 가하지 않았으며, 오히려 각종의 용매를 변화시켜 처리할 때 높 은 비중의 NC 를 얻을 수 있었다. 그는 이와 같이 얻어진 화약 울 압력봄베를 이용한 연소 실험에 의해 평행충으로 연소한다는 것과 또한 연소시간이 추전제 화약 두께에 따라 결정된다는 것을 확인하였다. 따라서 추전제 그레인의 전체 연소시간은 두께를 변
화시킴으로써 조절할 수 있고, 추진제 화약의 비바시티 (viv a c ity) 상수 (coeff icien t of the viv a c ity) 는 틀 )ma x 으로서 봄베 (manometr i c ) 내에서 결정될 수 있다(p는 봄베압력, t 는 연소시 간) . 이 같은 개 념 에 의 해 충분한 v i vac ity를 갖도록 추전제 장약을 포에 사용함으로써 화포의 표준화 (sta n dardiz a - ti on) 를 이룰 수 있었다. Vi ei l le 화약은 프랑스로 도입되어 B 화 약이라는 이름으로 알려지게 되었다. V i e ill e 는 이 화약을 제작 할 때 두 종류의 NC 를 사용하였다. 그 하나는 콜로이드상의 면 (cott on ) CP2 로서 에 데르, 알코올 혼합액 에 용해 시 킨 화약 반죽 이며, 또 하나는 에테르, 알코올에 불용인 CPI 면으로서 이를 CP2 화약에 섬유상으로 침투시켜 만들었다. 러시아에서는 Mendele y ev 가 파이 로셀룰로오스 (pyro cellulose) , 죽 에 테 르와 알 코올 혼합액에 용해되는 비교적 니트로 농도가 높은 (12 . 5 %N) 셀룰로오스로부터 무연화약이 제작되어 1892 년경 러시아 해군에 서 사용하였다. 이러한 형식의 니트로셀룰로오스는 미국에서도 군용으로 채 택 되 어 단기 화약 (sin g le base po wder) 또는 단기 추진 제라는 이름으로 사용되기 시작했다 .I) 두번째 형태의 NC 추진제 (화약)는 1888 년 Alfr ed Nobel 에 의 해 발명된 것으로서 Nobel 은 니트로글리세린이 니트로셀룰로오 스를 녹이는 성질을 이용하였다 .9) 죽, 이미 언급한 바와 같이 휘발성이며 비폭발성 용매인 에데르나 알코올을 사용하는 대신 비휘발성(휘발성이 낮은) 니트로글리세린을 사용하였다. NC 대 NG( 니트로글리세린)의 비율은 45 : 55 정도였는데 NG 의 양이 적 어서 NC 를 완전히 용해시키기 어려운 점이 있었다. Abel 과
Dewar10) 는 NC 와 NG 양쪽 성분에 작용하는 아세톤 용매를 적 용하여 이 문제를 해결하였다. 그러나 이렇게 제조된 Briti sh Cord it e 라고 불린 제품은 영국연방 국가들 이의에서는 거의 사용 되지 않았다. 알코올, 에테르 등의 휘발성 용매를 사용했던 방법 은 일시적으로는 성공하였으나 제조상의 문제점으로 인해 점차적 으로 용매 없이 제조하는 방법이 더 큰 관심을 끌기 시작하였다. 휘발성 용매를 사용하지 않는 NG 화약 제조의 첫째 목표는 어 떻게 하면 NG의 양을 가능한 한 줄일 수 있느냐 하는 점에 있 었다. 적당한 형태의 NC 를 선택하고 비휘발성 용매를 가하여 RP-12 (RPC-12) 라고 불리 는 새 로운 형 태 의 NG 화약이 1912 년 제작되었다 .11) 이 화약은 1 차 세계대전 때 광범위하게 사용되었 다. 이와 같이 NC 계 추진제(무연화약)의 소비량은 세계대전을 통 해 엄청나게 증가하였지만 NG 생산에는 한계가 있었다. 러시아 와 독일에서는 NG 를 DNT 또는 TNT 등과 같은 방향족 니트 로화합물로 바꾸는 노력도 하였다. 이 방향족 니트로화합물들로 부터 만들어전 화약은 NG 화약에 비해 더 낮은 폭발온도를 갖 고 있어 침식과 섬광이 적은 이점도 있었다. 한편 NG 를 니 트로글리 콜 (nit ro g ly c o l) 로 대 치 시 키 려 는 시 도가 여러 번 있었으나 니트로글리콜의 휘발성으로 인해 탄도 성능이 불안정해침으로써 성공하지 못했다. 또한 DGDN(die t h yle ne gly c o l din i t ra te : nit ro dig ly c o l) 은 NG 보다 NC 에 대 한 더 좋은 용 매이므로 더 큰 장점을 갖고 있는 것으로 밝혀졌다. 이와 같이 용매 효과가 좋아지면 더 균일한 젤라틴 화약을 얻을 수 있고, 제조성도 유리해지며, NC 함량이 높아지면서 섬광을 감소시키 기 위한 각종 첨가제를 더 많이 가할 수 있어 추진제 (화약)로서 의 성능도 더 좋아질 수 있다.
NG 를 사용하지 않는 무용제 화약은 더 낮은 폭발열을 갖게 됨으로써 총신(포신)의 마모도 감소시키는 이점이 있다. Gallw it z12) 는 총신 마모에 대 한 폭발열 (heat of exp lo sio n ) 의 영 향을 연구하여 NG계 무용제 화약 (폭발열 : 950 kcal) 의 경 우 총신 이 1700 회 발사시험을 견디었으나 폭발열 820 kcal 의 성능을 갖 는 비슷한 화약으로 시험한 결과 3500 회를 견디는 것을 확인했 다. 따라서 폭발열을 130kcal 감소시키면 총신의 수명은 두 배 로 연장되는 것을 알 수 있었다. NG 를 사용해서는 폭발열 칼로 리가 더 이상 줄지 않았으나 DGDN 을 NG 대신 사용한 화약의 폭발열은 690 kcal 까지 떨어졌으며 총(포)신은 15000~17000 회까 지 견디었다. 이와 같이 NG 나 DGDN(n it rod ig l y col) 을 NC 와 혼 합한 추진제 (화약) 는 복기 추진제 (double base po wder/pr o p e llant) 라고도 불리게 되었다. 그후 화약 개 발이 계 속되 어 무화염 (flas hless) 화약 (장약) 이 개 발되었다. 이를 위해 세계 여러 나라에서는 단기 추전제에 방향 족 니트로화합물을 첨가하는 방안과 복기 추진제에 칼리영 (po ta s siu m sal t)을 첨가하는 방안들이 시험되었다. 그러나 황산 칼륨 2 %를 첨가시킨 디글리콜(복기) 화약은 약간의 섬광현상을 나타냈다. 2 차 세계대전 동안 독일과 영국에서는 DGDN 을 사용 하는 복기 추진제에 니트로구아니딘 (n it ro g uan i d i ne) 을 상당량 첨 가한 화약을 흔히 사용했으며, 이 화약이 독일에서는 구돌 (gud ol) 화약, 미국에서는 삼중기 추진제라고 알려지게 되었다. 2.2 물리적 성질 니트로셀룰로오스 자체의 비중은 1. 66 이나 니트로셀룰로오스
(NC) 가 에데르 • 알코올과 같은 용매에 분산된 세미콜로이드 NC 추전제의 비중은 1.5 4~1 .63 정도로 낮아진다. 이 비중 차이로부 터 NC 가 기포를 가지고 있음을 알 수 있으며, 그 기포 내에는 보통 공기 나 용매 가 들어 있다. Brunsw ig에 의 하면 NC 장약 100 g은 대 략 4~ 8 cm3 의 공기 를 함유하고 있다고 한다. 13) 추진제 장약의 밀도는 장약 그레인의 디멘존과 모양에 따라 변 화되며, 그레인 표면을 갈아내고 흑연으로 코팅하면 그레인 밀도 는 증가하게 된다. 추진제 밀도는 카트리지 케이스 혹은 화약 연 소실에 채워질 수 있는 충전량을 결정하게 된다. 죽, 장약 충전 을 최대로 하기 위해서는 추전제 밀도는 가능한 한 높아야 한다. 예를 들어 NC 추전제의 밀도를 0.770 에서 0.830 으로 증가시킬 수 있으면 총 (r ifl e) 의 장약은 2.65 g에서 3.2 g으로 증가되므로 결 과적 으로 포구 초속 (muzzle veloc ity) 과 사거 리 는 그만큼 증가하 게 된다. 어떤 경우는 화약의 밀도가 너무 높아질 때가 있다. 이 런 경우는 연소실이나 카트리지 케이스의 상당 공간이 비게 되므 로 점화시의 신뢰성이 떨어지고 연소 상태도 균일하지 못하게 된 다. Brunsw ig에 의하면 화약 밀도가 0.850 일 때 22000 회 발사시 험 중 0.07 %의 불발, 1.2 %의 지연점화 (han gfi re) 를 기록했으나 밀도를 0.820 으로 떨어뜨린 장약을 사용했을 때는 이러한 불량이 발생 하지 않았다. 13) 세미콜로이드상의 NC 추전제 (단기 추전제)는 어느 정도 흡습 성이 있어 물로 포화된 공기중에서 2~2.5 %까지 수분을 흡수하 는 성질을 보이지만 복기 추진제(콜로이드상)는 거의 홉습성이 없다. 이는 복기 추진제 내의 NC 가 매우 낮은 홉습성의 콜로이 드상을 이루기 때문인 것으로 , 설명된다. NG 대신 DNT 를 사용 하면 홉습성은 더욱 떨어지고, 이런 추진제들은 NH- 화약 (non hyg ros cop ic po wder) 이 라고도 알려 져 있다.
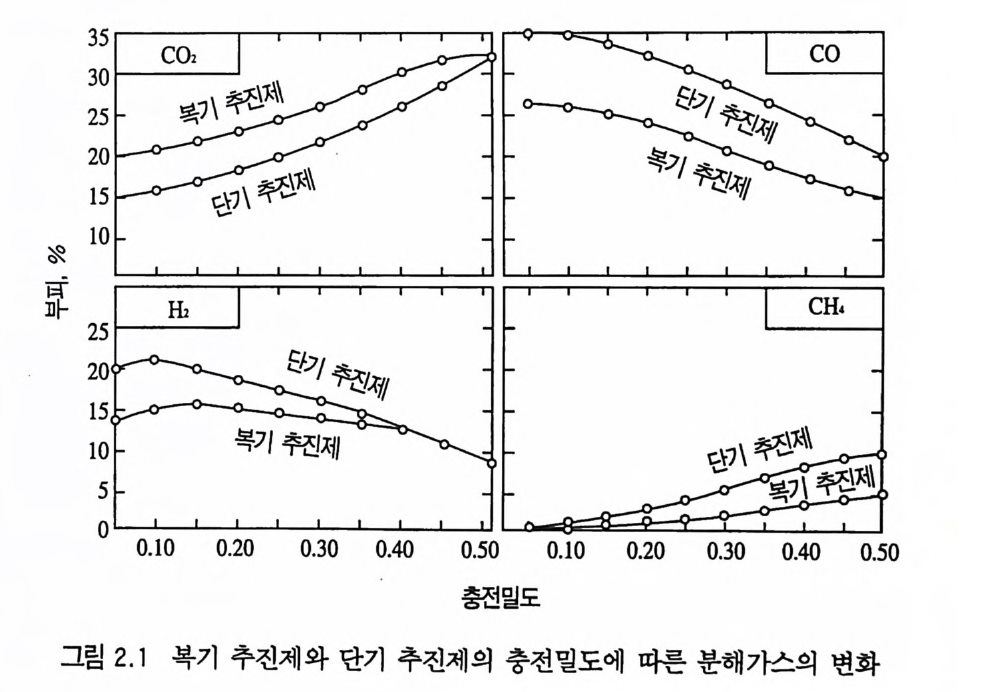
2.3 연소, 폭발 특성 2.3.l 분해 생성물 NC 추전제의 분해에 의해 나오는 생성물들은 그 원료들, 즉 NC, NG, 디니트로글리콜의 분해 생성물들과 매우 유사하다. NC 추진제 (단기)의 주요 분해 생성물들은 CO, H2, CO2, H20, N2 등이다. 복기 추진제의 경우에는 NG 로 안해 산소 함유량이 더 많으므로 완전연소 쪽으로 반응이 진행되면서 C 아와 H20 의 함량이 더 많아지게 된다. 이러한 NC 계 추진제의 분해에 의해 메탄가스나 시안화수소 및 탄소가 생성될 때도 있다. 분해 생성물의 조성은 추진제 연소 압력에 의해 크게 좌우되며, 연소압력은 추진제 장약 충전밀도에 따라 결정된다. 표 2.1 에는 충전밀도에 따라 변하는 분해 생성물 의 조성 예가 나타나 있으며, 충전밀도가 증가했을 때 CO2 와 CH4 양은 증가하며 CO 와 H2 의 양은 감소한다는 것을 알 수 있 다. 이상과 같은 결과는 다음 화학반응에서 보는 것처럼 압력 효과 에 따른 부피 감소 방향으로 반응(평형)이 이동하게 되며, 가스 가 냉각됨에 따라 평형은 오른쪽으로 이동하는 경향을 나타내는 것으로 설명될 수 있다.
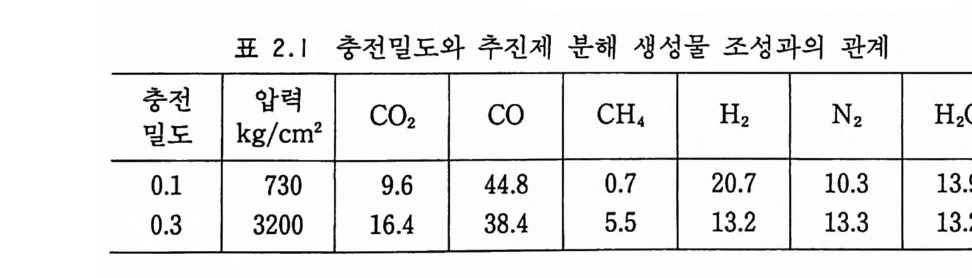 표 2.1 충전밀도와 추진제 분해 생성물 조성과의 관계
표 2.1 충전밀도와 추진제 분해 생성물 조성과의 관계
C0+3H2 ~ CH4+H20+57.8 kcal 탄이 발사되어 총구(또는 포구)를 향해 총신 내에서 이동함에 따라 온도와 압력은 대폭 감소하게 되므로 추진제 분해 생성물 역 시 크게 변하게 된다. Brunsw ig이 독일제 M/88 총을 사용하 여 탄 이동거리와 온도, 압력 측정을 한 결과는 다음 표 2.2 와 같다 .13) 상기와 같은 반응 의에 다음과 같은 분해반응도 일어날 수 있 으며 온도상승 효과에 의해서는 평형이 오른쪽으로, 압력에 의해 서는 왼쪽으로 평형이 이동된다고 볼 수 있다.
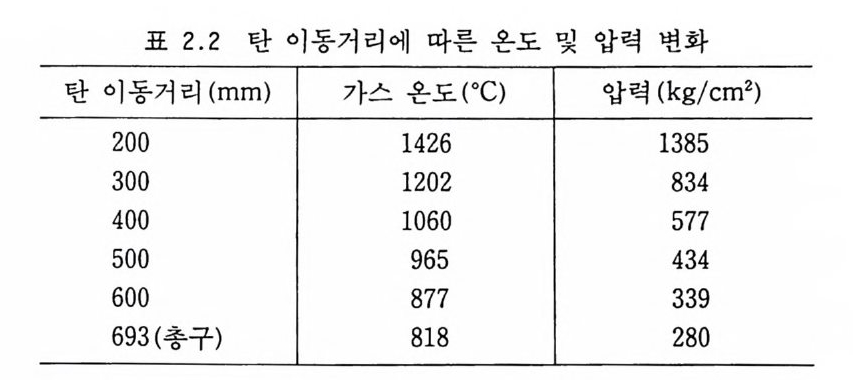 표 2.2 탄 이동거리에 따른 온도 및 압력 변화
표 2.2 탄 이동거리에 따른 온도 및 압력 변화
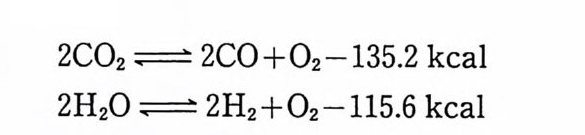 2C02 :;== 2CO + 02 —135 .2 kcal
2C02 :;== 2CO + 02 —135 .2 kcal
Po pp enber g와 S t e p han 은 위 와 갇은 시스템 에 서 • 압력 이 온도 의 영향보다 더 강하므로 탄이 총구를 향해 진행할수록 CO2 양 은 증가하고 CO 양은 감소한다고 설명하였다. 14) 또한 CO2/CO 의 비율은 탄이 이동함에 따라 다음 표 2.3 과 갇이 변한다고 보 고하였다.
 표 2.3 탄 이동거리에 따른 CO2 /C O 비율 변화
표 2.3 탄 이동거리에 따른 CO2 /C O 비율 변화
총신(포신) 내에서 일어나는 반응 중 다음과 갇은 일산화탄소 와 물의 반응은 매우 중요한 반응이다. co+H20 ~ C02+H2 이 반응은 발열반응이므로 온도가 상승함에 따라 평형은 왼쪽 으로 움직이게 된다(표 2.4). NC 계열 추전제 속에서 질산 (n it ra t e) 기로 존재하는 질소는 분 해반응 마지막 생성물로서는 질소분자 (N2) 로 완전히 변화되지만 압력이 낮은 경우 질소 중 일부는 질소산화물로 남게 된다. 어떤 경우는 총구(포구)로부터 나온 가스에서 암모니아 냄새를 말을 수 있는데, 이는 다음과 같은 반응에서처럼 뜨거운 분해가스가 냉각됨에 따라 약간의 압력이 걸려 있는 동안 일어나는 반응으로 서, 특히 가스 내에 철분이 섞여 있을 때 더 빨리 진행되는 반응 이다. 표 2.4 일산화탄소와 물의 반응에 미치는 온도의 영향
 온도 (°C) K [[CCOO]2 ][ H[H220]]
온도 (°C) K [[CCOO]2 ][ H[H220]]
N2+3H2 ~ 2NH3+22.0 kcal 다음 그립 2.1 은 반응 생성물 중 CO2, CO, H2, CH4 양에 대 한 단기 추전제의 충전밀도 영향을 나타내고 있다 .15) 이 그립에 서 복기 추진제 (cordit e) 는 NG 의 산소 함량이 더 높은 관계 로 완전연소 생성물 (CO 2 ) 이 더 많이 생성되는 것을 알 수 있다. 포에서 탄이 발사되고 폐쇄기 (breech block) 가 열린 후 폐쇄기 끝 (breech end) 을 통해 포신으로 가스가 나울 수 있는데 , 만일 폐쇄기 끝이 밀폐된 공간(해군 함정 포탑, 탱크, 콘크리트 특화점 등) 에 있으면 화포의 각 부품에 유독한 가스로 작용하게 된다. 따라서 공기를 불어넣어 이 유독가스를 제거하는 것이 필요하다. Kn ig h t와 Wal t on 은 함정용 화포를 대상으로 밀폐된 공간에서 생성되는 분해가스들을 조사하였다 .16) 32k g의 추진제 장약을 점
 35
35
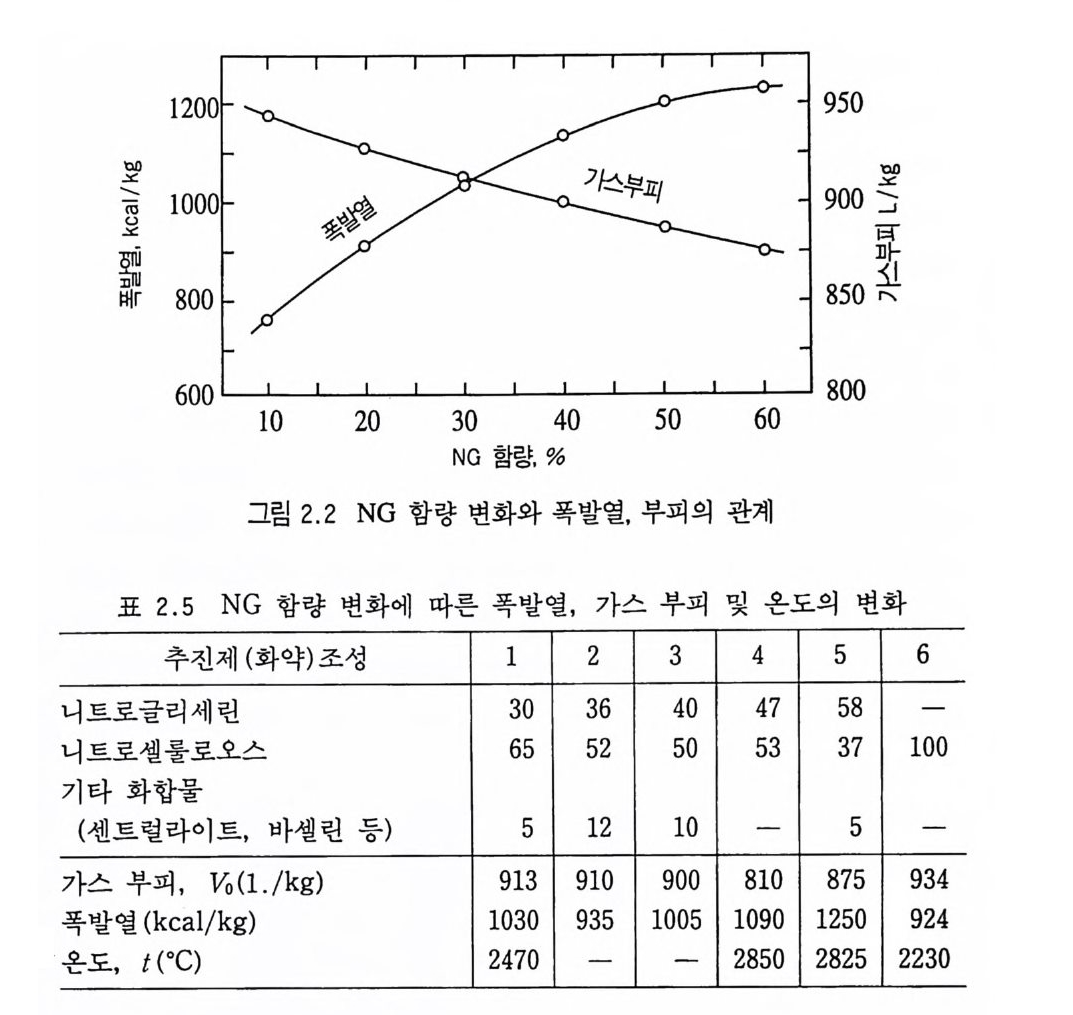
화시킨 후 10 초 후 25m3 의 밀폐 공간에서의 가스 조성은 NO 1% N02 7% CO2 17 % co 28 % H2 8% CH4 2% N2 37 % 였다. 20 초 뒤 화약의 연소가 끝난 후 밀폐 공간 속으로 깨끗한 공기 롤 주입하였을 때 뜨거운 분해가스는 새 공기와 접촉하여 다시 폭발적인 연소를 하였으며, 이 두번째의 연소 후 가스 조성은 N02 1 % CO2 8 % co 9% 02 12% N2 67% 였다. 이 가스 성분 중에 포함된 N 아와 CO 는 인체 및 실험장비 에 매우 유독한 가스들이었다. 2.3.2 폭발열과 분해가스의 부피 및 온도 폭발열 (heat of exp lo sio n ) 은 주로 추진제 (화약, 장약) 의 조성 에 의해 결정된다. 죽, 단기 추전제는 NC 의 질소 함량에 의해, 복 기 추진제는 NG 의 함량에 의해 결정된다고 볼 수 있다. NG의
 950
950
함량 변화에 따른 복기 추전제의 폭발열과 가스 부피가 그림 2.2 에, NG 함량 변화에 따른 폭발열, 가스 부피, 온도의 변화가 표 2.5 에 나타나 있다 .13) 일반적으로 복기 추전제는 단기 추전제보다 더 높은 폭발열을 발생하므로 그 분해 생성물 온도 역시 더 높아지게 된다. 따라서 복기 추진제가 포신을 더 많이 마모 (eros i ve) 시키고 더 많은 섬광 울 발생시킨다. 복기 추진제의 폭발열과 섬광 온도를 낮추기 위
해 비폭발성 물질인 바셀린을 첨가하거나 니트로구아니딘과 같은 연소온도가 낮은 화약을 가하기도 한다. 단기 추전제나 복기 추전제와 같은 NC 계열 추전제들은 같은 상(p hase) 에 있는 균일(균질) 추전제들이므로 연소 불꽃도 균일 하게 나타나며 연소 방향에 따라 일차원 구조가 된다. 연소표면 에서 분해가스 생성물들의 분자들은 산화제 및 연료와 마주쳐 서 로 혼합된다 .17) 다음 그립 2.3 에는 10, 20, 30 기압에서 복기 추 전제가 연소할 때의 불꽃 사전이 나타나 있다. 추전제가 연소될 때의 발광(l um i nous) 불꽃은 연소표면으로부터 일정거리 떨어진 곳에 위치하며, 압력이 증가하면 라미노우스 불꽃은 연소표면으 로 가까이 움직이게 된다. 추전제 연소표면과 라미노우스 불꽃 사이에는 투명한 부분이 있는데 이룰 다크 영역 (dark zone) 이라 고 부른다. 이 다크 영역의 두께는 불꽃충과 그 거리가 같으며, 이 다크 영역은 압력이 증가(죽 연소속도의 증가)함에 따라 감소
 (A) (B) (C)
(A) (B) (C)
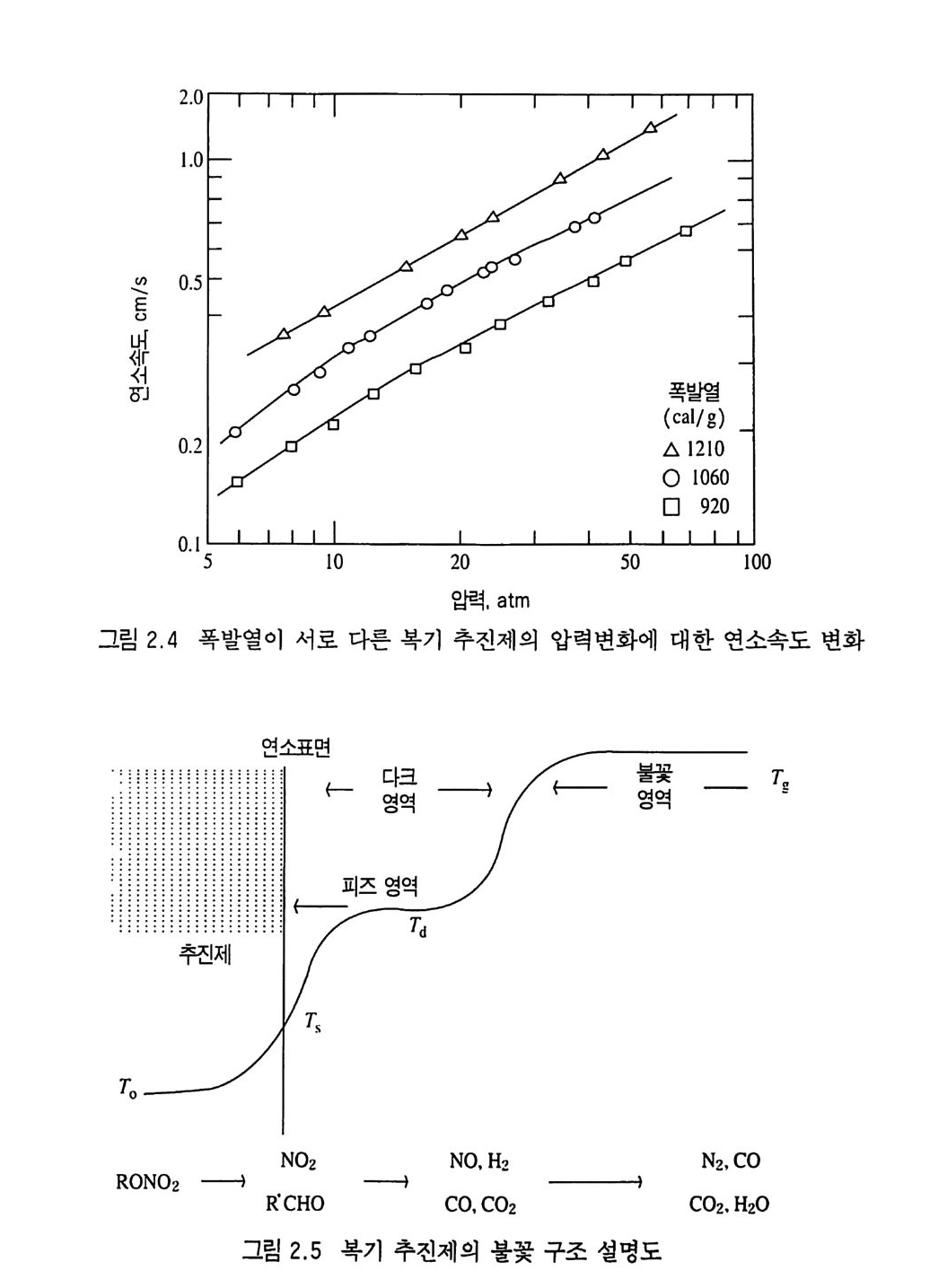 2.0
2.0
한다. 예 를 들어 이 두께 는 10, 20, 30 기 압에 서 각각 1.3, 0.33, 0.14 cm 이 며 이 때 의 추진제 연소속도는 각각 0.22, 0.31, 0.4 0 cm/sec 이다. 세 종류의 복기 추전제에 대한 연소속도가 그림 2.4 에, 불꽃 구조가 그림 2.5 에 나타나 있다. 17) V i e ill e 는 추전제 연소속도와 압력과의 관계에 대해 다음과 같 은 간단한 지수식을 제안하였다 .8)
 |I :::::::::::l |
|I :::::::::::l |
 v=kpn
v=kpn
추진제는 그레인 모양에 따라 연소표면이 결정되며, 평행충 (pa rallel la y er) 으로 연소되므로 추진제 그레 인 모양은 연소 모 드, 추력 내지는 화력에 결정적인 영향을 주게 된다. 추진제 그 레 인 모양 중 중립 형 (neutr a l, 연소 도중 연소시 간에 따른 연소 표 면적이 거의 일정한 경우), 점증형 (pro g res siv e , 연소시간에 따른 연 소 표면적이 증가하는 경우), 점감형 (de gr ess i ve, 연소 표면적이 시 간에 따라 감소하는 경우) 등의 예가 그림 2.6 에 나타나 있다.
니트로셀룰로오스계 추전제의 연소(폭발)분해 생성물의 온도는 추진제의 충전밀도에 따라 결정되며 충전밀도가 증가함에 따라 온도는 중가하게 된다. 충전밀도 변화에 따른 연소속도 변화의 예가 다음 그림 2.7 과 같이 나타나 있다. 이 그림에서 복기 추전 제인 Ballis t i te, Cord it e 에 비해 NG 함량이 적은 NC 화약(단기) 이, 즉 산소 함유량이 적은 추전제가 충전밀도에 따른 온도 증가 율이 급격한 것을 알 수 있다. 13) 탄의 포구속도 (muzzle velo city)는 추전제 장약의 폭발열에 따 라 크게 영향을 받으므로 추전제의 폭발열 데이터는 매우 실제적 인 중요성을 갖고 있다. 즉, 추전제 제조 공정시 일정한 간격을
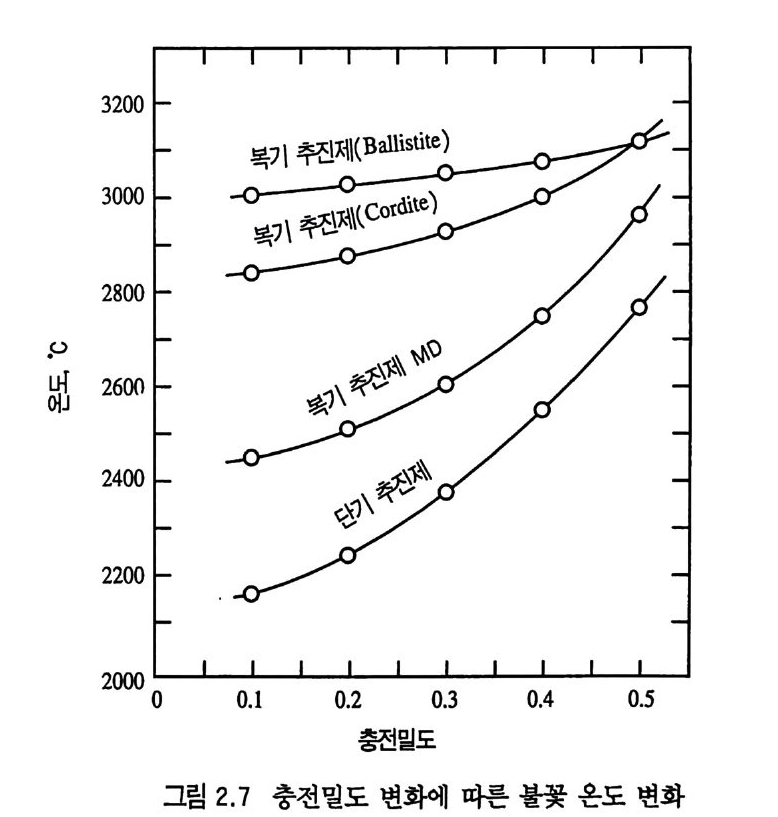 3200
3200
두고 제조되는 추진제 시료의 폭발열을 시험하여 만일 폭발열이 기준값을 벗어나면 추진제 장약 양( 量 )을 표준 포구속도에 맞도 록 조정하여 제작하여야 한다. 이에 관련된 실험식은 CQ = K 라는 식으로 표현된다. 여기에서 C 는 추전제 양, Q는 폭발열, K 는 추진제 종류에 따른 탄의 고유한 일정 상수로 표시된다. 예 를 들어 4.3 k g의 추전제 가 820 kcal/ k g의 폭발열을 갖고 탄에 충전된다면, 같은 포구속도를 내기 위해서는 같은 계열의 추전제 로서 폭발열 590 kcal/ kg 추진제 는 6.0 k g이 필요하게 된다. Ta y lor 는 영국의 단기 추전제, 미국의 복기 추진제 등 NC 계열 추전제들에 대해 폭발 분해열, 가스 부피, 연소속도에 관련된 데 이터를 다음 표 2.6 과 같이 정리하였다 .18)
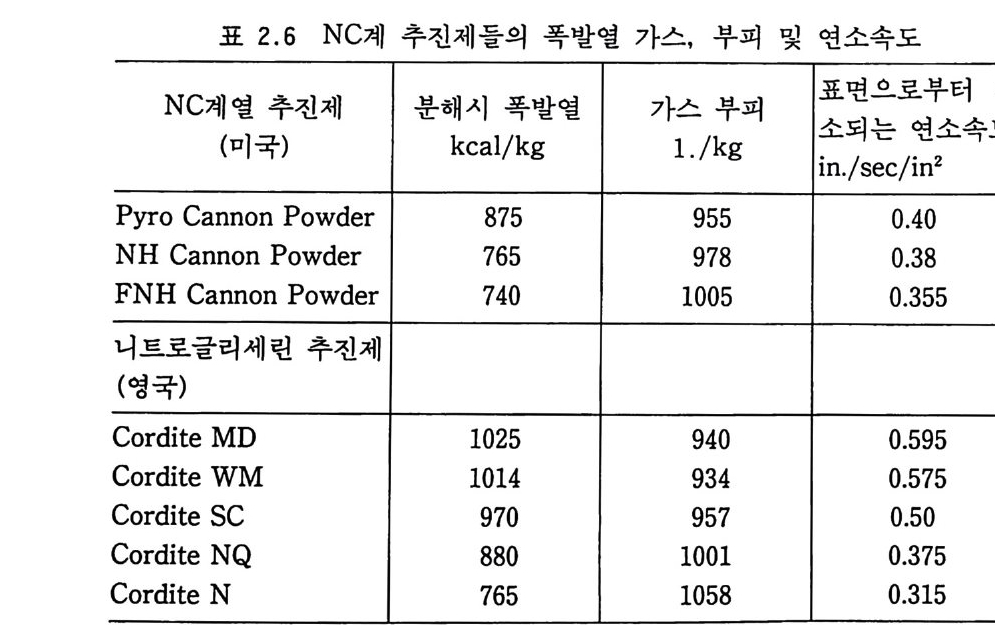 표 2.6 NC 계 추진제들의 폭발열 가스, 부피 및 연소속도
표 2.6 NC 계 추진제들의 폭발열 가스, 부피 및 연소속도
2.3.3 충격 및 마찰 감도 NC 계열 추진제는 충격에 비교적 낮은 감도 (sens iti veness) 를 갖고 있어 탄환의 충격에 의해서도 쉽게 점화되지 않기 때문에 전쟁 도중 군인들아 안전하게 조작하고 사용할 수 있었다. 그러 나 마찰에 의해서는 갑작스런 점화 혹은 폭발 현상을 나타내기도 하는데, 특히 추진제가 뜨거울 때면 더 예민하게 반응하는 성질 이 있다. 추전제 제작공정 도중 일어나는 많은 사고가 마찰에 의 해 일어났다고 볼 수 있다. 예를 들면 추전제의 건조공정 중 건 조기 내를 통과한 뜨거운 추전제가 마찰로 인해 폭발한 일들이 있었다. 특히 건조기를 통과한 추전제는 정전화 (elec t r ifi ca ti on) 되 어 있어 건조기에서 추전제를 꺼낼 때 방전으로 인한 폭발사고가 일어나기 쉬우므로 완전히 냉각될 때까지 기다려야 한다. 일반적 으로 단기 추전제는 복기 추전제보다 마찰에 더 예민하고, 복기 추전제는 충격에 더 예민한 것으로 알려져 있다 .19) 2.3.4 폭굉 감도 Kas t에 의하면 NC 단기 추전제는 매우 강한 기폭제 (피크린산 50g 또는 테트릴 100 g)로 기폭시킬지라도 찰 폭굉되지 않는 것으 로 알려져 있지만 일단 폭굉되면 1000~1800m/sec 의 속도로 폭 굉 된다고 보고하였다. 20) Urbanski, Galas21) 등에 의하면 NC 계열 추전제는 피크린산 20 g에 의해 폭굉속도 3800~7000m/sec 로 폭굉이 진행되며, 이 때 폭굉속도는 추전제 충전밀도에 따라 변화하고, 특히 박편형 추진제의 경우는 폭굉파가 박편에 대해 수직으로 전파하느냐 또
는 수평으로 전파하느냐에 따라 폭굉속도가 결정된다고 밝혔다. 다음 표 2 . 7 에는 폭굉속도에 대한 실험치가 강철파이프 26/30 mm, 피크린산 20 g을 점화개시제(기폭제)로 사용한 경우의 결과 들이 나타나 있다. 만일 추전제 박편 (str ip ) 이 나 추진제 튜브가 포신 축선 (axis ) 방향으로 놓여지면 폭굉이 훨씬 어렵게 일어나므로, 폭굉시키기 위해서는 더 강한 기폭력(점화력)이 필요하다. 다음 표 2.8 에서 의 결과들은 20 g의 피크린산을 기폭제로 사용하고 26/33mm 파 이프에서 실험한 결과이다. Dau tri che 는 1913 년 NC 계 추진제 BM17D2 (두께 44 mm) 는 피 크린산 50 g에 의해 폭굉되어 6560~7200m/sec 의 폭굉속도를 나 타냈다고 했으며, Burlot (1 920~1926) 역시 비슷한 결과를 발표 했다. Burlo t은 추전제가 낙하하는 총알과 같은 물체에 의해서 는 폭연 (defl ag rat i on ) 이 일 어 났으나 1200 m/sec 이 상의 속도를 지닌 총알 (ca li ber 8mm, 7.5 g)에 의해서는 폭굉되었다고 보고하 였다. 22) 2. 3. 5 점 화성 (ign it ab il ity) 복기 추진제의 점화온도는 대략 180°C 이고 단기 추전제의 점 화온도는 대략 2oo·c 정도이다. NG 가 함유된 NC 계 추전제를 직접 불꽃으로 점화시키는 것은 위험하고 또 의미가 없는 방법이다. 실제로는 간접적인 방법, 죽 흑색화약을 착화제 (Pr i mer) 로 점화시키는 방법 등을 사용하고 있 다. NC 계열 추전제는 쉽게 정전화 (elec t r ifi ca ti on) 되므로 순간 방
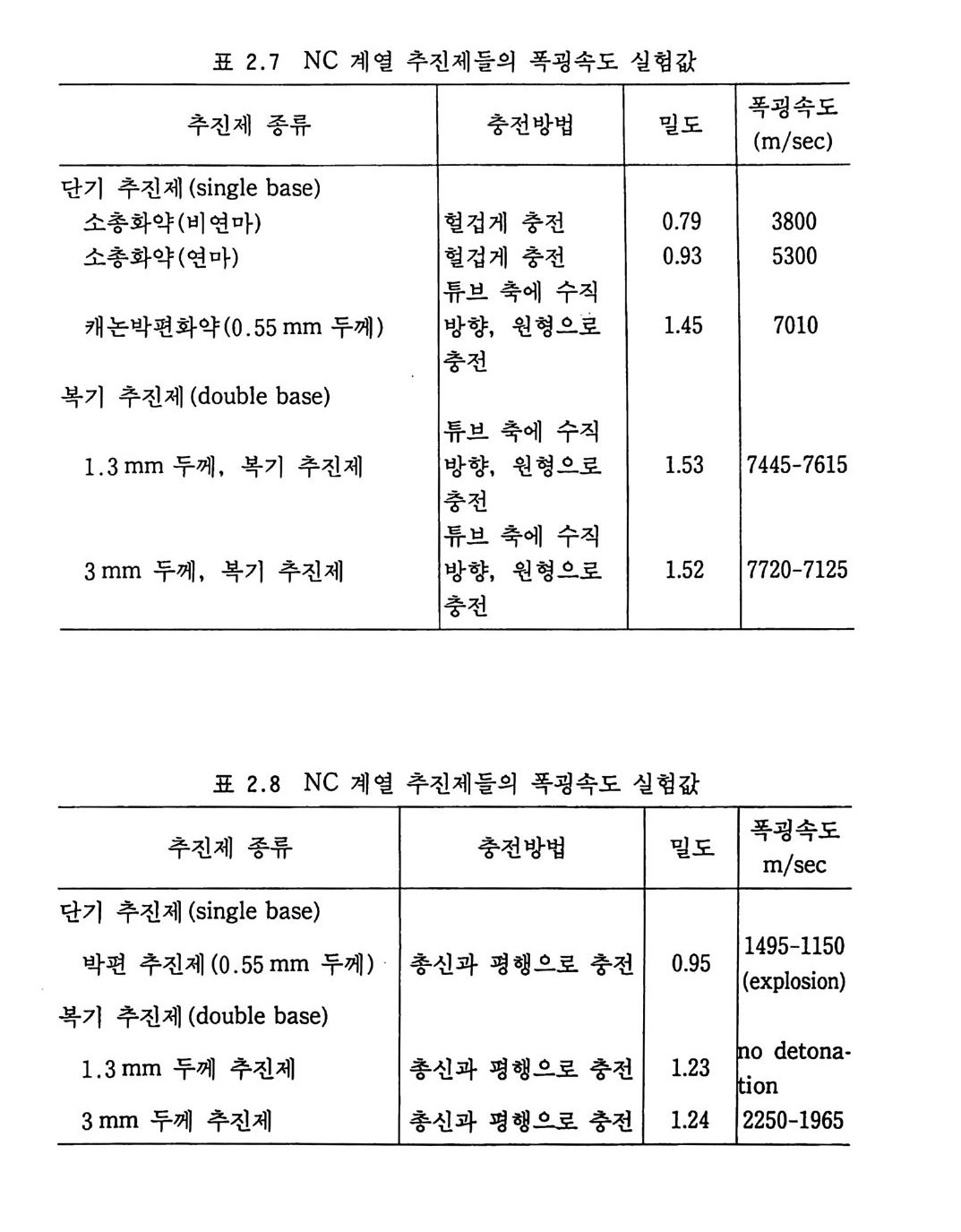 표 2.7 NC 계열 추진제들의 폭굉속도 실험값
표 2.7 NC 계열 추진제들의 폭굉속도 실험값
전 (d i schar g e) 될 때 점화가 일어날 수 있다. 특히 건조할 때, 따 뜻하고 건조한 공기를 통과시킬 때 또는 마찰에 의해서 정전기 발생이 일어난다. 이 추전제들은 목재와의 마찰이나 추전제 그레 인의 연마(p o li sh i n g)시에도 강한 정전기가 발생한다. Nash 의 보 고 19) 에 의 하면 NC 계 추전제 (EC 화약) 는 65 ·c 의 공기 가 통과할 때 정전기가 발생하며 추진제가 건조할(될)수록 더 정전기가 많 이 발생한다고 다음 그립 2 . 8 과 같이 발표하였다. 이 그림 2.8 의 실험 결과에 의하면 수분함량 1 %가 넘으면 쉽 게 정전화되지 않지만 그보다 건조해지면 정전기 발생이 300V 까지 올라가는 것을 알 수 있다. 콜로이드 계열 추진제는 더 강 하게 정전화 현상이 일어난다. 예를 들면 알코올과 에테르의 혼 합액으로 젤라틴화시킨 피로셀룰로오스(pyr ocellulose) 추전제를 50°C 로 가열하면 뜨거운 공기의 영향으로 인해 3200V 까지 정전 기가 발생한다. 15°C 로 냉각시키고 찬공기로 5 분 동안 통과시킨 후에는 2200V 로, 20 분 뒤에는 900V 로 떨어지게 되며 그후에는 거의 일정한 볼트 수준을 유지한다. 만일 2300 V 로 정전화된 추 진제를 찬 공기로 냉각시키지 않으면 오랜 시간 동안 2300V 를 그대로 유지하게 된다. Nash 는 19) NC 계열 추전제 (EC 화약)가 20000V 까지 충전된 후 방전시키면 추진제가 점화되는 것을 실 험적으로 확인하였고, 이를 근거로 추전제 건조공정 도중의 정전 기(수천 볼트) 발생보다는 훨씬 더 높은 전위차의 정전기 발생이 있어야 방전시 점화될 것으로 결론지었다. 그러나 실제로는 건조 기에서 뜨거운 추전제를 꺼낼 때 정전기의 방전에 의해 많은 사 고가 일어났으며, 이는 각 추전제(화약)별로 마찰과 정전기에 대 한 감도와 발생량이 다르기 때문으로 해석된다. NC 계열 추전제를 연마(p o li sh i n g)할 때 일어난 많은 폭발사고 들 역시 정전기 발생 및 방전에 의해 일어난 것으로 해석되고 있
고, 따라서 연마공정 중 사용하는 드럼과 같은 장비들은 어스 (earth ) 하여 사용하는 것 이 필수적 이 다. 프랑스 화약기술협회의 통계 23) 에 의하면 점화를 포함한 모든 폭발사고의 75 %는 완전 건조된 화약(추전제)에 의해 일어났으 며, 그 중 41 %는 마찰에 의한 정전기 발생에 의해, 34 %는 추 진제의 불안정 및 기타 미지의 원인에 의해 발생되었다고 발표하 였다. 또한 모든 폭발사고의 나머지 25 %는 건조되지 않은 일정 용매를 포함한 추전제에서 일어났는데 그 중 19.5 %는 마찰에 의 한 정전기 발생으로, 나머지 5.5 %는 용매의 정전화, 방전으로 일어났다고 발표하였다. 일반적으로 정전기 발생은 모든 추진제 (화약)류 사고 원인 중 대략 60 % 정도를 차지한다고 볼 수 있 다.
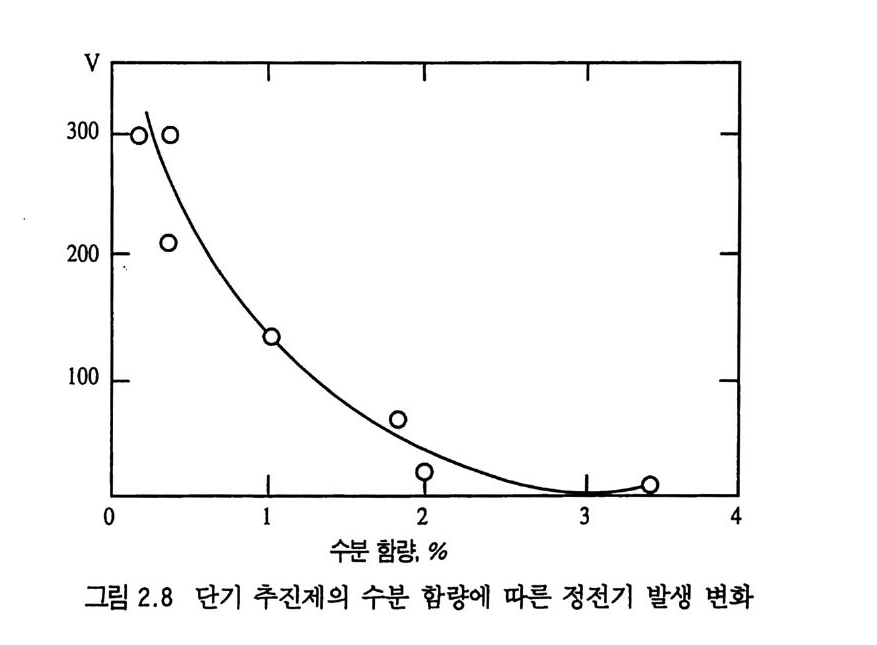 v
v
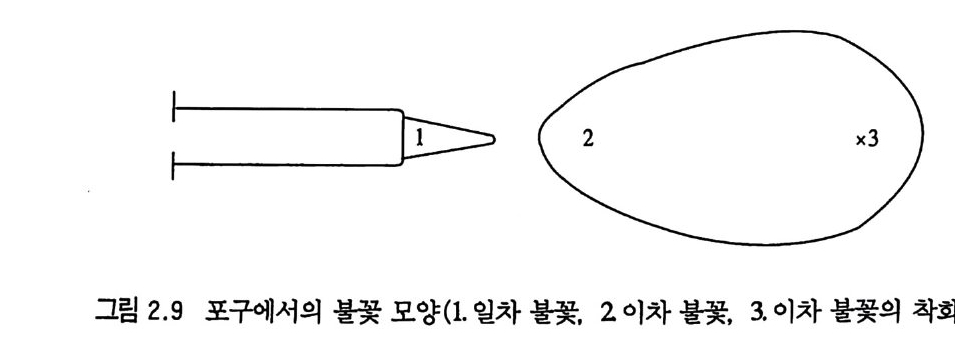
2.4 섬광 발생 및 섬광 억제 포(총)를 발사하면 대부분 섬광이 발생되어 그 발사 위치를 나 타내게 된다. 섬광의 불꽃 모양은 대개 다음 그림 2.9 와 같다. 이 그립에서 검붉은 색깔의 가연성 가스로 이루어전 일차불꽃 (1) 은 공기와 혼합되어 가연성 혼합물이 된다. 이 혼합물의 온도 가 충분히 높을 경우 포구로부터 멀리 떨어전 위치 (3) 에서 연소 내지는 폭발(또는 폭굉)될 때도 있다. 이러한 가스 혼합물의 폭 발은 밝은 불꽃을 동반하게 되 며 이 를 이 차불꽃 (secondary fl ame) 이라고 부른다. 이차불꽃은 타원형 모양으로 나타나며 매 우 먼 거리에서도 식별이 가능하다. 이 불꽃의 크기와 섬광도는 포의 구경에 따라 달라진다. 예를 들면 30 cm 포에서 나온 불꽃 은 50 m 길이롤 갖고 있어 발사 위치로부터 50 km 떨어진 지점 에서도 식별할 수 있다. 보통 다음과 같은 두 가지 요인들이 이 차불꽃 형성에 주로 영향을 준다고 알려져 있다. 첫째는 추전제 장약이 폭발(작동)된 후 생성된 분해가스 중 가 연성 혼합물을 만들 수 있는 가스 조성이며, 둘째는 포신의 분해 가스 온도이다. 그러나 가스조성 중 혼합가스의 폭발을 억제하 는 물질이 존재하면 이차불꽃을 일으키는 가능성이 감소될 수 있 다. 이차불꽃 폭발에 관련된 가장 중요한 가스 성분들은 수소,
 二
二
일산화탄소, 메탄 등으로서 공기와 혼합되면 폭발성 혼합물이 된 다. Roszkowsk i2 4) 에 의하면 이러한 가스와 공기와의 혼합물에 대 한 폭발한계 (exp lo sio n lim i t) 는 H2 9.2 ~ 68.5 % co 13~77.6 % CH4 5.5 ~ 13.2 % 라고 보고하였다. 이 결과로부터 메탄은 그 중 가장 좁은 폭발한 계를 갖고 있으므로 메탄가스를 되도록 많이 발생하도목 탄을 발 사헌다면 이차불꽃은 나타나지 않게 된다. 따라서 추전재 장약의 충전밀도를 높이면 포(총)신 내의 압력이 높아져 메탄가스 생성 량이 많아지므로 이차불꽃의 발생 확률이 줄어들게 된다. 이차불 꽃을 줄일 수 있는 다른 방법은 추진제 장약의 폭발 분해 생성물 내에서 불활성(비연소성) 가스인 탄산가스와 질소가스의 농도를 증가시키는 것이다. 그 중 질소가스의 질소 함량은 추전제의 질 소 조성에 의해 제한받게 되므로 추전제 조성을 그대로 유지하면 서 조절 가능한 가스는 탄산가스의 함량이다. 높은 압력과 낮은 온도는 탄산가스의 생성을 촉진한다고 알려져 있다. Coward 및 Hartw ell 등은 메탄-공겨 혼합물의 폭발한계가 그 혼합물에 탄 산가스나 질소가스를 첨가하면 좁아진다고 보고하였고 ,25) 대략 탄산가스 50 %를 포함하는 메탄-공기 혼합물은 비폭발성 성질을 나타낸다(그립 2.10). 메 탄-공기 혼합물에서 수증기가 상당량 존재 하면 탄산가스를 첨가시킨 경우와 비슷한 폭발억제 현상을 나타내지만 수증기 양 이 너무 적을 경우에는 가스 혼합물의 폭발 가능성을 오히려 증 가시켜 주기도 한다. 이 현상은 습도가 높은 대기 중에서 탄을 발사했을 때 이차불꽃이 더 쉽게 나타나는 것으로도 확인할 수
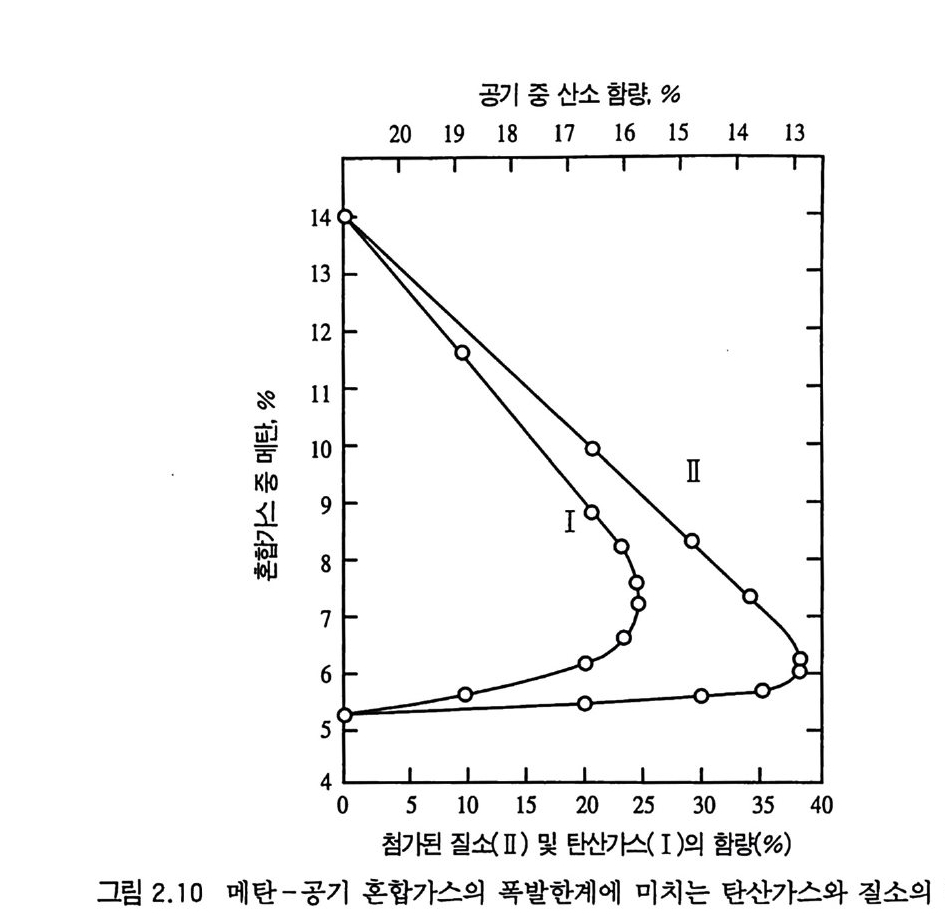 공기증산소함량, %
공기증산소함량, %
있다. 또한 추진제 장약이 폭발 연소될 때 불완전하게 분해되어 발생 하는 질소산화물 (N02) 의 소량이 메탄 _ 공기 혼합가스에 섞 여 있으면 이차불꽃 생성 확률을 증가시킨다고 알려져 있다. 추진제 가스의 온도는 폭발열과 가스 조성에 의해 결정되므로 폭발열이 클수록 가스 온도는 울라가며 이차불꽃은 더 쉽게 생성 된다. 그러므로 낮은 폭발열을 지닌 추전제의 경우 가스 혼합물 의 온도가 점화온도보다 낮으면 이차불꽃은 생성되지 않게 된다. 메탄-공기 혼합가스 시스템에서 폭발 분해 생성물의 점화온도 는 대략 다음과 같은 범위에 있다.
H2 390 ~ 620 °C co 610~725 °C CH4 730 ~ 790 °C 이 점화온도 데이터로부터 메탄가스가 더 많이 생성되는 방향 으로 추전제 장약의 폭발 연소반응의 평형상태를 옮겨 주면 이차 불꽃을 억제하는 데 유리한 것을 알 수 있다. 많은 실험 데이터로부터 추전제의 분해가스 생성반응시 방출된 열량이 어떤 한계값에 도달하지 않으면 이차불꽃에 의한 섬광은 발생하지 않는다고 보고되었다. 이러한 폭발열 효과는 사용된 화 포(총)의 종류(포신의 직경, 길이 등)에 따라서도 달라진다. 따 라서 무섬 광 (flas hlessness) 은 NG 와 같은 고에 너 지 성 분 대 신 니 트로구아니던 (n it ro gu an i d i ne) 혹은 DNT 등을 대신 사용하거나 바셀린, 센트럴라이트와 같은 비폭발성 물질을 첨가시킴으로써 얻을 수 있다. 이차불꽃 섬광을 억제하는 방법으로 이차불꽃 연소(폭발)반응 의 점화반응을 억제시키는 물질을 사용할 수 있다. 예를 들면 칼 륨 이 온 (po ta s siu m ion ) 은 이 러 한 성 질을 지 닌 물질로서 1908 년 Dau t r i che 에 의해 처음으로 적용되었다 .26) 이때부터 질산칼륨은 추진제 장약의 섬광 억제제로 흔히 사용되고 있다. Fauveau 등은 알칼리 금속이나 알칼리토류 금속들의 염화물 들을 추전제 장약 성분으로 첨가시키면 이차불꽃이 억제될 수 있 음을 발견하였다 .27) 이러한 물질들의 섬광 억제작용은 분해가스 의 온도를 떨어뜨린 효과가 아니라 이차불꽃의 폭발반응을 방해 하는 역할에 의한 것으로 밝혀졌다. 그 이유는 고온에서 많은 분 해 열 (heat of decomp o sit ion ) 혹은 증발열을 필요로 하는 금속영 이나 염화칼슘과 같은 알칼리토류 염둘은 이차불꽃 반응을 막지
못한다는 것으로 알 수 있다. 역으로 염화칼리 같은 대표적인 섬 광 억제제는 높은 증발열도 갖고 있지 않으며 총신 내의 온도에 서 분해되지 않는 물질이다. Pre tt re 는 일산화탄소와 공기의 혼합가스에 분산된 염화칼리는 이 혼합물의 점화온도를 상승시키지만, 수소와 공기의 혼합물의 점화온도에는 영향을 주지 않는 것을 발견하였다 .28) 영화칼리는 인덕션 기간(가스 혼합물의 가열이 시작된 시점에서 폭발이 일어난 시점까지의 시간 간격)을 길게 해준다고 알려져 있 다. Pease 는 내부벽을 염화칼리로 도포한 용기 내에서 수소와 산소의 혼합가스를 반응시킨 결과 염화칼리가 도포되지 않은 용 기에서의 반응에 비해 반응(폭발)이 상당히 억제되었고 인덕션 기간은 일천 배 이상 증가되었다고 보고하였다 . 2 9 ) 칼리 이온은 자유원자인 H, O 및 OH 라디칼이 불활성(i ner t) 분자로 조합되도 록 하여 폭발 연쇄반응을 차단시킴으로써 폭발을 억제한다고 설
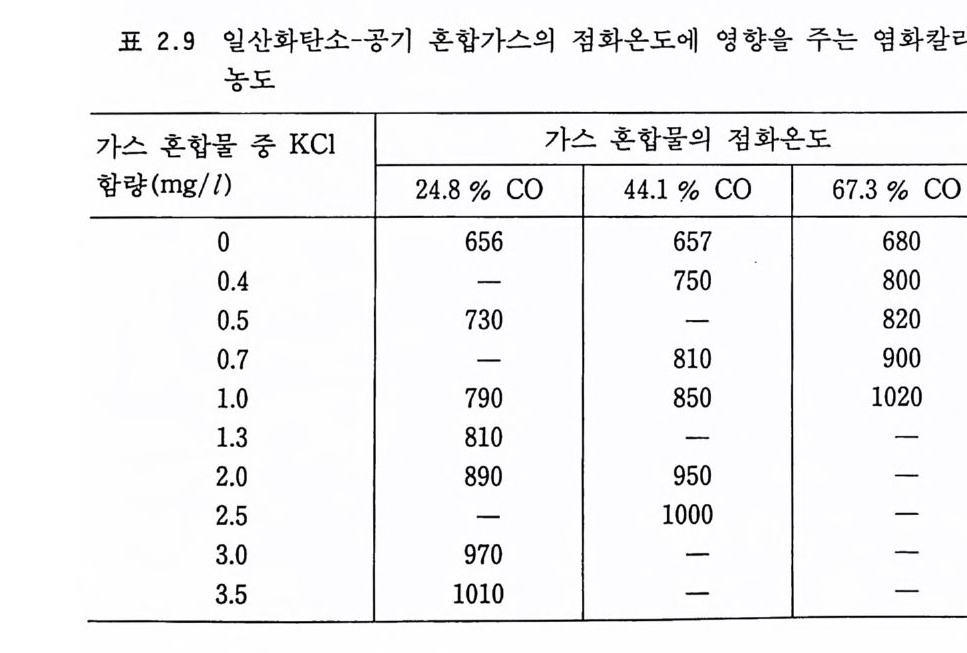 표 2 . 9 일산화탄소 - 공기 혼합가스의 점 화온도에 영 향을 주는 염 화칼리 의
표 2 . 9 일산화탄소 - 공기 혼합가스의 점 화온도에 영 향을 주는 염 화칼리 의
명된다. 수소와 산소의 자유원자 및 OH 라디칼은 다음과 같은 연쇄반응을 일으킨다고 설명되고 있다. H 2 +02 一 20H OH+H 2 一 H20+H H+02 一 OH+O OC+OH+2O 一H — -OcHo +2H+ H 수소와 산소 혼합물의 불꽃 스펙트럼을 조사하였더니 수소원자 와 OH 라디칼이 이 반응에 참여하고 있음을 알아냈다. OH 라디 칼이 수소와 산소 사이에 일어나는 반응의 중간체로서 존재하고 있다는 것은 이 가스들을 1000 °C 이상으로 가열한 혼합물의 흡 수 스펙 트럼 (absorpt ion spe c tr u m) 에 의 해 서 , 또한 1500 °C 이 상 으로 가열한 수증기의 흡수 스펙트럼에서도 증명되었다 .30 , 31 , 32 , 33) Nor ri sh 와 Por t er 에 의해 고안된 스펙트로스코피 (kin e cti c absor- ption spe c tr o scop y) 에 의 한 가스폭발 반응 경 로 조사방법 은 매 우 특기할 만한 일이었다 .34) 이 방법에 의해 Norri sh 등은 수소와 산소의 반응계에 대해, 산소 분위기 속에 있는 탄화수소의 연소 에 대해 또한 물이 최종 생성물 중의 하나인 또 다른 반응들에 의해 OH 라디칼이 반응 중간체로 존재한다는 것을 확인하였 다 .34,35 , 36) 칼륨 이온은 다음과 같이 연쇄반응을 차단 (break i n g)시 키는 반응을 촉진시킨다고 보여진다. H+H 一+ H2 H+OH 一 H20 o+o 一 02 co+o 一 CO2
칼륨 이외의 다른 종류의 알칼리 금속영들은 이차불꽃울 억제 하는 데 큰 효과가 없는 것으로 밝혀졌다. 포탄 발사시의 섬광을 제거하기 위해 실제로는 두 가지 방법이 사용되고 있다. 죽, 니 트로구아니딘이나 DNT 를 소량의 황산칼륨과 함께 추진제에 첨 가시키거나, 칼륨영과 같은 특별한 섬광 감소제를 추진제 장약에 첨가시키는 방법이다. 그러나 대부분의 섬광 억제 방법들은 스모 크 (smoke, 유연의 가스) 발생을 촉진시킨다. 이러한 관점에서는 니트로구아니딘을 첨가시키는 것이 그 중 가장 좋은 방법으로 보 여전다. 니트로구아니딘은 스모크를 크게 증가시키지 않고 섬광 억제 작용을 할 수 있기 때문이다. 추진제 장약이 연소된 후 생성된 연소성 가스는 가끔 후방섬광 (backfl as h) 현상을 일으키는 수가 있다. 이는 포의 폐쇄기 (breech block) 가 열려 있을 때 포신에서 생성된 공기 一 가스의 흐 름에 의해 나타나는 현상으로서 바람이 연소 방향 반대로 불 때 일어난다. 공기와 혼합된 뜨거운 분해가스의 점화는 총신에 남아 있는 연소 잔여물 (smoulder i n g rema i nder) 로 인해 일어난다. 어떤 때는 탄 발사 후 연소가 정지되지 않고 폐쇄기를 연 후 계속해서 연소되는 수가 있다. 이러한 후방섬광 현상은 포 주위의 사람들 에게 매우 위험하며, 특히 발사하기 위해 쌓아놓은 포탄둘을 점 화시킬 위험성도 있다. 그러나 이차불꽃 섬광의 경우처럼 칼륨염 울 추전제 장약에 첨가시킴으로써 후방섬광 현상도 억제할 수 있 다. 대구경 포에서는 탄 발사 직후 포신을 통해 찬 공기를 불어 넣어 주거나 물을 흘려보냄으로써 이러한 후방섬광을 억제하기도 한다.
2.5 스모크 단기 추전제나 복기 추전제 등의 NC 계 추진제 (장약, 화약)를 무연 (smokeless) 이라고 하는 것은 실제로 부정확한 표현이며 약 간의 연막성 (slig h tl y smoky ) 이 라고 부르는 것 이 맞는 표현이 다. NC 계열 추진제 장약 연소시 나오는 연기는 그 주성분이 수증 기이다. 총이나 소구경 포 발사시에는 무연이거나 거의 무연성이 라고 할 수 있다. 그러나 대구경 포는 상당한 양의 연기를 발생 한다. 포신 내 부 혹은 탄띠 (driv i n g band) 에 서 찢 겨 진 금속 성 분 이 추전제 연소가스 성분에 섞이면 스모크의 원인이 된다. 앞 절에서 언급했던 섬광을 억제하기 위한 대부분의 방안들은 스모크 생성량을 증가시킨다. 예를 들면 칼륨영은 흰색의 스모크 를, 방향족 니트로 화합물은 염소될 때 불완전 연소 상태의 탄소 로 인한 검은 색깔의 스모크롤 배출한다. 니트로구아니딘만이 스 모크롤 크게 증가시키지 않는 첨가제이며, 착화제로 사용되는 혹 색화약 역시 연소시 상당량의 스모크롤 배출한다. 2.6 침식 탄을 발사하는 빈도가 많아짐에 따라 추전제 장약으로 인해 총 신(총강)은 마모되기도 하고 부식되기도 한다• 발사 횟수가 크게 증가하면 그 마모율이 매우 높아지며, 특히 대구경 포에서 그 현 상이 많이 나타나 결국 탄의 정확도가 크게 감소된다. 이와 같은 추전제 장약에 의한 침식은 무엇보다도 그 불꽃 온도에 따라 좌 우된다. 복기 추전제는 폭발열이 높으므로 단기 추진제보다 더 높은 불꽃 온도를 갖기 때문에 더 큰 침식현상을 나타낸다. 복기 추전제의 폭발열을 감소시키기 위해 섬광 억제를 위해 사용하는
바셀린, 센트럴라이트와 같은 불활성 물질들을 첨가하거나 낮은 폭발열을 지닌 DNT 등을 첨가시켜 침식을 감소시키기도 한다. V i e ill e 는 8) 다음 그립 2 .11 과 같이 직 경 1.3 mm 의 금속 플러 그 오리피스 (or ifi ce) 가 부착된 완전 밀폐형 압력 봄베 (manometr i c bomb) 내에서 침식현상에 대해 광범위한 연구를 하였다. 추전제 장약 연소 후 오리피스를 통해 빠져나가는 뜨거운 가스는 플러그 를 침식시키므로 실험 전과 후의 풀러그 무게를 칭량함으로써 침 식 정도를 알 수 있다. 죽, 추전제 장약의 침식 효과는 풀러그의 무게 감량으로 계 산될 수 있다. V i e ill e 는 1000 kg /c m2 이 하의 압력은 침식에 거의 영향이 없다는 것을 밝혔으며, 1000 kg /c m2 이상 2000k g /cm2 로 압력을 높임에 따라 침식이 증가되었고 2000 k g /cm2 에 서 4000 k g /cm2 로 압력 을 높인 결 과 압력 증가에 따른 침식 효과가 별로 나타나지 않았다고 보고하였다. 금속의 침식은 주로 그 용융점에 따라 변한다. 여러 종류의 금속들에 대 해 추진제 장약 l g당 오리피스로부터 감소되는 침식량(침식된 금 속의 부피 , mm3) 은 다음과 같다•
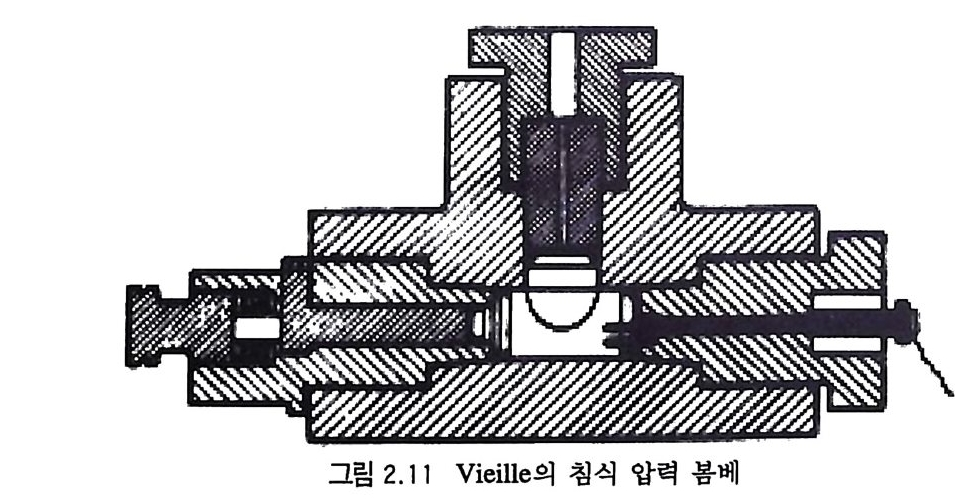 그립 2.11 V i e ill e 의 침식 압력 봄베
그립 2.11 V i e ill e 의 침식 압력 봄베
백 금 (pla ti nu m) 59mm3 백금-이 리듐(p la ti num- i r i d i um) 74mm3 놋쇠 (brass) 326 mm3 아연 (zin c ) 1018 mm3 V i e ill e 는 이와 같이 금속이 침식되는 원인을 금속이 용융된 후 용융된 물질이 튀어나가는 현상으로 해석하였다. V i e ill e 의 실험결과는 표 2.10 과 같다. 이 표에서 추진제 장약 폭발시 낮은 연소온도 때문에 침식이 약하게 나타난 니트로구아니딘의 경우가 특기할 만하다. 이상과 같은 분해가스의 온도나 가스의 압력 등 물리적인 영향 이의에도 침식에는 화학적인 영향도 관계된다. Monn i는 NC 계 추전제 에 탄소 (charcoal) 를 첨 가하면 침 식 현상이 감소한다는 사
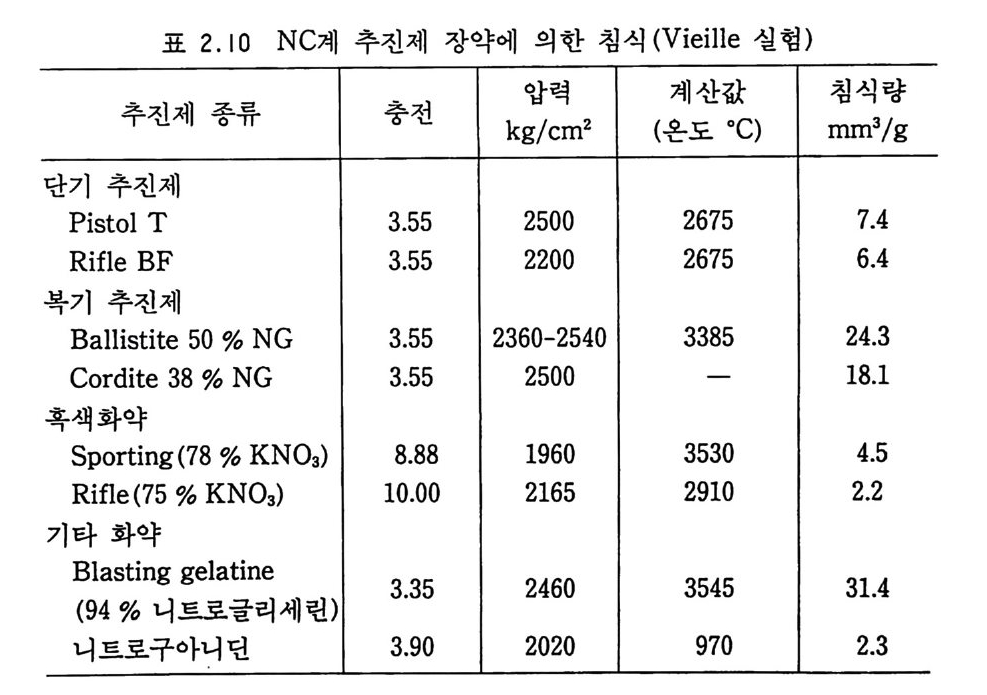 표 2.10 NC 계 추진제 장약에 의한 침식 (Vi ei ll e 실험)
표 2.10 NC 계 추진제 장약에 의한 침식 (Vi ei ll e 실험)
실을 발견하였다 .37) 그 이유는 탄소를 여분으로 가해 줌으로써, 고온에서 일어나는 다음과 같은 탈탄화 (CO2 의 영향)되는 강철울 보호하여 오히려 탄화시키는 작용을 하기 때문인 것으로 설명된 다. 강철을 탈탄화 (decarboniz a ti on ) 시 키 면 금속의 가공도가 증가 되며, 그 기공 속으로 가스들이 흡착되어 침식작용을 강렬하게 만든다. 왜냐하면 고온, 고압의 가스가 기공에 들어가면 기공을 확장시켜 결국 기공을 터뜨리게 되기 때문이다. C02+C 一 2CO 결론적으로 침식을 감소시킬 수 있는 요인들은 다음과 같다. 첫째 낮은 압력, 둘째 낮은 폭발(연소)온도, 셋째 추전제 장약 의 균일한 점화, 균일한 연소 및 완전 연소, 넷째 연소 분해가스 성분 중 수소가스 함량이 가능한 한 높고, 일산화탄소와 이산화 탄소 함량은 가능한 한 낮을 것 등이다. 침식을 극소화하기 위해 서는 700 k g /c 쿄 정도의 폭발열 및 21oo ·c 정도에서 연소하는 추진제 장약을 사용하는 것이 바람직하다고 알려져 있다. 2.7 추전제의 안정성 2.7.1 안정성 NC 계열 추전제가 제작되고 실제로 적용된 이후 그 안정성 (s t ab ility)에 관해 수많은 실험들이 행해졌다. NC 추전제의 안 정성에서 특이한 점은, NC 를 알코올-에테르 혼합액에 부분적으 로 녹여 얻은 NC 추진제의 안정성은 그 원료인 NC 자체보다 못한(떨어지는) 화학적 안정성을 지녔다는 것이다•
V i e ill e 는 NC 자체와 NC 추전제를 110 °c 로 가열하여 실험한 결과 0.04 cm3 NO/hr/ g r 의 산화질소가스를 발생 하는 탈니트로화 반응을 하는 데 비해, 이 NC 로부터 제작된 추진제는 0.1 0 ~0.1 5 cm3 NO/hr/ g r 의 거 의 두 배 속도로 탈니 트로화 반응을 일으키 는 것으로 보고하였다. 이와 갇은 NC 계열 추전제의 시간에 따른 불안정성 증가(탈니 트로화 반응)를 해결하기 위해 각 국가별로 많은 노력을 기울여 왔다. V i e ill e 는 많은 실험을 조직적으로 하였으나 그는 매우 잘 못된 결론을 얻었다. 죽, 그는 오랫동안 저장되어 분해반응이 시 작된 것으로 보이는 추전제들을 조사하였다. 그는 이 오래된 추 전제들이 새로 제조된 추진제에 바해 용매의 잔류량이 적다는 사 실을 중시하였다. 새로이 제조된 추진제는 1 % 정도의 용매를 함유하고 있으나 오래된 추전제는 1 %보다 훨씬 적은 양의 용매 를 함유하고 있었다. 이 사실로부터 V i e ill e 는 추전제의 안정성 은 함유한 용매의 손실에 의해 감소되며, 따라서 추진제의 안정 성은 화학적으로 유사하면서 덜 휘발성인 용매를 가함으로써 개 선될 수 있다고 결론내렸다. 실제로 그 당시 (1896 년경) 2 %의 아 밀 (am y l) 알코올을 가해중으로써 안정성은 개선되었다• 그러나 10 년 뒤 (1906 년), 이 추진제 역시 분해되는 현상이 나타났으므로 알코올의 양을 8% 로 증가시켜 가하게 되었다. 이 추진제들을 AM 으로 표시하였는데 이는 아밀 알코올 함량에 따라 AM2, AM8 등으로 분류하였기 때문이다. 1905 년 일본의 전함 〈미카사〉 호에서 영국제 복기 추진제(염화 수은을 포함한 장약)가 폭발하였다. 1907 년에는 프랑스 전함 〈J ena 〉에 저장되었던 탄약이 폭발하는 참사가 발생하여 프랑스 롤 위시한 세계 각국 여론이 이 사고에 대한 항의소동이 있었다. 이러한 함정에서의 탄약 폭발 사고에 대한 원인 규명 조사가 계
속되는 동안 1911 년에는 1906 년 제조된 아밀 알코올 8 %를 포함 하는 추전제 장약이 전함 〈 L i ber t e 〉 를 폭파시켜 버렸다. 따라서 추진제 장약을 저장하는 동안 분해반응이 일어나는 것은 잔류 용 매가 증발해 버린 때문이 아니라는 점이 분명해졌다 .38) 이와 같은 사고들로부터, 니트로셀룰로오스가 완벽하게 정제되 었을지라도 부분적으로든 전체적으로든 콜로이드상이라면, 완전 히 안전하다고는 할 수 없다. 그후 계속된 조사에 의해 상온보다 높은 온도 상태에 있는 추전제의 안정성에는 여러 가지 요인들이 관계된다는 것을 알 수 있었다. 예를 들어 75~80 °C 의 온도에 있는 추전제의 안정성을 조사한 결과 추진제 웨브 두께 (web t h ic kness) 가 커질수록 안정성은 더욱 높아지는 것을 알 수 있었 는데 이와 같은 관계는 Brunsw ig이 그린 그림 2.12 에 나타나 있 다 .13) 커브 1 은 가느다란 튜브형 추진제 장약 (3.7cm 화포)의 무 게 손실이며, 커브 II 는 약간 두꺼운 장약 (7.5cm 화포), 커브 Ill 은 더 두꺼운 장약 (15cm 화포)의 경우이고, 커브 W 는 매우 두꺼운 장약 (20cm 화포)인 경우, 커브 V 는 가장 두꺼운 장약 (30cm 화
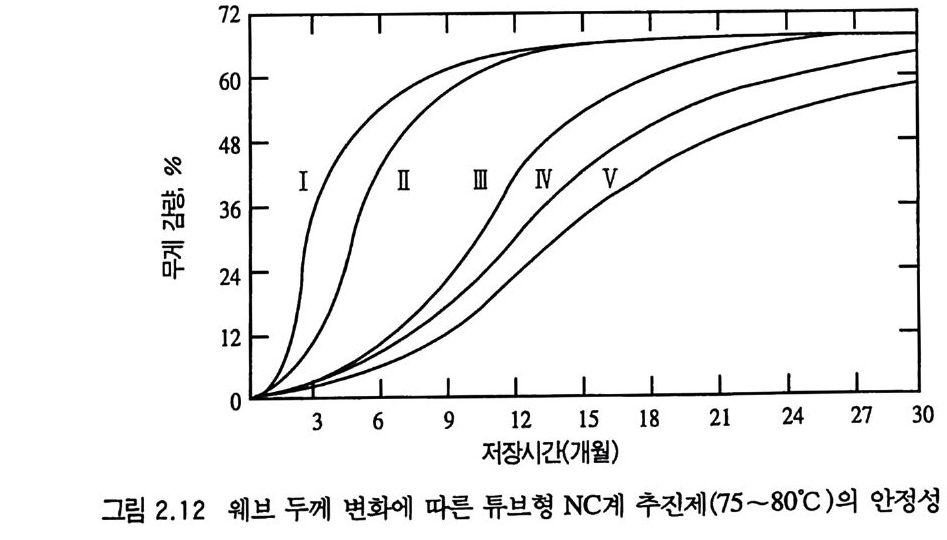 48
48
포)의 경우이다. 가장 가느다란 장약의 경우 2 -1t 개월 뒤 25 %의 무게 손실이 있었으며 웨브 두께가 두꺼워지는 순서에 따라 같은 양의 무게 손실 (25 %) 이 각각 4, 9, 10¾, 12¾ 개월 후에 일어났 다. NC 자체보다 NC 추진제의 안정성이 더 낮아지는 원인은 추 전제에 잔류하는 용매의 함량과 용매로 인한 산화반응 생성물 때 문으로 설명할 수 있다. 즉, 가느다란 그레인의 추진제는 무게에 대한 표면적 바율이 높기 때문예 산화반응이 더 강하게 일어난 다. 잔류 용매는 산화반응에 의해 분해 생성물을 더 많이 만들게 되며, 이 물질들은 추전제 속의 NG 또는 NC 를 분해(파괴)하는 작용을 하게 된다. 다음 표 2.11 에는 공기가 추전제 안정성에 해 로운 영향을 주는 실험 결과로서, 사용된 추진제는 NC 계열의 단기 추전제이며 알코올과 에데르의 용매에서 압출 성형된 제품 이다. 용매가 추전제 내에서 일으키는 반응에 대해 많은 연구가 이루 어졌으며 결국 아밀 알코올은 NC 가 분해될 때 생성되는 질소산 화물들에 의해 아밀 아질산염 및 질산염으로 변환된다고 밝혀졌 다. 이렇게 생성된 물질들(아밀 유도체들)은 발레르산 및 아밀 발
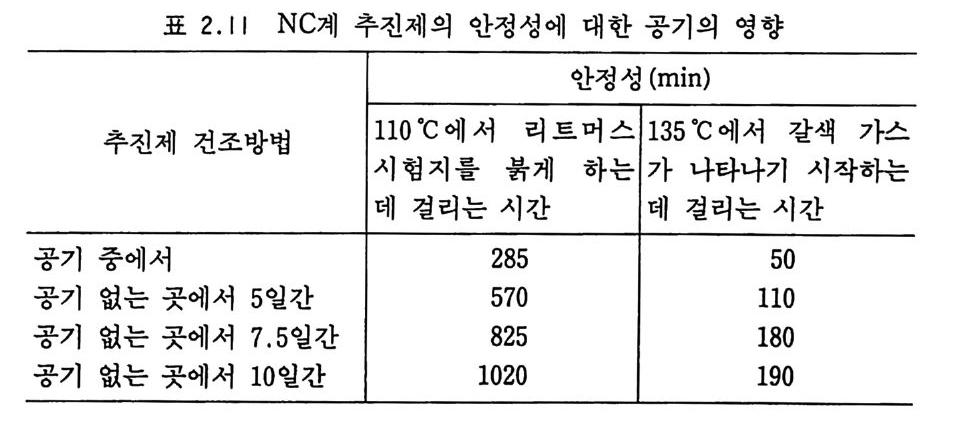 표 2.11 NC 계 추진제의 안정성에 대한 공기의 영향
표 2.11 NC 계 추진제의 안정성에 대한 공기의 영향
레레이트로 산화되는 것으로 보고되었다 .39,40) 이상과 감은 실험 이의에도 많은 연구자들이 추전제의 분해현 상에 대해 연구하였으며, 그 결과 실제로 추전제의 분해반응에 가장 중요한 영향을 주는 요인은 온도라고 밝혀졌다. NC 계 추 진제를 고온에서 저장할 때는 추진제의 분해속도가 NC 혹은 NG 자체와 별로 다르지 않다. 온도를 올리게 되면 추전제의 분 해반응이 더 활발해지며 질소는 NO 와 N02 의 질소산화물로, 탄 소는 CO 및 CO2 로 분해된다. 수소는 주로 물 (H20) 로 분해되지 만 온도가 증가할수록 물의 생성 속도는 감소하게 된다. 그러나 상온이나 상온 근처의 온도에서 추전제를 저장하면 분해 메커니 즘이 상당히 다르게 나타난다. 이때는 -ON02 기가 산화제로서 분자 상호간의 산화반응에 관계 하며 대 기 중의 산소와 잔류 용매 역시 산화반응에 참여하게 된다. 열대성 기후에서 사용되는 추전제는 온대성 기후에서 사용되는 추진제보다 더 높은 안정성을 필요로 하게 된다. 따라서 해군용 추전제는 열대성 지역에서의 사용빈도가 많으므로 높은 안정성을 요구하고 있다. 대기 중의 수분은 추전제 안정성에 해로운 영향을 준다고 알려 져 있다. S t orm 은 65.5 °C 에서 400 일 동안 보관시 별다른 분해반 옹을 하지 않은 상당히 안정된 추진제가 수증기로 포화된 대기압 내 같은 온도 (65.5°C) 에서는 175 일 지날 때부터 분해반응이 시작 되었다고 보고하였다 .41) 분해반응에 대한 수분의 영향은 홉습성 이 있는 NC( 단기) 추진제에서 특히 심각하며, 홉습성이 비교적 적은 복기 추전제에는 그 영향도 비교적 적다• Brunsw ig 13) 에 의 하면 튜브형 복기 추진제는 46 °C 에서 6 개월 동안 저장했을 때 건조한 조건에서와 습한 조건에서 모두 추전제 무게 손실이 동일 했다고 발표하였다. 또한 수분방지제인 바셀린을 포함하는 복기
추진제 (cord it e 형)는 습한 상태에서의 안정성이 건조한 곳에서와 같았다. Brunsw ig에 의하면 복기 추진제에 3 %의 바셀린을 첨 가하면 수분의 영향을 막을 수 있다고 보고하였는데, 바셀린을 포함하는 추전제는 46°C 의 습한 분위기 속에서 6 개월 동안 저장 하였어도 무게 손실이 없었으나 보통 추전제는 같은 조건에서 7. 1% 의 무게 손실을 나타낸 것으로 알려졌다. 그러나 바셀린을 포 함하는 단기 추전제는 그 내탄도적 특성이 너무 낮아지므로 바셀 린을 첨가하는 방법은 단기 추진제에 대해 사용해 볼 의미가 없 다고 판단된다. 질소산화물은 추진제의 안정성에 매우 해롭게 작용하며 탈니트 로화 반응 (denit ra ti on ) 을 촉전시 킨다. V i e ill e 는 다음 표 2 .12 와 갇이 이산화질소를 포함하는 40 °C 공기 중에서 보관한 단기 추 진제의 저장시험을 통해 이산화질소의 영향을 조사하였다 .8) Bru i n 과 Pauw 등은 추진제의 안정성에 대해 광범위한 조사를 한 결과 NC 계 추전제의 분해가스들 중 특히 질소산화물과 수증 기는 110 °c 에서 추전제의 분해속도를 상당히 증가시키는 것을 발견하였다 .42) 이것은 분해가스 생성물을 흡수하는 흡수제를 시 험장소에 장치하여 분해가스 생성물을 제거하면 그 무게 손실 기
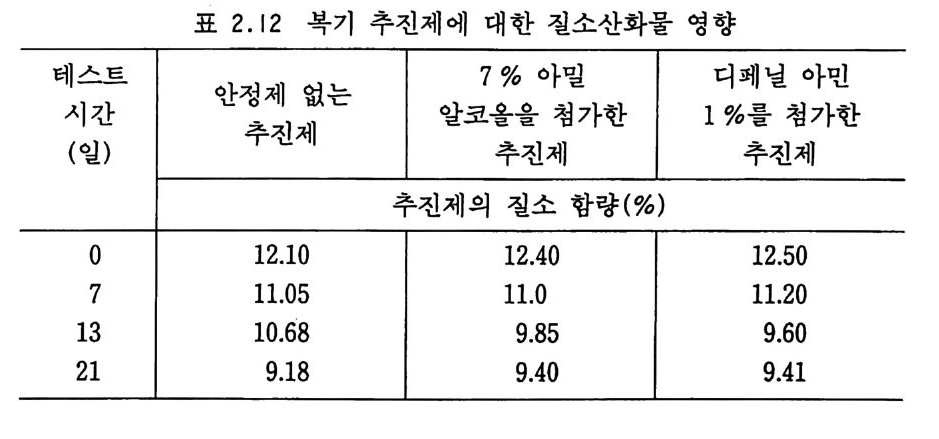 표 2.12 복기 추진제에 대한 질소산화물 영향
표 2.12 복기 추진제에 대한 질소산화물 영향
간이 길어지는 현상으로부터 증명되었다. 그림 2.13 에는 추전제 의 분해 생성물(가스)을 흡수할 수 있는 여러 종류의 흡수제 (absorbin g ag e nt) 들을 장치 하고 110 °C 에 서 단기 추전제 의 안정 성을 시험한 결과이다. 커브 VIII 은 흡수제가 없는 경우로서 가장 빨리 분해가 일어났다. 화약의 분해를 가장 효과적으로 막아준
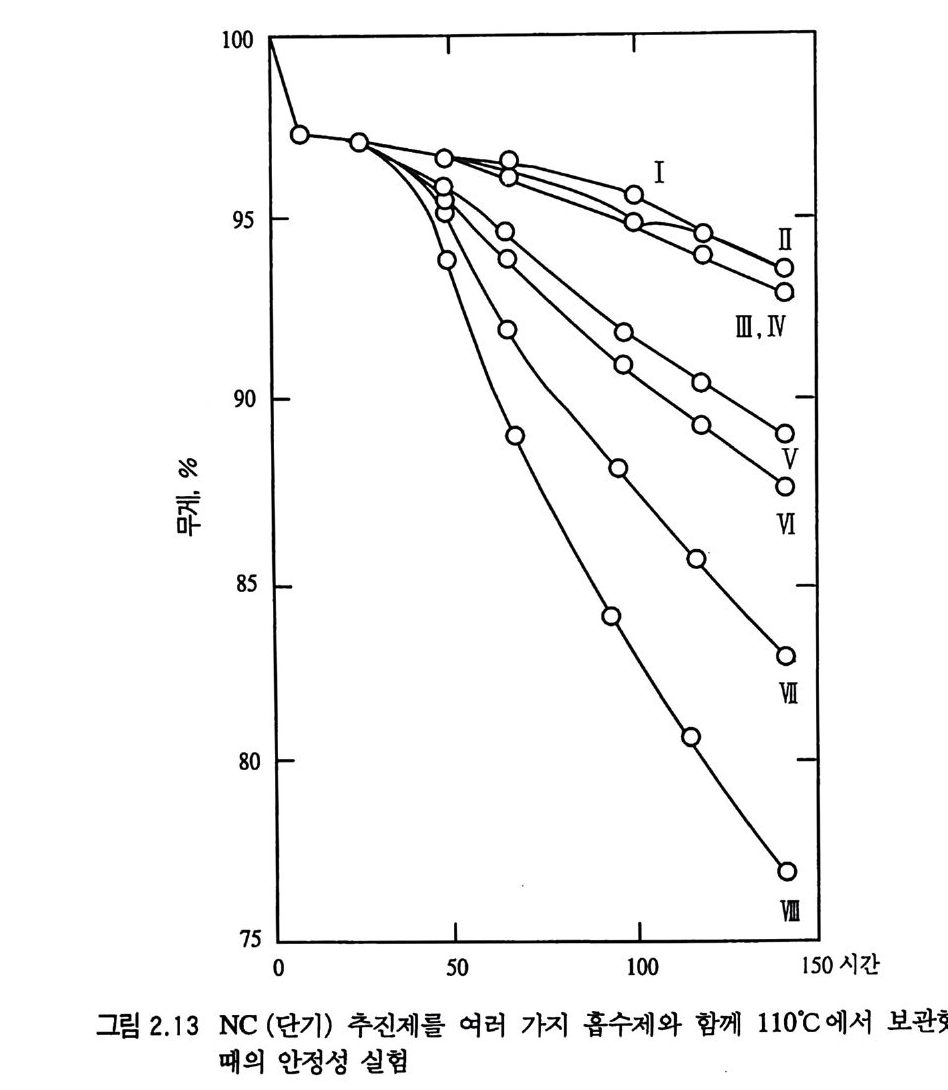 100
100
물질은 무수인산(p hos p hor ic anh y dr i de) 을 사용한 커브 I (물과 결 합), 산화칼슘을 사용한 커브 Il (물 및 N02 와 결합), 카바마이트 (carbami te) 를 사용한 커 브 Ill (NO 파 결 합) , 활성 탄소를 사용한 커브 1V( 물 및 N02 와 결합)였다. 그러나 탄산소다룰 사용한 커브 V (물 및 N02 와 결합하고 CO2 를 발생), 바셀린을 사용한 커브 W (질소산화물을 흡수), 무수황산구리를 사용한 커브맨(물과 결합)의 경우는 그 효율이 좋지 않았다. 이와 같이 추진제가 분해되어 질 소산화물을 발생하기 시작하면 추전제의 탈니트로화 반응은 안정 제의 유 • 무에 관계없이 급격히 빨라지게 된다. 또한 영산이나 황산 증기 같은 산성물질들 역시 추진제의 분해 반응에 비슷하게 작용하여 분해를 촉진시킨다. 만일 추전제가 분 해되어 생성된 분해물질(산성)이 깨끗한 추전제와 섞이게 되면 깨끗한 추전제가 분해되기 시작한다. NC 계열의 추진제가 분해 되 어 나온 분해 물질들은 개 미 산(fo rm ic aci d) , 히드록시 파이루빅 산, 히드록시부틸산 및 옥살산 등을 포함하고 있는 것으로 밝혀 졌다. 그러므로 추진제 내에 수분 함량이 증가할수록 분해속도는 빨라지게 되며 분해 생성물(산성)들은 알칼리성 안정제들을 파괴 한다. Brunsw ig은 다음과 같은 실험을 하였다. 13) 죽, 박편형 추진제 (30 g)를 유리판 위에 올려놓고 그 가운데에 분해 중인 튜브형 추전제 (산성 분해물질을 포함한) 소량을 울려놓은 뒤 시계집시로 덮고 수증기가 포화된 공기 속에 방치하면, 하루 이틀 지나면서 튜브형 추전제는 점성이 있는 반죽상 물질로 변화되고, 한두 주 일 후 박편형 추전제는 점차적으로 분해되어 가운데 튜브형 추전 제를 중심으로 원형의 모양을 그리면서 분해가 계속되는 것을 볼 수 있다. 따라서 산성의 분해물질들, 특히 질소산화물들을 중화 제거시키기 위해 어떤 추전제에는 중탄산소다 (NaHC03) 를 추진
제 조성으로 첨가하는 경우도 있다. An g el i와 Jo lles 등은 추진 제 내 의 아질산나트륨 양을 알아내 기 위 해 color i me t r i c 법 (추전제 를 물로 추출한 후 아닐린과 디메틸아닐린으로 반응시킴)을 확립하 였으며, 이 방법을 추전제의 분해도 (de g ree of decom p os iti on) 를 계산하는 방법으로 사용했다 .43) 아질산염의 양은 보통 추진제 100 g당 0~145m g을 나타냈다. J olles 와 Socc i 역 시 복기 추진제 내 아질산나트륨 양의 분포 도를 조사하였으며, 그 결과 튜브형 추진제의 내부와 표면에서의
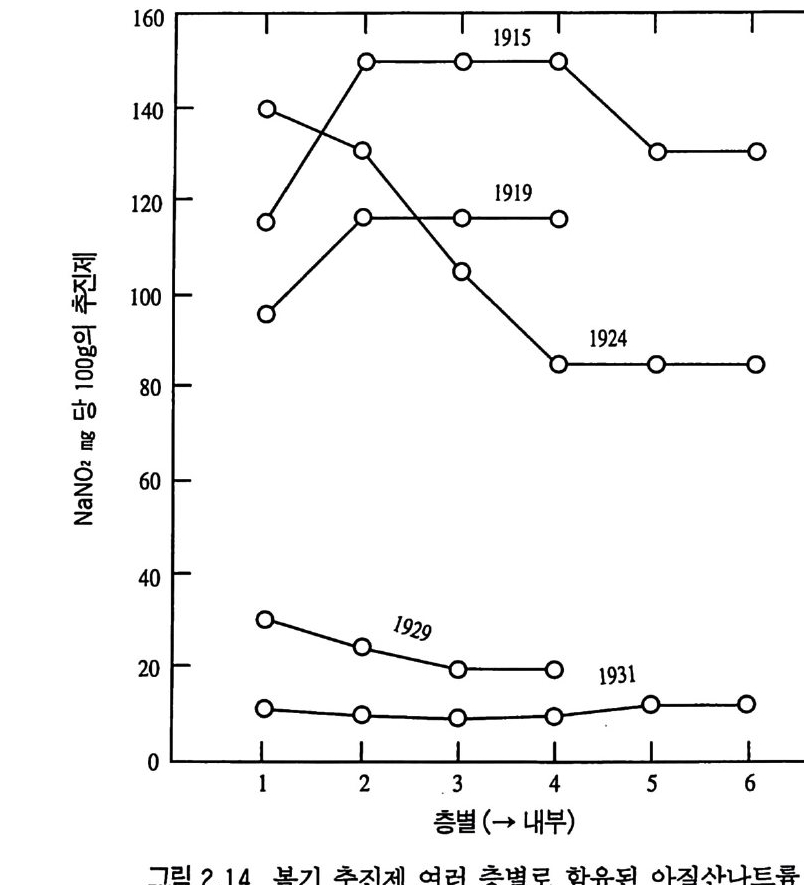 160 1915
160 1915
아질산염 분포도는 서로 다르게 나타났다 . 4 4) 제작된 지 오래되지 않은 추진제의 경우는 표면 아질산염 양이 내부보다 높았으나, 오래된 추진제일수록 내부의 아질산염 함량이 표면에 비해 점점 더 많아지는 것을 알 수 있었다(그림 2.14). 가열에 의해 산화질소 (N204~2N02) 를 발생하는, 죽 분해되 고 있는 추진제는 상온에서 약간의 열을 방출하고 있음이 Urbanski 등에 의해 확인되었다 .39) 그러나 본격적으로 분해되기 직전인 이 단계에서는 산화질소를 충분히 발생하지 않기 때문에 상온에서 큰 발열효과는 나타내지 않는다. 추전제의 발열반응을 시험하는 방법으로 silv e red vessel t es t가 있다. 이 시험은 추진제 시 료를 80 °C 로 가열하여 발열반응이 나타날 때까지 걸리는 시간을 측정하는 방법이다. 죽, 발열반응으로 인해 주위온도 (80°C) 보다 2 °C 정도 온도 상승이 관찰될 때까지 걸리는 시간을 측정하는 데, 우수한 추전제는 이 시험에서 500 시간을 견디기도 한다 (500 시간 동안 발열 분해반웅이 나타나지 않음). 82 °C 로 온도가 오르기 직전이나 직후 질산 혹은 아질산이 추진제로부터 분해되어 발생 하며, 가끔씩은 82°C 로 온도가 상승한 후 폭발되기도 한다. 복기 추전제를 안정제 없이 듀어 (dewar) 용기 내에서 95 °C 까 지 가열한 결과 de Bru i n 은 다음과 갇은 온도상승 데이터를 얻 었다 .45)
 경과시간
경과시간
강한 영기들은 이미 언급한 바와 같이 NC 계 추진제의 안정성 에 좋지 않은 효과를 나타낸다. 예를 들어 피리딘{pyri d i ne) 계 화
합물들은 NC 를 분해시키며, 가열할 경우는 110 °C 근처에서 분 해반응이 강해져 폭발 현상을 일으키기도 한다 .46) 제 1 차 세계대 전 이후 NC 계 추진제의 안정성에 대한 바닷물의 영향이 조사되 었다. 죽, 추전제를 바닷물 속에 수년간 담가 방치하였으나 추전 제의 안정성 변화나 분해반응 같은 흔적을 찾을 수 없었으며, 이 는 바닷물 일정 깊이 아래에는 바닷물의 온도가 낮기 때문인 것 으로 해석되었다. 햇빛은 추진제의 분해를 촉진시키는 원인이 되고 있다. Berth e lot 동은 여러 종류의 안정제를 포함하는 추전제에 대한 햇빛의 영향을 조사하였다 .47) 그 결과 아밀알코올로 안정화된 추 진제는 디페닐아민을 사용한 경우보다 햇빛에 더 강한 것으로 나 타났으며 햇빛의 영향에 의해 디페닐아민은 분해되는 것으로 밝 혀졌다. 따라서 NC 계 추진제는 그 모든 제조 단계에서 햇빛에 노출되지 않도록 주의하여야 하며, 추전제 제조공장의 모든 창문 온 가능한 한 북쪽으로 향하도록 설계하고, 가능한 한 단파장의 빛을 차단시킬 수 있도록 푸른색이나 노란색깔의 페인팅을 하고 있다. 2.7.2 안정성 시험 NC 계 추전제의 안정성 시험은 NC 자체의 안정성 시험과 대 부분 유사하며, 그 원리는 추전제를 가열하여 상온에서의 분해속 도보다 그 분해 반응속도를 빠르게 하는 데 있다. V i e ill e 는 1 시 간 동안 추전제를 가열하는 것은 대략 다음과 갇온 조전으로 분 해시키는 것과 비슷하다고 발표하였다 .48) U) 75 °C 에서 24 시 간 동안 가열 보관. ® 60 °C 에서 7 일 동안 가열 보관.
@ 40 °c 에서 30 일 동안 가열 보관. NC 추전제 제조공장에서 실제로 사용되는 검사 방법 중의 하 나는 추진제 제조 로트(l o t)별로 500 g씩을 취하여 완전 밀폐된 용기 속에 넣고 30~50 °C 로 유지시키는 것이다. 이때 각 용기 속에 들어 있는 추전제 표면 위에 메틸 바이올랫 시험지 (crys t a l v i ole t과 rosan ili ne 으로 착색된 시험지)를 놓아두면, 추전제가 분해 됨에 따라 시험지의 색깔이 변하는 것을 관찰할 수 있으므로 제 조 로트별로 추진제의 안정성을 시험 비교할 수 있다. 추진제의 안정성을 비교하는 또 다른 방법은 특별히 제작된 상자 속에 추 전제를 넣는다. 이 상자에는 배기밸브 및 튜브가 연결되어 상자 내 에서 추전제 분해로 발생한 가스들이 요오드칼리 (KI) 및 전분 용액이 들어 있는 용기를 통과하도록 장치되어 있다. 추전제 분 해로 인해 이산화질소 가스가 발생하면 KI/ 전분 용액 색깔 변화 로 죽시 감지할 수 있다. 이상과 같은 방법들 이의에도 추전제의 분해, 안정성 시험을 하는 간단한 방법들은 다음과 같다. (D Abel 열시험 (Kl- 전분 시험지하에서 75~80 °C 로 가열) @ 134.5 ·c 에서 추전제 (단기)롤 보관, 안정성을 시험하여 만일 이 온도에서 45 분 이내에 산화질소 가스를 생성하거나 5 시간을 견 디지 못하고 폭발하면 사용할 수 없는 추진제로 폐기 처리한다. 이 시험은 메틸 바이올렛 시험과 병행 적용될 수 있으며, 이 경우 30 분 또는 일정 기준 시간 이상은 그 시험지의 색깔 변화가 없어야 한다. ® 복기 추전제는 120 ·c 에서 상기 단기 추진제와 마찬가지로 산 화질소 가스의 분해 생성물이 45 분 이전에는 나타나지 않아야 하 며, 5 시간 이내에 폭발되지 않아야 하는 등의 조건이 있다. 마찬가 지로 30 분 이내에 메틸 바이올렛 시험지의 색깔 변화가 없어야 한 다.
® 푸른 리트머스 시험지 존재하에 110 °C 에서 가열(환원성)시켰 을 때 10 시간 이내에는 리트머스 시험지가 적색으로 변하지 말아야 한다. @ 110 °c 의 온도로 가열하는 Vi ei lle48 ) 시험에서는 추진제 시료를 매일 10 시간씩 가열하거나 리트머스 시험지에 적색이 나타날 때까 지 가열한 후 나머지 14 시간 동안 공기를 통과시킨다. 이러한 과정 울 매일 반복하여 리트머스 시험지의 적색 착색이 1 시간 이내에 생 길 때까지 계속한다. 이 시험과정을 예로 들면 다음과 같다.
 첫째 일, 10 시간
첫째 일, 10 시간
이렇게 계산된 x 시간이 70 시간 이상이 되면 안정성이 좋은 추 전제로 분류될 수 있다. 추전제 공장 내에서 정량분석법은 공정검사를 위해 자주 사용 되는 방법은 아니지만 Ber g mann 과 J unk 에 의해 고안된 방법, 49) 죽 추전제에서 발생된 산성물질의 양 (NO 로 계산)을 적당한 염 기로 적 정 (titra ti on ) 하여 분석 , 계 산하는 시 험은 흔히 사용되 고 있다. 이때 132 °C 에서 2 시간 동안 가열하여 발생된 NO 의 양 은 추전제 l g당 2.5 cm3 의 NO 함량을 초과하지 않을 것을 요구 하고있다. Jol les 등에 의하면 추진제 내의 아질산나트륨 양을 비색법으 로 결정하는 것은 화약의 안정성을 추측하는 데 도움이 될 수 있 는 방법이며, 그 결과는 열시험 결과와도 잘 일치한다고 보고하 였다 .43,44> Toneg u tt i 등은 이 시험이 120 °C 의 메틸 바이올렛 시
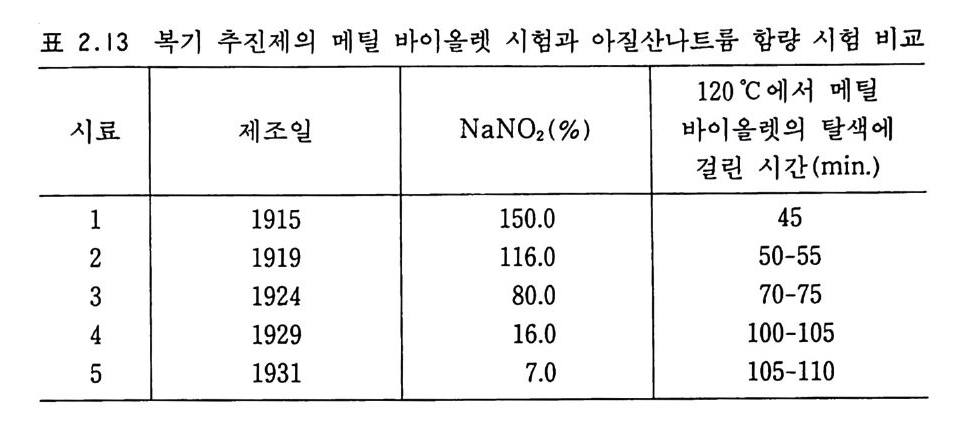 표 2.13 복기 추진제의 메틸 바이올렛 시험과 아질산나트륨 함량 시험 비교
표 2.13 복기 추진제의 메틸 바이올렛 시험과 아질산나트륨 함량 시험 비교
험결과와도 잘 일치한다고 보고하였다 .50) J olles 와 Socci에 의한 Tonegu tt i 시험결과와의 일치성 및 비교 데이터가 다음 표 2.13 과 같이 나타나 있다. 44) 2.7.3 안정제 2. 7. 3.l 디페닐아민 NC 계 추진제(단기 추진제)를 안정화시키는 데 크게 기여한 것 은 Alfr ed Nobel 에 의해 제안된 디페닐아민 (d ip hen y lam i ne) 을 추 전제에 첨가하는 방법이었으며, 이 방법은 독일에서 처음으로 실 용화되었다 .51) 독일에서 디페닐아민을 추진제에 적용한 것은 비 밀리에 행해졌지만 1896 년경 프랑스에서는 독일에서 제조된 추진 제에 2 %의 디페닐아민이 포함되어 있음을 알아냈다. 그러나 프 랑스에서는 디페닐아민이 너무 강한 염기성이므로 추전제에 첨가 되면 NC 를 가수분해시키기 쉬울 것으로 간주하였다. 그럼에도 불구하고 프랑스의 전함 Lib e rte 호의 참사가 일어나자 안정제로 서 디페닐아민 사용을 시도하게 되었고, 1911 년 그 성능이 입증
되었다. 추전제에 안정제를 첨가하지 않은 경우, 아밀알코올을 가한 경 우 및 디페닐아민을 첨가한 경우에 대한 결과는 다음과 같다. 죽 75°C 의 온도, 건조한 공기 중에서 2% 디페닐아민울 함유한 추 진제는 같은 온도와 건조한 공기 중에서 같은 시간 동안 가열 보 관한 8 % 아밀알코올울 함유한 추전제가 발생시킨 가스 양의 1/ 4 만을 발생하였다. 110°c 의 온도에서는 디페닐아민을 함유한 추 진제는 아밀알코올울 함유한 추진제보다 약 2.5 배 더 안정한 것 으로 증명되었다. 1.5 % 디페닐아민을 함유한 추전제는 75 °C 의 건조 공기 속에서 가열 보관될 때 512 일 만에 분해되기 시작하는 반면 2 % 아밀알코올울 함유한 추전제는 같은 조건에서 122 일이 지날 때부터 분해되기 시작하였다. 75 °C 의 습한 공기 속에서는 2 % 디페닐아민을 함유한 추진제는 8 % 아밀알코올을 함유한 추 진제에 비해 약 4 배 정도 더 늦게 분해되었다. Ber g er 는 40~110 °C 의 온도 범위에서 추진제 (단기)의 분해도 (deg ree of decomp o sit ion ) 를 다음과 같이 정 의 하였 다. 52) 즉, 새 로 이 제조된 추전제가 방출한 열량과 일부 분해된 추진제가 방출하 는 열량을 각각 결정하고, 그 열량 차이로부터 그 추전제가 분해 될 때 방출되는 열량을 계산하는 방법이다. 이와 같은 방법으로 Ber g er 는 안정제가 없는 추진제 (AT), 아밀알코올울 함유한 추 진제 (AM), 디페닐아민을 함유한 추진제 (D) 를 조사하여 다음 그 립 2.15 와 같은 커브롤 얻었다. 각 커브들은 추전제의 분해가 처 음에는 느린 속도로 진행되다가 어떤 단계에서 빨라지는 특성들 을 나타냈다. Ber g er 의 의견은 안정제가 들어 있는 D 추전제는 분해반응에 대한 저항력이 있으므로 완만한 커브가 오랫동안 계 속되며 AT 나 AM 추전제들은 오랜 시간이 되기 전에 급격한 분 해반응으로 전환되는 것으로 설명하였다.
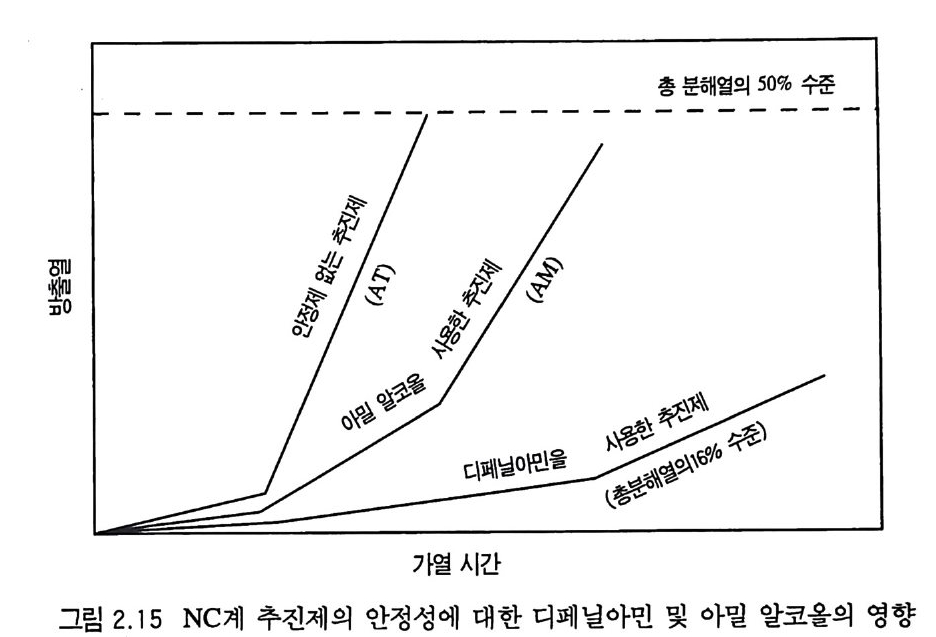 ----------- - -------충분-해열-의- 50%- 수-준 -
----------- - -------충분-해열-의- 50%- 수-준 -
여러 가지 연구 결과에 의하면 디페닐아민은 니트로셀룰로오스 를 가수분해시킬 정도로 강한 염기성은 아니지만 니트로셀룰로오 스의 부분적 분해 반응, 불순물에 의 한 분해반웅, 또는 잔여 용매 의 산화반응으로부터 발생하는 산성의 분해물질들을 충분히 중화 시킬 수 있을 정도의 강한 영기성을 갖고 있는 것으로 알려졌다. 또한 디페닐 키 아민의 영기성은 추진제 내에서의 함량이 5 %를 초 과할 때는 추진제 안정성을 오히려 해치는 것으로 증명되었다. 따라서 최적의 안정제 함량은 추전제의 1~2.5 %의 범위이고, 디 페닐아민을 사용할 때라고 밝혀졌다. Demou gi n 과 Landon 은 단기 추전제의 안정성을 조사하였는데 1.0 2~7.8 % 디 페 닐아민을 포함하는 추전제를 110 °c 로 가열 보 관하면서 시험하였다 .53> 160 시간 동안 가열 보관 후 추진제로부 터 NC 를 분리하여 NC 내의 질소 함량을 실험적으로 결정하였 다. 실험 결과 표 2.14 에 의하면 디페닐아민이 7.8 %를 함유한
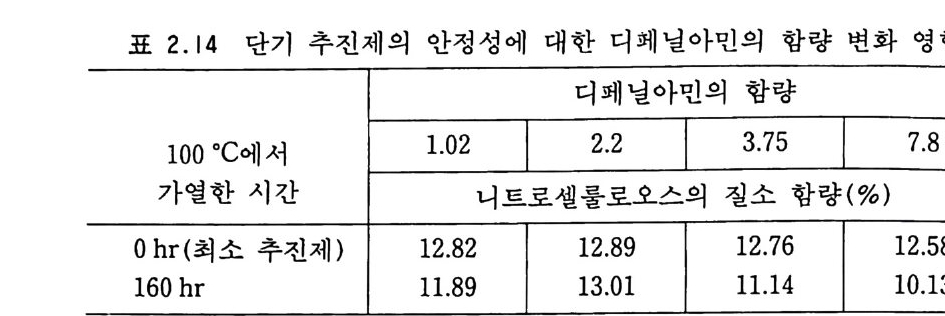 표 2. 14 단기 추진제의 안정성에 대한 디페닐아민의 함량 변화 영향
표 2. 14 단기 추진제의 안정성에 대한 디페닐아민의 함량 변화 영향
추진제 시료의 경우 160 시간이 경과했을 때 NC 중의 질소 함량 변화가 가장 심한 것으로 나타났다. 디페닐아민의 과다한 양은 추전제 장약으로서의 성능에 해롭게 작용하므로 일반적으로는 소총용 추진제에는 0.5~1 %의 디페닐 아민을 첨가하는 반면, 화포용 추전제에는 1.5 ~2 %를 첨가하여 사용하는 것으로 알려져 있다. 디페닐아민은 니트로글리세린과 갇은 에스테르 화합물들을 가 수분해시키므로 니트로글리세린을 포함하는 복기 추진제에는 사 용할수가 없다. 디페닐아민은 안정제로서만이 아니라 추전제 내에서 일어나는 산화반응 및 분해반응의 지시제로서도 작용한다. 오래 전부터 디 페닐아민을 함유한 추전제는 그 색깔이 초록 내지는 푸른 색깔에 서 어두운 푸른색, 갈색, 노란색 또는 검은색까지 변화하는 현상 으로부터 그 추전제가 어떠한 상태에 있는지를 감지하였었다. 예 를 들면 많은 양의 용매를 포함하고 있거나 50~60 °C 의 건조 공 기로 말렸든지 하면 추전제는 검푸른색으로 색깔이 변한다. Desmaroux, 54> Marqu eyr o l 등은 55,56) 이 색 깔의 변화가 용매 인 에데르 및 대기 중의 산소로부터 형성된 과산화물이 디페닐아민 울 산화시켜 일어난 것으로 보고하였다. 또한 금속을 일부 함유 하고 있거나 철, 구리, 아연 갇은 금속과 접촉해 있으면 추진제
는 푸른색을 띠게 된다. 추진제 내에 존재하는 금속조각 주위에 는 푸른 색깔의 흔적이 생기게 되며, 금속과 접하는 추진제 표면 에도 푸른 색깔이 나타나게 된다. 추전제의 색깔이 어둡게 변했다고 추진제 내에 분해반응이 일 어났다고 취급할 수는 없지만 디페닐아민이 어떤 종류의 화학반 응을 일으켜 새로운 화합물로 변화되었다는 것은 확실하다. 따라 서 이와 같은 색깔의 변화가 있는 추전제는 실제 적용할 수 없도 록 규정되어 있다. 추진제에서 디페닐아민이 안정제로 작용하는 메커니즘을 파악 하기 위한 연구들이 발표되었다• 그 중 Marqu ey ro l 등은 산화 제의 작용, 특히 에테르로부터 생성된 과산화물의 작용으로 인해 디페닐아민은 다음과 같이 테트라페닐히드라진( I ) 및 디페닐페 나전( Il )를 생성한다고 보고하였으며 페나진( Il )가 추진제를 검 은색으로 변화시킨다고 하였다 .56)
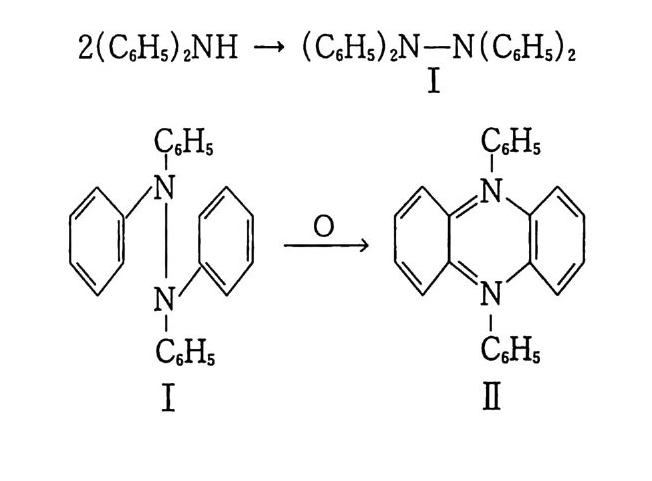 2(門C 晶c )2_N晶門NH~ H |--|-+_ N( C一。 _晶 c )I2 N-I N>(Ccs|C<_HHsS)s晶广 I 2I
2(門C 晶c )2_N晶門NH~ H |--|-+_ N( C一。 _晶 c )I2 N-I N>(Ccs|C<_HHsS)s晶广 I 2I
Dav i s 와 Ashdown 은 추전제에서 디페닐아민이 다음과 같은 반응을 거 친다고 보고하였다. 57)
 O仁〉-N-l부 H〈O그 m
O仁〉-N-l부 H〈O그 m
상기 반응에서 생성되는 N_ 니트로소디페닐아민 (III) 은 디페닐 아민과 거의 마찬가지로 안정제로 작용하는 것이 실험적으로 증 명되었다. 또한 이러한 물질들은 추전제를 강하게 착색하지는 않 는 것으로 밝혀졌다. Dav i s 와 Ashdown 은 추전제 속에 있는 이 러한 물질들을 검출하는 방법을 제안하였다. 죽, 만일 추전제를 알코올로 추출하여 그 속에 디페닐아민이 들어 있다면 과황산암 모늄 (ammon i um p ersul p ha t e) 과 반응시킬 때 푸른 색깔로 변하게 되지만, N- 니트로소디페닐아민은 과황산암모늄과 반응하여 착색 되지 않는다. 따라서 그 추출물을 과황산암모늄으로 처리하여 나 타난 색깔의 강도는 디페닐아민의 농도에 따라 좌우된다. 한편 N- 니트로소디페닐아민은 전한 황산과 반응하여 강한 푸 른 색깔을 띠게 된다. N- 니트로소아민에 대한 또 다른 시험은 그 알코올 추출액을 a- 나프틸아민 (a-na p h t h y lam i ne) 의 알코올 용 액으로 가열 처리할 때 오렌지 색깔로 변하게 된다. 추전제가 분 해되어 생성된 질산을 포함한 산 (m i neral a ci d) 에 의해 N- 니트로 소 화합물은 재배열되어 p-니트로소디페닐아민 (IV) 가 된다. 이
물질은 쉽게 산화되어 p-니트로디페닐아민 (V) 가 된 후 니트로 화 반응을 계속 거쳐 디니트로 유도체인 (VI) 및 (Vll), 트리니트 로 유도체인 (Vlll) 을 추전제 내에서 생성하게 된다. Dav i s 와 Ashdown 은 57) 65 oc 에서 240 일 동안 밀폐용기 내에 보관하였던 추전제로부터 2, 4, 4' -트리니트로페닐아민을 분리하 였다. 디페닐아민이 N_ 니트로소디페닐아민으로 완전히 전환된 추진제는 보관상 문제가 없지만, 만일 N-니 트로소디페닐아민이 사라지고 니트로 화합물로 완전히 변화된 경우는 안정제가 없는 상태이므로 더 이상 저장할 수 없는 것으로 취급되고 있다. 디페닐아민의 니트로 유도체가 생성된 추전제는 갈색 또는 적 황색의 색깔을 띠게 된다. Schroeder 에 의하면 단기 추진제 내 에서 디페닐아민이 반응하여 변화된 화합물들을 크로마토그래피 (s ili ca) 로 분석한 결과 디페닐아민은 핵사니트로디페닐아민으로 변화될 수 있다고 발표하였으며, 만일 추전제가 71 °C 에서 258 일 동안 저장되면 디페닐아민의 1/2~2/3 는 니트로 화합물로 변환된 다고 하였다 .58) 2.7.3.2 무기물계 안정제 1867 년 Abel 은 NC 는 산 속에서 분해되는 성질을 지니고 있는 것으로 알고 있었으며, 추진제, NC, NG 속에 있는 불순물에 의해 분해 생성된 산성물질을 중화시키기 위해 탄산소다를 가해 야 한다고 제안하였다 .59) 그러나 추전제 내에 2% 이상의 탄산 소다를 가하면 추진제의 안정성에 매우 해로운 반응이 일어난다. 따라서 탄산소다 대신 알칼리성 이 약한 중탄산소다룰 사용하는 방법이 시도되었다. 그 결과, 중탄산소다 1 %를 첨가한 추전제 는 안정성에 전연 영향이 없었으나 바셀린을 함께 첨가한 경우에 는 안정성이 확실히 개선되었다. Brunsw ig은 중탄산소다와 바셀
린을 함유하고 있는 복기 추진제가 20 년간 어떤 분해반웅 없이 안정한 반면, 중탄산소다 없이 바셀린만을 첨가한 추진제는 현저 한 분해 현상을 나타냈다고 보고하였다. 탄산칼슘을 NC 에 가해 주면 추전제의 안정성을 개선하기는 하지만 탄산칼슘의 양을 상당량 사용할 때만 그 효과가 뚜렷하게 된다. Brunsw ig은 탄산칼슘울 0.1 % 함유한 추진제 를 94 °C 의 밀폐 용기 내에 보관했을 때 4 . 5 시간까지는 분해가 없었으나 그 후 산화질소가스가 발생되기 시작하여 20 시간 뒤에는 무게 손실 이 19.7% 에 달하였다고 하며, 같은 계열의 추전제에 탄산칼슘울 6% 첨가시킨 경우에는 갇은 온도 94 °C 에서 200 시간까지도 무게 손실이 0.4 % 정도였다고 발표하였다 .13) 그러나 비연소성 물질인 탄산칼슘울 대량으로 추진제에 첨가시키면 탄도적 성능에서의 손 실이 크므로 탄산칼슘을 안정제로 사용하는 것은 바람직하지 않 다. 탄산마그네슘 역시 탄산칼슘과 비슷한 작용을 하는 것으로 알려져 있다. 제 2 차 세계대전 중 독일에서는 산화마그네슘 (M g O) 을 추전제에 첨가하여 사용하였다. 0.25 %의 산화마그네 슘울 추전제에 사용하면 그 안정성이 상당히 개선되는 것으로 보 고되었으며, 산화마그네슘은 무기화합물 안정제 중 가장 효율이 높은 것으로 알려져 있다. 2. 7. 3. 3 유기화합물 안정제 디페닐아민 이의에도 많은 종류의 영기성 유기화합물들이 추진 제, 화약류의 안정제로 사용될 수 있는 가능성이 검토되었다. 아 닐린 (a nili ne) 은 제 1 차 세계대전 중 디페닐아민의 공급이 부족했 울 때 가끔 사용되었다. 그러나 아닐린은 염기성이 너무 강하므 로 추진제의 안정제로서는 효과가 거의 없었다. 하지만 디페닐아 민과 비슷한 구조를 가전 카바졸 (carbazole) 은 안정제로서 좋은
효과를 나타냈다. 프랑스의 Mar q ue y rol 은 15 년 동안 각종 안정 제들의 효과에 대해 실험하였다(표 2.1 5 ).GO) 그는 아밀알코올과 디페닐아민 이 의에 N- 니트로소디페닐아민(디페닐니트로사민), 카바졸, 디페닐 벤즈아마이드, 니트로나프탈렌 및 나프탈렌 등도 시험하였다. 시 험용 추진제는 40 °C, 60 °C, 75 °C 에서 각각 보관되었으며 추진 제가 강렬하게 분해되어 산화질소 가스를 발생할 때는 실험을 중 지하였다. 캄포 (cam p hor) 와 부틸프탈산과 같은 비휘발성 용매 (젤라틴화 물질)는 추전제의 안정화 효과가 있는 것으로 밝혀졌으며, 특히 우레 아 치 환체 (centr a li te, carbami te) 및 우레 탄 유도체들도 안정 제로서의 효과가 있는 것으로 판명되었다. 예를 들어 센트럴라이 트는 추진제 가 분해 될 때 니트로화 (n it ra ti on) 됨 으로써 니트로 유 도체가 생성되는 것으로 밝혀졌으며, 이는 니트로기가 센트럴라 이트의 방향족 고리 (r i n g)에 치환되어 도입되는 반응으로 해석된 다. Lecorche 및 J ov i ne t은 복기 추전제 속에 있는 카바마이트
 二NH
二NH
(cen t ral it e) 는 스팀에 의해 휘발성 물질과 비휘발성 물질로 변환 되는 것을 보고하였다 .61) 이때 휘발성 물질은 p-니트로페닐에틸 니트로자민( I )이며, 비휘발성 물질은 디니트로카바마이트( Il )이 다. 바셀린, 캐스터오일, 송전유 등과 같은 유기물질들도 추진제의 안정화 효과가 있는 것으로 보고되었다. Brunsw ig은 추진제의 무게 손실 인자, 죽 단위 시간당 추전제의 무게 감소량 (:『)과 추진제의 안정성울 비교하여 다음과 같은 데이터를 발표했다 .13) 이때 무게 손실 인자는 면화약과 NG 를 10 : 8 비율로 제조한 추 전제를 사용했고, 용매는 아세돈으로 사용했을 때 얻어진 값들이 다(표 2.16). Urbanski 등은 많은 실험을 통해 방향족 니트로 화합물들이 NC 계열 단기 또는 복기 추진제의 안정성을 개선하는 것으로 보고하였다 .62> 13.4 % N 을 포함하는 NC 를 120 °C 에서 5 시간 동 안 가열하면 p H=2.28 을 나타내지만 9.1 %의 p-니트로톨루엔을 가하면 p H=2.89 가 되고, 9.1 %의 2, 4- 디니트로톨루엔을 가하면 p H=3.17 이 되며, a- 트리니트로톨루엔을 같은 양 가했을 때는 p H=3.34 가 되었다. 일정 부피 속에서 같은 종류의 시료들울 134.5 °C 에 서 가열했을 때 109 mmHg 분해 압력 (분해 생 성물 가스 에 의한)을 나타낼 때까지 걸리는 시간을 측정한 결과, 순수한 NC 에 의해서는 32.5 분, NC 에 9.1 %의 p_니트로톨루엔이 첨가된 경우에는 44.5 분, NC 에 9.1 %의 2, 4- 디니트로톨루엔이 첨가된 경우에는 48.5 분, NC 에 9.1 %의 a- 트리니트로톨루엔이 첨가된 경우에는 52.5 분이 소요되었다. 그러나 이와 감은 니트로 화합물 들은 NG 안정성에는 전연 영향을 주지 않았다. 표 2.15 에는 니 트로나프탈렌의 안정화 작용도 나타나 있으며 디니트로 및 트리 니트로 화합물들도 안정제 역할을 하는 것으로 밝혀졌다.
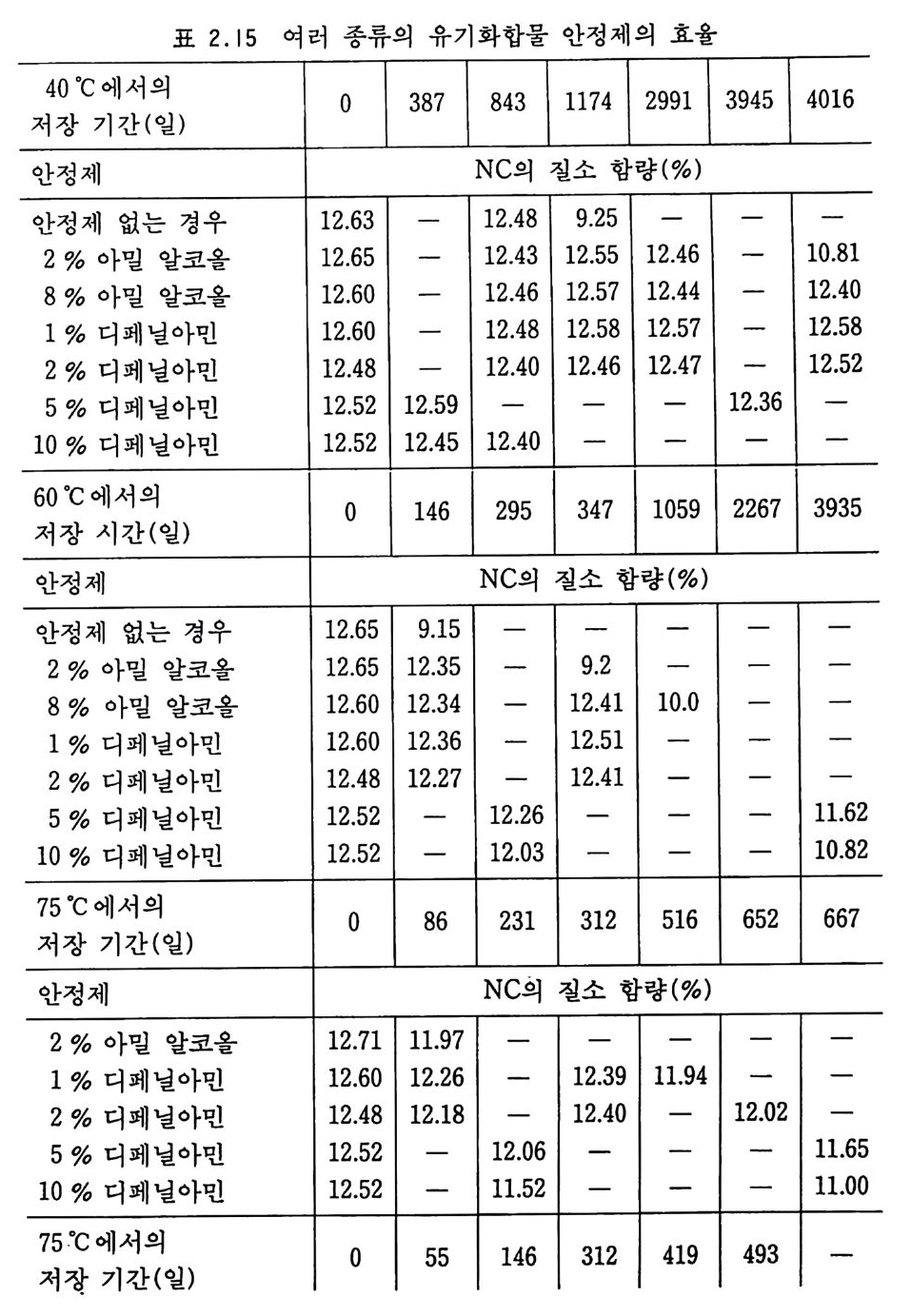 표 2.15 여러 종류의 유기화합물 안정제의 효율
표 2.15 여러 종류의 유기화합물 안정제의 효율
 안정제 NC 의 질소 함량(%)
안정제 NC 의 질소 함량(%)
2.8 로켓용 복기 추진제의 물리적 및 서베일런스 특성 추진제 그레인의 물리적 • 기계적 특성은 총 • 화포용 추전제에 서도 중요한 성질이나 로켓용 추전제로 사용되는 경우에는 제작 후 보관기간 동안 또는 발사 • 연소되는 동안 로켓 성능이 더욱 큰 영향을 주게 된다. 예를 들어 추전제의 기계적 성질이 좋지 않아 추진제 그레 인 에 균열 (crack) 또는 변 형 (defo r mati on ) 이 생 기면 로켓 발사시 탄도 성능에 심각한 영향을 주게 되며, 심할 경우 로켓 모터가 폭발해 버리는 수도 있다. 추진제의 물리적 • 기계적 특성으로 인해 나타나는 몇 가지 문제점들은 다음과 갇 다 .63) ® 온도 사이클링 (tem p . cyc l in g ) 추진제가 충전된 추전기관 모터 혹은 저장용기 등이 적도 근처 의 고온지대에서 북쪽의 한랭한 지역으로 이동되면 추진제 그레 인의 의부 온도가 변화되므로 추전제 그레인에 가해지는 힘이 발 생한다• 이 힘은 추진제를 열적으로 팽창시키거나 축소시키려는 방향으로 작용한다. 이때 추전제 그레인은 낮은 온도에서는 딱딱 하게 변화되므로 균열될 가능성이 있고 높은 온도에서는 소프트 (so ft)해지므로 그레인이 변형될 위험이 있다. 케이스 결합 (case bonded) 형 추진제 그레인의 경우에도 케이스의 열팽창 계수와 추전제의 열팽창 계수가 다르므로 그레인과 케이스 집착 부분에 응력 집중 현상이 일어나 접착 부분이 균열될 가능성이 높아진 다. ® 그 레 인 봉괴 (collaps e ) 카트리지 (car t r i d g e) 에 충전된 복기 추전제의 경우 그레인 외부
를 통해 연소가스 (화영 ) 가 흐르지 않도록 · 모터 후방 (aft end) 추 진제와 케이스 사이를 막았을 때 일어날 수 있는 문제로서 발사 초기 연소가스가 발생하는 p or t면적으로 인해 모터 앞부분과 뒷 부분 사이 에 상당한 압력 차이 (pr essure drop ) 가 발생 한다. 이 압 력 차이로 인한 힘을, 죽 추진제 그레인을 변형시키려는 이 힘을 추전제 그레인이 견디지 못하면 추전제 연소 도중 추전제 그레인 이 완전 붕괴되면서 폭발해 버리는 현상이 일어난다. ® 케이스 결합형 추진제 그레인의 경우 추전제가 연소하면서 압력이 증가하면 케이스는 팽창하게 되는 데, 이때 추진제도 동시에 팽창하여야 한다. 그런데 추전제가 케 이스와 함께 팽창할 수 없는 기계적 성질을 지니고 있으면 그레 인에 균열이 발생하면서 폭발하게 된다. 따라서 추전제 그레인은 충분한 신축성을 가져야 정상적인 연소를 하게 된다. 이와 같이 비교적 간단한 예로써 세 가지 문제점들을 나열하였 으나 실제로는 더 복잡하면서도 잘 규명되지 않는 여러 가지 요 인이 복합적으로 일어나는 경우가 많다. 따라서 추전제 그레인에 요구되는 물리적 • 기계적 특성의 기준치를 간단히 정의하기는 매 우 어렵지만 일반적으로 카트리지 충전추전제는 케이스 결합형 추전제보다 훨씬 딱딱한 성질을 지녀야 하며 케이스 결합형 추전 제는 모터가 작동되는 어떠한 조건하에서도 최소 50p si 이상의 강도 (str e ng th) 와 15 % 이 상의 연신율을 가져 야 한다고 경 험 적으 로 알려져 있다. 복기 추진제의 물리적 • 기계적 성질들은 NC 의 점탄성 특성으 로부터 설명될 수 있으나 폴리머 성분인 NC 의 선형 (line arity ), 결정성 (crys t a l l ini t y), 극성 (po larity ), 가교결합성 및 가소제와의 관계 등을 고려해야 한다• 추전제 그레인 설계 및 제작을 위해
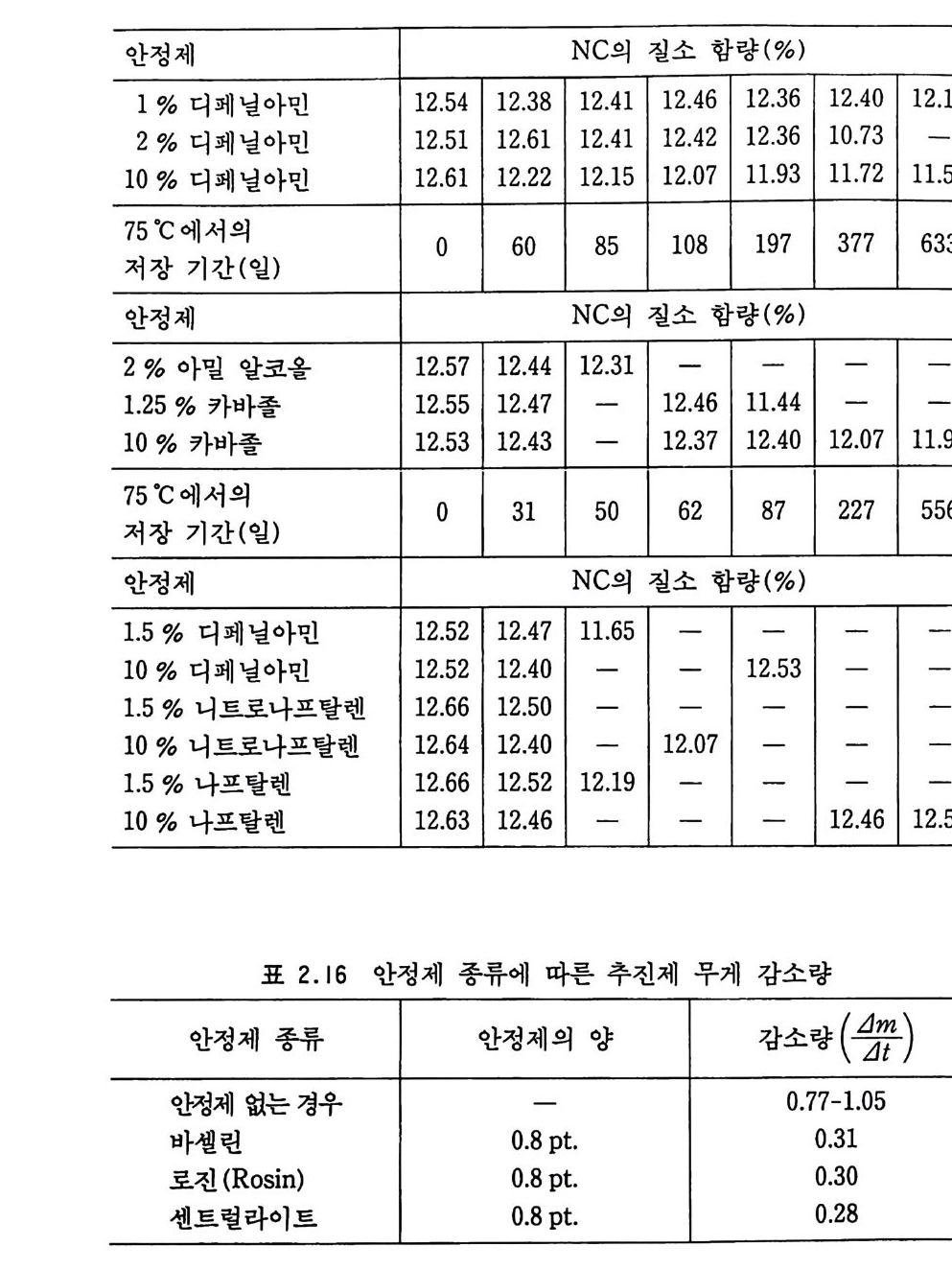
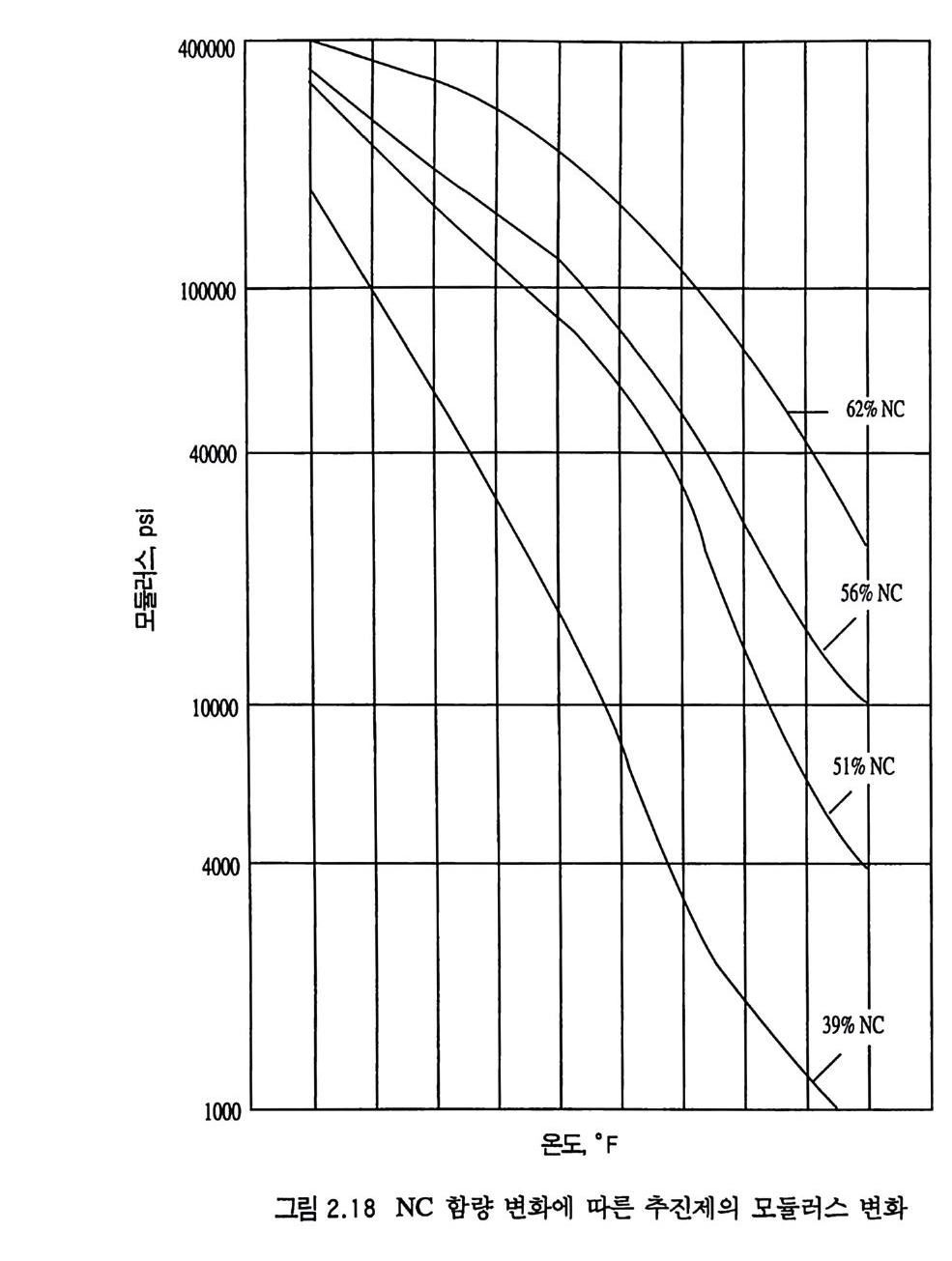
가장 간편하게 이용할 수 있는 유용한 데이터는 추전제의 스트레 스-스트레인 커브에서 얻어지는 최대 인장강도, 변형률 및 탄성 계수 등이다. 이 성질들은 측정온도와 변형속도에 따라 변하므로 수많은 실험을 통해 분석 확인해야 한다 .64 , 65) 2.8.1 추전제의 성분이 물리적 성질에 주는 영향 2.8.1.1 니트로셀룰로오스 (NC) 의 영향 NC 는 복기 추진제의 주요 성분으로서 추전제 메트릭스 구조 를 이루는 역할을 하고 있으므로 NC 의 함량은 추전제의 물리 적 • 기계적 성질에 큰 영향을 주게 된다. 그림 2.16, 그림 2.17, 그립 2.18 은 주조에 의해 제작된 복기 추진제의 경우 NC 함유 량 변화에 따른 추진제의 기계적 성질 변화를 보여주고 있다. 이 실험 결과들을 종합해 보면 NC 의 함량을 증가시킬수록 기 계적 성질의 커브들이 오른쪽 방향, 죽 고온 쪽으로 움직이므로
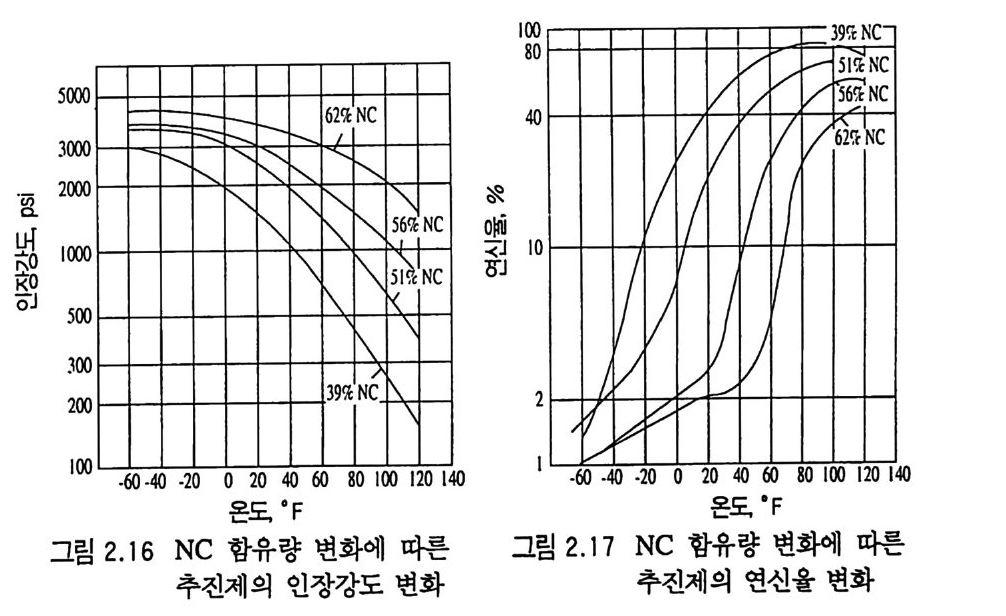 !Sdf버OJKO 血血血血5000 `` ``- \`` `~\ `\\ \ \\\6、 2 I' . t l\\\\ N C , , <\55\6~ \ % 1 IN N CC 凌.函BR 썹1 0
!Sdf버OJKO 血血血血5000 `` ``- \`` `~\ `\\ \ \\\6、 2 I' . t l\\\\ N C , , <\55\6~ \ % 1 IN N CC 凌.函BR 썹1 0
 400000
400000
같은 온도에서는 NC 함량이 많을수록 인장강도와 탄성계수 (modulus) 는 증가하고 스트레인은 감소한다고 볼 수 있다. 그림 2.17 로부터는 스트레인이 급격하게 감소하는 구간의 온도 범위를 읽을 수 있는데 그 낮은 쪽 온도를 취성 (brit tle) 온도라고 부르 기도 한다. 이때 똑같은 시편을 유전손실률 (d i elec t r ic loss fac to r ) 방법 으로 측정 한 이 차전 이 (second order tra nsit ion ) 온도는 그 취 성 온도와 잘 일치한다고 보고되었다 .66) 일반적으로 생각할 수 있는 것으로 NC 의 니트로화 정도 혹은 셀룰로오스의 길이 등이
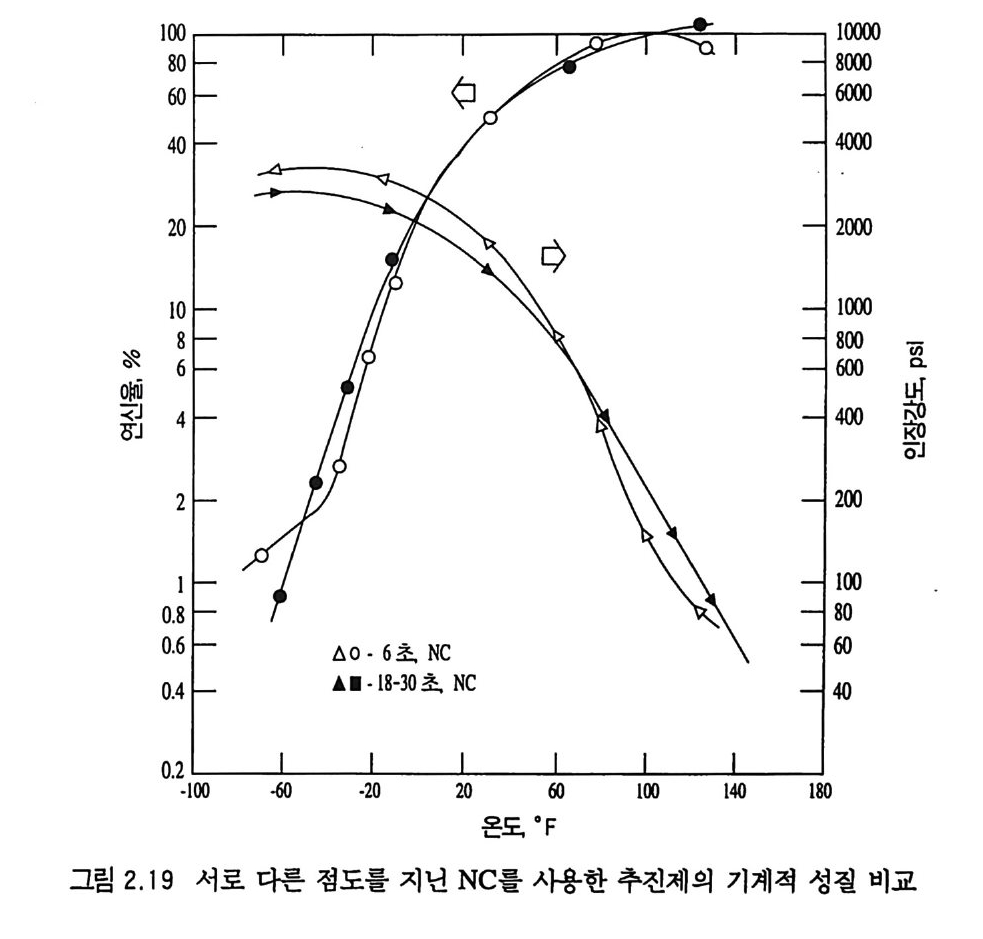 100 IOOXl
100 IOOXl
추전제의 기계적 성질에 큰 영향을 줄 것으로 가정하였으나 그립 2.19 와 같은 실험 결과로는 큰 영향을 주지 않는 것으로 밝혀졌 다. 즉, 4 배 정도의 서로 점도가 다른 NC 를 사용한 추전제의 기계적 성질이 별로 큰 변화가 없음을 알 수 있다. 그러나 NC 자체의 특성이 변하면 추전제의 기계적 성질은 크 게 변화된다. 예를 들어 그립 2 . 20 에서와 같이 NC 를 가교결합 화하면 고온으로 갈수록 연신율은 감소하고, 탄성계수는 증가하 며, 인장강도는 별 영향이 없다. 가교결합도 (de g ree of crossli nk -
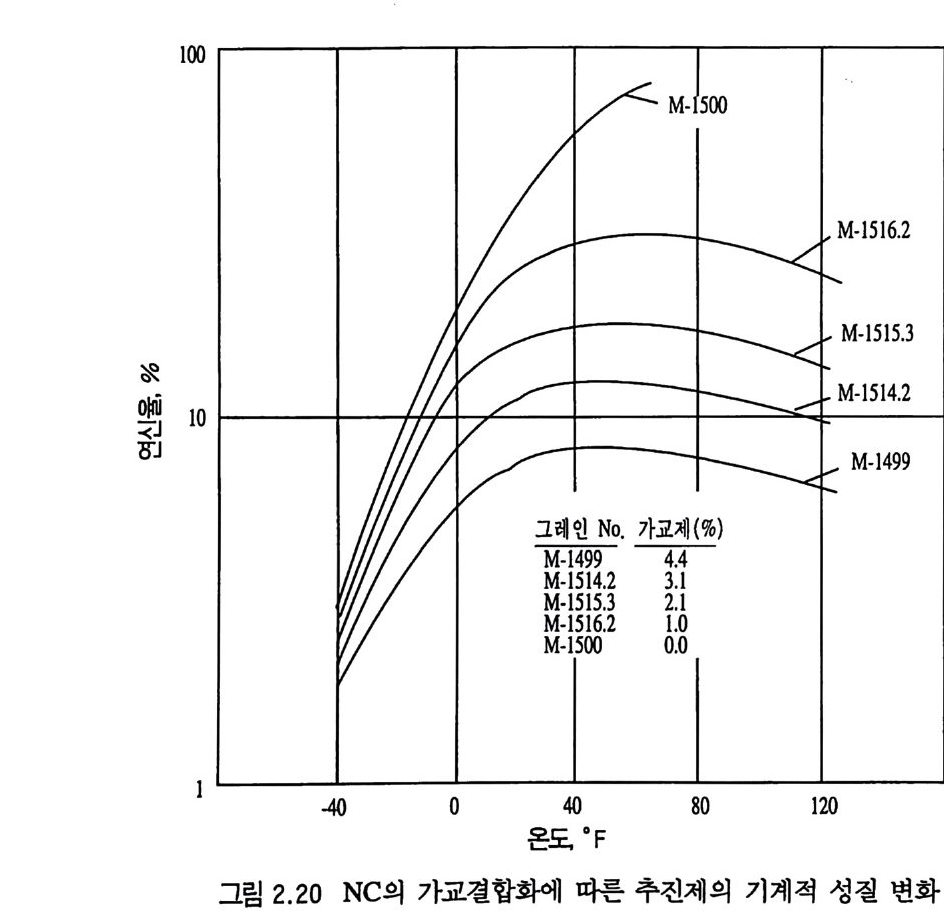 100
100
i n g)를 떨어뜨리게 되면 고온에서의 변형 또는 흐름(fl ow) 성질 을 감소시킬 수 있고, 저온에서의 성질은 그대로 유지시킬 수 있 는 이점이 있다. 이러한 가교결합은 NC 의 수산기와 반응할 수 있는 디이소시아네이트, 무수카르복실산 및 금속영 등의 2 개 관 능기가 있는 물질들을 첨가시킴으로써 쉽게 만들 수 있다 .67) 2. 8.1 . 2 가소제의 영향 가소제의 화학적 구조 및 사용하는 함량은 NC 계열 추전제의 물성 에 상당한 영 향을 준다. 66) NC 와 가소제 (ine rt 가소제 ) 의 혼 합물을 저온에서 실험한 결과 가소제는 NC 의 2 차 전이온도에 큰 영향을 주는 것으로 나타났다. 보통의 고분자 물질인 경우 긴 사슬 (lon g chain ) 구조를 지 닌 가소제 가 방향족 (aromat ic) 구조의 가소제보다 2 차 전이온도를 더 낮은 온도 쪽으로 이동시킨다고 알려져 있으나 NC 계열 추진제의 경우 가소제의 함량이 매우 낮으므로 (10 % 이하) 가소제의 화학구조에 따른 영향은 별로 없 는 것으로 알려져 있다. 그러나 가소제 자체를 다른 종류의 가소제로 바꾸면 매우 다른 성질을 나타냈다. 예를 들어 그림 2.21 에 나타난 바와 같이 40 % NC 를 함유한 추전제 조성 중에 서 NG 를 DGDN (die t h y l e ne gly c o l din i t ra te ) 으로 바꾸면 온도 범 위 에 따라 다음과 같은 영 향 이 있다. 첫째 저온 범위에서는 연신율의 차이가 별로 없는 대신 에 인장강도 및 탄성계수 값들이 떨어지며, 둘째 고온 범위에서 는 연신율이 감소하고 인장강도는 NG 추진제와 유사하며 탄성 계수는 증가하는 경향을 나타낸다. 따라서 DGDN 은 온도에 따 른 물리적 • 기계적 성질의 요구조건에 대해 NG 보다 더 잘 접근 시킬 수 있는 가소제이며, 특히 케이스 결합형 추전제 그레인 제 작시 더 유리한 원료라고 볼 수 있다.
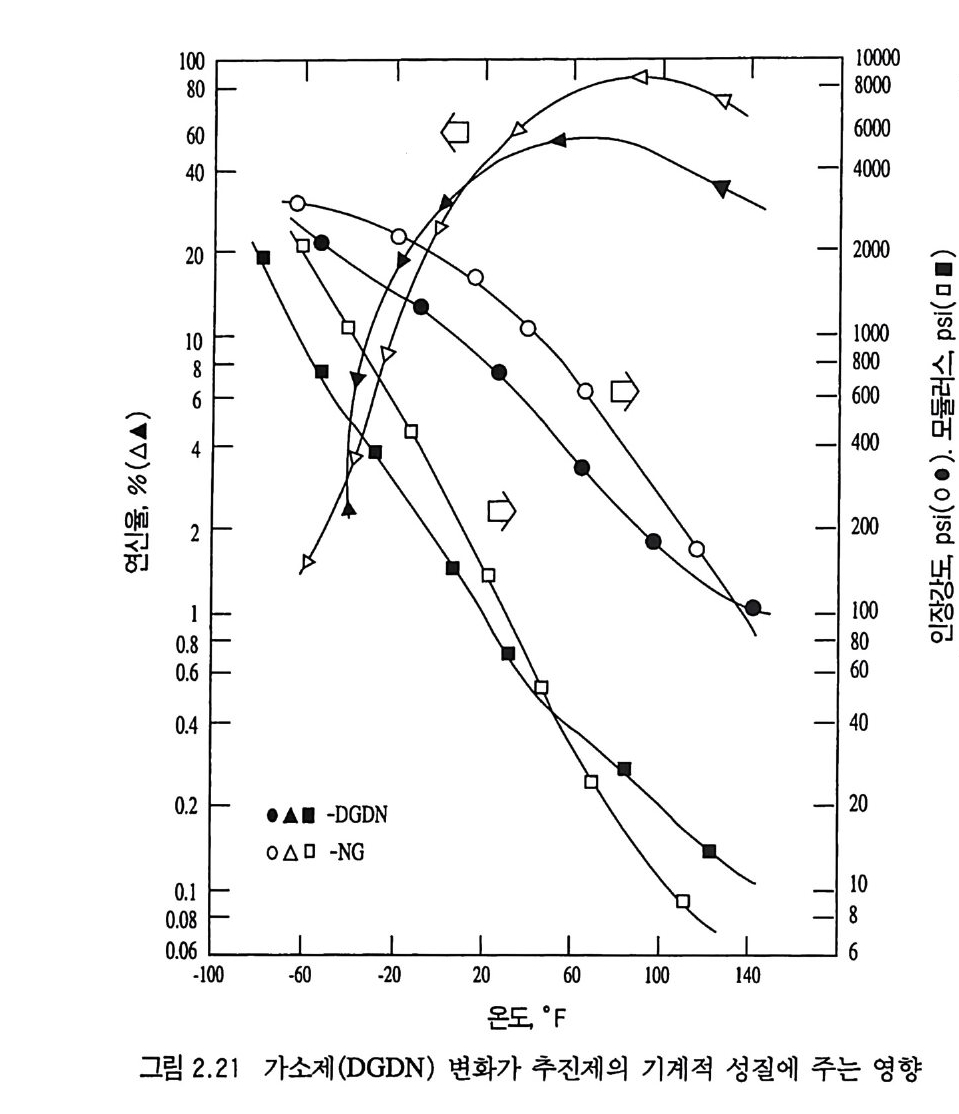 00806040 10000
00806040 10000
2. 8. 2 서 베 일 런스 (surveil lan ce) 트..., 入。1 로켓용 복기 추진제의 수명을 평가하는 데는 몇 가지 방법이 있다. 68) 첫 째 로는 안전저 장수명 (safe sto r ag e life) , 둘째 는 안전사 용수명 (safe use life) , 셋 째 로는 작동수명 (usefu l life) 이 다• 안전저
장수명은 추전제가 보관되는 도중 자동점화시까지의 시간 또는 추전제의 분해가스 (decom p os ti on fu me) 가 생성될 때까지의 시간 으로 정의될 수 있다. 안전사용수명은 로켓이 추전제 결함으로 인해 잘못 작동하여 인명이나 장비에 피해롤 주지 않을 수 있을 때까지의 추전제 사용기간으로 표현될 수 있으며, 따라서 어떠한 로켓이나 장비든 안전수명이 경과한 추진제를 장착한 상태로 운 용 • 작동해서는 안 된다. 작동수명은 가장 엄격한 조건을 충족시 킬 수 있는 수명으로서 추진제를 장착한 장비 (시스템)가 신뢰성 있게 잘 작동할 수 있는 기간이라고 정의될 수 있다. 이 작동수 명이 지난 로켓을 발사했을 경우 안전하게 발사될 수 있을지는 모르나 그 로켓을 사용할 가치는 이미 상실했다고 판단해야 한 다. 2. 8. 2.l 안전저장수명 복기 추진제의 원료 성분 중에는 열역학적으로 불안정한 성분 둘이 있다. 따라서 이러한 불안정한 원료들의 자동 촉매 작용에 의한 분해반응을 방지해 주는 것이 추전제 조성 개발시 고려해야 될 중요한 점이다. 추진제의 분해속도와 관련된 안전저장수명에 대해 많은 연구가 이루어졌으며, 안전저장수명은 로켓 모터의 수 명을 평가하는 중요한 요소 중의 하나가 되었다. 그러나 안전저장수명은 거의 대부분 가속화 (accelera t ed) 방법 에 근거를 두고 산출된 수명이라는 점을 항상 유의해야 한다. 다 음 표 2.17 에는 일반적으로 적용되는 추전제의 시험 종류가 나열 되 어 있다. 이 러 한 시 험 들의 결과를 의 삽 (extr a p o lat ion ) 함으로써 복기 추진제의 안전저장수명은 일반적인 저장 조건하에서 대략 30 년 이상인 것으로 계산되고 있다 .69) 따라서 복기 추전제가 로 켓에 충전되어 있을 경우 로켓 구조 내의 다른 부위의 결함이나
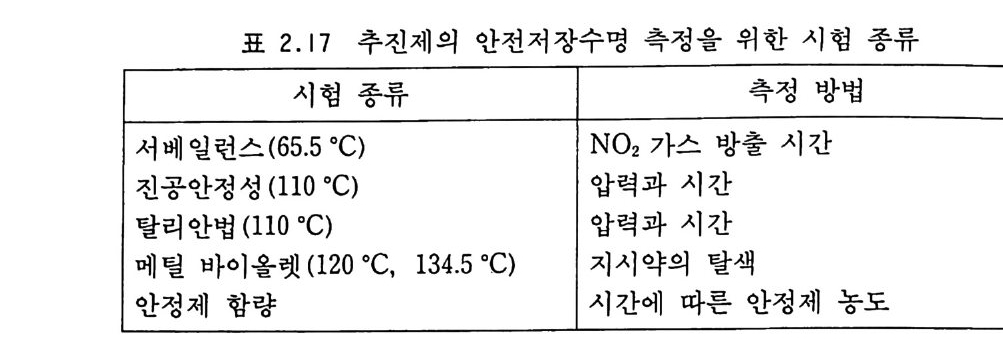 표 2.17 추진제의 안전저장수명 측정을 위한 시험 종류
표 2.17 추진제의 안전저장수명 측정을 위한 시험 종류
로켓 설계상의 결함이 없다면 일단 충전된 추전제는 매우 오랜 기간 사용될 수 있다고 보여진다. 2. 8. 2. 2 안전사용수명 안전사용수명은 로켓 추전기관 모터가 충전되어 있는 추전제로 인 하여 심 각한 오작동 (malfu n cti on ) 을 나타내 지 않을 때 까지 의 시간(수명)을 의미한다. 일반적으로 추전제 결함으로 인한 추전 기관 모터의 오작동은 추진제와 케이스 사이의 인히비터(i n h i b it or) 의 잘못이나 추전제 자체의 결함(예, 균열)으로 인해 일어 난다고 알려져 있다. 추진제 내부의 그레인 균열과 같은 결함은 추전제가 분해됨에 따른 분해가스의 발생 및 추진제의 물리적 • 기계적 특성의 저하 와 같은 복잡한 메커니즘으로 인해 일어난다고 볼 수 있다 .70) NC 및 NG, 질산에스테르 등 추전제에 사용되는 원료들의 분해 반응 메커니즘과 그 분해반옹 속도에 대해 많은 연구결과들이 보 고되어 있다 .71,72,73) 죽, 추진제 그레인으로부터 분해가스가 발생 하여 그레인 내부로 서서히 확산해 들어가면서 그레인 내부의 압 력을 증가시키는 동시에 NC 의 분해롤 일으키면 폴리머 사슬이 파괴되고 추진제의 물리적 • 기계적 특성이 저하되므로 추진제 그 레인 내에 균열이 발생한다고 설명되고 있다 .74,75) 추전제 그레인
내부의 균열을 예측하기 위한 간편한 가속 시험방법은 2 인치 정 사각형 추진제 덩어리를 80°C 에서 저장하면서 내부 균열이 관찰 될 때까지 X- 선 검사를 하는 것이다. 이와 같은 국한 조건에서 의 시험방법에서 복기 추진제는 보통 2 주일 이상의 균열 수명 (crackin g li ves) 을 갖고 있는 것으로 알려져 있다. 또 다른 극한 시험방법은 온도 사이클링 (cy cl in g ) 시험에 의해 추긴제의 균열이 나 깨지는 현상을 예측하기도 한다. 추진제의 내부 또는 의부의 균열이 매우 심각한 현상이긴 하지 만 안전사용수명을 결정하는 현상은 오히려 내열 인히비터의 품 질 저하에 의해 결정된다고 보고되어 있다. 다음 표 2.18 에는 추 진제의 균열과 인히비터의 결함에 관한 효과가 비교되어 있다. 이 데이터들은 76) 고온에서 로켓을 보관하면서 시험한 결과로서 불합격 (reje c ti on ) 점 10,000 개가 축적될 때를 안전사용수명으로 결정하였다. 이 결과로부터 고온에서보다 상온에서의 인히비터 결함 효과가 훨씬 우세하게 나타남을 알 수 있는데, 그 이유는 추진제 그레인 속의 가소제가 인히비터 쪽으로 이동 (m igr a ti on) 하여 접착부위에 결함을 발생시키는 영향 때문으로 해석되고 있 다. 이 데이터는 케이스 결합 (case bonded) 모터의 결과는 아니 지만 케이스 결합 모터의 경우도 비슷할 것으로 예측된다. 표 2.18 의 결과에 의해 그 추전제를 27°C 에서 보관한다고 가정할 경우 안전사용수명을 계산하면 35 년 (10,000/0.032/24/365) 이다. 그러나 이와 갇은 고온 가속 저장시험은 노화 성능 또는 수명 평가와 관련시켜 설계하거나 모터의 성능을 평가할 때는 매우 세 심한 주의가 필요하다. 왜냐하면 이러한 고온에서의 가속 노화 시험 결과는 대부분 실제 상황과 비교해 볼 때 그 결과가 매우 다른 경우가 많기 때문이다. 고온에서 발생하는 결함들이 상온조 건에서는 나타나지 않는 경우가 많으며, 그 반대로 상온 저장시
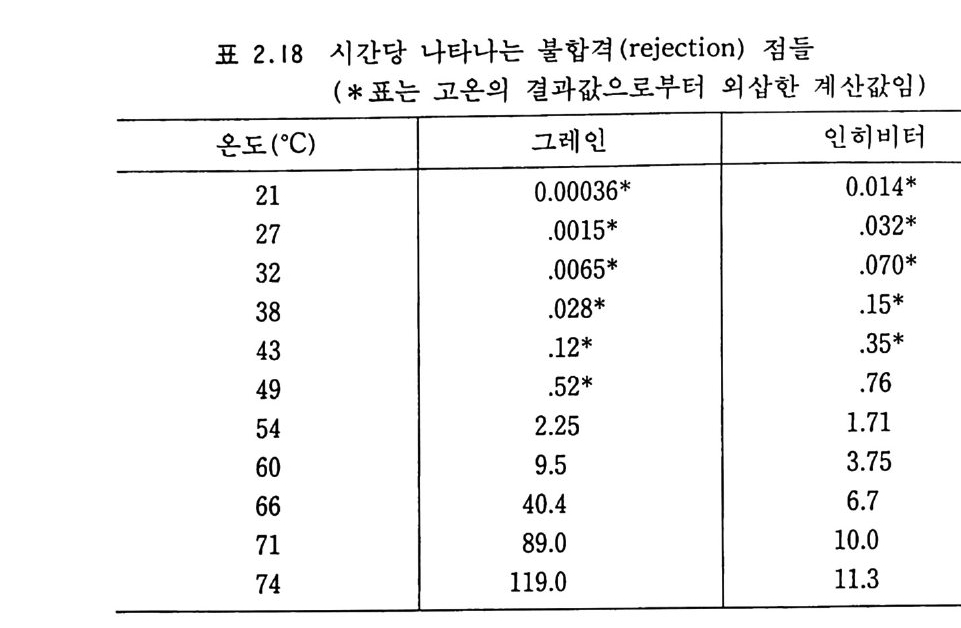 표 2.18 시간당 나타나는 불합격 (reje c ti on ) 점 들
표 2.18 시간당 나타나는 불합격 (reje c ti on ) 점 들
나타나는 결함이 고온에서는 나타나지 않는 경우가 혼하기 때문 이다. 따라서 이러한 시험 결과들로부터 의삽 (ex t ra p ola ti on) 범 위를 최소한으로 줄이기 위해 온도를 너무 높이지 않고 40 °C 정 도로만 가열하고 더 오랫동안 보관 시험하는 방법이 많이 채택되 는 경향이 있다. 2. a .2.3 작동수명 로켓을 설계 제작한 후 오랫동안 보관해 두면 시간이 경과함에 따라 로켓 모터의 성능 저하가 서서히 일어난다. 이때 로켓의 성 능이 원래 설계된 목적대로 작동할 수 없다고 판단되는 점까지 성능 저하가 일어나면 그 로켓의 작동수명은 끝났다고 보아야 한 다. 그러므로 작동수명은 기술적인 면에서 또한 전술적인 면에서 어떠한 요인에 의해 결정되는지, 그것을 검토하는 것은 매우 중 요한 일이다. 작동수명을 결정하는 중요한 요인들 중에는 총추
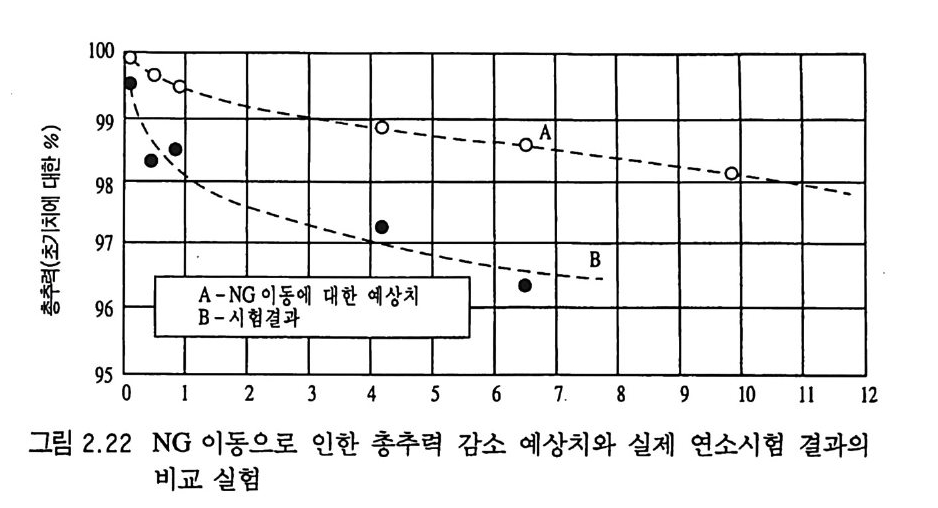 gJ_써¢(l吉KkkO4)―oi흙 1090 H9\ \• 9’’•8 9`BA- `7--- N`시9 kG 험 6-이. 결`- 등 -과에 -대--한 - 예1.상0 - 치- `-..-. ---II-- - --()• ...-A ,--- - B- - ---- ( .... ~-· -
gJ_써¢(l吉KkkO4)―oi흙 1090 H9\ \• 9’’•8 9`BA- `7--- N`시9 kG 험 6-이. 결`- 등 -과에 -대--한 - 예1.상0 - 치- `-..-. ---II-- - --()• ...-A ,--- - B- - ---- ( .... ~-· -
력, 연소시간 및 추력 프로그램 등이 있다. 이러한 요인들로부터 로켓이 얼마나 멀리, 얼마나 빠르게 비행할 것인가가 결정된다. 총추력은 추진제의 화학적 조성, 그 중에서도 NG 함량에 의해 크게 좌우된다. 그러나 NG 는 시간이 경과함에 따라 기화 (vap o riz a t ion ) 또는 이 동 (mi grat i on ) 에 의 해 추전제 로부터 손실 될 수 있으며 특히 이동 현상의 영향이 더 큰 것으로 알려져 있 다 .77) 그러나 로켓 모터의 성능 저하는 이동 현상만으로는 완전 하게 설명되지 못하고 있다. 예를 들어 로켓 모터의 성능과 NG 의 이동 현상과의 관계를 실험적으로 비교한 결과 그림 2.22 에서 보듯이 잘 일치하지 않고 있다. 죽, NG 의 이동 현상만으로 총 추력의 성능 저하를 설명하기에는 부족한 점들이 있다. 이 그림에서 A 커브는 인히비터를 화학적으로 분석하여 이동된 NG 양을 계산하여 예상한 총추력 곡선이고, B 커브는 실제로 지 상 연소시험에 의해 얻은 총추력이다. A 와 B 커브가 서로 찰 일 치하지 않는 이유는 추력에 영향을 주는 다른 요인이 있는 것으 로 설명할 수밖에 없다. 추전제 조성과 관련된 사항으로서 추력
과 관련된 요인은 추전제 연소속도를 조절하기 위해 사용된 소량 의 첨가물로 인한 것이 있다. 이러한 소량의 첨가물들은 특정 압 력 범위에서 추전제의 연소속도를 증가시키는 데 사용된다. 그러 나 이 촉매둘은 촉매 자체 혹은 추전제 조성 중 다른 원료에 약 간의 변화가 발생하면 촉매로서의 성능에 크게 손상을 입게 되며 그 결과 설계된 성능 프로그램으로부터 크게 이탈된 성능을 나타 내게 되는데, 일반적으로는 더 낮은 추력을 나타내게 된다. 이 상과 감이 로켓 모터 의 작동수명 (usefu l life) 또는 안전수명 (safe life)을 추진제 그레인의 결함이나 성능에 중점을 두어 설명 하였으나, 사실상 로켓 모터의 결함은 추전제 그레인 이의의 구 성 요소들, 죽 점화기, 모터 내열재, 압력 실 (sea l) 등과 같은 다른 부품들이 노화 (a g e i n g)에 더 취약한 상태에 있는 것으로 알 려져 있다. 2.9 NC 계열 추전제의 제작 NC 를 주요 성분 내지는 기본골격으로 하는 NC 계열 추진제 ―단기, 복기, 삼중기 등의 추전제는 대부분 총, 화포, 박격 포 및 소형 로켓 등의 추진제로 적용되고 있다. NC 계열 추전 제를 제작하는 공정은 다음 표 2.19 에서와 같이 용매를 사용하거 나 용매 를 사용하지 않으면서 압신 (extr u sio n ) 또는 주조 (casti ng ) 하는 공정들로 분류될 수 있다. 78) 추진제가 연소될 때, 가능한 한 균일한 연소가 일어날 수 있도 록 하기 위 해 추전제 내 의 NC 를 골고루 분산 (collo i d i n g) 시 켜 주 는 작업이 필요하다. 또한 NC 이의의 각 원료들을 기계적인 혼 합, 롤링 공정 등을 거치게 하여 더 균일한 배합이 일어나도록
 표 2.19 추진제 용도에 따른 주요 공정 예
표 2.19 추진제 용도에 따른 주요 공정 예
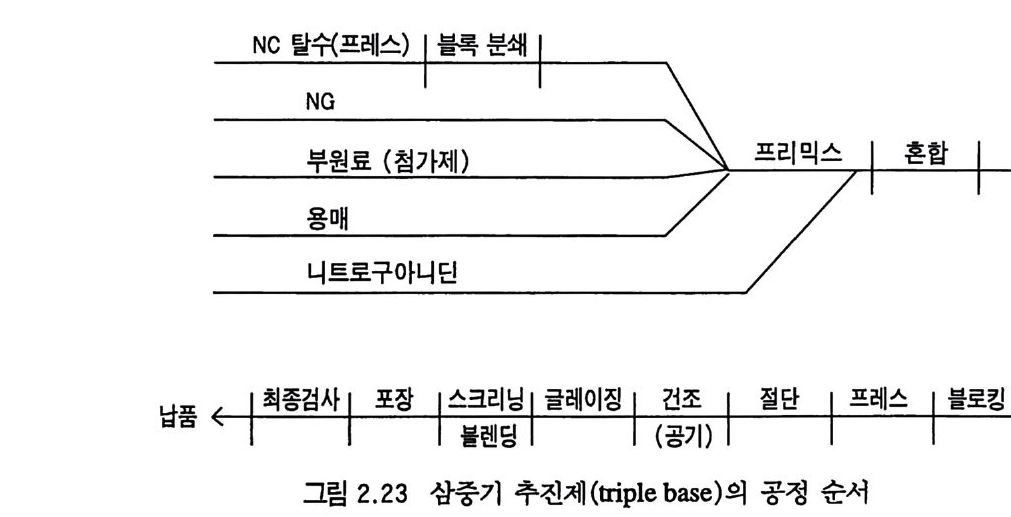
한다. NC 계열 추전제의 제작은 전통적으로 배치 (ba t ch) 식 공정 울 사용하여 단위 공정(설비) 사이에 충분한 안전거리를 둠으로 써 우연히 (또는 고의적으로) 일어날 수 있는 위험에 대비하여 추 전제 제작 전 공정의 파괴(연소 및 폭발)를 방지하도록 설계되어 있다. 2. 9. l 용제 압신공정 (solvent extr u sio n pro cess) 용제 압신공정법은 추전제의 모든 원료들을 균일하게 분산시켜 주기 위해 용매 혹은 가소제를 가하고, 가열하면서 교반을 한 후 압신시켜 원하는 크기와 모양으로 추전제를 제작한 후 따뜻한 공 기를 통과시켜 건조하는 방법이다. 이때 추전제 그레인의 두께 (web) 가 0.5 인치 이상의 두꺼운 경우는 건조시간이 오래 걸리며, 건조 도중 그레인의 수축에 의해 그레인 내에 균열이 발생할 수 있으므로 0.5 인치 이상의 두꺼운 추진제 그레인 제작에는 사용하 기가 어려운 공정이다. 일반적으로 사용되는 용매들은 단기 추전 제의 경우 에데르-알코올, 복기 추진제의 경우 아세톤-알코올, 삼중기 (다기) 추진제의 경우 역시 아세톤-알코올의 혼합용매들을 사용한다.
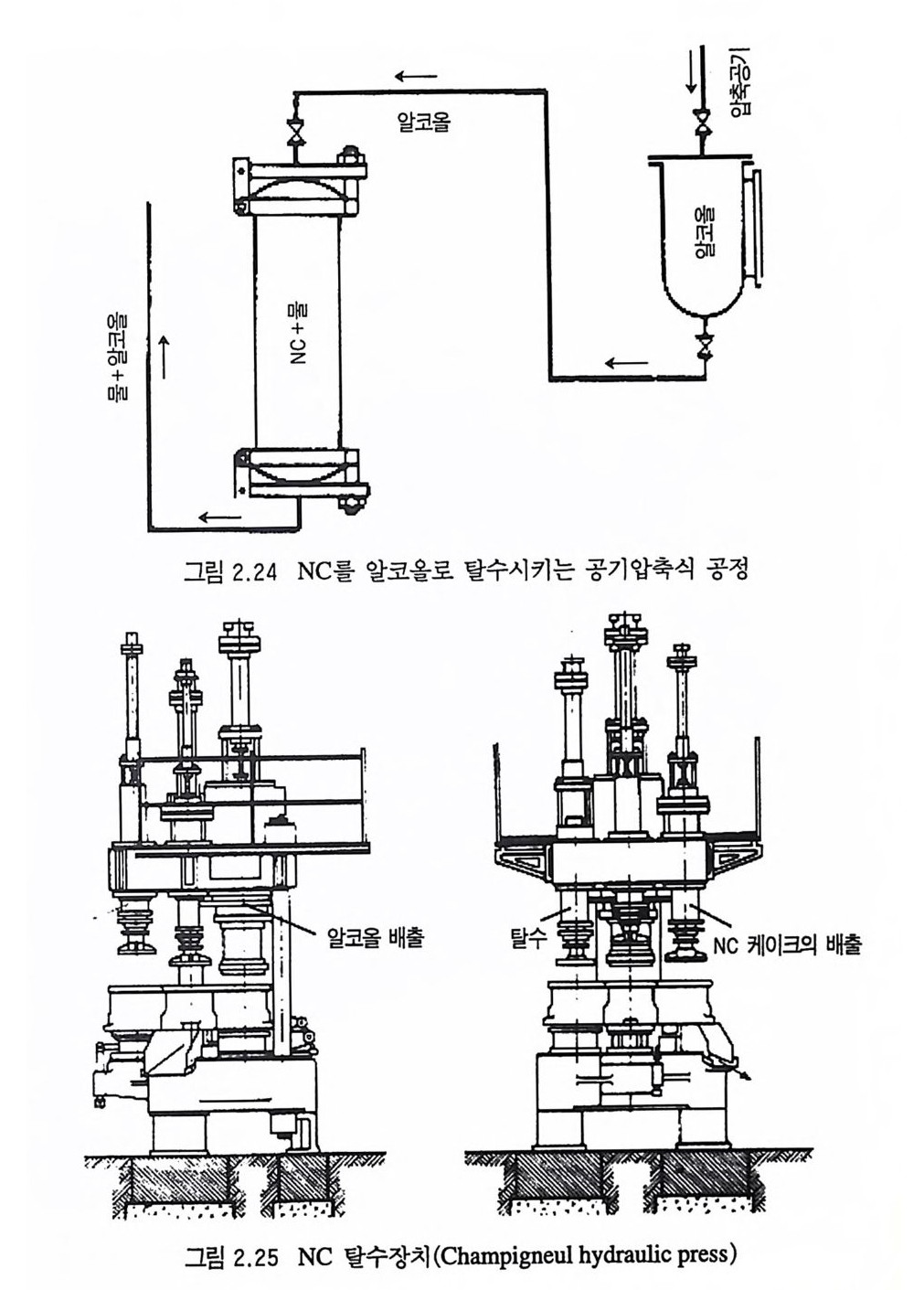
복기 추전제와 삼중기 추진제의 제조공정은 서로 유사하나 단 기 추전제는 혼합공정과 건조공정에서 약간의 차이가 있다. 삼중 기 추진제의 대표적인 제조공정은 그림 2.23 과 같다 .78) NC 공장 에서 운송된 NC 는 대개 20~30 %의 물을 포함하고 있으므로 탈 수 프레스를 사용하여 물을 일부 제거한 후 NC 와 같은 양의 알 코올울 NC 에 가한 후 고압으로 압축 (wrin g ing 또는 pre ssin g ) 하 여 NC 블록을 만든다. 이 블록은 30~40 %의 알코올을 포함하 게 된다. 이 공정에 의해 NC 중의 물이 알코올로 치환된다. 이 탈수공정에서 블록에 잔류하는 알코올의 양은 탈수 압착기의 압 력과 니트로셀룰로오스 (NC) 의 성질에 따라서 다르게 나타난다. 죽, NC 가 목재 (wood) 셀룰로오스에 서 제 조된 것 이 면 면 (cott on ) 에 서 제조된 것보다 알코올에 더 찰 스웰 링 (swell ing ) 하므로 더 많은 양의 알코올과 물을 포함하게 된다. 이러한 압착 탈수장치 예가 그림 2.24 에 있으며 유럽 지역에서 흔히 쓰이는 수압 탈수장치 (Champ igne ul 프레스)는 그립 2.25 와 같다 .I)
 NC 탈수(프레스) | 볼록 분쇄
NC 탈수(프레스) | 볼록 분쇄
 (
(
 그림 2 . 26 NC 블록을 부수는 Macerato r
그림 2 . 26 NC 블록을 부수는 Macerato r
용매 (알코올) 를 포함하고 있는 이 블록은 Macerato r (그립 2.26) 에 의해 다시 깨뜨려진 후 NC 가 너무 큰 덩어리로 엉켜 있 는 것 같은 덩어리들(입자들)울 가려낸다 .l) 그 다음 단계인 혼합공정은 보통 2 단계로(삼중기인 경우) 나누 어져 있다. 첫번째 혼합공정 단계는 프리믹스 단계로서 NC 및
 그림 2.27 프리 믹스에 사용되는 Schrader 혼합기
그림 2.27 프리 믹스에 사용되는 Schrader 혼합기
 그림 2. 28 수평 형 혼합기 의 예 (Wemei;- pflei d e rer kneader)
그림 2. 28 수평 형 혼합기 의 예 (Wemei;- pflei d e rer kneader)
각 첨가제들(안정제, 내탄도 특성 조절을 위한 촉매 등)을 NG 와 혼합하는 공정이다. NG 와 혼합될 때의 공정 안정성을 고려하여 교반기의 날과 날 사이, 날과 벽 사이의 간격이 커서 혼합물에 가해지는 기계적 힘(충격 또는 마찰 및 압력 등)이 작은 혼합기(그 립 2.27) 를 사용한다. 이 프리믹스 단계에서 니트로구아니딘을 가하여 혼합하며, 필요시에는 알코올울 추가로 가할 수도 있다. 혼합공정의 두번째 단계는 시그마형 교반날이 장치된 혼합기에 서 각 원료 성분들이 완전히 혼합되도록 충분한 시간 동안 혼합 한다. 사용되는 혼합기는 수평형(그립 2.28) 혼합기와 최근 많이 사용되는 수직형 혼합기 (그립 3.31) 가 있다. 혼합시간은 각 추전 제별로 다르나 보통 2~3 시간 정도이며, 혼합시의 온도는 상온보 다 높은 30~50 °C 부근에서 혼합한다. 혼합이 끝난 혼합반죽은 냉 각된 후 블록 프레스 (bloc pre ss) 에 의 해 고압 (수압)으로 압축 되어 원통형 블록으로 만들어전다 •78)
 2
2
이렇게 만들어전 블록을 압신장치 (사출기, ex t ruder) 에 옮겨 원 하는 추전제 그레인 형상으로 압신 제작한다. 압신기의 압력은 보통 2000 psi 이상이며, 압신기에 장치된 다이 (d i e) 는 원하는 그 레인 크기보다 약간 크게 제작해야 그 다음의 건조공정시 추진제 수축 후의 크기와 원하는 그레인 크기가 같아질 수 있다. 듀브형 추전제 그레인을 얻기 위한 다이 설계 모양이 그림 2.29 에 있다. 이 그림 에 는 일공 튜브형 (sin g le pe rfo r ate d tub e) 그레 인을 얻기 위한 다이가 표시되어 있는데 이 다아는 두 부분 (1 및 2) 으로 나 뉘어 있다 .79) 부분 1 은 그레인 튜브의 의경을 결정하는 다이이 며, 부분 2 는 그 중앙에 부착된 스틸선의 굵기에 따라 그레인 듀 브의 내경을 결정하게 된다. 그립 2.30 에는 다공 듀브형 (multip e rfo r ate d tub e) 추전제 그레 인을 얻기 위 한 다이 설계 모
 그림 2.30 다공 튜브형 그레인 제작용 다아
그림 2.30 다공 튜브형 그레인 제작용 다아
양들이 있다. 이와 같은 압신공정은 매우 위험한 공정이므로 안전에 각별히 유의해야 한다. 특히 프레스(p ress) 의 실린더 내에 들어간 추진 제 반죽에 공기가 채워지지 않도록 주의해서 작업해야 된다. 만 일 고압의 압력에 의해 공기와 용매(휘발성)의 가스 혼합물이 압 축 (ad i aba ti c) 되면 국부적으로 순간적인 점화가 일어나 작업중인 추진제가 폭발해 버릴 수도 있기 때문이다. 이렇게 압신공정에 의해 제작된 구멍이 뚫려 있는 추전제는 절 단기 (cu tt er) 에 의해 설계된 그레인 크기에 따라 절단되는데, 그 다음 공정인 건조시에 수축 (shr i nka g e) 되는 것을 감안하여 절단 한다. 건조공정시에는 추전제의 조성 및 추전제의 크기, 추전제 그레인의 모양, 휘발성 용매의 함량, 건조장치의 특성 등에 따라 건조시간과 건조기를 통과하는 공기의 온도를 결정한다. 건조된 추전제 그레인을 소량의 탄소(gr a p h it e) 분말로 처리하 여 추진제의 밀도, 흐름 특성 및 정전기 발생과 같은 성질들을
 NC I 탈수(프레스)
NC I 탈수(프레스)
개선시키기도 하고, 디니트로톨루엔과 같은 가소제로 그레인 표 면을 코팅시켜 추진제의 초기 연소속도를 조절(감소)할 수도 있 다. 표면처리가 끝난 추진제에서 이물질이나 덩어리들을 제거하 는 스크리닝 (screenin g ) 공정을 거친 후 블렌딩 (blend i n g)울 한 다. 추전제는 로트(l o t)별로 그 탄도적 특성 등이 균일해야 하므 로 요구된 로트의 크기에 따라 매우 주의해서 추전제 그레인들이 균일한 블렌딩이 될 수 있도록 조업해야 한다. 이상과 같이 제조되는 삼중기 추진제와 복기 추전제의 제조공 정은 원료 중 니트로구아니딘을 첨가하는 것과 첨가하지 않는 점 울 제의하고는 거의 다른 점이 없다. 그러나 단기 추진제의 경우 는 니트로구아니딘 의에 NG 도 첨가되지 않으므로 혼합공정이 상당히 다르다(그림 2.31). 죽, 혼합기 내의 온도를 더 낮은 온 도(예, 25°C 이하)에서 혼합시간도 더 단축하여 조업한다. 또한 어떤 경우(어떤 종류의 추진제)는 마카로니 (Macaroni) 스트랜드를 만들어 주는 공정을 하기도 한다. 이 공정은 혼합이 끝난 추전제 를 마스레이터로 잘게 부순 뒤 블록으로 만들어 압신 프레스와 비슷한 프레스를 사용하여 국수가락 모양의 스트랜드로 뽑아주는 스크리닝 공정이다. 이러한 스크리닝 공정을 통해 추진제 혼합물
 추진재 I I 공기
추진재 I I 공기
내부에 섞여 있는 공기, 찰 섞이지 않은 덩어리들, 이물질 등이 제거될 수 있고 추전제에 혼합된 각 원료 성분들의 분포가 균일 해지며 NC 의 콜로이드 분산을 증가시킬 수 있고 추전제의 밀도 도 증가되는 장점이 있다. 단기 추전제는 혼합공정시 알코올 이의에도 휘발성 용매들, 즉 에테르 혹은 아세톤 등을 추전제 종류에 따라 다르게 사용한다. 따라서 추전제 그레인의 압신, 성형, 절단공정 후 건조시에 용매 둘의 회수 및 제거공정이 필요하다. 용매의 회수공정은 주로 두 가지 방법을 사용하고 있다. 첫째는 용매기체를 냉각 또는 압축 에 의 해 응축 (condensat ion ) 시 키 는 방법 이 며 , 두번째 방법 은 특 수물질에 용매기체를 흡착시키는 방법이다. 그림 2.32 는 용매기 체를 냉각, 응축시켜 용매를 회수하는, 오래 전부터 사용된 장치 이다 .80) 상기와 같은 방법 이외에 제 1 차 세계대전 중 프랑스에서는 크
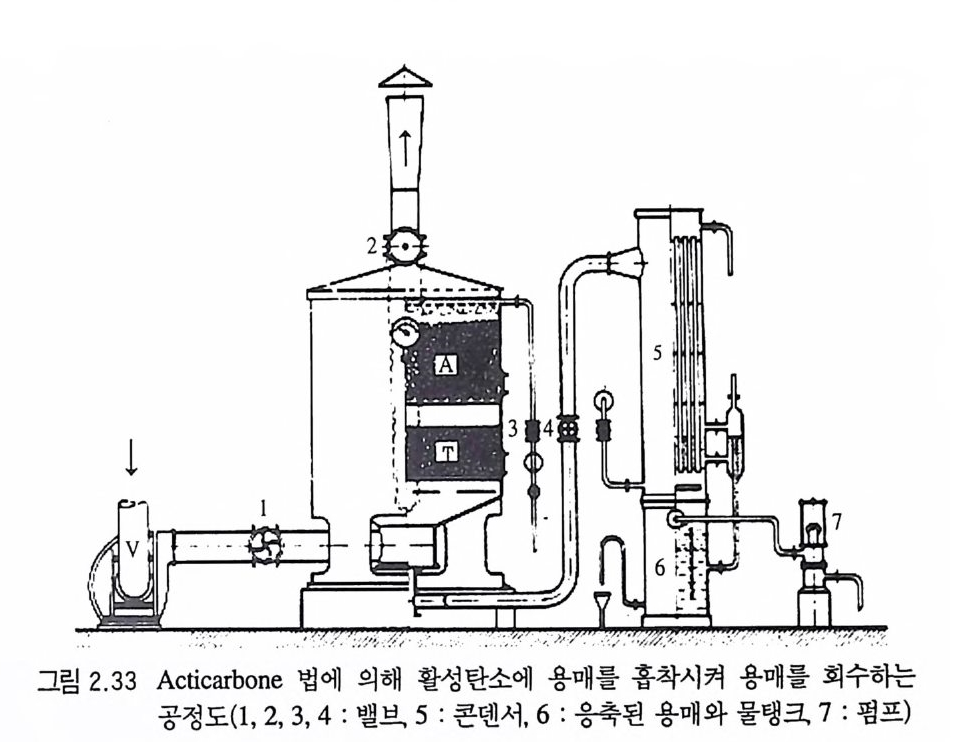 수
수
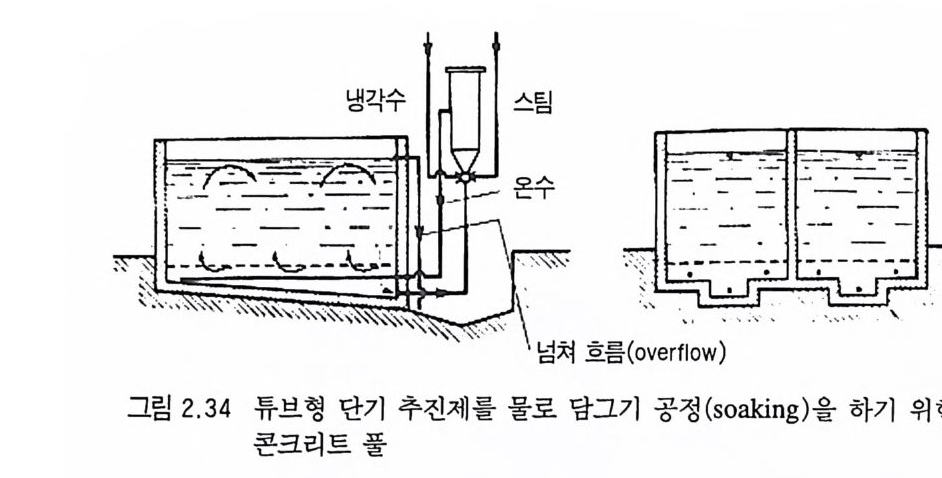
레졸 (cresol) 을 사용하여 알코올과 에데르를 흡수 회수하는 공정 이 개 발되 었다. 그후 활성 탄소 (acti va te d charcoal) 를 이 용하여 용 매 기체를 흡착 (adsor pti on) 하여 회수하는 공정이 개발되었다. 최 근에 사용되는 용매(알코올과 에데르) 회수공정은 그림 2 . 33 (Ac tic arbone 법 ) 과 같다. 1) 이상과 같이 단기 추전제를 선건조(p redr yi n g)시켜 일부 용매 를 회수한 후 추진제 그레인을 상압(대략 80°C) 이나 감압(대략 50~60 °C) 에서 건조시킨 후 다시 추전제 그레인을 물 속에 담가서 (soakin g ) 그레 인 속의 용매를 제거 하는 공정을 한다. 81) 이때 담 그기 (soak i n g) 공정은 주로 그레인에 알코올과 에데르가 함유된 추전제일 경우 적용되지만 아세톤과 같은 용매를 함유한 추전제 의 경우(예, 복기 추진제)에는 그레인으로부터 NG 성분을 일부
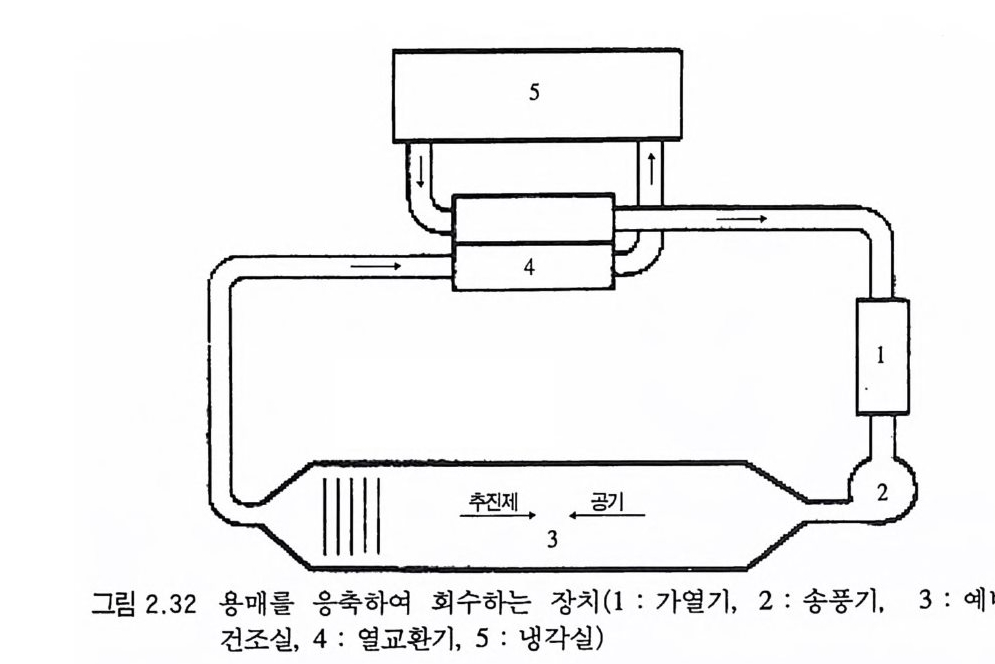
 三广玉三
三广玉三
녹여내므로 적용할 수가 없다. 추전제 그레인을 물 속에 담가두 면 그레인 내부의 알코올과 에데르 용매들이 물 속으로 서서히 확산 이동되어 제거되므로 공기 중에서 건조시킬 때 나타나는 그 레 인 표면의 딱딱해 지 는 (hardenin g ) 현상을 막을 수 있다. 이 러 한 담그기 공정은 상온의 물 (15~30 °C) 혹은 뜨거운 물 (50~80 ° C) 속에서, 그림 2.34 와 같은 콘크리트 수조를 일반적으로 사용 한다. 이와 같이 담그기 공정에 의해 용매가 제거된 추진제 그레인을 상온보다 높은 온도 (50~60°C) 에서 건조시켜 그레인 표면의 물을 제거한다. 건조된 추진제는 표면처리(글레이징), 스크리닝, 블렌 딩 공정을 거친 후 최종 검사 후 납품하기 위해 저장된다. 2. 9. 2 무용제 압신공정 (solventl e ss extr u sio n pro cess) 알코올, 에데르 혹은 아세톤과 같은 용매를 사용하지 않고 NC 를 주성분으로 하여 제작되는 무용제 타입의 추전제는 주로 복기 (double base) 추전제를 제조하는 데 많이 적용되고 있다.
또한 작은 그레인의 추진제를 제작할 경우 용매를 사용하여 제작 하면 건조공정 중 수축으로 인한 그레인 크기 변화가 추전제 형 상과 특성에 좋지 않은 영향을 줄 때, 또는 건조공정을 채택하기 어려운 추전제 조성인 경우 더 오랜 기간 동안 추전제의 탄도적 성질을 유지(마량의 용매가 시간에 따라 감소됨으로 인한 성능 변화 룰 원치 않는 경우)하고 싶은 추진제를 제조할 때 적용되는 공정 이다 .82) 용매를 사용하지 않는 점을 제의하고는 NC, NG 등 원료 자 체의 제조, 처리 공정은 앞장(용제 압신공정)에서 언급한 바와 거 의 유사하다. 복기 추진제의 경우, NC 는 가소제로서 작용하는 NG 및 물에 불용인 각종 첨가제들과 함께 뜨거운 물 속에서 술 러리 상태로 혼합된다. 이 슬러리 혼합물을 원심 분리장치로 옮 겨 대부분의 물을 제거하면 25~30 % 정도의 물을 포함하는 5 인 치 정도의 두께를 지 닌 반죽 (pa ste ) 혼합물이 된다. 이 혼합물 반죽을 면으로 만든 백 속에 넣어 건조기에서 보관하여 수분 함 량 10 % 정도로 건조시킨다. 건조되는 동안 추전제 반죽 속의 NC 는 가소제를 흡수하여 부분적으로 젤라틴화된다. 건조된 추 진제 반죽은 블렌더로 옮겨져 약 10~20 분 정도 블렌딩된 후 롤 링 (roll ing ) 공정으로 들어가게 된다. 롤링 공정은 보통 두 단계 로 나뉘어 있는데 첫번째 롤링은 50~60°C 에서 건조되는 공정이 며, 두번째 롤링은 젤라틴화를 위한 공정이다. 롤링 단계별로 롤 과 롤 사이의 간격, 온도, 롤 통과 횟수 등을 잘 조절하여 작업 한다. 롤 통과 횟수가 많을수록 젤라틴화가 많이 일어나므로 그 다음 압신공정시 압력이 더 높아지게 된다. 그림 2.35 는 젤라틴 화를 위한 롤링 공정 작업을 보여주고 있다. 이와 같이 롤링 공 정을 거친 추진제 반죽은 수분 함량이 0.5 % 이하로 떨어지게 되 며 각 원료 성분들은 매우 균일하게 분산된다. 이 롤링 공정은
 .... ’ .. ·’ ’ · ’ ·‘ · 1
.... ’ .. ·’ ’ · ’ ·‘ · 1
추진제 반죽 내에 NG 성분이 들어 있으므로 추전제의 연소, 폭 발을 대비하여 원격 조종으로 작업하는 것이 일반적이다 .78) 롤링 공정에서 나온 추전제 (shee t모양)는 압신기의 프레스로 들어가기 위해 우선 프레스 크기에 맞도록 적당한 크기로 절단된 다(이때 카펫 롤링과 같은 장치를 사용하기도 한다). 10 만 ps i 이상 의 압력하에 작동되는 수평 또는 수직형 압신기를 사용하여 원하 는 크기와 모양으로 추전제를 사출한다• 압신기에는 추진제 사출 온도를 조절하기 위한 가열 재킷 (hea ti n gj acke t) 및 추진제 내의 공기를 제거하기 위한 진공 펌프가 연결되어 있다. 압신기에 장 착된 다이의 크기와 모양에 따라 사출되는 추전제 그레인의 기하
학적 모양이 결정되며, 설계치에 맞도록 길로틴 (Gu ill o ti ne) 과 같 은 절단기를 사용하여 추진제를 절단한다. 그림 2.36 은 추전제를 압신기로부터 사출하는 장면이며 그림 2.37 은 튜브형 그레인을 절단하는 길로틴 절단기이다 .1,78) 이와 같은 압신공정에서 압신기의 압력을 균일하게 (unif orm ) 가하여야 사출되는 추전제 그레인의 모양이 일정하고 품질이 균 일하며 깨끗한(매끄러운) 표면을 갖게 된다. 압신압력에 영향을 주는 요인들은 첫째로 추진제 조성 중 질소 함량 (NC 의 N %), 탄소 (grap h it e) 나 산화마그네 슘 첨 가제 (압신기 의 실 린더 와 int e r nal 마찰을 감소시킬 수 있는 첨가제) 존재 여부 또는 추전제의 젤라틴 화를 중전시킬 수 있는 첨가제 함유량 등이며, 둘째는 압신기로 주입되기 전 롤링 공정시 롤링 통과 횟수 및 겔라틴화된 정도, 셋째로는 압신기에 걸린 추전제의 양과 다이의 설계 모양(추진제 가 사출되는 op e nin g 면적), 넷째로는 압신기 내의 추전제 온도 등이다. 또한 사출된 추전제 그레인의 표면은 압신 사출 속도가 너무 빠르거나 추진제 온도가 높으면 균일하지 못하고 그레인 성 능이 좋지 않게 되며, 추진제 조성 중 질소 함량이 너무 높거나 낮을 경우에도 추진제 표면에 좋지 않은 영향을 주게 된다. 상당히 뜨거운 온도에서 작업하는 압신공정에서는 가끔 연소, 폭발과 같은 사고가 발생한다. 이러한 폭발 사고의 정확한 원인 온 파악되고 있지 않으나 추진제와 실린더 사이, 추전제와 추전 제 자체의 마찰 또는 추전제 반죽 내에 존재하는 소량의 공기가 압축됨으로 인한 마찰열 및 정전기 발생 때문으로 추측하고 있 다. Fleur y는 피스톤에 의해 가해진 힘 (work) 이 열에너지로 전 환됨으로써 국부적 인 과열 (overhea ti n g)로 인한 추진제 의 접화에 의해 폭발이 일어난다고 설명하였다 .83) 무용제 압신기에서 일어나는 연소, 폭발에 대한 메커니즘을,
 그림 2.36 추진제를 압신기 (ex tru der) 로부터 사출하는 장면
그림 2.36 추진제를 압신기 (ex tru der) 로부터 사출하는 장면
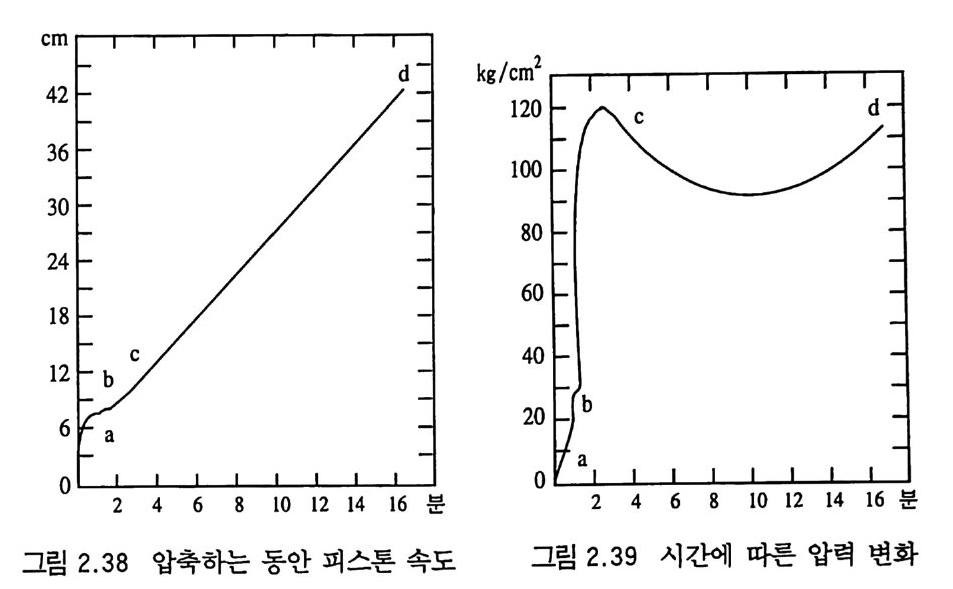 cm
cm
압축시키는 피스톤의 속도와 수압 변화 등과 함께 Fleur y는 다 음과 같이 실험 분석하였다. 죽, 그림 2.38 에는 피스톤 속도, 그 립 2.39 에는 시간에 따른 압력 변화, 그립 2 .4 0 에는 피스톤의 가 속도와 일량 (work) 이 나타나 있다 .83) 그립 2.38 에는 ab 구간과 cd 구간의 서로 다른 피스톤 속도 구간이 나타나 있다. 첫번째 구간 ab 에서는 피스톤 속도가 상당 히 높았다가 전환구간인 be 를 거쳐 속도가 일정하게 유지되는 cd 구간으로 연결된다. 그립 2.39 에는 b 접에서부터 2 분까지 압 력이 급격히 증가하는 커브롤 나타내고 있다. 그립 2.40 에서는 커브 P 가 처음 30 초 동안에 급격히 상승했다가 순식간에 0 으로 떨어진 후 많은 양의 열을 발생하고 다시 증가하는 곡선을 나타 내고 있다. 이는 피스톤이 움직이기 시작하여 추진제를 압축하기 시작한 초기에는 압력이 너무 낮아 다이를 통해 추전제 사출이 이루어지지 않고 있다가 압력이 더 높아지는 순간 추진제가 사출 되는 현상 (P 커브가 0 으로 떨어지는 접)과 일치한다고 볼 수 있다.
kgm /cm 2 P1 46 125 - l()() - 75 -
P 50 - 25 - v963 v I 0 2 4 6 8 10 12 14 16 분 그림 2.40 피스톤 일량 (work) 과 가속도 (P : 피스톤 일〔w ork 〕, v : 가속도)이러한 압신공정에서 대부분의 사고(폭발)가 그립 2.39 와 그립 2.40 의 b 점에서 발생하였다. 이 b 점에서는 압력은 최대값보다 훨씬 아래에 있으나 피스톤 속도는 매우 빠르므로 피스톤의 일 (work) 로부터 열에너지로 전환된 에너지 양이 매우 크다는 것을
알 수 있다. 따라서 추진제를 압신할 때 주어지는 압력하에 가장 적당한 피스톤 속도를 결정하는 것이 필요하며 과도한 양의 열이 발생하지 않도록, 특히 압축 초기 단계에서의 피스톤 압축 속도 를 유의해서 설계해야 한다. 압신기에 의해 사출된 후 길로틴으로 절단된 추진제 그레인은 약간 높은 온도에서 아닐링 (annea li n g)된다(아닐링 온도는 추진제 조성 특성에 따라 결정한다). X - ra y에 의한 추전제 그레인의 내부 에 있는 기포, 기포의 크기 및 모양 등 비파괴 검사를 한다. 일 반적으로는 추전제 그레인 표면에 적당한 두께 및 크기로(설계값 에 맞도록) 셀룰로오스아세테이트 혹은 에틸셀룰로오스를 코팅 (입히는)하는 내열처리 작업을 한다. 특히 로켓용 추진제 그레인 의 경우에는 대부분 인터널(i n t ernal) 연소를 하기 때문에 이 내 열처리 공정 (restr i c ting 또는 inh ib i t ion 공정)이 필수적이다. 상기와 같은 혼합, 롤링, 압신사출 공정에 의해 추전제 그레인 울 제작하는 방법 이의에 최근에는 공정이 더 간단하고 장비의 가격, 인건비, 추전제의 균일성 면에서 더 유리한 스크루 압출기 룰 사용하는 공정이 개발되어 적용 중이다 .84,85,86) 이 스크루 압 출장치의 내부설계 개략도가 그립 2.41 에 나타나 있다 .87,88)
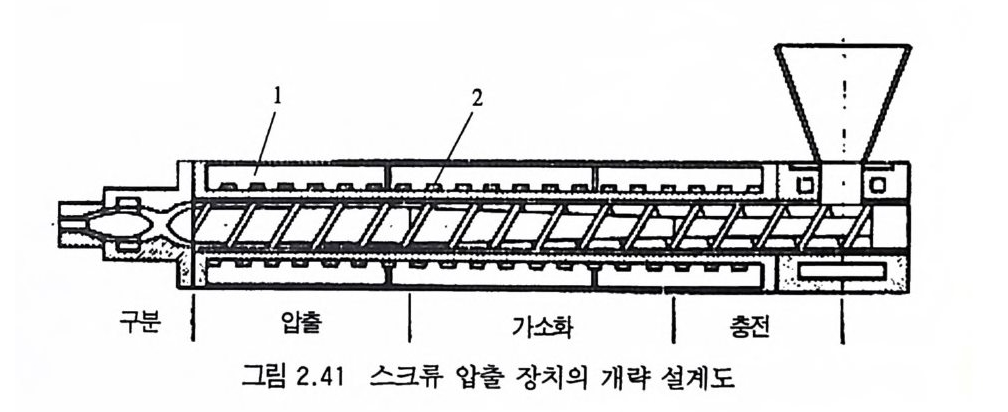 압출 | 가소화 | 충전
압출 | 가소화 | 충전
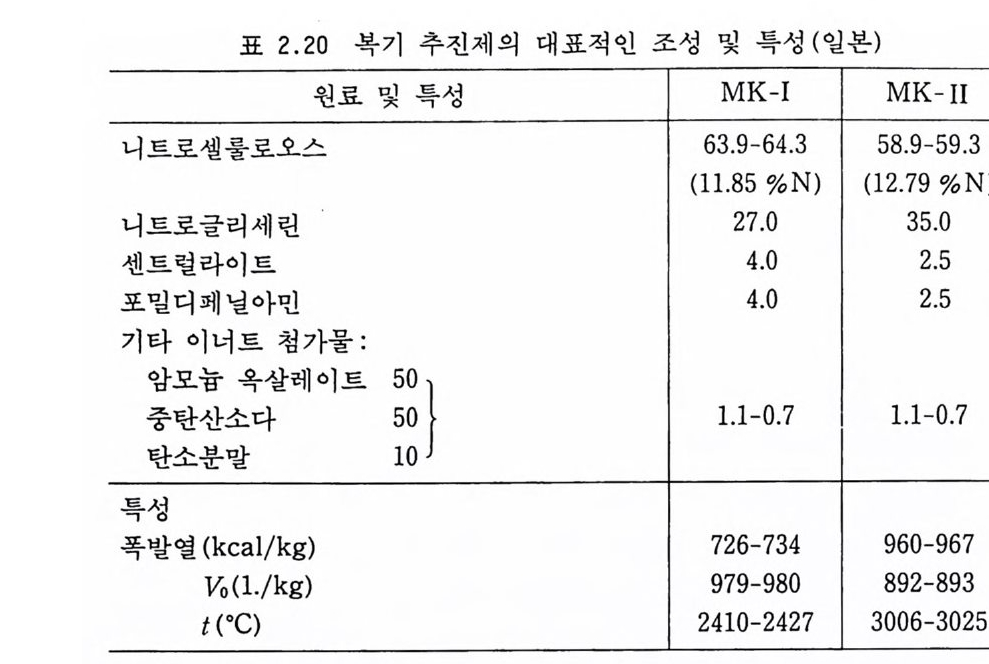 표 2.20 복기 추진제의 대표적인 조성 및 특성 (일본)
표 2.20 복기 추진제의 대표적인 조성 및 특성 (일본)
이와 같은 무용제 압신공정은 소형의 튜브형 그레인을 대량으 로 제작할 때 매우 효율이 높은 공정이지만, 대형 그레인을 제작 하기 어렵다는 점과 케이스 결합형 추진제 (case bonded g ra i n) 를 제작할 수 없다는 단점이 있다. 일본에서 제작되는 무용제 압신 공정에 의한 대표적인 복기 추전제의 조성과 특성이 표 2.20 에 나타나 있다 .89) 2. 9. 3 주조공정 (casti ng pro cess) NC 계열 추전제(특히 복기 추진제의 경우)를 주조공정에 의해 제작하면 가장 다양한 크기 및 모양의 추전제를 제작할 수 있으 므로 설계된 형상대로 추전제 그레인을 얻을 수 있다. 이 주조공 정은 다른 공업에서 사용되는 주조공정과 그 공정기술 내용이 크
게 다르지 않다. 단지 고체 부분과 액 체 부분을 별도로 주입 한다 는 점과 고체 부분이 NC 를 주성분으로 하는 추전제 입자(알갱 이)라는 점이 다르다. 주조공정은 보통 2 단계로 나뉘어 있다 .63,90,9 1. 92) 첫째 단계는 이 미 언급된 용제 압신공정에서 사용되는 추전제 그레인 제조 방법 과 같은 방법 으로 주조용 NC 그레 늘, 죽 NC 알갱 이 (casti ng p owder) 를 제작하는 단계이며, 두번째 단계에서는 주조용 추진 제 사이에 가소제를 채워넣어 가소제가 NC 입자 속으로 확산되 면 NC 입자들과 가소제가 어우러져 추전제 그레인이 한 몸체 (mono lit h i c) 로 변하게 된다. 이 두번째 과정에서는 화학적 반응 은 일어나지 않으며 단지 물리적 변화만 일어나게 된다. 가소제 로서는 NG 와 같은 고에너지 물질울 흔히 사용한다. 이러한 주조공정법에 의해 로켓 추전기관 모터에 사용할 추진 제 그레인을 주조하는 몰드 (mould) 가 그림 2.42 에 있다. 로켓 모터가 작동할 때는 추진제의 연소에 의해 모터 내부에 고열이 발생하므로 이 고온의 화염으로부터 케이스를 보호할 수 있도록 추진제 그레인 외벽을 내열재로 라이닝(li n i n g)해야 한다. 내열재 재료로서는 셀룰로오스아세데이트, 에틸셀룰로오스 또는 열에 강 하고 추진제와의 접착력이 좋은 물질을 선택한다. 그 두께는 추 전제의 연소시간, 추전제의 연소온도, 화염이 케이스에 노출되는 시간 등을 고려하여 결정한다. 내열재를 추진제 그레인에 라이닝하는 방법은 추진제 그레인을 먼저 제작하고 그레인 위에 라이닝하는 방법과, 내열재를 먼저 제작하고 그 속에 추전제를 주조하는 방법이 있다. 제작 편의상 그레인이 먼저 제작되는 압신공정의 경우는 그레인 위에 에틸셀 룰로오스 같은 내열재로 라이닝하는 방법을 사용하고 있고, 주조 공정법에서는 그립 2 . 42 에서와 같이 내열재를 미리 제작하여 몰
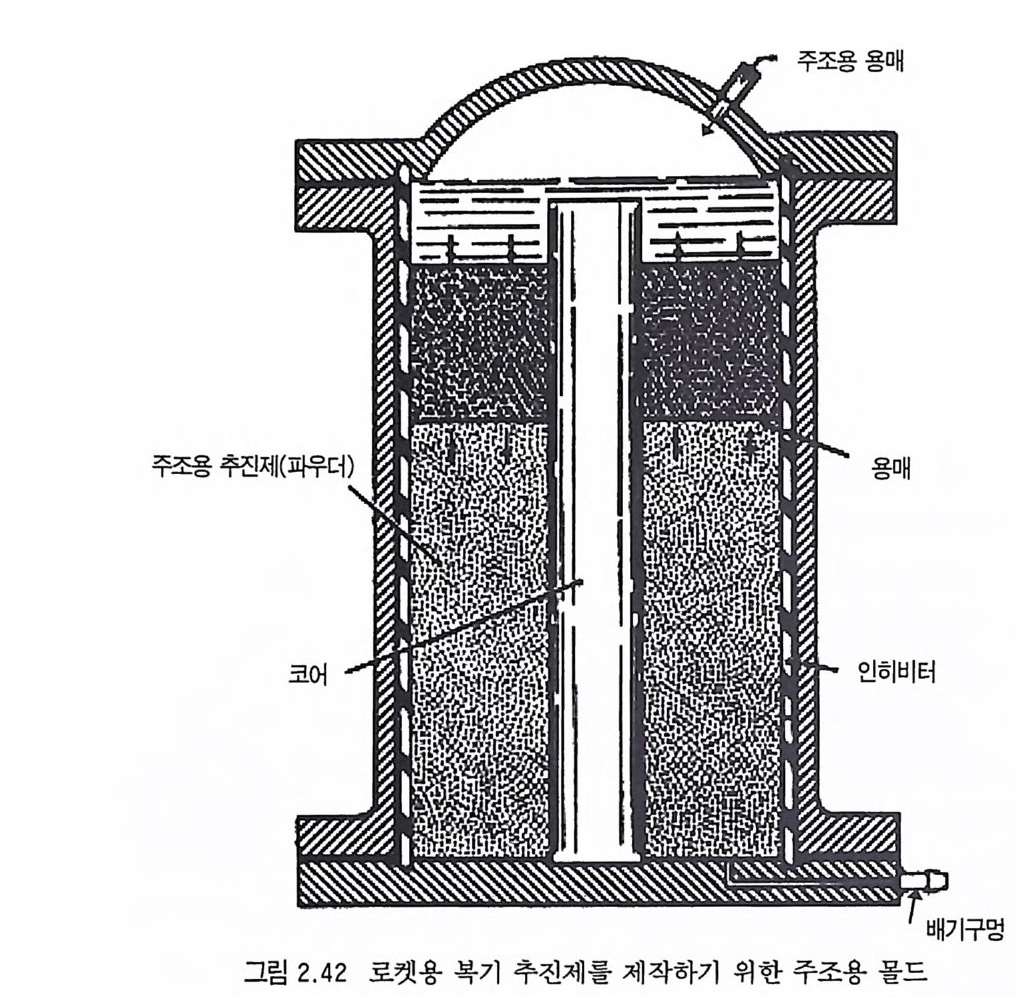 그립 2.4 2 로켓용 복기 추진제 를 제작하기 위한 주조용 몰드
그립 2.4 2 로켓용 복기 추진제 를 제작하기 위한 주조용 몰드
드 내부에 장착하고 추진제를 주조하는 방법을 채택하는 것이 일 반적이다 .1 , 63) 그림 2.42 의 모터 몰드의 2/3 정도를 주조용 추전제 (주조용 gr anule) 로 채운 뒤 몰드 내부의 공기를 뽑아내 전공 상태로 만 든 다음 가소제 (NG 또는 액체 상태의 고에너지 물질)를 느린 속도 로 주입한다. 가소제 주입은 위에서부터 아래쪽으로 하는 방법과 아래로부터 위쪽으로 주입하는 두 가지 방법이 있다. 가소제 주
입이 끝난 몰드는 완전히 밀폐된 후 오븐(약 60°C) 에 옮겨져 경 화된다. 경화되는 동안 가소제는 주조용 추진제로 확산되어 추진 제 알갱 이 가 스웰 링 (swell ing ) 되 며 일부는 녹아들어 가기 도 하여 몰드 내부의 추진제 덩어리(그레인)는 균일한 상태가 된다. 이러 한 주조공정법은 공정속도가 느린 단점은 있지만 대형의 그레인 을 제작할 수 있고 복잡하고 까다로운 모양의 그레인도 제작 가 능한 이점이 있다. 상기와 같이 압신공정에 의해 제작된 주조용 추진제를 사용하 여 주조하는 방법 대신에 NC 를 직접 가소제 및 부원료들과 혼 합한 후 모터 몰드에 주조하는 슬러리 (slurry) 주조공정이 개발되 어 사용되고 있다. 슬러리 주조공정에서는 섬유상 NC(fi br ous) 보다는 밀도가 높은 알갱이상 NC 를 주로 사용한다. 우선 NC 를 NG 와 같은 가소제와 시그마형 교반혼합기 (예, 그림 2.20 또는 그 림 3.3 1 참조) 안에서 혼합 분산시킨다. 액체 상태의 부원료(첨가 제)들을 가하여 교반시킨 후 혼합기 내를 진공으로 하여 휘발성 분을 제거한다. 고체 성분의 부원료(알루미늄, 과염소산암모늄, RDX 등의 고체 분말) 를 첨가하고 교반한다. 혼합이 끝난 슬러 리 혼합물을 모터 몰드에 그림 2.34 와 유사한 방법으로 주조한 후 오븐에서 경화시킨다. 이와 같이 제작된 추진제 그레인의 메트릭 스는 NC 로 이루어 져 있으므로 흔히 니트라졸(nit rasol) 이 라고 부 르며 NC 대 신에 PVC (po ly vin y l chlorid e ) 를 사용하여 제 작된 추 진제 는 플라스티 졸 (pla sti so l) 이 라고 부른다. 미국의 J.P .N 로켓용 추전제의 조성을 예로 들떤 다음과 같 다 .93) NC(l2.2 % N) 51. 5 % NG 43.0 % DEP (die t h ylp h th a late ) 3.25 %
카바마이트(센트럴라이트) 1.0 % 황산칼리 1.25 % 왁스 0.08 % 카본블랙 0.2 % 수분 0.6 % 〔참고문헌〕 1) T. Urbanski, Chemi stry and Technolog y of Ex plo siv e s, Vol. III, Eng lish Ed., Perga mon Press, New York (1985) . 2) C. F. Schonbein , Sit zu ng sb er. Natu r f ors h. Ge. Basel, 7, 27 (1846) . 3) J.H . Pelouze, Comp t. rend. 23, 809, 837, 861, 892 (1846) . 4) Lenk von Wolf sb urg, accordin g to S. J. Romocki, Geschic h te der Ex plo siv s to f f e, B d. II , Op pe nheim , Berlin (1896) . 5) E. Schult ze , Deuts c he Industr ie-Z tg . 10, III, 1865; Das neue chemi s- che Schie s sp u lver, Berlin (1865) . 6) Hartig , Unte r suchung e n uber den Besta n d und die Wi rk ung e n der explo siv e n Baumwolle, Braunschweig (1874) . 7) M. von Dutt en hofe r, accordin g to H. Brunswig , Das rauchlose Pulver, de Gruy ter , Berlin - Leip z ig , 1926; Brit . P at. 6022 (1887) ; 8776 (1902) . 8) P. Vi el ill e, Mem, Poudres 3, 9, 177 (1890) ; 4, 256 (1891) ; 7, 19, 30 (1894) ; 11, 157 (1901) ; 15, 61 (1909 -1910) ; Z. ge s. Schie s s-u . Sp r eng - stof f w . 6, 181, 303, 441, 464 (1911) . 9) A. Nobel, Brit. Pat. 1471 (1888) . 10) F. A. Abel and Dewar, Brit. Pat. 5614 (1889) . 11) C. Claessen, Ger. Pat. 256572 (1910 一 1913) . 12) H. Gallw ttz, Di e Geschutz l adung, Heereswaff en amt , 1944, accord- ing to Technic al Rep o rt PB 925, U.S. Dep t. of Commerce, Washi n-
gton . 13) H. Brunswig , Das rauchlose Pulver, de Gruy ter , Berlin - Leip z ig (19 26) . 14) 0. Pop pe nberg and Ste p h an, Z. ge s. Schie s s-u . Sp re ng s to j f w. 4 , 281, 305, 388 (1909) . 15) Andrew Novle, Proc. Roy. Soc. (London) A 76, 381, 512 (1905) ; 78, 218 (1906) . 16) H. C. Knig h t and D. C. Walto n , Ind. Eng. Chem. 18, 287 (1926) . 17) K. Kuo, Fundamenta l s of Solid Prop el lant Combustio n , AIAA Inc., New York, N.Y., U.S.A., 27 .:-29 (1984) . 18) J. Tayl o r, Solid Prope l lants and Exoth e rmi c Comp os it ion s, Newnes, London (1959) . 19) H. Nash, Army Ordnance 11, 293 (1931) ; Mem. art ill. fran c. 12, 765 (1931) . 20) H. Kast, Z. ge. Schie s s-u . Sp re ngs to ff w . 15, 196 (1920) . 21) T. Urbanski and T. Galas, Wi ad . Techn. Uzbr. 34, 501 (1939); Z. ges . Schie s s-u . Sp re ng st o ff w . 34, 103 (1949) . 22) E. Burlot, Mem. artil l. fi'an c. 23, 183 (1949) . 23) Comi te Sc ien ti fiqu e des Poudres. 24) J. Roszkowski, Z. Phys i k . Chem. 7, 485 (1891) . 25) H. F. Coward and Fl J. H art we el, Safe ty in Mi ne s Research Board, London 19 (1926) . 26) H. Dautr i c h e, Comp t. rend. 146, 535 (1908) . 27) J. Fauveau and le Pair e , Mem. pou dres 25, 142 (1932-1933) . 28) M. Prett re, Mem. pou dres 25, 160, 169, 531 (1932-1933) . 29) R. N. Pease, ]. Am. Chem. Soc. 52, 5106 (1930) . 30) K. F. Bonhoeff er and H. Reic h ardt, Z. phy s ik. Chem. A139, 75 (1928) . 31) L. J. Avramenko and V. N. Kondraty ev , Zh. eks p. teo r. fiz. 7 , 842 (1937) . 32) V. N. Kondraty ev , Ki ne tik a Khim i ch eskik h ga zovy k h reakts ii: Izd.
Akad. Nauk SSSR , Moskva (1958) . 33) J. Dwy er and 0. Oldenberg, ]. Chem. Phys . 12, 351 (1944) . 34) R. G. W. Norris h and G. Porte r , Natu r e 164, 658 (19 49) ; Proc. Roy. Soc. (London) A210, 439 (1952) . 35) R. G. W. Norri sh G. Porte r and B. a. Trush, Proc. Roy. Soc. (London) A216, 165 (1953) ; A227, 423 (1955) . 36) R. G. W. Norris h , XVI Cong res de chim i e Pure et Ap pliqu e, Paris, 1957, Ex pe ri en ti a Supp l. VII, 87 (1957) . 37) Accordin g to V. Recd, Z. ges . Schie s s- u. Sp re ngs t of f w . 1, 285 (1906) . 38) A. Buis s on, Le pro bleme des pou dres, Dunod & Pin a t, Paris (1913) . 39) W. Swi et o s lawski, T. Urbanski, H. Caiu s and M. Rosin s ki, Rocznik i Chem. 17, 444 (1937) . 40) W. Swi et o s lawski and J. Salcewic z, Rocznik i Chem. 14, 621 (1937) . 41) C. Sto rm, Army Ordnance 9, 230 (1929) . 42) G. de Bruin and P. F. M. de Pauw, N. V. Konin k lijk e Nederlands- che spring s t oj f enfa b r iek en 3, 4 (1926) ; 6 (1927) ; 8 (1928) ; 9 (1929) . 43) A. Ang e li and Z. E. Jol les, G. Chim . Ind. ed Ap pl. 14, 65 (1932) . 44) Z. E. Jol les and M. Socci , G. Chim . Ind. ed Ap pl. 15, 113 (1933) . 45) G. de Bruin , N. V. Konin k li jke Nederlandsche Sp ring s to j f enfa b ri e- ken 5 (1927) . 46) A. Ang e li, At ti reale accad. Lin z ei, Roma 23, 20 (1919) . 47) D. Berth e lot and Gaudechon, Comp t, rend. 153, 1220 (1911) . 48) P. Vie i l le, Mem. pou dres 5, 81 (1892) . 49) E. Bergm a nn and A. Jun k, Z. ang ew . Chem. 17, 1022 (1904) . 50) M. Toneg utti and B. Debenedett i, Ann. Chim . Ap pli ca ta 22, 627 (1932) . 51) A. Nobel, Ger. Pat. 51471 (1889) . 52) E. Berge r, Bull. soc. chim . France [사 , 11, 1 (1912) .
53) P. Demoug in and M. Landon, Mem. pou dres 26, 273 (1934 一 1935) . 54) J. Desmaroux, Mem. pou dres 21, 238 (1924) . 55) M. Marqu eyr o l and H. Muraour, Memo. pou dres. 21, 259 (19 24) . 56) M. Marqu ey ro l and P. Lorie t t e, Mem. pou dres 21, 277 (1924) . 57) T. L. Davis and Ashdown, Ind. Eng. Chem. 7, 674 (1915) . 58) W. A. Schroeder, E. W. Malmberg, L. L. Fong , K. N. Trueblood, J. D . Landerl and E. Hoerge r, Ind. Eng. Chem. 41, 2818 (1949) . 59) F. A. Abel, Trans. Roy. Soc. 157, 181 (1867) . 60) M. Marqu erol, Mem. pou dres 23, 158 (1928) . 61) H. Lecorche and P.L. Jov in e t, Mem. pou dres 23, 147 (1928) ; Comp t. r end. 187, 1147 (1928) . 62) T. Urbanski, B. Kwi at k o wski and W. Mi lad owski, Przemy sl Chem. 19, 22 (1935) . 63) S. S. Penner and J. Ducarme, The Chemi str y of Prop el lants , AGARD Combusti on and Prop u lsio n Panel, Paris, France (1959) . 64) Eley, D. D., and Pepp e r, D. C., Trans. Farad. Soc. 43, 559-91 (1947) . 65) Doy le , G. J., a nd Badge r, R. M., ]. Ap pl. Phys . 1 9, 373 -7 (1948) . 66) Ni el sen, J. M., Ernsberge r, F. M. , U . S. Gov t. Inte r nal Rep o rt (1953) . 67) Newman, S., Drechsel, P. D., U . S. Gov t. I nte r nal Rep o rt (1957) . 68) Boy a rs, C., ARS Jou rnal 29, 148 (1959) . 69) Boy a rs, C., and Goug h , W . G., Anal. Chem. 27, 957-61 (1955) . 70) Rosser, R. J., U. K. Gov t. R ep o rt (1949) . 71) Levy , J. B., ]. Am. Chem. Soc. 76, 3254-7, 3 790-3 (1954), 77, 2015 -6 (1955) . 72) Gelernte r , G ., Brownin g , L. C., H arris , S . R., Mason, C . M., J. Phys . Chem. 60, 1260-4 (1956) . 73) Phil lips, R. W., Orlic k , C. A. and Ste i n b erge r, R., J. P hy s. C hem. 74)5 9D, 1e0s3m4 a-r9ou x(1, 95C5o)m . p t, rend. 194, 1649 —51 (1932) .
75) Giz y c k i, J. F. V., Chem. Ztg . 74, 649-51 (1950) . 76) Ivin s , H., Keller, P. R., Kesti ng , L. W., Moore, W. J., U. S. Govt. Inte r nal Rep o rt (1953) . 77) Cruis e , R. D., Moon, E. L., Tayl o r, C. A., U. S. Govt. Inte r nal Repo r t (1958) . 78) V. Lind ner, Exp lo siv e s, Prop e llants , Theory And Practi ce, V. S. Arnament Command, Denver, N. J. (1978) . 79) T. S. Yeg o rov, Proiz v odstv o bezdym nog o piro ksil in o vog o po rokha, Moskva (1935) . 80) L. Vennin , E. Burlot and H. Lecorche, Les pou dres et exp lo sifs , Berange r, Paris - Li e g e ( 1932) . 81) V. V. Sukhin s kii (1892-1894) , accordin g to V. A. Boldy ry e v and S. A. Brouns, Kratk i i kurs tek hnolog ii po rokha, Gosnate k hiz d at, Moskva-Lenin g rad (1932) . 82) A. Davenas, Solid Rocket Prop ul sio n Technology , Perga mon Press Inc., Oxfo r d (1993) . 83) G. Fleury, Mem. Prodress 24, 49 (1930-1931) . 84) Gim ler, J. R., Solventl es s extr u sio n of double-base pro p el lant pre p a red by a slurry pro cess. Unit ed Sta t e s Pate n t 4-29 8-55 2 (1981) . 85) Muller, D. and Ste w art, J., Twi n screw extr u sio n for the pro duc- tion of sti ck pro p e llants . jo urnal of Hazardous Mate r i als , 9, 47-62 (1984) . 86) Olsson, M., Screw extr u sio n of double-base pro p el lants . ICT Jah res- tag ung Karlsruhe (1981) . 87) G. Schenkel, Schneckp re ssen fur Kunsts tof f e, Hanser, Mi lnc hen (1959) . 88) K. Wrobel and J. Luczaj, Wy tla czanie two rzyw sztu c ny ch , PWT, Warszawa (1961) . 89) BIOS/]AP/PR/1292/Rep o rt. Jap a nese Prop e llants - Research on Non-volati le Solvent Powders, H.M.S.0., London.
90) 20th Meeti ng Bulletin , Joi n t - A rmy -N a vy -A i r- F o rce-ARPA-NASA Panel on Phys i c a l Prop er tie s of Soli d Prope l lants , 1961, Riv e rsid e , Cali forn ia , Vol. I and II, Joh n's Hop k in s Univ e rsit y (1963) . 91) D. E. Boy n to n and J. W. Schoweng a rdt, Chemi ca l Eng ine eri ng Prog re ss 59, 81 (1963) . 92) F. A. Warren, Rocket Prop el lants , Rein h old, New York (1958) . 93) M. E. Serebrya kov, Vnutr e nnaya ballist ika , Oborong iz, Moskva (1962) .
제 3 장 혼합형 추진제 매우 오래 전부터 사용된 것으로 알려진 흑색화약은 제 2 차 세 계대전 전까지만 해도 로켓에 사용된 유일한 고체 추진제였다. 흑색 화약은 탄소 (charcoal) 의 함유량이 상당히 높고 연소속도가 느린 화약이다. 흑색화약은 해안가와 배 사이에 로프롤 연결(운 반)시키기 위한 소형 로켓이나, 구조용, 신호용 및 그와 유사한 목적의 산업용 소형 로켓 추진제로 현재까지도 사용되고 있다. 흑색화약이 제 2 차 세계대전 이후 로켓용 추진제로서 거의 사용되 지 못하는 첫 째 이 유는 그 비 추력 lsp ( spe c i fic im p u lse) 가 40 ~ 80 sec 정도로서 현재 사용되는 무연 복기 추전제의 180~220sec, 혼합형 추진제의 230~250sec 와 바교해 볼 때 너무 낮은 성능을 갖고 있기 때문이다. 1,2,3,4) 흑색화약 이후 등장한 복기 추전제는 한동안 로켓 추전제로 사 용되어 왔으나 대형 로켓으로 사용하는 데는 몇 가지 문제접이 드러났다. 죽, 복기 추전제로서는 대형 추진제 그레인을 제작하 기가 어렵고, 제작이 가능한 경우라도 설비 무자비가 너무 높아
경제성에 문제점이 있다. 또한 니트로글리세린을 원료로 사용해 야 하므로 제작공정 중 폭발, 연소 위험성이 크며 더욱이 복기 추진제는 그 안정성을 주기적으로 시험해야 하는 번거로움이 있 다. 특히 복기 추진제는 그후 개발된 혼합형 추전제에 비해 비추 력 (Is p)이 20~30sec 낮은 수준에 있다. 이와 같은 이유로 인해 최근에는 복기 추진제 대신 고체 산화제가 폴리머 메트릭스에 충 전된 혼합형 추진제가 개발되어 로켓용 추진제로 적용되고 있 다 .2,3,5) 혼합형 추진제 성분 중 고체 산화제는 산화제의 양, 산 화제의 입자 크기 및 분포도 등에 따라 추전제의 기계적 성질에 큰 영향을 주므로 고체 산화제의 물리 화학적 상태는 여러 가지 제약 조건이 따르게 된다. 고체 충전율을 높여 내탄도 특성이 우 수한 추진제를 제조하기 위해서는 추전제 제작시 원료들을 혼합 한 혼합물 반죽 (pa ste ) 의 유동성 (flui d i t y, 또는 점도) 이 나쁘지 않 은 조건하에, 또한 바인더 성분이 추전제 속에서 불연속 상태가 없는 균일한 기계적 성질을 유지시키는 조건하에, 가능한 한 고체 성분의 충전도를 높이는 것이 필요하다. 일반적으로 산화제의 평 균 입자 크기를 두 종류 또는 세 종류로 선택하여 바인더 성분과 혼합하고 있다. 3,5) 로켓용 추전제는 보통 다음과 같이 나누어진다. ® 주조 및 경화(중합)에 의해 제작되는 혼합형 추진제(예, PPG 와 과염소산암모늄의 혼합물) ® 주조형 복기 추전제 (예, NG/NC 를 주원료로 하는 추진제) ® 혼합형 개 선 복기 추진제 (Comp o sit e Modif ied Double Base 추전제) 위의 ®와 , ®의 복기 추진제류는 이미 제 2 장에서 언급하였으므 로 이 장에서는 생략하기로 한다. 로켓용 추진제로서 현재 가장 많이 사용되는 ®항의 혼합형 추진제는 프리폴리 머 (pre p o lym er)
를 바인더의 기본 원료로 선택하여 경화제와 화학반응(경화)을 시켜 고무상의 점탄성 고무상 물질로 얻어진다. 추진제 제조공정 중 경화반응이 일어나기 이전 고체 산화제 및 각종 첨가제들을 바인더에 혼합시키므로 산화제나 각종 첨가제들을 혼합한 상태는 밀가루를 반죽한 상태와 비슷한 성질을 갖게 되며, 고체 분말로 인해 그 점도가 매우 높다. 따라서 높은 점도를 지닌 이 반죽 혼 합물 내에 포함된 각 원료의 분포가 균일해야만 경화된 후 만들 어진 추진제의 성질이 균일하게 된다. 또한 그 혼합 반죽상태가 좋지 않으면 다음 단계인 주조공정을 진행하지 못할 경우도 있으 므로 반죽 혼합물의 유동성 (rheolog ica l pro p e rt y) 은 혼합 및 주조 공정에서 가장 중요한 성질이라고 볼 수 있다 .5,6,7) 산화제로서는 질산암모늄, 질산칼리, 질산나트륨, 과염소산칼 리, 과염소산암모늄 등이 흔히 사용되고 있다. 고체 추진제는 탄 성체 혹은 플라스틱 성질을 가지고 있으며, 이러한 성질을 유지 시키는 바인더 성분이 연소성 물질로서 또한 추전제 강도(기계적 성질)를 유지시키는 물질로 작용한다. 추진제 연소시의 방출 에 너지를 증가시키기 위해 알루미늄과 같은 금속 분말을 첨가하기 도 하며, 연소 또는 점화 상태를 좋게 하기 위해 탄소 분말을 첨 가하기도 한다. 혼합형 추전제의 또 다른 장점 중에는 그 원료들의 가격이 비 교적 저렴한 점, 추전제를 오랫동안 보관해도 분해반응이 쉽게 일어나지 않고 안정적인 점, 제작공정이 비교적 안전한 접 등이 있다. 일반적으로 추진제의 연소시 배출되는 가스의 평균 분자량 이 가능한 한 낮아야 그 추진제의 추력이 높아질 수 있다. 따라 서 가능한 한 작은 원자량을 지닌 원소로 이루어전 원료들을 추 전제 원료로 사용하는 것이 더 높은 비추력을 얻기 위해 유리하 다고볼수 있다.
3.1 추진제 그레인
로켓 추전제 그레인들은 그립 3.1 및 그림 3.2 와 같이 여러 가  지 모양으로 설계, 제작되고 있다. 추전제는 점화제에 의해 연소 가 시작되며 연소되는 면적에 따라 배출되는 추력 및 압력의 크 기가 결정된다. 따라서 추진제 그레인 모양과 불꽃에 노출되는 면적을 조절하기 위해 내열 라이너 혹은 연소 억제제 (inh ib i t ing a g en t)로 추진제 표면의 일부를 연소 불꽃으로부터 차단시키기도 한다. 왜냐하면 많은 종류의 로켓 추진기관은 추진제가 연소하는 도중, 죽 추진기관이 작동하는 동안 추전기관 내부의 연소압력을 균일하게 유지시키는 것이 바람직하기 때문이다. 추진제 그레인 설계에 따라 연소 표면적을 조절하는 가장 간단한 경우인 원통형 및 스타형 (sta r shap e d) 추진제 그레인의 추력, 연소시간 곡선아 그림 3.3 에 나타나 있다, 1,8)
지 모양으로 설계, 제작되고 있다. 추전제는 점화제에 의해 연소 가 시작되며 연소되는 면적에 따라 배출되는 추력 및 압력의 크 기가 결정된다. 따라서 추진제 그레인 모양과 불꽃에 노출되는 면적을 조절하기 위해 내열 라이너 혹은 연소 억제제 (inh ib i t ing a g en t)로 추진제 표면의 일부를 연소 불꽃으로부터 차단시키기도 한다. 왜냐하면 많은 종류의 로켓 추진기관은 추진제가 연소하는 도중, 죽 추진기관이 작동하는 동안 추전기관 내부의 연소압력을 균일하게 유지시키는 것이 바람직하기 때문이다. 추진제 그레인 설계에 따라 연소 표면적을 조절하는 가장 간단한 경우인 원통형 및 스타형 (sta r shap e d) 추진제 그레인의 추력, 연소시간 곡선아 그림 3.3 에 나타나 있다, 1,8)
증乙병 프로그레시브 및 드그레시브 約
® 器
©o: 릴a 霜1 0 썬 빙 .鬱
그림 3.1 추진제 그레인 설계
126
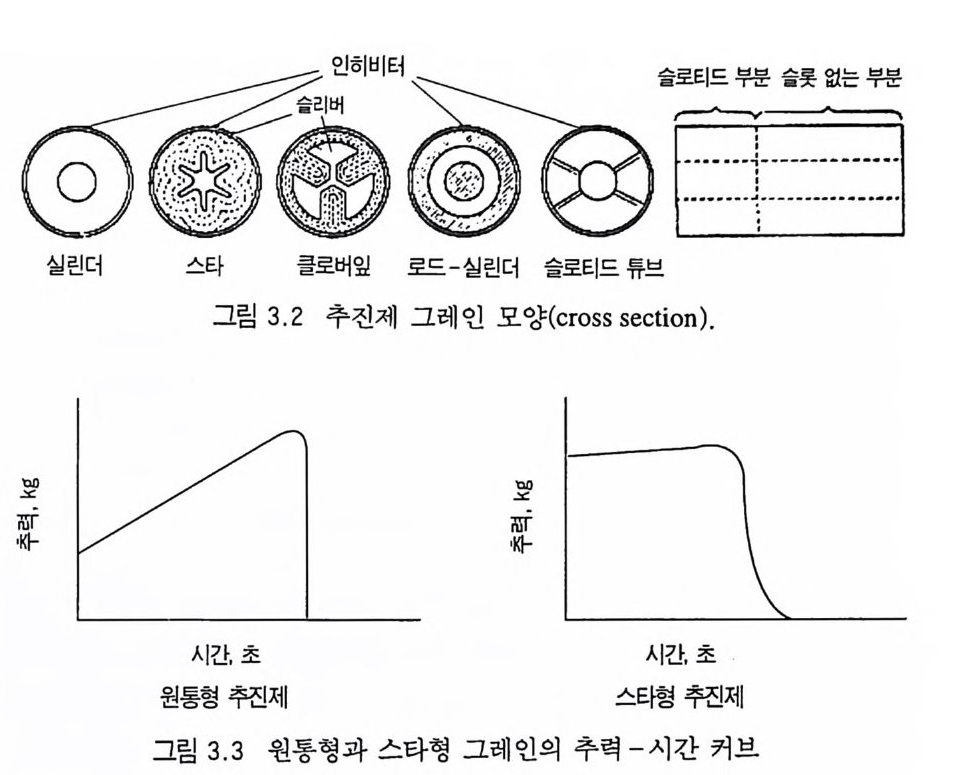 실린더 스타 `출클로인버히잎 비 터 로드-실린더 슬로티드튜브슬 二로티드 부분 슬롯 없는 부분
실린더 스타 `출클로인버히잎 비 터 로드-실린더 슬로티드튜브슬 二로티드 부분 슬롯 없는 부분
현재 사용되고 있는 많은 종류의 고체 추진 로켓은 추전제가 연소실, 죽 연소 케이스 벽에 접착되어 있다. 일반적으로 추전제 와 케이스 벽 사이에 내열 라이너를 사용하고 있으며, 라이너가 접착 작용을 하는 동시에 추진제 연소시에 발생하는 뜨거운 연소 가스로부터 케이스를 보호해 주는 역할도 하고 있다. 추진제 연 소는 추진제 그레인의 빈 공간에서부터 일어난다. 다음 그림 3.4 에 는 이 와 같은 케 아 스 접 착 모터 (case-bonded moto r ) 의 그립 과 그 모터 내부 중 위험한 부위를 나타냈다. 그 중 a 부분은 모터 의 앞끝쪽 부분으로서 추전제와 내열 라이너에 응력이 집중되는 부분이므로 추전제가 들어 있는 모터를 장기 보관시 그레인에 균 열이 나타나거나 혹은 집착 불량 등으로 인해 모터 작동이 실패
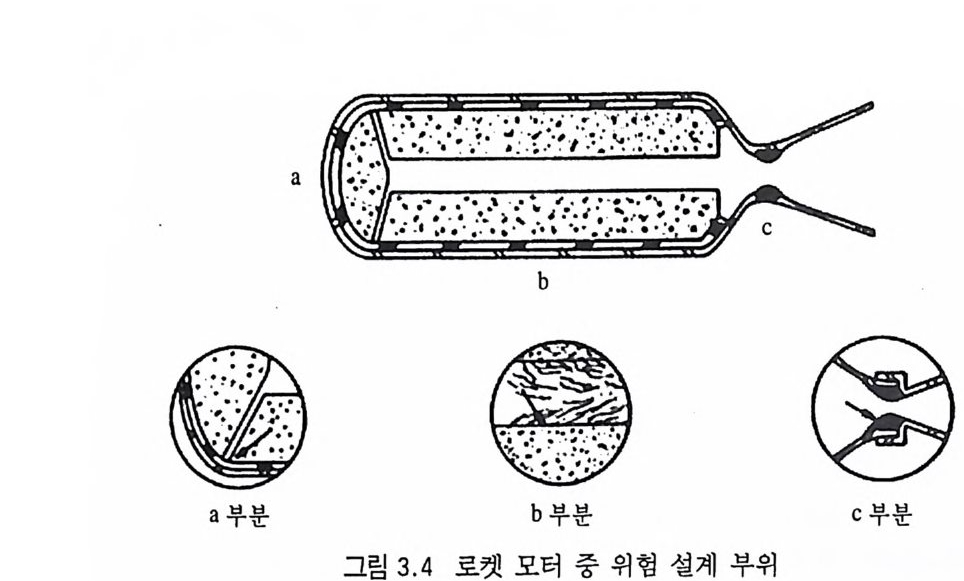 a ((l::,':: \:..-:: ,: . :,`.: •- •. .:. . •.:. ,• ..국 ...,·,. .- ... \ • >;.: • ., ..`,' ., . . ,f.`.- .; . 미. g . ,, . '.•.•. • ,r gr. : ’,.~. .、. ?r . `.. · `:· I'4.: V ·. ` :. . ?:‘ ...:: . :. . '. ”• ; ?'. •
a ((l::,':: \:..-:: ,: . :,`.: •- •. .:. . •.:. ,• ..국 ...,·,. .- ... \ • >;.: • ., ..`,' ., . . ,f.`.- .; . 미. g . ,, . '.•.•. • ,r gr. : ’,.~. .、. ?r . `.. · `:· I'4.: V ·. ` :. . ?:‘ ...:: . :. . '. ”• ; ?'. •
할 수 있는 위험 부위이다. 마찬가지로 뒤끝쪽 부분도 응 력이 집 중되는 부분이므로 위험한 부위이다. 추전제와 라이너에 작용하 는 응력은 이상과 같이 그레인 모양이나 부위에 따라 발생하지 만, 세 종류의 상이한 재료(추전제 • 내열재 • 케이스)를 사용하는 데서 오는, 죽 추전제 공정상의 이유로 인해서도 일어난다고 볼 수 있다. 죽 추전제의 주조온도, 경화온도 및 보관, 작동온도가 다르므로 추전제의 부피 변화로 인한 응력이 발생하며, 서로 다 른 재료들이 집착되어 있으므로 각 재료의 상이한 열팽창 성질로 인해서도 응력이 발생한다. 현재에는 추진제 제작기술의 발전으 로 경화공정으로 인해 일어나는 응력은 상당히 해결되었다고 볼 수 있다. 일반적으로 추전제의 열팽창 계수는 스틸(케이스) 재료 의 열팽창 계수의 10~15 배 정도로 큰 것으로 알려져 있다. 또한 추전기관 모터의 작동온도는 ― 55°C~+70°C 이므로 각 온도별 구간에서의 추전제, 라이너, 케이스 재료 특성을 유의해서 시험, 분석할 필요가 있다 .2,9) 만일 저온에서 추전제가 그 점탄성을 상실하여 너무 딱딱하게
변화되는 성질을 갖고 있으면 열적 수축에 따른 응력에 의해, 또 는 추전제가 점화되는 순간 급격한 압력 상승으로 압축 응력이 발생하여 추진제가 갈라질 경우에 추전기관 모터 작동은 실패할 우려가 있다. 그립 3 . 4 의 b 부분은 추전제의 연소 표면으로서, 추진제 내에 균열 혹은 기포 등이 있으면 추진제의 연소면적이 급격히 증가하게 되며 연소면적의 증가는 연소압력 증가를 가져 오게 되고, 압력 증가는 추진제의 연소속도를 빠르게 하는 효과 를 가져오므로 결과적으로는 추전제 그레인의 폭발적인 연소 현 상이 일어나게 되어 추진기관 모터가 폭발하는 것과 같은 모터 작동의 실패가 일어난다. 따라서 추전제 내에 들어 있는 기포 혹 은 균열등의 유무 및 크기와 연소될 때에 예상되는 압력 증가, 보관 도중 균열, 기포의 크기, 변화 등에 유의해야 한다. c 부분 은 노줄목 (nozzle th roat) 부위로서 추진기관 작동시 추전기관 내 에서 가장 큰 열 전달 속도가 있는 부분이며 연소가스에 의한 마 모 및 침투 현상이 있는 주요 부위이다. 이 노즐목은 흑연, 텅스 텐과 같은 내화금속 (re fr ac t or y meta l ) 또는 탄소-탄소 (carbon/ carbon) 복합재료 등으로 설계 , 제 작한다. 3.2 추진제의 에너지 및 연소속도 로켓 추전기관 모터 내부에서 추전제가 연소될 때 이 모터 내 부는 일종의 화학 반응기라고 볼 수 있다. 모터 내에서 추전제가 연소하여 가스로 변화되면 연소가스는 매우 빠른 속도로 노즐을 통해 배출되는 동시에 추전기관 모터는 앞으로 전전하는 추력 (힘)을 얻게 된다. 이와 갇이 발생한 에너지, 죽 추력 (im p u lse) 은 추진제 조성으로부터 열화학적 방법으로 계산되거나 추진기관
모터 연소 실험에 의해 측정되기도 한다. 일반적으로 혼합형 추 진제는 연소열이 대 략 1000~2000 ca l/g의 수준에 있다. 비추력 fs p는 추전제 단위 무게당 발생된 에너지(힘)로서 다음과 같은 식 에 의해 계산된다.
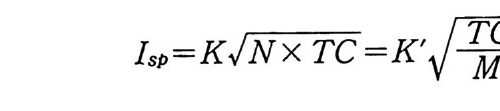 fs p =K /N言~ =K' ff
fs p =K /N言~ =K' ff
여기에서 N 은 추전제 무게당 생성된 연소가스의 몰수이며, M은 연소가스의 평균 분자량, TC 는 추진기관 모터 연소실 내 에서의 추진제 연소온도(평형온도), K 및 K' 는 상수이다. 이 식으로부터 생성된 연소가스의 분자량이 작을수록, 연소온 도가 높을수록 비추력이 증가하는 것을 알 수 있다. 추진제 원료 중 고분자 구조인 바인더 성분에 따라 연소가스의 성질이 결정되 므로 바인더의 분자량 및 그 화학적 특성은 추전제의 성능(비추 력)을 결정하는 가장 중요한 결정 요소 중의 하나이다. 비추력과 관련된 연소 온도 (TC) 를 울리기 위해서, 첫째 추진제 조성 성분 둘의 생성열은 가능한 한 높아야(LJ H>O) 하며, 둘째 연소 생성물 의 생성열은 가능한 한 낮은 값을 나타내어야 하고(LJ H
3.3 혼합형 추진제의 성분과 역할 혼합형 추전제는 세 종류의 기본 성분으로 구성되어 있다. 첫 째는 추진제의 연소에 필요한 산소를 공급해 주는 고체 성분인 산화제, 둘째는 추전제 연소시 높은 에너지를 방출시키는 금속 (환원제)연료 분말, 셋째는 바인더, 죽 폴리머 메트릭스이다. 폴 리머 성분은 연소성 물질의 주요 공급원이며 추전제 그레인 형상 울 유지시키는 데 필요한 각 성분들의 결합작용(결합제)을 한다• 3. 3. 1 바인 더 (bin d er) 3.3.1.1 바인더의 역할 이미 설명한 바와 같이 바인더는 혼합형 추전제의 주요 성분으 로 몇 가지 역할을 하고 있다. 첫째는 추전제를 탄성체의 메트릭 스로 형성하여 넓은 범위의 작동 온도하에 노출되어 있는 추전제 그레인이 만족할 만한 기계적 성질을 유지시키도록 하는 역할을 하고 있다. 일반적으로 추진제 내에서 바인더의 함량이 증가할수 록 추전제의 기계적 성질은 더 좋아진다. 바인더 함량이 추전제 의 12~20 % 수준일 때 매우 양호한 기계적 • 물리적 특성을 얻 울 수 있다고 알려져 있다. 추진제의 기계적 성질은 바인더와 고 체 분말의 상호특성 및 관계에 따라 다르지만, 바인더는 고체 산 화제 분말 및 금속 분말을 기계적 • 화학적으로 결합시켜 질긴 고 무상의 추진제 덩어리를 만드는 작용을 하여 추전제의 의부 및 내부에서 가해지는 열적 및 기계적 옹력을 견딜 수 있게 하고 있 다. 따라서 바인더는 추진제의 기계적 특성을 좌우하는 가장 큰 역할을 하는 성분이라고 볼 수 있다. 또한 추전제의 바인더는 모터 케이스 금속 부분을 추진제와 연
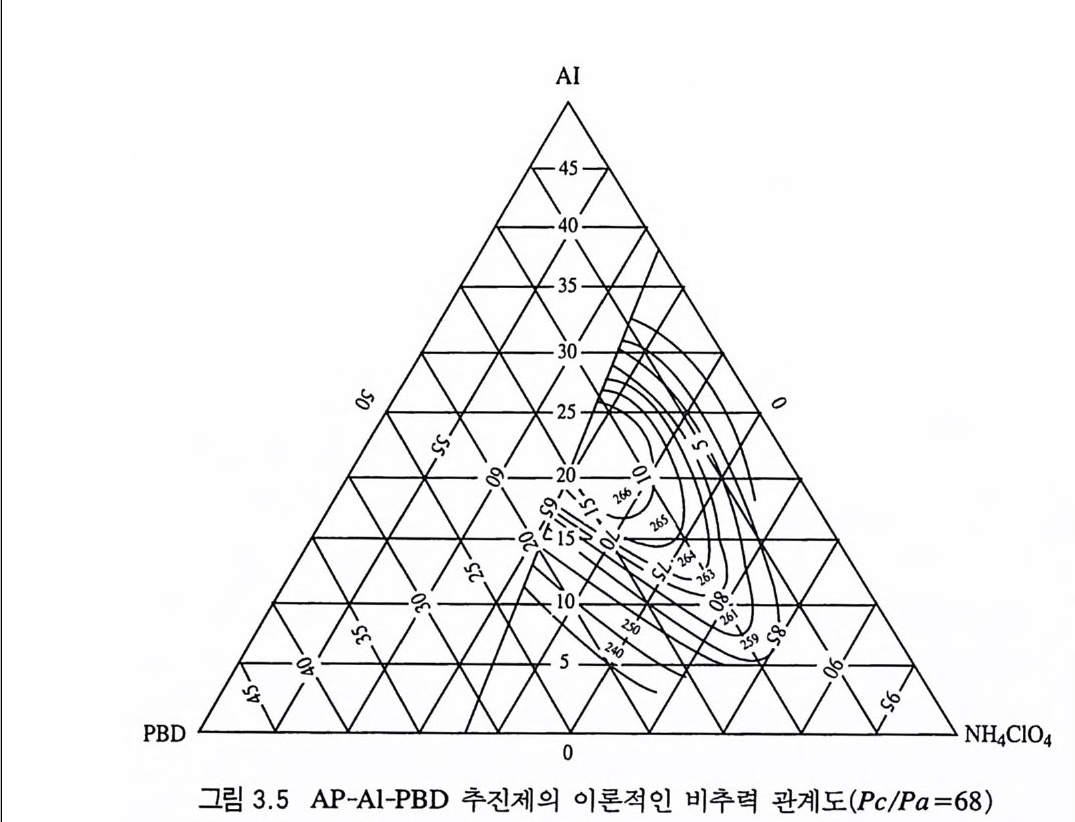 AI
AI
결 • 접착시키는 내열재 혹은 내열 라이너와의 결합(접착)작용도 한다. 바인더의 또 다른 역할로서는 추진제 연소시 추력 발생에 필요한 연소성 물질로 작용하는 것이다. 추진제 비추력은 그림 3.5 에서 보이는 것처럼 바인더, 산화제, 금속연료 함량에 따라 영향을 받는다. 따라서 특정 바인더 시스템에서 최대의 비추력을 얻을 수 있는 바인더 함량(%)이 최고의 기계적 성질을 얻을 수 있는 조성이 아니므로 추전제 그레인 설계자로부터 요구된 기계 적 성질을 맞추어 줄 수 있는 범위 내에서 최대의 비추력을 얻을 수 있도록 추진제 조성을 선택해야 한다 .11 , 12)
3.3.1.2 바인더의 종류 및 주조가능 시간(p o t life) 혼합형 추전제 제조에 최초로 도입된 바인더는 아스팔트, 폴리 이소부틸렌, 폴리영화비닐 (PVC) 과 같은 열가소성 바인더였다. 열가소성 레전을 가열용융(어떤 경우에는 swe lli n g)시킨 후 가소 제, 산화제 및 금속연료 분말 및 각종 첨가제를 가하여 혼합시켜 주조 냉각하는 제조 방법에 의해 열가소성 혼합형 추전제를 제조 하게 된다. 그러나 이러한 추진제는 그 기계적 성질이 좋지 않고 제조 단계 중 매우 높은 온도로 가열해야 되는 단점 및 냉각시 응축 효과가 너무 커서 대형 그레인 제작에 어려운 점들이 있다. 표 3 . 1 에 PVC 계 추진제의 대표적인 조성이 있다. 이러한 열가 소성 추진제는 그 분자 구조가 선형 (line ar) 고분자이다 .13)
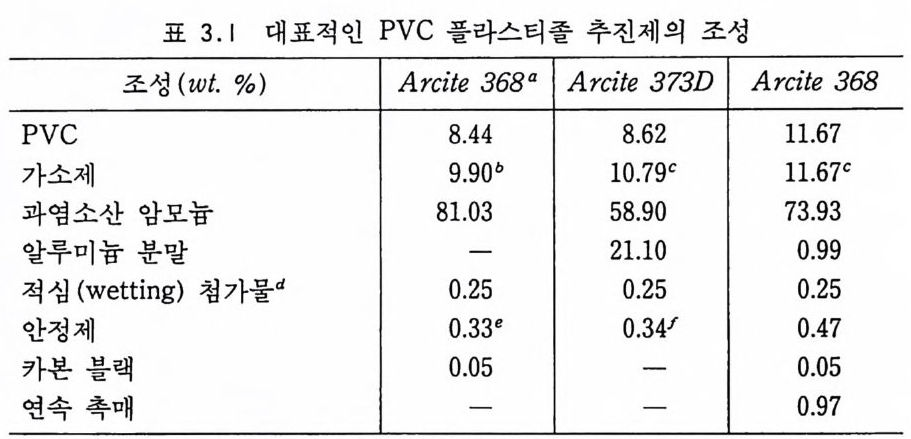 표 3.1 대표적인 PVC 풀라스티졸 추진제의 조성
표 3.1 대표적인 PVC 풀라스티졸 추진제의 조성
a Arci te : a reg ist e r ed tra demark of At la nti c Research Corp . b Plastic ize r : dib u ty l sebacate . c Plastic ize r : 2-eth ylh exy l adip a te . d Glyc erol monooleate , pe nta e ryt hri t ol dio l eate 및 dio c ty l sodiu m sulfo s uc- ci na te. 의 1 : 1 : 1 의 혼합물 • Ferro 1203, Fer:r o Corp. f BC 74 와 XE 82 의 혼합물 (1 : 1) Advance Chemi ca l Co.
최근 혼합형 추진제에 적용되는 바인더는 대부분 가교 결합형 (crossli nk ed) 바인더들이다. 표 3 . 2 에 가교 결합형 추전제의 제 조 특성들을 비교하였다 .9) 표 3.3 에는 대표적인 바인더용 폴리 머들의 구조들을 표시하였다 .13) 그림 3.6 에는 선형 폴리머와 가교 결합형 폴리머의 구조를 비 교하였는데 위쪽 그림이 선형, 아래쪽 그림이 가교 결합형 구조 이다. 선형 폴리머 구조는 긴 사슬을 가지고 있으나 서로 연결되 어 있지 못하여 폴리머 분자가 서로 미끄러지는 경향을 갖고 있 으므로 기계적 성질이 크리프 (cree p) 현상을 보여준다. 반면 가 교 결합된 폴리머는 분자간의 연결이 3 차원 망상 구조형으로 이 루어지게 되므로 탄성을 갖게 되고 의부로부터 하중을 받았을 때 라도 그 의부 하중이 없어지면 원래의 위치로 되돌아오는 복원력 이 있다. 그러나 3 차원 구조가 너무 완벽하게 이루어지게 되면 그 폴리머 구조는 딱딱하고 부스러지기 쉬운 성질을 지니게 된 다. 가교 결합 폴리머의 기계적 성질은 가교 결합 숫자와 댕글링
 표 3.2 바인더의 종류에 따른 추진제 제작 기술
표 3.2 바인더의 종류에 따른 추진제 제작 기술
(dang li n g ) 사슬의 숫자에 의 해 좌우된다. 그림 3 . 7 은 이 와 같은 바인더 구조 네트워크 (ne t work) 의 움직임을 보여주는 것으로서 맨 윗부분의 그림은 변이 이전의 상태이며, 아래쪽 그림은 응력 이 가해졌을 때의 폴리머 분자 구조로서 가교 결합 부분과 댕글 링 사슬이 확실히 나타나고 있다. 따라서 좋은 기계적 성질, 죽 강하고 질기며 탄성이 좋은 추전제를 얻기 위해서는 바인더의 가 교 결합 양(숫자)을 결정하는 것이 중요하다. 바인더가 경화제와 반응하여 가교 결합이 생성될 때, 죽 폴리 머가 생성될 때 젤화(g ela ti on) 과정이 진행된다고 볼 수 있다. 따라서 젤화점이 있으며, 이 젤화점을 기준으로 점성 유체로부터 탄성 젤 (elastic g el) 로 변화하게 된다. 젤화가 되기 전에는 적당 한 용매에 용해되거나 녹여지기도 하는데 젤이 생성된 후에는 잘 녹지도 않으며 용해도는 급격히 감소한다. 추진제를 제조할 때는 설계된 그레인 형상으로 주조된 후 젤화가 일어나도록 해야 한 다. 따라서 추전제는 각 원료들을 혼합한 후 주조공정에 필요한 몇 시간 동안(이 시간을 po t life라고 부름)은 주조할 수 있을 정도 로 충분히 낮은 점도를 유지해야 한다. 바인더의 경화반응, 죽 프리폴리머와 경화제의 화학반응은 젤화점 이후에도 계속된다고 볼 수 있으며, 추진제의 기계적 성질이 일정하게 유지되면 그 경 화반응이 종료되었다고 볼 수 있다. 이와 같은 가교 결합형 바인 더 로 서 는 PS (po lys u lfid e ) , PU (po lyu reth a ne) , PBAA (cop o lym e r of buta d ie n e and acryl ic aci d) , PBAN (cop o lym er of buta d ie n e and acryl ic ac id acryl o nit ri l e ) , CTPB (carboxy l- te r mi na te d po lyb u ta d ie n e) , HTPB (hy d roxy -ter mi na te d po lyb uta d ien e) , CTBN (carboxy -ter mi na te d po lyb u ta d ie n e acryl o nit ril e ) 등이 있다. 5)
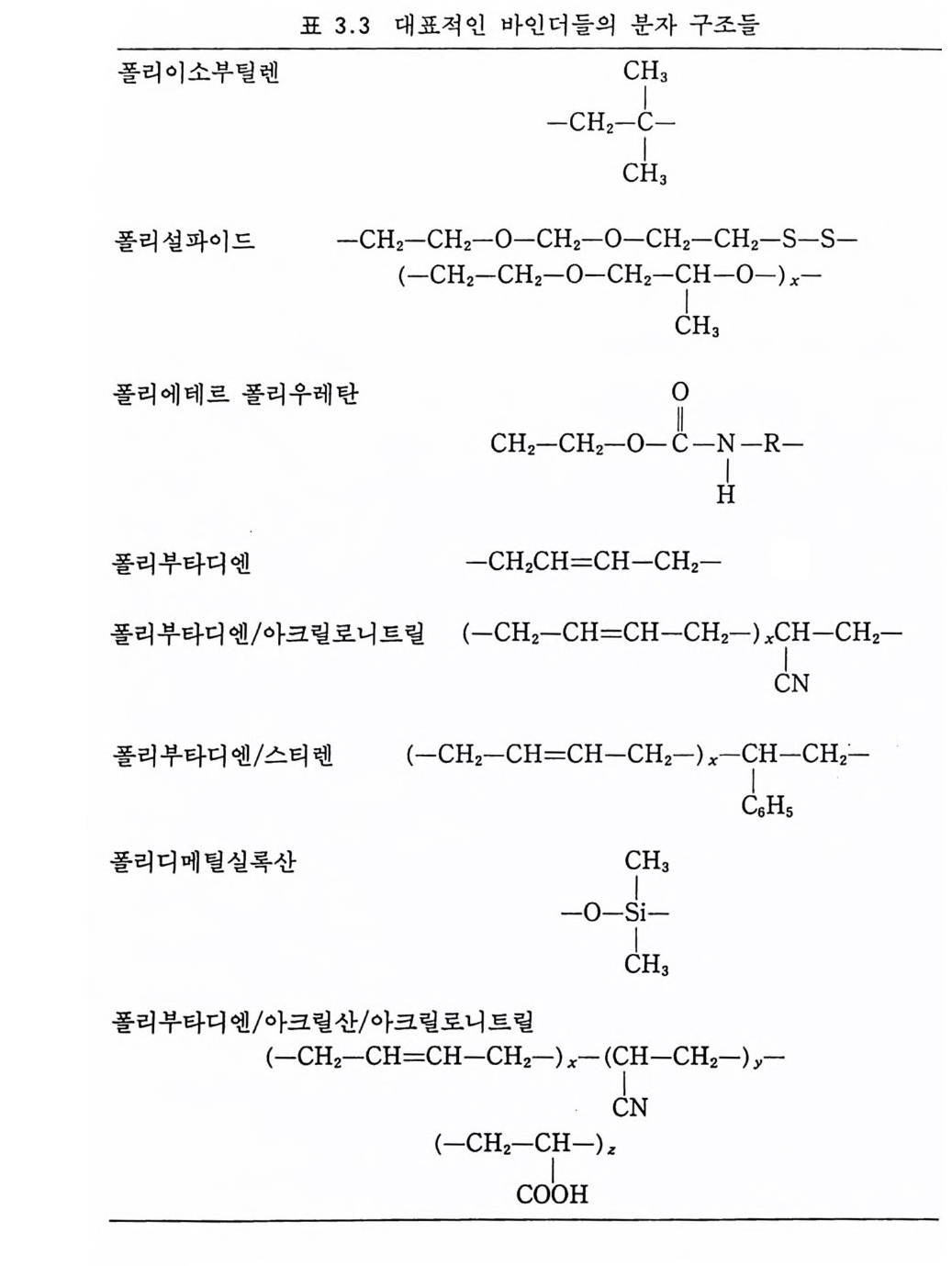 표 3.3 대표적인 바인더들의 분자 구조들
표 3.3 대표적인 바인더들의 분자 구조들
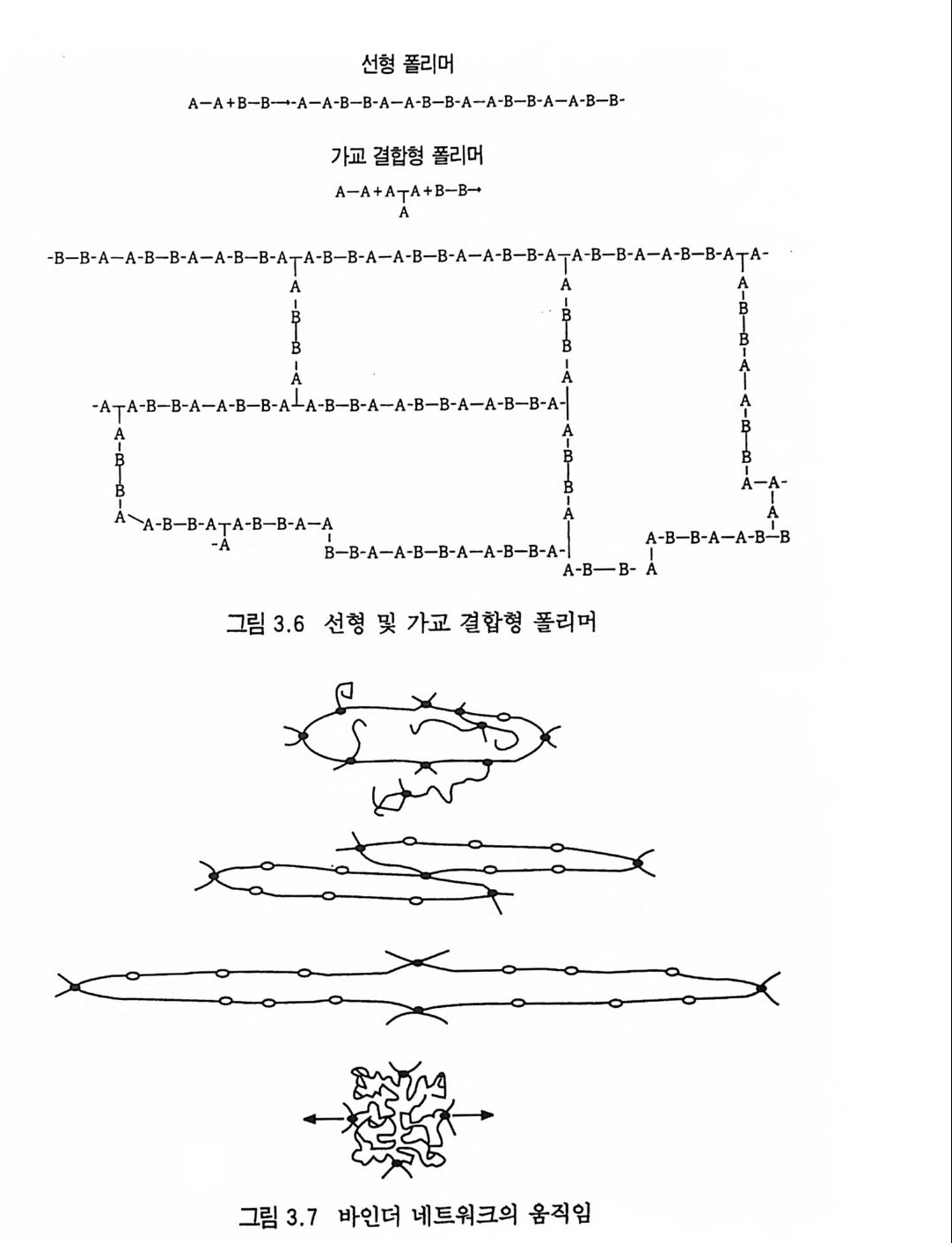 선형풀리머
선형풀리머
3. 3.1 . 3 아스팔트 추진제 혼합형계 추전제의 바인더로서 최초로 사용된 것은 아스팔트 였으며, 과염소산칼리 75 %를 아스팔트 25 %와 혼합하여 제조하 였다. 제조공정을 보면 고분자량의 아스팔트를 약간 가열하여 연 화시킨 후 과염소산칼리와 액체 탄화수소의 혼합물과 혼합시켜 모터에 충전시키는 방법이었다. 그러나 이렇게 제조된 아스팔트 추진제는 기계적 성질이 좋지 않았다. 탄화수소유를 첨가함으로 써 바인더의 특성 및 공정 특성(흐름성)을 어느 정도 개선한 조 성을 얻을 수 있었으나 실제로 탄성이 있는 추진제를 얻지는 못 하였다. 따라서 아스팔트 추진제는 실용화되지 못했으며 그후 아 스팔트 대신 폴리이소부틸렌, PVC 동의 열가소성 수지, 불포화 폴리에스테르, 스티렌 (s ty rene), 아크릴 등을 바인더로 적용하여 라디칼 중합을 시킴으로써 물성이 개선된 추진제들을 얻을 수 있 었으나 실제로 추전기관 모터에 적용된 경우는 드물었다. 3. 3.1 . 4 PS 계 추진제 로켓 및 유도무기의 사정거리가 증대되고 그 추진기관 모터가 대형화되면서 추진제 그레인의 스트레인 (s t ra i n) 특성이 우수한 추진제가 요구되고 있을 때 PS 계 추진제가 JP L( Je t Prop u lsio n Labora t or y)에서 개발되었다• 죽, 미국 J PL 에서는 1946 년 Th i okol 사의 LP-3 PS 액 체 폴리 머 에 과염소산암모늄 (혹은 과염 산칼리)을 혼합시킨 후 P- 퀴논디옥심의 산화성 경화제로 경화시 킴으로써 가교 결합 바인더로 이루어진 추전제를 최초로 제작하 였다. 이 액체 PS 폴리머는 디클로로에틸포르말, 트리클로로프 로판과 나트륨 p ol y sul fi de 를 반응시킴으로써 합성되었다. P- 퀴 논디옥심은 경화반응에 의해 P- 페닐렌디아민 또는 그 유도체 등 으로 환원된다 .14)
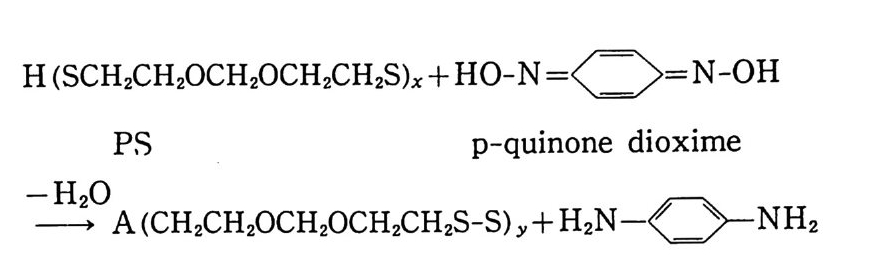 H (SCH2CH20CH20CH2CH2S)x + HO-N =〈二二〉= N-OH
H (SCH2CH20CH20CH2CH2S)x + HO-N =〈二二〉= N-OH
Thio k ol LP-3 액 체 폴리 머 는 평 균 분자량이 1000, 점 도는 7 ~ 12 포아즈 (po is e ) , 비 중은 1.27 정 도이 며 , 상기 경 화반응식 중 말단기 A 는 그 숫자가 매우 적지만 최종 중합 반응시 치환되는 기이거나 혹은 수산기 (h y drox y l) 인 것으로 알려져 있다. 3.3.1.5 우레탄 (ure t hane) 계 추진제 로켓 추전기관 모터의 크기가 점차 대형화되면서 PS 계 추전제 보다도 더 높은 내탄도적 특성과 더 우수한 기계적 성질을 지닌 추전제가 요구되고 있던 1950 년대 중반 폴리우레탄 추전제가 새 로이 개발되었다. 폴리우레탄 추진제 바인더는 수산(히드록실)기 를 갖고 있는 유기물질을 이소시아네이트 (NCO) 와 반응시킴으로 써 고무상 메트릭스 구조를 만든 것으로서, 이 메트릭스 속에 산 화제, 금속연료 분말 및 각종 첨가제를 혼합 분산시키면 우레탄 추전제가 제조된다. 이 메트릭스의 기본 구조는 폴리에데르, 폴 리에스데르, 혹은 폴리에데르와 폴리에스테르의 혼합물로서 우레 탄 메트릭스는 연소에 의해 추력을 낼 수 있도록 필요한 연소가 스 양을 공급해 줄 뿐만 아니라 추전제의 기계적 강도를 좋게 해 주는 역할을 한다. 특히 최근 들어 수산기를 말단기로 갖는 부타 디엔계 HTPB 바인더가 개발되었으며 그 기계적 특성, 내탄도 적 특성 및 제조공정상의 이점들로 인해 현재 가장 많이 적용되 는 중요한 바인더 시스템이다.
® 우레탄 반응 우레탄 반응은 거의 정량적으로 반응이 전행되며 적당한 촉매 에 의해 반응속도 조절이 용이하고 추전제로 적용시 기계적 성질 울 조철할 수 있는 적당한 히드록실 화합물(수산기를 포함한 폴리 머)의 합성이 가능하므로 추전제에 적용하기가 매우 좋은 반응이 다. 우레탄 반응은 간단해 보이는 반응이지만 그 반응 메커니즘 은 아직까지 완벽하게 해석되지 못하였다. Baker 등은 이 반응 이 알콕사이드 (alkox i de) 이온이 이소시아네아트기의 탄소원자에 공격함으로써 개시되는 것으로 제안하였다.
 R— (N-) ;-;-; (C+) ;-;-; (—0 ) + (H+ ) …(O -)R =R— N -C=O
R— (N-) ;-;-; (C+) ;-;-; (—0 ) + (H+ ) …(O -)R =R— N -C=O
우레 탄 추진제 의 바인더로서는 디올 (d i ol) 또는 트리울(tri ol) 이 사용되 며 , 그 중 흔히 사용되는 프리 폴리 머는 골격 (backbone) 구조에 따라 다음과 같이 폴리에스데르, 폴리에테르, 폴리부타디 엔계 등이 있다.
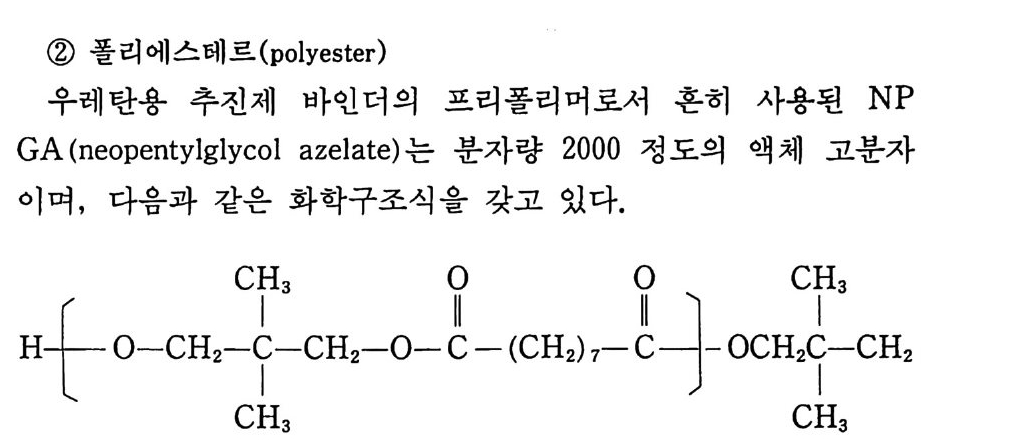 ® 폴리에스테르(p ol y es t er)
® 폴리에스테르(p ol y es t er)
NPGA 이의에도 몇 가지 폴리에스테르들이 우레탄 바인더 조 성으로 채택되었으나 폴리에스데르 자체의 점성도가 높은 관계로 고체산화제 충전을 많이 할 수 없음으로 인해 비추력이 낮아지는 결정적인 단점을 갖고 있다. 폴리에스데르의 높은 점성도는 폴리 에스데르의 분자량 분포도가 너무 넓게 분포된 때문으로 설명되 고 있다. 16,17,1 8 ) ® 폴리에테르(p ol y e th er) 폴리에데르 디올 중에서 가장 많이 사용되는 프리폴리머는 PPG (po ly (1, 2-oxyp ro p yle ne) dio l ) , B-2000 (po ly ( 1, 2-oxy bu ty le ne) dio l ) 로서 PPG, B-2000 이 라고 부르며 두 폴리 머 모두 secon- dary 히드록실기를 말단기로 포함하고 있다. Poly( 1,4-buty le ne) d i ol 은 pr im ary 히드록실기를 말단기로 갖기 때문에 PPG 나 B -2000 에 비해 더 반응성이 빠른 프리폴리머로 알려져 있다. 이와 같은 폴리에테르계 프리폴리머들은 추전제 이의의 일반용으로도 많이 사용되므로 저렴한 가격으로 대량공급이 가능하며, 이 폴리 머들의 낮은 점성도 및 경화제와의 적당한 경화속도 등은 추진제 충전에 좋은 이접들이다. 폴리에데르는 산소를 흡수하여 과산화
물을 생성하게 되어 온도가 상승하며, 분해에 의해 탄소 사슬 구 조가 파괴되는 문제점이 있으나, 방향족 아민 화합물 계통의 산 화 방지제를 소량 첨가하면 쉽게 해결될 수 있다. PPG, B-2000 은 유리 전 이 점 ( T8 : gla ss tra nsit ion tem p e ratu r e) 이 매 우 낮고, 이 프리폴리머들로 제조된 추진제를 노화시험한 결과 그 기계적 성질에 큰 문제접이 없어 여러 종류의 유도탄, 로켓 추진기관 모 터에 적용되었다 .6) ® 폴리부타디 엔(p ol y bu ta d i ene) 부타디엔을 주골격 (backbone) 으로 하고 말단에 히드록실기를 갖는 HTPB (hy d roxy ter mi na te d po lyb uta d ie n e) 는 1970 년 대 이 후 개발되어 최근까지 로켓 추진제로 가장 많이 적용되는 프리폴리 머 중의 하나이다. 그 중 R-45M 으로 불리는 HTPB 는 수평균 분자량이 2000 내지 3000 정도로서 낮은 점도를 갖고 있으므로 높은 고체 충전도가 가능하며 PPG 또는 CTPB 계 추전제보다도 비추력이 높고, 낮은 유리 전이점 (Tg ) 및 저온에서의 양호한 기 계적 성질과 저온 내탄도 성능이 우수하고, 노화 성능이 우수한 추전제로 제작되고 있다. 19,20) HTPB 는 말단에 히드록실기를 갖 고 있으므로 우레단 계열 추전제의 바인더용 프리폴리머로 분류 하였지만 그 주요 골격이 부타디엔이므로 다음에 설명하는 부타 디엔계 추전제용 프리폴리머로서 분류되어 비교 검토될 수도 있 다 .6) 이상과 같이 히드록실기를 분자 내에 갖는 프리폴리머를 경화 시키기 위해 사용되는 경화제 (cura ti ve) 로는 다음 표 3.4 와 같이 TDI, HDI, IPDI, N-100 등과 같은 디 이소시 아네 이트 등이 있 다. 이러한 디이소시아네이트들은 TDI 를 제의하고는 모두 지방 족 화합물에 속해 있다. 그 이유는 방향족 이소시아네이트 화합
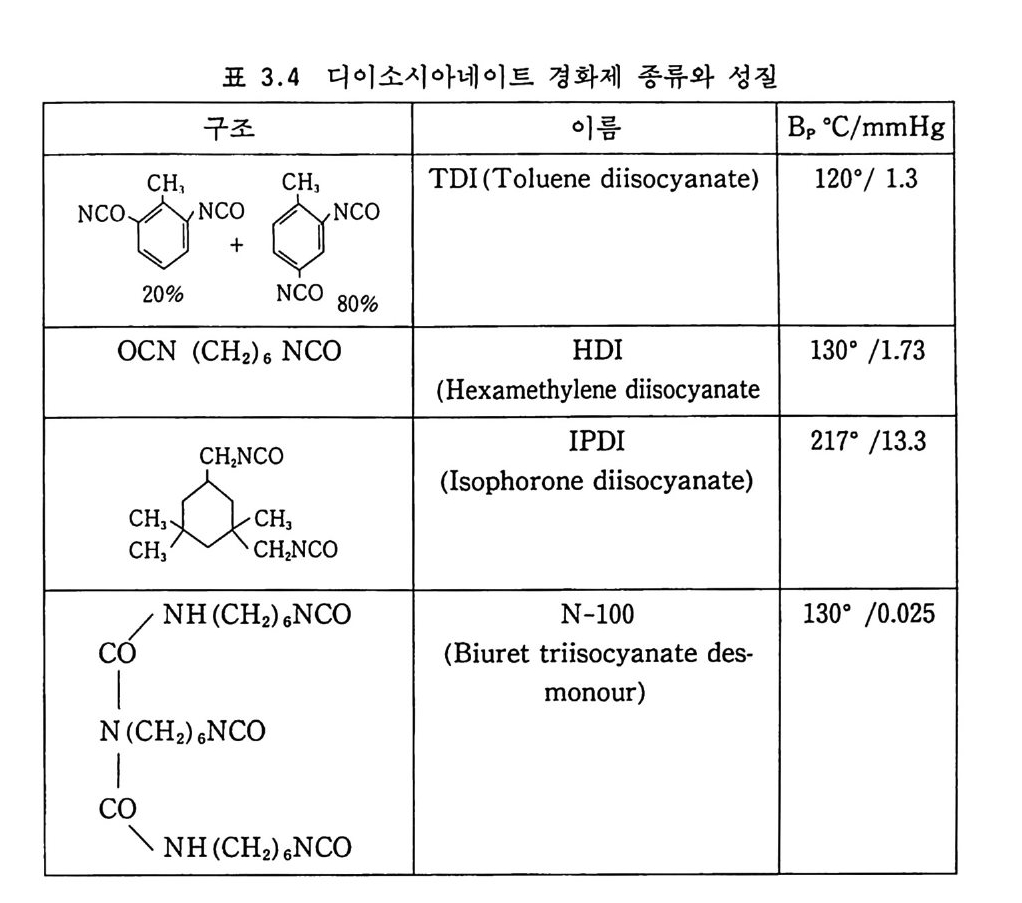 표 3.4 디이소시아네이트 경화제 종류와 성질
표 3.4 디이소시아네이트 경화제 종류와 성질
물들을 사용하면 경화 반응속도가 너무 빨라져서 추전제 제조시 추전제 페이스트의 주조가능 시간이 너무 짧아지는 문제접이 있 기 때문이다• 3.3.1.6 부타디엔계 추진제 주 골격을 부타디엔으로 하는 프리폴리머 중 가장 흔히 사용되 는 것으로는 PBAA, PBAN, CTPB, CTBN, HTPB 등이 있으 며, 그 중 HTPB 는 앞의 항에서 언급한 바와 같이 말단기를 히 드록실기로 갖는 우레탄계 추진제용 프리폴리머로 분류, 설명하 였다. 21)
(D PBAA (buta d ie n e 및 acryl ic ac i d 의 cop ol ym er) PBAA 는 부타디엔계 프리폴리머 중 로켓 추진제에 적용된 최 초의 프리폴리머로서 부타디엔과 아크릴산의 액체 공중합체이다. PBAA 는 유화 중합반응에 의해 합성되며 분자량 3000, 평균 작 용기수(fu nc ti ona lity) 2 정도의 프리폴리머이다. PBAA의 관능 기 들은 체 인 (chain ) 에 랜 덤 (random) 하게 분포되 어 있으며 분자당 관능기 수의 범위가 넓기 때문에 이러한 프리폴리머는 관능기가 없는 분자 (nonfu n cti on al) , 한 개 가 있는 분자 (monofu n cti on al) , 두 개가 있는 분자 (d ifu nc ti onal), 여러 개가 있는 분자(p ol yf un cti on al) 들의 혼합물이라고 볼 수 있다. 따라서 PBAA 로 제조된 바인더 및 추전제의 기계적 성질은 재현성이 좋지 않다. PBAA 프리폴리머는 점도가 낮기 때문에 그 이전의 프리폴리머들에 비 해서는 더 많은 양의 고체 충전을 할 수 있는 장점도 있었지만 추전제로 제조되었을 때 낮은 기계적 성질 및 저장 도중 후경화 현상 등으로 인해 최근에는 로켓 추전제에 거의 적용되지 않고 있다 .5,6) ® PBAN(buta d ie n e, acryl ic acid 및 acr y lon itril e 의 ter po l ym er) PBAA 계 추전제의 좋지 않은 기계적 성질과 저장 특성 동은 PBAN 과 같은 부타디엔, 아크릴로니트릴, 아크릴 산의 삼원공 중합체 (ter -po l ym e r) 를 적 용함으로써 개 선되 었다. PBAA 제 조시 사용한 방법과 같은 에멀션 중합방법으로 제조된 이 액체 삼원공 중합체인 PBAN 에 의해 제조된 추진제는 PBAA 에 비해 아크릴 로니트릴이 주입되어 재현성이 좋게 되었고 니트릴 고무에서처럼 표면에서의 산화 작용이 감소하여 표면이 딱딱하게 굳어지는 경 향도 줄어들게 되었다 .22) 다음 표 3.5 에서 보는 것처럼 PBAN 추진제는 표면에서 굳어지는 경향이 니트릴이 포함되지 않은 CTPB 추전제에 비해 훨씬 작은 것을 알 수 있다 •23)
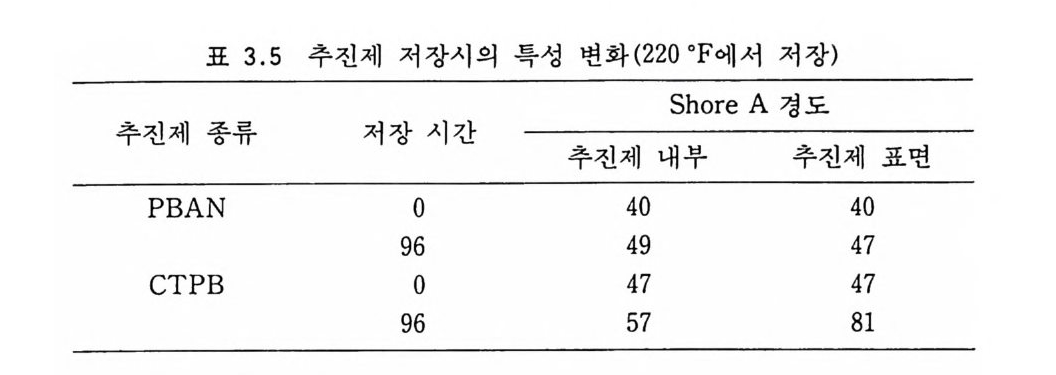 표 3.5 추진제 저장시의 특성 변화 (220 °F 에서 저장)
표 3.5 추진제 저장시의 특성 변화 (220 °F 에서 저장)
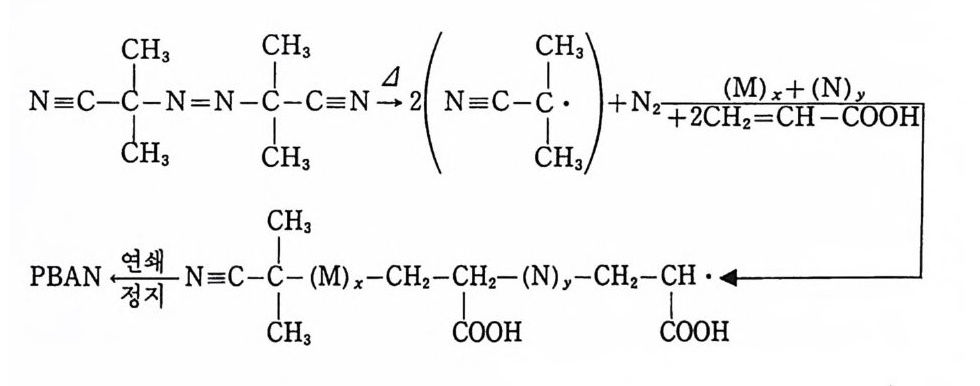 N =C-\;C3H3 N =N-\HC-H33C =N 12(N=cC\H)3+ N2 +2Ck쁜 않 _N ty0 0H
N =C-\;C3H3 N =N-\HC-H33C =N 12(N=cC\H)3+ N2 +2Ck쁜 않 _N ty0 0H
1957 년 PBAN 이 개발된 이후 많은 종류의 유도탄 • 로켓 추전 기관 모터에 PBAN 추진제가 충전되었으나 최근 들어서는 PBAN 보다 성능이 우수한 CTPB, CTBN, HTPB 등의 추전제 들이 개발됨으로써 그 사용량이 점차 줄어들고 있는 추세에 있 다. 그러나 PBAN 추진제는 그 원료가 저령한 점, 풍부한 제작 경험 및 추전기관 적용 경험과 신뢰성으로 인해 미국의 위성 발 사체 부스터 로켓을 위시한 여러 로켓에 아직까지도 사용되고 있 다. PBAA 와 PBAN 의 자유 라디칼 개시제에 의한 에멀션 중합반 응은 다음과 같은 화학식으로 표시될 수 있으며, Mx 는 부타디엔 의 x 몰수, N y는 아크릴로니트릴의 y 몰수를 의미하며, PBAA 의 경우 y =O 이다.
@ CTPB (carboxy l ter mi na te d po lyb uta d ie n e) 말단기에 카르복실기를 갖는 부타디엔 (CTPB) 프리폴리머는 라디칼 중합 또는 리튬을 개시제로 하는 음이온 중합법에 의해 합성되었으며, 평균 분자량 3000~5000 의 작용기수가 거의 2 정 도인 프리폴리머이다. PBAA 나 PBAN 보다 더 우수한 기계적 성질 및 더 높은 비율로 고체 충전이 가능한 장점으로 인해 HTPB 계 추진제가 개발되기 전까지는 높은 기계적 성질을 요구 하는 추전기관에 많이 적용되었다 .24,25) 라디칼 중합반응에 의한 CTPB 합성 반응은 다음과 같이 glu ta ric ac id 과산화물을 개 시 제 로 하는 방법 과 아조 화합물 (4, 4'-azobis - 4-cy a nop e nt an oic a ci d) 을 개시제로 하는 방법이 사용되고 있다 (Mx 는 부타디엔 분자 의 x 몰수, R 은 C3H6 를 의 미 한다) . 26,27)
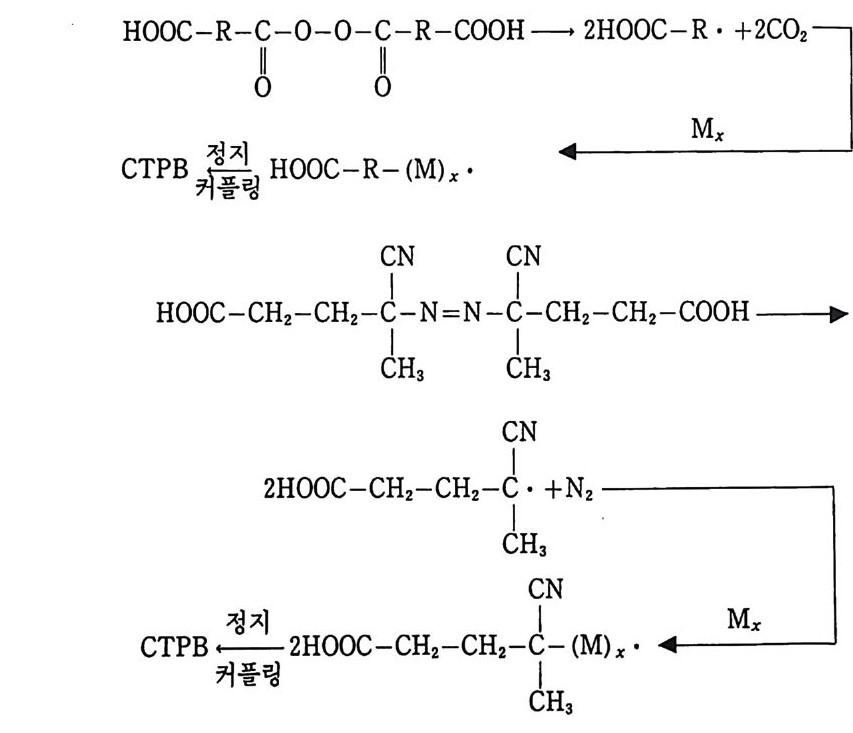 CHTOPOBC _晶R ― C3 H_OoO_Co-_RC\(-MR)_xC·O OH 一E 2HO 二OC-R • +二2C02
CHTOPOBC _晶R ― C3 H_OoO_Co-_RC\(-MR)_xC·O OH 一E 2HO 二OC-R • +二2C02
리튬울 이용한 음이온 중합반응에 의한 CTPB 합성방법은 다 음과 같이 진행되어 라디칼 중합에 의해 얻어전 CTPB 보다 훨씬 좁은 범위의 분자량 분포도를 갖고 있다 .28)
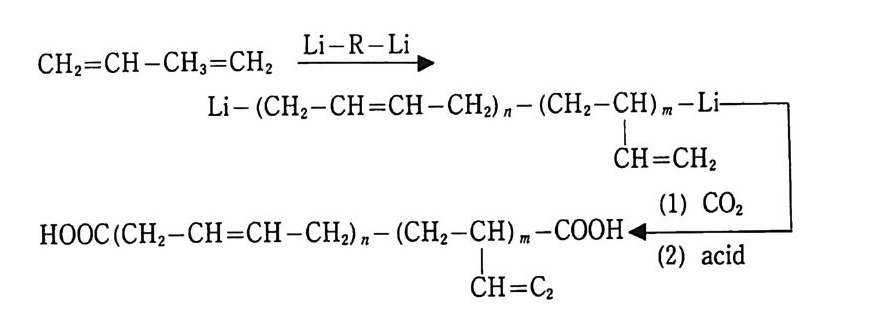 CH2=CH- C HJ = CH2 ~Li_ R— Li
CH2=CH- C HJ = CH2 ~Li_ R— Li
@ CTBN (carboxy l ter mi na te d po lyb uta d ie n e acryl o nit rile) CTPB 프리폴리머와 같이 말단기에 카르복실기를 갖도록 하고 PBAN 처럼 아크릴로니트릴을 주 골격에 넣어중으로써 CTPB 의 장점과 PBAN 의 장점을 살리기 위해 합성된 CTBN 프리폴리머 를 사용한 추전제는 HTPB 추진제와 거의 같은 시기에 미국 NOS 에서 개발 적용되었다. CTBN 프리폴리머는 점도가 HTPB 보다 높으므로 고체 충전율이 낮아서 비추력이 HTPB 보 다 약간 떨어지는 단점이 있는 반면 추전제 제조시 수분에 대한 영향이 적으므로 품질관리상의 장점, 우수한 기계적 성질과 접착 력, 우수한 열안정성이 있다. HTPB 계 추전제와 경쟁적인 입장 에 있는 추진제의 특성을 지녔으나 가격과 비추력이 떨어져서 범 용적으로는 적용되지 못하고 일부 로켓 추진기관 모터에 적용되 고 있다• 29,30) 이상과 같이 카르복실기를 포함하는 PBAA, PEAN, CTPB, CTBN 등과 같은 프리폴리머에 대한 경화제로는 아지리딘 (az i r i d i ne) 계 화합물과 에폭시계 화합물의 두 종류가 대표적으로 사용되고 있다. 다음 표 3.6 에는 아지리딘계 경화제가, 표 3.7 에
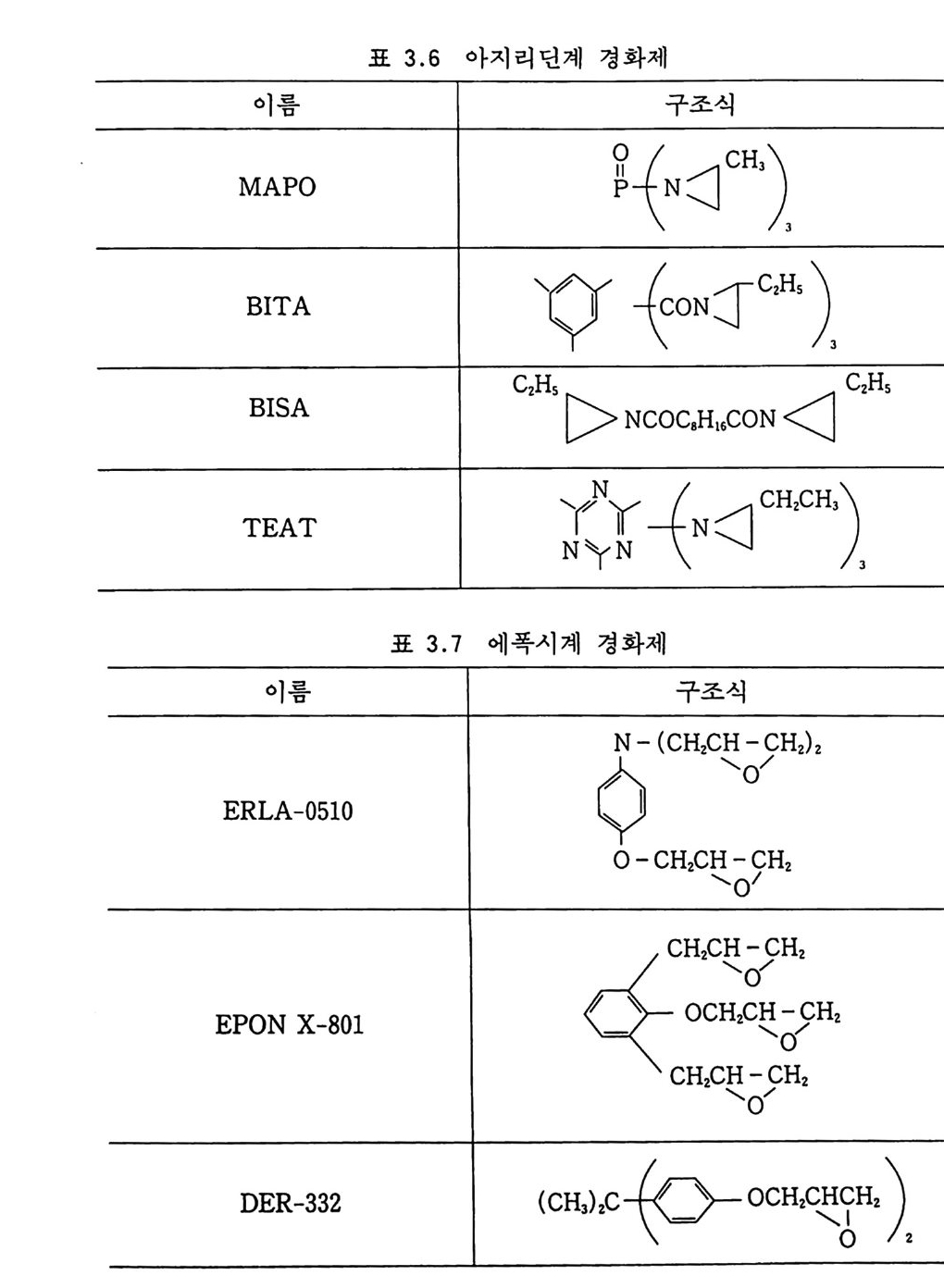 표 3.6 아지리딘계 경화제
표 3.6 아지리딘계 경화제
는 에폭시계 경화제가 나타나 있다. 카르복실산울 포함하는 프리폴리머 중 CTPB, CTBN 등은 이 작용기 성 (bif un cti on al) 성 질을 주로 갖고 있으므로 3 차원 망상구 조의 가교결합 구조를 만들어주기 위 해 서 다작용기 성 (multif un - cti on al) 아지리딘 또는 에폭시를 사용해야 한다. 그러나 다작용 기성 경화재를 사용하게 되면 산화제 존재하에 여러 종류의 부반 응을 일으키는 동시에 경화제 자체의 단중합반응도 일어나게 된 다. 그립 3.8 에서 보는 것처럼 아지리딘 화합물 BITA 를 경화제 로 사용했을 때는 주반응인 아미도에스테르 (am i does t er) 생성 반 응 이의에도 단중합 반응 및 옥사졸린으로의 자리옮김 반응도 일 어난다 .31,32)
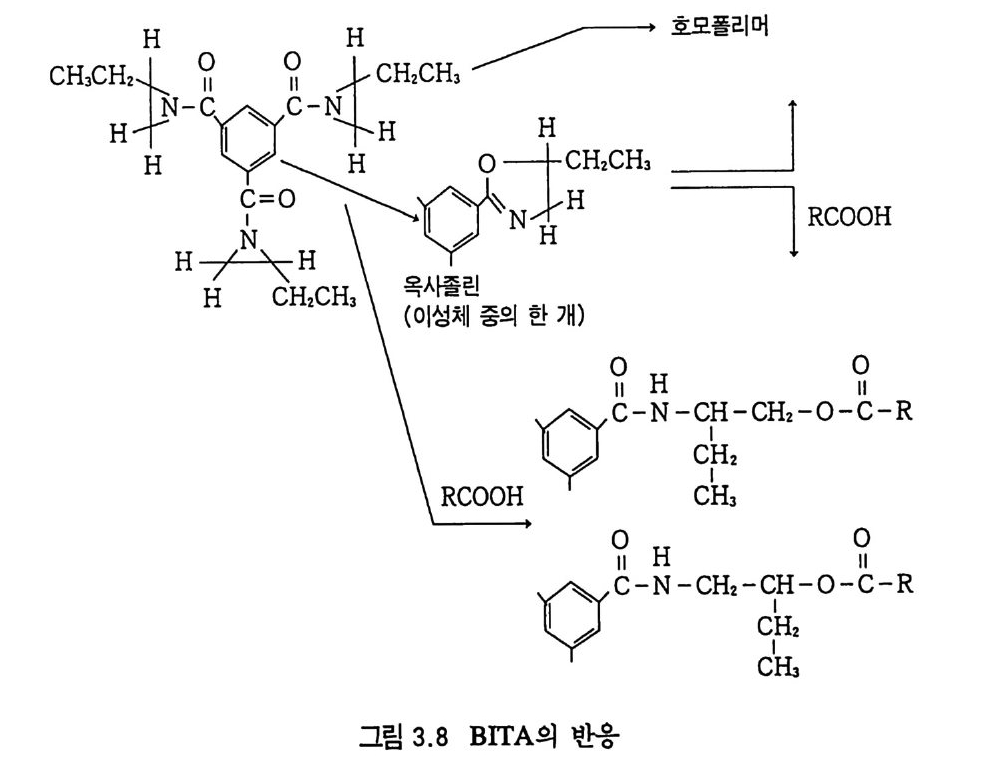 H l-l _.,-—~ 함 2 풀리머
H l-l _.,-—~ 함 2 풀리머
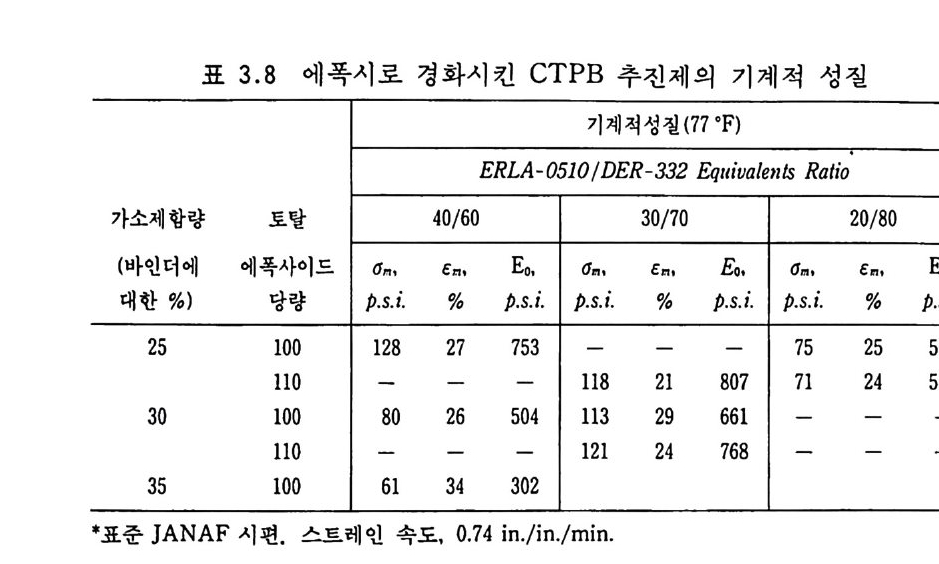 표 3.8 에폭시로 경화시킨 CTPB 추진제의 기계적 성질
표 3.8 에폭시로 경화시킨 CTPB 추진제의 기계적 성질
에폭시를 경화제로 사용하면 프리폴리머의 카르복실산기와 반 응하여 폴리에스데르를 형성하는 망상구조의 가교 결합반응이 일 어나지만 대부분의 에폭시 화합물들 역시 여러 종류의 부반응에 참여하게 된다. 부반응이 비교적 적게 일어나는 에폭시계 경화제 로는 표 3.7 에서 보여주는 ERLA-0510, EPON-X-801, DER -332 등이 주로 사용되고 있다. 또한 산화제 (과염소산암모늄) 존 재하에서는 그 부반응 종류 및 속도가 변화되므로 경화제 사용량 변화(당량 변화)에 따른 추진제의 기계적 성질을 시험하여 경화 제 사용 수준을 결정할 필요가 있다(예, 표 3.8). ® HTPB(hy d roxy l term i na te d po lyb uta d ie n e) 전항의 우레탄계 추전제(폴리부타디엔)에서 언급한 바와 같이 HTPB 프리폴리머는 부타디엔을 주 골격으로 하고 말단에 히드 록실기를 갖고 있으며 추전제에 적용했을 때 기계적 성질, 내탄 도적 성질 등 각종 성질이 우수하고 가격도 저령하여 최근 가장 많이 사용되고 있다.
3.3.1.7 고에너지계 추진제 전항까지 언급한 HTPB, CTPB, CTBN, PEAN 등 프리폴리 머 이의에 최근에는 추진제의 에너지(비추력)를 더 높이기 위한 고에너지 추진제 개발 연구가 활발히 진행되고 있으며 동시에 추 전제 연소시 배기가스를 무연화시키려는 연구도 하고 있다. 이러 한 고에 너 지 추진제 의 바인더 로는 다음과 같은 GAP (po lyg ly c idy l azid e ), Poly BAMO/NMMO 등이 시도되고 있다 (BAMO : bis -azid o meth y lo xeta n e, NMMO : nit ra to m eth y l- meth y lo xeta n e) . 3·6•33>
 \CH,- 三广
\CH,- 三广
3. 3. 2 산화제 (oxid i z i n g age nt) 혼합형 추진제 조성 중 가장 많은 함유량을 차지하는 산화제는 바인더와 혼합시 바인더와 상용성 및 안정성이 있어야 하고, 그 가격이 저령하여야 하며, 품질이 균일하고, 원료로서 공급이 안 정되어 있어야 한다. 그 의에 다음과 같은 조건을 만족해야 한 다. 첫째 연소할 때 바인더 및 금속연료와 반응하여 가능한 한 많 은 숫자의 가스분자를 배출할 수 있어야 하며, 둘째 가능한 한 밀도가 높아야 한다. 셋째 생성열이 가능한 한 낮아야 한다.
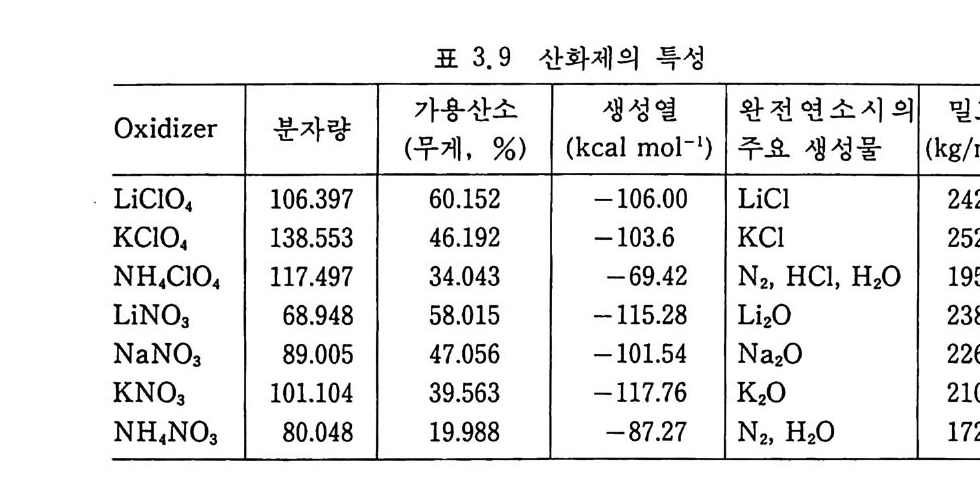 표 3.9 산화제의 특성
표 3.9 산화제의 특성
표 3.9 에는 혼합형 추진제에 사용될 수 있는 여러 종류의 산화 제 특칭이 나타나 있다. 그 중에서 과염소산암모늄 (ammon i um pe rchlorate ) 이 가장 혼하게 사용되고 있으며 , 가스발생 기 나 저 연 소속도 추전제용 산화제로는 질산암모늄이 가끔 사용되고 있다. 그러나 질산암모늄은 추전제 보관 또는 작동 온도 범위에서 상변 화(p hase chan g e) 를 일으키는 문제점이 있어 최근에는 상변화롤 억 제 한 PSAN (ph ase sta b il ize d ammoniu m nit ra te ) 을 대 신 이 용하 고 있다. 34,35,36) 산화제로서는 상기 와 갇은 무기 영 (ino rga nic salt) 을 사용하는 대신에 최근에는 니트라민 (n it ram i ne) 계 물질을 사용하기도 한 다 .2,3) 즉, 그립 3.9 와 갇은 RDX 혹은 HMX 와 같은 물질을 사 용한 니트라민계 추전제는 연소될 때 할로겐 물질과 같은 유연의 유독성 배기가스가 적은 무연 혹은 저연 추전제로 적용될 수 있 으며, 특히 고에너지 바인더와 함께 사용될 때는 비추력 (Is p) 성 능을 높일 수 있는 장점이 있으므로 점화시의 안정성, 연소속도 성능 등 몇 가지 문제점들을 해결하면 매우 우수한 추전제로 사 용될 수 있다. 37,38) 혼합형 추전제 조성 중 고체 산화제가 차지하는 비율은 보통
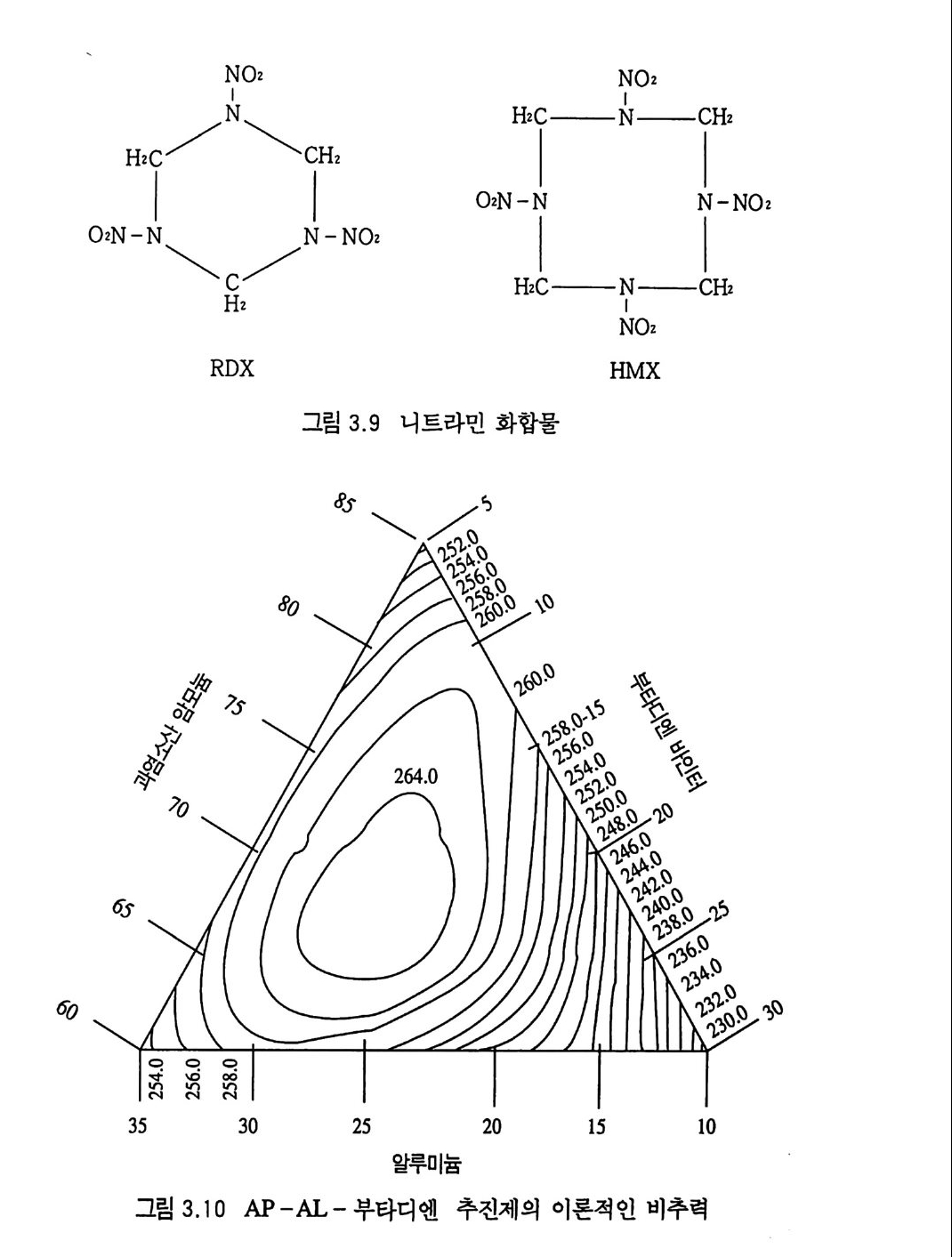 OzN -NI \ 요N願/ NI - N02 OzNHH-NIIzz CC--NN뿐\。-- 2CC HHNIIz-z N Oz
OzN -NI \ 요N願/ NI - N02 OzNHH-NIIzz CC--NN뿐\。-- 2CC HHNIIz-z N Oz
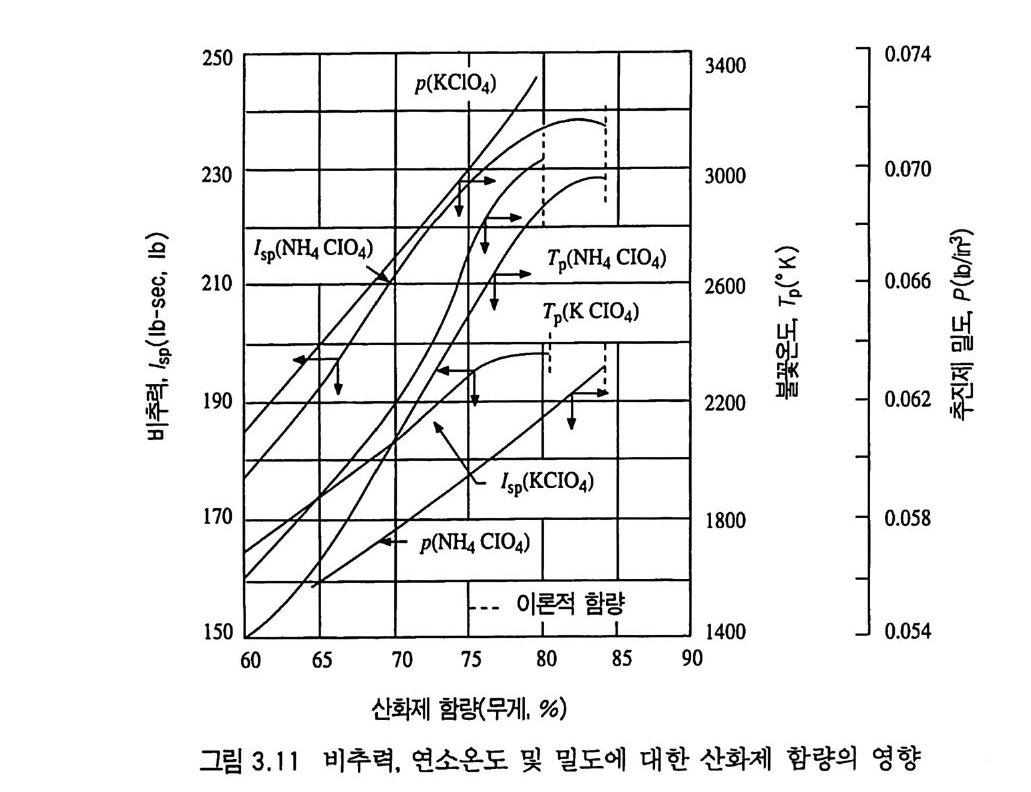 250 3400 0.074
250 3400 0.074
70~90 % 정도로서 추진제의 여러 가지 특성에 직접적인 영향을 주게 된다. 사용되는 산화제의 종류, 양, 산화제 분말의 입자 모 양 및 크기, 입도 분포도 등은 추진제의 제조 공정성 (점성도 및 유변성), 비추력과 같은 연소 특성, 물리적 및 기계적 성질에 큰 영향을 주게 되는데 그 중 연소 특성 및 물리적 기계적 성질에 관련된 사항은 추후 해당되는 장에서 좀더 자세히 언급하기로 하 고 여기에서는 그 결과만 약술하였다. ® 산화제 함량에 따른 비추력 변화는 과염소산암모늄을 산화 제로 사용한 경우 바인더 및 알루미늄과의 상호 관련된 효과가 그립 3.10 과 같이 나타났으며, 산화제 종류 및 양의 변화에 따른 비추력 변화(예)는 그립 3.11 과 같이 나타났다 .39) 일반적으로 산
화제 함량이 증가할수록 비추력은 증가하여 폴리부타디엔계 추전 제의 경우 산화제가 80~90 %(wt .%) 부근에서 최대의 비추력을 나타내게 된다. 그러나 산화제 이의에도 그립 3.10 에서 보이는 바와 같이 알루미늄 함량과 바인더의 종류도 비추력에 큰 영향을 주고있다. ® 과염소산암모늄을 산화제로 사용한 추전제의 경우 산화제 함량이 많아질수록, 특히 작은 입자의 산화제 함량이 많아질수록 추진제의 연소속도는 빨라진다. 그림 3.12 에는 산화제로서 과염 소산암모늄과 과염소산칼리를 사용한 경우 산화제 함량 및 입자 크기에 따른 연소속도 (1000 p s i, 70°F) 변화를 보여주고 있다 .39) ® 고체 충전도(산화제 및 알루미늄 분말 함량)가 많아질수록,
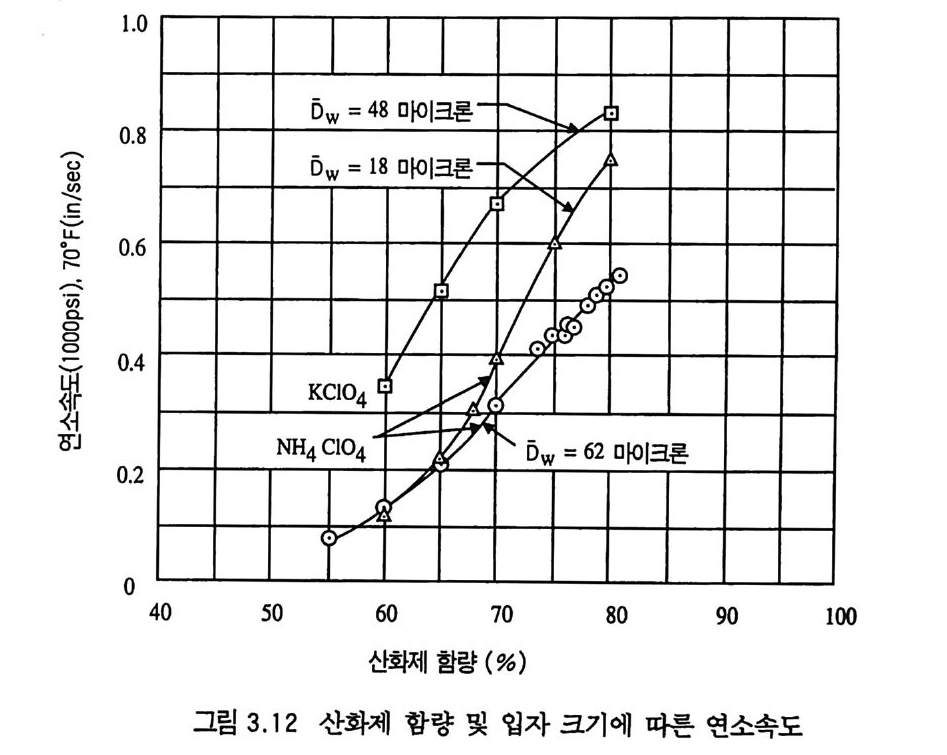 1.0
1.0
또한 입자 크기가 작은 고체 함량이 많아질수록 추진제 반죽 (p as t e) 의 점도는 높아지게 된다• 이때 고체 충전도 및 입자 크 기에 관련하여 고체 분말의 충전(쌓임, pa ckin g ) 상태 또한 매우 중요한 영향을 주게 된다. 죽, 충전 고체 분말들이 단일입도모드 (monomodal) 인 경 우보다는 이 성 분입 도모드 (bim odal) 또는 삼성 분입 도모드 (tri m odal) 가 되 면 충전분율 (pa ckin g frac ti on ) 이 높아 져서 그 점도가 낮아지는 효과가 나타난다. 그림 3 . 13 은 고체 충 전 함량과 b i modal 인 경우 점도 변화 곡선이며, 그립 3.14 는 고 체 충전도 65 % (vol.% )의 경우 고체 충전 분말의 삼성분입도모 드 혼합 비율에 따른 점도 변화 곡선이다 .40,41,42)
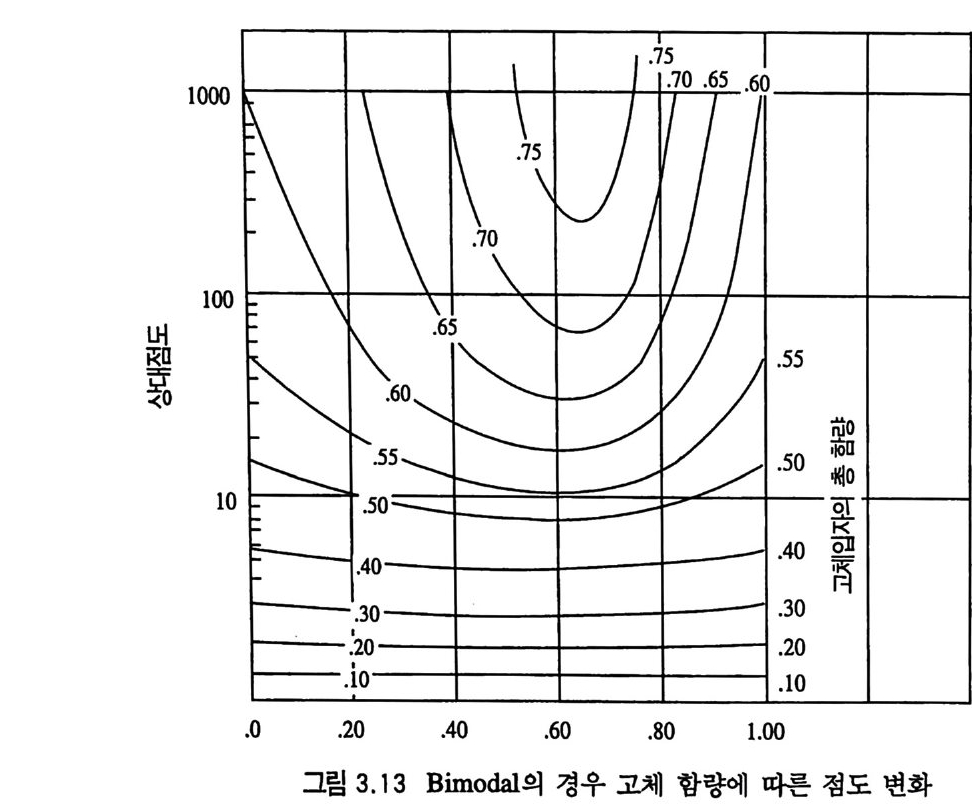 1000
1000
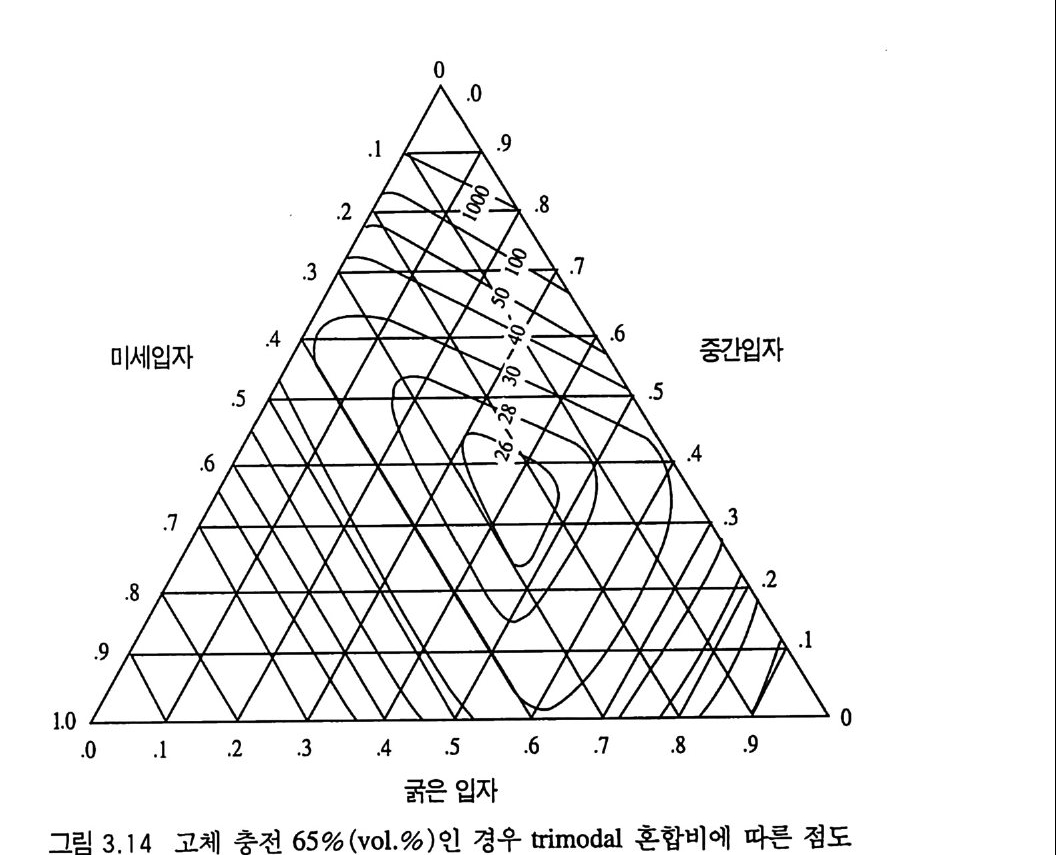 미세입자 증간입자
미세입자 증간입자
@ 고체 충전도 및 그 입자 분포도에 따라 추전제의 기계적 성 질은 크게 영향을 받게 된다. 일반적으로는 고체 충전 함량이 증 가할수록 추진제의 인장강도 및 영률(y oun g 's modulus) 이 증가하 며, 작은 입자의 고체 분말 함량이 증가할수록 추진제의 인장강 도가증가한다. 그림 3.15 에는 NaCl 입자 크기에 따른 PPG계 추전제의 응력 -신장률 (s t ress-s t ra i n) 변화가 나타나 있다. 죽, 42 % 충전도에서 서로 다른 크기의 입자들을 충전제로 사용하여 비교한 결과 충전 입자 크기가 작아질수록 웅력은 크게 증가하고 신장률 및 커브의 모양도 변하게 된다. 5,6)
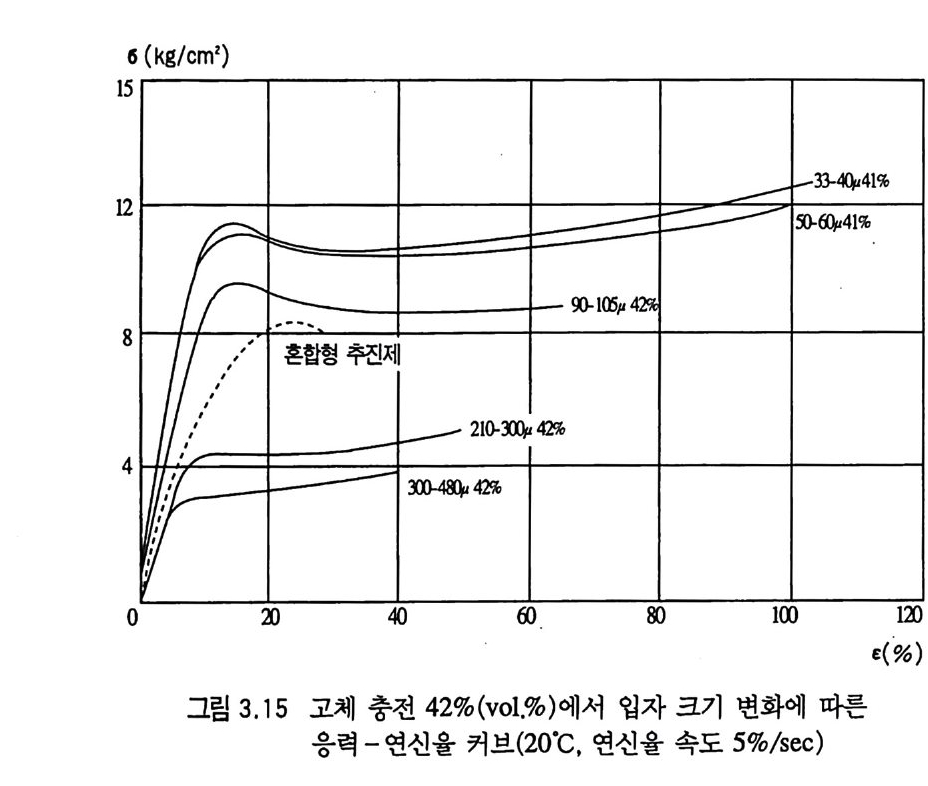 6 (kg/c m')
6 (kg/c m')
3. 3. 3 금속연료(금속환원제) 금속연료는 추전제 연소시 비추력을 상승시키는 동시에 연소온 도를 울리는 효과가 있다. 또한 금속연료의 밀도가 바인더나 산 화제에 비해 훨씬 높으므로 금속연료 함량이 증가하는 만큼 추전 제 밀도를 증가시키게 된다. 금속연료로 사용될 수 있는 물질로 는 리튬 (L i), 베릴륨 (Be), 알루미늄 (Al), 마그네슘 (M g), 지르코 늄 (Zr) 등이 있는데 산화제와 접촉했을 때의 안정성 및 가격 등 의 면에서 알루미늄이 가장 많이 사용되고 있다. 그립 3.16 은 알루미늄 또는 마그네슘울 바인더 (25 %)와 산화 제 (75 %)의 조성에 5, 10, 15 %(wt.%)로 가했을 때의 비추력 변
화곡선이 다. 43) 그립 3 .10 의 바인더 (PBD) , 산화제 , 알루미 늄의 조성과 비추력 관계로부터 비추력이 최대치를 얻는 영역은 알루 미늄 함량이 대략 20 % 정도 부근일 때라고 볼 수 있다. 그러나 예를 들어 바인더 함량(예, 10 %)을 일정하게 유지한다고 가정했 울 경우 산화제와 알루미늄 함량은 알루미늄이 20~24 %, 산화
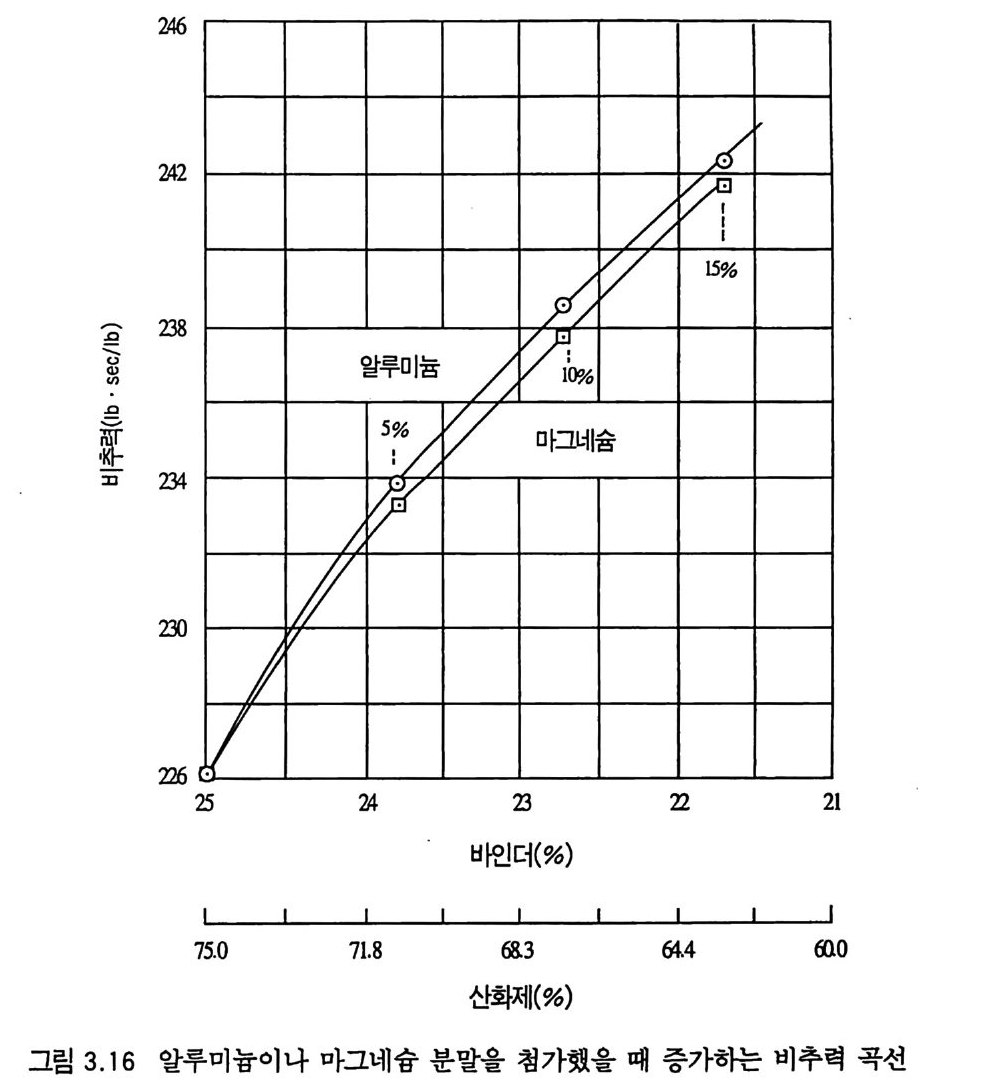 246
246
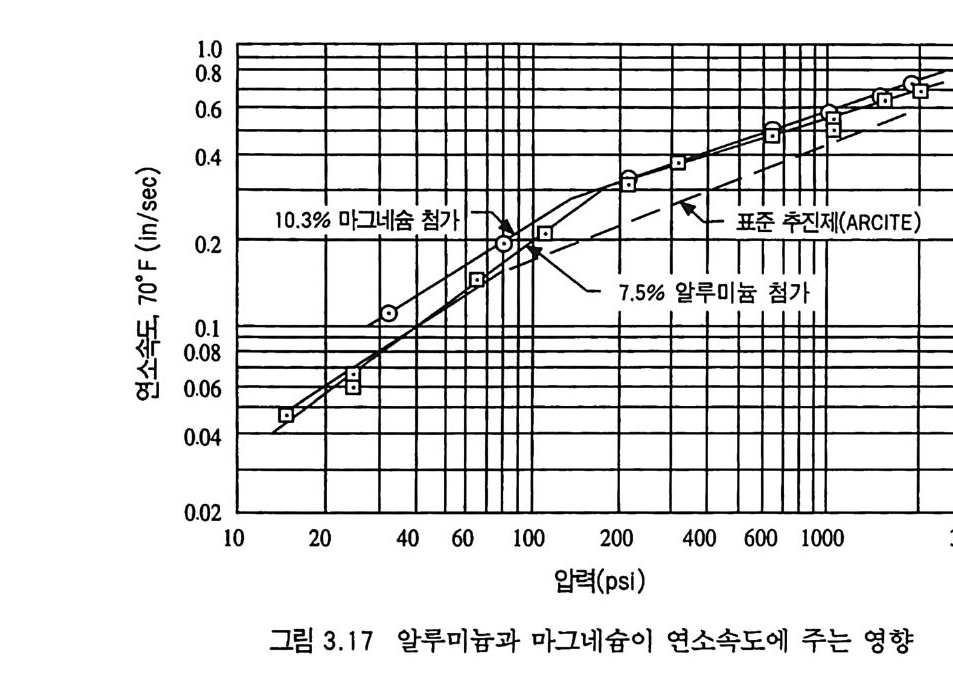 1.0
1.0
제 (과영소산암모늄) 66~70 % 범위에서 최대의 비추력을 얻을 수 있는 것을 알 수 있다. 이때 산화제 밀도(1. 95 g /cm3) 보다는 알루 미늄 밀도 (2.70 g /cm3) 가 더 높으므로 추진기관 모터 내에서 부피 당 얻을 수 있는 추력을 계산하면 밀도가 더 증가될 수 있는 알 루미늄 함량을 높이는 것이 유리하게 된다 . 그러나 알루미늄의 입자 모양과 크기에 따라 추전제의 공정성, 죽 페이스트 (uncured) 의 점도가 변화되므로 공정성을 함께 검토하여 조성을 결정하여야 한다. 추전제에 입자가 작은 알루미늄이나 마그네슘 분말을 가해 주 면 추전제의 연소속도는 약간 증가하고 압력지수는 약간 감소하 게 된다. 그립 3 . 17 에는 325 메쉬 이하의 알루미늄과 마그네슘 분말을 Ar cit e 사 추전제 (PVC 계 추전제 ) 에 가했을 때 의 압력 /연 소속도 곡선이 나타나 있으며 그립 3.18 의 커브는 마그네슘 분말
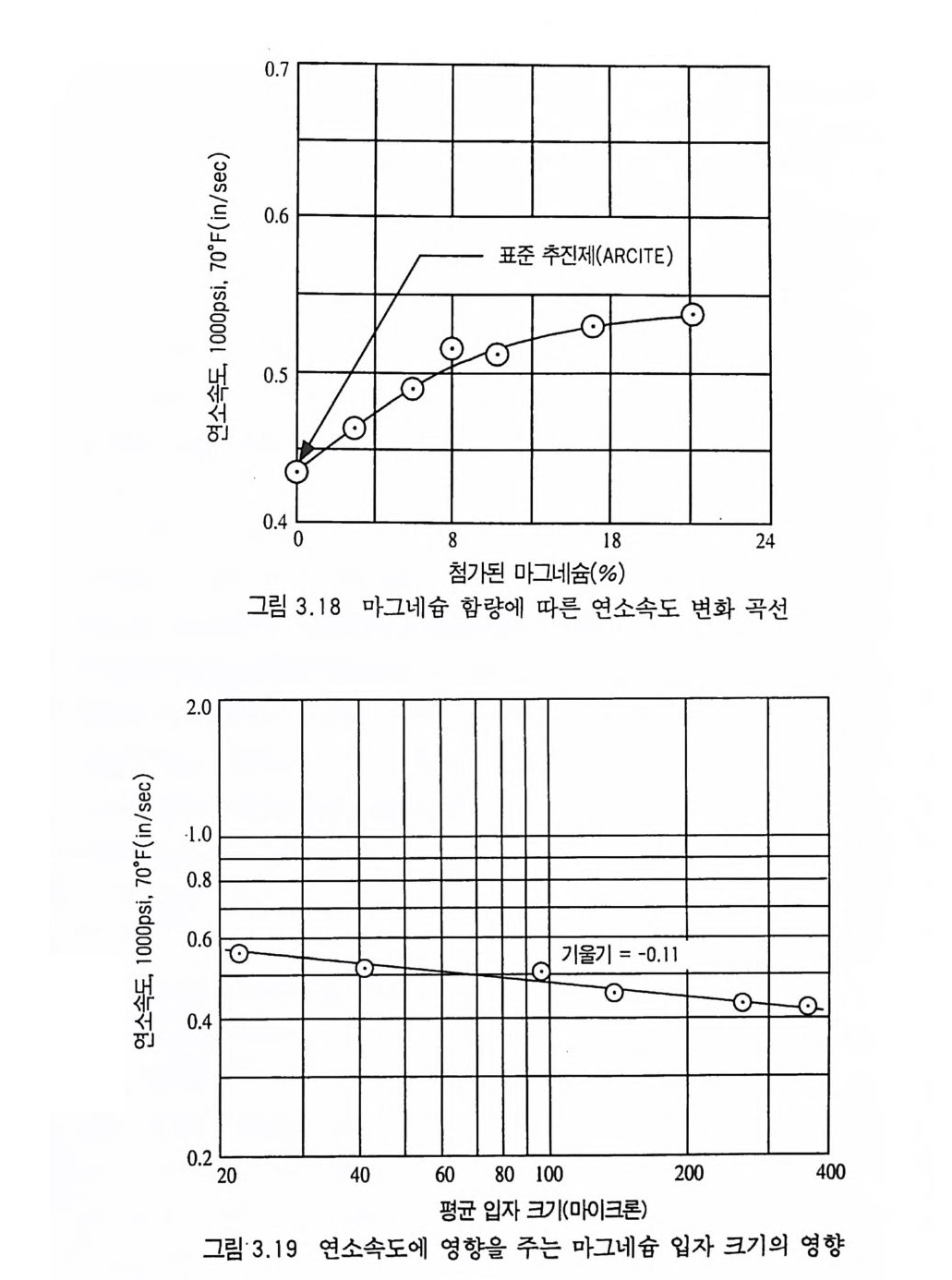 0.7
0.7
함량에 따른 연소속도 영향이다. 산화제 입자 크기에 따른 연소 속도 변화와 마찬가지로 금속분말의 입자 크기가 작아지면 추진 제 연소속도는 약간 감소하는 경향이 있다 .39) 3. 3. 4 첨가제 혼합형 추전제의 기본 원료는 메트릭스 구조를 형성하는 바인 더, 산화제 및 금속연료의 세 종류이나 이 밖에도 추진제의 물리 적 기계적 성질, 연소 특성 혹은 공정성을 개선하기 위해 첨가해 주는 각종 첨가제들이 사용되고 있다. 3.3·4.1 가소제 고분자 물질인 바인더에 가소제를 첨가하면 바인더와 산화제 및 금속연료 분말을 혼합시킬 때 그 혼합물 페이스트의 점도를 떨어뜨려 공정성을 좋게 하며 주조가능 시간을 증가시킬 수 있 다. 가소제를 첨가하면 추전제가 경화된 후 기계적 성질이 개선 되므로 영률을 감소시 키고, 신율 (elong a ti on ) 을 증가시 키 며 , 유리 전이점 (T g)을 떨어뜨리는 개선 효과도 있으며 특히 저온에서의
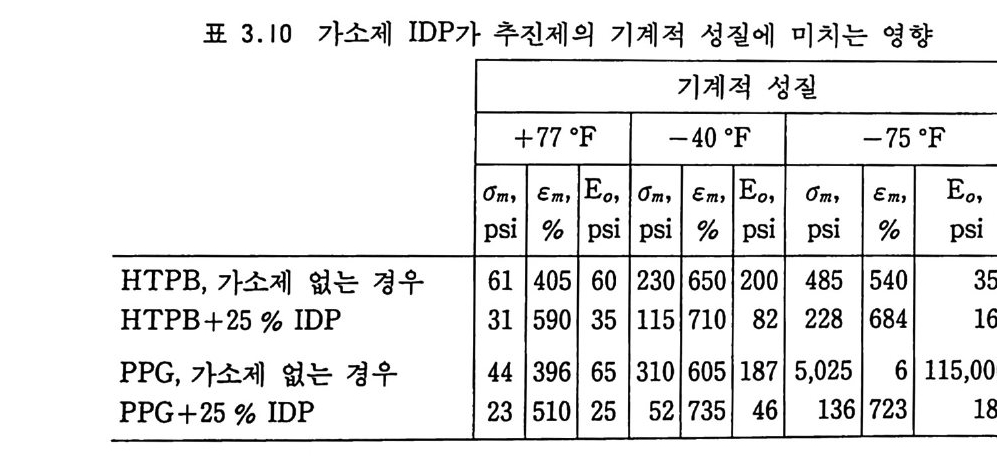 표 3.10 가소제 IDP 가 추전제의 기계적 성질에 미치는 영향
표 3.10 가소제 IDP 가 추전제의 기계적 성질에 미치는 영향
기계적 성질이 크게 향상된다고 알려져 있다. 가소제는 보통 바 인더의 30 %(wt .%) 이상을 초과하지 않는 범위에서 사용되는 경 우가 대부분이다. 표 3.10 에는 HTPB 추전제와 PPG 추전제에 서 가소제를 첨가한 경우와 첨가하지 않은 경우, 기계적 성질에 미치는 영향을 비교하였다 .5 , 6) 3. 3. 4. 2 경 화촉매 (cure cata l ys t ) 추전제 혼합기 (m i xer) 에서 추진제의 모든 원료들이 충분히 혼 합된 후 경화반응이 진행됨과 동시에 추전제 페이스트의 점성도 가 증가하기 시작한다. 계속 이어지는 주조공정에서는 주조에 필 요한 충분히 낮은 점도 및 충분한 주조가능 시간이 필요하며, 주 조공정이 끝나면 경화공정으로 이어지게 된다. 경화공정은 낮은 온도에서 가능한 한 짧은 시간 내에 경화반응을 완료시키는 것이 경제적으로 유리하다. 따라서 경화반웅 속도를 빨리 진행시키기 위해 경화촉매를 프리폴리머와 경화제의 화학반웅 형식에 맞추어 첨가시키고 있다. 그러나 경화반응 속도가 너무 빨라지면 주조가 능 시간이 짧아져서 주조공정을 진행시킬 수 없게 되므로 충분한 주조가능 시간(대형 추진기관 그레인의 경우 대략 4~8 시간)을 유지 한 후 경화반응 속도가 빠르게 진행되는 반응 형식이 유리하다. 3. 3. 4. 3 연소속도 촉매 추진제 연소속도에 영 향을 주는 요인들은 다음과 같다. ® 추전제의 조성 중 바인더, 산화제, 금속연료의 함량 ® 산화제 분말의 입자 모양, 크기 및 분포도 ® 금속연료(알루미늄) 분말의 입자 모양, 크기 및 분포도 추전기관 모터에 사용될 추전제의 각종 성질, 죽 물리적 기계적 성질, 비추력, 제조공정성에 적합한 추진제의 조성 (for mulati on )
작업이 이론적 실험적으로 결정된 후 얻어진 추전제의 연소속도 가 모터에 적용하기에 적합하지 않은 연소속도인 경우 다시 연소 속도 증감을 위해 추전제 조성 조절 작업을 하게 되는데, 이때 다른 조성 및 특성을 거의 그대로 유지하면서 연소속도만을 변화 시킬 수 있는 방법이 연소속도 촉매를 사용하는 것이다. 연소속 도 촉매는 바인더 종류 및 산화제 종류에 따라 그 효과가 매우 다르지만, 과염소산암모늄을 주산화제로 사용하는 추전제에서는 일반적 으로 산화철 (fer ric oxid e ) , 산화크롬 (chromi c oxid e ) , 크롬
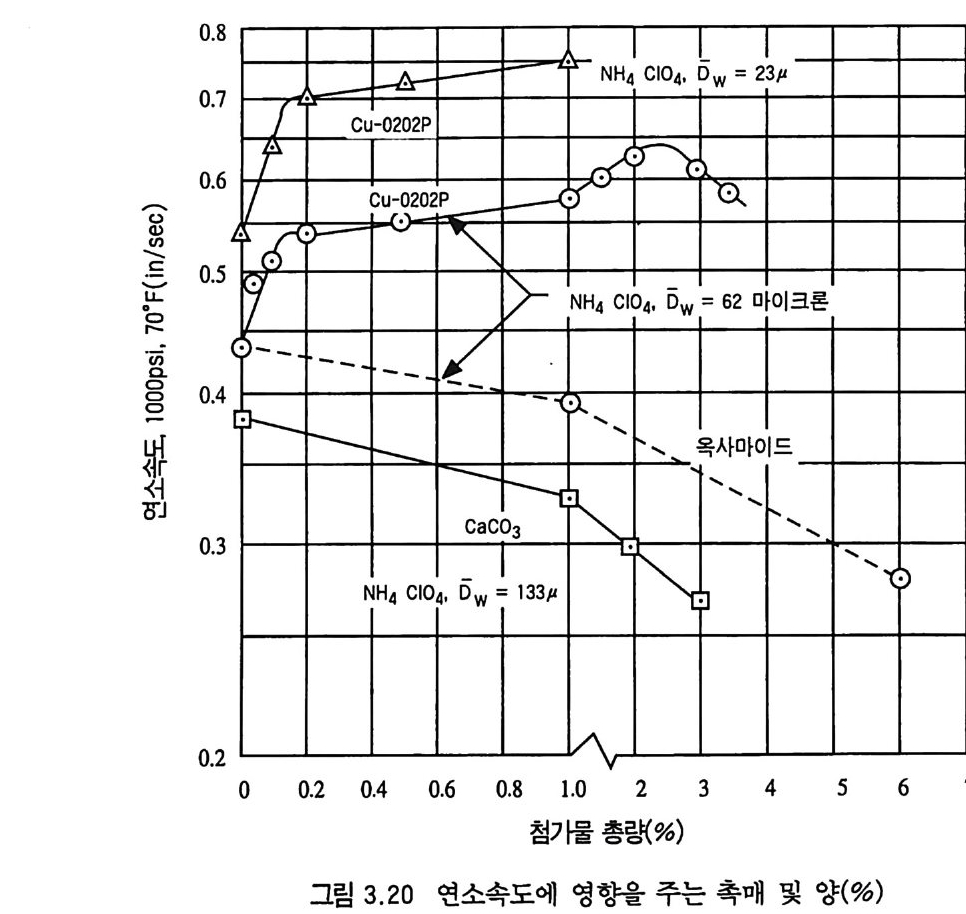 0.8
0.8
산구리 염 (cop pe r chromi te) , 산화마그네 슘 (mag n esiu m oxid e ) 등 의 금속 산화물을 첨가하면 연소속도가 증가하며, 옥사마이드 (oxami de ) 및 탄산칼슘 (calciu m carbonat e) 은 연소속도를 감소시 키는 작용을 한다. 그립 3.20 에는 연소속도 촉매 양에 따른 연소 속도의 증감에 대한 영향이 나타나 있다 .39) 그 중 크롬산구리영 이 연소속도 증가 효과가 가장 우수하고 압력지수도 감소시키는 우수한 촉매인 것으로 나타나 있으나 부타디엔계 추전제와 같은 어떤 종류의 추전제에 대해서는 노화 작용을 가속화시키는 결접 도 있는 것으로 알려져 있다. 3. 3. 4. 4 결 합제 (bondin g ag e nt) 추전제 속에 있는 바인더와 고체 충전제 (분말) 사이에 결합력 이 있느냐 없느냐 하는 것은 추진제의 기계적 성질에 큰 영향을 주게 된다. 만일 이 결합력이 강해지면 추전제의 인장강도는 크 게 증가하게 된다. 그립 3.21 에 나타나 있는 고무 탄성체의 경우 바인더로서 작용하는 고무에 유리 알맹이와 에폭시계 고체 입자 를 충전시킨 스트레스-스트레인 커브를 살펴보면, 고무 바인더와 결합력이 없는 유리 알맹이 충전 시스템에 비해 결합력이 있는 에폭시계 입자 충전 물질의 응력 곡선이 훨씬 높게 나타나 있다. 죽, 유리 알맹이에 비해 에폭시 물질이 충전제로서의 강화 (rein f o r cin g ) 효과가 뚜렷하다고 볼 수 있다. 44,45) 이와 같이 충전물질의 강화 효과를 크게 하기 위해, 죽 바인더 와 충전 고체 입자와의 결합력을 증진시키기 위해 결합제를 첨가 해 준다. 추전제에 결합제를 사용하여 바인더와 충전 고체 입자 와의 결합력을 강화시킨 예가 그림 3.22 에 있다• 이 예에서 두 추진제 모두 팽창 (d il a t a ti on) 하지만 같은 부피 팽창에 소요되는 응력이 결합제를 사용한 추전제에서 훨씬 높게 나타나며, 결합제
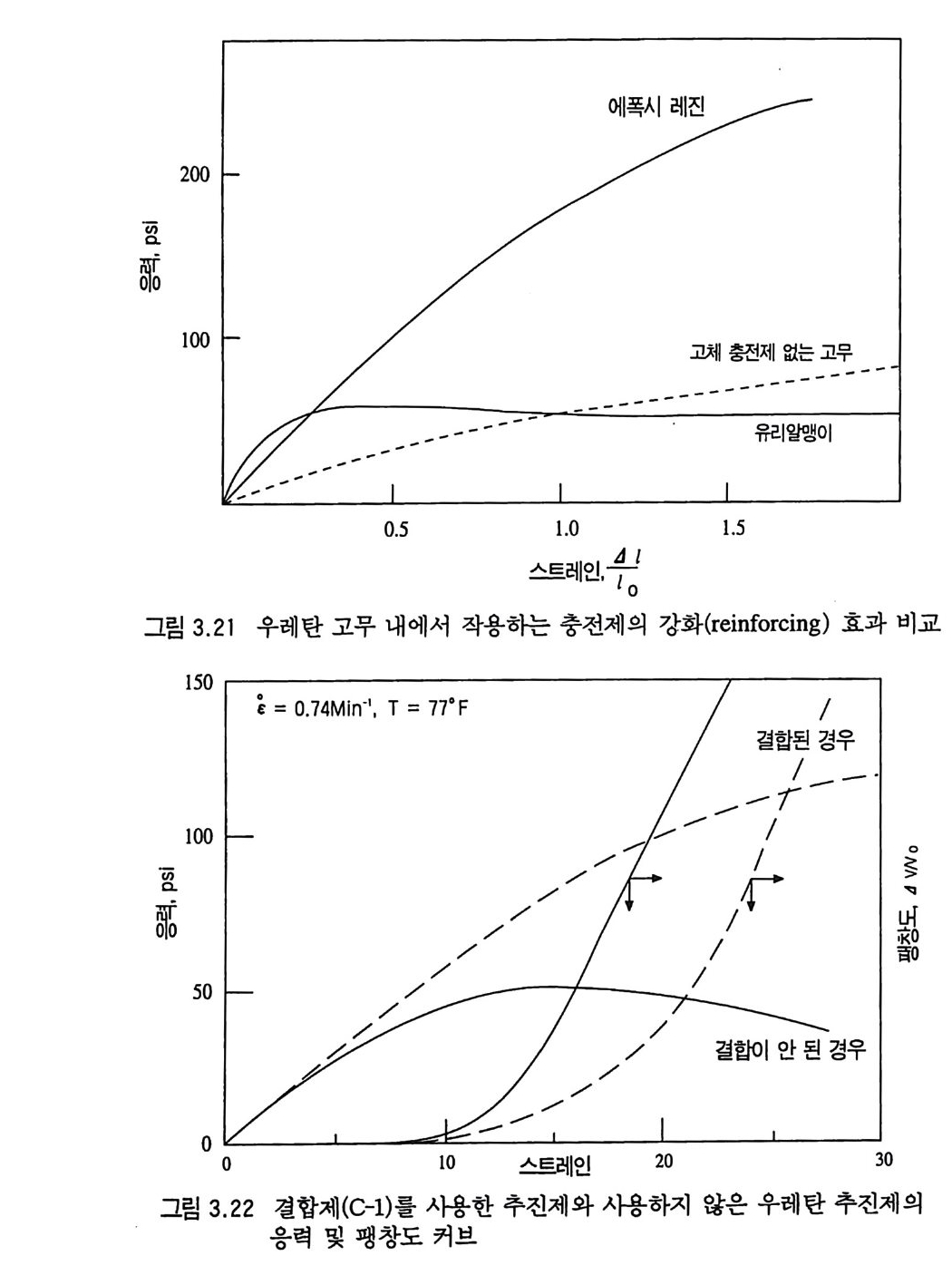 200
200
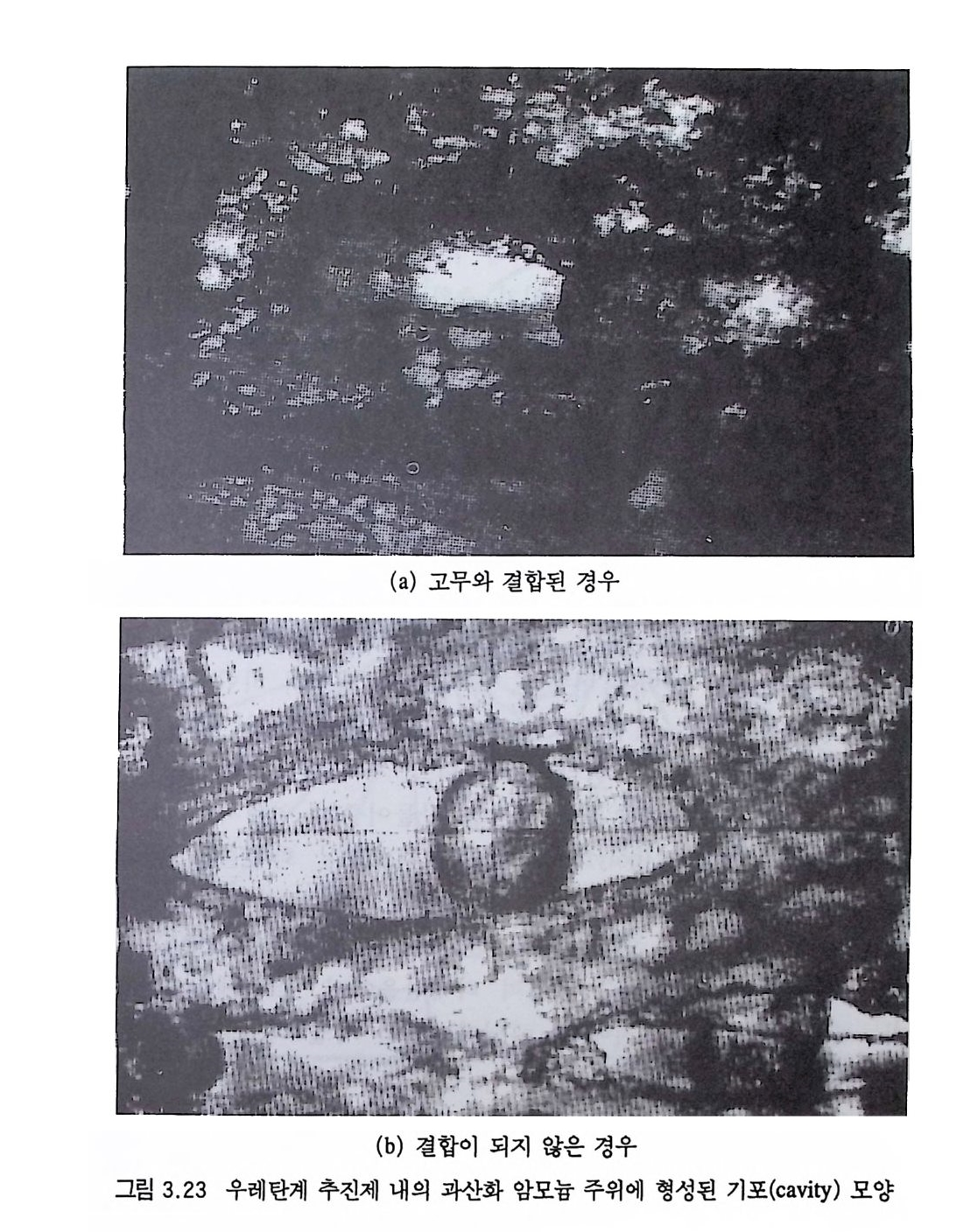 (a) 고무와 결합된 경우
(a) 고무와 결합된 경우
를 사용한 추전제는 부피가 크게 팽창하는 스트레인 20~30 범위 에서도 응력 증가가 계속되고 있다 .46 , 47,48) 그림 3.23 은 이러한 두 종류의 추전제에 응력을 계속 가하여 생긴 기포 (cav ity)의 모양으로서 위쪽 그립의 결합이 잘된 산화 제(과염소산암모늄) 입자는 바인더와 섞여 결합되어 있으나, 아래 쪽의 결합력이 없는 경우에는 입자 주위에 기포가 뚜렷하게 구별 되어 강화효과가 전연 없는 것으로 나타나 있다. 부피가 팽창할 때 충전 입자에 작용하는 응력은 입자 모양에 따라 다르며, 바늘 모양의 입자가 가장 낮은 응력이 작용한다. 그림 3 . 24 에는 구형 입자에 작용하는 응력을 기하학적으로 계산한 결과이며 최대의 응력은 2.3 배로서 입자 표면에서 바인더 쪽으로 약간 떨어전 점 에 응력이 집중되는 것으로 계산되었다• 이상과 같이 결합제는 추전제의 물리적, 기계적 성질에 중요한 영향을 주게 된다. 결합제를 추진제에 사용하면 첫째로, 충전 입 자 주위에 튼튼한 피막을 만들며, 둘째로 이 껍질과 바인더 메트 릭스 사이에 강력한 결합력을 만들어주는 역할을 하게 된다. 이 때 입자 주위에 형성된 피막은 메트릭스 자체보다도 더 질기고 강해야 한다. 이렇게 튼튼한 피막을 만들어주기 위해서는 보통 저 분자량의 다관능기 (po lyf un cti on al) 인 히 드록실 화합물을 폴리 이소시아네이트로 반응시켜 주면 바인더 메트릭스보다 더 강력한 코팅 물질을 얻을 수 있다. 이때 결합제는 반드시 다관능기여야 만 결합제로서 작용할 수 있으며 디에탄올아민, 트리에탄올 아 민, 데트라키스(히드록시에틸) 에틸렌디아민 계열 화합물들이 혼 히 결합제로 사용되고 있다. 결합제가 충전 입자 주위에 피막을 형성하는 것은 그립 3.25 에 서와 같이 현미경으로 확인할 수 있다. 이 그립에서는 과염소산 암모늄을 아지리딘계 결합제로 코팅시킨 후 아세톤에 일부 용해
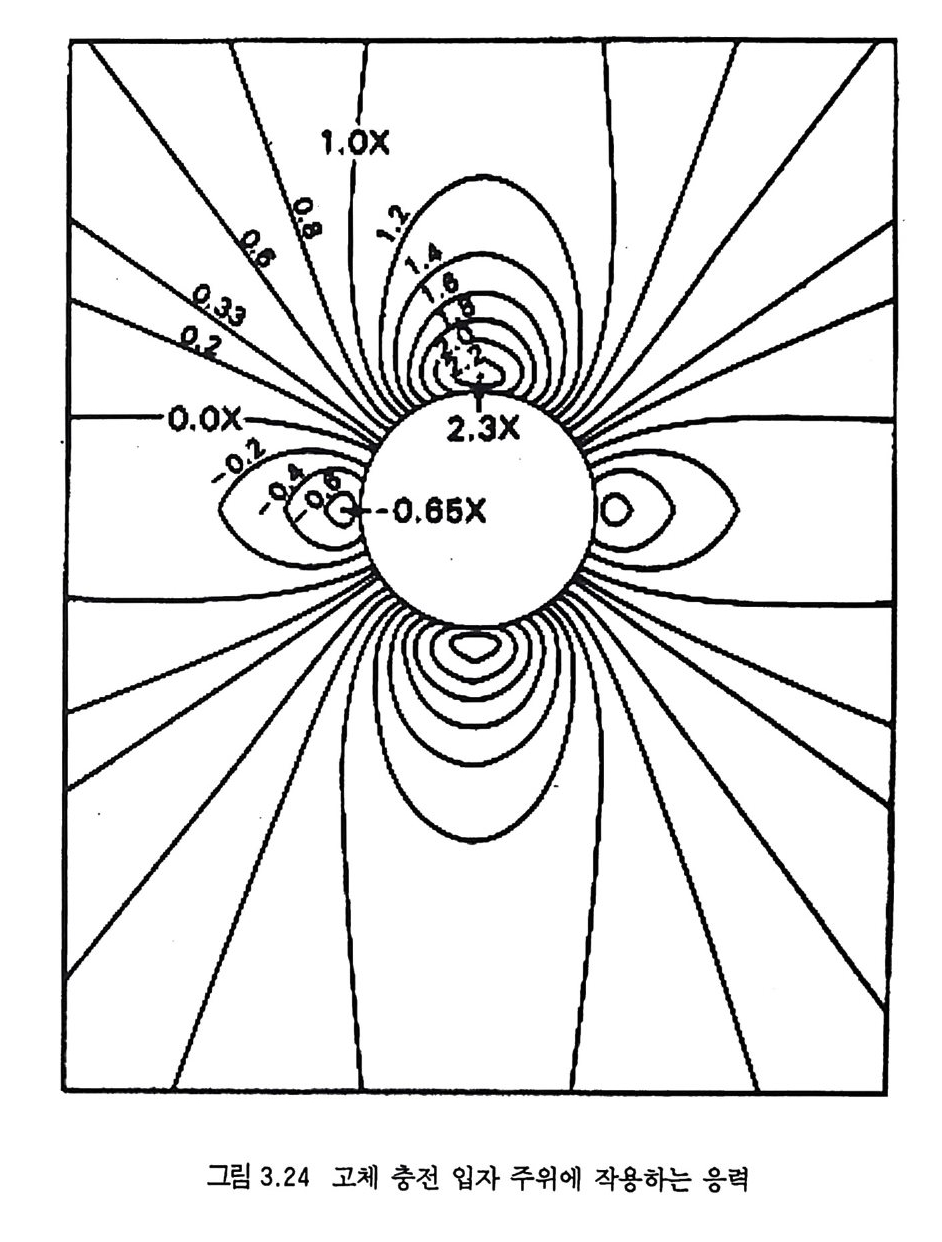 그림 3.24 고체 충전 입자 주위에 작용하는 응력
그림 3.24 고체 충전 입자 주위에 작용하는 응력
 广 ·三 _三三二三_ _\ _. ―一~:-·~군_.\ \一._、
广 ·三 _三三二三_ _\ _. ―一~:-·~군_.\ \一._、
시켜 현미경으로 촬영하였다. 결합제는 결합제가 흡수 코팅되어 야 할 충전 입자 표면과 어느 정도 친화력이 있어야 한다. 영기 성 화합물들은 과염소산암모늄이나 니트라민 화합물에 의해 화학 적 흡착이 일어난다. 죽, 결합제의 극성이 강하면 산화제 입자에 대한 천화력 및 흡수력이 강해지므로 결합제로서의 역할이 강력 해진다고 볼 수 있다. 결합제 역할에 관련된 메커니즘으로 또 다른 설명은 결합제가 바인더 메트릭스(용액)로부터 고체 입자 표면 위에 침전된다는 것으로서, 니트로셀룰로오스와 같은 고분자 결합제를 사용한 경 우가 그 대표적인 예로 설명된다. 이 메커니즘에 의하면, 결합제 는 바인더에 대한 용해도가 어느 정도 낮아야만 메트릭스로부터 그 일부가 쉽게 빠져나와 고체 입자 표면으로 이동하여 흡착된다 는 것이다. 따라서 니트로셀룰로오스 혹은 부틸 초산셀룰로오人 등을 결합제로 사용하면 바인더 상으로부터 일부분이 고체 표면 에 이동하게 되 어 추진제 의 물성 (modulus) 에 크게 영 향을 주게 된다. 그러나 바인더에 완전히 불용인 결합제를 사용하게 되면 결합제로서의 기능이 상실된다. 예를 들어 바인더에 불용인 C-1 울 사용한 우레탄 추전제의 경우 결합제로서의 효과가 거의 나타 나지 않았다(표 3.11). 결합제는 대부분 극성을 지닌 저분자량 화합물로서 니트릴, 아 미드, 우레아와 같은 관능기 (fun c tion al gr ou p)들을 갖고 있다. 결합제는 대부분 액체 상태이므로 혼합공정시 균일하게 분산시키 는 공정에 문제점이 전혀 없다. 고분자성 결합제일지라도 바인더 에 완전 용해된 다음 균일 분산된 후 혼합공정을 거치면서 경화 제가 가해져 경화반응이 일어날 때 결합제가 점차 바인더 상으로 부터 불용화되면서 폴리머 피막을 고체 입자 주위에 형성하게 된 다. 예를 들어 아지리딘계 결합제는 과염소산암모늄과 반응을 일
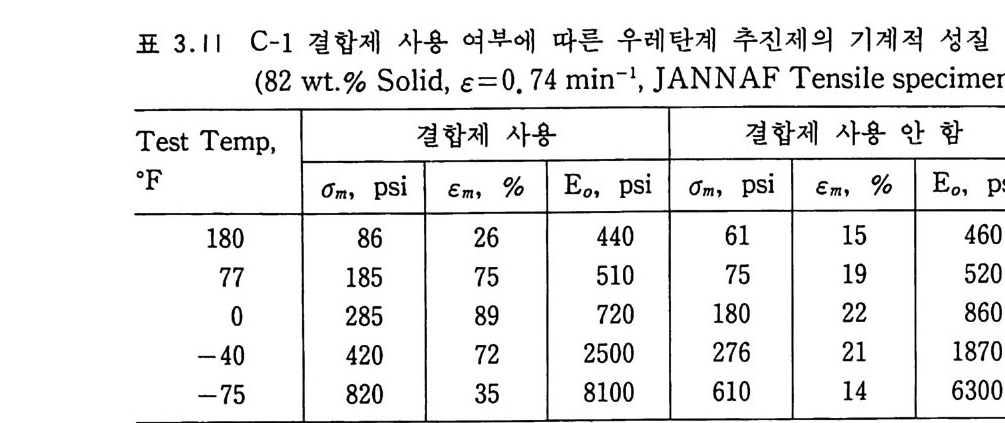 표 3.11 C-1 결합제 사용 여부에 따른 우레단계 추진제의 기계적 성질
표 3.11 C-1 결합제 사용 여부에 따른 우레단계 추진제의 기계적 성질
으켜 과영소산암모늄 표면에 피막을 형성하게 된다. Ober th는 현재까지 알려진 결합제들을 다음과 같이 여섯 종류로 분류하여 설명하였다 .6) ® 알카놀아민 (alkanolami ne ) 알카놀아민계 결합제는 폴리우레탄계 추전제에 적용된 것으로 서 산화제에 대한 천화력은 다음과 같이 과산화암모늄 표면에서 의 반응으로부터 나온 것이다.
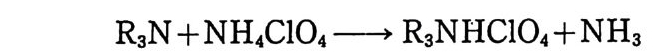 R3N + NH4Cl04 一 R3NHC104 + NH3
R3N + NH4Cl04 一 R3NHC104 + NH3
이와 같은 반응에 의해 과염소산암모늄 표면에 결합제의 흡착 충이 형성되며, 이 충은 디이소시아네이트(경화재)와의 반응에 의해 딱딱한 피막으로 변한다. 이러한 결합제들은 효과가 좋고 사용하기가 간편하며 가격도 저렴한 반면, 암모니아 발생으로 인 한 기포 생성의 단점도 있다. ® 중성 화합물 중성 화합물 결합제로는 히드록실기 (OH) 를 포함하는 아미드
또는 우레아 계열 화합물이 대부분이며 BHEGA, C-1, 및 그 유도체들이 있다. 이 결합제의 천화력은 그 극성으로 인한 것이 므로 HMX, RDX, KP, Al, 유리 등과 같이 반응성 이 낮은 고체 입자의 결합제로 사용된다. 이러한 중성 화합물 결합제들은 추전 제 노화 특성이 우수하고 저온에서의 특성이 특히 우수하며 알카 놀아민보다 폭넓게 적용될 수 있으나 특정 바인더 종류에서는 알 카놀아민보다 결합제로서의 효과가 크게 떨어질 때도 있다.
 ® 폴리아민(p ol y am i ne) 계 결합제
® 폴리아민(p ol y am i ne) 계 결합제
와 같다. 그러나 TEPA 중 반응하지 않고 남아 있는 아민기들 은 바인더 내에 있는 디이소시아네이트 경화제와 반응하게 된다. 이 경우 결합제 내의 아민기가 모두 과영소산암모늄과 반응하여
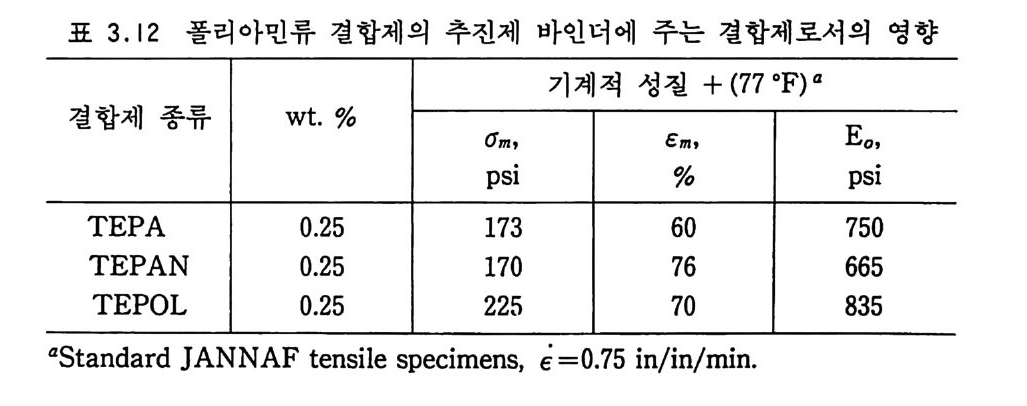 표 3.12 폴리아민류 결합제의 추진제 바인더에 주는 결합제로서의 영향
표 3.12 폴리아민류 결합제의 추진제 바인더에 주는 결합제로서의 영향
소모되어 버리면 디이소시아네이트와 반응할 아민기가 없으므로 피막을 형성하여 바인더 메트릭스와 연결할 수가 없게 된다. 따 라서 폴리아민의 아민기 숫자와 과산화암모늄의 p H 는 결합제로 서의 역할에 큰 영향을 주게 된다. 최근에는 TEPA 를 아크릴로 니트릴 등과 같은 다른 화합물과 반웅시켜 TEPAN 이나 TEPANOL, TEPOL 등을 합성하여 효과가 다론 결합제롤 사 용하기도 한다. ® 아지리단계 결합제 아지 리 딘은 과염소산암모늄의 영 향으로 단중합 (homop o lym e r- i za ti on) 하는 성질이 있으므로 과염소산암모늄과 접촉하면 입자 표면에 직접 피막을 형성하게 된다. 아지리딘 화합물 구조는 다 음과 같으며, X 는 메틸아지리딘기를 나타낸다 . 4 9) 이상과 같은 아지리딘 결합제의 구조에는 히드록실기, 아지리 딘기, 카르복실기를 포함하고 있는데, 그 중 아지리딘기나 카르 복실기를 다른 종류의 치환기들로 반응시켜 치환시키면 여러 종
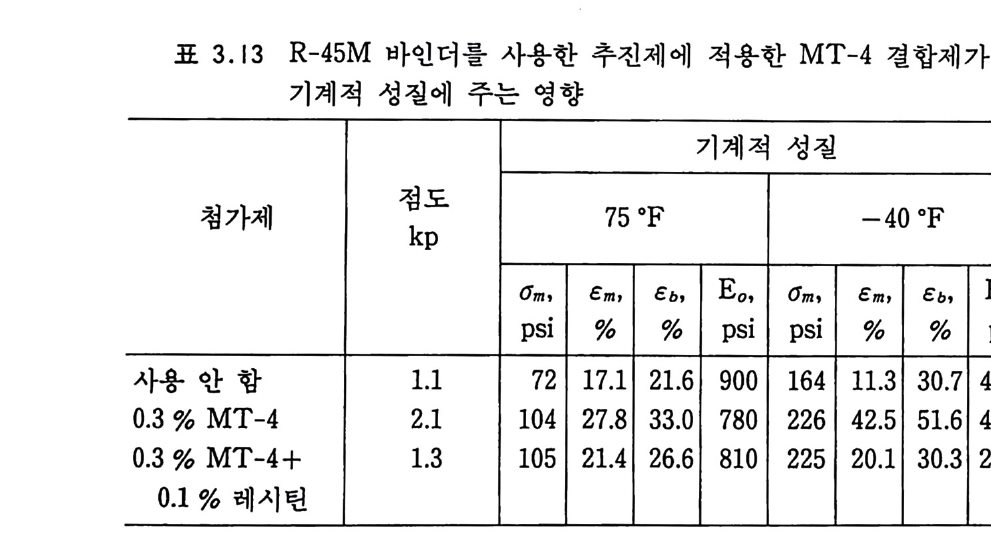 표 3.1 3 R-45M 바인더를 사용한 추진제에 적용한 MT-4 결합제가
표 3.1 3 R-45M 바인더를 사용한 추진제에 적용한 MT-4 결합제가
류의 유도체들이 합성될 수 있다. 예를 들어 MAPO, 아디핀산, 타타르산 (tar ta r ic aci d) 을 반옹시 켜 얻은 물질들의 혼합물을 MT -4 라고 부르는데, 이 혼합물은 우레탄계 추진제의 결합제로서 혼 히 사용되는 물질이다. 표 3.13 에는 MT-4 를 적용한 추진제의 기계적 성질이 나타나 있다 .50) 아지리딘 결합제의 구조 중 히드록실기는 결합제로서 작용시 필수적인 기가 아니라는 주장도 있었다. 그 이유는 히드록실기가 없는 HX-752 라는 아지 리 딘 화합물이 R-45M (HTPB 의 상품명 ) 바인더 추전제에서 결합제로서의 역할이 가능하기 때문이다. 추전제 메트릭스와 산화제 입자 표면 피막 사이에 연결되는 일 차결합 형성 메커니즘에 관해 최근까지도 토론이 계속되고 있다. 예를 들어, Brol i ne 에 의하면 HX-752 결합제는 히드록실기와 다음과 같이 반응할 수 있다고 하면서 이 반웅이 일차결합의 주 반웅이라고 설명하였다 .51)
RCH20H +JC』H HH3 — 2 /N COR' 一 RCH,0-CH,_ C』 HH3- NHCOR' CH2-CH2 R-{/C HI2 + OCN— R '-— R — N N| -R' \CH2 \/ Azir i d i n e Isocy an ate Irnid a zoli do ne
그러나 Immoos 와 Bowen 등은 과량의 과염소산암모늄 산화제 가 존재할 때는 단중합 반응이 주반응인 것을 주장하였다. 아지 리딘은 이소시아네이트와 반응하여 이미다졸리돈을 생성하므로 이를 통하여 우레탄 메트릭스와 피막과의 직접 결합은 가능한 것 으로 보인다. 아지리딘계 결합제는 추전제 공정에 적용하기 간편하고 강력한 결합제로서의 역할을 하지만, 그 가격이 너무 높고 독성이 강한 단점으로 인해 점차 그 사용량이 감소하는 추세에 있다. ® 셀룰로오스 유도체들 니트라민계 추전제 혹은 니트로글리세린을 가소제로 하는 추전 제에서는 니트로셀룰로오스 계통의 물질들이 결합제로 자주 사용 된다. 그러나 니트로글리세린을 가소제로 사용하는 추진제의 조 성 결정시 특히 결합제 선택에 있어 주의하여야 할 점은 첫째로 니트로글리세린이 염기성에 매우 예민하므로 분해될 위험성이 있 으며, 둘째로 HMX 및 RDX 와 같은 니트라민 고체 입자 표면 의 극성이 니트로글리세린과 다를 바가 없으므로 결합제가 결합 하려는 힘이 니트라민 표면이나 바인더 상이나 비슷하다는 점과, 셋째로 대부분의 저분자량 결합제는 니트로글리세린에 용해되어 버린다는 점이다. 따라서 이러한 추진제의 경우에는 저분자량 결 합제보다는 셀룰로오스와 같은 고분자 물질의 결합제가 더 효과 적으로 작용한다고 볼 수 있다. ® 산화제 입자의 프리코팅(p recoa ting )52> 흔히 사용하는 방법은 아니지만, 추전제 원료 혼합시 결합제를 가하여 바인더 메트릭스와 산화제 입자 결합력을 강화시키는 방 법 대신 추전제 혼합공정에 들어가기 전 산화제 표면을 프리코팅
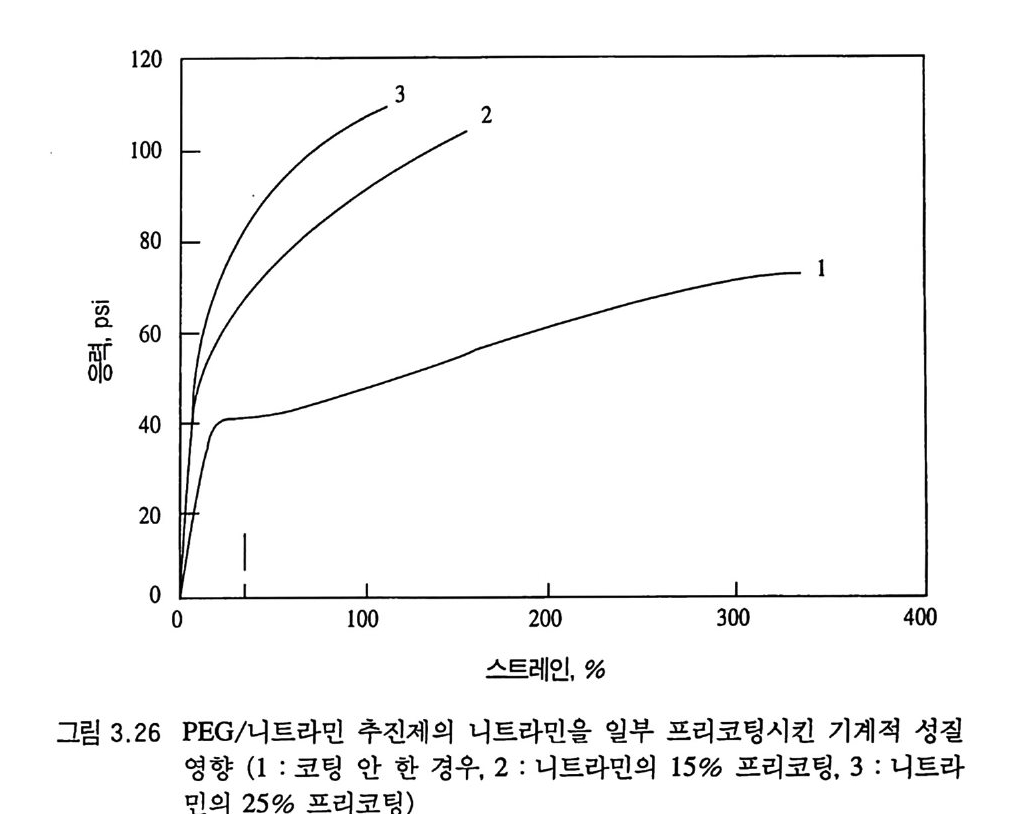 120 3
120 3
하는 방법을 사용할 때도 있다. 죽, CMDB 추진제의 경우 과염 소산암모늄 분말을 트리에탄올아민으로 처리한 후 PAPI 와 같은 이소시아네이트로 반응시켜 산화제 입자 표면에 피막을 만들어준 다. HMX 와 같은 니트라민 역시 부탄디올과 PAPI 를 반응시켜 그 표면에 피막을 형성시킨 후 추전제 혼합공정에 사용하면 그림 3.26 경우처럼 그 기계적 성질이 크게 개선된 결과를 얻을 수 있 다. 이러한 프리코팅 방법은 코팅시키는 재질의 화학적 특성을 고려하여 잘 선택하면 추전제 바인더에 방해받지 않으면서 코팅 할 수 있는 접과 산화제 입자 표면에 피막 형성 물질을 다양하게 바꿀 수 있으므로 결합제를 바인더에 주입시키는 방법보다 더 좋
온 방법으로 평가되지만, 프리코팅 공정 경비가 증가하는 단점으 로 인해 혼하게 사용되지는 않고 있다. 3.3.4.5 기타 첨가제 다른 물질과 마찬가지로 추전제도 시간이 지나가면서 노화되어 기계적 성질이나 연소속도 동의 각종 성질이 변하게 된다. 추전 제의 각종 특성둘은 일정기준 범위 안에 있는 동안에만 사용할 수 있도록 추전제 그레인이 설계되어 있다. 그러나 추전제의 종 류에 따라 어떤 추진제는 시간이 흐름에 따라 더 딱딱해지며 또 다른 추전제는 더 연하게 변하기도 한다. 이러한 노화현상은 추 전제 바인더의 화학적인 특성에 따라 좌우되므로 바인더의 변질 현상을 가능한 한 억제하기 위해 안정제 혹은 산화방지제 및 가 수분해 방지제를 첨가한다. 또한 어떤 종류의 추진제는 추진제 원료혼합 공정시 혼합 도중 상당량의 거품이 생기는 경우가 발생하는데, 이 거품을 전공으로 제거하면 추진제 조성이 변화되어 경화된 추전제의 성능에 영향 울 주게 되므로 거품 방지제를 미리 첨가하여 거품 발생을 방지 한다. 3.4 추진제 제조 추전제 원료들은 주 혼합기에서 혼합공정을 거치기 전에 전처 리 공정을 한다. 죽, 바인더의 폴리머와 첨가제를 서브믹스 단계 로 혼합시킨 후 다시 금속연료와의 균일한 혼합을 위해 프리믹스 를 한다. 그후 주 혼합기에 옮겨져 산화제와의 혼합공정으로 이 어진다. 주입되는 원료들은 모두 철저한 품질관리에 의해 통제되
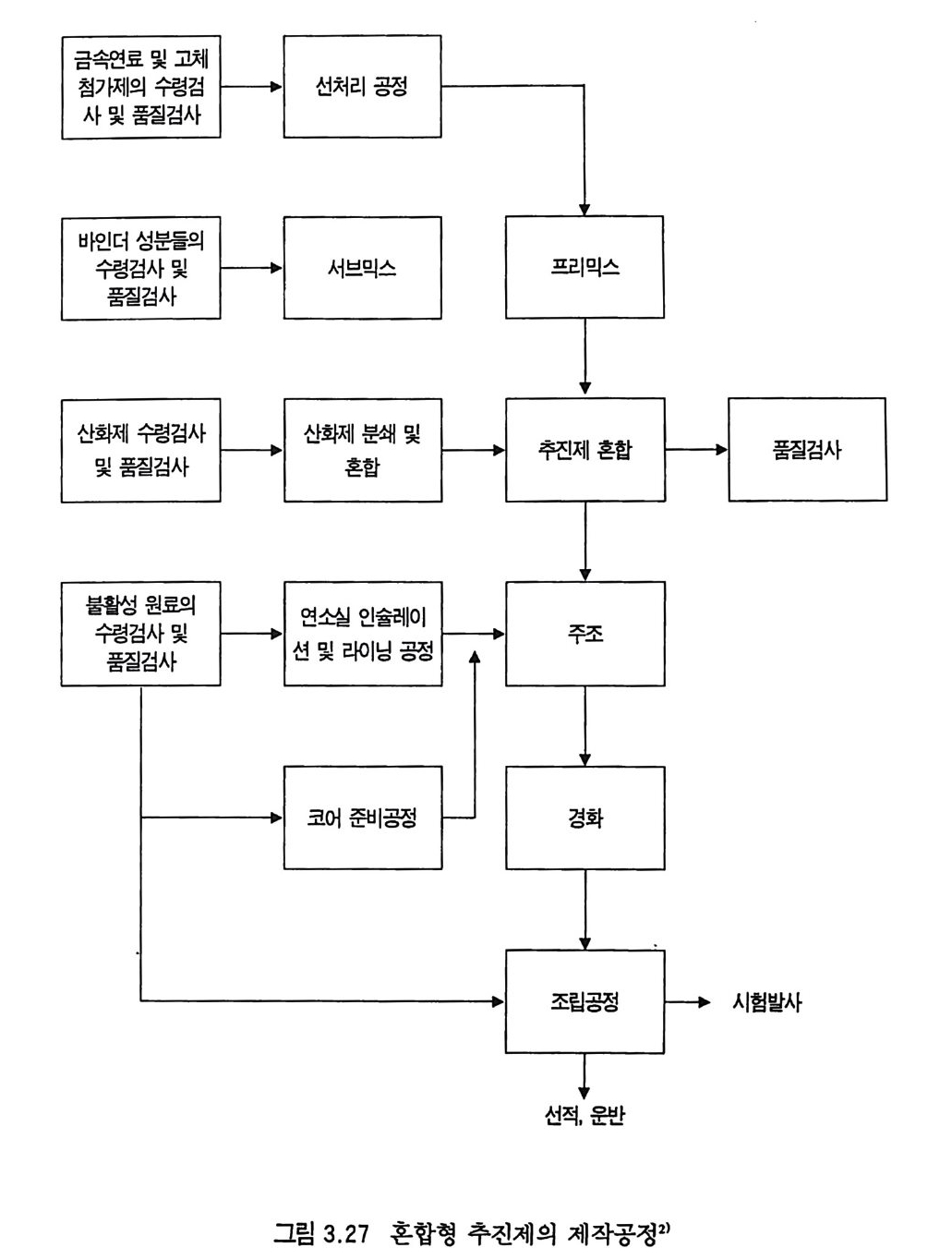 금속견료및고체
금속견료및고체
어 요구되는 특성과 규격을 만족해야 된다. 특히 산화제인 과염 소산암모늄의 경우에는 그 화학적 순도만이 아니라 입자 분포도 와 입자 모양이 추전제의 제조 특성, 기계적 성질, 내탄도적 성 질에 민감하게 영향을 주므로 철저한 품질관리가 요구된다. 그림 3.27 에서 보이는 공정도와 같이 추진제 혼합시 추진제 페이스트 (pa ste , 반죽)의 점도와 페이스트의 연소속도 등 몇 가지 데이터 를 신속히 분석하여 그 다음 공정인 주조공정을 계속할지에 대한 판단을 내 리 며 , 주조공정 및 코어 링 (corin g ) 공정 후 오븐 속에 서 경화공정을 거치게 된다. 경화가 끝나면 코어를 제거하는 작 업을 한 후 추진기관 조립작업으로 이어진다. 이와 같이 조립된 추진기관 모터들은 로트별로 주어전 품질관리 계획에 따라 발사 시험에 의해 그 성능이 합격되면 납품하는 작업이 진행된다 .3,5) 이상과 같이 로켓 추진기관 모터에 추전제를 제조 충전시키는 주요 공정은 연소관 준비 공정, 산화제 준비 공정, 추진제 혼합 공정, 주조공정, 경화공정, 마감공정 및 검사 등으로 분류할 수 있다 .53) 3.4.1 연소관 준비 공정 로켓 추진기관의 연소관은 추전제 그레인 모양을 제작하는 몰 드 역할을 한다. 케이스 내부는 매우 깨끗하게 유지하며 추전제 가 잘 접착될 수 있도록 해야 한다. 케이스 내부에 이물질이나 녹이 슨 부분이 있을 경우에 대비하여 와이어브리싱 또는 샌드불 래스팅 (sand blas ti n g)을 한 후 트리클로로에틸렌과 같은 강력한 용매 를 사용한 탈유 (deg rea sin g ) 공정 을 한다 (그림 3 . 28) . 54> 대부분의 로켓 연소관에는 추전제가 케이스에 직접 집착되도록 주조하지 않고, 연소관 내부 표면에 라이너와 내열재를 먼저 도
 i'
i'
포한다. 내열재는 추진제 연소시 연소불꽃의 높은 온도로부터 연 소관을 보호하기 위해 도포하며, 라이너는 추전제와 연소관 사이 또는 추진제와 내열재 사이의 접착력을 증진시키는 작용과 함께 추진기관 모터 운반과 같은 작업이 있을 경우 의부로부터의 충격 이나 진동이 추진제 그레인에 직접 전달되는 현상을 완화, 완충 시키는 역할을 겸하고 있다. 따라서 라이너 재료로서는 추전제에 사용된 바인더와 유사한 바인더를 선택하고 추진제와의 접착력도 좋고 연소관과의 접착력도 우수한 성질을 지녀야 한다. 이와 갇 이 연소관 준비가 끝나면 추전제 그레인 모양에 따라 설계 제작 된 코어(맨드릴) 준비를 해야 한다. 코어는 추진제를 주조하기 전 미리 연소관에 장착할 수도 있고 경우에 따라서는 추전제를 먼저 주조한 후에 코어를 장착 (co ri n g)할 수도 있다. 3. 4. 2 산화제 준비 분쇄공정 전술한 바와 갇이 산화제의 입자 크기, 모양 및 입자 분포도는 추전제의 여러 성질에 큰 영향을 주므로 산화제를 분쇄하는 공정 은 주의해서 운전해야 하며, 특히 산화제가 작은 입자로 분쇄되 는 과정 중 산화제에 가해지는 충격과 작은 입자들의 폭발 위험 성에 대비하여 극히 조심스러운 작업이 필요하다. 또한 산화제로 사용되는 과염소산암모늄이나 질산암모늄 등은 홉습성이 매우 크 므로 분쇄 공장 내의 엄격한 습도 조절이 요구된다. 분쇄된 산화 제는 장시간 보관시 케이킹 현상이 일어나므로 특별 포장으로 처 리된 경우 이의에는 오랫동안 보관하지 않는 것이 일반적이다. 그립 3 . 29 에 는 애 토마이 저 (ato m i ze r) , 펄 버 라이 저 (pu lveriz e r) 및 풀루이 다이저 (flui d i z e r) 등의 장비 를 사용하는 산화제 분쇄공정 이 나타나 있다. 분쇄된 산화제는 반드시 그 수분 함량, 입자 크
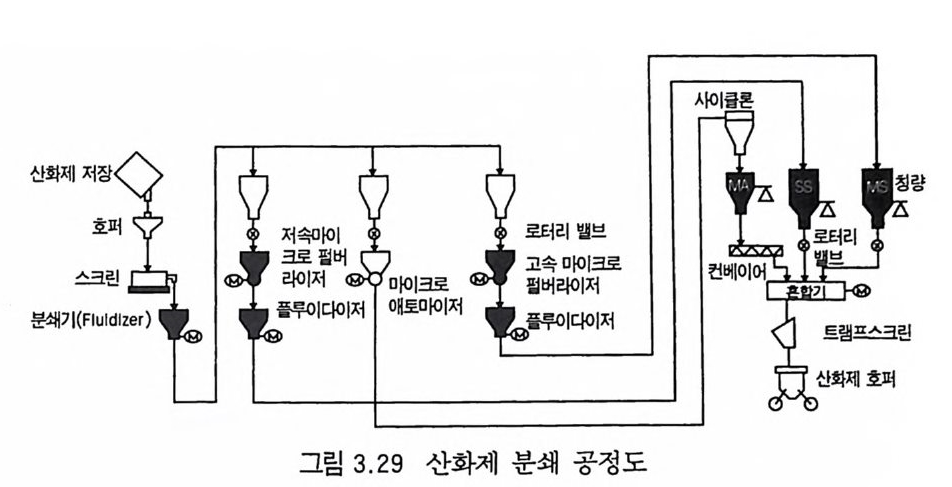 산화제저장
산화제저장
기 및 분포도를 분석하여 주어전 규격 범위 내에 있는지 확인한 후 추진제 원료로서 혼합공정에 사용된다. 산화제 입자 크기 및 분포도는 채 (sie v e) , 서 브시 브사이 저 (sub-sie v e-siz e r) , 기 타 입 도 분석장치를 이용하여 분석한다 .53,55) 3.4.3 혼합공정 전술한 바와 같이 혼합공정의 처음 단계는 바인더로 사용되는 폴리머와 연소속도 촉매 및 안정제와 같은 첨가제들을 혼합시키 는 서브믹스 (sub-m i x) 한 후, 다시 금속연료 분산(알루미늄 분말) 을 잘 시켜 주는 프리믹스(p re-m i x) 를 한다(그립 3. 30 ). 53,56) 프리믹스가 끝나면 산화제 및 경화제와 혼합하는 주 혼합공정 이 이루어진다. 혼합기로서는 혼합량의 크기에 따라 여러 종류의 혼합기 가 사용되 며 , 수평 형 (horiz o nt a l) 또는 수직 형 (verti ca l) 의 두 종류가 있다. 최근에는 혼합 효율이 우수한 수직형 혼합기가 더 많이 사용되는 경향이 있다(그립 3.31). 혼합기에는 가열 • 냉 각 • 진공 • 산화제 및 촉매 주입구, 혼합 도중 연소폭발을 감지하
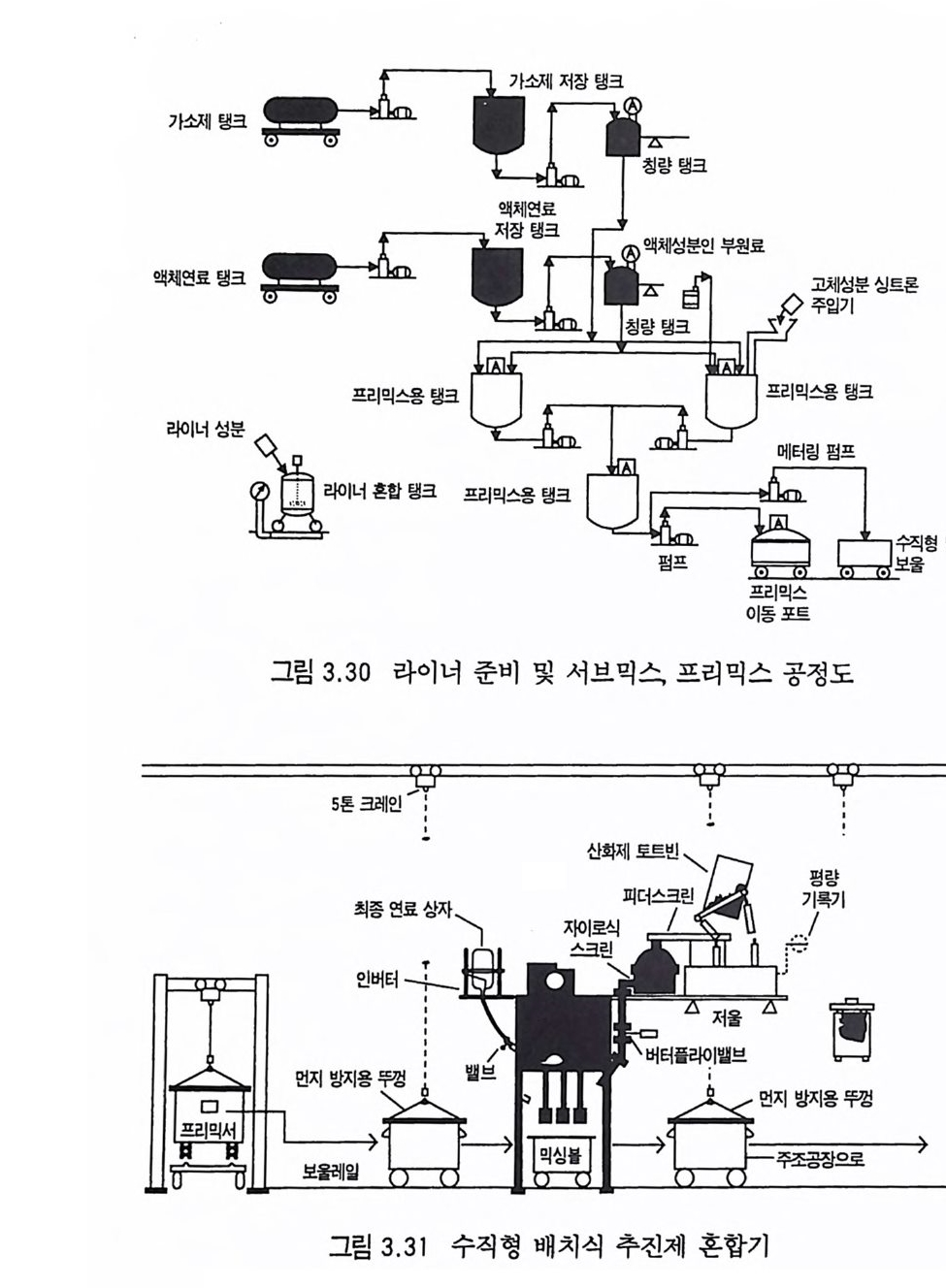 가소재탱크
가소재탱크
여 소화하기 위한 센서 등이 부착되어 있다. 이러한 배치식 혼합 기에서의 혼합 시간은 추진제 혼합량 및 조성 종류에 따라 차이 가 있지만 대개 1. 5~6 시간 정도를 필요로 한다. 추전제 혼합기 한 배치의 추진제 양으로는 추전기관 모터 그레 인을 완전히 채우지 못할 정도로 대형의 추전기관인 경우에는 추 전제 혼합 충전을 연속공정식으로 작동하기도 한다. 이러한 연속 식 공정에서는 그립 3.32 에서 보는 바와 같이 산화제 공급, 연료 프리믹스 공급 및 경화제 공급 라인이 혼합기로 연결되어 l~2 분 간의 혼합 후, 진공 속에서 가스가 제거되면 곧바로 주조공정으 로 연결된다. 53,5 7 ,5 8) 또 다른 연속식 혼합공정은 복기 추전제에서 사용되는 퀵믹스 (qu ic k -mi x) 공정에서와 같이 제트 혼합기를 사용하는 방법이므 로 위험성이 낮은 공정이다(그립 3.33). 최근에 개발되어 사용되 는 또 다론 연속공정식 혼합공정은 그림 3.34 와 같이 고체 성분
 산화평제량감 량 二-----댄연 감료량프밸브리평二 믹량스 Y 4----- 』 경화제저장
산화평제량감 량 二-----댄연 감료량프밸브리평二 믹량스 Y 4----- 』 경화제저장
(산화제 및 알루미늄 분말)과 액체 성분을 모두 공기압에 의해 공 기압 혼합을 하는 방법으로서, 원료들은 다공성 튜브를 통해 공 기 압에 의 해 이 동됨 과 동시 에 난류 혼합 (tur bulent mi xi n g ) 이 진 행된댜 따라서 혼합 속도가 빠르면 혼합 효율이 높은 이점이 있 다.
 AP 분산기
AP 분산기
3.4.4 주조공정 혼합공정이 끝난 추진제 반죽은 주조공장으로 옮겨져 라이너와 인슐레이션이 미리 도포된 로켓 연소관에 주조된다. 주조(충전) 공정은 일반적으로 전공하에 주조하여야 추진제 내의 기포를 최 소로 줄일 수 있다. 추진제 반죽의 점도가 낮은 경우에는 주조에 어려운 점이 없으나, 점도가 높으면 주조 시간이 길어지며 주조 공정에서 실패할 수도 있으므로 전동을 주면서 질소가스로 압력 을 가하여 주조하는 경우도 있다. 코어링 공정은 주조가 끝난 후 코어를 장착하거나, 또는 주조 하기 전 추전기관 모터 내에 코어를 미리 장착하는 두 가지 방법 이 있다. 추전제 반죽의 점도가 낮은 경우는 코어롤 미리 장착하 는 것이 유리하지만, 코어의 크기와 연소관 크기, 추전제 반죽이 주조될 입구의 크기 등을 검토하여 코어 공정의 순서를 정한다. 3.4.5 경화공정 주조, 코어링 공정이 끝난 모터를 경화 오본으로 옮겨 가열 경 화한다. 경화 온도와 경화 시간은 추진제 그레인 설계시 요구된 특성에 부합되도록 면밀하게 조절되어야 한다. 경화되는 추전제 표면의 경도 (shore hardness) 를 시간별로 검사하여 추진제의 경화 가 정상적으로 이루어지고 있는지 확인하기도 한다. 경화가 끝나 면 상온으로 냉각시켜 코어를 제거한다• 3.4.6 마감공정 및 검사 경화가 끝나고 코어가 제거된 추전제를 설계된 치수에 맞도록
과량이나 여분의 추전제를 깨끗하게 절단하는 마감 처리 공정을 한 후, 추진기관 모터 내의 추진제, 라이너, 인슐레이션에 주입 된 원료들의 분석 시험 결과들을 재검토한다. 또한 추진기관 모 터를 비파괴 시험실로 옮겨 X-ra y나 초음파에 의한 추진제 그레 인 내부의 기포, 균열, 추전제-라이너-인슐레이션-연소관 사이의 접착 불량 등을 면밀히 조사한다. 또한 모터 추전제 주조시 별도 로 시편을 받아 모터에 충전된 추진제와 같은 경화 오븐에서 경 화시킨 후 기계적 성질 및 내탄도 성질 등을 시험하여 추진기관 모터에 충전된 추전제의 성능을 평가한다 .48 , 53) 3.5 추전제의 기계적 성질 추전제가 충전된 추진기관 모터는 보관, 운송, 발사되는 여러 환경 분위기에서 열적 순환(th ermal cyc l in g ), 조작, 진동, 점화 압력 및 가속화 등의 영향을 받게 된다. 추진기관 모터 내에 충 전되어 있는 추진제는 이러한 영향으로부터 충분히 견딜 수 있는 물리적, 기계적 성질을 보유하고 있어야 한다. 또한 오랜 시간이 흐름에 따라 추진제의 물리적, 화학적 변질 또는 변화가 나타나 므로 추전제의 노화 현상에 대한 이해와 함께 추진제의 실패 평 가 기준 설정과 관련된 기계적 성질 연구 및 측정은 매우 중요하 다. 기계적 성질을 측정하는 가장 일반적인 시험은 그림 3.35 와 갇은 미국의 JA NAF 인장시험 (일축시험) 시편에 의한 것으로서, 이 시 편은 다이 (die ) 절단방법 이 나 주조방법 또는 밀 링 (mi lling ) 에 의해 제작될 수 있다. 그러나 그 중 가장 정교한 시편은 밀링 에 의해 제작된다. 이 JA NAF 시편에 의한 기계적 성질 시험은 추전제의 품질관리, 추전제의 조성 실험 등에 적용되고 있다. 그
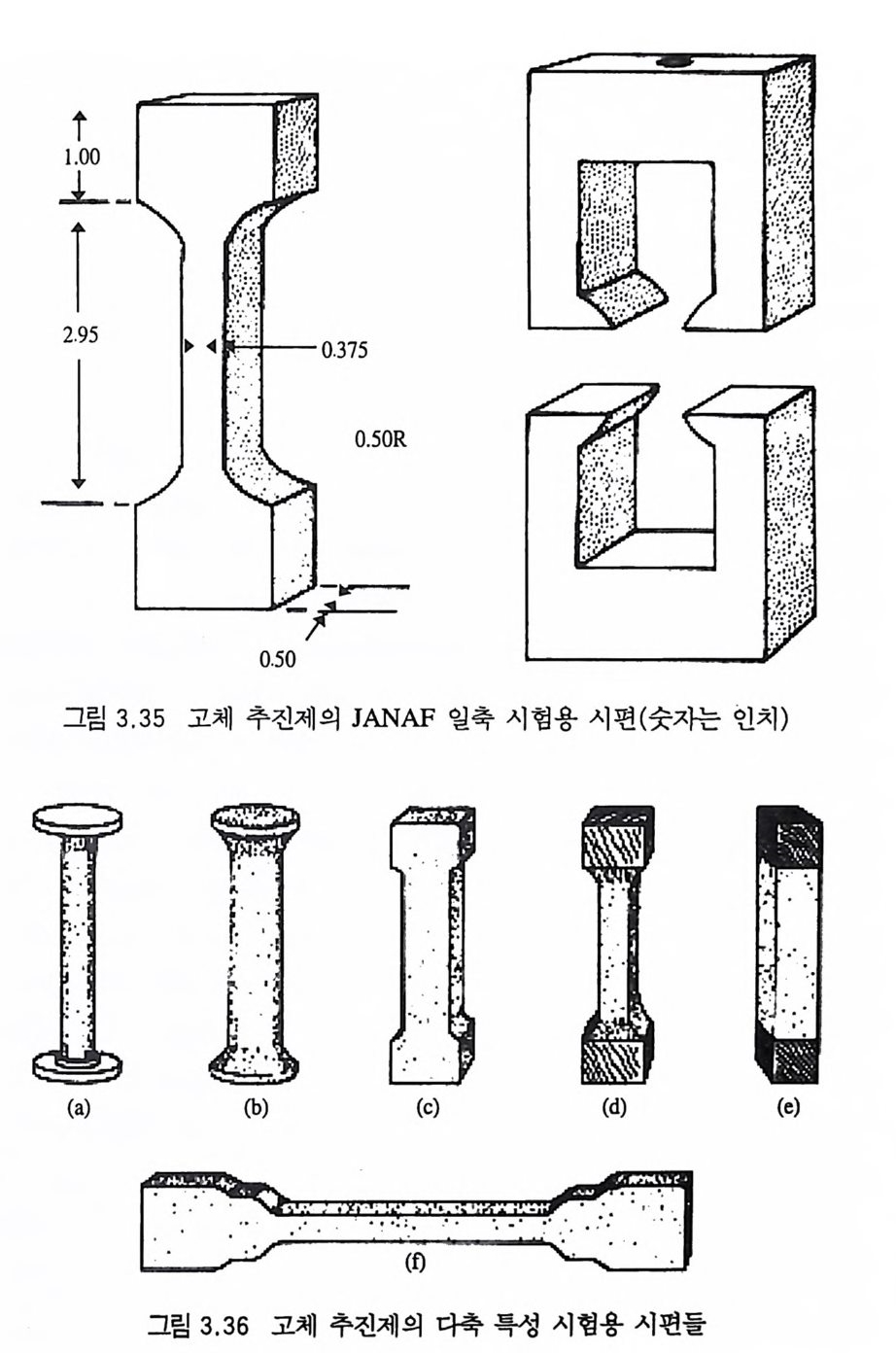 _ 4f _-
_ 4f _-
러나 고체 추진제의 기계적 성질을 더 충분히 이해하기 위해서는 추진제 의 다축 (multia x ia l ) 특성 시 험 이 필요하다. 다축 시 험 시 사 용되는 시편들은 그림 3.36 과 같은 시편들이 사용된다 .48,59,60) 고체 추진제에 주기적으로 가해지는 하중에 대한 영향을 알기 위해 동적 시험이 사용되고 있으며, 그 대부분은 사인파 (s i nuso i dal) 적인 방법으로 하중을 주어 시험하고 있다. 이러한 동적 시험 측정은 구조물의 진동 분석, 추진제의 점탄성 성질 , 추진제 의 전동연소 (osc illa ti ng combusti on ) , 충격 파의 내 부 감쇠 (att en uat ion ) 및 피 로수명 (fatigue life) 연구에 매 우 유용한 데 이 터로 적용된다. 동적 시험 방법은 짧은 시간 동안 변형률을 1 % 이하 저전폭으로 하는 시험 방법과 파괴가 일어날 때까지 고전폭 으로 충분한 시간 동안 변형을 주는 방법이 있다. 추진기관 모터의 실패 기준(fail ure cr it er i a) 이 정해지면 이로부 터 추전기관 모터의 작동과 취급시 예상되는 안전도 마전 (sa fety mar gi n) 을 예측하게 되며, 또한 추진기관 모터가 비정상적 작동 을 자주 일으킬 수 있는 하중 범위를 정할 수 있으므로 실패 기 준은 매우 중요한 의미를 갖고 있다. 실패를 정의하는 데는 여러 가지 방법이 있다. 작동이라는 의미에서 정의되는 실패는 모터 압력, 연소시간, 연소속도 등의 성능이 예측, 설계된 것과 벗어 날 때를 의미한다. 또 다른 실패의 정의는 추진제 그레인에 생긴 균열(눈에 보이는), 시편의 파괴, 스트레스-스트레인 곡선상의 최대 응력점 증가 등을 포함하는 의미이다. 실패라는 것은 본질 상 통계적인 방법이지만 예측 단계에서는 반드시 고려되어야 할 사항이다. 고체 추진제에는 두 종류의 실패 기준이 있다. 그 첫번째 기준 은 실험실적 시험과 실제 모터 실패의 관계로부터 도출되는 기준 이며, 두번째는 분석적 특성인 실패표면(fail ure sur fa ce) 에 의해
기하학적으로 표현되는 기준이다. 그 중 첫번째 방법은 고체 로 켓 산업에서 전통적으로 흔히 사용된 방법이다. 두번째 방법은 일반적이며 수학적으로 정밀한 방법이지만 실험의 어려움 때문에 예를 들어 어떤 기준은 단순장력에 의한 일축 방향 변이 능력에 근거를 두고 있는 반면, 또 다른 기준은 이축 방향의 장력 조건 울 고려해야 하는 어려움성이 있다. 에너지 기준 역시 적용되어 왔으며 에너지 기준과 실패와의 상호 조합도 제안되었다. 또 하 나 중요하게 생각해야 할 점은 반복되는 열적 순환 및 전동에 의 해 손상 (dama g e) 상태나 부위가 축적된다는 점이다. 오랫동안 보관을 하면 많은 양의 에너지 성분을 포함하고 있는 고체 추진제가 심각하게 열화 (de t er i ora ti on) 되어 각 성분들이 상 호간 혹은 대기와 반응하여 추전제의 화학적 변화가 일어나므로 추진제의 기계적 및 내탄도적 성질에 치명적인 영향을 주게 된 다. 이와 같은 노화 (a g e i n g)에 의해 일어나는 현상으로서는 혼합 형 추진제의 경우 가소제의 이동 및 증발, 가스 발생 , 가교 결합 및 체인의 파괴 등이 있으며, 복기 추진제의 경우에서는 원료 성 분의 분해에 따른 추전제 감도 (sens iti v ity)를 증가시키는 물질들 이 생성된다. 이러한 화학적 노화와는 별도로 추전제의 기계적 성질 저하에 영향을 주는 기계적 열화 및 물리적 요인들도 고려 해 야 한다. Fr i es 와 J ohns 가 정 리 한 추진제 노화시 물성 저 하에 영향을 주는 물리적 및 화학적 요소들은 표 3.14 와 같다 .61) 복기 추전제와 혼합형 추진제는 노화 특성이 서로 다르다. 복 기 추전제의 경우 노화는 곧 안정성에 관련된 사항이므로 복기 추전제를 사용하는 로켓에서는 안전보관수명이라는 말을 흔히 사 용했었다. 혼합형 추전제의 경우 노화현상은 추진제가 딱딱하게 굳어지는 현상, 부스러지는 현상 및 접착성의 변화로서 나타나므 로 비파괴 시험이 매우 중요한 시험 항목이다. 추진제를 개발하
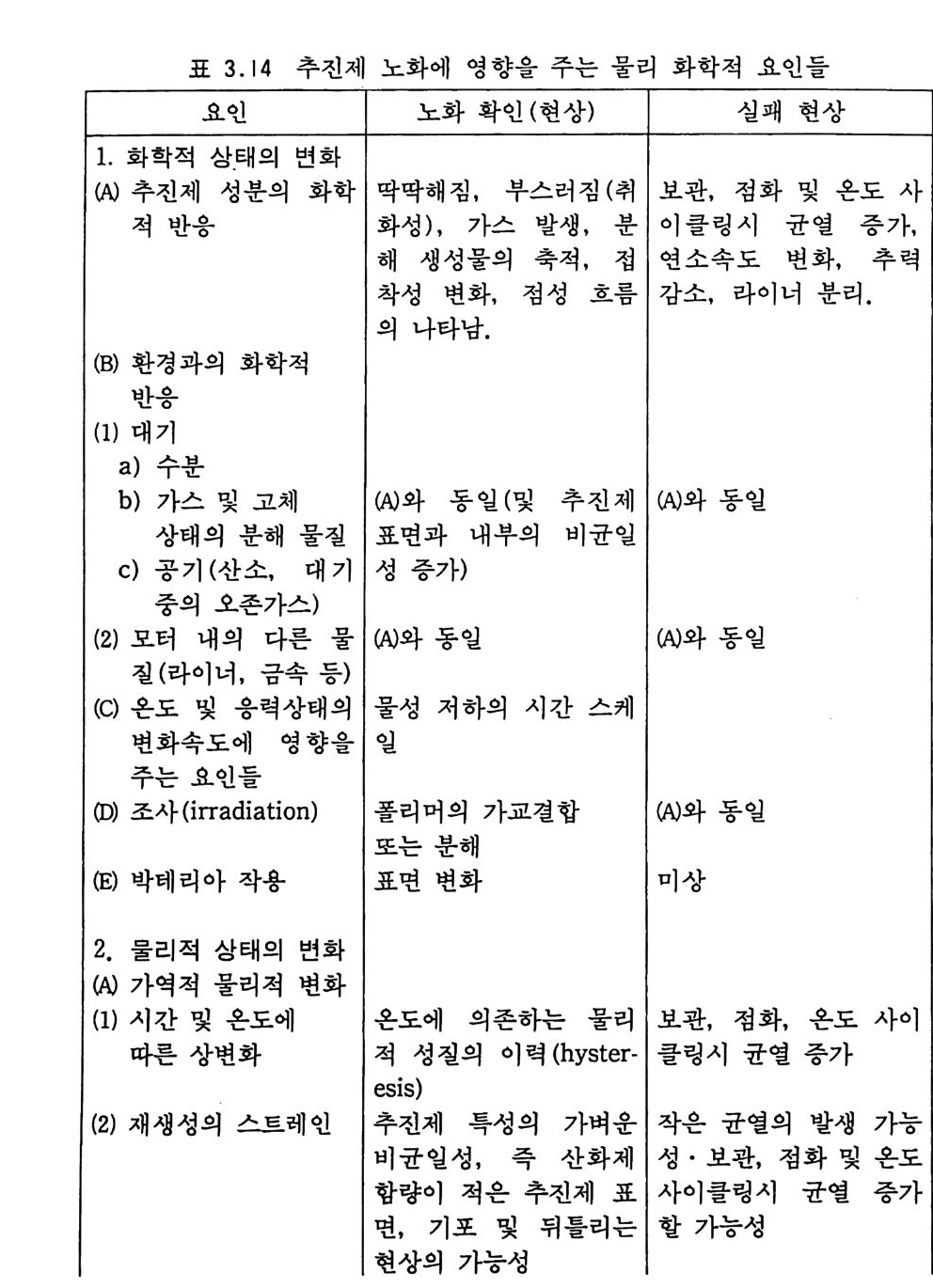 표 3.14 추진제 노화에 영향을 주는 물리 화학적 요인들
표 3.14 추진제 노화에 영향을 주는 물리 화학적 요인들
 (B) 비가역적 물리적 변
(B) 비가역적 물리적 변
여 사용하기 전 추진제를 상온에서 10~20 년간 보관하여 노화 특 성을 완전히 확인한 후 추진기관 모터에 적용한다는 것은 그 시 간이 너무 길고, 경제적인 방법도, 현명한 방법도 아니다. 따라 서 추진제의 노화현상을 이론적으로 또한 물리 화학적인 실험에 의해 규명하는 것이 필요하다. 실험적으로는 온도를 높여 수행하 는 가속노화 시험, 환경변화 시험 및 바인더나 산화제별 구조 변 화 시험 등을 병행함으로써 추전제의 노화 특성을 예측하기도 한 다. 3.6 추전제의 연소 및 내탄도 특성 고체 추전제의 연소에 관해서는 Summerfie l d, Becks t ead 를 비롯한 많은 사람들이 연소 모델 및 연소 메커니즘에 관해 발표 하였다. 이들에 의하면 추전제의 종류에 따라, 죽 추진제에 포함 된 산화제, 바인더 종류 및 금속물질의 존재 여부에 따라 연소현 상이 달라지는 것으로 보고되었다. 본문에서는 혼합형 추전제에 가장 많이 적용되고 있는 과영소산암모늄을 포함하는 추전제를 예로 들어 그 연소현상을 간단히 언급한 후, 추전제의 연소속도 방정식을 다루었다.
3.6.l 연소현상 산화제로서 과염소산암모늄을 포함하는 혼합형 추진제에서 과 염소산암모늄이 차지하는 비율은 보통 70 %(wt . %) 이상이므로 산화제의 연소 특성이 추전제의 연소 메커니즘에 큰 영향을 준다 는 것은 당연하다. 과염소산암모늄 자체를 연소시켜 얻은 연소표 면을 SEM(Scannin g Electr o n M ic rosco p e) 으로 조사한 결과 과염 소산암모늄은 연소될 때 그 연소표면에 녹는 부위(용융충)가 있 음이 발견되었다. Hi gh to w er 및 Pr ic e 는 이 용융충의 깊이가 연소압력 증가에 따라 감소하는 것을 확인했고, 이 용융충에서는 기 체 상과 응축상 (condensed ph ase) 사이 에 반응이 일어 나고 있는 것으로 결론지었다 .62,63,64) 그후 이 용융충에 대한 많은 연구 발 표가 있었으며, Guir a o 등은 불균일 반응이 이 용융충에서 일어 나는 것울 가정으로 한 과염소산암모늄 연소이론을 발표하였 다 .65) 바인더의 연소는 바인더 성분만이 아니라 비화학적인 요소들, 죽 연소시의 열전달(전도, 대류, 복사), 화합물의 증발 및 액화 과정 등에 의해 변화된다. 따라서 바인더 성분, 즉 폴리머의 연 소는 매우 복잡하여 가스상 반응 또는 표면 및 응축상에서의 반 응과 같은 여러 가지 형식의 반응을 포함하게 된다. 가스상 반응 은 일반적인 탄화수소 연소시의 확산 불꽃 경우와 유사하며, 응 축상 반웅은 표면에서의 반응을 포함하게 된다. 부표면에서의 반 옹은 고체상의 분해반웅으로서 증발되기 이전 분해로 인해 일어 난다. 표면반웅은 액체 표면과 고체 챠르 (char) 표면 두 종류의 반응으로 나누어질 수 있는데, Cohen 등은 CTPB, HTPB, PBAN, PU 등의 바인더를 표면에서 열분해시킨 결과 넓은 범위 의 가열 속도 및 압력 범위에서 챠르 표면 형성과 함께 연소표면
에서 용융물이 비등하는 현상이 나타났다고 보고하였다 .66) 추진제 연소는 연소표면 위 응축상에서의 반응을 중요하게 다 루는 학설과 무시하는 주장도 있다. 가스상 모델들은 질량 및 에 너지 수지식에 근거를 두고 있다. 고체 및 연소의 보존 방정식을 처음 선형화한 후 적당한 경계 조건에 의해 해를 구하였다. 이때 표면에서의 연소속도는 아레니우스 형식의 열분해 법칙에 의해 서술하였다. 이러한 접근 방식은 주로 J ohnson 과 Nachbar,57> Summerfie ld 68> 등에 의해 시도되었으며, 이들은 연소면의 평면 (p lanar) 이 액상이 없는 균일한 상태라고 가정하였다(실제로는 용 융충을 포함하고 있음). 이러한 가스상 모델들은 연소속도 경향을 예측할 수 있지만 연소속도에 중요한 영향을 주는 입자 분포에 대해 설명할 수 없으며, 또한 바인더의 변화, 표면온도, 응축상 에서의 열손실, 연소속도의 온도 계수, 촉매 영향, 압력지수 등 울 예측할 수도 없다. 한편 고체상 모델들은 고체상에서의 열손 실, 연소속도의 온도 계수, 촉매 영향 등을 예측할 수 있으나 압 력의 영향을 예측할 수 없는 단점이 있다. 연소 모델 중 처 음 발표된 것은 Nachbar 및 Parks 에 의 한 모 델 (sandwic h columnar dif fusi o n flam e) , 죽 〔그립 3 . 37 의 (a) )로 서 추전제가 연료 및 산화제의 얇은 박편으로 이루어전 것으로 가정한 모델이다 .69) 고체와 기체 사이의 계면에 액상이 없는 것 으로 가정되었으며, 이 모델에 의해서는 압력이 연소속도에 주는 영향이 예측될 수 없었다. Summerfie l d 등은 그림 3.37(b) 와 같 은 입 상확산모델 (gran ular dif fus io n flam e) 모델을 발표하였는데 , 이는 산화제와 연료 성분이 열분해된 기체성분들의 연소반응으로 생성된 화염 에너지가 연소표면으로 전달된다고 가정하였다 .68) 이때 총괄적인 연소속도 ’는 확산 불꽃 에너지의 전달에 의해 또는 균일한 기체 반웅들에 의해 결정된다. 이 경우도 연소표면
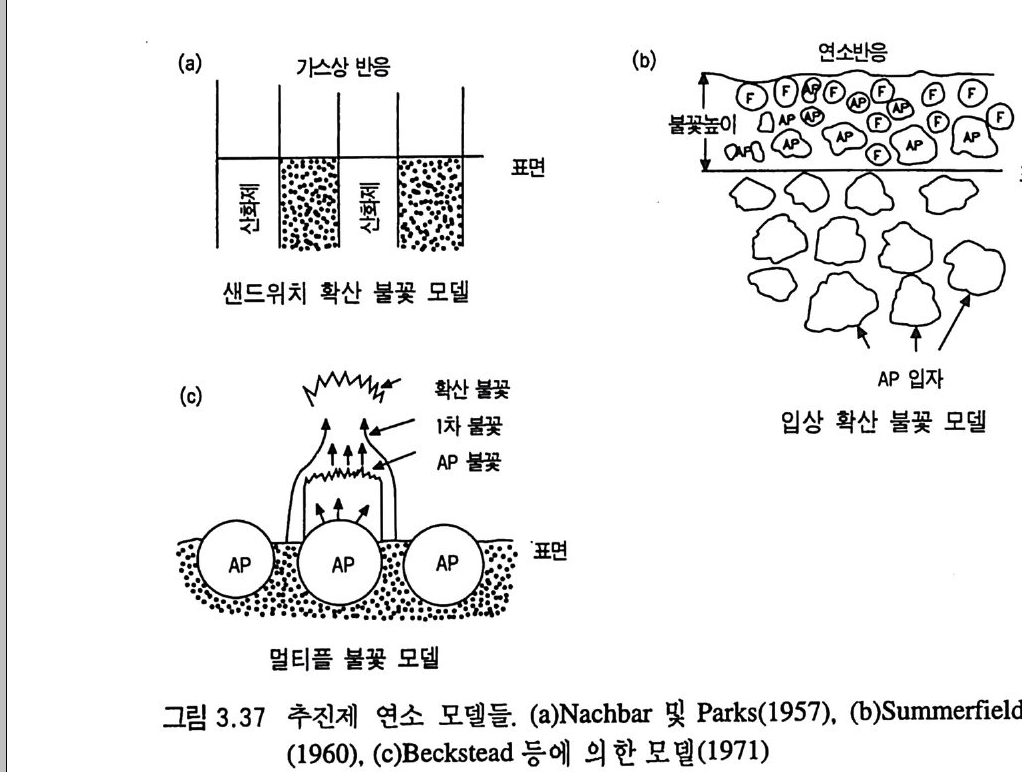 (a) 가스상반응 (b) 몽연소반응
(a) 가스상반응 (b) 몽연소반응
은 액상이 없는 것으로 가정하였으며 다음과 같은 연소속도 식이 유도되어진다. 죽, p /r=a+bp 21a( 실험상수 a, b 는 각각 균일한 기 체들의 반응속도 및 확산속도에 의해 결정된다). 이 이론은 lOMPa 이하에서는 비교적 잘 일치한다고 알려져 있으나 가정 조건이 너 무 많은 것이 단점이다. Beckste a d 등은 통계적인 방법에 의한 다중 불꽃 모델을 그림 3.37(c) 와 갇이 제안하였다 .70) 이 모델은 ® 열전도에 의한 바인 더와 산화제의 예열 과정이 있고, ® 바인더와 산화제는 홉열반 옹에 의해 열분해가 되며, ® 분해 생성물들은 응축상에서 발열 반응 형식의 반응이 일어나며, ® 기체 생성물들은 연소표면 위 의 기체상에서 반옹한다는 가정을 하고 있다. 이때 낮은 압력하
에서는 세 종류의 서로 다른 불꽃들을 고려하게 되는데 이는 일 차 불꽃, 죽 바인더와 산화제 분해 생성물 사이의 불꽃, 산화제 불꽃 및 일차 불꽃과 산화제 불꽃 사이의 확산 불꽃 등 세 종류 이다. 이 모델은 산화제 충전, 추전제 그레인의 초기 온도, 추전 제 그레인의 평균온도 및 불꽃 거리 등이 연소속도에 주는 영향 울 예측할 수 있다. Becks t ead 는 이 모델을 확장하여 복기 추전 제에도 적용하였고, 그후 추진제 연소시 연료와 산화제가 서로 다른 연소표면 온도를 갖고 있다고 가정한 이론을 발표하였다 .7 1) 그후 이와는 약간 다르지만 유사한 연소 모델들이 Glic k ,12> Condon,73) Renie 7 4) 등에 의해 발표되었다. 그들은 충전제 입자 들을 크기 에 따른 그룹들로 분류하여 프티 앙상블 (pe ti t ensemble) 이라는 접근 방식을 택하였다. Ki ng 역시 비슷한 접근 방식을 택하였으나 응축상과 확산 불꽃에서의 열발산을 고려한 것이 특 이한 점이었다 .75> S t rahle 에 의해서는 추진제로 대표되는 충전 배드 (pa cked bed) 를 통과하는 불꽃의 이 동을 계 산하는 통계 적 인 모델이 제 안되 었다. 76) 니트라민 추전제의 경우는 · 산화제 성분으로 과염소산암모늄 대 신 들어 있는 고체 성분인 RDX 혹은 HMX 의 니트라민 물질이 홉열에 의해 먼저 용융된다 .77) 그러나 RDX 와 HMX 의 용융점 (RDX : 200 °C, HMX : 280 °C) 은 가열속도에 따라 변화되므로 용 융될 때 발생하는 추진제 분해 가스의 축적에 의해 영향을 받게 된다. HMX 분해는 통상 추진제 표면 이하 0.02~1mm 정도에 서 시작된다. HMX 자체 분해반응은 홉열반응이지만 실제로는 50~100 cal/ g의 열량을 방출하는 발열반응이다. 570 °K 정도의 표면온도에서 기화가 일어나서 대략 0.2mm 두께인 피즈(fi zz) 영역 (1050~1750 °K) 에서 연소가 시작된다. 피즈 영역 다음 단계 로는 다크 영역 (dark zone) 의 연소로 이어지며, 다크 영역에서의
온도는 피즈 영역에서 상승했던 온도 수준으로 거의 일정하게 유 지된다. 마지막 연소는 다크 영역 꼭대기에서 일어나는 라미노우스 (lum i no us) 불꽃으로서 대 략 1650 ~2200 °K 정 도의 온도 범 위 에 있다. 이때 압력을 증가시키면 다크 영역 거리가 점차 짧아지게 되므로 다크 영역이 없어질 수도 있다. 만일 니트라민계 추진제 에 과염소산암모늄을 첨가시키면 다크 영역이 사라진다. 따라서 과염소산암모늄은 연소속도의 압력지수를 낮추는 작용을 한다는 점과, 라미노우스 영역으로부터 열전달을 다크 영역이 차단시키 는 작용을 한다는 점을 고려하면, 고압에서의 압력에 대한 연소 속도 변화는 다크 영역 때문이라고도 할 수 있다. 라미노우스 불 꽃 영역이 연소표면에 직접 붙어 있는 추전제는 저압에서의 연소 속도 민감도가 더 낮아지는 좋은 점이 있다. 그립 3.38 에 두 종 류의 추전제, 죽 과염소산암모늄이 포함된 추전제와 포함되지 않 은 추진제의 불꽃 구조가 나타나 있다 .78 , 79)
 파J벼fc g富可R g可젊 To fc벼J 可f젊OcTO
파J벼fc g富可R g可젊 To fc벼J 可f젊OcTO
복기 추전제 경우에는 연소표면에서 발생된 가스들은 산화제와 연료가 분자 형태로 서로 혼합되어 있는 상태이다. 라미노우스 불꽃은 연소표면으로부터 어느 정도 먼 위치에 존재하고 있으나 압력이 증가하면 그 거리가 좁혀진다. 복기 추전제의 연소 메커 니즘은 이미 언급한 니트라민 추전제와 정성적으로 매우 유사하 다. 복기 추진제의 메트릭스에 니트라민 화합물을 첨가하면 불꽃 온도가 상승하므로 비추력이 증가하게 된다. 그러나 니트라민이 첨가된 복기 추전제의 불꽃 구조는 매우 다르게 변한다. 또한 과 염소산암모늄을 첨가한 복기 추진제의 다크 영역은 없어지게 된 다. 추진제 연소 연구는 특히 미국의 Prin c eto n , Purdue, Penn. sta t e , Georgi a Tech, 프랑스의 ONERA 및 일본의 방위 청 제 3 연구실 등에서 활발히 연구되고 있다. 3·6·2 추전제의 연소속도 방정식 액체 추전기관에서는 연료와 산화제의 소비속도는 연소실로 주 입되는 연료 주입속도 및 산화제 주입속도와 일치한다. 고체 추 전제의 경우에는 추전제 그레인이 일단 점화된 후에는 그레인의 연소를 조절할 방법이 거의 없다. 고체 추전제의 소비속도는 추 진제 고유의 연소속도 특성과 추전제 그레인 설계에 의해 영향을 받게 된다. 추전제 그레인 설계는 추전제 표면과 연소실 압력의 함수이며, 추전제 고유의 연소속도는 추진제의 조성에 따라 결정 된다. 추전제 고유의 연소속도는 이미 언급한 바와 같이 바인더, 산화제, 산화제의 입도 분포, 고체 함량, 연소속도 촉매의 농도 에 의해 변화된다. 노즐을 통해 배출되는 연소가스의 속도 (md) 는 연소실 압력에 비례한다.
 md=Cv At Pc (1)
md=Cv At Pc (1)
연소실 내에서의 가스 발생 속도가 노즐로 배출되는 가스 방출 속도를 초과하지 않을 때, 죽 n 값이 1 보다 작은 경우에만 로켓 모터는 정상적으로 작동한다(그립 3.39). 로켓 모터가 작동하는 연소실 압력 범위에서는 가스 방출 속도는 연소실 압력에 비례하 며 그림 3.39 에서 점선으로 표시되어 있다. 노줄은 연소실 압력 이 적당히 유지될 수 있도록 설계되어 있다. 앞의 식 (4) 에 의해 가스의 생성은 노즐을 통해 방출되는 가스 의 양과 정확하게 균형이 맞춰져 있으며 연소실 내부는 일정 압 력으로 유지되게 된다. 연소실 압력이 순간적으로 상승하거나 하 강하는 경우, 추진제 압력지수가 l 이하인 추전제를 사용했으면 자동으로 보정되며, 압력지수가 l 이상인 추진제를 사용했으면 폭발 또는 소화되는 현상이 나타난다. 일반적으로는 압력지수가 0.8 이하의 추진제를 사용하면 모터 설계시 큰 문제점은 없다고 알려져 있다. 그러나 n 값이 증가할수록 연소실 압력 편차가 커 짐으로 인해 연소실 의벽을 강화해야 하고 연소실 무게가 무거워 지는 단점이 나타나므로 압력지수가 가능한 한 낮은 추진제를 사 용하는 것이 추진기관 모터 설계에 유리하다. 추진제의 연소속도는 Crawf or d 봄베 또는 스트랜드 버너 (str and burner) 라고 부르는 장치를 이 용하여 측정 한다. 이 스트 랜드 버너에는 연소실 내부의 압력을 조절하기 위한 질소가스 공 급 라인이 부착되어 있다. 또한 추진제 시편의 연소시에 연소시 간을 측정하는 타이머가 연결되어 있고 연소시에 발생되는 기체 압력 상승을 최대한 방지하기 위한 서지 (surge ) 탱크가 연결되어 있다. 스트랜드형 추전제 시편에는 접화선과 퓨즈(연소 start 및 s t o p)를 연결하여 추전제 스트랜드 시편의 연소시간을 측정한다 (그립 3 . 40) . SO)
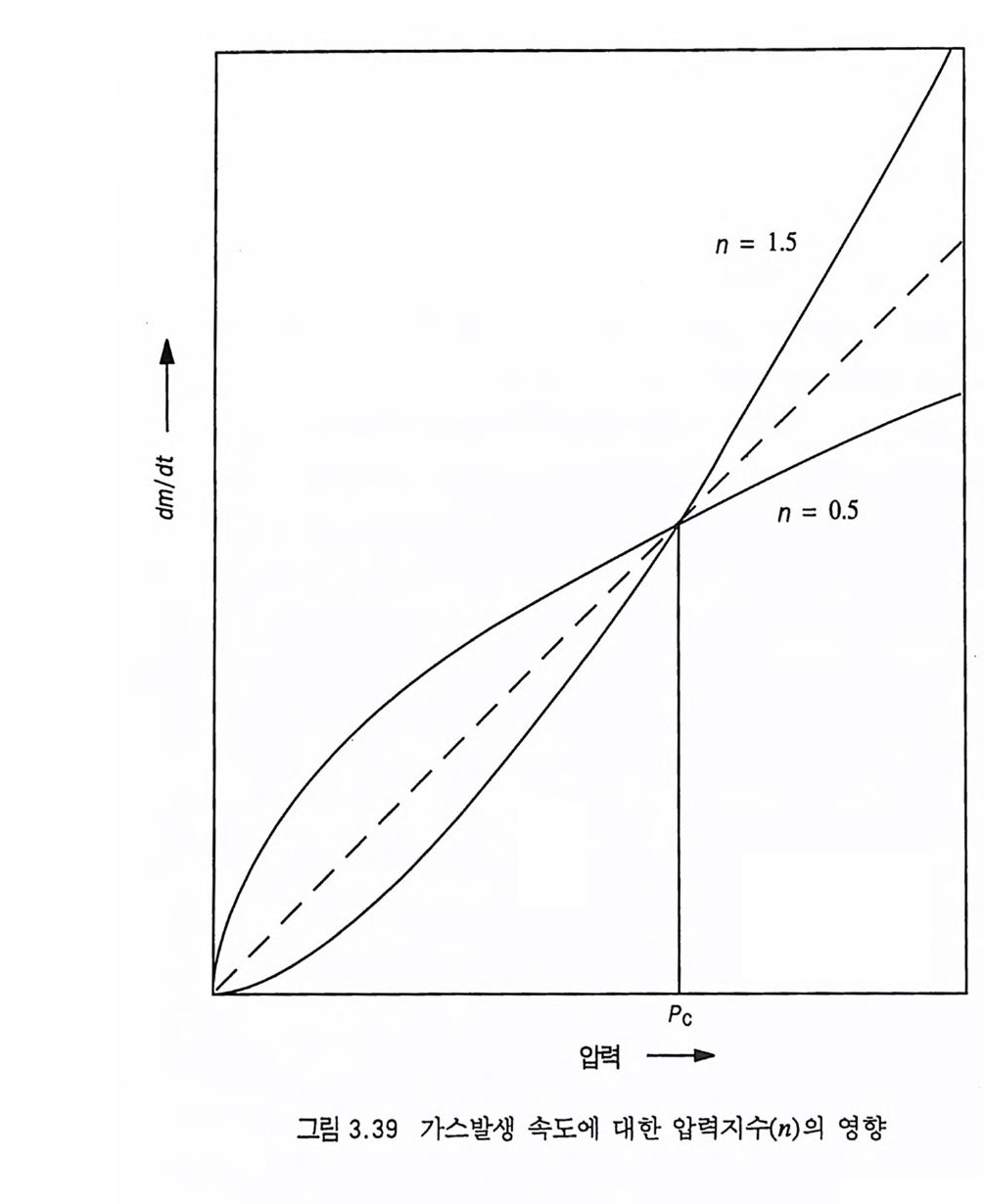 n = 1.5
n = 1.5
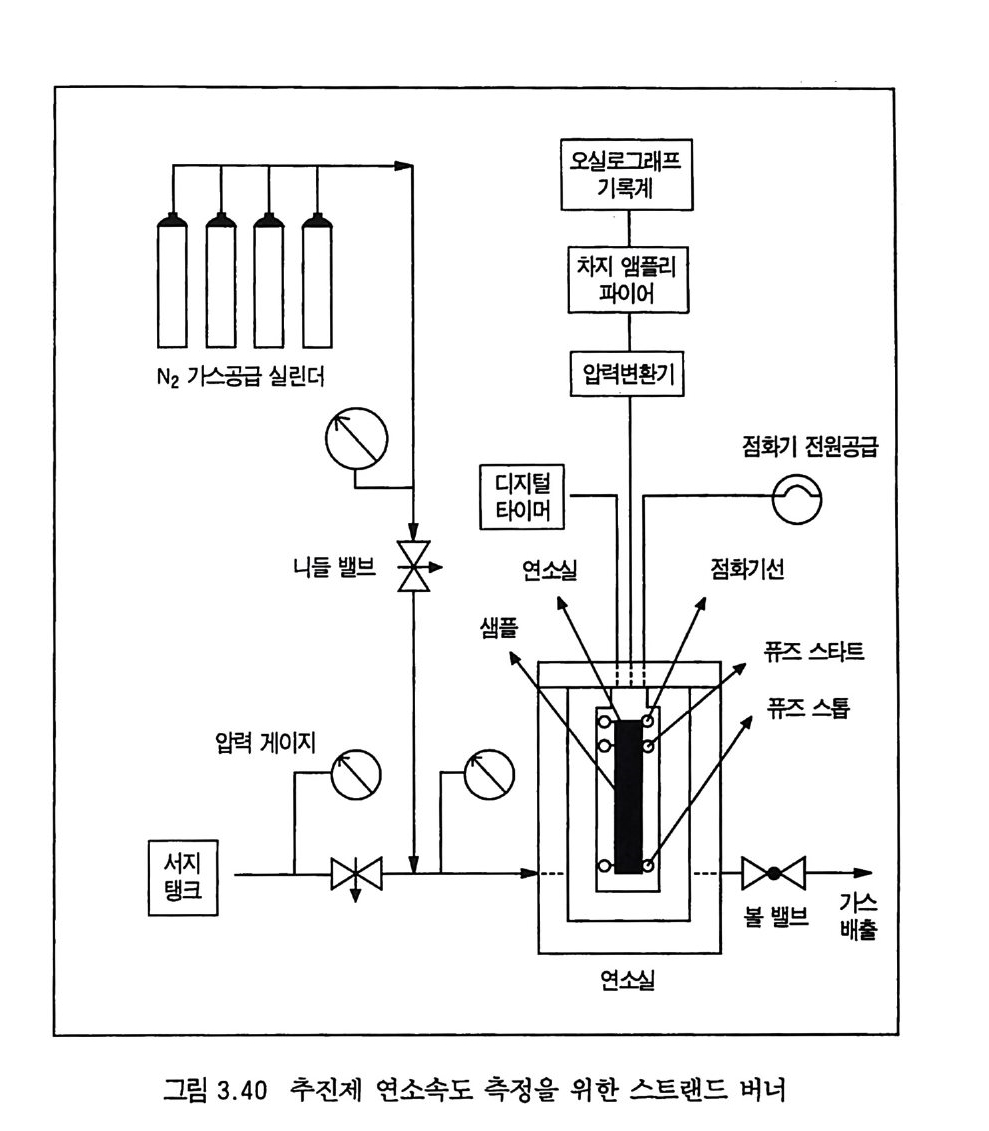 N2 가스공급실린더
N2 가스공급실린더
3.6.3 침식 연소 추진기관 모터 내에서 추진제가 연소되는 속도는, 연소표면과 평행으로 흐르는 고속 가스에 의해 영향을 받게 된다. 침식 연소 는 가스의 속도가 가장 높은 노즐 근처에서 제일 크며, 침식 현
상으로 인해 추전제 연소속도는 증가하게 된다. 추진제의 침식 상수는 c= r'/rc。 (r’ 은 침식이 있는 추진제 연소속도, ro 는 스트랜드 추진제 시편으로부터 얻어진 연소속도)와 같이 정의된다. 침식 연 소는 저연소속도 추전제, 특히 그레인의 내경이 작은 경우 연소 초기 에 심 하게 나타난다. 3.6.4 온도 민감도 추전제 연소속도는 추전제가 연소되기 전 추진제 자체의 온도 에 따라 영향을 받게 된다. 죽, 추진제 자체의 온도가 높은 상태 에서 연소될수록 더 빠른 속도로 연소하게 됨으로써 더 높은 연 소압력을 발생하게 된다. 한편 추력은 연소실 압력에 바례하므로 높은 온도에 있는 추전제를 연소시킬수록 더 높은 추력을 얻게 된다. 일반적으로 전술용 로켓이나 유도탄이 운용되는 작동 온도 범위는 -50~70°C 부근이므로 추전기관 모터는 작동온도 범위 중 가장 낮은 온도에서 최소한의 추력을 낼 수 있도록 설계되어 야 한다. 반대로 사막지대같이 대기 온도가 높은 곳에서 운용되 는 경우는 연소실 압력이 상당히 높아질 수 있는 경우를 대비하 여 설계해야 하므로 연소관의 두께가 두꺼워지게 되고 무거워지 므로 유도탄이나 로켓의 추진기관 성능에 악영향을 끼치게 된다. 예를 들어 (8) 식과 같이 표시되는 온도 민감도 7rk 값이 0.5 %/°F 정도인 추진제의 경우 150 °F 에서의 연소실 압력은 -50 °F 에서의 연소실 압력보다 2 배 정도로 높은 압력으로 작동하게 된다. 이 경우 탄도 조정과 같은 몇 가지 중요한 특성들이 추전제 연소속 도의 온도 민감도에 의해 큰 영향을 받게 된다. 또한 모터의 추 력 역시 온도 민감도에 의해 약간의 영향을 받게 된다 . 즉, 뜨거 운 추진제 그레인은 차가운 그레인보다 더 높은 에너지를 갖고
 있으므로 엔탈피 차이는 다음과 같이 표시된다.
있으므로 엔탈피 차이는 다음과 같이 표시된다.
 Pc=[-%강 ]占 (9)
Pc=[-%강 ]占 (9)
이와 같은 정의 (식)들에 의해 6p 는 추전제 연소속도(스트랜드) 데이터로부터 구해질 수 있으며, 'Ir k 는 모터의 연소시험에 의해 얻어지게 된다. 'Ir k 값을 구하기 위한 모터 연소시험 방법은 모터 의 제작 시험 비용이 너무 많이 소요되므로 일반적으로 추진제 개발 마지막 단계에서만 적용되고 있다. 식 (10) 의 'Irk 와 6 p의 관 계식으로부터 'Irk 를 스트랜드 데이터에 의해 구할 수 있으나 이 렇게 하여 계산에 의해 얻어전 'Irk 값들은 실제 모터 실험에 의해 얻어진 'Ir k 값들에 비해 높게 계산된다고 알려져 있다 .81) 온도 민감도는 매우 중요한 추전제의 특성임에도 불구하고 아 직까지 추전제 조성 원료 인자들과의 관련성에 관해 깊이 연구된 예가 거의 없으며, 단지 대부분의 추전제들의 6p 값들은 0.05~0. 5 %/°F 근처 에 있다고 알려 져 있다. 복기 추전제 의 6p 는 0.3 %/ OF 수준에 있는데 HMX 를 그 추진제에 첨가하면 6p 는 낮아지지 만 압력지수가 증가하여 결국 'Irk 값을 낮아지게 하는 효과를 얻 을 수 있다. 복기 추전제의 경우 납화합물들이 첨가되면 압력지 수가 0 (zero) 되는 범 위 (pla te a u) 혹은 마이 너 스 값을 갖는 범 위 (mesa) 가 나타날 수 있다. 이 때 플래 토 (pla te a u) 내 에 서 는 6p 와 T마 매우 작은 값들을 나타내지만 플래토 범위 밖에서는 아무 런 영향을 주지 못한다. HMX, AP, Al 등을 첨가시키면 납화 합물들의 효과를 상쇄시켜 정상적인 온도 민감도 특성을 나타내
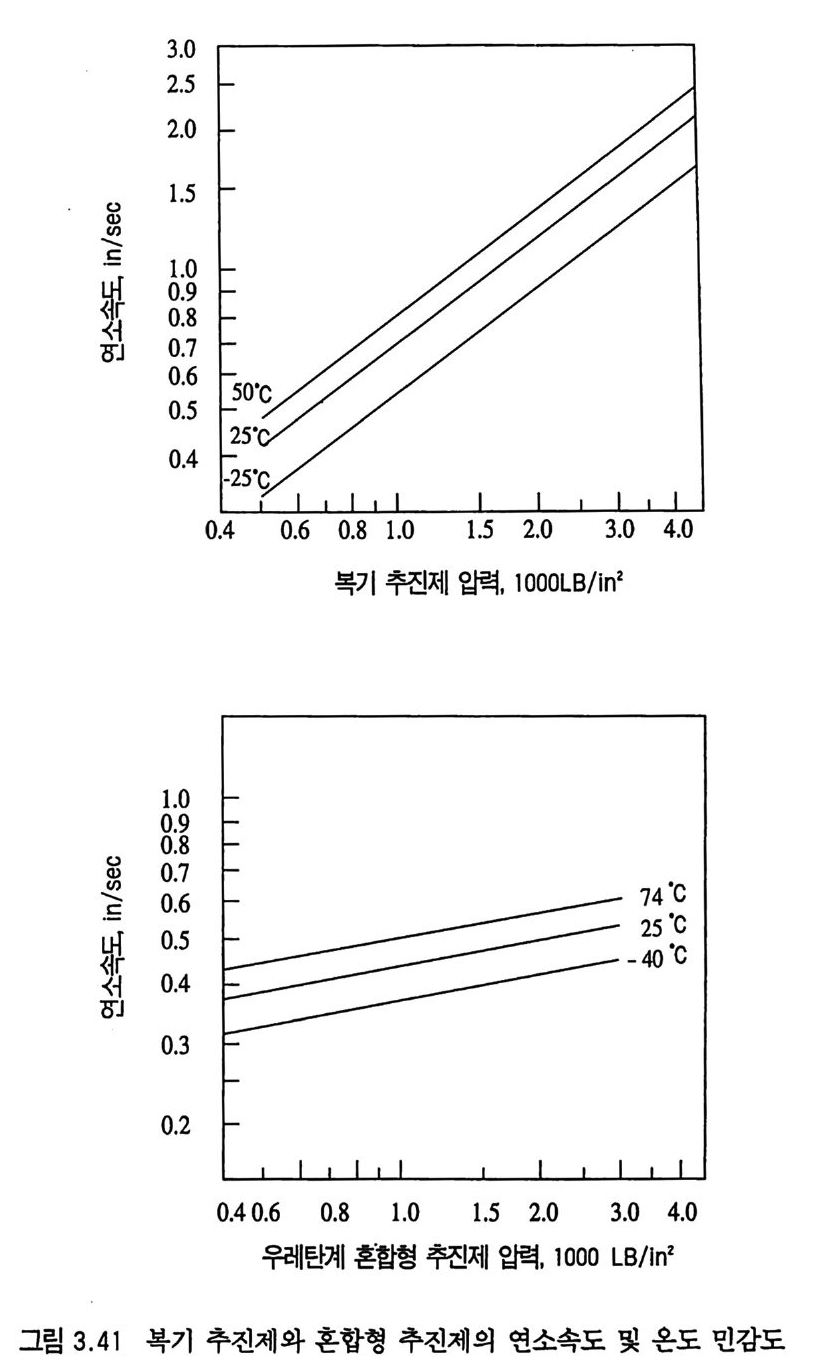 3.0
3.0
게 된다. HTPB- 니트라민 추진제의 경우 연소속도와 온도가 증가함에 따라 6 p가 낮아지는 경향이 있으며 6p 값은 0.06~0.2 %/°F 정도 를 나타낸다. 그러나 이 조성은 n 값이 높으므로 7f k 값 역시 높은 상태에 있으며, 일반적으로 니트라민으로서 RDX 를 사용한 추진 제가 HMX 를 사용한 추진제에 비해 온도 민감도가 약간 높은 것으로 알려져 있다. 과염소산암모늄을 포함하는 우레탄 추진제의 경우 6p 는 산화 제의 입도 분포도 넓이에 따라 낮아지는 것으로 알려져 있으나 산화철 (Fe2 이 촉매를 첨가하면 과염소산암모늄 입도 분포도의 효과를 감소시키는 경향이 있다. 산화철을 사용한 고연소속도 추 진제의 경우에는 6p 가 0.3 %/°F, 7rk 는 0.1 %/°F 정도로 얻어졌 다고 보고되었다. 보통 연소속도를 증가시키는 첨가제(촉매)를 사용하면 6 p를 낮아지게 하는 영향이 있다고 하지만, 이때 7fk 값 이 동시에 낮아지지는 않는다고 보고되었다. 또한 과염소산암모 늄 일부를 알루미늄으로 대체하여 첨가시키면 6 p를 증가시키는 경향이 있다고 보고되었다. 그림 3.41 에는 복기 추전제와 혼합형 추전제에 대한 연소속도 온도 민감도 효과가 나타나 있다. [참고문헌〕 1) T. Urbanski, Chemi stry and Technology of Exp lo siv e s, Vol. III, Eng li sh Ed., Perga mon Press, New York (1985) . 2) Y. M. Ti m nat, Advanced Chemi ca l Rocket Prop ul sio n , Academi c Press Inc. (1987) . 3) A. Davenas, Solid Rocket Prop ul sio n Technology , Perga mon Press, New York. (1993) .
4) V. Lind ner, Exp lo siv e s-Prop el lants , Theory and Practi ce, V. S. Armament Command, Dover N. J. (19 78) . 5) R. F. Gould, A vances in Chemi st r y Serie s , P rop el lants Manufa c tu r e, Hazards, and Testin g , Amer. Chem. Soc. (1959) . 6) A. E. Oberth , CPIA Public a ti on No. 469, Pr inc iple s of Solid Prope l lant Develop m ent (1987) . 7) 민경주, 홍명표 , 노만균, 고분자과학과 기술, 1(4), 211 (1990). 8) MATRA Defe n se DAST/PC 28/no 1917, Rocket Moto r Tech-nology , MATRA Defe n se Comp a ny, 1993. 9) K. Klag e r and J. M. Wrig h ts o n, Recent Advances in Solid Prop el- !an t Bi n der Chemi stry, Perga mon Press Inc. (1967) . 10) R. T. Holzmanu, Chemi ca l Rockets and Flame and Exp lo siv e s Technology , Marcel Dekker Ltd. (1969) . 11) S. F. Sarner, Prop el lant Chemi stry, Rein h old, New York (1966) . 12) B. Sie g e l and L. Schie l er, Energe ti cs of Prop el lant Chemi stry, Wi ley , New York (19 64) . 13) K. E. Rumbel, Poly ( vin y l chlor ide) Plastis o l Prop el lants , Advances in Chemi st r y Serie s (No. 88) , ACS, Washin g ton D. C., p p. 36-66 (1969) . 14) W. F. Arendale, Chemi stry of Prop el lants on Chemi ca lly Crosslin - ked Bin d ers, Advances in Chemi st r y Serie s (No. 88) , ACS, Washin - gton D. C., p p. 67-83 (1969) . 15) J. H . Baker, et al, ]. Chem. Soc., 713 (1947) ; 9, 19, 28 (1949) . 16) R. J. Farris, CPIA Publica ti on 94 U, 1, 105 (1965) ; Trans. Soc. Rheol. 12, 315 (1968) . 17) K. C. Frisc h, S. L. Reege n, and W. V. Floutz , J Polym e r Sc i., 5 A, 35 (1967) . 18) N. G. Gay lo rd, Polye t h e rs, Part I II, I nte r sci en ce, New York (1962) . 19) 홍명표, 노만균, 대 한화학회 지 , 27, 58 (1983) . 20) 홍명표, 최영보, 노만균, 문탁진, Polym e r(Korea), 11(1) 49 (1987) .
21) E. J. Mastr o li a and K. Klag e r, Solid Prop el l ants Based on Poyb u ta ,d ie n e Bi nd ers, A dvances in Chemi st r y Serie s (No. 88) , ACS, Washin g ton D. C., p p. 122-164 (1969) . 22) Americ a n Cy a nami de Co., The Chemi stry of Acry lo nit rile (1959) . 23) CPIA, IRCPG Solid Prop el lant Behavio r Manual, Feb. (1968) . 24) 황갑성 , 노만균, 윤여 길, 학술논문집 (국방과학연구소) , Vol. I (2) , 346 (1981) . 25) 이범재, 노만균, 학술논문집 (국방과학연구소), Vol. I (2), 397 (1981) . 26) M. B. Berenbaum, U. S. Pate n t 3,235,589 (1966) . 27) U. S. Naval Ordnance Test Sta ti on , Mi nn esota Mi ni n g and Manufa c tu rin g Co., Fin a l Rep o rt, Contr a ct N123 (60530) 50116A (Ju ne 1967) . 28) C. A. Uraneck, U. S. Pate n t 3,135,716 (1964) . 29) C. Gotz m er and N. Seid e n, JA NNAF Prop u lsio n Meeti ng , Vol. II , CPIA Publica ti on 300, pp. 541-55 3 (1979) . 30) M. Houser and C. Gotz m er, Prop u lsio n Revie w , NWC TP 6200 -0 9, 31 (1981) . 31) D. E. Joh nson and A. J. DiM i lo, Aeroje t - G enal Corp. , Rept . AFRPL-TR-66-40 (Feb. 1966) . 32) E. J. Mastr o li a, K. W. Bil ls, and C. B. Frost, Aeroje t - G eneral Corp ., Re pt. BSD- TR -66-28, 1, 162-166 (1966) . 33) 노만균, 임 유진, 한국항공우주학회 지 , 22 (2) , 114 (1994) . 34) I. B. Mi sh ra, JA NNAF Prop u lsio n Meeti ng , CPIA No. 377, pp. 25 -36 (1983) . 35) W. Eng el and P. Charbit , ]. Therm. Anal., 13, 275 (1978) . 36) J. S. Ing ma n, G. J. Kearley and S. F. A. Kett le, ]. Chem. Soc., Faradary Trans., 78, 1817 (1982) . ]. Mol. Str u ctu r e, 79, 319 (1982) . ]. Phy. Chem., 86, 4007 (1982) . 37) T. L. Bog gs , The Thermal Behavio r of RDX and HMX . In Kuo and Summerf ield , pp. 121-175 (1984) .
38) R. A. Fif er, Chemi stry of Ni tra te Este r s and Ni tra mi ne Prope l- !ants . In Kuo and Summerfie ld , pp. 177-23 7 (1984) . 39) K. E. Rumbel, J. H. Grover, M. Cohen, and A. C. Scurlock, Army -N avy -A i r Force Soli d Prop el lant Meeti ng , 9th . (1953) . 40) R. J. Farris, Trans. Soc. Rheol., 12, 281 (1968) . 41) B. J. Alley and J. W. H. Dy k es, CPIA Publica ti on No. 168, Vol. II, 223 (1968) . 42) H. C. Brin K man, ]. Chem. Phys . 20, 571 (1952) . 43) A. C. Scurlock, K. E. Rumbel, and M. L. Ric e, U. S. Pate n t 3,107, 186 (1963) . 44) R. J. Farris, CPIA Publi cat i on , 61U, Vol. I , 291 (1964) . 45) R. J. Farris, CPIA Publi cat i on , 94U, Vol. I , 105 (1965) . 46) A. E. Oberth , Rubber Chem. Technol, 40, 1337 (1967) . 47) A. E. Oberth and R. S. Bruenner, Trans. Soc. Rheol. 9 (2), 165 (1965) . 48) F. N. Kelly, Soli d Prop el lant Mechanic a l Prop e rtie s Testi ng , Fail ur e Crite r ia , and Ag ing , Advances in Chemi st r y Serie s , (No. 88) , ACS, Washin g ton D. C., pp. 188-24 3 (1969) . 49) C. 0. Parker and T. E. Ste v ens, Technic a l Re po rt No. S-205, Redsto n e Research Laborato r ie s , Rohm and Hass Co., (1969) . 50) H. C. Allen, M. T. Cucksee, ana H. S. Wi lliam s, CPIA Publica it on No. 196, Vol. I , p. 693 (1970) . 51) B. M. Broli ne , Ac yla zir idin e Reacti vit y in the Lin e r and Prope l- !ant Envir o nments , Boein g Aerospa c e Co., P.O. Box 3999, Seatt le, WA 98124. 52) H. C. Dehm and E. C. Braak, CPIA Publica ti on No. 141, Vol. II , p. 65 (1967) . 53) G. A. Fluke, Comp os it e Solid Prope l lant Processin g Techniq u es, Advances in Chemi st r y Serie s (No. 88) , ACS, Washin g ton D.C., pp. 165-186 (1969) . 54) P. R. Dy k str a , Thio k ol Astr o naut, 3, No. 2, 5 (1961) .
55) F. A. Warren, Rocket Prop el lant, Rein h old, New York, 1958. 56) J. C. Chap m an, Thio k ol Astr o naut, 1, No.1 , 3 (19 59) . 57) Chemi ca l & Eng ine ers News, 42, 50 (1964) . 58) Thio k ol Astr o naut, 3, No.1 , 20 (1961) . 59) CPIA Publica ti on SPIA/PPB (1956, 1957) . 60) ICRPG Solid Prop el lant Mechanic a l Behavio r Manual, CPIA, Joh ns Hop k in s Ap plied Phy si c s Laborato r y, Sil ve r Sp ring , Md. (1963) . 61) M. De Frie s and R. H. Joh ns, At lan ti c Research Corp. Rep. RTD-TDR-63-1106 (1963) . 62) J. D. High to w er and F. W. Pric e, Combustio n of Ammoniu m Perchlorate , 11th . Sy m p . (Int. ) an Combusti on , p. 463, W illiam s and W ilki n s , Baltim ore (1967) . 63) R. H. W. Waesche and J. Wenog rad , The eff ects of Pressure and Addit ives on the Ki ne ti cs of Decomp os it ion of AP, UARL Rep. WSCI-67-8 ( 1967) . 64) R. Frie d man and J. B. Levy , Research on Soli d P rop el lant Combus- tion , Fin a l TR AF 49 (638) , 913 (1961) . 65) C. Guir a o and F. A. Wi lliam s, A Model For AP Defl agr a ti on Betw e en 20 and 100 atm , A/AA ]. 9, 1345 (1971) . 66) N. C. Cohen, R. W. Flemi ng , and R. L. Derr, Role of Bi nd ers in Solid Prop el lant Combustio n , AIAA J. 12, 212 (1974) . 67) W . E. Joh nson and W. Nachbar, Defla gra ti on Li m i ts of a Mono- Prope l lant wi th Ap pli ca ti on to Ammoniu m Perchlorate , 8th . Sy m p . (Int. ) on Combusti on , p. 678 W illiam s and W ilki n s , Baltim ore (1962) . 68) M. Summerfie ld , G. S. Suth e rland, M. J. Webb, H. J. Tbak, and K. P. Hall, Prog re ss in Astr o nauti cs and Rocketr y, Vol. I , p. 141, Academi c Press, New York (1960) . 69) W. Nachbar and T. M. Parks, A Sandwi tch Burner Model for the Comp os it e Soli d P rop el lant, LMSO 2191, AFOSR TN DD 132 -417
(1957) . 70) M. W. Beckste a d, R. L. Derr, and C. F. Pric e , The Combustio n of Soli d M onop ro p el lants and Comp os it e P rop el lants , 13th . Sy m p . (Int. ) on Combusti on , p. 1049, The Combusti on Insti tut e , Pit tsb urgh (1971) . 71) M. W. Beckste a d, AIAA ]. 18, 980 (1980) . 72) R. L. Gl ick , On sta t i sti c a l analys i s of comp os it e soli d pro p el lant combusti on . AIAA ]. 12, 384 (1974) . 73) R. L. Glic k and J. A. Condon, Sta t i st i ca l analys i s of monodis p e r se solid pro p el lant combusti on : ste a dy sta t e . Proc. 13th . JA NNAF Combustio n Meeti ng ; CPIA Puhl. 281, Vol. II , 313 (1976) . 7754)) MJ. . P K. .R Kenii ne g , , JA. AIA. AC oPndapone ,r a8n0d — R1.1 2 O6s b(o19rn8,0 )A . IAA ]., 17, 877 (1979) . 76) W. C. Str a hle, AIAA ]., 16, 843 (1978) . 77) T. L. Bog gs. Thermal Behavio r of RDX and HMX, Chap ter 3 of Fundamenta l s of Prop el lant Combusti on Publi sh ed by AIAA, New York, N. Y. (1984) . 78) T. L. Bog gs, Fi na l Repo r t NWC TP 5514, Jul y 1973, Naval Weap o ns Cente r , Chin a Lake, Calif ., U.S.A. 79) T. L. Bog gs, R. L. Derr, and M. W. Beckste a d, AIAA ]. Vol. 8, 370 (1970) . 80) 임유진, 노만균, 이승무, 화학공학(한국화학공학회지), 22, 83 (1984) . 81) N. S. Cohem and D. A. Flanig a n, CPIA Publica ti on No. 390, Vol. 3, 243 (1984) .
제 4 장 니트라민계 원료 이미 언급한 니트로글리세린과 니트로셀룰로오스는 다시 설명 할 필요 없이 추전제, 화약류에서 가장 혼하게 사용되어 온 원료 둘이다. 그러나 1940 년대의 제 2 차 세계대전 이후 사용되기 시작 한 RDX 를 시작으로 RDX 와 유사한 니트라민계 원료들이 그 에 너지가 높은 장접으로 인해 큰 관심을 끌기 시작했으며, 1960 년 대에는 현재 적용되고 있는 고에너지 물질로서는 가장 에너지가 높고 가장 밀도가 높은 HMX 가 등장했다. RDX 및 HMX 는 추 진제나 화약의 일부 성분으로 니트로글리세린, 니트로셀룰로오스 와 함께 혹은 다론 폭발물 원료들과 혼합되어 사용되기도 하며 단독으로 바인더와 함께 혼합하여 사용되기도 하였다. RDX, HMX 이의에 많은 니트라민계 원료들이 추진제 원료로서 연구 되고 있으며 적용시의 탄도적 성능, 기계적 성질, 물리적 성질, 제조 공정성, 안정성 및 원료 가격에 이르기까지 각종 시험을 거 치고 있다. 많은 니트라민계 화합물들 중에는 위와 같은 특성 중 에서 몇 가지를 만족하더라도 나머지 성능들을 충족시키지 못하
는 경우가 많기 때문에 현재 가장 많이 쓰이는 원료는 RDX 와 HMX 두 종류이다. RDX 에 비해 HMX 는 각종 성능(내탄도적 성능, 폭발력)면에서 훨씬 우수한 것으로 알려져 있으나 HMX 의 높은 가격 및 HMX 제조공장 유무 등으로 인해 국가별, 회사별 로 그 선호도가 다른 것으로 알려져 있다. 1,2,3) 4.l 니트라민의 화학적 성질 니트라민의 화학 구조를 살펴보면, 니트라민 계열 화합물은 질 소 원자에 니트로기 (N02) 가 결합된 물질들이다. 학술적으로는 모든 니트라민 화합물들은 NH2N02 라는 무기물질인 니트라민의 유도체로서 분류된다. 만일 NH2N02 중 한 개의 수소 원자가 알킬 (alky l) 또는 아릴 (aryl ) 그룹으로 치 환되 면 1 차 니 트라민 (pri m ary nit ra mi ne ) , 두 개 의 수소 원자가 치 환되 면 2 차 니 트라 민 (secondary nit ra mi ne ) 으로 부론다.
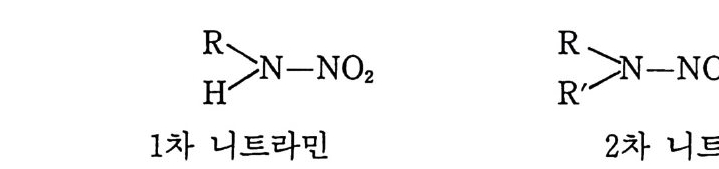 HR ,,-)N — N02 RR..'.. ,..._)_N — N02
HR ,,-)N — N02 RR..'.. ,..._)_N — N02
니트라민 유도체 중 R 그룹 대신 아실 (ac y l) 기 또는 술포닐 (sulph o ny l) 기 가 치 환되 면 니 트라마이 드 (nit ra mi de ) 라고 부른다. 니트라민의 구조에서 니트로기의 질소 원자와 아민기의 질소 원자가 결합되어 있다는 것은 니트라민을 수소로 환원시키면 히 드라진 (h y draz i ne) 이 생성되는 것으로 증명된다. 이때 2 차 니트 라민 역시 해당하는 히드라전 유도체를 생성하게 된다. 1 차 니트 라민계 화합물들은 다음과 같은 이성질체를 갖고 있다.
 R— N\。- 2H R ― NNI O一 OH
R— N\。- 2H R ― NNI O一 OH
 C6H5NN| O— OH
C6H5NN| O— OH
이 니트로소히드록실아민 역시 환원되면 해당되는 히드라전 유 도체를 생성한다. 그러나 1 차 니트라민의 물리적 성질은 니트로 소히드록실아민과 서로 다르며, 예를 들어 페닐 니트라민은 페닐 니트로소히드록실아민과 그 성질이 전혀 다르다.
1 차 니트라민 속에 N-N 결합이 존재한다는 것은 이 화합물들 이 디아조 (d i azo) 화합물의 산화반응에 의해 생성된다는 사실로 부터 확인된다. 또 다른 증거로서는 니트라민 화합물이 니트라이 트 (n it r it e) 기가 아닌 니트로기를 포함하고 있으며, R-N-ONO 의 구조를 갖고 있지 않음으로 인해 니트로우스 에스테르는 알칼리 에 매우 불안정하나 니트라민은 알칼리에 비교적 안정한 성질을 지니고 있는 것으로부터 증명된다. 이상과 같이 화학적 성질에 근거하여 증명된 니트라민 구조는 X-ray 분석에 의해서도 확인되었다. 특히 Whit mo re, Costa i n e 등은 니트라민이 다음과 같은 평면구조(p lanar) 를 갖고 있으며, 결합 각도 및 원자간 결합 길이는 그림 4.1 과 갇다고 보고하였 다 .4,5) 물, 알코올 등에 녹인 니트라민은 넓은 자의선 흡수띠 (U.V. absor pti on) 를 나타내며, 최대파장은 225~240 µ에 있다. Jon es, Thorn 등은 1 차 니트라민인 에틸렌 디니트라민(그립 4.2) 및 2
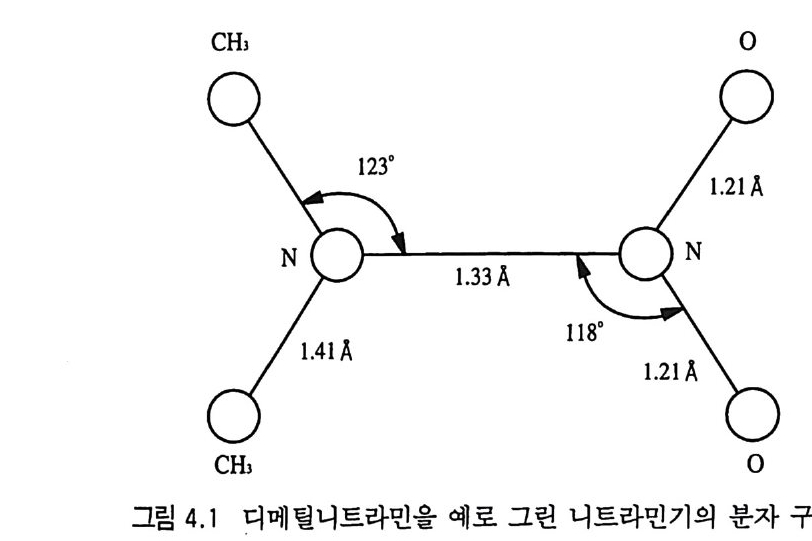 CH1 。
CH1 。
차 니 트라민 2,5- 디 니트로 -2,5- 디 아자핵 산 (그림 4 . 3) 의 흡수 커브 롤 보고했다 .6) J ones 와 Thorn 은 60 여 종류의 니트라민 화합물 들의 U.V. 흡수 스펙트럼을 조사하여 니트라민의 홉광 (ex ti nc tion ) 계수에 관한 홍미로운 법칙을 발견하였다. 이 법칙에 의하 면 1 차 니트라민기를 포함하는 화합물들은 최대 홉수 커브 상에 서 €max 7000 정도의 값을 나타내며 만일 1 차 니트라민기의 숫자 가 한 분자 내에 n 개이면 €max = 7000 n 으로 n 에 비례하여 중가 한다. 이 실험식은 n 이 1~3 의 범위에서 잘 일치한다. 2 차 니트 라민의 경우는 €max = 5500 이며 2 차 니트라민기 숫자가 분자 내 에 n 개이면 cmax=5500 n 의 값을 갖게 되며 n 이 1~4 의 범위에 서 유효한 것으로 보고되었다. 만일 분자 내에 1 차 및 2 차 니트 라민기를 모두 포함하고 있을 경우 cmax 는 각각의 니트라민기 숫 자에 7000 및 5500 을 곱한 값의 합계가 된다. 니트라민의 I.R 스 펙트럼은 일반적인 니트로 화합물과 동일한 주파수를 나타내는 것으로 알려져 있다. Bellam y에 의하면 니트라민의 니트로기는 일 반적으로 비대칭 1587~1530 cm-1, sym metr i c 1292~1260 cm-1 의
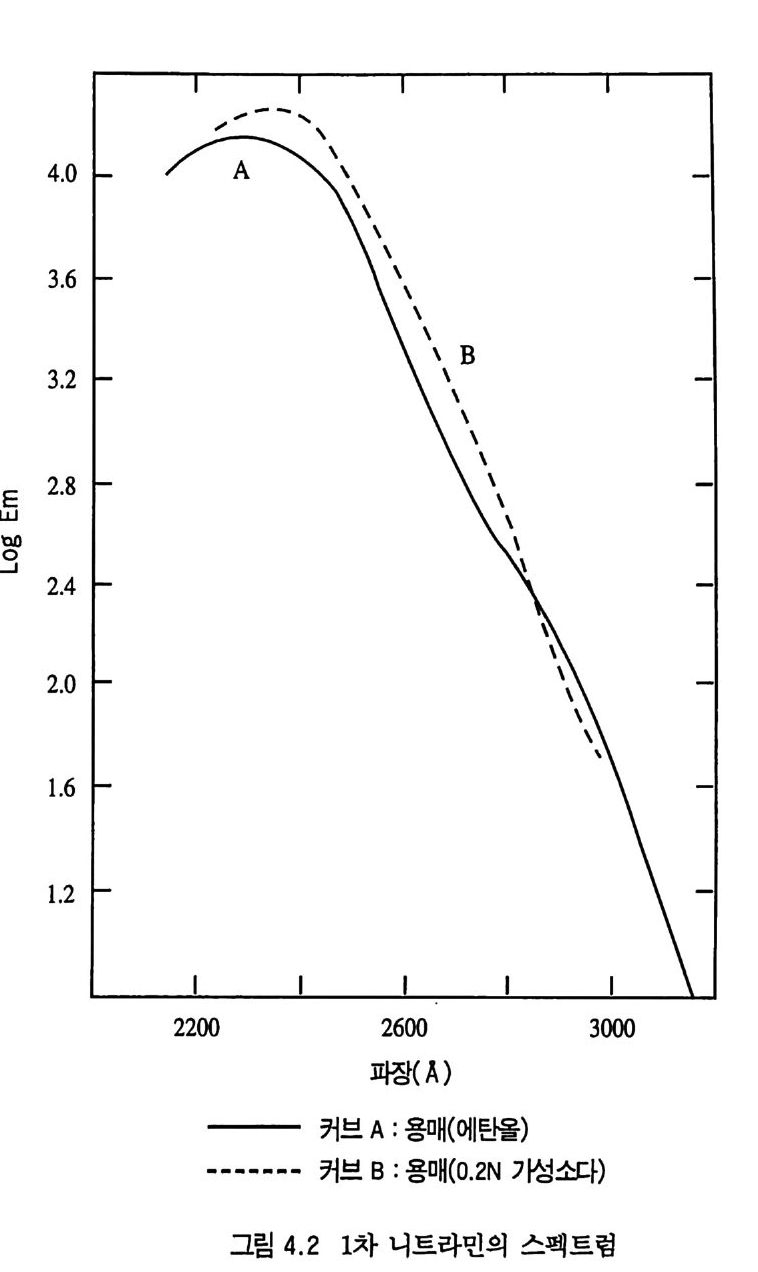 / / --`` `\
/ / --`` `\
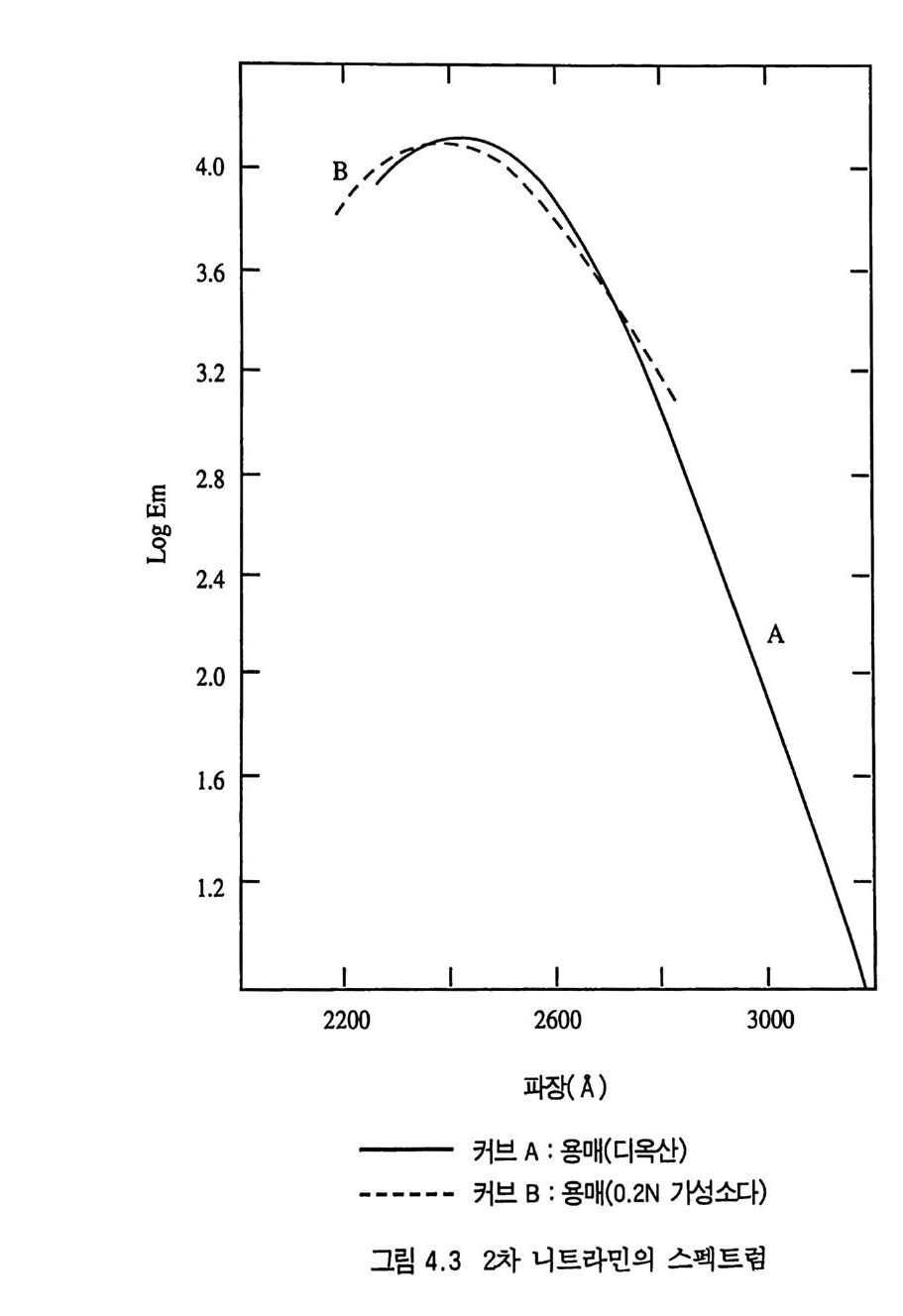 4.0
4.0
진동 주파수를 갖고 있다고 보고하였다. 7,8,9) 그러 나 Lieb er 및 Kumler 에 의 하면 니트로구아니 던의 경 우는 비대칭 주파수가 이보다 훨씬 높은 1655~1620cm-1 의 범위에 있 다고 보고하였다. 1 차 니트라민은 알칼리와 반응하여 영 (sal t)을 생성하는 산성을 나타내 며 니트라마이 드 (nit ra mi de ) 는 카르복실산보다 더 큰 산성 이 다. 예 를 들면 니 트로우레 탄은 개 미 산 (for mi c aci d) 보다 더 강 한 산성을 나타내고 있다. 일반적으로 1 차 아민(p r i mar y am i ne) 은 벤젠 용매 속에서 암모 니아와 반응하여 암모늄염을 생성한다. 따라서 Han t zsch 는 1 차 및 2 차 니트로파라핀처럼 1 차 니트라민 (1) 역시 유사산{p seudo a ci d) 이고 토오토메르(t au t omer ic) 산 (2) 형태로 반응하는 것으로 가정하였다 .10)
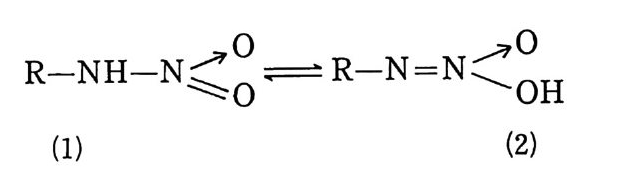 R-NH— N`~ {~o0 ~ R— N= N \/~°OH
R-NH— N`~ {~o0 ~ R— N= N \/~°OH
이러한 가정은 일반적으로 인정되고 있지만 실험적으로는 많은 증거가 나타나 있지 않다. Euler 는 암모늄염 생성 속도를 측정 했을 때 Han t zsch 가 믿었던 것처럼 속도가 느리지 않은 사실로 부터 토오토메르 현상에 대해 의심을 표현하였다. 11) 사실상 디메 틸니트라민의 이성질체인 0 ―알킬 유도체, 죽 O- 메틸-메틸니트 라민 (3) 이 얻어질 때까지는 니트라민이 산 형태로 존재한다는 것 을 확인할 더 이상의 증거가 없었다. 12)
 CH3-N=N _' ,,,;,OO CH3 (3)
CH3-N=N _' ,,,;,OO CH3 (3)
이 화합물 (3) 은 40 % 황산에 의해 가수분해되어 두 분자의 알코올이 생성된다.
CH 나 N = N< 。 一 N20 + 2CH30H 노 -O--H-1! 「iH~: --ocH3
 니트라민의 특칭 중의 하나는 니트라민이 황산에 의해 쉽게 분
니트라민의 특칭 중의 하나는 니트라민이 황산에 의해 쉽게 분
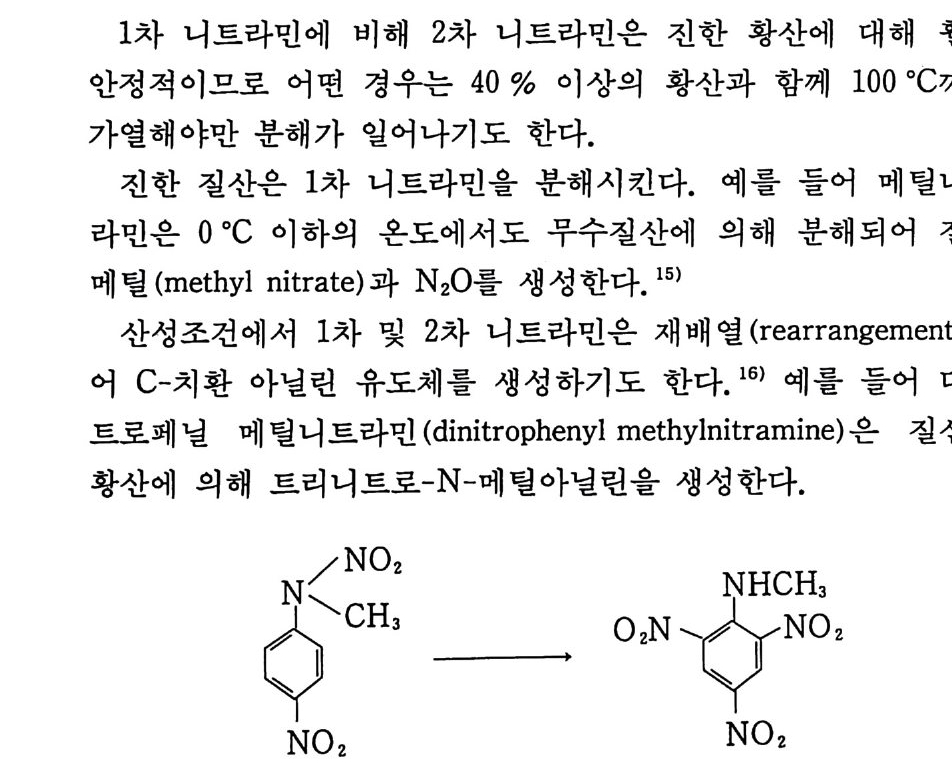 1 차 니트라민에 비해 2 차 니트라민은 전한 황산에 대해 훨씬
1 차 니트라민에 비해 2 차 니트라민은 전한 황산에 대해 훨씬
Hu g hes 와 In g old 는 이러한 재배열을 다음과 같은 방법으로 설명하였다 .17)
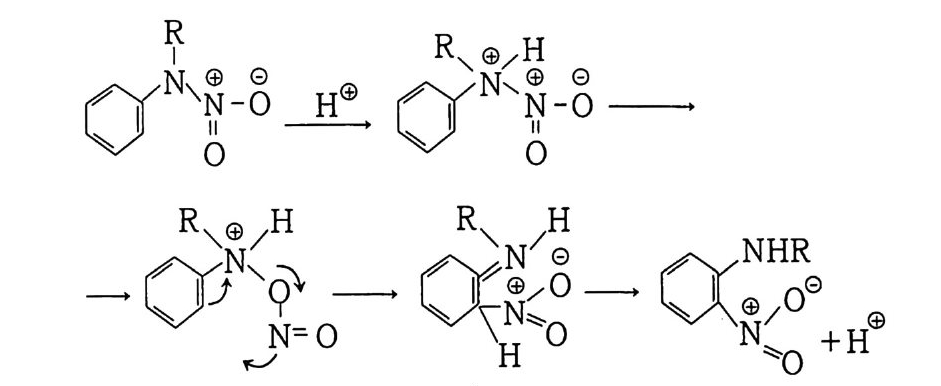 R R\@/H
R R\@/H
이와 같은 재배열 반응은 데트릴(t e t r y l) 을 제조할 때 매우 중 요하다. 대부분의 니트라민은 (aroma tic이든 a lip ha ti c 이든) 페놀과 함께 가열하면 탈니트로화 반응이 일어나며 황산이 존재할 때 반 응은 더 잘 진행된다. 예를 들어 테트릴은 다음과 같이 반응한다.
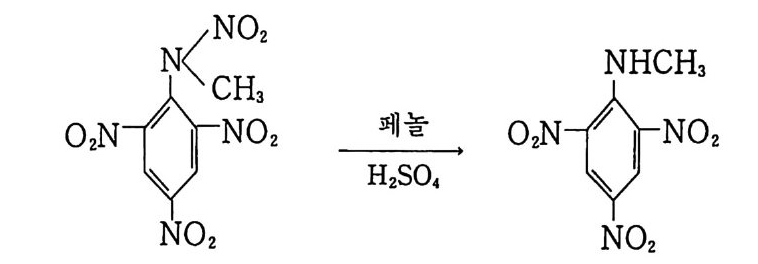 NHCH3
NHCH3
진한 황산에 용해된 니트라민은 디페닐아민과 반응하여 푸른 색깔을 나타낸다. 1 차 니트라민과 황산이 반응하면 질산을 분리 시킴으로써 분해반응이 계속된다. 대부분의 니트라민들은 알칼리 에 비교적 안정하여, 예를 들면 1 차 니트라민은 뜨거운 20 % KOH 용액 속에서도 분해되지 않는다 .13) 한편, Barrott , Gi llibr and 등에 의하면 대부분의 1 차 니트라민은 가성소다 0.8 ~8% 용액으로 95°C 에서 처리하면 다음과 같이 분해된다고 보
고하였다. 18,19)
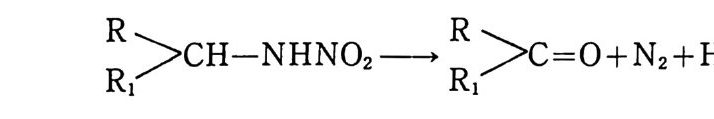 RRI >CH ― NHN02 一 R~I >c=O+N2+H20
RRI >CH ― NHN02 一 R~I >c=O+N2+H20
이와 같은 반응에서 필요한 알칼리의 농도는 R 및 R' 기의 특 성에 따라 변한다. 이 기가 더 천전자성일수록 니트라민은 더 산 성이므로 분해반응은 쉽게 일어나게 된다. 2 차 니트라민은 가성 소다 수용액에 의해 분해될 수 있으나 니트라민 종류에 따라 가 성소다 농도를 적절히 변화시켜야 분해반응이 일어날 수 있다. Van Erp 등은 다음과 같은 메커니즘에 의해 분해반응이 일어남 을 발견하였다 .20)
 RN -/..N.......0cH22 • R1 —~ RH2— oNj = CH-R'+HN03
RN -/..N.......0cH22 • R1 —~ RH2— oNj = CH-R'+HN03
니트라민을 환원시키면 반응물질에 따라 여러 종류의 다른 물 질들이 생성된다. 강력한 환원반응에 의해서는 N-N 결합이 파 괴되어 아민과 암모니아가 생성되며, 약한 환원제에 의해서는 히 드라전과 감은 여러 가지 화합물들이 생성된다.
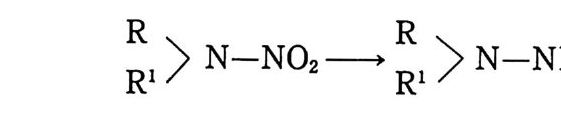 RR1 >N -N02 一 RR1 >N — NH 2
RR1 >N -N02 一 RR1 >N — NH 2
이와 같은 히드라진으로의 환원반응은 정량적으로 진행되므로 니트라민 분석에도 이용될 수 있다. Cop e , Barab 등에 의하면 다음과 같은 환원반응이 FeCI2 + HCI 에 의해 진행되므로 화학 분석에 적용될 수 있음울 보여주었다 .21)
 >N ― N02 一 >N H+NO
>N ― N02 一 >N H+NO
방향족의 1 차 니트라민 화합물은 니트로스산 (n it rous a ci d) 과 반응하여 질산디 아조늄염 (dia z oniu m n it ra t e) 을 생 성 한다 .22) S t evens 에 의하면 이 반응은 환원반응의 일종으로 다음과 같이 진행된다고 한다 .23)
 ArNH-N02 + HN02 一 Ar— N < NN 0O2 브요 ~[iA rNrNH2 ]- N나 ONO+ H춘 N+0 H32 0
ArNH-N02 + HN02 一 Ar— N < NN 0O2 브요 ~[iA rNrNH2 ]- N나 ONO+ H춘 N+0 H32 0
Franch i mon t는 1 차 니트라민이 포름알데히드 및 2 차 아민 (pip er i d i ne) 과 반응하여 아미노메틸니트라민 (4) 를 생성한다고 보고하였다.
1 차 니트라민은 디아조메탄과 반응하여 N- 메틸 또는 0- 메틸 유도체를 생성한다. 메틸니트라민은 디메틸니트라민으로, 페닐니 트라민은 페닐 0 - 메틸니트라민 (3a) 로 변환된다.
 C 晶 N=N< O。 CH3 (3a)
C 晶 N=N< O。 CH3 (3a)
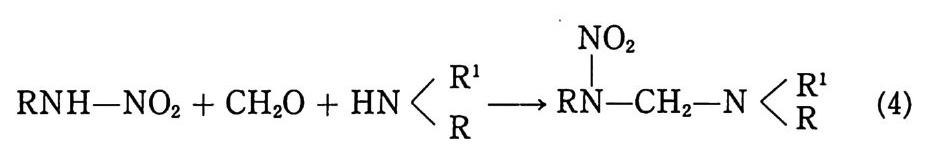 RNH— N 02 + CH20 + HN
RNH— N 02 + CH20 + HN
Woodkocke 등은 위와 같은 종류의 반응에서 히드록시메틸 화합물의 중간체가 다음 (5) 와 같이 생성되는 것으로 보고하였 다 .24) Lamberto n 등은 다음과 같이 수용액 내에서의 니트라민의 평 형을 설명하였으며, 이 갇은 히도록시메틸 유도체 (6) 은 산성에 서 더 안정적이므로 중성에서는 평형이 왼쪽으로 옮겨진다고 하
였다 .25)
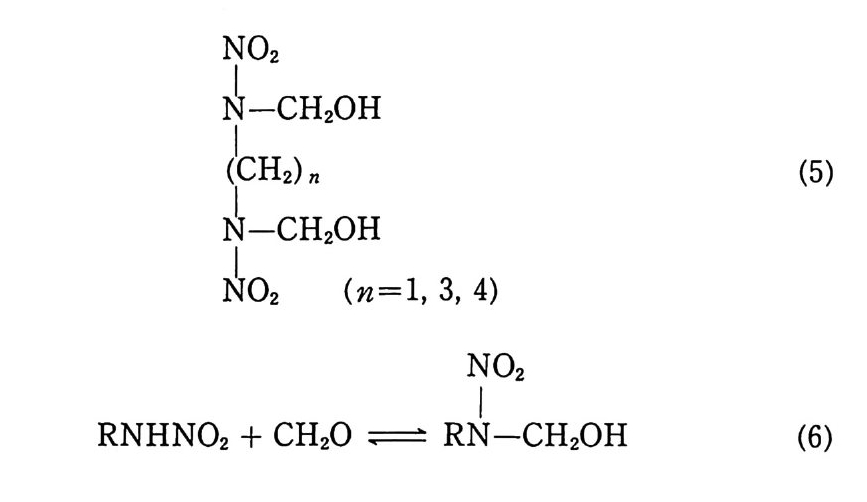 ~02
~02
 4·2 니트라민의 합성
4·2 니트라민의 합성
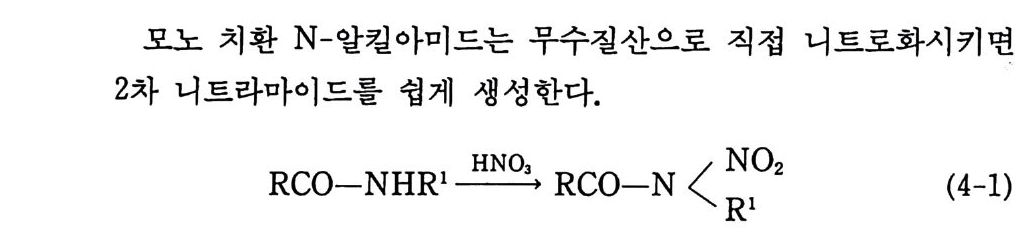 모노 치환 N- 알킬아미드는 무수질산으로 직접 니트로화시키면
모노 치환 N- 알킬아미드는 무수질산으로 직접 니트로화시키면
1 차 아민을 니트로화시키는 직접법도 위의 반응에 근거를 두고 있다. 죽, 아민을 아실화시켜 1 차 아미드를 만든 후 상기 반응
(4-1) 과 같이 니트로화시키고 다시 아실기를 제거하기 위한 가수 분해를 시키는 것이다. 일반적으로 앞의 (4-1) 과 같은 반응은 N- 에 치환체가 없는 1 차 아미드의 경우 성공적이지 못할 때가 많 으며, 대부분 니트로 반응 도중 분해되기 쉽다. 1 차 사슬족 아민 (pr im ary alip h ati c ami ne ) 역시 진한 질산 속에서는 분해된다. 1 차 방향족 아민들은 뜨거운 질산으로 처리되며 복잡한 반응을 거 치게 된다. 안트라퀴논, 피리던, 티아졸(t h i azole) 의 아민 유도체 들의 아민기들은 예의적으로 질산 혼합물에 의해 분해된다. Scholl 은 /3-아미노 안트라퀴논의 다음과 같은 니트로화 반응을 보고하였다 .26)
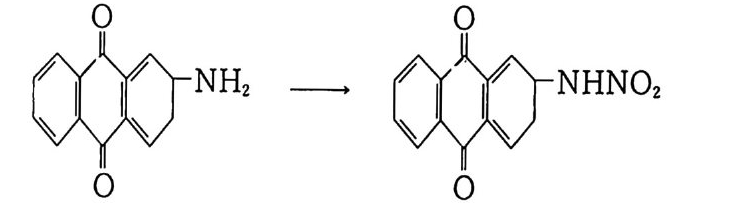 :NH2 一 ¥NHNO,
:NH2 一 ¥NHNO,
Chic h ib a bin 등은 피 리 딘의 아미노 유도체 역 시 니트로화되는 것을 발견하였다 .27) 예를 들면 질산과 황산의 혼합산은 a- 아미 노피리딘을 a- 니트라미노피리던으로 변환시킨다. Ganap a th i 등 은 아미노티아졸 (a i m i no thi azole) 계 화합물들이 상기 질산, 황산 의 혼합산을 2 배로 과량 사용했을 때 니트라민 형성 반웅이 일어 나는 것을 발견하였다. 28)
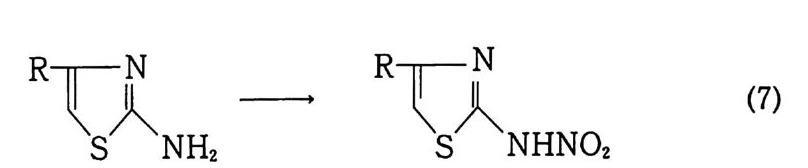 R~s·J : . NH 2 I R 亡s〔 NHN02 (7)
R~s·J : . NH 2 I R 亡s〔 NHN02 (7)
니트로화시킬 때 질산을 화학양론적으로 사용하면 상기 화합물 (7) 이의에도 아래와 갇이 링 (r i n g)에 니트로화되는 화합물 (8)
울 상당량 얻을 수 있게 된다. Wrig h t 등에 의하면 물 10~30 %를 포함하는 니트로화 혼합액으로 아미노티아졸울 처리하면 화 합 (8) 이 얻어진다고 보고했다 •29)
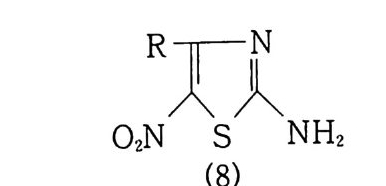 R[ [
R[ [
Or t on 은 링 내에 니트로화하기 어려운 아닐린 유도체들은 초 산 및 무수초산으로 처리될 때 N- 니트라민을 생성하기 쉬운 것 을 발견하였다 .30) 예를 들면 2,4,6- 트리브로모아닐린으로부터 다 음과 같은 니트라민이 합성되었다.
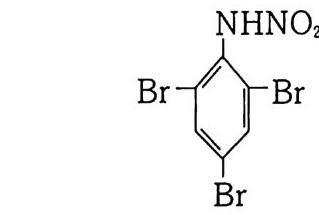 NHN02
NHN02
2 차 아민의 니트로화 역시 위와 동일한 방법으로 초산과 무수 초산에 의해 다음과 같이 진행된다.
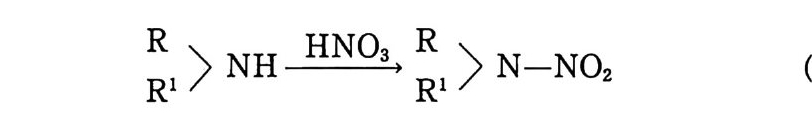 :1 >N H 쁘~ :l >N — N 02 (4-2)
:1 >N H 쁘~ :l >N — N 02 (4-2)
여기에서 2 차 아민 >NH 기가 >N-N02 로 변환되는 것은 언 제나 쉽게 진행되는 반응이 아니다. Frauchim ont 등은 31) 만일 —>CNOH - 가) 니이트미도로기화의 반 성응질을을 일 가으지키고지 있않으는며 반(면예를 만들일면 아-C미O도-기N의H 특징 (-CO-NH-CH2- )을 갖게 되면 쉽게 니트로화된다고 발표
하였다. Wrig h t 등은 이와 같은 점들을 이용하여 >NH 기를 지 닌 매우 약한 염기성 아민은 같은 기를 가진 강한 영기성 아민보 다 더 쉽게 니트로화되는 현상을 발견하였다. 강한 영기는 니트 로니화트하로기화 위반해응 이촉 매를일어 필난다요.로 하예는를 반들면어 약 R염— 기C는H 2 —촉 매N H 없-C이H도2 -R(R = CN, COOH, CONH2) 의 아민들은 영기성이 약해져 있 으므로 비교적 쉽게 니트로화된다 .32 , 33) 어떤 종류의 2 차 아민은 혼산에 의해 쉽게 니트로화된다. 예를 들어 DINA (die t h a nalnit ra mi ne din i t ra te ) 는 디 에 탄올아민을 질산, 무수질산 및 염화아연의 혼합액으로 니트로화하여 얻어전다. 4.2.2 간접법 4,2.2.1 질산아민의 탈수에 의한 합성 1 차 아민 (3) 이나 2 차 아민 (4) 의 니트로화 반응에 적용되는 질산아민의 탈수에 의한 니트라민의 합성방법은 특히 니트로구아 니던, 니트로우레아의 제조에 이용된다.
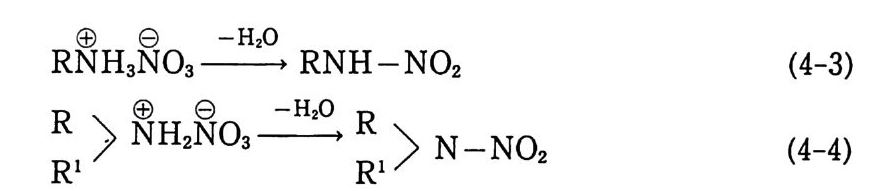 RN© H3N8 03 -H20 ) RNH-N02 (4-3)
RN© H3N8 03 -H20 ) RNH-N02 (4-3)
이러한 방법은 Bamberge r 등에 의해 디메틸니트라민과 니트 로피페리딘 합성에 적용되었으며, 이때 무수초산을 탈수제로 사 용하였다 .34> Wrig h t 동은 무수초산 이의에 염산(혹은 영화아연) 울 첨가하면 니트라민 수득률이 크게 높아져, 디메틸니트라민의 경우는 65% 까지 수득률이 증가되는 것을 발견하였다 .32,35) 이와
같이 염산울 가하면 아민의 영기성을 감소시켜 니트로기가 더 쉽 게 도입되는 것으로 해석될 수 있다. 또한 질산아민의 탈수반웅 으로서 전한 황산으로 처리하는 방법이 있으며, 이는 공업적인 방법으로서 1 차 아민으로부터 니트로구아니딘을 제조할 때 이용 하는 방법이다. 4.2.2.2 아실화 (ac y la ti on) 에 의한 1 차 아민의 니트로화 반응 1 차 아민을 아세틸기나 옥실릴 (ox y l y l) 기로 아실화시켜 생성된 2 차 아민을 반응식 (4-1) 과 같이 질산으로 처리한 후 다시 알칼 리로 가수분해시키면 1 차 니트라민을 얻을 수 있다.
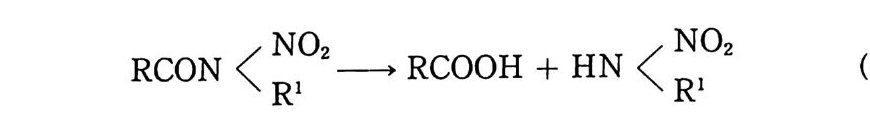 RCON
RCON
클로로포메이트 (chloro fo rma t e) 로 1 차 아민을 처리하는 것과 갇이 우레탄을 중간체로 하는 반응도 흔히 이용되는 방법이다. 이때 얻어지는 N- 치환 우레탄 N- 수소원자를 치환 니트로화한 후 알칼리 가수분해시키면 1 차 니트라민과 염기로 이루어진 영 (sal t)을 얻게 된다. 이 염을 산으로 처리하면 니트라민을 얻을 수 있다. 이러한 형식의 반응은 다음과 같은 메틸아민의 니트로 화 반응으로 설명될 수 있다 (4-6). 36,37)
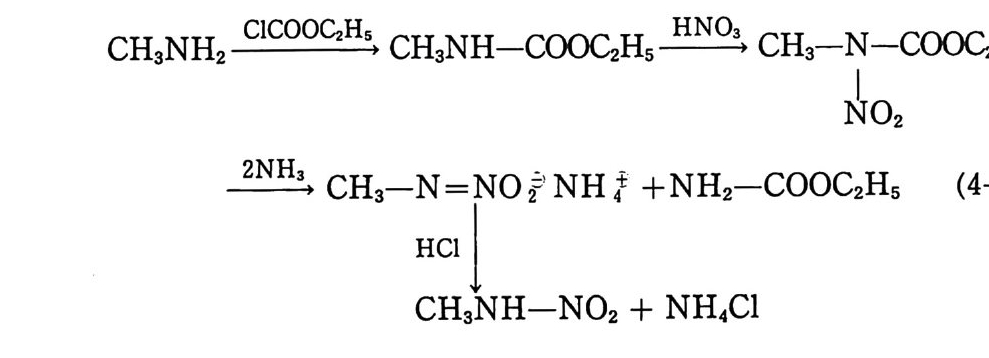 CH3NH2~ CH3NH ― cooc 晶쁘 cH3_N ― C 야 EH5
CH3NH2~ CH3NH ― cooc 晶쁘 cH3_N ― C 야 EH5
4. 2. 2. 3 클로라민 (chloram i ne) 으로부터의 합성 Ber g에 의해 제안된 이 방법은 클로라민에 질산은을 반응시키 는 것이다 .38)
![]() RNHCl + Ag N 03--+ RNHN02 + Ag C l
RNHCl + Ag N 03--+ RNHN02 + Ag C l
Wrig h t 등은 무수초산 존재하에 질산으로 클로라민을 처리하 여 니트라민을 합성하는 새로운 방법을 보고하였다 .39) 예를 들면 다음과 같이 디클로라민 (9) 로부터 sec- 부틸니트라민 (11) 을 만 드는 것이다.
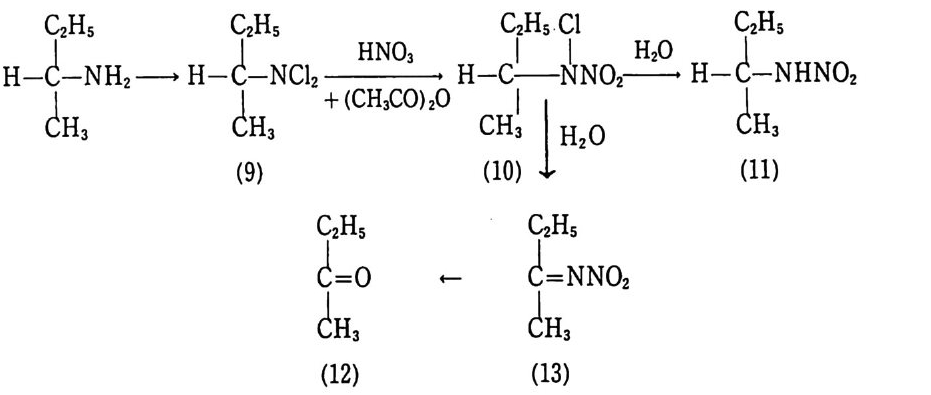 C2Hs C2Hs C2Hs Cl C2Hs
C2Hs C2Hs C2Hs Cl C2Hs
이 반응에서 중간체 (10) 은 불안정하여 물에 의해 가수분해되 면 화합물 (11) 및 (13) 을 생성하게 되나, 화합물 (13) 역시 불 안정하여 부탄온 (bu t anone-2) (12) 로 분해된다. 중간체로서 클로라민이 생성된다는 것은 앞에서 언급한 바와 같이 아민을 니트로화시킬 때 염산이 촉매로서 작용한다는 것을 설명해 주는 것이다. 이에 대해 Wr ig h t는 다음과 같은 메커니즘 으로 해석하였다 .40)
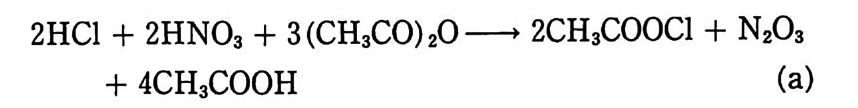 2HC1 + 2HN03 + 3 (CH3C0 ) 20 一 2CH3COOC1 + N203
2HC1 + 2HN03 + 3 (CH3C0 ) 20 一 2CH3COOC1 + N203
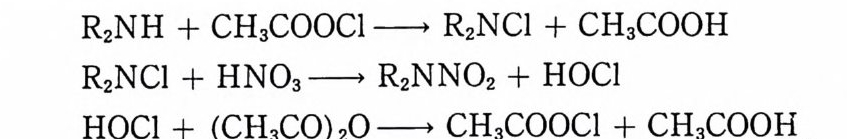 R2NH + CH3COOCI 一 R2NCI + CH3COOH (b)
R2NH + CH3COOCI 一 R2NCI + CH3COOH (b)
이와 갇이 염산은 질산 존재하에 반응하여 염화초산 (a) 를 생 성하며, 이 염화초산은 클로라민 (b) 를 생성하고 다시 니트라민 (c) 로 니트로화된다. 아민의 니트로화 메커니즘은 아직까지 충분 히 해석되지는 않았지만 Wrig h t 등은 2 차 아민, 즉 2 차 아미드 형태의 아민기가 질산과 중간 콤플렉스를 형성함으로써 진행되는 반응 메커니즘을 제안하였다 .33,41) 이는 N-N 결합이 다음과 같은 반응식에서와 같이 생성된 후 HOX 를 잃어버리는 반응으로부터 해석될 수 있다.
상기 반응에서 촉매 반응시에 X 는 영소 (Cl) 를 의미하며, 비촉 매 반응에서는 수소원자 (H) 를 의미한다. 그러나 이와 같은 제안 (4-7) 은 니트로늄 (NO 리 이온의 영향을 무시하는 단점을 나타내 고 있다. 이러한 점을 보완하여 Lamber t on 은 또 다른 변칙적인 반응 메커니즘을 제시하였다 .42) 죽, 다음과 갇이 반응식 (4-8) 에 의해 니트라민을 생성하면서 반응식 (4-9) 와 같이 질산염이 생성 되는 과정으로 설명하였다.
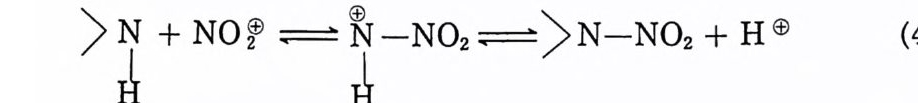 〉 N+NO 운 =8 ― N02 주〉 N-N02+H $ (4-8)
〉 N+NO 운 =8 ― N02 주〉 N-N02+H $ (4-8)
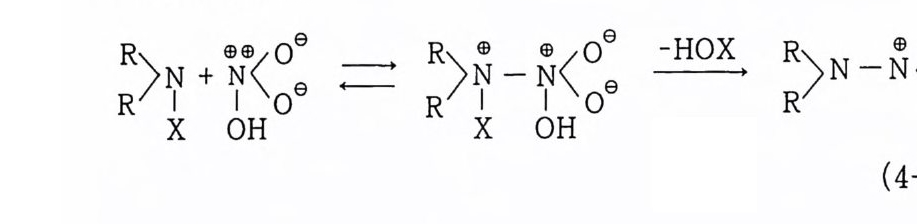 R\R/N |+X _없00 ee’ I R\R/e N IXoN H/\ _eo0 e0- H OX I RR> N -N< °O
R\R/N |+X _없00 ee’ I R\R/e N IXoN H/\ _eo0 e0- H OX I RR> N -N< °O
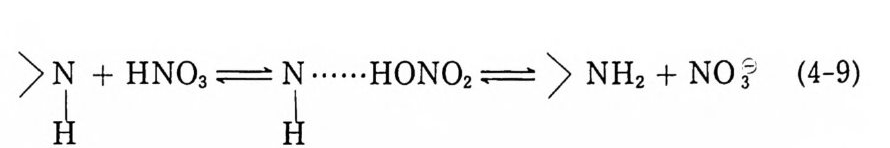 >NA + HN03 = AN … . . HQ N 02 = >N H2 + NO ? (4-9)
>NA + HN03 = AN … . . HQ N 02 = >N H2 + NO ? (4-9)
니트로화를 하기 어려운 강 염기가 있을 경우 반응 (4-9) 는 반 응 (4-8) 보다 우세 하게 진행 된다. 또한 반응 (4-9) 는 가역 반응으 로 일어날 가능성도 있다고 Lamber t on 은 보고하였다.
 4.2.2.4 니트롤리시스에 의한 합성
4.2.2.4 니트롤리시스에 의한 합성
니트롤리시스는 다음과 같은 반응 (4-11) 처럼 알코올을 거치지 않고 진행될 수 있으나 이때는 알킬 양이온과 N03- 이 반응하여 질산에스테르가 생성된다.
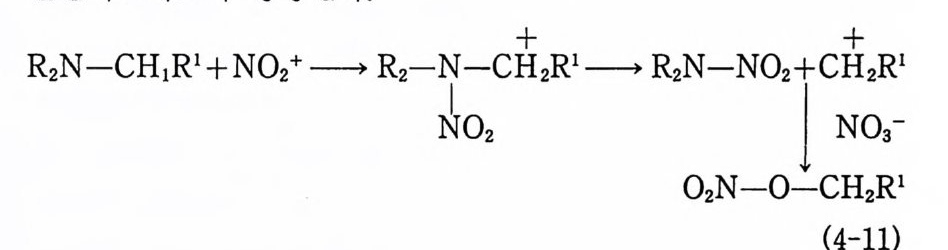 R2N— CH1 R1 + N02+ ― ➔ R2— \N。— 2 CH+2 R1 一 R20N2 ―N N— 002—+j CC NHH+20 潔R3_1
R2N— CH1 R1 + N02+ ― ➔ R2— \N。— 2 CH+2 R1 一 R20N2 ―N N— 002—+j CC NHH+20 潔R3_1
위의 반응식에서 제안된 것처럼 아미드를 니트로화시키면 니트 라민 또는 니트라마이드가 생성되는 반웅이 (4-12a), (4-12b) 경우처럼 일어날 수 있다.
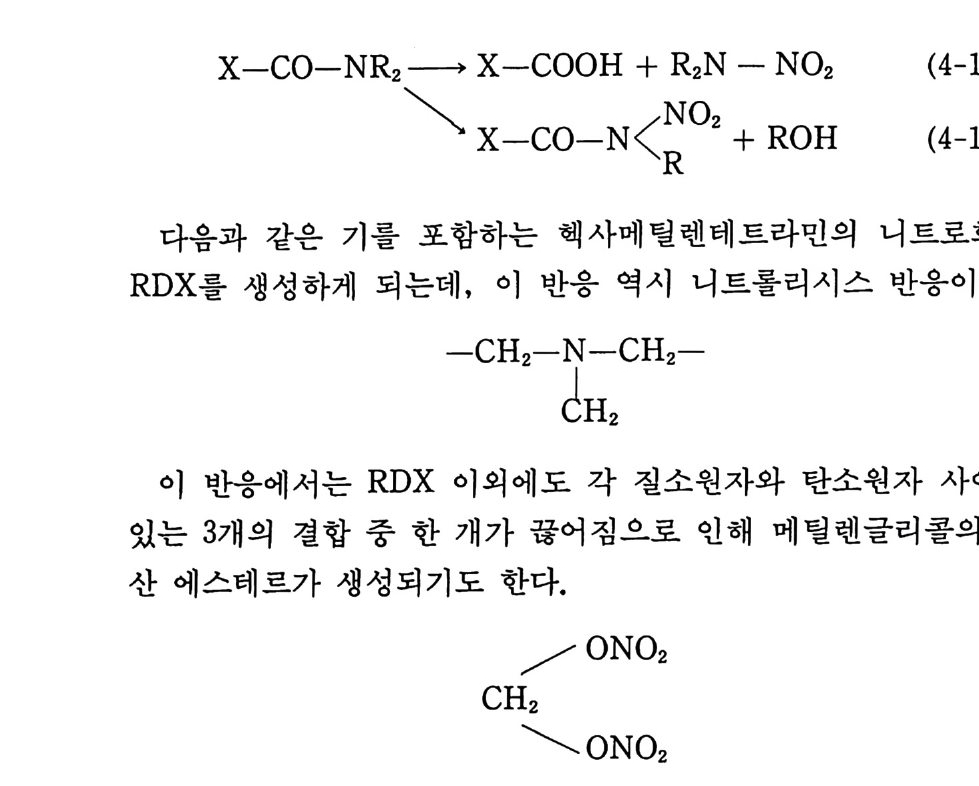 X-CO-NR2 一- X-COOH + R2N -N02 (4-12a)
X-CO-NR2 一- X-COOH + R2N -N02 (4-12a)
4. 3 RDX RDX, 핵소겐, T4, 시클로나이트로 알려진 이 화합물의 화학 명은 시클로트리메틸렌트리니트라민, 1, 3, 5- 트리니트로핵사히드 로-씹-트리아전 또는 1, 3, 5- 트리니트로 -1, 3, 5- 트리아자씨클로핵 산으로서 매우 중요한 추전제 및 폭발물 원료이다 . RDX 는 제 2 차 세계대전 중 고성능의 폭발력을 필요로 하는 폭발물에 사용된 가장 중요한 원료였으며 특히 폭굉제나 착화제 또는 테트릴 (tet r y l ) 을 대 치 할 수 있는 물질로서 부스터 (booste r ) 에 도 적 용되 었다. 또한 RDX 는 TNT 와 혼합된 폭발물로서 핵소라이트, 준 액 체 (semi -liqu id ) 및 용융 폭발물이 라고 부르는 폭발물로서 광범
위하게 사용되었다. 이의에도 RDX 는 TNT 및 알루미늄과 혼합 된 폭발물로서, 풀라스틱계 혼합 폭발물 또는 질산암모늄과 혼합 된 폭발물로 사용되 었다. RDX 는 Lenz 에 의 해 처 음으로 합성 되 었다. 44> 1899 년 Hennin g 의 45) 특허에 의해 질산 헥사메틸렌테트라민을 질산으로 니트로화 하여 RDX 를 얻는 방법이 발표되었으나, 그 당시에는 RDX 롤 폭발물로 생각하지 않고 단지 의약품으로 규정했었다. 그후의 특 허에서 RDX 를 무연 추진제에 적용하는 방법이 제안되었다 .46) 1921 년 Herz 는 Henn i n g의 방법을 개선하여 질산염 상태가 아닌 핵사메틸렌테트라민 자체를 니트로화시키는 방법을 제안 발표하 였다 .47> Hale 은 헥사메틸렌데트라민을 니트로화시켜 RDX 를 제 조하고 폭발물로서의 특성을 발표하였다 .48) 그후 RDX 에 대한 폭발물로서의 우수한 특성들이 계속 발표되 었으며, 특히 방향족 니트로 화합물에 비해 크게 떨어지지 않는 화학적인 안정성을 갖고 있는 점과 TNT, 피크린산과 같은 방 향족 니트로 화합물을 훨씬 능가하는 큰 폭발력을 지닌 것이 큰 장점으로 알려졌다. PETN 과 비교하면 폭발력은 서로 비슷한 수준이나 RDX 가 화학적으로 더 안정하며 기계적인 충격에 덜 예민하여 폭발물로서 다른 것과 비교될 수 없을 정도로 RDX 가 우수하였다. 전쟁이 전행 중이던 어수선한 분위기 속에서도 화학 공업이 발달되었던 영국, 미국, 캐나다, 소련, 특히 독일에서는 더 경제적이고 더 안전한 새로운 RDX 합성 방법을 찾기 위해 노력하였다. 전쟁이 끝난 후 독일에서 개발된 방법이 일반적으로 알려지게 되었으나 영국, 캐나다, 미국 등에서의 제조 방법도 독일 방법과 근본적으로 크게 다른 점이 없었던 것으로 밝혀졌다. 이러한 방 법들을 살펴보면 RDX 는 여러 형태의 포름알데히드와 니트라미
노기의 공급원인 질산암모늄 혹은 질산 핵사메틸렌데트라민과의 반응으로부터 합성되어진다. 제 2 차 세계대전 중에 독일은 매월 7000 톤의 RDX 를, 미국은 매월 15,000 톤의 RDX 를 생산하였다 고한다. 4.3.1 물리적 성질 RDX 는 사방정 (orth o rhomi bi c ) 결정의 흰색물질로서 굴절률 nD 는 a=l. 57 75 ; /3=1.59 66 ; r= l. 6015 이다. 49,50) 녹는점은 실험자 마다 약간씩 차이가 있으나 202~207 °C 부근에 있다. RDX 의 (비he 열at은 o f 0f.3o0 r mcaal t/i o gn °)C 은 이 며— 96연 k소ca열l 은 2285 Kcal/k g이 다. 생 성 열 /kg , 죽 -LJH , = -21. 3 kcal/ mole 의 홉열성이고 이로 인해 높은 폭발력을 나타낸다고 볼 수 있다. Edwards 에 의하면 여러 온도에서의 RDX 증기압은 다음과 같 다고 보고되 었다. 51)
 110.0 °C 4.08 X 10-s cmHg
110.0 °C 4.08 X 10-s cmHg
이 값들은 아래와 같은 경험식과 일치한다.
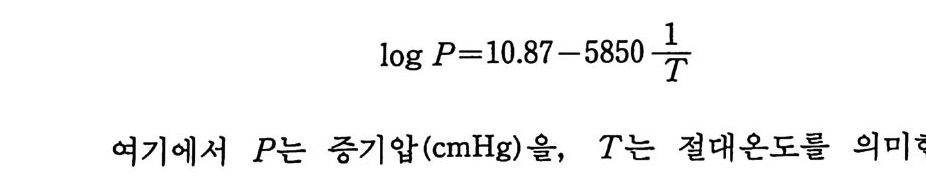 log P=l0.87— 585 0 i1,
log P=l0.87— 585 0 i1,
RDX 결정의 비중은 1.820 이며 압축될 때 압력에 따른 밀도는 표 4.1 과 같다. 또 왁스와 같은 둔감제를 가하여 마찰을 감소시 킨 경우는 2000 k g /cm2 의 압력하에서 1. 73 의 밀도가 얻어전다고 한다. RDX 는 물에 거 의 불용이 나 Ma j r ic h 는 RDX 의 물에 대 한 용해도를 o· c 에서 0.01 %, 100 ·c 에서 0.1 5 %로 보고하였다. 52) RDX 는 전한 질산에 용해되지만, 황산 속에서 70 % 이상의 농 도가 되면 분해된다. RDX 는 대부분의 유기 용매에 약간씩 용해된다. 유기 용매에 대한 용해도(1 00 g 용액당의 RDX g수) 데이터 (표 4.2) 에서 나타난 바와 같이 최선의 용매는 아세톤이다 .53) 또 다른 종류의 유기 용 매에 대한 RDX 의 용해도가 표 4.3 에 나타나 있다. RDX 는 이 황화탄소에는 불용이나 뜨거운 페놀이나 아닐린에는 용해된다. Urbanski 등은 RDX 가 용융된 니트로 방향족 화합물, 치환된 우레 아 유도체 화합물들 및 캄포 (camp h or) 등에 의해 표 4 . 4 와 같이 공융 (eu t ec ti이점을 나타내면서 용해되는 것을 발견하였 다. 54)
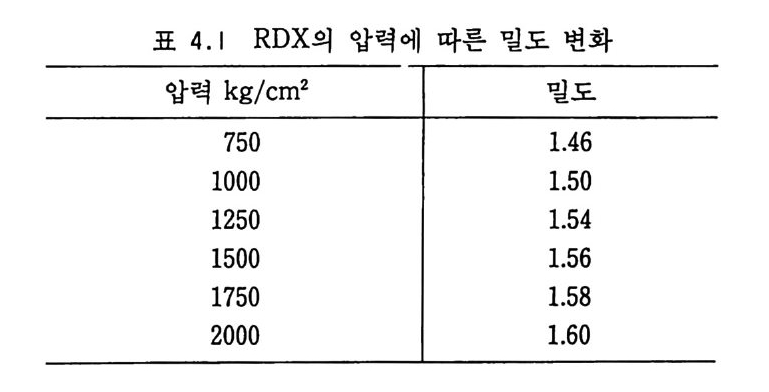 표 4.1 RDX 의 압력에 따른 밀도 변화
표 4.1 RDX 의 압력에 따른 밀도 변화
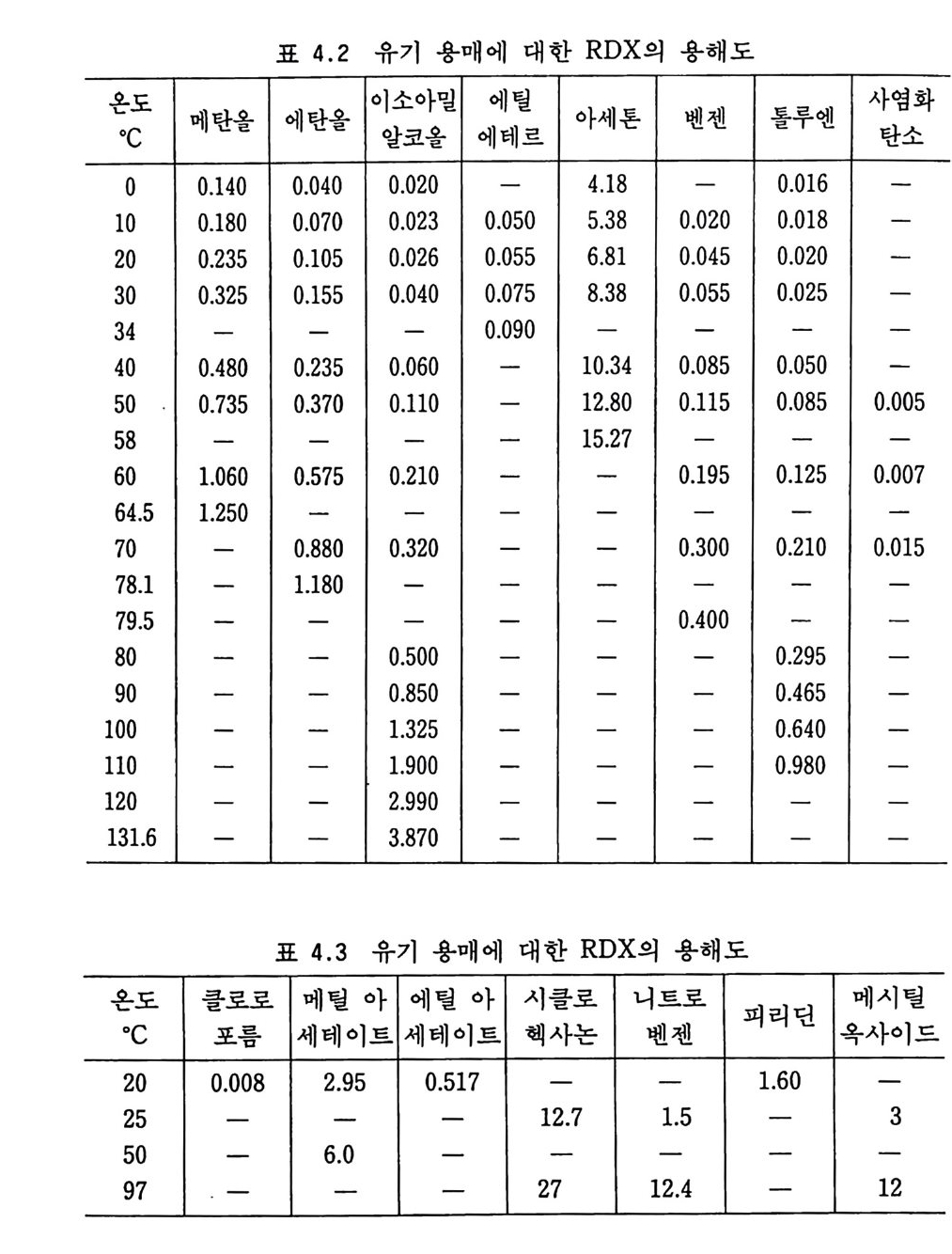 표 4.2 유기 용매에 대한 RDX 의 용해도
표 4.2 유기 용매에 대한 RDX 의 용해도
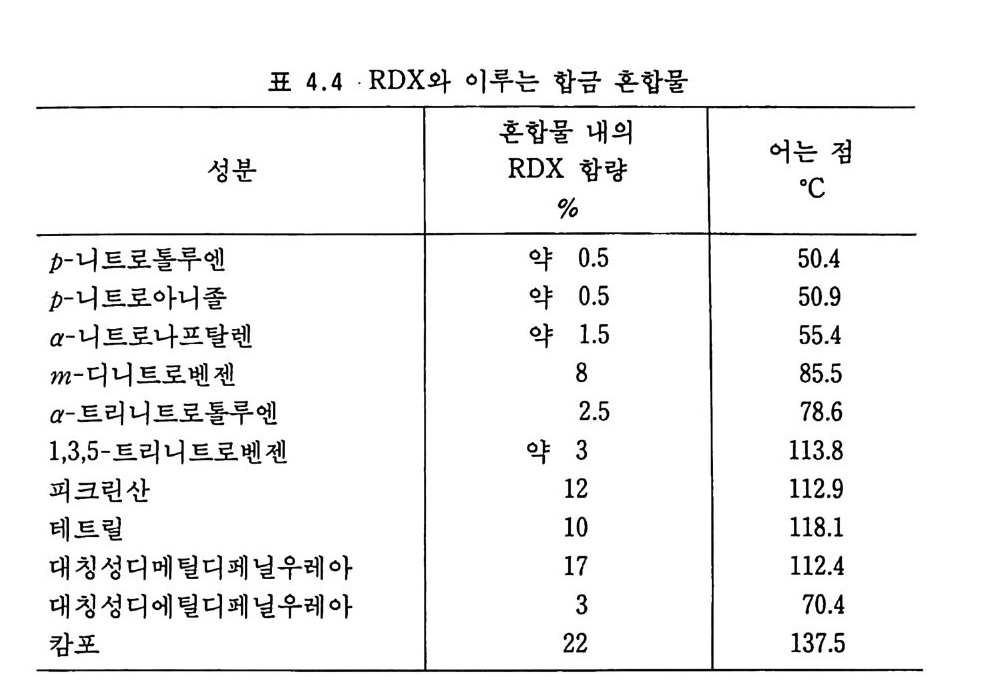 표 4.4 RDX 와 이루는 합금 혼합물
표 4.4 RDX 와 이루는 합금 혼합물
4.3.2 화학적 성질 Herz 가 가정했던 RDX 구조 (1 4) 는 그후 많은 실험을 통해 옳은 것으로 확인되었다 .47) 또한 핵사메틸렌데트라민을 니트로소 화하여 얻은 시클로트리메틸렌트리니트로소아민 (15) 를 40 % 질 산으로 산화시키면 RDX 를 생성하므로 (15) 의 니트로소아민으로
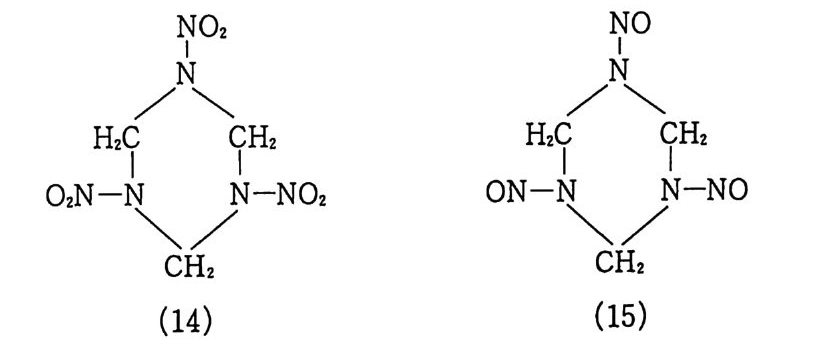 02NH-2NCI/\
02NH-2NCI/\
부터 산화반응에 의해 RDX 가 얻어전 것은 RDX 가 (14) 의 구조 를 갖고 있는 또 다른 증거가 된다. 이 방법은 RDX 생성물에 옥토겐 (HMX) 과 같은 불순물이 포함되지 않은 화학적으로 순수 한 RDX 를 얻는 방법이다. 그러나 다음과 같은 N,N,N- 트리클로로-시클로트리메틸렌트 리아민 (16) (헥사메틸렌테트라민을 h yp ochlorous 산으로 처리하여 얻 음)과 같은 트리아진 링 화합물로부터 RDX 를 얻는 방법은 실패 하였다고 보고되었으며 ,55) 이 화합물 (16) 을 아질산은으로 반응 시킨 결과 역시 RDX 합성에는 실패하였다.
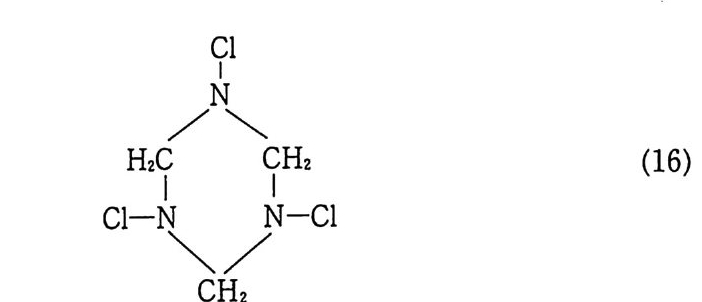 cl_N/\c
cl_N/\c
Vernazza 는 RDX 가 전한 황산에 의해 25 °C 에서 질소와 포름 알데히드로 분해되며, 그 반응은 다음과 같이 일어난다고 발표하 였다 .56)
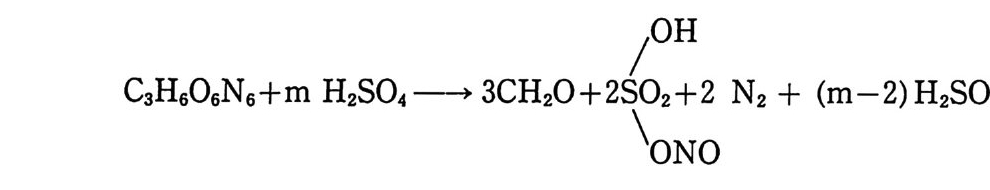 C3H606N6+m H2so4 ―겁 CH20+2SI\0 OO2HN+2 O N2 + (m-2) H2S04
C3H606N6+m H2so4 ―겁 CH20+2SI\0 OO2HN+2 O N2 + (m-2) H2S04
그러나 S i mecek 은 Vernazza 의 반응식 에 의문을 표시 하고 20 ~40 °C 에서 진한 황산에 의한 RDX 분해는 다음과 같은 반응일 것으로 보고하였다. 57)
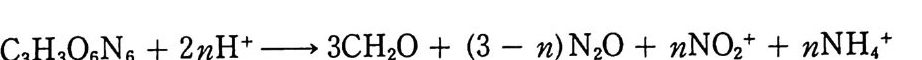 C3H306N6 + 2nH+ 一 3CH20 + (3 —n) N20 + nN02+ + nNH4+
C3H306N6 + 2nH+ 一 3CH20 + (3 —n) N20 + nN02+ + nNH4+
또한 S i mecek 은 이 분해반웅의 메커니즘을 다음과 같이 제안 하였다.
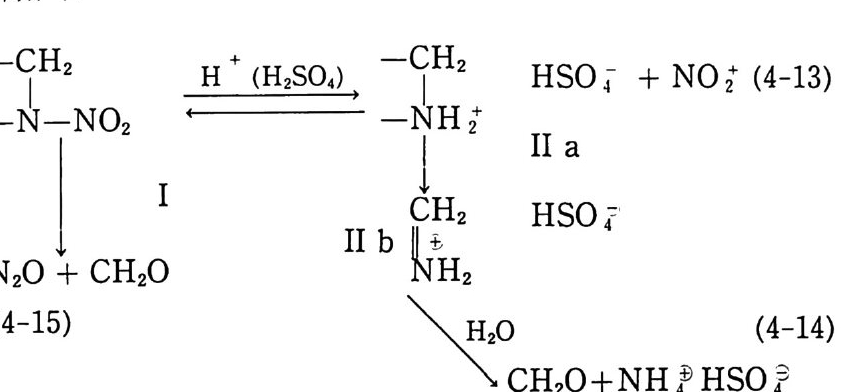 __c Hz―} |NN 0I202H0H4 + (H 2 S0 4) ——CNl| H H 2 { IHI Sa 0 i + N0 ; (4-13)
__c Hz―} |NN 0I202H0H4 + (H 2 S0 4) ——CNl| H H 2 { IHI Sa 0 i + N0 ; (4-13)
반응식 (4-13) 에 의하면 전한 황산이 RDX 로부터 니트로늄 이 온을 분리해 내고 황산 시클로트리메틸렌트리아민 (II a) 이 생성 된 후 Sch iff베이스의 황산염 (II b) 으로 변화된다. 이 황산염은 반 옹 (4-14) 와 같이 포름알데히드와 황산암모늄으로 가수분해된다. 마찬가지 방법으로 황산에 의해 영향을 받지 않는 RDX 분자 들은 반응 (4-15) 와 같이 진행되어 포름알데히드 및 니트로우스 산화물을 생성한다. S i mecek 은 RDX 의 분해시 포름알독심 (CH2=NOH) 이 포름알 데히드 존재하에서 생성되므로 N20 가스가 니트로메터 내에서 분리되지 않으므로 니트로메터에 의한 니트라민 분석 결과가 낮 은 값을 나타낸다고 결론지었다. 황산 속에 물이(특히 물이 1~15 해%)반 응존이재 하매면우 R느DX려 의진 다분.해 s를o 3 촉가진스시키를며 포, 함물하이는 없질을산 은경 우R는DX 분를 분해시키지 않는다. Somlo 에 의하면, 60 °C 에서 가성소다 4 % 용액으로 처리하면
5 시간 뒤 RDX 는 완전히 분해되었으며, 전한 가성소다 용액으로 처리했을 때는 같은 온도에서 2~4 시간 뒤에 완전히 분해되었다 고 발표했다 .58) 또한 그는 RDX 분해 생성물로부터 질산염, 아 질산염, 유기산, 암모니아, 질소, 포름알데히드 등을 검출하였 다. J ones 는 19~44.9 °C 의 온도 범위에서 메틸알코올 용액에 있는 리튬 또는 나트륨 메톡실레이트 등의 알칼리에 의한 RDX 의 분 해 반응속도에 관한 연구를 발표하였다 .59) 이 연구에서 그는 여 러 종류의 지시약을 사용하여 산으로 적정(폴라로그래픽법)함으로 써 니트로늄 이온 농도를 결정하여 분해과정을 연구하였다. J ones 는 이 분석법을 사용하여 메독실레이트 음이온 (OCH 더의 영향으로 진행되는 RDX 분해를 다음과 같이 4 종류의 반응식으 로 제안하였다 •60)
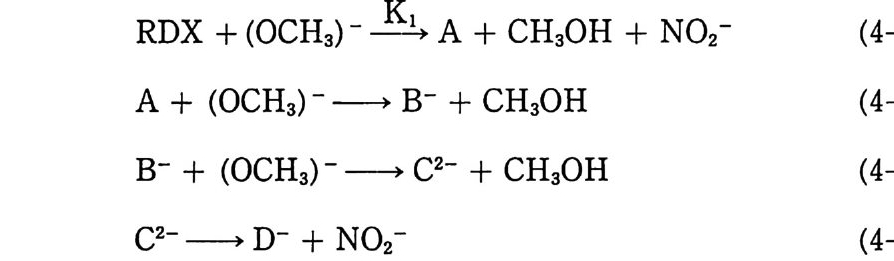 RDX + (OCH3) ~K一1 A + CH30H + N02- (4-16)
RDX + (OCH3) ~K一1 A + CH30H + N02- (4-16)
위 의 반응 (4-17) 과 (4-18) 은 매 우 빨리 진행 되 며 , K1 과 K2 는 반응 (4-16) 과 (4-19) 의 반응상수, c2- 는 강한 염 기 , B- 와 D- 는 약 염기이다. 또한 A, B 및 C 는 이론적인 중간체를 가정한 것이다.
 J ones—dd 에xt =의K 하1면(a,— 3반x1응 ) ( b속— 도X1는) + K2(X1 급) (4-20)
J ones—dd 에xt =의K 하1면(a,— 3반x1응 ) ( b속— 도X1는) + K2(X1 급) (4-20)
초 농도이다. 시간 t가 지난 후 x 는 N02- 이온의 전체 몰 농 도, X1 은 반응 (4-16) 의 N02- 몰 농도이 며 X2 는 반웅 (4-19) 의 N02-농 도이 다. J ones 는 다음과 갇이 t 시 간 후의 식을 제 안하 였다.
 —dd—Rt =K1R(a— 3 b+3R) (4-21)
—dd—Rt =K1R(a— 3 b+3R) (4-21)
 테트릴 203 °C
테트릴 203 °C
이들의 점화온도 결정시에는, 150 °C 부터 가열을 시작해 10°C/ m i n 의 속도로 가열하였다. 이의에도 Majr i c h , 52> Toneg utti, 53> Becker64) 등 많은 저자들이 RDX 가 펜트리트나 테트릴보다 더
RDX 는 펜트리트(p en t hr it e) 보다 열에 더 안정하며, 데트릴의 용융점 (130~132 °C, 138~140 °C 등)보다 훨씬 높아 테트릴보다 안정성이 훨씬 크다고 알려져 있다 .61> Avo g adro 에 의하면 RDX 의 점화온도는 215 °C 로서 펜트리트(점화온도 185 °C) 보다 높다. Urbanski 둥은 이 물질들에 대해 다음과 같은 점화온도를 보고 하였다 .62)
안정하다고 보고하였다. Rober t son 은 용융점 이상 213~299 °C 범위에서 일어나는 RDX 의 분해는 1 차 반응으로 진행된다는 것 울 발견하였다. 65) 죽, 시료의 반이 분해되는 213 °C 에서는 410 초 가 소요되었으며, 259°C 에서는 0.25 초가 걸렸다. 이때 가스 상 태의 분해 생성물은 주로 N2, N20, NO, CO 및 CO2 를 포함하 고 있었다. Rober t son 에 의하면, 이 온도 범위에서 RDX 의 열 분해 반응에 대 한 활성 화 에 너 지 E 는 47,500 cal, log B = 18 . 5 임 을 밝혔다. 액체상에서의 반응속도는 용융점 아래에 있는 고체상 에서의 속도보다 10 배 정도 빠른 것으로 보고되었다. Urbanski 등은 RDX 및 RDX 와 TNT 혼합물의 안전성에 대해 연구한 결 과를 다음과 같이 발표하였다. 66) CD RDX 의 샘플을 120 °C 에 서 62 시 간 동안 가열 하거 나, 110 °c 에서 36.5 시간 가열 후 다시 120 °C 에서 60 시 간 가열했을 때 그 분해 생성물은 산성물질을 포함하고 있지 않았다. @ 134.5 °C 탈리안 (Ta li an i) 시험에서 RDX 는 데트릴과 유사한 성질을 나타냈다• 이때 RDX 를 24 시간 동안 가열했을 때 11. 5 mmH g의 압력을, TNT 의 경우는 16mmH g를 나타냈다. @ 80 % RDX 와 20 % TNT 혼합물은 좀더 빠른 속도로 분해 되어 24 시간 후 39mmH g를 기록했는데, 이는 RDX 가 부분적으 로 용융되었기 때문인 것으로 해석되었다. 그러나 이 혼합물은 데트릴보다 더 안정한 것으로 나타났다. Tabouis 등에 의하면 80°C 의 Abel 열 시험을 거친 RDX 는 반복시험을 견딜 수 없을 정도로 전분-요오드 시험지를 매우 빨리 변색시키는 것으로 보고 되었다. 이것은 RDX 결정 내에 존재하는 미량의 질산 때문인 것으로 보인다. 가열하게 되면 불순물 질산이 RDX 결정으로부 터 분리되어 그 시험을 반복할 수 없을 정도로 시료를 변질시키 는 것으로 해석된다. 그러나 0.035 % 이상의 질산을 함유한
RDX 가 Abel 열 시험을 합격할 수 있으므로 미량의 질산일지라 도 가능한 한 제거하는 것이 바람직하다고 보고되었다. Ma j r i ch 는 빛이 RDX 에 거의 영향을 주지 않는다고 보고하였 다. 그러나 자의선 영향으로 RDX 의 흰색이 엷은 황색으로 변화 되지만 다른 성질은 아무 변화가 없는 것으로 알려져 있으며, 특 히 다른 종류의 질산에스데르와는 달리 빛에 의해서도 NO 혹은 N02 가스의 발생이 없는 것이 특징이다 .67) 4.3.3 폭발 특성 RDX 는 폭발하여 CO, CO2, H20, N2, H2 가스 등을 방출하 면서 분해된다고 알려져 있으며 폭발 후 냉각시킨 가스의 조성은 다음과 같다.
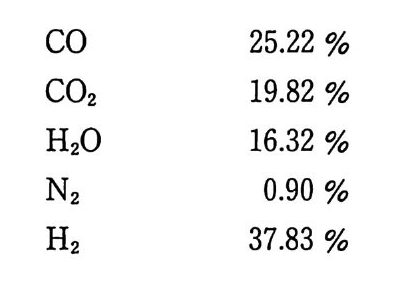 co 25.22 %
co 25.22 %
Avo g adro 는 RDX 폭발에 대해 다음과 같은 상수들을 이론적 으로 계산하였고 또 실험적으로 확인하였다.
 폭발열 1390 kcal/ kg
폭발열 1390 kcal/ kg
폭발열 (heat of exp lo sio n ) 에 대 한 또 다른 보고는 위 의 데 이 터
보다 약간 낮은 1370 kcal/ kg 68 > 및 1359.5 kca l/ k g 69) 을 보여 주었 다. Ap in 등은 밀도 변화에 따른 폭굉 열 (heat of deto n ati on ) 및 가 스 부피 (ga s volume) 를 조사하였다. 70) RDX 의 충전밀도가 0.5 에 서 1.78 g /cm3 까지 증가함에 따라 폭굉 열은 1290 에서 1510 kcal/ k g으로 증가하였으며, 반면 가스 부피는 730 에서 630 !/k g으로 감소하였다. 충전밀도에 따른 폭굉열 변화는 그림 4.4 에서 나타 낸 것처럼 선형적이다. 이때 분해가스 생성물의 조성 역시 충전 밀도 변화에 따라 변화되었으며, 충전밀도가 증가할 때 탄산가스 의 함량이 증가하는 것으로 보고되었다• Ap in 등은 충전밀도가 1. 10 일 때 RDX 의 분해 반응식은 다음 과 같다고 가정하였다.
 1500
1500
 표 4.5 RDX 의 폭굉속도
표 4.5 RDX 의 폭굉속도
RDX 의 폭굉속도에 관한 보고들이 표 4 . 5 에 나타나 있으며 그 값들은 펜트리트보다 높은 것으로 보고되었다. RDX 는 펜트리트보다 충격(i m p ac t)에 덜 예민한 것으로 알려 져 있는데, 다음 값들은 10 % 및 50 %의 확률로 폭발이 일어나 기 위한 충격 에너지의 비교이다 .71)
 10% 폭발 50% 폭발
10% 폭발 50% 폭발
Izzo 등은 RDX 와 테트릴 사이에 충격 예민성이 별 차이가 없 는 것으로, 죽 2 kg 무게추를 사용한 낙하실험에서 RDX 는 30 ~32cm, 데트릴은 40cm 를 나타냈다고 보고하였다 .72) RDX 는 테트릴보다 쉽게 폭굉되지만 펜트리트보다는 폭굉되기가 쉽지 않
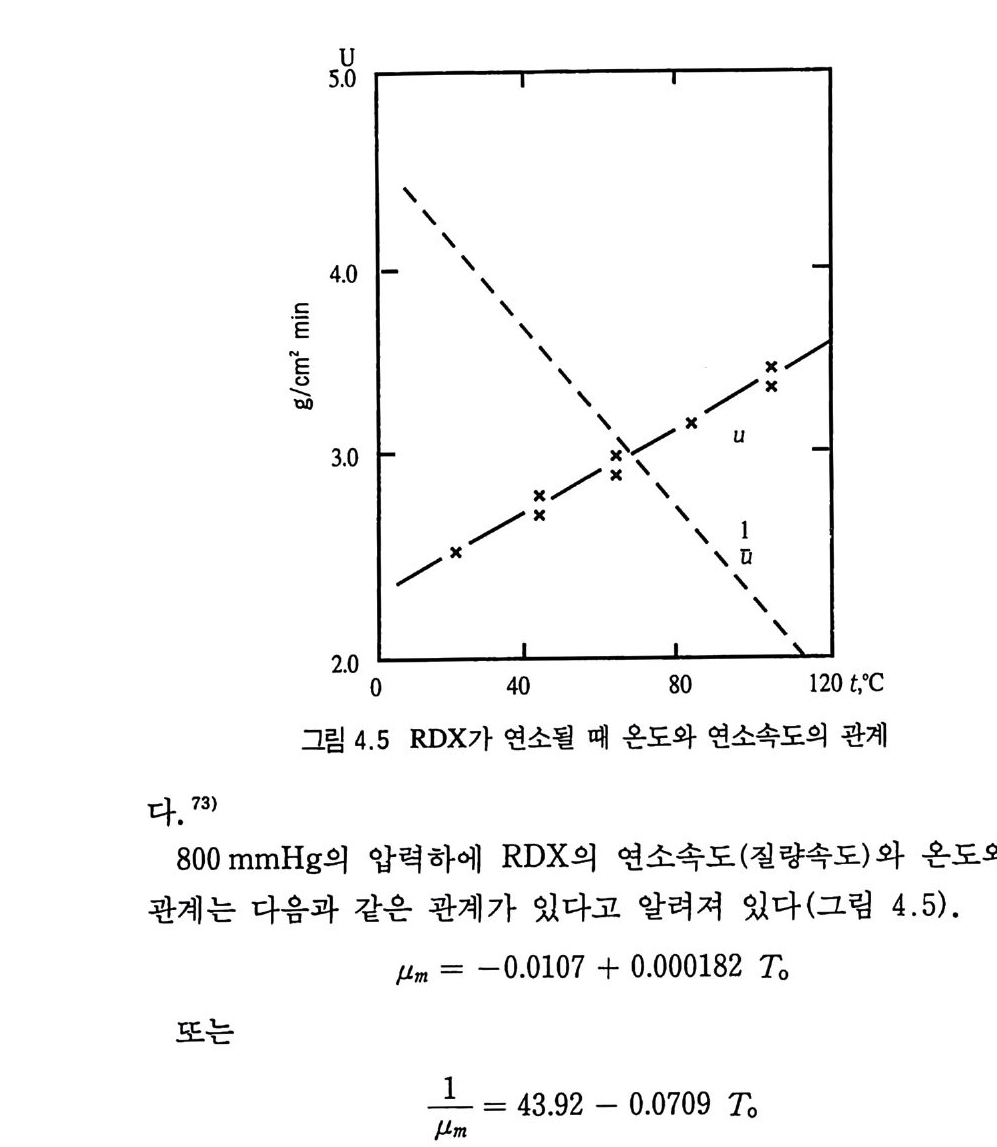 uo
uo
4.3.4 RDX 제조 RDX 제조는 일반적으로 다음과 같이 다섯 가지 방법으로 구 분될 수 있다.
4.3.4.1 헥사민과 질산의 반응 헥 사민 (hexami ne , hexameth y le nete t r a mi ne ) 과 질산을 반응시 켜 RDX 를 제조하는 방법은 가장 오래되고 가장 간단한 방법이다. 이때 사용되는 질산은 비중 1. 50~ 1. 52 의 질소산화물이 없는 전한 질산으로 25~30 °C 에서 반응시킨 후 찬물에 반응액을 쏟아넣어 RDX 를 분리한다. Hale 에 의하면 이때의 반응은 다음과 같이 진행된다고 알려져 있다 .48)
 (CH2) sN4 + 4HN03 一 (CH2 • N • N02) 3 + 3CH20 + NH4N03
(CH2) sN4 + 4HN03 一 (CH2 • N • N02) 3 + 3CH20 + NH4N03
 그러나 실제로는 위의 두 반응이 동시에 일어나는 것으로 보인
그러나 실제로는 위의 두 반응이 동시에 일어나는 것으로 보인
 (CH2) sN4 + 6H20 一 4NH3 + 6CH20 (4-26)
(CH2) sN4 + 6H20 一 4NH3 + 6CH20 (4-26)
(4-26) 식 및 (4-27) 식 반웅들 이의에도 다론 부반응둘이 일어 나서 RDX 이의의 물질들을 생성하기도 한다. 그 이유는 핵사민 울 질산으로 니트로화시키는 것이 니트롤리시스 (n it rol y s i s) 에 의 한 단계적 열화 (de gr ada ti on) 반응이기 때문이다. 즉, 이 반응은 아민의 니트로화 반응으로서 질소와 탄소 원자 사이의 결합을 단 계적으로 깨뜨리는 반응을 포함하고 있기 때문이다. Hi rs t 및 Vroom 등에 의하면 핵사민의 질산화물 (17) 은 질산 에 의해 니트롤리시스 반응이 일어나 다음과 같은 물질 (18) 을 생 성 한다. 75,76)
 /\ceHHz\/N/z_He\H\z |_z0NoH
/\ceHHz\/N/z_He\H\z |_z0NoH
이러한 니트롤리시스 반응이 계속되면 질소와 탄소 사이의 결 합이 깨지게 된다. Wrig h t 및 M y ers 의 실험 결과에 의하면 결 합 (A) 가 가장 쉽게 끊어져 가정된 화합물 (19) 를 생성하며 ,77) 이 화합물 (19) 는 B 위치에서 니트롤리시스 반응이 일어나 또 다른 가정 화합물 (20) 과 이미 알려진 화합물 (21) 을 생성하는 것으로 보고되 었다. 77,78,79,80)
 CI H-2 -NNeI Ho?—A i CI H2 C| H2— 뿐N 1 F \/CH20N02
CI H-2 -NNeI Ho?—A i CI H2 C| H2— 뿐N 1 F \/CH20N02
앞의 화합물 (20) 의 결합 C 에서 니트롤리시스 반웅이 일어나 면 RDX 가 생성되며, 결합 D 에서 니트롤리시스 반응이 일어날 경우는 사슬 화합물이 생성된다. 만일 결합 E 및 F 에서 니트롤 리시스가 일어나면 화합물 (19) 로부터 다음과 같이 사슬이 열려 진 메틸니트라민 종류인 1,9- 디니트록시 -2,4,6,8- 테트라니트로 -2,4, 6,8, -데 트라자노난 (22) 이 생 성 된다.
 CH2— NN0— 2 C H2 — NN0— 2 C H2 — NN0— 2 C H2 — NN0— 2 C H2 (22)
CH2— NN0— 2 C H2 — NN0— 2 C H2 — NN0— 2 C H2 — NN0— 2 C H2 (22)
이와 같은 물질의 생성은 니트로화 반웅온도가 낮아지면 더 잘
일어난다. 그러나 이러한 물질들은 충격에 매우 예민하고 또 불 안정한 화합물들이므로 RDX 에 불순물로 포함되는 것은 바람직 하지 못하다. S i n g h 에 의하면, H2N03+ 의 영향으로 헥사메틸렌데트라민은 (19 ) 화합물을 생성하며, 이는 다시 가수분해에 의해 (19a) 가 생성되거나 계속된 헤테롤리시스 반응에 의해 (19 b) 및 (19c) 가 생성된다. 81) 화합물 (19c) 는 니트롤리시스 반응에 의해 RDX, 메틸니트라민, 포름알데히드를 생성한다.
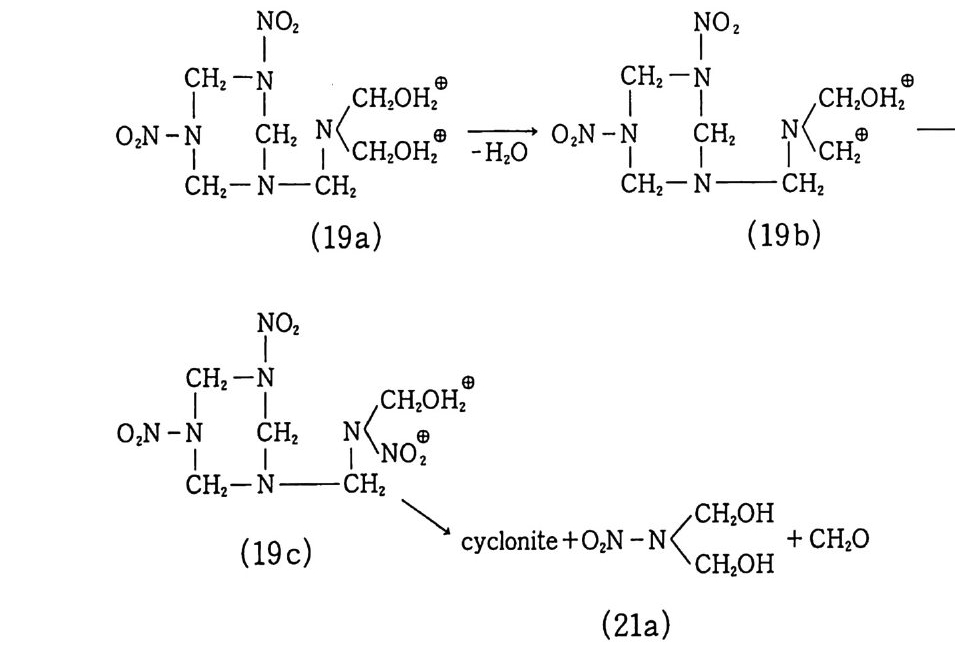 NOz N02
NOz N02
이와 같은 반웅 생성물 중에는 다음과 같은 구조의 화합물 (23) 시클릭 에데르 (c y clic e t her) 가 포함된 것으로 알려져 있는 데, 이 화합물 (23) 은 화합물 (20) 의 결합 D 및 G 위치에서 니 트롤리시스 반응 및 이 니트롤리시스에 의해 생성된 두 개의 알
코올기의 탈수 (deh y dra ti on) 반응에 의해 생성되었거나, 또는 화 합물 (20) 의 니트롤리시스 반응에 의해 직접 생성된 것으로 설명 되고 있다. 이 화합물 (23) (3, 5-din i t ro -1-oxa-3, 5-dia z acyc l ohex- ane) 은 뜨거운 물에 용해되어 냉각시킬 때 결정으로 석출되며 그 용융점 은 97 °C 이 다. 화합물 (18) 을 니트롤리시스시키면 팔각형 구조의 HMX 라고 불리는 화합물 (24) 와 RDX 를 생성하게 된다.
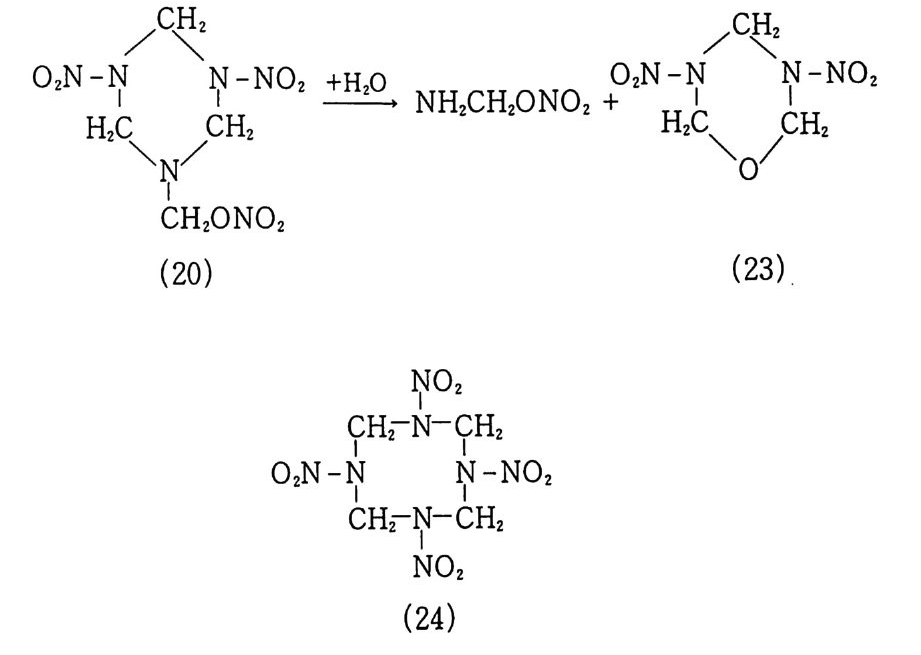 /c ~ /옛
/c ~ /옛
니트로화 중간 생성물로서 화합물 (18) 이 존재한다는 확증은 Wrig h t 등이 폐산으로 암모니아를 중화시켜 RDX 양의 5~12 % 정 도로 분리 되 어 얻은 물질 (25) 인 DPT (1, 5-endometh yle ne-3, 7, -din i t ro- 1, 3, 5, 7 -te t r a zacyc l o-octa n e) 에 의해 확인되 었다.
 C「NC HN 三0 2 C『H 2m .p. 2二13°N:HN20 2
C「NC HN 三0 2 C『H 2m .p. 2二13°N:HN20 2
그러나 이 물질을 계속해서 니트롤리시스시키면 화합물 (24) 로 변환되며, 결합 G 나 H 에서 니트롤리시스 반응이 일어나면 충격 에 매우 예민한 성질을 지닌 사슬이 열려진 화합물 (22) 를 생성 하기도 한다. Wrig h t 등에 의하면 DPT 역시 질산 (99 %)과 질산암모늄에 의 해 3, 7-din i t ro -3, 7-dia z a-1, 5-dio x acy c loocta ne (m. P. 263 ~264 ·c )를 생성한다. 78)
 CH2-0-CH2
CH2-0-CH2
한편 Wrig h t 등은 헥사민의 K 및 M 결합의 니트롤리시스로 부 터 1-di (hy d roxy m eth y l) -ami n ometh y l -3 , 5-din it ro -1, 3, 5- tri a z acy cl ohexane(26) 중간체를 생성하는 것으로 보고하였다. 82) Wrig h t 등은 위의 반응에서 중간체 화합물 (26) 이 존재한다는 것은 P 와 R 결합들이 니트롤리시스하여 얻어질 수 있는 물질 (22) 가 분리됨으로써 증명된다고 보고하였다. 화합물 (20) 이나 (26) 의 히드록실기를 아세틸화하기 위해서 니트롤리시스한 반응
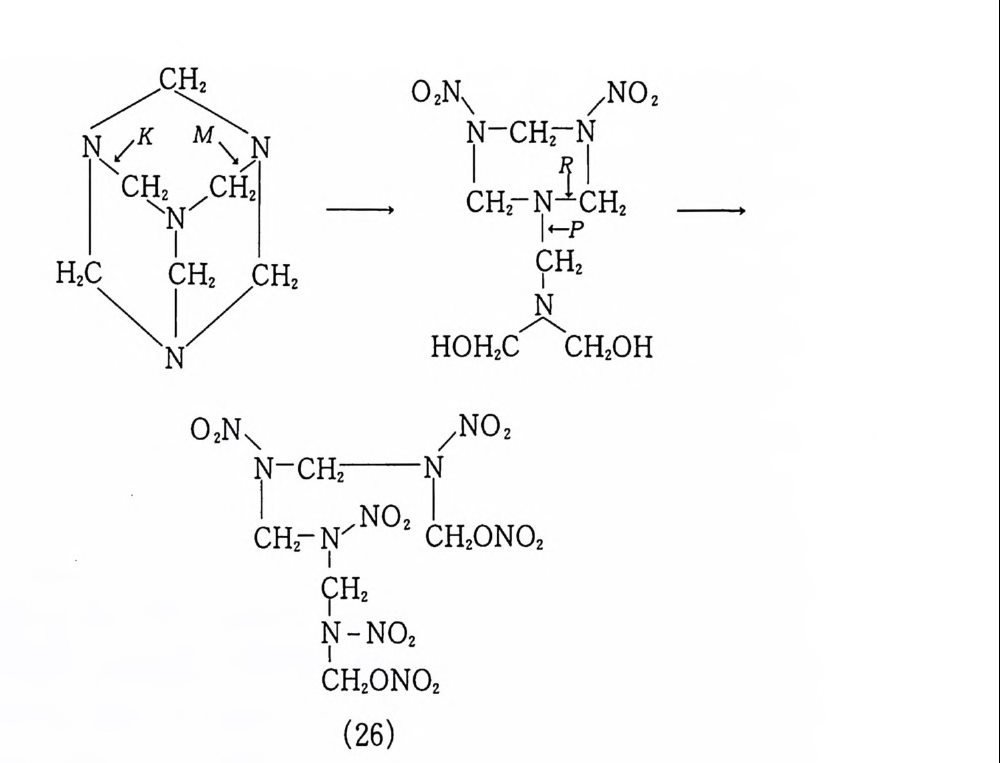 。 Z\ON/HNN02 |_H cHHzo
。 Z\ON/HNN02 |_H cHHzo
액을 —55 °C 까지 냉각한 후 一 25 °C 이하의 온도를 유지시키면 서 무수초산울 가하였다. 이렇게 반응시킨 결과 1-aceto x y -7 - nit ro xy -2 , 4, 6-tr i n i t ro -2, 4, 6-t r i azahe pt ane(27) 을 얻을 수 있었는 데, 니트롤리시스 반응액을 무수초산에 가했을 경우에는 디아세 틸 유도체 인 1, 7-dia c eto x y -2 , 4, 6-tr i n i t ro- 2, 4, 6-tr iaz ahep tan e (28) 을 생성하였다.
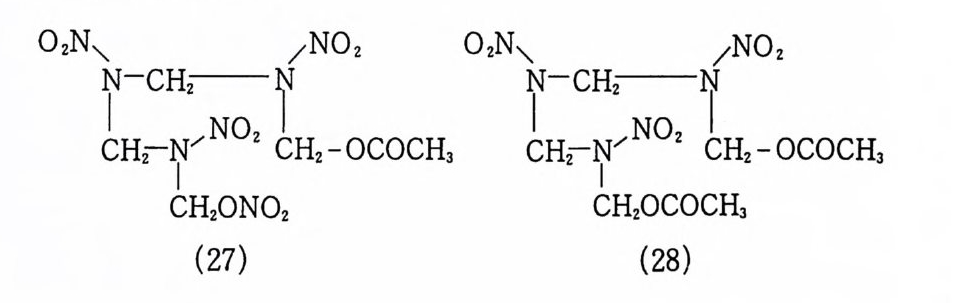 02N-尸CH _ C广H N2| / N02C?/H N2 0-2O COCH3 °2N\N나 _「CH NI2 / N02 N鬪/N 0 -O2 COCH3
02N-尸CH _ C广H N2| / N02C?/H N2 0-2O COCH3 °2N\N나 _「CH NI2 / N02 N鬪/N 0 -O2 COCH3
만일 핵사민 이질산화물 (17) 을 매우 낮은 온도 (-40 °C) 에서 니트롤리시스시키면 RDX 가 아닌 디니트로 유도체의 이질산화물 (14a) (3, 5-din i t ro -3, 5-dia z ap ipe rid i n i u m n it ra t e) 를 얻게 된다. 75)
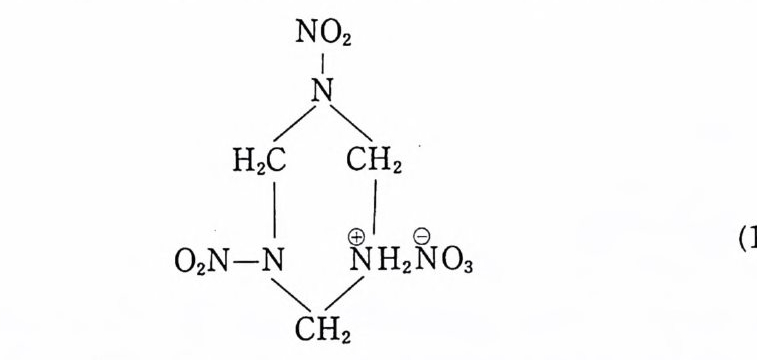 N~2
N~2
Vroom 및 W i nkler 는 위의 물질 (14a) 를 -20 °C 에서 무수질 산 75 %에 녹인 후 -20~-15 °C 의 온도에서 얼음물을 가하여 정 제하였으며, 그 용융점은 98~99°C 를 나타냈다 .76) 이 화합물은 저온에서 구조 (20) 의 결합 C 에서 니트롤리시스 반응이 일어남 으로써 질산아민과 알코올이 만들어전 후 화합물 (14a) 가 생성 되는 것으로 설명되고 있다. 이 화합물 (14a) 는 불안정하며 뜨 거운 물에 의해 분해되어 포름알데히드, 산화질소 및 질산암모늄 울 발생한다.
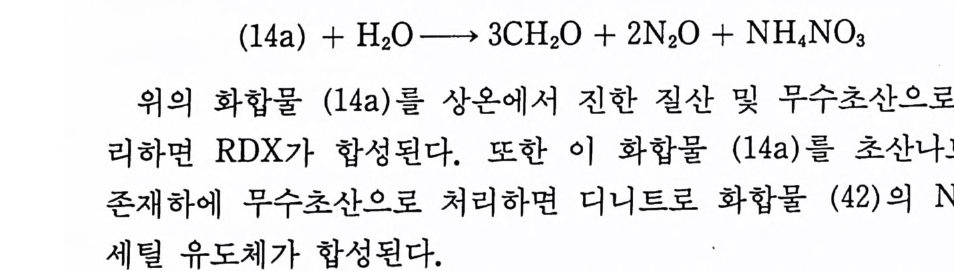 (14a) + H20 一 3CH20 + 2N20 + NH4NOa
(14a) + H20 一 3CH20 + 2N20 + NH4NOa
Vroom 과 W i nkler 는 (14a) 를 헥사민의 니트롤리시스에 의한 RDX 생성반웅의 중간체로 생각했으나, Wrig h t 등은 (14a) 와 같은 물질이 니트롤리시스 반응이 일어나는 과정 중에서는 중간
체로 존재할 수 없다는 것을 증명하였으며 ,82) Wrig h t 등은 물질 (14a) 는 반응 용액이 몸은 경우에 물질 (20) 과 (26) 의 결합 C 및 P 가 니트롤리시스됨으로써 생성된다고 제안하였다. Dunnin g 등에 의하면 이질산헥사민을 -30 °C 의 온도에서 니트 로화시킨 후 에탄올로 처리하면 물질 (19) 의 B 결합에 니트롤리 시스가 일어난, 다음과 같은 에테르계 물질 (14b) 및 사슬형 화 합물 (22a) 가 분리되었다고 발표하였다 .83)
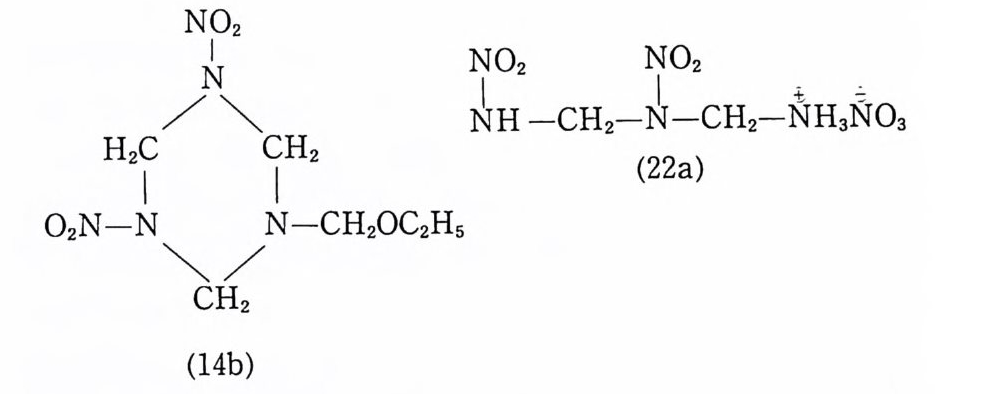 /N:\ NNI 0H2 -C_H.. . . 2 -NN_L 0— 2C __ H_ 2-N_-1,_H __3 N--= 0 3
/N:\ NNI 0H2 -C_H.. . . 2 -NN_L 0— 2C __ H_ 2-N_-1,_H __3 N--= 0 3
또한 위 와 같은 저 온 반응에 서 비 시 클릭 (bic y c lic ) 화합물 (14c) 가 니트로화 반응 생성물에서 분리 확인되었으며, 반면 (14b) 화합물을 무수질산으로 -30 °C 에서 니트로화시키면 (14d) 와 같은 에테르계 물질이 합성된다.
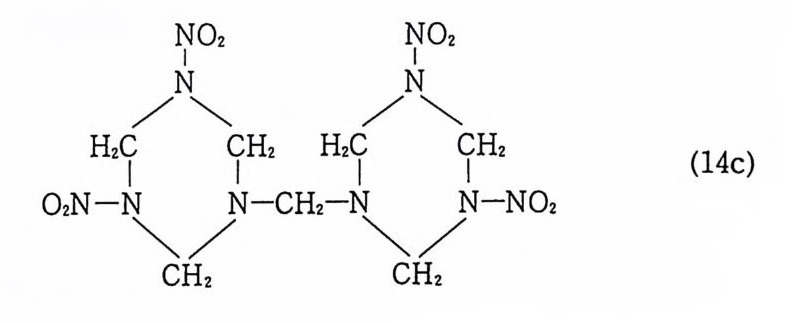 02NH-2NC|\/ CNNHI\/ 0 22 CN| H — 2 CH2 H— 2NCI\/ CNNHI\/ 0 22 CNI H -2N 02 (14c)
02NH-2NC|\/ CNNHI\/ 0 22 CN| H — 2 CH2 H— 2NCI\/ CNNHI\/ 0 22 CNI H -2N 02 (14c)
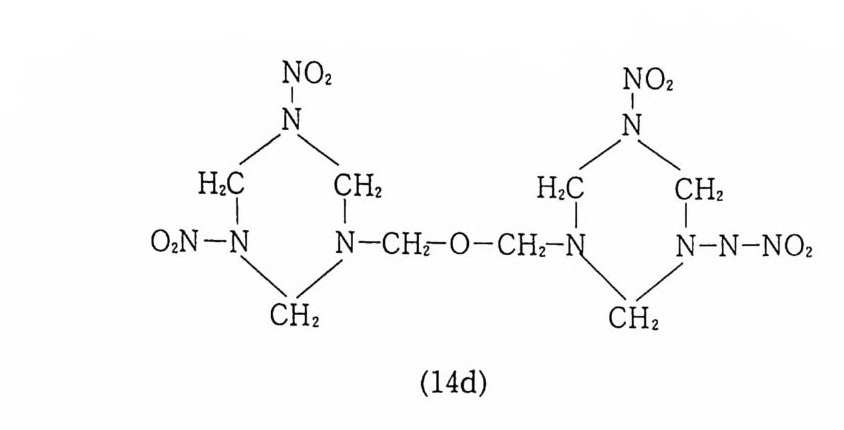 /N討\ ° 2 /NN|\ 0 2
/N討\ ° 2 /NN|\ 0 2
위와 같은 화합물들 (14b), (14c), (14d) 를 상온에서 니트로 화시키면 RDX 를 얻을 수 있으며, (22a) 역시 무수초산 및 파라 포름알데히드와 반응시킬 때에도 RDX 가 합성된다. Karpu kin 등에 의하면 핵사민의 니트롤리시스 반응시에는 다 음과 같이 트리히드록시 메틸아민 질산에스데르 (29) 의 생성이 병 행된다고 하였다 .84)
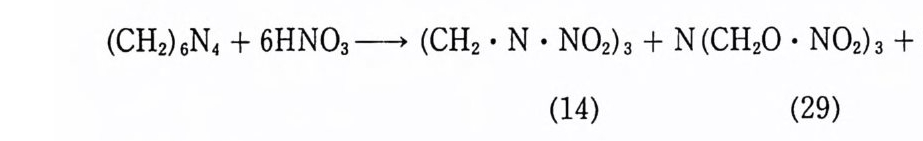 (CH2) 6N ◄ + 6HN03 一 (CH2 • N • N02) 3 + N (CH20 • N02h + 3H20
(CH2) 6N ◄ + 6HN03 一 (CH2 • N • N02) 3 + N (CH20 • N02h + 3H20
이는 화합물 (VI) 의 C 결합에서 니트롤리시스 반응이 일어난 경우로 생각된다. (21) 이나 (22) 와 에스데르들은 불안정하여 쉽 게 분해되나, 헥사민으로부터 끊어져나온 포름알데히드는 무수질 산 속에서 불안정한 질산메틸렌글리콜 (30) 을 생성한다.
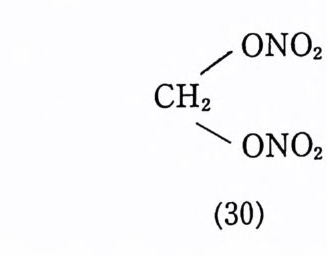 /ON02
/ON02
헥사민을 니트로화하고 물로 처리하여 RDX 를 침전시킨 후에 도 (21), (22), (25), (27) 과 같은 불안정한 화합물들로 인하여 발열반응 형태의 여러 가지 반응둘이 폐산 속에서 일어난다. 이 러한 반응들은 어떤 경우 폭발을 일으키기도 하므로 RDX 의 제 작을 시도하던 초기단계(제 1 차 세계대전 직후) 당시에는 폐산에 의한 위험성이 RDX 제조공정 중 가장 위험한 공정으로 분류되 었었다. 또 폐산 속에 이러한 부산물들이 존재하므로 폐산울 보 관하는 것은 매우 위험한 공정으로 취급했었다. 이러한 부산물들 은 RDX 에도 불순물로 섞여지므로 RDX 의 안정성을 떨어뜨리는 나쁜 효과도 나타냈다. 이와 같은 부산물들을 제거하기 위해 이물질들을 매우 엄격히 통제된 조건하에 화학적으로 분해시키는 방법을 취했다. 예를 들 면 니트로화가 끝난 혼합물을 뜨거운 물에 쏟아넣음으로써 분해 반응이 일어나도록 하였다. 죽, 물의 양과 온도를 조절하여 질산 50~55 %, 온도 70~90 °C 로 유지시키면 매우 순도가 높은 RDX 가 침전되며 불안정한 부산물들이 모두 분해되면서 N02 가스를 발생하였다. 이러한 방법은 가스제거 (deg a ssin g ) 공정이라고 불 렸다. 헥사민의 니트로화 반응에 의한 RDX 수득률은 부반웅으로 인 해 많이 감소되어 75~80 % 정도〔반응식 (37) 및 (38) 에 의한 계 산〕이므로 핵사민 100 k g으로부터 110~119 k g의 RDX 를 얻게 된다. Dunnin g 등은 0°C 에서 질산 농도를 변화시키면서 헥사민의 니트로화 반응속도를 연구하여 표 4.6 및 그림 4.6 과 같은 결과 룰 얻었다 .85)
Vroom 등도 헥사민의 니트로화에 의한 RDX 제조 반응속도 에대해 0°C 에서 질산 농도를 변화시키면서 연구하여 다음과같 은 결론을 발표하였다 .76)
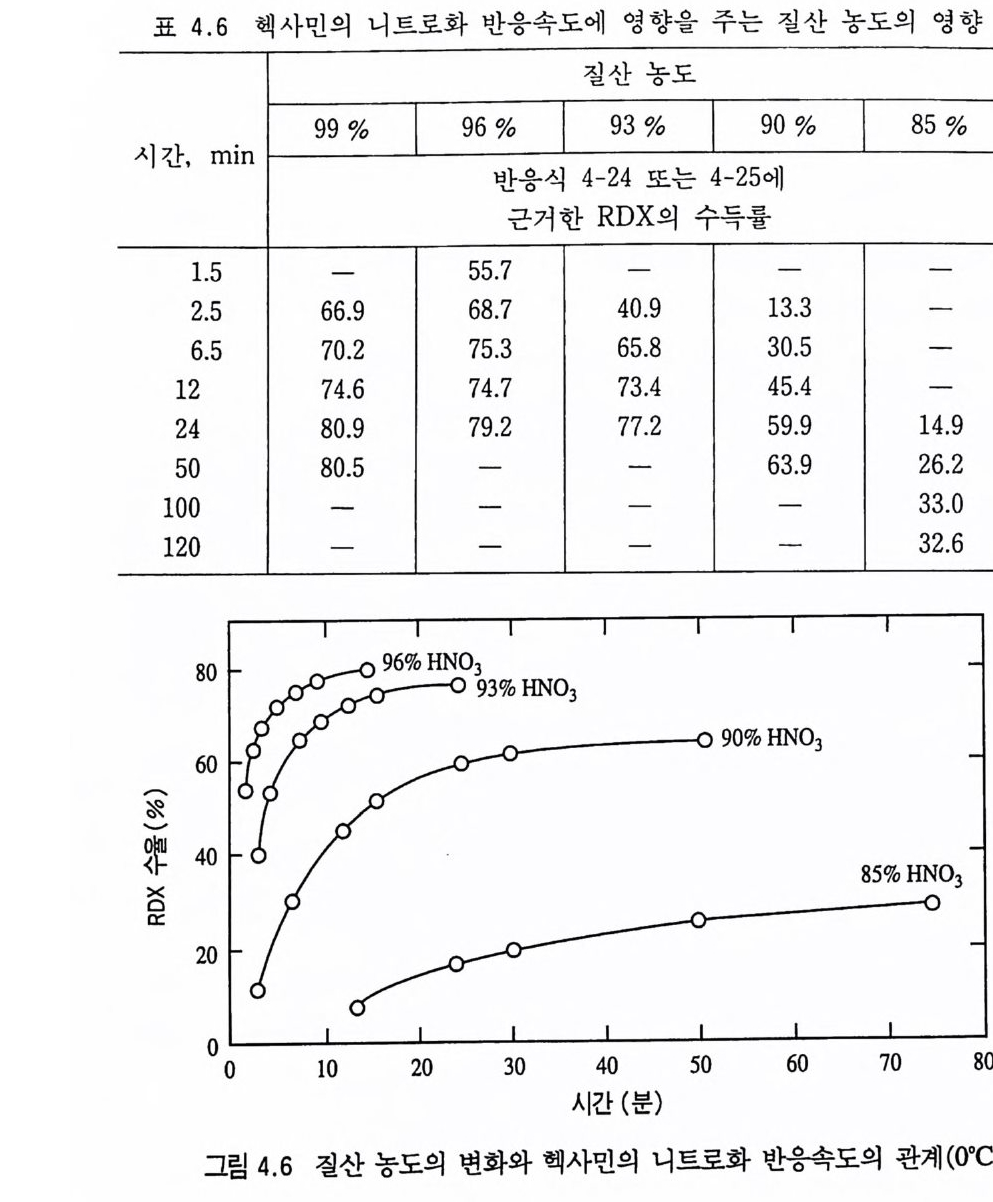 표 4.6 헥사민의 니트로화 반응속도에 영향을 주는 질산 농도의 영향
표 4.6 헥사민의 니트로화 반응속도에 영향을 주는 질산 농도의 영향
첫째, 헥사민을 기준으로 계산된 최대 수득률 80 %가 질산 농 도 88~97 %를 사용했을 때 얻어졌다(그립 4.7 ) . 최대 수득률을 위한 질산 대 헥사민의 최소의 몰 비율 (molar ra ti o) 은 97 % 질 산 사용시 26 : 1 로부터 88 % 질산 사용시 110 : 1 까지 증가하였 다. 둘째, 니트롤리시스 반응속도는 질산 대 헥사민의 몰 비율이 증가함에 따라 크게 증가되었으며, 최대 수득률에 필요한 비율보 다 더 높아지는 구간에서도 반응속도는 계속 증가되었다(그립 4.8 ) . 이 경우 0 °C 에서 85~96 %의 질산을 사용한 경우 다음과 같은 경험식을 얻게 되었다.
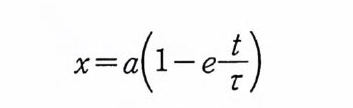 x=a(l- 국)
x=a(l- 국)
영국에서 발표된 데이터에서는 핵사민 1 k g을 RDX 로 니트로
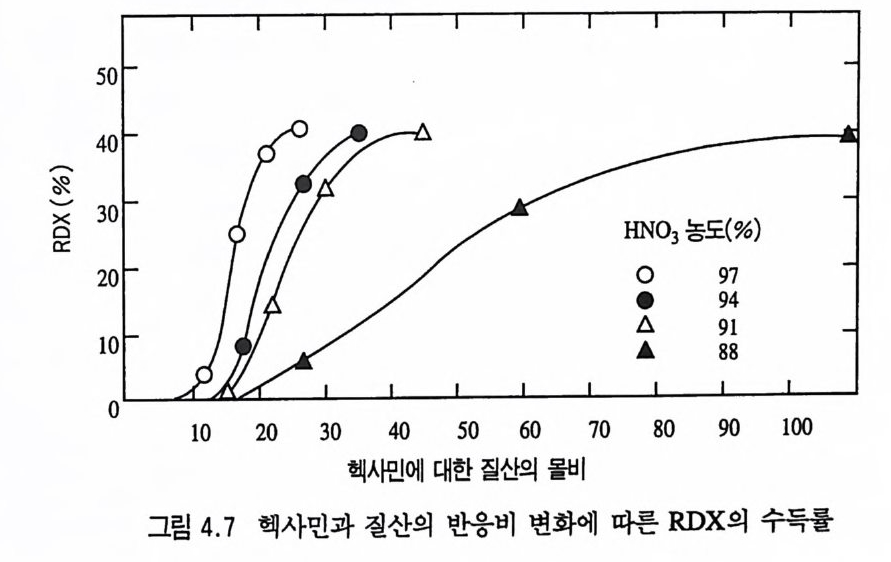 50
50
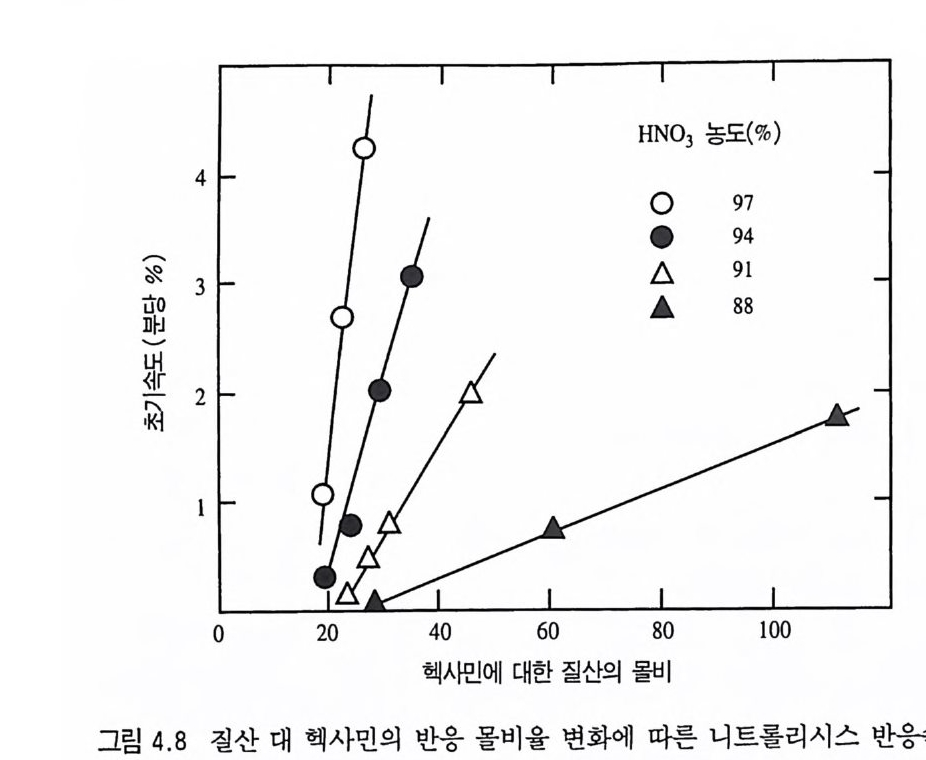 ~ 34 十卜 』 T f H•~。AN0 3 농도989(187 % )
~ 34 十卜 』 T f H•~。AN0 3 농도989(187 % )
 화하는 열량 (hea t of n it ra ti on) 은 277 kcal/k g이라고 발표되었으
화하는 열량 (hea t of n it ra ti on) 은 277 kcal/k g이라고 발표되었으
는 - LJ H=15.7 _ kca l/ mole 의 열량이 발생된다. 이러한 근거로 저 자들은 헥사민이 니트로화되어 RDX 로 생성되는 중간단계로서 이질산헥사민이 생성된다고 주장하였다. Dunnin g 및 Mi la rd 등 은 헥사민이 RDX 로 니트로화되는 니트로화 열량과 질산 농도의 관계를 그림 4 . 9 와 같이 보고하였다 .85) 이 그래프에서는 20°C, -35.5 °C 에서의 헥사만의 니트로화 열량과 -35.5 °C 에서의 이질산 헥사민의 니트로화 열량이 나타나 있다. 20 °C 및 -35 .5 °C 에서 질산의 농도를 변화시키면서 니트로화하 는 동안의 온도 변화는 그립 4 .1 0 과 같이 보고되었으며, 특히
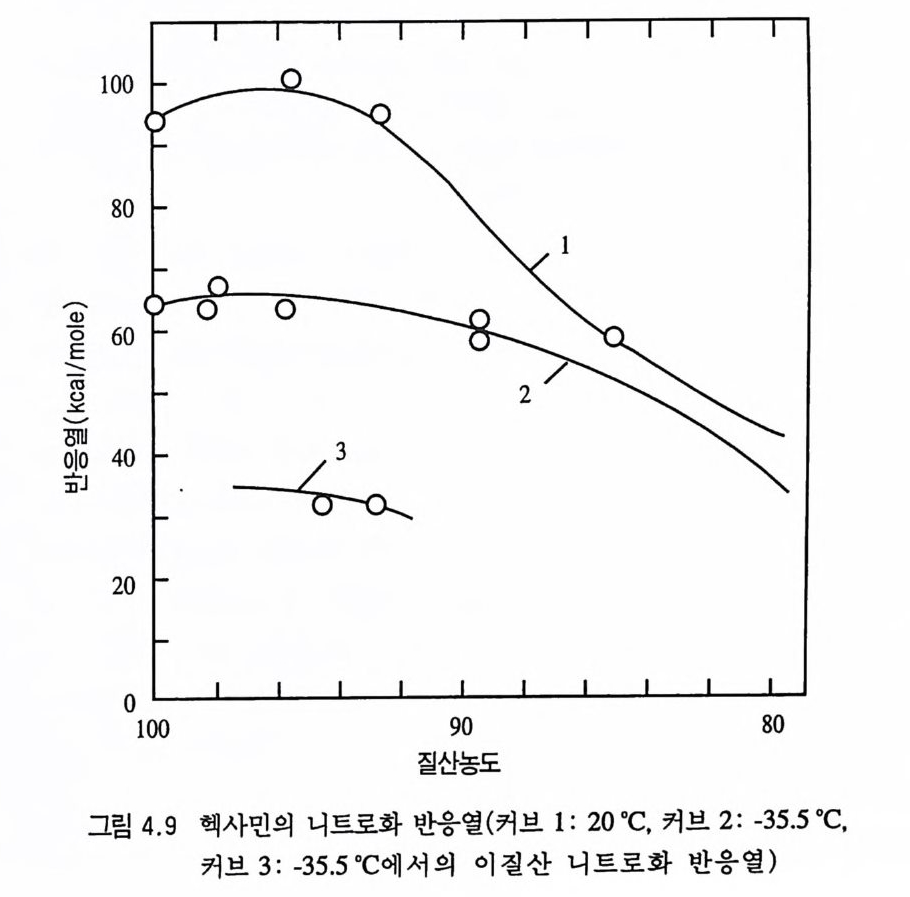 100
100
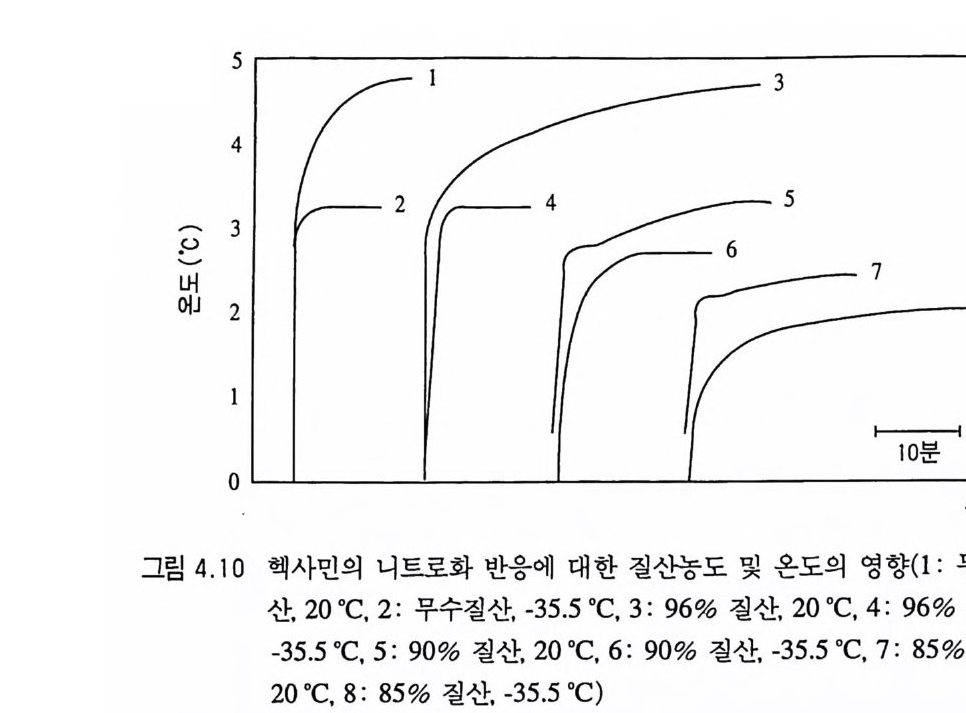 5
5
-35.5°C 에서 니트로화하는 경우의 커브가 심하게 변화하고 있음 울 알 수 있다. 85 % 질산으로 -35.5 °C 에서 니트로화할 때 발생 되는 열량은 이질산헥사민 생성과 일치하고 있으므로, 이 단계에 서는 반응이 정지된다는 것을 나타내고 있다. Kir s ch 등은 헥사민의 니트롤리시스에 의한 RDX 제조반응에 대한 초산의 영향을 보고하였다 .87) 이 실험은 1 °c 및 30 °C 에서 초산과 헥사민의 몰 비율을 변화시키고 동시에 질산과 헥사민의 비율을 26 : 1 에서 81 : 1 까지 변화시키면서 반응시켰다. 그 결과 초산은 반응속도 및 RDX 수득률을 감소시켰다(그림 4.11). 그러 나 초산에 대한 핵사민 농도가 가장 붉은 용액 (10.5 : 1 의 몰비)을 사용했을 때에도 RDX 의 수득률은, 질산 대 헥사민 몰비 48 : 1 의 최대 수득률인 80 %에 근접하였다. 초산이 없을 경우에는 몰 비가 26 : 1 일 때 같은 수득률이 얻어졌다. 따라서 질산의 일부가
초산과 반응하여 소비되므로 니트롤리시스에 참여하는 질산의 양 이 줄어든다고 결론지울 수 있다. 핵사민의 니트로화 후 남은 폐산을 이용하는 방법이 몇 가지 있 다. 즉, ® 질산울 증류해 내고 남은 질산암모늄을 타목적으로 사용하 는방법, ® 폐산을 암모니아로 중화시켜 질산암모늄을 제조하는 방법, ® 폐산을 이용하여 이질산헥사민을 제조하고, 이질산헥사민으 로부터 RDX 를 합성 하는 방법
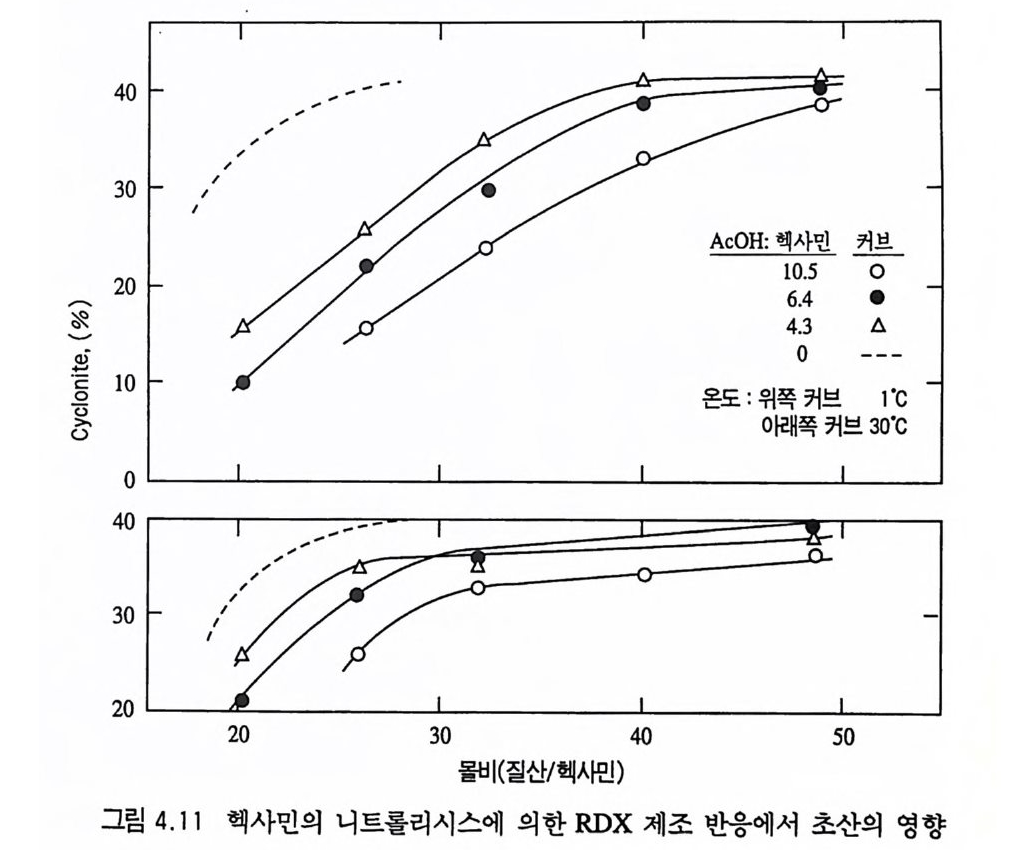 40 I ,,.
40 I ,,.
등이 있다. 상기 ®항의 방법에서 이질산염을 RDX 로 니트로화하는 반응 은 상당히 어려운 반응으로서 남아 있는 이질산염의 일부분이 변 화되지 않고 생성물에 존재하게 된다. 이러한 이질산화물은 안전 한 물질이 아니므로 RDX 에 불순물로 섞이는 것은 바람직하지 못하다 .88) 그러나 RDX 제조를 위해 이질산핵사민을 질산암모 늄, 무수초산과 반응시키는 제조방법아 채택되기도 한다 .89) 이탈 리아의 Avig ilia n a 공장에서는 폐산 (45 % NH03) 을 이용하는 완 전히 다른 공정을 사용하고 있다. 죽, 냉각시킨 혼합액에서 RDX 를 여과한 후 남은 폐산을 증류하여 포름알데히드를 회수하 는 방법이다. 그러나 이 경우 가스제거공정을 거치지 않은 폐산 울 증류하는 것은, 저온 진공하에서 증류할지라도 이 공정은 매 우 폭발 위험이 높은 공정이라고 할 수 있다. CD Brid g w ate r 공정 a 니 트로화 공정 영 국 B ri dg wa t er 에 있는 공장에 서 생 산하는 이 방법은 니트로화 반응기에 질산과 핵사민이 연속적으로 공급 되며 생성물과 폐산은 연속적으로 반응기로부터 빠져나오게 된 다 .90) 스테인리스 스틸로 제작된 반응기는 길이 90cm, 폭 32 cm, 높이 80cm 정도로서 그 중간에 있는 벽들에 의해 세 개의 방으로 나뉘어 있으며, 각 방에는 고속으로 회전하는 교반기가 각각 설치되어 있다. 그 첫번째 방은 세 개의 동심냉각코일(직경 16mm, 냉각 면적 l. 85m2) 이 설치되어 있고, 나머지 두 개의 방 에는 단선 코일들이 설치되어 있다. 반응시 발생하는 가스는 반 응기 천장에 부착된 파이프롤 통해 흡수탑을 거친 후 밖으로 배 출된다. 첫번째 방에서 나오는 파이프에는 발생 가스 색깔을 관 찰하기 위 한 관측 유리 (sig h t gla ss) 가 장치 되 어 있다. 만일 가스
 그림 4.12 RDX 의 제조 반응기 90)
그림 4.12 RDX 의 제조 반응기 90)
의 색깔이 갈색일 경우는 반응기 내의 내용물을 반응기 아래에 설치되어 있는 물랭크로 죽시 배출시켜야 한다. 상단에 보관된 핵사민은 스크루컨베이어를 통해 첫번째와 두번째 반응기로 보내 진다. 헥사민의 공급 방법은, 두번째 반응기로 공급되는 양온 첫 번째 반응기로 공급되는 양의 1/4 정도가 되도록 양을 조절한다. 니트로화 반응기로 공급되는 핵사민 공급속도는 시간당 56~170 kg 정도에서 조절된다. 직경 2.5cm 파이프를 통해 첫번째 방(반 웅기)으로 공급되는 질산의 무게는 헥사민 양의 12 배 정도이다. 니트로화 반응기의 첫째와 두번째 방의 온도는 25°C 이하로 조 절되도록 충분히 냉각시켜 주어야 한다. 38°C 로 유지하는 세번 째 방은 따뜻한 물을 코일에 통과시켜 줌으로써 온도를 조절한 다. 반응기의 일반적인 모양이 그림 4.12 에 있다. 산과 반응 생 성물은 바닥으로부터 58cm 에 위치한 세번째 방 옆에 설치되어 있다. 반응기 아래에 설치되어 있는 물탱크는 직경 7.5cm 파이 프롤 통해 반응기와 연결되어 있으며, 물탱크 내에는 반응기로 부터 반웅액이 배출될 때 발생하는 질소산화물과 반응시키기 위 한 우레아 (urea) 수용액이 채워져 있다. b 희석 (dilu ti on ) 공정 니트로화 반응기 에서 나온 액 체 는 아래 쪽에 설치된 희석기 (d il u t er) 로 들어가서 불안정한 부산물들은 분 해되고 RDX 결정은 침전된다. 그립 4.13 에 있는 것과 같은 희 석기는 265X60x115cm 의 크기로서 4 개의 방으로 구분되어 있 고 각 방에는 교반기 (195 r. p .m) 와 가열 코일이 설치되어 있다. 희석기에는 55 % 질산울 연속적으로 가하여 65 cm 높이까지 채 운다. 질산농도가 너무 높을 경우는 이 농도를 유지시키기 위해 물을 가하여 조철하며, 희석기 내부는 가열 코일을 이용하여 그 온도를 75°C 로 유지시킨다.
 ··_' I
··_' I
희석하는 동안 발생된 많은 양의 질소산화물들은 벤틸레이터 (Venti lat o r ) 나 스팀 이 젝 터 (ste a m eje c to r ) 에 의 해 냉 각탑으로 보 내져 20~30°C 로 냉각된다. 높이 3.3m 의 냉각탑 의부에는 직경 38mm 의 스틸 파이프가 장치되어 있으며, 냉각수에 의해 냉각탑 이 냉각되면 희석기로부터 보내진 증기 중의 상당량이 응축
 .` i`
.` i`
c 여과공정 질산에 분산되어 있는 RDX 는 직경 7.5cm 파이 프롤 통하여 스데인리스 스틸 케이스에 들어 있는 연속식 전공 여과기로 보내진다. 여과기의 케이스는 질소산화물을 흡수탑으로 배기시킬 수 있도록 감압 (suc ti on) 시스템에 연결되어 있다. 여과 하여 얻어진 생성물은 기계적인 방법으로 다른 여과기로 옮겨져 가능한 한 산성이 없어지도록 냉각수로 깨끗이 세척된다. 기계적 스크레 이 퍼 (scrap e r) 를 사용하여 여 과기 로부터 젖은 (wet) RDX 를 제거하고 트럭에 의해 정제공정으로 보내진다. d 정제공정 정제공정에 도착한 RDX(crude) 는 그 결정 크기
가 균일하지 않으며 0.1~0.2 %의 질산을 포함하고 있다. 이룰 정제하기 위해 물에 RDX 를 분산시킨 액체를 감압으로 스테인리 스 스틸 재질의 디스크 밀 (d i sk m ill)로 옮겨 RDX 결정 크기를 미세하게 만든 후 밀 (m ill)을 통과해 나온 RDX 서스펜존을 목재 로 된 통 (bo ili n g va t)으로 옮긴다. 이 통은 높이 250 cm, 직경 240 cm 의 크기로서 밑바닥이 경사져 있으며 교반기 및 스팀 공 급 파이프가 장치되어 있다. 또한 이 통에는 두 개의 밸브가 부 착되어 있는데 아래쪽 밸브는 통의 내용물을 쏟아낼 때, 위쪽 밸 브는 액 체 를 따를 때 (decanti ng ) 사용한다. 각 통은 대 략 2500 k g의 RDX 를 채울 수 있다. 물에 분산된 RDX 는 통 속에서 3/4 시간 정도 방치된 후 물은 위쪽 밸브를 통해 제거된다. 물을 제 거한 후 다시 찬물을 가하고 내용물을 교반한 후 다시 방치하고 물을 제거하는데, 이같은 공정을 3 회 정도 시행한다. RDX 를 90 ~100°c 로 12 시간 정도 세척하고 액체를 제거한 후, 젖은 상태 의 RDX 는 여과기로 보낸다. 여과된 RDX 는 10 % 정도의 물을 포함하고 있으며, 젖은 상태로 혹은 건조된 후, 화약 및 추진제 공장으로 운반된다. 이 공정에 의해 RDX 1000k g을 제조하기 위해서는 833k g의 헥사민과 8779 k g의 질산이 필요하지만 3482 k g의 55 % 질산 및 3429 k g의 질산이 흡수탑으로부터 회수되므로, 질산의 순수한 소 비량은 1868k g이 필요하게 되며 동시에 질산울 농축시키기 위한 황산 490 k g이 추가로 필요하다. ® SH 제조공정 독일의 Schnurr 가 1937~1938 년 사이에 개발한 이 방법은 〈 SH-me t hod 〉라고 했으며 이렇게 얻어전 RDX 를 〈 SH-salz 〉라 고 불렀다 .74)
이 방법은 작동 용량 1.5 m3 의 배 치 (batc h ) 식 니 트로화 반응기 룰 사용하여 헥사민 1 에 대해 질산 8 의 비율에 맞도록 헥사민을 99 % 질산에 가하여 반응시킨다. 반응기의 온도는 붉은 영수로 냉각되는 냉각 코일에 의해 5~10 °C 로 조절되며, 이때 니트로화 에 필요한 시간은 대략 1 시간 정도이다. 니트로화 반웅기에서 나온 내용물들은 연속적으로 별도로 배열 된 반응기를 통과하는 2 시간 동안 니트로화 반응이 완결되며, 그 반웅온도는 15~20°C 로 유지된다. 반응이 끝난 혼합물은 여섯 개의 희석기 (d il u t er) 로 주입되어 70~75 °C 의 온도로 유지 된다. 첫번째 희석기는 3m3 의 용량을, 그후 네 개의 희석기는 1.5m 3, 마지막 희석기는 3m3 의 용량을 보유하고 있다. 희석기 내의 질 산 농도를 50 %로 유지시키도록 물을 가해야 하며, 이 농도에서 RDX 는 침전이 잘 일어난다. 산에 분산된 RDX 는 50, 35, 20 °C 의 순서로 유지되는 냉각기들로 운반된다. 냉각된 RDX 는 진공 여과기에 의해 산으로 분리되면 다시 물로 세척한 후 아세톤 용 매를 이용하여 결정화, 정제된다(처음에는 가압솥 내에서 3 . 5 기압, 140 °C로 2 시간 동안 물로 끓이는 정제 방법을 사용했으나 그 폭발 위험성으로 인해 사용이 중단되었음). 희석기에서 발생된 질소산화물은 사이클론 분리기로 보내지며 이때 분리되어 떨어지는 액체는 흡수탑에서 50 % 폐산을 60 % 질산으로 농축시키는 데 사용된다. 1000 k g의 RDX 를 이 방법으 로 제조하기 위해서는 831~840 k g의 핵사민과 7100 k g의 99 % 질산을 필요로 한다. 그러나 5200k g의 질산이 회수되어 원래 농 도의 질산으로 농축됨으로써 산의 순 소비량은 1700k g이 된다. 또 다른 데이터에 의하면 RDX 1000k g당 원료 소비량은 880kg 의 핵사민과 6800~7760 k g의 99 % 질산이다.
이 원료에 의해 질산 1720~1850 k g이 상기 반응에서 사용되며 질산 5080~5850 k g이 98 % 질산으로 회수되었다고 한다. 독일에서의 RDX(SH-Salz) 의 사양 (s p e cifi ca ti on) 은 다음과 갇 다. ·녹는점 200 °C 이상 ·발화온도 215~230 °C • Ap pa rent densit y 700 g/1. • 100 °C 에서 5 시 간 동안 건조했을 때 의 무게 감량 0.1 % 이하 • 120 °C 에 서 Abel 시험지가 10 분 동안 경시 열시험 변화 없어야 함 (20 분 후 약 간의 착색은 허용됨) 이 사양에 의하면 끓는 물로 추출한 수용액은 중성 반응을 나 타내야 하며 CI-, SO/-, NOa- 등의 이온을 포함해서는 안 된 다. 단지 암모니아 및 포름알데히드의 극소량만 허용되고 있다. 또한 RDX 생성물은 아세톤에 0.1 % 이상의 불용물울 포함해서 는 안 되며, 아세톤 용액은 0.2 % 이상의 산 (HN03 로 계산된 양) 을 포함해서는 안 된다. 그리고 물로 침전시킨 후의 아세톤 용액 은 S042-이 온을 포함하지 말아야 한다. 고폭발물 화약에 사용될 RDX 는 0.75mm 메쉬 채를 통과해야 하며, 폭굉제용 RDX 는 0.60mm 메쉬 채를 통과해야만 한다. 필 요한 경우 RDX 는 몬탄 (mon t an) 왁스와 혼합시킬 수 있다. 4.3.4.2 헥사민, 질산 및 질산암모늄의 반응 이 방법은 Kno ff ler 에 의 해 개 발되 어 K-me th od 라고도 알려 져
있다 .74> K-me t hod 는 핵사메틸렌테트라민이 여섯 개의 메틸렌기 를 갖고 있으며, 네 개의 아민기를 가지고 있으므로 2 분자의 RDX 를 얻기에는 아민기가 두 개 부족한 점을 이용하여, 그 부 족한 두 개의 아민기를 질산암모늄으로 공급하기 위한 다음과 갇 은 니트로화 반응을 이용하고 있다.
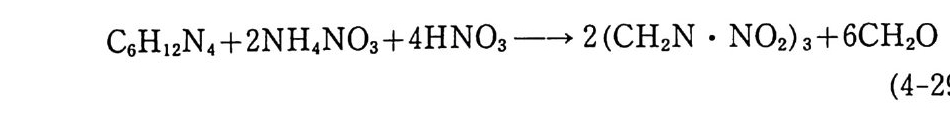 C6H12N4 + 2NH4N03 + 4HN03 _-2 (C H2N • NO 사 3+ 6CH20
C6H12N4 + 2NH4N03 + 4HN03 _-2 (C H2N • NO 사 3+ 6CH20
따라서 이 반응에서는, 사용된 헥사민을 기준으로 계산하면 이 론 양보다 더 높은 수득률이 얻어지게 된다. 이 니트로화 반응은 대략 80°C 의 높은 온도에서 진행되므로 매우 유리한 점이 있다. 왜냐하면 질산암모늄이 없는 질산과 헥사민의 혼합물은 25 °C 근 처의 온도가 되면 폭발 위험이 있는 반면 질산암모늄이 함께 섞 여 있는 앞의 반응 혼합물은 폭발 위험 없이 원하는 반응 온도까 지 안심하고 가열할 수 있기 때문이다. Elsn ig에 있는 공장의 경우를 보면, 용량 500l 니트로화 반응 기 속에서 헥사민 1 에 대해 질산 8 의 비율로, 질산암모늄은 그 이론적인 양에 맞추어 헥사민과 질산암모늄을 99 % 질산에 가하 면서 반응시킨다. 반응기 내 온도는 15 °C 가 유지되도록 냉각시 키며, 니트로화 반응이 끝난 후 그 밀쪽에 설치된 반응기 속으로 반응 혼합액을 내려보낸다. 이 반응기에는 여러 개의 수직 파이 프가 장치되어 있어 이 파이프 속으로 뜨거운 물을 통과시켜 반 응기 내 온도를 상승시킨다. 대략 80 °C 까지 가열하여 30 분간 80 oc 를 유지하여 반응 (4-29) 의 질산암모늄의 반응이 완결되도록 한다 .76) 이 혼합물은 다시 반응기 밀의 통으로 옮겨져 20°C 로 냉각된다. 이때 얻어지는 RDX 는 수득률 90 % 정도로서 다음과 같이 결정화된다. 죽, 폐산에 분산되어 있는 주 생성물 RDX 를
회전 여과기에서 여과한 후 물로 세척하고 탄산나트륨 5 % 용액 으로 중화시킨 후 아세톤에서 결정화시킨다. 그러나 이와 갇은 방법에서는 폐산 속에 질산암모늄이 상당량 포함되어 있으므로 폐산을 이용하는 것이 매우 어렵다. 폐산을 황산과 함께 증류함으로써 질산울 회수하는 방법은 농축공정에서 각 장치 부위들, 죽 증류 칼럼 (columm), 파이프, 밸브 등에 질 산암모늄을 석출시키기 때문에 실제로 적용하기가 어렵다. 따라 서 질산을 회수하기 위해 다음과 같은 방법들이 개발되었다. 죽, RDX 를 여과 제거한 폐산울 -12°C 까지 냉각시키면 삼질산암모 늄 (ammon i um tri n i t ra t e) 결정이 석출되므로, 이 삼질산암모늄을 원심분리기로 분리하여 니트로화 반응기로 순환 공급한다. 여과 액 폐산은 10 %의 RDX 의 상당량의 질산암모늄을 포함하고 있 으므로 다음과 같은 방법으로 RDX 를 회수하고 HN03 는 질산암 모늄으로 전환시킨다. 폐산을 3 而 용량의 컨테이너에 주입시키고 암모니아 가스로 중화하면서 온도는 70°C 를 유지시킨다. 이때 생성된 RDX 는 진 공여과기로 분리하고, 여과액을 냉각시키면 질산암모늄 중 2/3 정도가 결정으로 석출된다. 질산암모늄을 원심분리기로 분리 제 거시키면 여과액 속에는 소량의 이질산헥사민과 질산암모늄이 남 아 있게 된다. 이 여과 용액을 농축시킨 후 니트로화 반응기로 순환시켜 재사용한다. 이 공정으로 1000 k g의 RDX 를 제조하기 위 해 서는 480~500 k g의 핵사민, 4800 k g의 질산암모늄, 8600 k g의 질산이 필요하 다. 그러나 3600k g의 질산암모늄 및 7200k g의 질산이 회수되므 로 이 공정에서 소비되는 질산암모늄은 1200k g이고, 질산은 1400 k g이다.
4.3.4.3 포름알데히드, 질산 및 술파민산의 반응 술파민산, 포름알데히드 및 질산으로부터 RDX 를 제조하는 방 법은 1934 년 Wol fr am 에 의해 개발되어 〈 W-me t hod 〉라고도 하 였고, 이 방법으로 얻은 RDX 를 〈 W-salz 〉라고도 했다 .74) 이 W-me th od 의 기본이 되는 반응은, 술파민산 칼륨염을 포름알데 히드와 축합 (condensa ti on) 반응시킨 생성물〔화합물 (31) 의 Schif f base) 울 질산으로 니트로화시키는 화학반응이다. 무수의 황산화 물 (S03) 및 암모니 아로부터 출발하는 합성반응 단계는 다음과 같다.
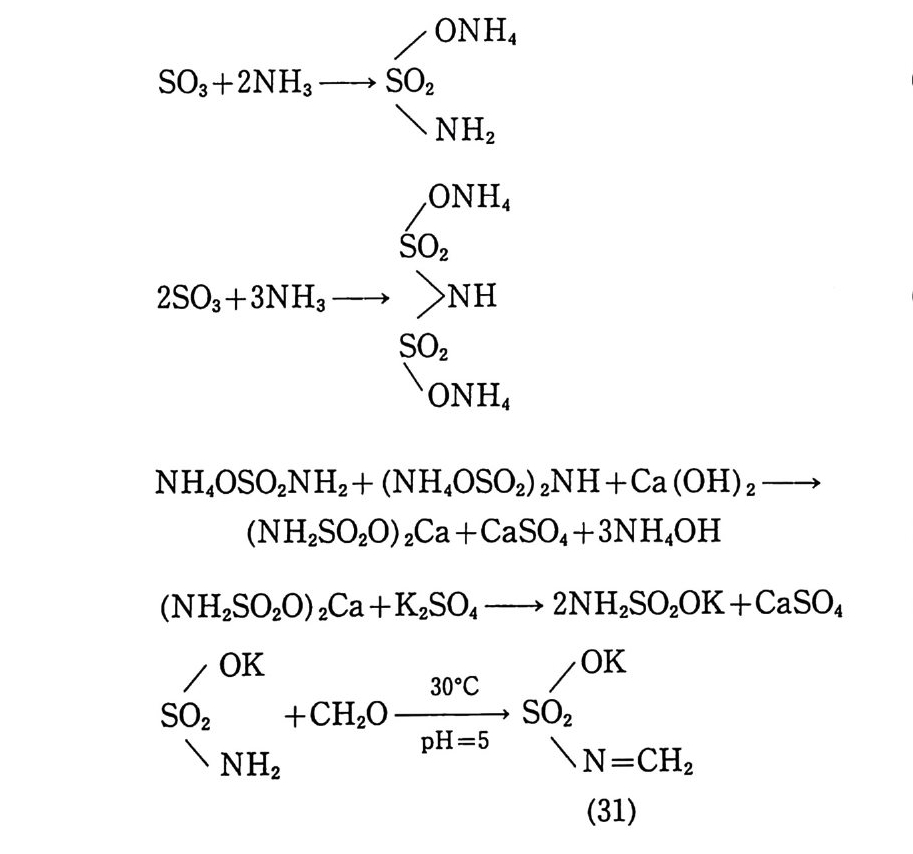 /ONH4
/ONH4
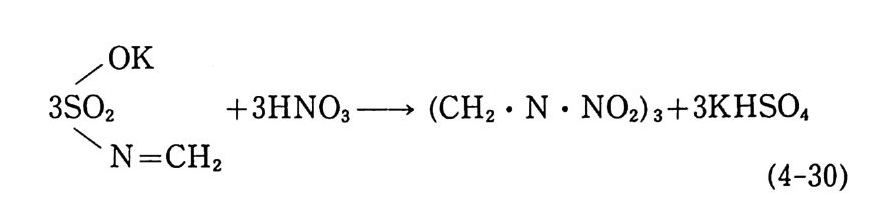 / OK
/ OK
위의 반응 (C) 에서와 같이 술파민산 칼슘 제조시 발생하는 암 모니아는 재순환되어 (a) 및 (b) 의 반응공정에서 다시 사용된 다. 술파민산 칼슘은 매우 쉽게 용해되므로 반응 (d) 에 의해 잘 녹지 않는 칼륨염으로 변환된다. Bi nn ie 등은 상기 반응둘의 메 커니즘을 연구하였으며, x- 선에 의한 결정 격자 분석 방법에 의 해 화합물 (31) 은 시 클릴트리 머 인 1, 3, 5-t ri a z acy cl ohexanetr i - sulph onic a ci d-(31a) 이며 다음과 같은 구조를 가지고 있다고 보 고하였다.
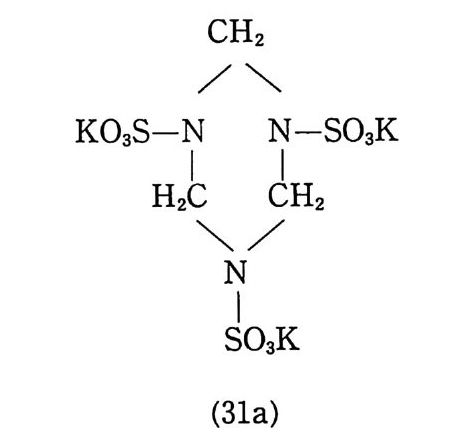 CH2
CH2
이 화합물 (31a) 를 완전 무수의 조건 (80 % 질산 및 20 % SOa) 하의 질산으로 처리하면 술폰 (sul p hon) 기가 니트로기로 치환 (80 % 수득률)되면서 RDX 가 생성된다. 이 반응에서는 니트로화 혼합물 속에 황산이 존재함에도 불구 하고 RDX 가 분해되지 않는데, 그 이유는 반응 혼합물 속에 so3
가 과량으로 존재하고 있으므로 반응액이 완전 무수 (anh y drous) 상태에 있기 때문이다. 독일 Krummel 에서는 화합물 (31) 을 니트로화하기 위해서 다 음과 갇은 니트로화 혼합액을 사용하였다 .74)
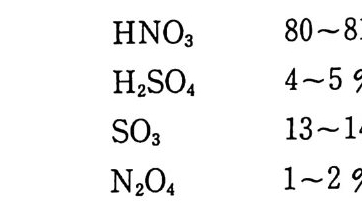 HN03 80~81 %
HN03 80~81 %
이 니트로화 혼합액은 99 % 질산과 무수황산으로부터 제조되 었으며 화합물 (31) 을 30°C 에서 이 혼합액에 가한다. 니트로화 하는 동안 발생하는 열(대략 500kcal/ kg RDX) 은 냉각 코일에 의 해 제거되며, 생성된 RDX 는 부분적으로는 용해되어 있고, 부분 적으로는 고체 상태로 분산되어 있다. RDX 생성물을 고체 상태 로 완전히 침전시키기 위해서는 물을 가한 후 진공여과기로 분리 한다. 이때 폐산의 조성은 다음과 같다.
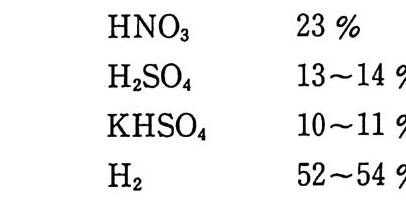 HN03 23 %
HN03 23 %
RDX 를 물로 세척 하고 잔류된 산울 다시 5 % 탄산소다 용액 으로 중화시킨 후 RDX 를 재결정시킨다. 최초에는 니트로벤젠 용매로부터 RDX 를 재결정시켰는데, 니트로벤젠의 휘발점이 너 무 높아 위험성이 있었으며 한 차례의 폭발사고가 있은 후 재결 정 용매는 아세톤으로 바뀌 었다. 폐 산은 탈니트로화 (denit ra t ion ) 하며 KHS04 를 회수한다. 이와 같은 방법으로 얻어진 RDX 수
득률은 사용한 포름알데히드 종류에 따라 80~90 % 정도이다. 그러나 이 방법은 다른 제조방법에 비해 큰 이점이 없으므로 제 2 차 세계대전 이후는 사용되지 않고 있다. 4.3.4.4 파라포름알데히드, 질산암모늄 및 무수초산으로부터 의 합성 이 제조 방법은 Ebele 에 의해 개발되었으며 독일에서는 E -me t hod 라고 알려져 있다 .74) 이와 똑같은 제조방법이 독립적으 로 캐나다의 Ross 등에 의해 1940 년 개발되었다 .91) 이 제조공정 에서 포름알데히드와 질산암모늄은 무수초산에 의해 탈수반응이 일어나므로 다음과 같은 반응식처럼 RDX 를 생성한다.
 3CH20+3NH4N03+6(CH3CO)2 一 (CH2N • NO 硏
3CH20+3NH4N03+6(CH3CO)2 一 (CH2N • NO 硏
Wrig h t 및 W ink ler 등은 이 반응이 두 단계 의 반응과정 을 거 치고 있다고 보고하였다 .92,93) 그 첫번째는 포름알데히드와 질산 암모늄으로부터 무수초산에 의해 핵사메틸렌테트라민이 합성되는 단계이다.
 6CH20 + 4NH4N03 + 3 (CH3C0) 20 一 CsH12N4 + 4HN03 +
6CH20 + 4NH4N03 + 3 (CH3C0) 20 一 CsH12N4 + 4HN03 +
 두번째의 반응은 니트롤리시스 단계이다.
두번째의 반응은 니트롤리시스 단계이다.
는 단계에서는 안전하지만, 이 반웅이 발열반응이므로 폭발을 일 으킬 위험성이 있다. 반응액들을 서로 혼합 가열시키면 반응이 너무 급격히 일어날 가능성이 있다. 반응 혼합액에 불화보론 (boron fluo rid e ) 을 첨 가하면 개 시 (ini t iat i on ) 반응을 촉진시 키 면서 안정성을 좋게 한다. 이상과 같은 공정에서 RDX 이의에도 HMX(24), N- 아세틸 유도체 (43) 및 RDX 보다는 불안정하지만 많은 사슬형 니트라민 화합물들이 부산물로 생성된다. 따라서 이 방법에 의해 얻어진 RDX 는 다른 방법에 의해 제조된 RDX 보다 비교적 낮은 용융점 (190~195 °C) 을 갖게 되며, 안정성이 떨어지게 된다. 불화보론을 첨가하여 반응시킨 경우는 부산물의 양이 감소된다. 부산물로 생성된 사슬형 화합물들의 대부분은 RDX 에 비해 초 산에 잘 용해되므로 폐산으로부터 RDX 를 여과함으로써 RDX 와 부산물이 섞이는 것을 어느 정도 막울 수 있다. 부산물을 순환 재사용하면 그 중 상당 부분이 주반응인 무수초산 및 질산암모늄 으로 처리되는 과정에서 RDX 로 변환될 수 있다. Bonb i n g en 의 공정에서 생산되는 공정 라인을 예로 들면 다음과 같다 .74,94) 알 루미늄이나 스테인리스 스틸 재료로 만든 반응기(용량: 1. 2m3) 를 무수초산으로 채운 후 0.4 %의 BF3 를 가한다. 무수초산을 60 ~65 °C 로 가열 유지시키면서 질산암모늄과 파라포름알데히드를 서서히 가한다. 온도가 높은데다가 불화보론의 영향으로 인해 반 웅은 죽시 시작되며 열이 발생된다. 열이 발생하면 죽시 가열 (heati ng ) 을 정 지 함과 동시 에 냉 각시 키 면서 반응온도를 60 ~ 65 °C 로 유지시킨다. 반응액을 가하는 데는 대략 ' 6 시간 정도의 시간이 필요하다. 반웅이 끝난 후 반웅액의 온도를 20°C 로 냉각시키고 침전된 RDX 는 진공여과기에 의해 분리시킨다. 부산물은 폐액 (여과액)에 섞여 제거된다.
의 반반응응기기 한 로 부개터로 부얻터어 대진략 8q1o6 0k kg g 의정 도R의DX 가R D얻X어 는지 며가,압 솥여러 (a ut개 o- clave) 에 넣고 140 °C 로 가열함으로써 안정화된다. 폐산으로부터 초산을 제거하기 위해 증류공정을 거치며, 증류 하는 도중 RDX 및 부산물이 반죽상으로 밑바닥에 가라앉게 되 므로 사이폰 (s yp hon) 을 사용하여 연속적으로 반죽을 제거하여야 한다. 이렇게 제거된 반죽 덩어리 중 대략 80 % 정도는 무수초 산에 용해하여 반응기로 순환시켜 RDX 제조 반웅기에서 재사용 되며, 나머지 20 % 정도의 양은 질산암모늄과 혼합하여 값싼 저 성능 화약으로 사용된다. 무수초산은 인산에 틸 (e t hy l ph osph ate ) 촉매 를 사용하는 Ke t ene 법에 의해 증류 초산으로부터 제조된다. 진공여과기에서 RDX 를 세척할 때 첫번째 세척수는 평균 20 %의 초산울 포함하 고 있으므로 이 (20 %) 초산은 초산에틸울 사용하여 추출 회수한 다. 실제 공장 규모에서의 RDX 수득률은 포름알데히드 기준으로 63~65 % 정도인 반면 실험실에서는 80 %까지의 수득률이 얻어 진다. 따라서 1000 k g의 RDX 를 얻기 위해서는 다음과 같은 원 료가 필요하다. 파라포름알데히드 630~650 kg 질산암모늄 1800 kg 무수초산 5000~5100 k g(불화보론 함유) 불화보론 19 kg 폐산으로부터는 RDX 및 부산물 반죽 110kg 무수초산 4150 ~ 4200 kg
이 회수된다. 따라서 RDX 1000 k g당 무수초산의 소비 량은 800 k g에 달하게 된다. 독일 데이터에 의하면 이와 같은 방법으로 제조된 RDX 제품은 다음과 같은 성분을 포함하고 있는 것으로 보고되었다. 93,5 % RDX 6.0 % HMX 0. 5 % 초산 유도체 (42) 4.3.4.5 이질산헥사민, 이질산암모늄 및 무수초산으로부터의 제조 이 방법은 앞의 4.3 .4.1과 4.3.4 .4 방법 을 조합시 킨 방법으로서 1941 년 미국 Bachmann 에 의해 처음으로 개발되었으며, 1943 년 에는 독일의 Ko ffl er 에 의해 별도로 개발되었다. 독일에서는 이 방법이 KA 법으로 알려져 있다 •74 , 95) 이질산헥사민 (hexam i ne d init ra t e) 은 무수초산하에서 이질산암모늄 (ammon i um d i n it ra t e) 과 반응한다. E 법과는 달리 파라포름알데히드를 메틸렌기의 공급원 으로 사용 않는 대신 RDX 의 메틸렌기는 헥사민 분자로부터 공 급되며 아미노기는 질산암모늄으로부터 공급된다. 질산 (HN03) 은 헥사민과 조합해서 이질산암모늄 형태로 반응에 참여하게 된 다.
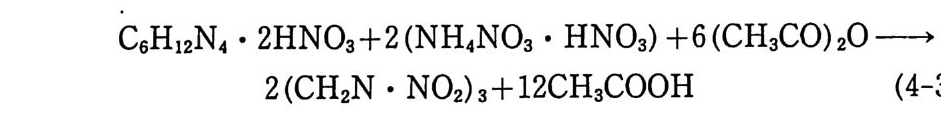 C6H12N4 • 2HNOa + 2 (NH4NOa • HNOa) + 6 (CHaC0) 20 一
C6H12N4 • 2HNOa + 2 (NH4NOa • HNOa) + 6 (CHaC0) 20 一
이 방법에 의한 RDX 수득률은 이질산헥사민 중의 CH2 기를 기준으로 계산했을 때 75~80 % 정도이다. E 법과 비교하여 KA 법의 이점이라고 볼 수 있는 것은 RDX 화학구조 내의 아민기의
일부분과 메틸렌기가 무수 (deh y dra t ed) 형태의 핵사민으로부터 공급된다는 점이다. 반면 파라포름알데히드를 사용한 E 법에서는 탈수반응이 필수적이다. KA 법에서는 E 법에서보다 적은 양의 무 수초산이 사용된다. Bobin g e n 공장에서는 15 °C 이하의 온도에서 헥사민과 50 % 질산을 반응시켜 이질산핵산울 제조한다. 이때 사용되는 질산은 펜타에리트리톨의 니트로화 공정시 나오는 폐산울 이용하게 된 다. 또한 NH4N03 • HN03 는 질산암모늄과 진한 질산을 같은 몰 비율로 반응시켜서 얻는다. 이전 반응기에서 RDX 를 여과한 여 과액 1871 k g과 무수초산 325 k g울 반응기 내 에 서 혼합하고 40 ~50 °C 로 반응 혼합액을 유지하면서 23.9 k g의 이질산암모늄과 이질산핵사민 22k g씩을 5 회에 걸쳐 반웅기에 가한다. 다시 무수 초산 271 k g을 가한 후 이질산암모늄과 이질산헥사민을 위에서 가해 준 양만큼 다시 다섯 번 가한다. 이때 이질산헥사민을 약간 과량으로 가해 주는 것이 더 효과적이라고 알려져 있다. 반웅액 의 마지막 부분을 가한 후 반응기 내의 혼합물 온도를 60 °C 로 가열한 후 30 분 동안 유지시킨다. 이때까지의 반응은 대략 4 시간 이 소요된다. 다시 20 °C 로 냉각시켜 석출되는 RDX 를 여과한 다. 대략 70~71 %의 수득률로 얻어전 RDX 의 용융점은 188 ~190·c 근처이다. 여과액 중 일부분은 재순환되어 반웅기에서 다시 반응되며, 나머지 여과액을 증류하면 반죽 모양의 불순물 함량이 높은 RDX( 용융점 : 160°C) 가 얻어진다. 이 불순물 함량 이 높은 RDX 혼합물은 반응기로 보내져 RDX 제조를 위해 다 시 사용된다. 여과된 RDX 를 물로 세척하면 30 % 초산 수용액 울 얻게 되며, 이 초산액을 초산에틸 용매로 추출하여 진한 초산 을 회수하게 된다. 초산에틸의 소모량은 RDX 100kg 정도가 필 요하다. 회수된 전한 초산으로부터 85 %의 무수초산이 Kete n e
법에 의해 얻어전다. KA 법에 의해 1000k g의 RDX 를 얻기 위해서는 다음과 같은 원료가 필요하다. 400 kg 헥사민 430kg 질산암모늄 680kg 99 % 질산 2400 kg 초산 (무수물) 그러나 초산 1950 k g이 회수되므로 초산의 순수 필요량은 450 kg 정도이다. 제 2 차 세계대전 도중 Bachmann 등은 위의 방법 과 거의 동일한 RDX 제조공정을 개발하였다 .95,95,97> Bachmann 의 제조원리는 헥사민 니트로화 반응과 E 법을 결합하는 것이었 다. Bachmann 공정에 의한 공장 규모의 설비가 1941 년 말부터 운영되기 시작했으며, RDX 가 생산되면서 초산의 재순환 사용 문제가 해결되었다. 미국에서는 주로 이 공정에 의해 RDX 를 제 조하고 있다. Bachmann 공정 에 서는 5~ 15 °C 로 반응온도를 유 지하면서 117 부분의 98 % 질산과 508 부분의 무수초산을 반응시 킨 다음 반응온도를 70~75 °C 로 울려준 후 다시 114 부분의 질산 암모늄과 192 부분의 이질산헥사민을 여러 부분으로 나누어 가한 다. 질산암모늄과 질산헥사민이 가해짐에 따라 열이 발생하면서 온도는 점차적으로 상승하게 된다. 이때부터는 가열을 정지하고 냉각기를 적당히 조절하여 반응온도를 73~78 °C 로 유지한다. 반 웅액이 완전히 주입된 후에도 가열하여 75°C 로 반응온도를 유지 하면서 15 분간 더 교반해 준다. 다시 60 °C 로 냉각한 후 여과하 며 초산과 물로 세척한다. 이렇게 얻어진 RDX 의 용융점은 203 ~204 °C 이며 수득률은 61~63 %(195~202 부분)이다. 만일 반웅 이 끝난 후 25 °C 까지 냉각시켜 여과하면 수득률은 70~73.5 %로
증가하지만 이 생성물 속에는 불순물로서 HMX 가 상당량 포함 되므로 용융점은 191~202 °C 로 낮아진다. 이와 같은 Bachmann 반응액과 헥사민의 반응에 의한 RDX 생 성 반응은 발열반응으로서 -L1H = 140 kcal/mole 의 반응열을 나 타냈다고 Gi lp i n 등은 보고하였다. 86) 질산헥 사민 (momonit ra te ) 과 Bachmann 반응액 의 경 우는 -L1H = 126 kcal/mole 의 반응열을, 이질산헥사민과의 경우는 ~L1H=118 kca l/ mole 의 반응열을 나타 내는 것으로 알려졌다. Elsnig 공장에서는 RDX 의 결정화를 다음과 같이 하고 있다. 즉, RDX 110 kg 정도를 밀폐된 탱크에 주입한다. 이 탱크 내부 는 모직 의 여 과천 (woolen filter cloth ) 으로 라이 닝 되 어 있고 교반 기가 장치된 1000l 의 용량이다. 대략 900l 정도의 아세톤 용매 롤 밀폐 탱크에 가하여 50°C 로 가열하며 RDX 를 용해시킨다. 용해된 RDX 용액은 여과천을 통과하여 탱크 아래쪽에 설치된 3000 l 용량의 탱크로 흘러내린다. 여과천은 10 시간 정도의 간격 으로 새것으로 교환한다. RDX 용액이 들어 있는 탱크 내부 온 도를 25 °C 로 유지 시 키 면서 5 분간에 걸쳐 1350 l 의 물을 가하게 되면 RDX 가 아세톤 용액으로부터 큰 입자의 결정 상태로 침전 되며 전공 여과장치를 사용하여 RDX 롤 분리한다. 석출된 RDX 입자 크기는 90 % 이상이 0.1 mm 이상이다. 한편, 아세톤 증기는 흡수기에 의해 회수된다. RDX 를 여과하 고 남은 물과 혼합된 아세톤 여과액은 증류공정을 통해 분리 정 제된다. 이 공정에서의 아세톤 손실은 RDX 100 k g당 7~8 % 정도이다. 이와 같이 얻어전 RDX 는 추진제, 화약 등의 공장으로 운반되 기 전 보통 둔감화 (desensit iza ti on , ph legm a ti za ti on ) 를 시 키 고 있 다. 독일 공장에서 사용하는 둔감화 공정은 다음과 같다.
가열 재 킷 이 나 냉 각 재 킷 이 장치 된 둔감화 탱 크(p hle g ma ti z i n g t ank) 에 450~500 l 의 물을 가하고 80~88 °C 로 가열한다. 170 rp m 정도로 교반시키면서 RDX 120 k g을 넣은 후 용융된 몬탄 왁스 (mont 1n wax) 를 RDX 무게 의 5 ~ 10 % 정 도 첨 가한다• 왁스 의 양을 적게 가했을 때는 온도를 더 높이 유지시켜 준다. 예를 들어 5% 의 왁스를 첨가한 경우는 대략 88°C 로, 10% 의 왁스를 사용한 경우는 80~82 °C 로 혼합액의 온도를 유지시킨다. 왁스를 모두 가해 준 후에는 곧 냉각수를 재킷에 통과시켜 혼합액 온도 롤 떨어뜨린다. 왁스를 둔감화시킨 RDX 는 전공여과기에서 여과 한 후 100°C 의 건조기에서 건조시킨다. 이와 같이 둔감화시키는 데 걸리는 시간은 대략 2 시간 정도이다. 또한 둔감화시킨 RDX 와 둔감화시키지 않은 RDX 를 구분하기 위해 둔감화시키는 탱크 내에 있는 RDX 에는 특정 염료를 가하기도 한다. 4.3.4.6 E 및 KA 법에 의한 RDX 합성 이론 RDX 의 구조와 유사한 구조를 지닌 여러 가지 부산물들이 생 성되는 반웅 조건들에 관하여 RDX 제조반웅 메커니즘과 관련하 여 검토되었다. 이러한 부산물들은 KA 법에서는 언제나 불순물 로 함유되어 나타나며, E 법에 의해서도 불순물로 RDX 와 섞이 게 된다. KA 법에서는 헥사메틸렌데트라민의 니트롤리시스 반응 이 일어나므로 생성된 알코올기는 무수초산으로 에스테르화된다 〔방법 (4.3.4.1) 에서는 무수질산으로 에스데르화됨〕. KA 법에 의 해 생성되는 가장 중요한 부산물은 거의 10 %의 수득률에 달하 는 HMX(24) 이다. 무수초산의 존재하에 이질산헥사메틸렌테트 라민은 생성물 (32) 로, 그 다음 단계에서는 (33) 으로 변환된다. 이 물질들은 물질 (18) 및 (19) 와 동일 계 열 화합물이 라고 볼 수 있다.
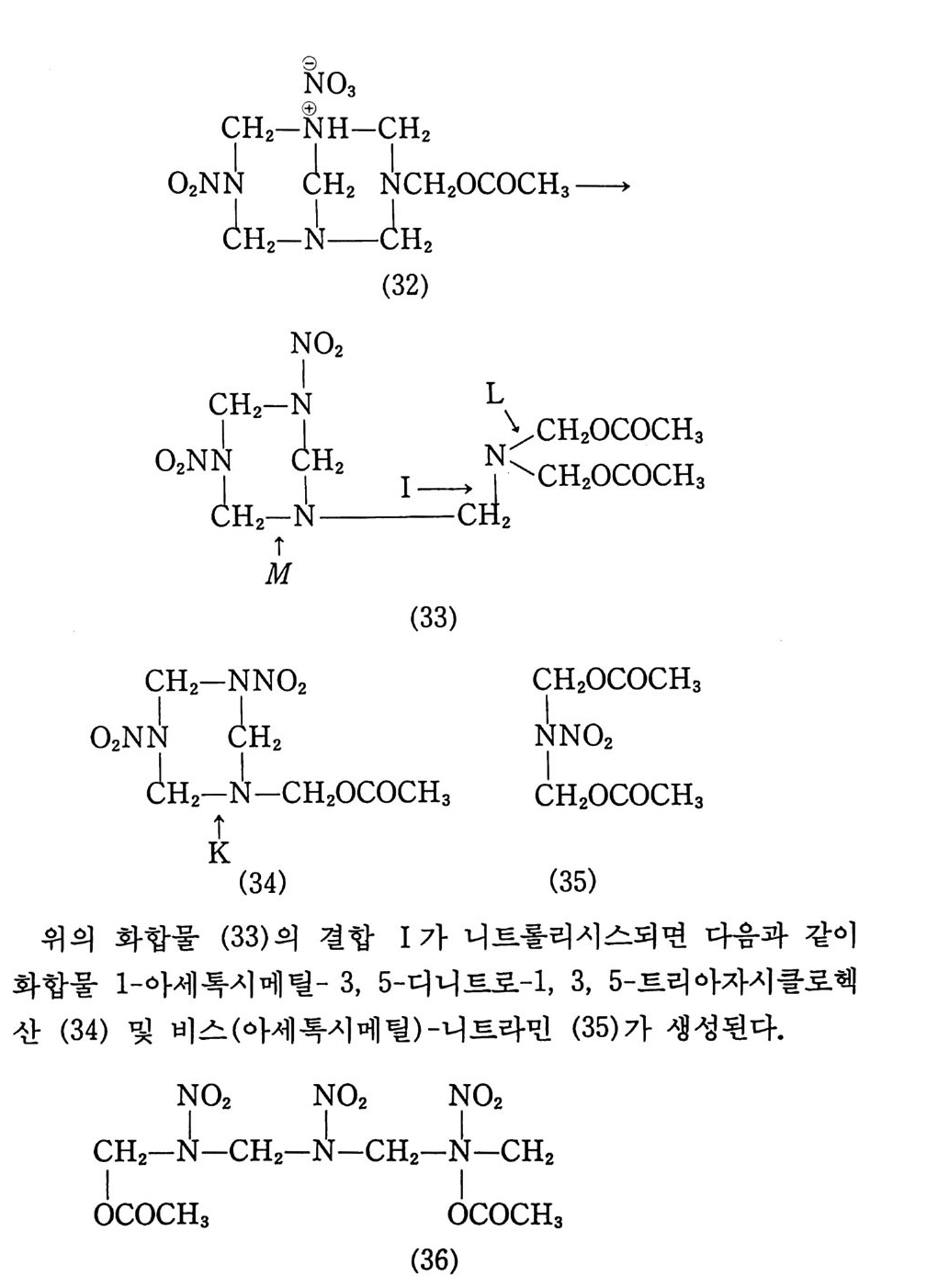 02NC야J H 2 2 —노 ~N詞H0 3 — CJ:HHc22H 2 0COCH 3一
02NC야J H 2 2 —노 ~N詞H0 3 — CJ:HHc22H 2 0COCH 3一
이 화합물 (34) 는 K 위치에 니트롤리시스 반응이 일어나 니트 라민에스테르인 (36) 을 생성한다. 이 화합물 (36) 은 E 법과 KA 법에 의한 RDX 제조시 상당량 합성되어지는데 RDX 에 비해 충격에 더 예민하고 안정성이 낮은 성질을 지닌 좋지 않은 불순물이다. Bachmann 등은 반응액을 비교적 낮은 온도 (0°C) 에서 교반한 후 곧 75°C 로 가열하게 되면 화합물 (36) 의 생성이 촉진됨울 보고하였다. 95) 한편, Wrig h t 등은 무수초산하에 (질산암모늄은 첨가하지 않은 상태에서) 헥사민을 질산으로 니트롤리시스하면 시클릭 (cy cl ic ) 화합물들의 생성이 감소되며 화합물 (36) 과 같은 사슬형 화합물 들의 생성이 촉진됨을 보고하였다 .98) 니트라민기들이 무수초산 존재하에 질산암모늄과 질산에 의해 합성된다는 것은 다음과 같 이 아미노메틸니트라민(니트라민과 포름알데히드로부터 얻어진 화합 물)을 질산과 질산암모늄 존재하에 55°C 에서 무수초산으로 처리 할 때 일어나는 반응에 의해 확인되었다 .99)
 NNN(IIIIC 0 HH_H_2 2) n 一 _NNN(I|_||C - 0—H 2C 2C-H)-H -2n2 一— -NN\/ \ ~_J_ /00 +ANCH 240N, OHaN, 5053° C
NNN(IIIIC 0 HH_H_2 2) n 一 _NNN(I|_||C - 0—H 2C 2C-H)-H -2n2 一— -NN\/ \ ~_J_ /00 +ANCH 240N, OHaN, 5053° C
화합물 (36) 은 니트롤리시스되어 에스데르 화합물인 1,5- 디아 세톡시 -2,4- 디니트로 -2,4- 디아자펜탄 (37) 을 생성한다.
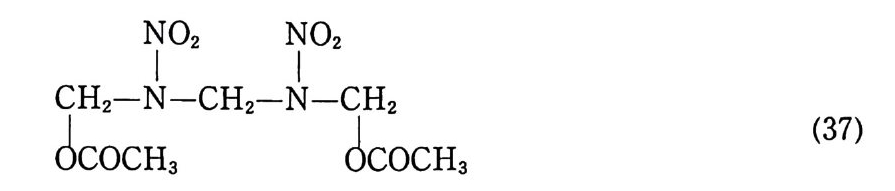 CdcHo2c-HNN30— 2C H 2-NN0— 2C dHc2o cH3 (37)
CdcHo2c-HNN30— 2C H 2-NN0— 2C dHc2o cH3 (37)
또한 화합물 (33) 의 화학결합 L 및 M 위치에 니트롤리시스 반응이 일어나면 용융점 182.5~183.5 °C 의 화합물 1,9- 디아세톡 시 -2,4,6,8- 데 트라니 트로 -2,4,6,8- 데 트라자노난 (38) 이 생 성 된다.
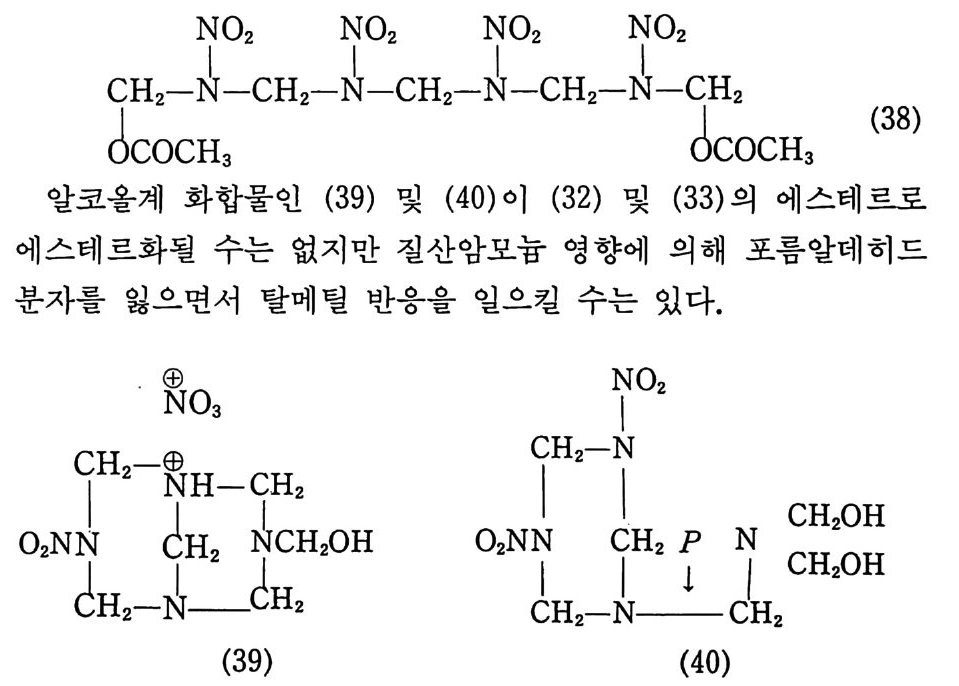 CH2-NN0— 2C H 2-NN0-2C H2— NN0-2 C H2-NN-0C2 H2 (38)
CH2-NN0— 2C H 2-NN0-2C H2— NN0-2 C H2-NN-0C2 H2 (38)
만일 상기 화합물 (39) 가 탈메틸화된 후 니트로화 반응이 계속 되 면 DPT (25) 가 합성 된다. KA 법에 의한 RDX 합성에서 질산암모늄은 필요한 숫자만큼의
니트라민기를 공급할 뿐만 아니라, (38) 및 (40) 형태의 중간체 화합물로부터 포름알데히드 분자들이 빠져나가는 반응이 일어나 도록 한다. 만일 알코올 화합물 (36) 이 에스데르화될 만한 반응 시간이 충분치 않으면 이 화합물은 P- 결합에 니트롤리시스 반응 이 일어나서〔화합물 (33) 의 경우처럼 L,M,I- 결합에서가 아닌〕 RDX 가 생성된다. 실험에 의하면 (36), (37), (38) 및 (22) 와 같은 사슬형 화합물들은 질산, 무수초산, 질산암모늄 존재하에 시클릭 화합물로 변환한다. 100) 파라포름알데히드, 질산암모늄 및 초산으로부터 RDX 를 합성 할 때는 주생성물인 RDX, HMX 의에 시클릭 구조의 화합물들 이 다음과 같은 화합물로 가정될 수 있는 중간물질인 메틸렌니트 라민 (41) 의 중합에 의해 부산물로 얻어진다 .101)
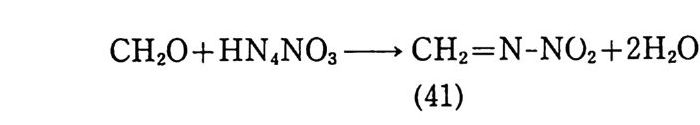 CH20+ HN4NOa —+ CH2=N-N02 + 2H20
CH20+ HN4NOa —+ CH2=N-N02 + 2H20
Wrig h t 등이 주장하는 또 다른 의견에 의하면 처음에는 헥사 메틸렌테트라민이 합성되고 그 다음 단계에서 니트롤리시스 반웅 이 일어난다고 한다. Wrig h t 등에 의한 이 가정은 E 법과 KA 법 의 반응 생 성물로부터 1- 아세토 -3, 5- 디 니트로 -1, 3, 5- 트리 아자시 클로핵 산 (42) 및 1- 아세 토 -3, 5, 7- 트리 니 트로 -1, 3, 5, 7- 테 트라아자 시클로옥탄 (43) 의 시클릭 화합물들을 분리하였다는 점 및 이 화 합물들은 질산과 무수초산으로 헥사민을 처리할 때 얻어질 수 있 다는 점들에 의해 그 실험적 근거를 두고 있다. 생성물 (42) 1- 아세토 -3,5- 디니트로 -1,3,5- 트리아자시클로핵산 (42) 및 생성물 1- 아세토 -3, 5, 7- 트리니트로 -l, 3, 5, 7- 테트라자시 클로옥탄 (43) 의 구조식은 다음과 갇다.
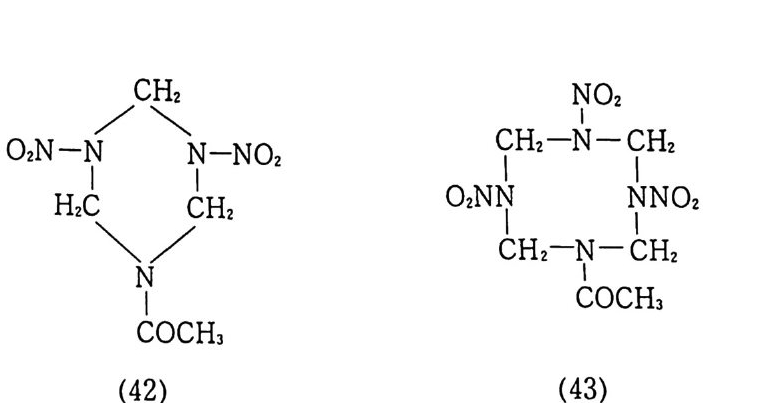 OzNH-2yNC/ C\Hz CN H-Nz Oz 02NNCCI H H22— -헌NNCI 0O—— 2 C CNCHIH HN32 2 0 2
OzNH-2yNC/ C\Hz CN H-Nz Oz 02NNCCI H H22— -헌NNCI 0O—— 2 C CNCHIH HN32 2 0 2
E 법 및 KA 법에서의 반응 생성물들은 또 다른 시클릭 N- 아세 틸 유도체들을 포함하고 있다. 죽, 1,5- 디아세토 -3,7- 엔도메틸렌 -1, 3, 5, 7- 테트라자시클로옥탄 (44) 및 1, 5- 디아세토 -3, 7- 디니트로 -1,3,5,7- 데트라자시클로옥탄 (45) 등이 있다.
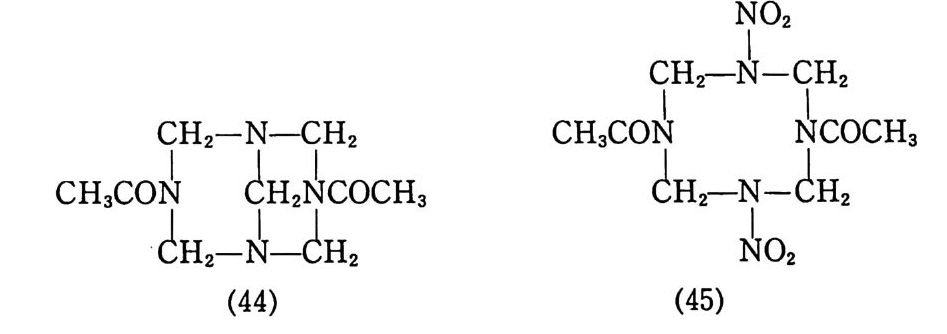 CH2— N- CH2 CH3COCNIH 2— -~N0- 2C NHI C2O CH3
CH2— N- CH2 CH3COCNIH 2— -~N0- 2C NHI C2O CH3
Marcus 등에 의하면 무수초산과 질산 영향하에 화합물 (43) 은 화합물 (38) 을 생 성 한다. 102> W ink ler 등은 E 법 에서 핵 사민 중간체 생성을 확인하였으며 동시에 RDX 제조반응 메커니즘을 조사하였다. 그들은 이질산헥사민 (din i t ra te ) 이 중간체로서 35 °C 에서 생성되는 것을 발견하였으며, 질산이 발생하는 동시에 이질 산헥사민의 니트롤리시스 반옹이 일어나므로 RDX 생성 메커니 즘은 핵사민을 질산으로 직접 니트로화하는 것과 유사하다고 보 고하였다. 이러한 관찰결과로부터, 과량의 포름알데히드가 RDX
의 수득률에 나쁜 영향을 미치는 이유를 설명해 주게 되었다. 특 히 W i nkler 는 초산 수용액 속에서 헥사민 용액에 포름알데히드 를 가하면 헥 사민 이 분해 (decomp o sit ion ) 되 며 , 그 분해 속도는 헥 사민에 대한 포름알데히드 비율에 의해 결정된다는 현상을 발견 하였다. 4.4 HMX HMX (Hi gh Meltin g Exp lo siv e ) (24) 는 Oc t o g en 으로 잘 알려 져 있 고, 화 학 명 은 cy c lote t r a meth y le ne-te t r a mi ne , 1, 3, 5, 7 -tetr a nit ro -1, 3, 5, 7- t e t razac y clo-oc t ane 으로서 흰색 결정 이 며 용융점이 276~277 °C 이다. HMX 는 몇 가지 종류의 결정 상태 로 존재하며 각 결정 상태별로 비중 및 충격에 대한 예민성 등이 서로 차이가 있다. 보통 사용되는 HMX 는 /3-결정형으로서 충격 감도가 가장 우수한 것이다(표 4.7). McCrone 에 의 하면 HMX 가 다형 태 성 (po lym orph ic ) 으로서 4 개 의 상태, 죽 I(/3 ), II (a), IIHr), IV(o) 형태들로 존재하고 있음 을 밝혔다. I , II, III 형태는 상온에서 안정하지만 HMX-IV 는 불안정하여 쉽게 다른 형태로 변환된다. 103) HMX-I (/3) 형태는 몇 가지 방법으로 제조될 수가 있다. Wrig h t 등은 뜨거운 아세톤이나 아세토니트릴 용매에 HMX 를 포화시킨 후 냉각하여 석출시키는 방법을 제안하였다 .l04) HMX -II(a) 형태는 HMX 를 70 % 질산에 용해시킨 후 냉각하여 침전 시켜 제조한다. HMX-III(r) 형태는 물을 포화시킨 시클로핵사 논 (c y clohexanone) 의 뜨거운 용매 속에 용해 된 HMX 용액으로부 터 용매를 스팀 증류 (s t eam d i s tilli n g)함으로써 얻어진다. 또한
HMX-1 을 180 °C 에서 승화시키면 HMX-IV 가 얻어진다.
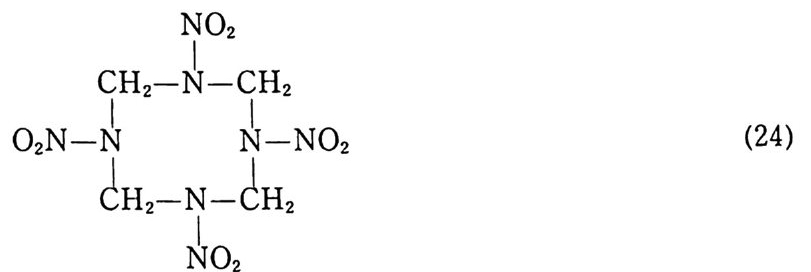 02N— CNH 2-N부 0— 2 CNH-2 N 02 (24)
02N— CNH 2-N부 0— 2 CNH-2 N 02 (24)
Wrig h t 등은 X- 선 회절, 적의선 스펙트럼 및 유전상수 (die l ectr i c consta n t) 등을 측정 하여 HMX 의 결정 상태 를 조사한 후, HMX 의 다형태성이라는 것이 실제로는 결정격자 내부에서 일 어 나는 형 태 변 형 (confo r mati on al modif ica ti on ) 이 라고 결론지 었 다 .104) HMX 는 RDX 와 마찬가지로 물에 불용이며 비홉습성이다. 유 기 용매에 대한 용해도 역시 RDX 와 비슷하며, RDX 나 HMX 는 그 화학적 반응성도 유사하다. 그러나 가성소다에 대한 반응성은 HMX 가 RDX 보다 작으므로 이 성질을 이용해 HMX 와 RDX 를 분리해 낸다. 즉, 가성소다 용매 내에서 HMX 와 RDX 혼합물 울 가열하면 RDX 는 분해되고 HMX 는 변화되지 않은 상태로 남도록 하여 분리한다.
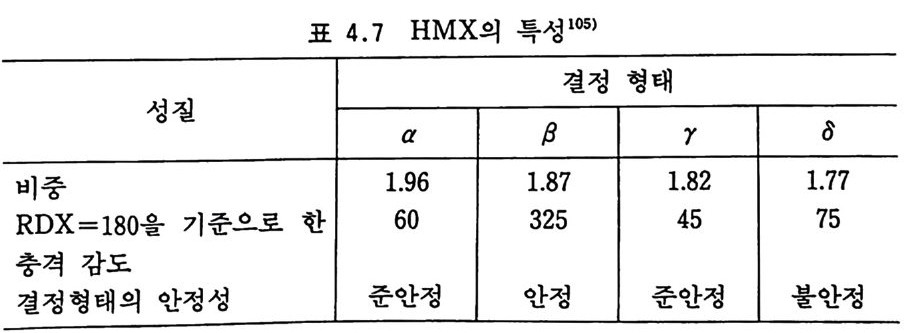 표 4.7 HMX 의 특성 105)
표 4.7 HMX 의 특성 105)
RDX 로부터 HMX 를 분리하는 또 다른 방법은 두 물질의 용 해도 차이룰 이용하는 것으로서, HMX 는 RDX 보다 55 % 질산 또는 2- 니트로프로판에 더 쉽게 용해되는 성질을 갖고 있기 때문 이다. 죽, HMX 와 RDX 혼합물을 질산 속에서 따뜻하게 가열 하고 여과하게 되면 HMX 가 풍부한 혼합물은 결정화되어 여과 액과 분리된다. 혼합액 (여과액)을 2- 니트로프로판으로 추출하면 RDX 는 2- 니트로프로판에 용해 추출되며, HMX 는 혼합액 속에 남게 된다. HMX 는 다시 70 % 질산을 사용하여 결정화시켜 정 제한다. HMX 를 많이 함유하고 있는 HMX 와 RDX 혼합물은 상기 KA 법에 의해, 죽 질산헥사민, 질산암모늄 및 무수초산으 로부터 제조하는 것이 최선의 방법이다. 특히 Bachmann 방법 에 따라 73 ~78 °C 범 위 의 온도에 서 HMX 와 RDX 혼합물을 생성시킨 후 25°C 로 냉각 여과하는 공 정이 HMX 의 함유율을 더 높이는 것으로 알려졌다• 이때의 혼 합물은 용융점 191 ~202 °C 를 나타내 며 대 략 10 %의 HMX 를 포 함하게 된다. Ep s te i n 등은 KA 법 에 의 해 생 성 되는 RDX 에 대 한 HMX 의 비율을 조사하였고, 이때 반응에 사용된 질산암모늄 양을 감소시 키면 RDX 의 양이 감소되고 상대적으로 HMX 생성량이 증가하 는 경향을 발견하였다 .106) 그 실험결과를 보면 그림 4 . 15 와 같이 질산암모늄 2 몰과 헥사민 1 몰의 비율로 반응시킬 때 HMX 생성 량이 최대값을 나타냈다. Bachmann 등은 15~30 °C 의 온도에서 초산과 무수초산 존재 하에 헥사민과 질산으로부터 화합물 (25) (DPT) 를 대략 20 %의 수득률로 제조하였다고 보고하였다 .97) 이 화합물을 질산암모늄 및 무수초산 속에서 60~65°C 의 온도하에 질산으로 니트롤리시 스하면 HMX 를 80 % 수득률로 얻게 된다.
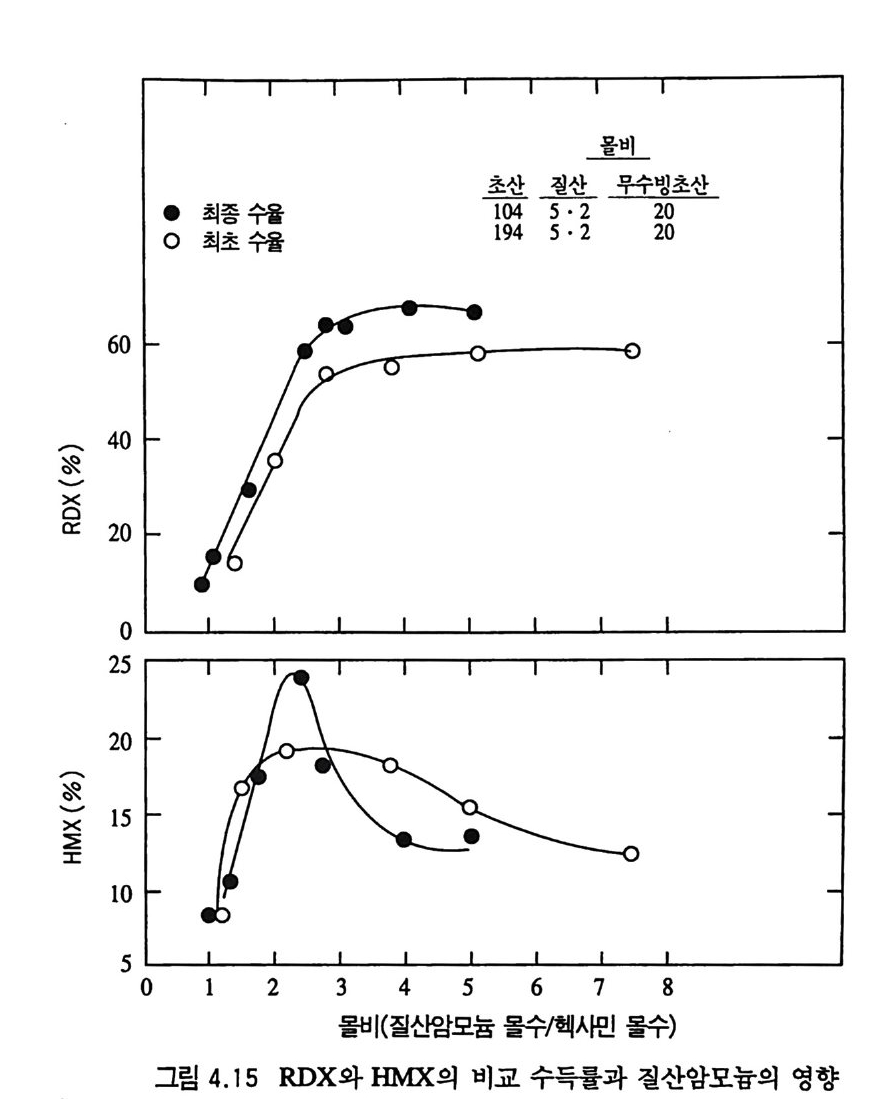 •°최최 종초수수율율 초1109산44 55질 ••산 22 몰비무 수22빙00초 산
•°최최 종초수수율율 초1109산44 55질 ••산 22 몰비무 수22빙00초 산
폭발물로서 HMX 는 RDX 보다 성능이 더 우수하댜 예를 들 면 접화온도의 경우 HMX 는 335 °C 에서 5 초 이내에 폭발하는 반면 RDX 는 260 °C 에서 폭발한다. 폭굉속도 역시 HMX 가 훨씬 빠른 것으로 보고되어 있다. 화학적 안정성 역시 HMX 가 더 우 수하다. 죽, 진공하에 HMX 는 120 °C 에서 40 시 간 동안 0.4 cm3
가스를, 150 °C 에 서 0.6 cm3 를 발생 하는 반면 RDX 는 각각의 경 우 0.9 c m3 및 2.5 cm3 의 가스를 발생 한다. 150 °C 에 서 의 안정 성 데이터를 보면 HMX 는 TNT 나 피크린산과 같은 수준의 안정성 울 나타내고 있다. Rober t son 에 의 하면 280 °C 이 상의 온도에 서 HMX 는 분해반응이 일어나며 그 활성화 에너지는 E=52kcal, log B=19.7 로 보고되었다• 65) 〔참고문헌〕 1) R. S. Sp e a and W. S. Wi lso n, MRL-R-850, Commonwealth of Austr a li a (1982) . 2) T. Urbanski, Chemi stry and Technology of Ex plo siv e s, Vol. III, Eng lish Ed., Perga mon Press, New York (1985) . 3) A. Davenas, Solid Rocket Prop ul sio n Technology Chap ter 11, Per- ga mon Press, Oxfo r d (1993) . 4) W. Costa i n and E. G. Cox, Natu r e 160, 826 (1947) . 5) F. J. Llewellyn and F. E. Whit mo re, ]. Chem. Soc. 1948, 1316. 6) R. N. Jon es and G. D. Thom, Can. ]. Research 27 B, 828 (1949) . 7) E. Lieb er, D. R. Leverin g and L. J. Patt er son, Anal. Chem. 23, 1594 (1951) . 8) G. S. Salya m on and N. G. Yaroslavskii , Sbornik Sta tei Obshch. Chim . 2, 1325 (1953) ; G. S. Salya m on and Ya. Bobovic h , ibi d . 2 , 1332 (1953). 9) L. J. Bellamy , The Infr a -red Sp ec tr a of Comp le x Molecules, Meth - uen, London (1958) . 10) A. Hantz s ch and F. E. Dollefu s, Ber, 52, 258 (1902) . 11) H. Euler, Ber. 39, 1607 (1906) . 12) M. I. Gil libr and and A. H. Lamberto n , ]. Cheml Soc. 1949, 1883. 13) H. van Erp , Rec. tra v. chim . 14, 40 (1895) .
14) H. J. Backer, Di e Ni tram i ne -Ahrens Sammlung 18, Stu t t - ga rt, 1912. 15) A. P. N. Franchir n ont, Rec. tra v. chim 29, 296 (1910) . 16) E. Bamberge r and K. Landste i n e r, Ber. 26, 490 (1893) . 17) E. D. Hug h es and C. K. Ing o ld, Qua rt . Rev. 6, 34 (1952) . 18) A. Hantz s ch and W. V. Metc a lf , Ber. 2 9, 1680 (1896) . 19) J. Barrott , M. I. Gil libr and and A. H. Lamberto n , ]. Chem. Soc. 1951 , 1282. 20) H. van Erp and A. P. N. Franchir n ont, Rec. tra v. chim . 14, 224 (1895) . . 21) W. C. Cop e and ]. Barab, ]. Am. Chem. Soc. 38, 2552 (1916) ; see also K. Lehrnste d t and 0. Zumste i n , Ber. 58, 2024 (1925) . 22) E. Bamberge r , Ber. 30, 1248 (1897) . 23) T. S. Ste v ens, accordin g to A. H. Lamberto n [42] . 24) D. Woodcock, ]. Chem. Soc. 1949, 1635. 25) A. H. Lamberto n , C. Lind ley, P. G. Owsto n and J. C. Sp ea kman, ]. Chem. Soc. 1949, 1641 . 26) R. Scholl et al., Ber. 37, 4427 (1904) ; R. Scholl and A. Krie g e r, ibi d . 37, 4681 (1904) . 27) A. E. Chic h ib a bin and B. Razorenov, Zh. Russ. Khim . Obshch. 47, 1280 (1915) . 28) K. Ganap a th i and A. Venkata r aman, Proc. I ndia n Acad. Sci. 22 A, 343 (1945) . 29) J. B. Dic k ey, E. B. Towne and G. F. Wrig h t, ]. Org. Chem. 20, 449 (1955) . 30) K. J. P. Orto n , ]. Chem. Soc. 81, 806 (1902) . 31) A. P. N. Franchim ont and J. V. Dubsky , Rec. tra v. chim . 36, 80 (1916) . 32) W. J. Chute , G. E. Dunn, J. C. Mackenzie , G. S. My er s, G. N. R. Smart, J. W. Sug gitt and C. F. Wrig h t, Can. ]. Research 26 B, 114 (1948) .
33) G. E. Dunn, J. C. Mackenzie and G. F. Wrig h t, ibi d . 26 B, 104 (1948) . 34) K Bamberge r and A. Ki rp a l, Ber. 28, 462, 535 (1895) . 35) W . J. C hute , K. G. Herrin g , L. E. Toombs and G. F. Wrig h t, Can. ]. Research 26 B, 89 (1948) . 36) A. P. N. ranchim ont, Rec. tra v. chim . 13, 308 (18 94) . 37) R. C. Bria n and A. H. Lamberto n , ]. Chem. Soc. 1949, 1633. 38) A. Berg, Ann. Chim . 3, 358 (1894) . 39) J. C . Mackenzie , G. S. My e rs, G. N. R. Smart and G. F. Wrig h t, Can. ]. Research 26 B, 138 (1948) ; G. S. My e rs and G. F. Wrig h t, ibi d . 26 B, 257 (1948) ; G. N. R. Smart and G. F. Wrig h t ibi d 26 B, 284 (1948) ; J. W. Sug gitt, G . S. My e rs and G. F. Wrig h t, ]. Org. Chem. 12, 373 (1947) . 40) G. F. Wrig h t in H. Gil m an's Orga nic Chemi stry, Vol IV, p. 951, J. Wi ley , New York (19 53) . 41) G. S. My e rs and G. F. Wrig h t, Can. ]. Research 22 B, 257 (1948) . 42) A. H. Lambert on , Qu art . Rev. 5, 75 (1951) . 43) R. P. Lins te a d, unp u bli sh ed; qu ote d by A. H. Lamberto n [42] . 44) F. Lenze, accordin g to H. Kast, Sp re ng- u . Zundstof f e, V i ew eg & Sohn, Braunschweig (1921) . 45) G. F. Hennin g , Ger. Pat. 104280 (1899) . 46) G. F. Hennin g , Ger. Pat. 298412, 298539, 299028 (1916) . 47) G. C. V. Herz, Brit. Pat. 145793 (1921) ; U.S . Pat. 1402693 (1922) . 48) G. C. Hale, ]. Am. Chem. Soc. 47, 2754 (1925) . 49) P. Terps tr a , Z. Kr ist. 64, 150 (1924) . 50) R. Hultg ren , ]. Chem. Phys . 4, 84 (1936) . 51) G. Edwards, Trans. Faraday Soc. 49, 152 (1953) . 52) A. Majr i c h , Mem. art ill. fra nc. 14, 127 (1935) . 53) T. Urbanski and B. Kwi at k owski, Rocznik i Chem. 13, 585 (1933) 54) T. Urbanski and I. Rabek-Gawronska, Rocznik i Chem. 14, 239 (1934).
55) M. Delep ine , Bull. soc. Chim . France [4] , 9, 1025 (1911) 56) E. Vernazza, At ti reale accad. sci . T or ino 70, I, 404 (1934/35) . 57) J. Sim eck, Chem. Lis ty 51, 1323, 1699 (1957) ; XVI Cong e rs Inte r -nati on al de la Chim i e Pure et Ap pliqu ee II , p. 310, Paris (1957) . 58) F. Somlo, Z. ge s. Schie s s-u . Sp re ngs t of f w . 35, 175 (1940) . 59) S. Ep s te i n and C. A. Wi nk ler, Can. ]. Chem. 29, 731 (1951) . 60) W. H. Jon es, ]. Am. Chem. Soc. 76, 829 (1954) . 61) M. Avog a dro, Mem. art ill. Franc. 10, 875 (1931) . 62) T. Urbanski and J. Pil lich , Wi ad . Techn. Uzbr. 43, 79 (1939) . 63) M. Toneg utti, Chim i ca l e ind ustr ia 17, 517 (1935) . 64) A. Haid , F. Becker and P. Drrtm ar, Z. ge s. Schie s s- u. Sp re ng- stof f w . 30, 66, 105 (1935) . 65) A. J. B. Roberts o n, Trans. Faraday Soc. 45, 85 (1949) . 66) T. Urbanski and W. Krawzy k , Wi ad . Techn. Uzbr. 45, 490 (1939) . 67) T. Urbanski and W. Malendowi cx z, Rocznik i Chem. 18, 856 (1938) . 68) M. Toneg utti, Z. ge s. Schie s s-u . Sp re ngs to ff w . 32, 93 (1947) . 69) L. Medard, XXVII Cong res Chim i e Industr ., Brussel, Chim i e et ind ustr ie, No. spe cia l, 81 (19 54) . 71) T. Urbanski, Przemy s l chem. 20, 117, 179 (1936) ; Z. ges . Schie s s-u . Sp re ng st of f w . 33, 41, 62. (1938) . 72) A. Izzo, Riv . art igli er i a e ge nio 373 (1932) . 73) K. K. Andreye v, Thermal Decomp os it ion and Burnin g of Explo - siv e s (in Russia n ) , Gosenergo iz d at, Moskva-Lenin g rad (1957) . 74) Techn ica l Rep o rt P .B. 925, General summary of Exp lo siv e Plants , D. A. G. Krummel, Duneberg, Chris t i an sta d t, U.S. Dep t. of Com-merce, Washin g ton , 1945; Techn ica l Rep o rt P.B. 262, RDX Manu- fac tu re in Germany, U.S. Dept . of Commerce, Washi ng ton , 1945; W. De C. Crate r , Ind. Eng. Chem. 40, 1627 (1948) . 75) E. L. Hirs t, A. Carruth e rs, W. J. Dunnin g , J. K. N. Jon es, H. D. Sp ri n ga ll et al., unp u bli sh ed, accordi ng to a. H. Lambert on [79] .
76) A. H. Vroom and C. A. Wi nk ler, Can. ]. Research 28 B, 701 (1950) . 77) G. S. My e rs and G. F. Wrig h t, Can. ]. Research 27 B, 489 (1949) . 78) W. J. Chute , D. C. Downin g , A. F. McKay, G. S. My e rs and G. F. Wrig h t, Can. ]. Research 27 B, 218 (1949) . 79) A. H. Lamberto n , Qu art. Rev. 5, 75 (1951) . 80) G. F. Wrig h t, Gil m an 's Orga nic Chemi stry, Vol. IV, p.9 83, J. Wil ey , New York (1953) . 81) K. Sin g h , ]. Sc i. Ind. Research (Ind ia ) A 15, 450 (1956) . 82) L. Berman, R. H. Meen and G. F. Wrig h t, Can. ]. Research 29 B, 767 (1951) . 83) K. W. Dunnin g and W. J. Dunnin g , ]. Chem. Soc. 1950, 2920, 2925, 2928. 84) P. P. Karp u kh in and W. N. Chety rk in , Trudy Kharkovsk. Khin . -Technol. Inst. Ki ro va 4, 143 (19 44) ; Chem. Abstr . 42, 5918 (1948) . 85) W. J. Dunnin g , B. Mi llar d and C. W. Nutt , ]. Chem. Soc. 1952, 1264. 86) V. Gil pin and C. A. Wi nk ler, Can. ]. Research 30 B, 743 (1952) . 87) M. Ki rs ch and C. A. Wi nk ler, Can. ]. Research 28 B, 715 (1950) . 88) T. Urbanski, unp u bli sh ed (1936) ; ref. [2] · 89) F. Grott an ell i, Chim i ca e ind ustr ia 20, 801 (1938) . 90) W. H. Sim mons, A. Forste r and R. C. Bowden, Ind. Chemi st 24, 530, 593 (1948) . 91) R. W. Schie s sler and J. H. Ross, Brit. Pat. 595354 (1947) ; U. S. Pat. 2434230 (1948) . 92) E. Aris t o f f , J. A. Graham, R. H. Meen, G. S. My e rs and G. F. Wrig h t, _ C an. ]. Research 27 B, 520 · (1949) . 93) A. Gi llies, H. L. Wi lliam s and C. A. Wi nk ler, Can. ]. Chem. 29, 377 (1951) . 94) Techn ica l Rep o rt P. B. 4272, Hexog e n Manufa c tu r e in Bobin ~ en, U. S. Dept . of Commerce, Washi ng ton , 1945; CIOS XXXII 8, G.m.b. H. zur Verwertu n g Chemi sc her Erzeug niss e, Fabrik Bobin g e n.
95) W. E. Bachmann and J. C. Sheehan, ]. Am. Chem. Soc. 71, 1842 (19 49) . 96) W . E. Bachmann, W. J. Horto n , E. L. Jen ner, N. W. Mac-Naug h to n and C. E. Maxwell, ]. Am. Chem. Soc. 72, 3132 (1950) . 97) W. E. Bachmann, W. J. Horto n , E. L. Jen ner, N. W. MacNaug h to n and L. B. Scott , ] . Am. Chem. Soc. 73, 2769 (1951) ; W. E. Bachmann, and E. L. Jen ner, ibi d . 73, 2773 (1951) ; W. E. Bachmann, E. L. Jen ner, a nd L. B. Scott , ibi d . 73, 2775 (1951) . 98) W. J. Chute , A. F. McKay, R. H. Meen, G. S. My e rs and G. F. Wrig h t, Can. ]. Researc h 27 B, 503 (1949) . 99) R. C. Bria n , F. Chapm an, a. H. Lamberto n , C. Lind ley, P. G. Owsto n , J. C. Sp e akman and D. Woodcock, Chemi stry & Industr y 223 (1949) . 100) F. Chap m an, ]. Chem. Soc. 1 949, 1631; F. Chap m an, P. G. Owsto n and D. Woodcock, ibi d . 1949, 1647. 101) e.g . E. E. Roberts , unp u blis h ed; ref. [2] • 102) R. A. Marcus and C. A. Wi nk ler, Can. ]. Chem. 31, 602 (1953) . 103) W. C. McCrone, Anal. Chem. 22, 1225 (1950) . 104) M. Bedard, H. Huber, J. L. Meye rs and G. F. Wrig h t, Can. ]. Chem. 40, 2278 (1962) . 105) W. H. Rink enbach in Ki rk & Oth m er Ency cl op ed ia of Chemi ca l Technology 6, 41, Inte r sci en ce, New York (1951) . 106) S. Ep s te i n and C. A. W ink ler, Can. ]. Chem. 30, 734 (1952) .
제 5 장 차세대 추전제와 고에너지 물질 고체 추진제는 오래 전부터 사용되던 흑색화약으로부터 니트로 셀룰로오스, 니트로글리세린, 니트로구아니딘 동의 원료를 주성 분으로 제 조된 단기 (sin g le base) , 복기 (double base) , 다기 (multi base) 추진제를 거쳐 최근에는 폴리머류의 바인더, 금속연료, 산 화제 등을 주성분으로 하는 혼합형 추전제가 개발되어 각종 총 • 포류 화약 및 전술 • 전략 유도탄, 로켓 추전제로서 또는 위성체 발사용 로켓 추전제로서 적용되고 있다. l,2) 1960 년대 이후 급격 히 발전한 고분자 및 정밀화학 기술에 힘입어 혼합형 추전제는 그 물리적 성질, 기계적 성질, 내탄도적 성질 등 각종 성능 및 제작 공정성이 개선되었다 .3) 이와 같이 각종 성능 및 제작성이 우수해침에 따라 대형 추전 기관용 추전제 제작이 가능해지고 대륙간 탄도탄과 같은 군사용 유도탄 추전제는 물론 위성발사 로켓용 추전제와 같은 민수용으 로서도 그 수요가 접차 증가하게 되었다. 그러나 추전제의 성능, 제작성 등의 기술이 크게 발전하였음에도 불구하고 다음과 같이
해결해야 될 문제점들이 남아 있다 .4,5) • 추전제 연소시 배출되는 유연의 배기가스로 인한 위치 노출 및 송수신 교란 • 추전제 연소가스로 인한 환경오염 • 고의 또는 우연한 사고에 대비한 둔감화의 필요성 • 더 높은 비추력을 갖는 고성능 추전제의 필요성 이상과 같은 문제점들이 해결된 추전제는 연소시 환경을 오염 시키지 않는 깨끗한 무연의 가스를 방출하며 그 추진제의 비추력 은 기존의 혼합형 추진제와 동등하거나 그 이상으로 높아야 하 며, 그 추진제가 충전된 추전기관 모터가 의도적이든 우연이든 사고(충격, 화재 등)를 당했을 때 폭발(폭굉)하지 않고 안전하게 소화시킬 수 있을 정도로 안전한(둔감한) 추전제이다. 이와 같은 이상적인 추진제를 제작한다는 것은 현재로서는 불가능한 것같이 생각되지만 새로운 고에너지 물질들이 개발되고 있고 추진기관 모터 설계, 제작, 연소 이론 등이 발전하고 있으므로 머지않아 이상형에 가까운 차세대 추전제가 등장할 것으로 추측된다. 본문 에서는 차세대 추전제 개발을 위해 연구되고 있는 기술을 크게 분류하여 무연 및 청정 추전제 기술, 둔감 추진제, 고에너지 물 질로 나누어 기술하였다. 추전제의 고에너지화 기술은 추진제를 무연으로 하든 둔감화를 하든 추진제 에너지를 높여야 하는 것은 공통된 목표 기술이므로 별도로 분리하지 않고 공통사항으로 필 요시마다 언급하였다.
5.l 무연 및 스카벤저 추전제 무연 추진제와 스카벤저 (scarveng e r : 청정) 추진제는 그 연소 배기가스가 무연이나 무연에 가까운 특성을 지녀야 한다는 점 및 추전제 조성을 개발하는 기술적 접근 방법이 서로 비슷한 점을 고려하여 함께 취급하였으나 그 필요성 및 목표는 완전히 다른 점에서 출발하였다. 죽, 무연 추진제는 연소가스로 인한 로켓 위 치 노출방지 및 유도조종 방해 방지 등의 전술 • 전략적 이유로 인해, 청정 추진제는 연소 배기가스로 인한 대기오염, 공해의 방 지를 위해 그 연구가 진행되었다고 볼 수 있다 .6,7) 니트로셀룰로오스를 주성분으로 하는 균일 추전제의 경우는 추 진제 속에 금속분말을 첨가하지 않으면 연소 배기가스의 주성분 이 일산화탄소, 이산화탄소, 수분, 질소산화물 등으로서 유색의 배기가스에 대한 염려가 크게 없었다. 그러나 내탄도 성능 및 물 리적 성질이 크게 향상된 혼합형 추전제의 경우는 바인더, 금속 분말, 산화제의 원료 조성에 따라 유연의 배기가스가 대량 배출 된다. 비추력을 230 sec 이상 (1000 psi a ) 올릴 수 있는 혼합형 추 진제의 산화제로는 과염소산암모늄을 대부분 적용하고 있는데 추 진제 연소시 과염소산암모늄이 분해 연소될 때 염화수소 가스를 방출한다. 염화수소 가스는 함께 방출되는 수분 및 대기 중의 수 분과 섞 여 스모그 유색 가스로 방출될 뿐만 아니 라 오존층 파괴 및 대기를 오염시키고 주변 동식물들을 크게 해치는 유독가스로 작용한다. 이러한 혼합형 추전제에 비추력을 더욱 높이기 위해 알루미늄 분말을 첨가하게 되면 산화알루미늄 (Al2 아)의 분말이 연소가스에 섞여 방출되므로 그 연소 배기가스의 색깔이 더욱 검 게 보인다.
추전제 연소시 배출하는 유연 배기가스의 수준을 비교할 때 혼 히 일차스모크라고 부르는 것은 산화알루미늄 분말과 갇은 고체 입자가 배기가스에 섞여 방출될 때의 것으로, 이차스모크는 과산 화암모늄 연소에 의해 발생되는 염화수소와 같은 할로겐화 물질 이 배기가스에 섞여 방출되는 유연의 배기가스를 의미하고 있다. 이와 같은 유연의 배기가스가 로켓이나 유도탄 추진기관 모터로 부터 배출될 때는 전술 • 전략적으로 몇 가지 심각한 문제점을 야 기시키게 된다. 죽, 추진제 연소로 인한 유연 배기가스 영향으로 인해 발사된 위치가 적에게 쉽게 노출되고, 유도무기의 경우 그 유연가스로 인해 비행 위치가 계속 노출되며, 유도무기 작동에 필요한 조종장치 작동이 간섭이나 방해롤 받게 된다. 그 의에도 발사대를 위시한 발사장비가 부식되는 문제점들이 있다. 추전제 조성 중 알루미늄과 같은 금속연료 성분을 제거하거나 극소화 (1~2 % 이하)하면 연소가스 중 일차스모크가 거의 없는 추진제를 만들 수 있는데, 이러한 추진제를 저연 (reduced) 스모 크 추진제라고 부르며, 연소가스 중에 염화수소 가스도 배출되지 않는 추전제 를 미 니 멈 (mi ni m um) 스모크 추전제 라고 부른다. 2,3) 한편, 1970 년대 위성운반체 공동기구에서 위성운반체 주변지역 의 환경공해가 심각할지도 모른다는 발표가 있은 후 로켓 발사시 발생되는 연소가스에 의한 환경 영향에 대한 연구가 몇 개의 그 룹에서 조사되어 왔으며, 그 결과 추전제의 산성 연소가스는 오 존층 파괴, 산성비, 독성가스, 공기의 질 저하 현상, 지구 온난 화 현상에도 크게 관계된다는 결론이 내려졌다. 1991 년 AIAA 워크숍에서는 추전제 연소가스로 인한 환경공해가 아직까지는 매 우 미미한 정도라고 발표했으나 실제로는 로켓 발사 직후 케네디 센터 발사장 지 역 주변 숲의 상당량의 수목과 바다, 하천의 고기 들이 떼죽음 하였음이 목격되었고, 그 지역 수목 숫자가 대폭 줄
어들고 있으며, 그 피해 지역이 발사장 지역으로부터 점차 확대 되고 있음이 보고되었다 .6,7) 현재는 위성발사체를 발사한 직후 주변 지역들(도로, 하천, 바다 등)을 중화액 및 물로 철저히 세척 하여 환경오염을 가능한 한 줄이기 위한 작업을 하고 있다. 한편, 지구 환경공해에 관한 법률이 미국을 위시한 선전국으로 부터 전세계적으로 점차 강화되고 있고 Green Round 와 같은 기 구를 통해서도 지구 환경공해 단속 규율은 더욱 강화될 것이 확 실하다. 미국은 오존층 보호를 위해 1995 년까지는 불소 및 염소 가 함유된 CFC (chlorofl uo ro carbon) 계 화합물을 사용 금지 시 키 기로 결정하였고, 미국 공군에서는 염소가 함유된 용매들을 미국 공군이 운영하는 모든 시설, 장비운영 용도로는 구매하지 않기로 결정하였다. 이와 같은 상황으로 볼 때 위성발사체 및 각종 군사 용으로 그 사용량이 증가하고 있는 고체 추전제 원료인 과염소산 암모늄은 지구 환경오염 방지 차원에서 계속 사용하거나 생산하 는 데 큰 제약이 따를 것으로 예상된다. 따라서 전 인류의 생존 에 관계되는 청정 추진제 또는 그 원료의 개발 필요성은 군수, 민수 어떤 면에서든 시급히 풀어야 할 문제이다. 이상과 같은 환 경공해 방지 및 군사적인 목적에 의해 고체 추전제의 연소 배기 가스를 무연화 또는 청정화시키는 연구는 현재 다음과 같은 두 가지 방법으로 전행되고 있다. 5. l. ] 스카벤저 추전제 스카벤저 추진제는 추전제 연소시 다음과 같은 화학반응이 일 어나는 현상을 기본으로 하여 개발되었다 .4) NH4CI04+ 알칼리 금속(또는 알칼리 금속 질산화물) 一 금속염화물+무연가스
죽, 과염소산암모늄과 동일 당량비율로 배합한 알칼리 금속이 과염소산암모늄 연소에 의해 생성된 공해물질인 영소화합물과 반 응하여 고체 분말 상태인 금속영화물로 떨어뜨림으로써 추전제 배기가스 성분 내에 공해 발생 원인인 영소화합물(영화수소)을 제거해 주는 방법이다. 이때 고체 분말 상태로 떨어지는 염화마 그네슘, 영화나트륨 등 금속염화물은 물에 쉽게 용해될 뿐 아니 라 지구상(땅 속)에 혼하게 존재하는 염화물이므로 공해에 문제 가없다. 이러한 스카벤저 추진제는 미국의 차기 위성발사체 부스터 (booste r ) 인 ALS (Advanced Launch Sy st e m ) 용으로 미 국 4 대 추 전제 제작사들이 각각 독자 개발하였으나 기존의 HTPB 계 과염 소산암모늄계 혼합형 추진제와 비교하여 비추력이 2~5 % 정도 낮으므로 그 적용이 어려운 상황에 있다. 그러나 표 5.1 에서 보 여주는 이러한 스카벤저 추진제는 염화수소 배출량이 1~2 %(무 게)에 불과하여 HTPB 과염소산암모늄 추전제의 염화수소 배출 량 20~22 %(무게)의 경우와 비교할 때 크게 개선되었다고 볼 수 있다. 표 5.1 에서 보는 것처럼 스카벤저 추전제에 사용한 물 질은 질산나트륨 또는 금속 마그네슘이며, 추전제 연소시 질산나
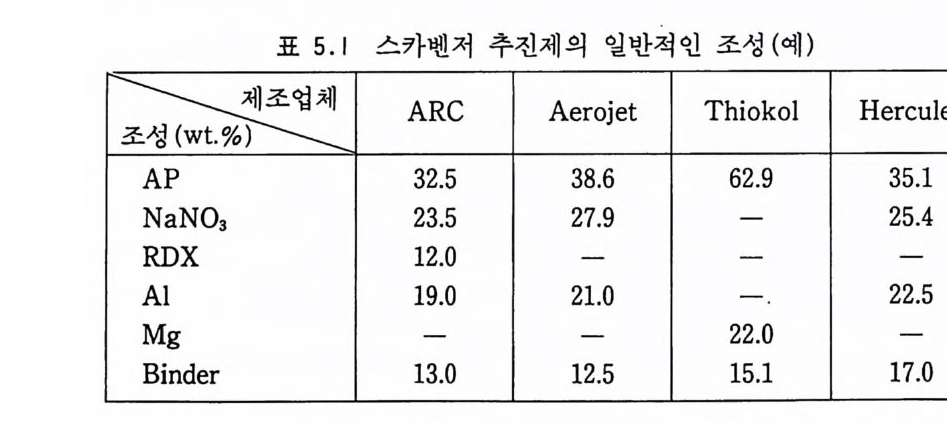 표 5.1 스카벤저 추진제의 일반적인 조성(예)
표 5.1 스카벤저 추진제의 일반적인 조성(예)
트륨은 염 화나트륨으로, 마그네슘은 염 화마그네슘으로 염 소 성분 이 변화되어 배출된다. Th i okol 사에서 개발한 마그네슘을 사용한 경우 연소가스 반응 메커니즘은 다음과 같이 진행된다.
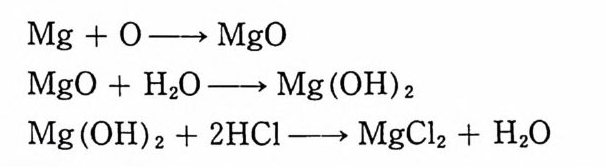 M g +O 一 M g O
M g +O 一 M g O
이와 같은 스카벤저 추전제의 제작비용은 일반적인 혼합형 추 전제와 거의 비슷한 수준이며, 제조공정 역시 통상 추전제의 제 조공정과 비슷하므로 제작공정 도중에 나오는 폐기물은 여전히 공해 발생 요인으로 남아 있게 된다. 스카벤저 추진제의 노화 특 성은 아직 완전히 구명되지 않았으나 신뢰성, 작동성 및 원료 수 급성 등은 기존 추전제와 비슷한 수준으로 평가되고 있다. 5.1.2 무연 추전제 고체 추전제의 연소 배기가스 내에 이차스모크 원인인 할로겐 화물, 특히 염화수소가스를 제거시키기 위해서는 추전제 조성 속 에, 죽 추전제 원료 성분 중에 할로겐(영소) 성분이 제거되어야 한다. 그러나 현재 혼합형 고체 추진제의 산화제로서 가장 많이 사용되고 있는 과염소산암모늄은 다량의 염소 성분을 함유하고 있으므로, 무연 추진제의 산화제로서 과영소산암모늄은 사용할 . 수가 없다. 또한 무연 추진제의 바인더 원료 성분에도 염소 성분 이 들어가지 않도록 원료를 선택해야 한다. 이와 같은 무연 추전 제 개발을 위해 수많은 종류의 조성과 원료들이 제안되어 왔다. 무연 추진제 개발을 위해 제안된 원료 중 바인더와 첨가제류에
염소 성분이 없는 원료를 선택하는 것은 쉽게 해결되었으나, 산 화제로서 과염소산암모늄을 대치할 수 있을 만큼 성능과 가격 면 에서 더 좋은 산화제가 아직까지는 실용화되지 않았다. 그러나 공해문제 해결, 민수 및 군용으로서의 무연 추진제용 산화제 개 발 필요성이 오래 전부터 있었던 관계로 최근 과염소산암모늄보 다 성능이 우수한 새로운 산화제들의 개발이 보고되고 있다 .2,4) 현재까지 사용되었거나 연구 중인 혼합형 무연 추진제로는 질 산암모늄을 산화제로 적용하는 방법, 히드록실 질산암모늄 (HAN -hy d roxy la mmoniu m nit ra te ) 을 질산암모늄과 공융 (eute c ti c) 혼합 물을 만든 후 PVA 로 처리하여 젤(g el) 추전제를 제조하는 방 법, 니트라민 계열 산화제 RDX, HMX 또는 최근 개발된 CL -20 을 적용하는 방법 및 암모늄디니트라마이드 (ADN) 와 같은 새 로운 산화제를 적용하는 방법들이 있다. 이상과 같은 방법들 중 질산암모늄을 산화제로 사용하는 방법이 가장 오랫동안 가장 광 범위하게 연구되어 왔는데, 그 이유는 질산암모늄은 연소시 염화 수소와 같은 유연성 가스를 배출하지 않으며, 가격이 매우 저렴 하여 원료로서의 공급이 매우 원활하다는 이점을 갖고 있기 때문 이다. 그러나 질산암모늄울 산화제로 적용한 결과 다음과 같은 문제점들이 발생하였다 .10,11,12) ® 질산암모늄을 적용한 추전제는 기존 혼합형 추진제에 비해 내탄도 성질이 상당히 낮아졌다. 죽, 비추력은 복기 추진제 수준 으로 저하되었으며 (200~220sec), 연소속도의 범위는 2~4mm/ sec (1000 psi a ) 이 하로 느려 졌고, 연소속도 압력 지 수는 0.5 이 상으 로 증가하게 되었다. ® 기계적 성질은 내탄도 성질보다도 더 많이 저하되어 160 p s i의 응력 (str e ss) 에서 10~20 %의 변형 (str a in ) , 80 ps i 응력 일 때는 30 %의 변형을 나타냈다.
® 과염소산암모늄과 동일 비율로 질산암모늄을 산화제로 섞어 충전할 경우 어떤 종류의 바인더에서는 추전제 반죽의 흐름성, 죽 충전 공정성이 좋지 않은 점둘이 발생하였다. ® 질산암모늄은 과염소산암모늄보다 더욱 습기에 민감한 관계 로 공정설비 및 원료 컨트롤이 어려운 점 등이 나타났다. ® 질산암모늄은 추진제 사용(작동) 온도 범위 (-50 °C~60 °C) 에 서 상변화를 일으켜 추진제의 밀도를 변화시키는 결정적인 단점 이 있다. 따라서 상변화를 억제하기 위해 상변화 억제물질을 질 산암모늄에 혼합 공융 (또는 결정 화) 시 켜 PSAN (Phase Sta b il ize d Ammoniu m N it ra t e) 을 제조하여 산화제로 적용하는 공정이 개발 되었다. PSAN 제조를 위해 첨가하는 상변화 억제제로는 Ni O , ZnO, KN03 등의 화합물이 주로 사용되는 것으로 알려졌다. 그 러나 PSAN 을 적용한다 할지라도 상변화 억제제 함량만큼 추진 제로서의 성능이 저하되는 단점은 피할 수 없다. 이상과 같은 문제점들로 인해 질산암모늄을 산화제로 적용하는 추진제는 아직까지 큰 전전이 없는 상태에 있으나, PSAN 을 사 용하는 추진제는 활발한 연구가 진행 중에 있으며 일부 소형 그 레인의 로켓에는 실제로 적용되고 있다고 알려져 있다. 한편, 1980 년대 초반부터 활발히 개발되기 시작하고 있는 새로 운 고에너지 폴리머와 에너지성 가소제들로부터 고에너지 바인더 룰 얻어 추진제에 적용하는 연구들이 진행되고 있으며, 이러한 고에너지 바인더를 무연 산화제인 PSAN 과 조합시켜 무연 추전 제를 개발하려는 노력도 시도되고 있다. 13,14,1 5 ) 그러나 GAP 과 갇 은 고에너지 프리폴리머를 추전제 바인더로 적용할 경우 추진제 의 에너지는 상당히 높아지지만 다른 특성들, 죽 기계적 성질, 제조성, 안정성 등이 완전히 해결되지 않고 있어 가까운 시일 내 에 실용화되기는 어려울 것으로 보인다 .16,17,18) 질산암모늄 이의
에 니트라민계 산화제인 RDX 또는 HMX 를 산화제로 적용하면 배기가스는 무연화될 수 있으나 내탄도적 성질, 물성, 점화성능, 안전성, 가격 등 여러 가지 문제점들이 발생한다. 특히 RDX 나 HMX 를 혼합하는 추진제는 안전성 면에서 추진제의 위험 등급 이 달라지게 되고 제조 공정시에도 위험 등급이 높아지는 단점이 있다. 최근에 개발되고 있는 ADN 및 CL-20 과 같은 새로운 고 에너지 물질은 그 성능 면과 배기가스 측면에서 매우 우수한 것 으로 알려져 있으며, 그 적용 제조공정 확립과 공급이 원활하게 되면 차세대 추전제 원료로 채택되어 사용될 수 있는, 가장 유망 한 신물질들이 다. 9,19,20) 5 . 2 둔감 (Insensit ive ) 추전제 추전제, 화약, 폭발물류는 매우 오래 전부터 제품의 종류, 용 도에 상관없이 그 물질 자체에 폭발, 연소의 특성이 있기 때문에 폭발 또는 화재의 위험성이 항상 문제시되어 왔으며, 이러한 물 질들 자체 및 그 제조공정에 이르기까지 안전관리에 국도로 세심 한 주의를 필요로 하고 있다. 이미 니트로글리세린이나 니트로셀 룰로오스 추진제에서 언급한 바와 같이 추진제, 화약류가 본격적 으로 연구되기 시작한 19 세기 말부터 20 세기 초반 이후 원인을 알 수 없는 이유로 인해 육·해 ·공, 죽 해상의 함정에서, 육상 의 탄약고에서 또는 군 운용 도중, 또는 비행기 내에서 수많은 인명, 물적 피해를 가져오는 사고가 빈번히 발생하였다. ◄ ,21) 이 와 같은 사고들은 그 사고 원인을 잘 알 수 없었기 때문에 그 다 음에도 계속되는 사고를 방지할 수 없었다고 보인다. 이 분야에 서 사고의 원인과 이유를 잘 알 수 없다는 것은 다른 과학 분야
와 달리 그 사고 원인이나 이유가 될 만한 물증들이 폭발과 연소 에 의해 대부분 사라져 버리기 때문이다. 제 1 차 및 제 2 차 세계대 전 이후 전세계적으로 군사 과학기술이 크게 발전함과 동시에 각 종 무기 생산량도 크게 증가하게 되었고, 이에 따라 탄약, 추진 제, 화약, 폭발물 등의 관리 잘못으로 인한 사고도 증가하고 있 고 그 피해 규모도 엄청나게 증대되는 경향에 있다. 1967 년 미국 해군의 항공모함에서의 어처구니없는 사고로 인명 (134 명)과 함정 이 손상을 입은 이후 미국 해군에서는 모든 탄약의 둔감화(I M : Insensit ive Munit ion ) 개발을 서두르기 시작하였으며, 미국 육 • 해 ·공군이 공동으로 참여하는 IM 개발사업 (IMAD) 이 적극적으 로 추전되고 있다 .22,23,24,25) 미국방성에서는 표 5.2 와 같은 IM 규 격 초안 (Draft MIL-STD-2105A) 을 제 정 하였다. 미 해 군은 IM 개 발사업과 관련하여 다음과 같은 기본방침을 정하였다 .4,8) ® 앞으로 개발하는 탄약류는 IM 규격을 만족해야 하며, ® 이미 배치된 탄약류들도 표 5.3 과 같은 15 종의 탄약류는 1995 년까지 IM 규격을 만족시키도록 재개발 배치한다. 표 5.2 에서 보여준 SD, Bl, Fl, SCJ 등의 특성은 충격감도 특 성과 관계된다고 알려져 있고 SCO, FCO 등의 Cook-Of f 특성 들은 연소특성과 관계된다고 알려져 있다. 최근 고폭화약 및 추 진제 원료로 흔히 사용되고 있는 RDX, HMX 등의 니트라민계 원료들을 적용한 추진제나 폭발물들은 · 현재 사용 중인 고체 함량 을 유지할 경우 SD, Bl, Fl, SCJ 등의 IM 규격을 만족시키기 어 려운 것으로 보고되고 있다. 로켓 추전제에 흔히 사용되고 있는 과영소산암모늄 (AP) 계 추진제는 폭굉성이 낮은 우수한 성능을 지녔으나 배기가스가 유연이고 독성이며 FCO, SCO 등의 Cook -Of f 특성이 IM 규격을 만족시키지 못한다. 또한 이미 언급했던 질산암모늄계 산화제를 적용한 추전제는 낮은 가격, 무연의 배기
 표 5 . 2 MIL-STD-2105A (초안) 에 의 한 IM 규격
표 5 . 2 MIL-STD-2105A (초안) 에 의 한 IM 규격
가스, 낮은 위험성으로 장점들이 있으나 낮은 성능, 상변화 문제 점, 공정성(홉습성), 물리적 • 기계적 성질 등의 문제점이 우선 해결되어야 한다. 다음 그립 5 .1 에 서 보여 주는 EMCDB (Elasto m er Modif ied Cast Double Base) 추전제는 질산암모늄계 추전제와 니트라민계 추전 제와의 중간적 위험 특성을 갖고 있는 것으로 나타났으며, 복기 추전제는 EMCDB 와 니트라민계 혼합형 추진제의 중간 정도의 위험 특성을 지닌 것으로 알려져 있다 .8) 표 5 . 4 에는 미국 육군에서 IM 규격을 만족하는 무연 추전제의 개발 구상을 정리한 것으로서 단기, 중기 및 차세대의 3 단계에 걸친 기술개발 계획 내용이다. 이 계획에 의하면 PSAN 의 적 용, GAP 과 RDX, GAP 과 새로운 고에너지 물질 CL-20 적용, ADN (ammoniu m din i t ra mi de ) 적 용 등이 계 획 되 어 있다. 특히 새 로운 고에 너 지 물질 (HEDM : Hi gh Energy Hi gh Densit y Mate -
 1| | 과염색 암모卽 P)
1| | 과염색 암모卽 P)
 표 5.4 (미)육군의 IM 규격 추진제 및 무연 추진제 개발 계획
표 5.4 (미)육군의 IM 규격 추진제 및 무연 추진제 개발 계획
r i al) 들은 기존의 전술용 추진제, 폭발물의 성능 개선은 물론 장 래 전술 • 전략무기 시스템에의 적용 및 우주용 로켓 기술개발 방 향에 큰 영향을 줄 것으로 예상되고 있다. 이 밖에도 과산화암모늄계 혼합형 추전제를 둔감화시키는 방안 도 검토되고 있다. 예를 들면 자체 소화성 성질을 주기 위한 바 인더나 첨가제의 개발 및 과염소산암모늄의 일부를 과염소산칼리 로 대치 충전하여 Cook-Of f 특성을 개선시키려는 방안 등이 그 것이다. 그러나 무기체계의 IM 규격에 대해 지적할 점은 IM 규격의 만족이 무기 시스템 전 구성품에 대해 적용된다는 것이다. 죽,
추전제 한 구성품만 IM 규격을 만족해서는 그 무기체계의 효과 가 불충분하며 점화기, 탄두, 추진제 각 구성품이 모두 IM 규격 을 만족해야 된다는 것이다. 5.3 고에너지 고밀도 물질 고에너지 물질은 밀도가 높고 에너지를 많이 방출할 수 있는 물질, 죽 단위무게 (부피)당 에너지를 많이 함유한 물질로서 그 대표적인 예가 추전제, 폭발물 및 그 원료들이 있다. 고에너지 물질은 일반적으로 그 분자 내에 (혹은 그 화합물 내에) 연료 성분 과 산화제 성분이 모두 포함되어 있다. 화학반응에 의해 산화제 성분은 전자 공여체로서 연료성분은 전자 수용체로서 작용하여 발열반응을 일으키게 됨과 동시에 고온의 가스를 다량으로 발생 하게 된다. 이 반응이 폭발적으로 매우 빨리 일어나는 경우에는 충격 파를 수반하는 폭굉 (deto n ati on ) 이 되 어 그 높은 압력 에 의 해 높은 파괴력을 갖게 되는 폭발물로 사용된다 .2,5,26) 만일 그 반응이 연소현상 (combus ti on 또는 de fl a gr a ti on) 으로 일 어나는 경우에는 연소반응에 의해 발생되는 다량의 가스로 인해 추전력을 갖게 되므로 추진제 또는 발사약으로 적용된다. 이 두 현상, 폭굉과 연소의 공통점은 고온의 가스가 급속도로 발생한다 는 점이다. 따라서 고온의 가스를 급속도로 많이 발생시키기 위 해서는(반응열이 크기 위해서는) 반웅 전 물질의 생성열과 반웅 후 생성된 가스의 생성열 차이가 가능한 한 커야 한다. 일반적으로 연소, 폭발반응 생성물에 CO2 나 H20 가 다량 함유 되어 있다는 것은 그 고에너지 물질은 연료로 작용하는 탄화수소 계 성분과 이를 산화시키기 위한 산화제 성분으로 구성되어 있다
는 것을 의미한다. 고에너지 물질은 이미 언급했던 혼합형 추진 제나 PBX 폭발물처럼 두 성분의 화합물로 이루어진 혼합물질인 경우도 있고, TNT 나 RDX 처럼 단일 화합물 분자 내에 두 성분 의 분자 구조가 포함된 단일 물질인 경우도 있다. 고에너지 물질이 로켓 추전제로 사용되는 경우는 이미 추진제 장에서 언급한 바와 같이 비추력(I s p) 값이 커야 유리하며, 발사 약으로 사용될 때는 IP(Imp e tu s ) 값에 따라 그 에 너 지 의 크기 가 평가되며, 폭발물의 경우는 그 파괴력이 폭속과 충격파 압력에 따라 좌우되므로 폭발물로 사용되는 고에너지 물질은 빠른 폭속 과 충격파 압력이 큰 물질이 요구된다. 폭속은 Vs:::(Tr• pp )112 의 관계식에 의존하며 이때 Vs 는 폭속, T f는 화영온도, PP 는 에너 지 물질의 밀도이다. 고에 너 지 물질 중 TNT (tri n i t ro to l uene) 는 제 2 차 세 계 대 전 전 까지는 가장 강력한 고에너지 물질로서 군사용 폭약과 산업용 폭 약으로 사용되었었다. TNT 는 석유 또는 석탄으로부터 분리 생 산되는 톨루엔을 원료로 하여 혼산(황산+질산)으로 니트로화시 키는 공정에 의해 저렴한 가격으로 생산되었다. 특히 TNT 는 상온에서 결정상 물질로 약 80°C 에서 용융되므로 고온수나 고온 수증기를 이용하여 비교적 안전하게 탄두, 폭발물을 원하는 형상 대로 주조할 수 있다. 현재도 TNT 는 군사용 및 산업용의 저렴 한 폭약으로 흔히 사용되고 있다. 제 2 차 세계대전 돌입시 TNT 와 니트로글리세린의 수요가 크게 증가하게 되었을 때 이 물질들 울 대신할 다른 고에너지 물질들의 필요성이 대두됨에 따라 세계 각국은 새로운 폭약 원료들을 찾기 시작하였다. 이때 영국과 독 일에서는 각각 독자적인 방법으로 암모니아와 포름알데히드에 질 산을 반웅시켜 TNT 보다 더 강력한 폭약 제조에 성공하였으며, 이 물질 이 바로 RDX (Research and Develop m ent Exp lo siv e ) 로 알
려진 시클로트리메틸렌트리니트라민이다 .21) 실상 RDX 는 20 세기 이전 1890 년경 의약품용으로 합성 개발되어 특허를 획득한 화합 물이었으나 폭약에 적용되기는 제 2 차 세계대전 이후이다. 한편, RDX 제조시에 소량의 불순물로 포함되어 제조되는 시 클로데트라메틸렌데트라니트라민 (HMX) 은 그 위력이 RDX 보다 더 높아 제 2 차 세계대전 직후 HMX 제조 방법에 대한 많은 연 구 개발이 진행되어 현재까지 각종의 고성능 고급 폭약으로 적용 되고 있다. 특히 1960 년대 이후 고분자 재료의 발달에 힘입어 RDX 나 HMX 의 분말을 폴리머와 혼합, 성형하여 고성능 화약 PBX (Plasti c Bonded Exp lo siv e ) 가 개 발되 어 흔히 사용되 고 있다• 그러나 최근 이와 같은 RDX 나 HMX 류보다 성능이 월등히 높 은 새로운 고에너지 물질에 관한 연구가 체계적으로 심도있게 전 행되어 몇 가지의 새로운 물질들이 개발된 것으로 알려지고 있 다. 이러한 새로운 고에너지 물질들의 연구는 미국의 경우 LLNL (Lawrence Liv e rmore Nati on al Laborato r y ) , NAWC (Naval Aeronauti ca l Weapo n Cente r ) , NSWC (Naval Su rfac e Warf ar e Cen- ter ), 육군, 해군, 연구소 및 각 대학교 연구실 등에서 많은 연 구 진척이 이루어진 것으로 알려졌다 .26,27 , 28 , 29) 새로운 고에너지 물질로서 개발되어 적용되기 위해서는 단위 질량당 함유한 에너지 및 밀도가 높아야 하고, 안전성 (마찰감도, 충격감도 등), 내열성 및 경시변화가 우수해야 하며, 제조과정이 안전해야 하고, 가격이 저령할 것 등이 요구된다• 이러한 요구 특성치들은 적용되는 폭약, · 발사약, 추전제에 따라 다를 수 있으 나 본질적인 요구조건은 동일하다. 화학기술· 이론의 발달에 따 라 고에너지 물질을 합성하기 전 이론적인 방법에 의해 고에너지 물질의 화학 구조 설계와 그 특성을 예측할 수 있으나 합성이 완 결되어 성공할 때까지, 죽 합성반웅 조건을 찾아낼 때까지는 수
십 년의 기간이 소요되기도 한다• 또한 그 반응이 현실적으로 불 가능한 경우에는 합성 자체를 포기하는 경우도 많다. 또 다른 경 우, 합성반웅이 어렵게 성공했을지라도 이론적으로 예측했던 성 질과 아주 다른 특성을 지닌 물질이 합성되는 경우도 있다. 더욱 이 이론적으로 예측이 매우 어려운 마찰감도나 충격감도 등의 안 전성 성질(예민성) 때문에 실용화를 포기하는 경우도 많다. 예를 들어 ENTA (eth y le ne tet r a nit ra mi ne ) 와 같이 간단한 구조를 지 닌 물질은 니트라민기를 (N-N02) 분자당 4 개나 가지고 있는 높은 에너지성 물질일 것으로 이론적으로 계산되었으나, 실제로 ENTA 를 합성하여 그 안전성을 시험한 결과 그 충격감도와 마 찰감도가 너무 커서 도저히 화약류 원료로 적용할 수 없었으며, 벤젠에 6 개의 니트로기를 치환시킨 HNB(hexan it robenzene) 의 경우는 대기 중의 수분과 반응하여 대기 중에 노출되면 연소반응 (자연발화)을 일으켜 사용을 포기하였다. 최근 미국에서는 고에너지 물질의 중요한 특성, 에너지 및 밀 도를 최대로 높이기 위한 HEDM( 고에너지 고밀도 물질, Hi gh Energy High Densit y Mate r ia l ) 개 발 프로젝트를 국방 연구개 발 분야의 핵심기술 과제 21 개 분야 중 한 분야로 채택하여 그 연구 개발에 온 힘을 쏟고 있다. 참고로 이러한 HEDM 에 두자하고 있는 미국의 연구개발비는 다음과 갇다. HEDM 을 사용하게 되면 전략·전술적인 유도 및 무유도 추진 기관으로부터 재래식 탄약, 탄두, 폭발물, 추진제에 이르기까지 그 에너지와 밀도를 증가시킬 뿐만 아니라 안정성, 안전도, 식별 성 (sig na tu r e), 독성 및 신뢰성 등에서 대폭적인 개선이 이루어질 수 있다. 예를 들면 유도탄 로켓의 사거리 증대 및 탄두 효과의 증가, 탱크 장갑판의 침투 능력 증가 (50 %), 전술 유도탄의 노 출 위험성 감소로 인한 안전성 향상 등의 개선이 기대되고 있다.
 연도 -11999817 1991 1992 1993 1994 1995 1996 1997
연도 -11999817 1991 1992 1993 1994 1995 1996 1997
따라서 HEDM 의 이용과 무기체계는 전략 • 전술적으로 매우 중 요시되며 DCTP 에 의하면 다음과 같은 무기체계에 사용 가능하 다고 보고되 었다. 22) • 전략 유도탄 추진기관(전략적 공격, 보복, 방어) • 전술 유도탄 추전기관(대공방어, 대함공격, 원거리 공격, 공중 후방차단, 근접 항공지원) • 운동 에너지 비행체 추전기관 • 궤도 전이체계 추진기관 • 진로 요구 발사체계 추전기관 • 전술 유도탄 탄두(대공방어, 대함공격, 원거리 공격, 공중 후방 차단, 근접 항공지원) • 어뢰탄두(해전) • 폭탄(근접 항공지원, 해상 전두, 공중 후방차단) • 성형 장약탄두 • 기뢰 (기뢰전) • 대응책 (해전, 기뢰탄두) • 핵 폭발장치 구성품 ·신관
또한 이와 같은 HEDM 물질이 사용될 때 무기체계에 끼칠 영 향은 다음과 같이 요약될 수 있다. • 무연 (Mi ni m um Sig n atu r e) , 고성 능, 고연소 속도의 유도탄용 추진제(전략 및 전술용) • 살상거리가 증대된 탄두 • 참수함 구조물 파괴를 극대화할 수 있는 차세대 수중 폭약 • 장갑 철판이나 견고 표적의 침투성 증대를 위한 고성능 성형 장약 • 우주선 발사 고속도 운동 에너지 살상 비행체 ·목표물 파괴나 유효 사거리가 증대된 저취약성 (Low Vulnerable) 총 • 포 추진제 • 매우 둔감하며 사고 위험에 안전한 핵 탄두 • 속도 및 성능이 증가된 새로운 점화 • 착화 작동장치 이와 같은 HEDM 을 개발하기 위한 노력은 크게 두 종류로 구 분될 수 있다. 첫째는 바인더 성분에 속하는 폴리머 (Polym er), 가소제 (Plasti ci a e r) , 경 화제 (Curati ve ) 및 기 타 첨 가제 (Addit ive ) 등을 고에 너지화하는 것 이 며 , 둘째는 산화제 (Oxid i z e r) 자체를 고에너지 고밀도 산화제로 바꾸는 방법이다. 그 중 산화제의 경 우 기존의 질산암모늄 (AN), 과염소산암모늄 (AP), TNT, RDX, HMX 등이 1960 년대 이전에 등장한 이후 더 이상의 큰 진척이 없는 상태에 있었다. 왜냐하면 산화제로서 사용될 수 있 는 화합물의 구조 모델링은 주로 다음과 같은 화학적으로 에너지 성 기(gr ou p)들을 지닌 물질로서 이미 20 세기 초반에 그 계산이 가능했었기 때문에 기존의 산화제 이상의 것들을 고안해 낸다는 것이 매우 어려웠기 때문이기도 하였다.
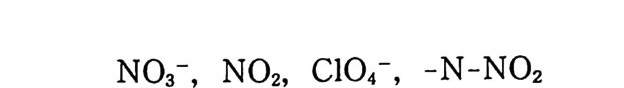 NOa-, N02, Cl04-, -N-N02
NOa-, N02, Cl04-, -N-N02
그러나 바인더에 속하는 폴리머의 경우는 1960 년대 이후 급격 히 발달한 석유화학공업 • 정밀화학 기술 발전에 힘입어 저에너지 폴리머인 HTPB, PEG, PBAN, PU, CTPB 등을 대치하여 에너 지화한 폴리머들이 고안되었다. 이러한 에너지성 폴리머를 사용 하게 되면 바인더에 의해 에너지 준위가 높아지므로 고체 충전도 가 낮아지게 되어 혼합 점도가 낮아지며 가공성이 좋게 되는 동 시에 추전제나 화약의 물성도 개선되는 좋은 효과가 나타나게 된 다. 예를 들면 GAP, BAMO, PGN, Poly- G 등의 새로운 폴리머 가 1970 년대 후반 이후 개발되어 시험 중 또는 무기체계에 채택 되어 사용 중이다. 일반적으로 이러한 에너지성 폴리머들은 주로 다음과 같은 기 (grou p ) 들을 갖도록 고안 개 발되고 있다. 14,15,16,17)
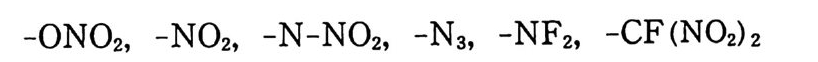 -ON02, -N02, -N-N02, -Na, -NF2, -CF (N02h
-ON02, -N02, -N-N02, -Na, -NF2, -CF (N02h
이와 같이 바인더(폴리머)에 대해서는 비교적 많은 발전이 있 었고 연구도 활발히 진행되어 왔으나 고체 산화제류로서는 RDX, HMX 이후 새로운 종류의 산화제 연구 결과가 발표된 바 가 없었다. 그러나 최근 들어 1988 년 CL-20 이라고 부르는 HNIW, 1990 년 합성된 ADN, TNAZ 등 몇 가지 새로운 고에너 지 고밀도 산화제류의 합성이 보고되어 관련된 학계 및 업계에 큰 충격을 주고 있다. 이러한 HEDM 에 속하는 산화제들은 이름 그대로 에너지가 높은 동시에 밀도가 높으므로 차기 세대에 적용 되는 모든 화약, 추진제, 폭발물 원료로서 군사적으로 또한 민수 용으로 광범위하게 사용될 매우 중요한 물질들이라고 볼 수 있 다. 1990 년대에 둘어서면서 문헌에 나타나고 있는 차기 세대의 산
화제로서 사용이 유력시되고 있는 HNIW(Hexa Nit ro Hexaazais - owur t z it ane) 는 기존의 RDX 나 HMX 에 비해 훨씬 조밀한 분자 구조를 갖고 있다. RDX 는 육각형, HMX 는 8 각형 구조이나 HNIW 는 5 면체의 새장 (ca g e) 구조이므로 그 밀도가 높다. 분자 내의 에너지성 N-NO 기 숫자도 RDX 는 3 개, HMX 는 4 개인 데 비해 HNIW 는 6 개를 갖고 있다. 기존의 RDX 는 밀도가 1.82 , HMX 는 1.90 , 과염소산암모늄 (AP) 은 1. 95 인 데 비해 HNIW의 밀도는 1. 98 로 AP 보다 더 높은 것으로 보고되었다. 또한 HNIW 는 폭속, 폭발력도 기존의 어떤 물질보다 월등히 높고 추 진제 산화제로 적용시 추전제의 비추력을 크게 높일 수 있는 동 시에 추진제 배기가스도 무연화시킬 수 있는 장점도 있다 .2,4,5,9) 최근까지도 고폭약에 사용되고 있는 물질로서 가장 성능이 좋 은 것으로 알려진 HMX 나 RDX 가 40 여 년 전 개발되어 적용된 것으로 미루어, HMX 및 RDX 에 비해 밀도 및 폭발 성능이 훨 씬 우수한 CL-20 의 등장은 폭발물 관련 업계나 학계에 매우 고 무적인 일로 받아들여지고 있다. 미국 DCTP 보고서에서 CL-20 의 중요성과 적용성에 대해 반복해서 강조한 이유는 그 성능을 비교해 보면 당연하다고 판단된다 .22) 특히 미국의 경우 미 육군, 해군, 공군에서 모든 탄약, 추진제, 화약류에 대해 심각하게 요 구하고 있는 둔감성 (Insens iti v ity)을 만족시킬 수 있는 유일한 방 법이 새로운 고에너지 고밀도 물질을 적용함으로써 해결될 수 있 다고 판단하고 있으므로 이 물질의 이용은 크게 기대되고 있다. 또한 CL-20 의 분해반응을 보면 RDX 나 HMX 의 경우 방출하 는 산소의 양이 없는 데 반해 CL-20 은 같은 조건하에 이용할 수 있는 산소가 상당량 방출된다. 19)
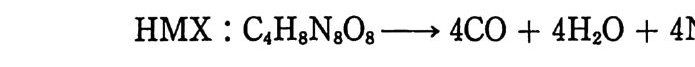 HMX : C4H8N808 一 4CO + 4H20 + 4N2
HMX : C4H8N808 一 4CO + 4H20 + 4N2
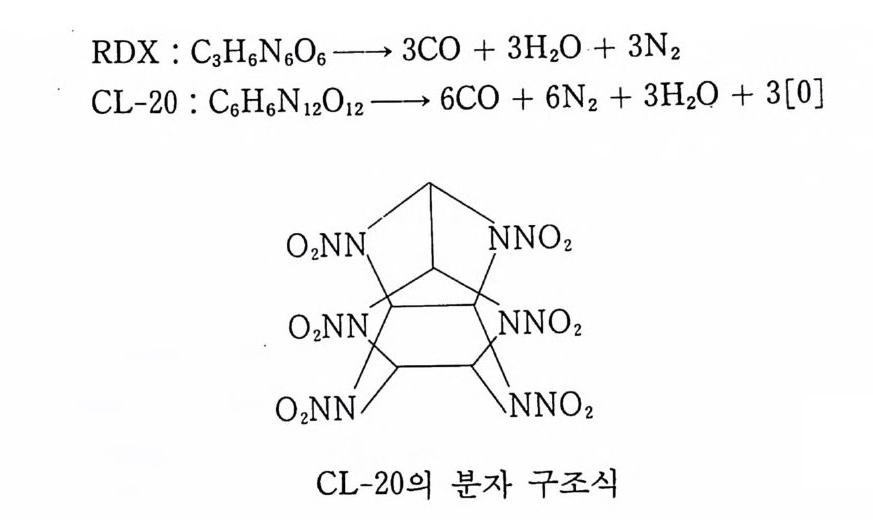 RDX : C3H6N606 _ 3CO 十 3H20 + 3N2
RDX : C3H6N606 _ 3CO 十 3H20 + 3N2
1989 년에 미국 SRI 에서 최초로 합성된 ADN(ammoniu m d i n it am i de) 은 이론적 및 소규모 실험적 연구에 의해 로켓 추전 제와 폭발물 원료로 매우 우수한 물질인 것으로 파악되었다. 현 재 미국에서는 Scale-up 공정 개발에 의해 파일럿 플랜트에 의 한 공정 및 적용 연구, 시범 프로그램들이 전행 중인 것으로 알 려졌다. ADN 은 고체 추전제(혼합형)의 산화제로서 그 용도가 크게 기대되고 있다. 왜냐하면 ADN 을 산화제로 적용시, 기존 사용 중인 과영산암모늄 사용시 배출되는 . 공해성 가스인 할로겐 화물이 전연 없다는 점과 비추력이 증가하는 장점이 있다고 알려 져 있기 때문이다. 따라서 군사 로켓용만이 아니라 위성발사체 대형 로켓용 추진제로서는 가장 적합하고 성능이 우수한 산화제 로 선택될 가능성이 있다. 또한 ADN 의 암모늄 이온을 칼리 이 온으로 대 치 한 KDN (po ta s siu m dit ra mi de ) 은 질산암모늄 (AN) 의 상 안정제로서도 작용한다고 알려졌다. 가격, 무공해성 등 여러 가지 면에서 좋은 점들을 지닌 AN 의 상을 안정시키고 그 내탄 도적 성질을 개선시킬 수 있는 PSAN(Phase Sta b il ize d AN) 이
개발되면 그 용도가 매우 다양해질 수 있으므로 KDN 이 AN 의 상 안정제로 작용하면서 동시에 그 내탄도적 성질을 개선시킬 수 있다면 더욱 우수한 추진제가 개발될 수 있을 것이다 .27) 원래 이 물질의 화학구조는 질소 원자에 두 개의 니트로 (N02) 기가 결합된 디니트라민으로서 일반적인 니트라민 (N-N02) 기의 합성도 쉬 운 것 이 아님 에 비 추어 디 니 트라마이드 (din i t ra mi de ) 는 매우 불안정하여 합성을 시도하지 않았던 계열의 화합물이었다. 그러나 이 ADN 의 합성이 알려진 후 디니트라마이드 이온의 안 정 성 이 다음과 갇은 공명 (Resonance) 구조에 의 해 기 인 한다고 알 려졌다 .5 , 9)
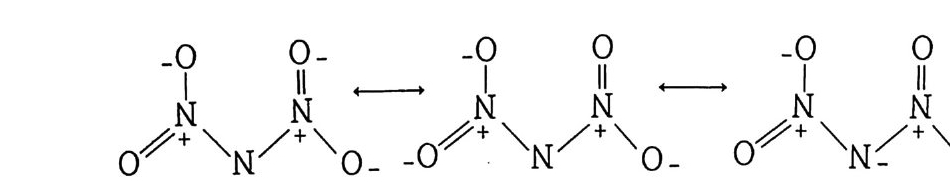 _o O- _o o -0/_N + OIIN+
_o O- _o o -0/_N + OIIN+
ADN 은 러시아에서 1970 년대에 합성하여 사용 중이라는 비공 식적인 보고가 있었으나 문헌상으로 최초로 나타난 것은 미국의 특허출원에 의한 다음과 같은 합성 방법에서이다 .27,30)
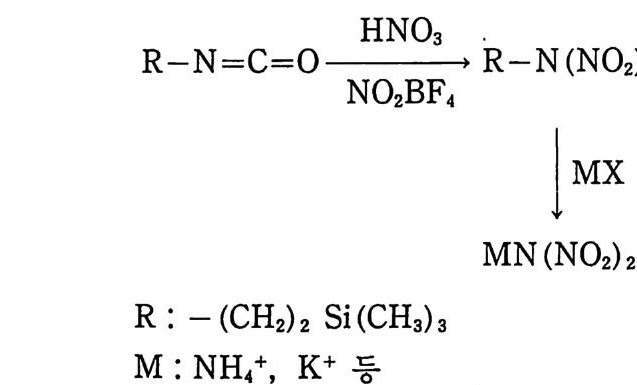 R-N=CN=H0N2OB0F34 - R· - N(jNM0X2) 2
R-N=CN=H0N2OB0F34 - R· - N(jNM0X2) 2
그후 최근에 발표된 합성방법들은 다음과 갇다. 죽, 암모니아
룰 출발 물질로 하거나 카바메이트를 원료로 하여 저온에서 니트 로화시키는 방법이며, 반응 중간물질인 디니트라마이드산을 암모 니아로 처리하면 ADN 이 얻어진다. 만일 암모니아 대신 다른 종 류의 아민 혹은 적당한 알칼리영으로 처리하면 각각 해당되는 디 니트라마이드염이 얻어질 수 있다 .31,32)
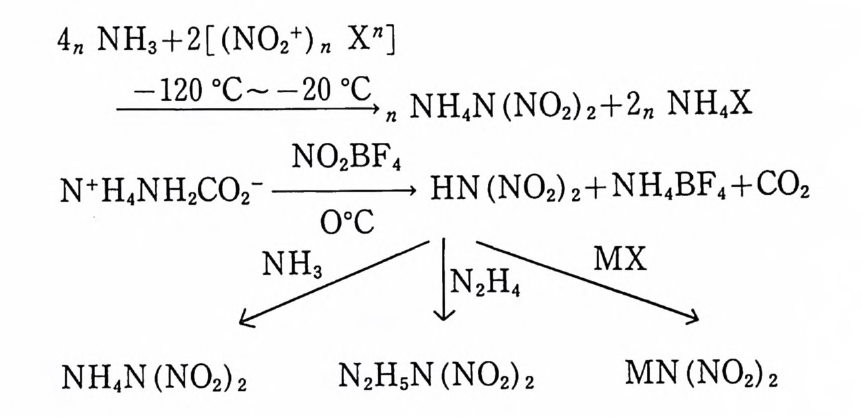 4n NH3+2[(NO 리 n X기
4n NH3+2[(NO 리 n X기
정육면체 구조를 가지고 있는 탄화수소 큐벤 (cubane) 은 1964 년 시카고 대학교의 Ea t on 에 의해 처음으로 합성되었으며, 그후 이 물질의 밀도가 1. 29 로서 군사용, 우주공간에서 사용되는 차기 세 대 추진기관 연료로 매우 유망하다고 판단되어 큰 관심을 끌기 시작하였다 .33) 최근 들어 큐벤 구조에 니트로, 아지도, 디니트로 등 에너지성 기 (gr ou p)들을 치환시켜 HEDM 으로 이용하면 그 밀도 및 에너지 면에서 현재까지 알려진 어떤 물질보다도 우수한 것으로 예측되어 미국에서는 활발한 기초 연구들이 집중되고 있 다. 그러나 아직까지는 값싼 공업적인 제조 방법이 개발되지 않 고 있어 실제 추진제나 폭발물 원료로 활발하게 적용되지는 못하 고 있으나 그 성능상의 높은 이점으로 미루어 볼 때 큐벤계 물질 은 차기 세대의 추진제, 폭발물의 주요 원료가 될 것으로 예측된 다. 34,35,36)
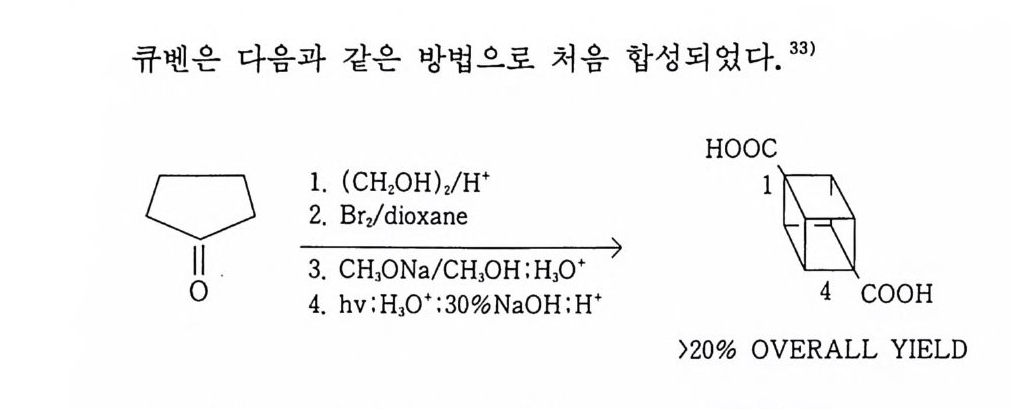 큐벤은 다음과 같은 방법으로 처음 합성되H었0다 .33o%) 0e \
큐벤은 다음과 같은 방법으로 처음 합성되H었0다 .33o%) 0e \
큐벤의 구조 및 물리화학적 특성은 다음과 같다.
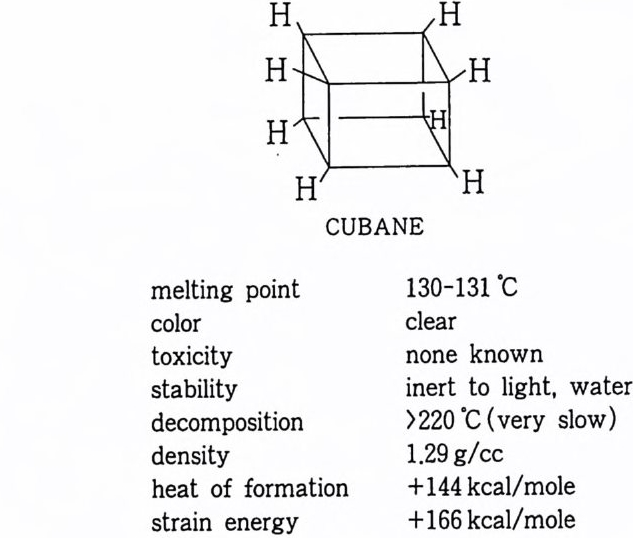 CUBANE
CUBANE
큐벤의 고에너지, 고밀도 성질을 이용하기 위해 큐벤 구조에 아민기, 니트로기, 디이소시아네이트기들을 도입하여 추전제나 폭발물에 적용하려는 연구가 활발히 진행되고 있다. 이상과 같은 기 (gr ou p)들이 도입된 화합물들의 합성반응은 다음과 같이 보고 되 었 다. 34,35,36,37,38,39) TNAZ 는 밀도가 RDX (l.80 ) 보다는 높고 HMX (l.90 ) 보다는 낮
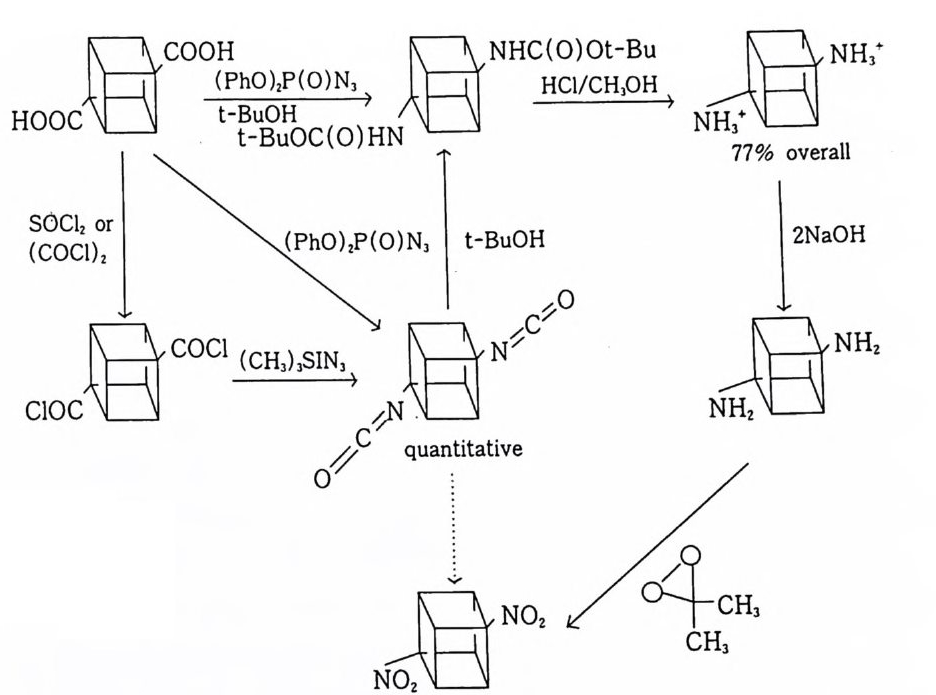 H麟OOC ' 言 l)~rl °°(P二h0),PH( ON)巳 Nl lt BNuH0CH二 N巳 l2Na:0 HH ;
H麟OOC ' 言 l)~rl °°(P二h0),PH( ON)巳 Nl lt BNuH0CH二 N巳 l2Na:0 HH ;
은 1.84 정도이나 성능(에너지)은 HMX 와 비슷한 수준의 고에너 지 물질이다. 그러나 안정성은 RDX 보다 훨씬 안정하고, 더욱이 용융점이 101 °C 로 낮으므로 TNT 처럼 스팀주조 화약으로 제작 될 수 있는 공정상의 이점이 있어 폭발물 원료로 적용하기가 매 우 유망한 물질이라고 볼 수 있다. 죽, TNAZ 를 저렴한 가격으 로 생산이 가능한 제조공정이 개발된다면 현재 각국에 세워져 있 는 방대한 규모의 TNT 공장 시설을 그대로 살리면서 TNAZ 롤 적용한 폭발물을 제조할 수 있는 장점이 생긴다 .4,5,9,23) TNAZ 는 다음과 같이 에피클로로히드린으로부터 몇 단계의 화학반응을 거쳐 합성된다고 보고되었으나 그 전체적인 수득률 (overall yiel d) 이 낮은 수준이고 따라서 그 제조 가격은 매우 높 은 수준에 있어 새로운 제조 방법에 대한 연구가 활발히 진행 중 에 있 다. 37,3 8 ,39)
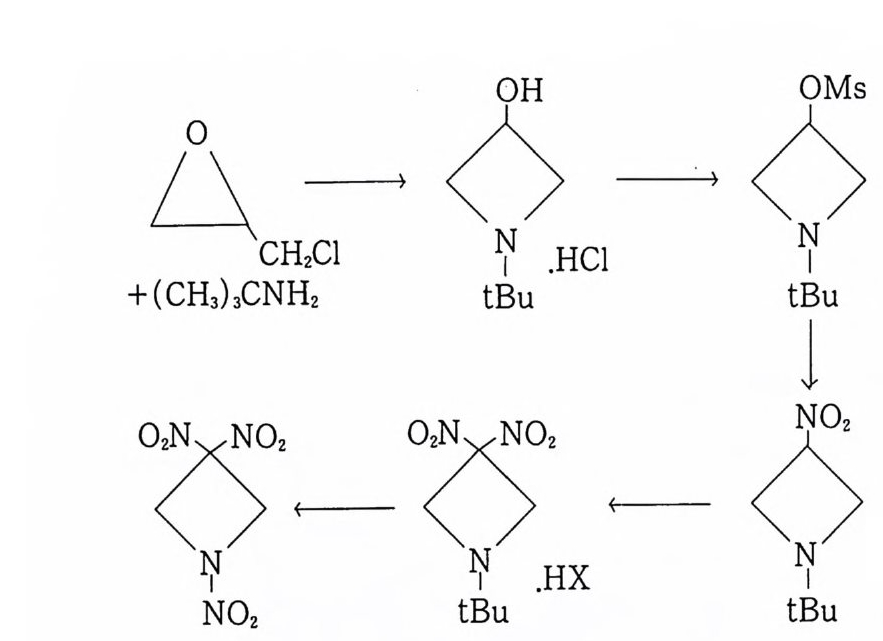 + 广( CHJ ) 3 C CNHH2 2C l 二tBNI u .H Cl :tNOB | uM s
+ 广( CHJ ) 3 C CNHH2 2C l 二tBNI u .H Cl :tNOB | uM s
이미 언급한 바와 같이 제 2 차 세계대전 이후 TNT 는 용융 주 조용 폭발물로서 가장 일반적으로 사용되어 왔으며, 결과적으로 TNT 를 80 °C 부근의 온도에서 용융시키고 주조하기 위한 수많 은 시설과 장치들이 세워져서 가동되고 있다. HAP(hyd roxy ammoniu m pe rchlorate ) 는 용융점 이 89 °C 로서 다른 산화제 와의 공융점 (Eute c ti c Poin t ) 을 85 °C 로서 낮출 경 우, HAP 의 높은 밀 도와 높은 산소량 (ox yg en balance) 을 생각할 때 TNAZ 처럼 TNT 에 사용하는 시설과 장비를 그대로 이용할 수 있는 상업적 으로 매우 유리한 조건이 있을 뿐만 아니라 매우 높은 에너지를 낼 수 있는 폭발물을 제조할 수 있게 된다 .5) 또한 HAP 자체의 높은 밀도 (2.0 7 g/c c) 와 높은 에 너 지 (산소 발생 량) , 낮은 용융점 을 이용하여 추전제나 화약의 원료로 적용하는 연구도 병행되고 있 으나 추진제 적용시는 과염소산암모늄과 마찬가지로 연소시 유독 성 염화수소가스를 방출하는 문제점이 있다.
[참고문헌] 1) Y. M. Ti m nat, Advanced Chemi ca l Rocket Prop ul sio n , Academi c Press Inc. (19 87) . 2) A. Davenas, Solid Rocket Prop ul sio n Technology , Perga mon Press Inc. (1993) . 3) A. E. Oberth , CPIA Publica ti on No. 469, Pr inc i ple s of Solid Prop e l- lant Developm ent (1987) . 4) A. Yamamoto , ]. Industr ial Expl o siv e s Soc., Jap a n , 1 (1) , 47 (1991) . 5) 노만균, 임 유진, 한국항공우주학회 지 , 22 (2) , 114 (1994) . 6) D. W. Doll and G. K. Lund, AIAA-91-2560, J. of Prop ul sio n and Power, Vol. 8, 1185-1191 (1992) . 7) R. R. Bennett , AIAA-3398, AIAA/SAE/ASME/ASEE, 28th joi n t Prop ul sio n Confe re nce and Exhib i t (1992) . 8) Proceedin g s of 1990 Joint Government / Industr y Sy mpo siu m on Insensit ive Munit ion s Technology , ADPA, March 13-14 (1990) . 9) D. H. Lie b enberg, R. w. Armstr o ng , and J. J. Gil m an, Str u ctu r e and Prop er tie s of Energe tic Mate r ia ls , MRL Sy mpo siu m Proceedin g s , Vol. 296 (1992) . 10) I. B. Mi sh ra, JA NNAF Prop ul sio n Meeti ng , CPIA No. 377, pp. 25 -36 (1983) . 11) W. Eng e l and P. Charbit , ]. Therm. Anal., 1 3, 275 (1978) . 12) J. S. Ing m an, G. J. Kearley and S. F. A. Kett le, ]. Chem. Soc., Faraday Trans., 78, 1817 (1982) . ]. Mol. Str u ctu r e, 79, 319 (1982) . ]. Phy. Chem., 86, 4007 (1982) . 13) K. Klag e r, His tor y of Bin d er Develop m ent in Comp os it e Prope l- !an ts , ICT Inte r nati on al Sy mpo siu m on Use of Plastic Mate r ia l s for Prop e llants and Exp lo siv e s, Karlsruhe, Germany (1982) . 14) Y. Oy umi, Thermal Decomp os it ion of Azid e Polym ers, ibi d , pp. 226 -231, (1992) .
15) T. Mi ya zaki, and N. Kubota , Energe t i cs of BAMO, Prope l lants , E.-cp l osiv e s, Py ro te c hnic s , Vol. 17, pp. 5 내, (1992) . 16) Y. Oy u mi , Y. Mi tar ai, and T. Anan, Mechanis m of Cata l yt ic iEbfi df e,c Vts ool.n 1A8, MppM. O19/5H—M 2X00 , C(o1m99p3 o) s. i t e Prop el lants Combustio n Rate s , 17) Y. Oy u mi , Y. Mi tar ai and H. Bazaki, Thermal Decomp os it ion of AMMO/AP Comp os it e P rop el lants , _ibi d . , V ol. 18, pp. 168-17 2, (1993) . 18) R. L. Wi ller, and R. S. Day, Poly ( Glyc id y l Ni tra te ) Revis ited , Proceedin g s for the Joi n t Inte r nati on al Sy mpo siu m on Comp a ti - bil ity of Plasti cs and Ot he r Mate r ia l s wi th Exp lo siv e s, Prop e llants , Py rot e c hnic s and Processin g of Prop e llants and Ing re die n ts , Oct. 1989, pp. 258-269. 19) D. G. Pati l, and T. B. Brill , Thermal De co mp os it ion of Energe t ic Mate r ia ls 59. Characte r i za ti on of the Resid u e of Hexanit ro- hexaazais o wurt zit a n e, Combustio n and Flame, Vol. 92, pp. 456-45 8, (1993) . 20) D. G. Pati l, and T. B. Brill , Thermal Decomp os it ion of Energe t ic Mate r ia ls 58. Chemi stry of Ammoniu m Dini t ra mi de Near the Burnin g Sur face Temp er atu r e, ibi d , Vol. 92, pp. 178-186 (1993) . 21) T. Urbanski, Chemi stry and Technology of Ex plo siv e s, Vol. III, Eng lish Ed. (1985) . 22) AD-A234900, The Depa r t me nt of Defe n se Cr itica l Technolo .훈' es Plan(U .S .A.), May, 1991 . 23) J. C. W. Chen and M. K. Rho, CPIA Publica ti on No. 481, pp. 331-332 (1988) . 24) J. C. W. Chen and M. K. Rho, CPIA Publica ti on No. 481, pp. 333 -340 (1988) . 25) J. C . W. Chen and M. K. Rho, CPIA Publica ti on No. 481, pp. 341 -371 (1988) . 26) H. J. P asman, ]. Industr ial Ex plo siv e s Soc., jap a n , 2 (2) , 94 (1992) .
27) S. Borman, C& E News, Jan . 17, 1994, pp. 18-22. 28) C. D. Bedfo r d, M. Chay k ovsky , M. K. Rho, P. R. Dave, F. Forohar, and R. Gil ar di, ADPA (A Joi n t Inte r natio n al Sym p os iu m and Exhib i - tion ), Oct. 5— 7 , 1992, pp. 306— 3 11 . 29) C. D. Bedfo r d, M. Chay k ovsky , M. K. Rho, P. R. Dave, F. Forohar, aenncde ,R N. SGWil aC rDdiD, , TAhipr rd i l A8n— n1u0a , l1 9R9&2 .D Info r mati on Exchang e Confe r- 30) J. C . Bott ar o, R. J. Schmi dt , P. E. Penwell and D. S. Ross, PCT Int. Ap pl. WO 91, 19,669 (1991) . 31) R. J. Schmi dt , P. E. Penwell, and D. C. Bomberge r, U. S. US 5,316, 749 (1994) . 32) J. C. Bott ar o, R. J. Schmi dt , P. E. Penwell, and D. S. Ross, PCT Int. Ap pl. WO 91, 19,670 (1991) . 33)S Po.c .E, .V Eoal.t o8 n 6,, papn. T 9.6 2W— . 9C64 o,l e,( 1J9r6. 4)T .h e Cubane Sys t e m , ]. Am. Chem. 34) P. E. Eato n , and G. Casta l di, Sys t e m ati c S ubsti tut i on on the Cubane Nucleus, Ami de Acti va ti on for Meta la tio n of ''Satur ate d Sys t e m s, ibi d , Vol. 107, pp. 724-72 6, (1985) . 35) P. E. Eato n , B. K. Ravi Shankar, G. D. Pric e, J. J. Pluth , E. E. GOirl gb. e rCt,h eJm. .A , lVstoe lr ., 4a9n, dp p0. . 1S8a5n—d u1s8,6 ,S y(1 n9 t8h 4e s) i.s of 1,4-D i'ni t ro cubane, ]. 36) P. E. Eato n , Fir s t, Second, and Thir d Generati on Hig h Energy Mate r i als fro m Cubane, Sta teg i c D efe n ce Research Init iative Off ice of Sc ien ce and Technology , N00014-86-K-0553, AD-A247559, Feb. (1992) . 37) V. R. Gaert ne r, ]. Org. Chem., 32, 2972 (1967) . 38) T. Okuta ni, T. Kaneko, and K. Masuda, Chem. Pharm. Bull, 22, 1490 (1974) . 39) Y. Oy umi, T. B. Brill , A. L. Rheng o ld, and T. M. Haller, ]. Phys . Chem., 89, 4317 (1985) .
찾아보기•용어
-1 가교 결합형 바인더 133, 136 간접법 229 개미산 61 결합제 165 경화공정 (혼합형 추전제) 187 경화 촉매 163 고체 충전도(혼합형 추진제) 155 공해성 16 균일 (균질) 추진제 13, 34 그레이닝 20 그레인 붕괴 79 금속 연료(환원제) 158 기계적 성질(혼합형 추전제) 188 기폭 11 기폭제 13, 39 길로틴 108 l 나프탈렌 75, 78 나프릴아민 72 난류 혼합 186 네트워크 136 노즐 15, 200 노줄목 129, 200 노화 191 뇌관 14 니트라민계 152, 197, 208, 215 니트라졸 115 니트로 글리콜 25 니트로 나프탈렌 75, 239
니트롤리시스 233 니트로디페닐아민 20, 73 니트로소디페닐아민 72, 75 니트로소히드록실아민 217 니트로 아니졸 239 니트로 우레탄 221, 229 니트로우스 에스테르 217 니트로 톨루엔 76, 239 니트로 파라핀 221 니트로 페닐에틸아민 76 니트로 프로판 294 니트로 피페리딘 229 C: 다축특성시험 189 다크 영역 34, 198 담그기 공정 103 둔감 추전제 312 둔감화 공정 285 디니트로 -2.5.- 디아자 핵산 218 디니트로벤젠 239 디니트로 카바마이트 76 디니트로 톨루엔 76, 101 디니트로페닐 메틸니트라민 222 디니트록시 데트라 니트로 데트라자 노단 251 디메틸 니트라민 221, 229 디메틸 페닐 우레마 239 디아세록시 데트라 니트로 테트라자 노난 289 디아조늄염 225 디아조 메탄 225
디에틸디페닐 우레마 239 디이소시아네이트 143 디페닐 아민 20, 64, 67, 77, 223 디페닐 벤즈아마이드 75 디페닐 페나전 71 근! 라미노우스 34, 198 라이너 (준비) 180 라이닝 113, 182 롤링 93 리뮴 158 E, 마감공정 187 마그네슘(분말) 158 마카로니 101 마찰감도 39 멀터플불꽃 196 메탄 46 메톡실레이트 242 메틸니트라민 225 메틸렌 니트라민 290 메틸 바어올렛 65, 88 모터 케이스 15, 127, 180 무게 손실 57 무섭광 47 무수인산 61 무용제 압신 104 무화염 화약 26 미니멈 스모크 306
닌 바인더 131 바셀린 34, 60, 78 박편화약 40, 61, 195 발광불꽃 34 방전 41 볼파우더 20 베릴륨 158 부탄온 231 부틸니트라민 231 부틸프탈산 75 분해생성물 28 분해속도 58 분해열 47 불꽃 영역 35 불화 보론 281 불활성 45, 48 블렌딩 101 비바시티 상수 24 비색법 66 비추력 17, 123, 130 人 산화 마그네슘 74 산화제 분쇄 182 삼성분 모드 156 삼중기 추전제 13~192 삽질산 암모늄 275 샌드위치 확산 195 서베일런스 86 서브믹스 178, 183
선형 고분자 133, 136 섬광 44, 55 센트럴라이트 78, 112, 116 셀룰로오스아세테이트 113 수분 방지제 58 수압탈수장치 94 수직형 혼합기 98 수평형 혼합기 98 술파민산 275 스모그 305 스모크 50 스웰링 94, 114, 133 스카벤저 추진제 305 스크류 압출 111 스크리닝 101 스타형 그레인 126 스트랜드 버너 201, 203 슬러리 주조 115 술로티드 126 시클로트리메틸렌트리니트소아민 239 시클로트리메틸렌트리아민 241 시클로헥사논 292 시클릭에테르 252 실패기준 190 。 아닐린 74 아미노메틸니트라민 225, 288 아미노안트라퀴논 227 아미노티아졸 228 아미노피리딘 227 아밀 알코올 • 55~77
아지리딘계 결합체 174 아지리딘계 경화제 147 아질산염 62, 66 안전도 마진 190 안전 사용 수명 86~92 안정성 54~64 암모늄 옥살레이트 112 압력봄베 52 압신 92~115 압력지수 130, 201 애토마이저 182 어스 43 에틸렌 디니트라민 217 에틸 아세테이트 20, 113 에폭시계 경화제 147 에피클로로히드린 329 연마 42 연막성 51 연소 모델 195 연소 불꽃 34 연소 생성물 130 연소속도 방정식 199 연소속도 촉매 163 연소 억제제 126 연소 잔여물 50 연소현상 194 연쇄반웅 49 열용량 130 영화칼리(륨) 48 염화칼슘 47 오리피스 52 오작동 88 옥사졸린 149 옥살산 61
온도 민감도 204, 208 온도 사이클링 79 우레아 75 우레탄 75 원통형 그레인 126 웨브(두께) 56 유동성 125 유연 16 유전상수 293 유전손실률 83 이성분 모드 156 이질산암모늄 282 이질산핵사민 257~282 이질산핵사메틸렌데트라민 286 이차 불꽃 44, 50 이차 스모크 306 인덕션 기간 48 인산에틸 281 일산화탄소 48 일차 불꽃 44 일차 스모크 306 입상 확산 195 x: 작동수명 86~92 저연 스모크 306 전방 스커트 15 전분 용액 65 정감형 36, 126 정증형 36, 126 접화 11 점화기 15 정전화 39, 43
젤라틴(화) 105 중립형 36, 126 중탄산소다 73, 112 지르코늄 158 지연 점화 27 직접법 (니트라민) 226 질산메틸렌글리콜 258 질산아민 229 질산헥사민 284, 294 * 초음파 검사 188 총강 14 총구 14, 29 총신 14 총추력 91, 130 추전장약 13, 14 추전제 그레인 15, 126 충격 39 충전분율 156 취성온도 83 침식 51, 203 구' 카르복실산계 143~150 카바마이트 61 카바졸 74, 78 칼륨이온 47, 50 캄포 75, 237, 239 코노실 126 코어 (준비) 공정 179, 187 콜로이드 계열 42
콜로이드상 56 큐벤 327 클로라민 231 클로로 포메이트 230 쿨로버잎 126 E 탄산나트륨 22 탄산 마그네슘 74 탄산 소다 61 탄산 칼슘 74 탄 이동거리 29 탄피 14 탈니트로화 55, 59 탈유 공정 180 탈탄화 54 테트라페닐히드라전 71 테트릴 223, 239, 247 튜브형 58, 99 트리니트로메틸 아닐린 222 트리니트로 벤젠 239 트리니트로 톨루엔 76, 239 트리니트로 페닐아민 73 트리브로모 아닐린 228 트리클로로시클로 트리메탈렌 트리 아민 240 포 파라포름알데히드 282, 290 파이로셀룰로오스 24, 42 팽윤 134 팽 창 (dil at a ti on ) 165
펠버라이저 182 페나전 71 페닐 니트라민 217, 225 페닐렌 디아민 138 페이스트 124, 160 펜트리트 243, 247 평행충 36 폐쇄기 31, 50 포구 속도 38 포구 초속 27 포름알독심 241 포밀디페닐아민 112 포탄 14 폭굉 11, 39 폭굉속도 39 폭굉열 246 폭발열 32, 38, 248 폭발한계 45 풀리 디메틸 실록산 135 폴리 부타디엔계 135~154 폴리 영화비닐(계) 133 폴리 설파이드 134~139 폴리 스티렌 135~138 폴리 아민계 결합제 173 폴리 아크릴로니트릴 135 폴리 에스테르 138, 142 폴리 우레탄 134~140, 150, 208 폴리 에데르 135, 142 폴리이소 부틸렌 133~138 프리믹스 98, 179 프리코팅 (산화제) 176 프리 앙상블 197 풀라스티졸 115 플래토 206
플루이다이저 182 피노실 126 피로수명 190 피즈영역 35, 198 피크린산 41, 239 _lo 형태 변형 293 헥사메틸렌데트라민 235~274
핵사민 249~262 화포(표준화) 24 확산 불꽃 196 활성탄소 61 후방 섬광 50 후방 스커트 15 흑색화약 11, 123 히드록시부틸산 61 히드록시파이루빅산 61
노만균 고려대학교 화학공학과를 졸업하고 동대학원에서 공학박사 학위 를 받았다. 국방과학연구소 추진제 실장, 추진기관 부장 및 로켓 추진제 공장장을 역임하였다. 미국(미해군 지상무기연구소, Aeroje t Co., Un iv. of Mass.) 과 프랑스 (S.N.P.E. Co.) 에서 추진제에 관한 연구를 하였다. 현재 국방과학연구소 책임연구원으로 재직중이다. 고체추진제 대우학술총서 자연과학 119 1 판 1 쇄 펴냄 __- 1998 년 2 월 25 일 지은이-노만균 펴낸이 __- 朴孟浩 펴낸곳-(주)던읍사 출판등록 1966. 5. 19. 제 16-490 호 서울특별시 강남구 신사동 506 대표전화 515-2000, 팩시밀리 515-2®7 값 20,000 원 ® 노만균, 1998 Printed in Seoul, Korea 화학공업 추진체 폭발물 KDC / 572.2 6 ISBN 89-374-3619-1 (94430) 89-'51 4 -3000-2 (세트)
대우학술총서 기다」 |(I I
1 소립자와 게이지 상호작용 김진의 2 동력학특론 이병호 3 질소고정 송승달 4 상전이와 임계현상 김두철 5 촉매작용 진종식 6 뫼스바우어 분광학 옥항남 7 극미량원소의 영양 승정자 8 수소화봉소와 유기봉소 화합물 윤능민 9 항생물질의 전합성 강석구 10 국소적 형태의 Atiy a h-sin ge r 지표이론 지동표 11 Mucop ol y sacchar id es 의 생화학 및 생물리학 박준우 12 천체물리학 홍승수 13 프로스타글라딘 합성 김성각 14 천연물화학연구법 우원식 15 지방영양 김숙희 16 결정화유리 김병호 17 고분자에 의한 화학반응 조의환 18 과학혁명 김영식

42 핵구조물리학 민동필 43 후리에 해석과 의미분 작용소 김도한 44 한국의 고생물 이 하영 45 질량분석학 김명수 46 금변론 박대현 47 생체에너지 주충노 48 리이만 기하학 박을룡 49 군표현론 박승안 50 비선형 편미분 방정식론 하기식 51 생체막 김형만 52 수리분류학 고철환 53 찰스 다윈 정용재 54 금속부식 박용수 55 양자광학 이상수 56 효소반응 속도론 서정현 57 화성암 성인론 이민성 58 확률론 구자홍 59 분자 분광학 소현수 60 벡터속 이론 양재현 61 곤충신경 생리학 부경생

85 비가역 열역학 이철수 86 등각장론 임채호 87 방사선생물학 남상열 88 석유지질학 아용일 89 베르누이 시헹의 통계적 분석 배도선·김성인 90 신경세포생리학 강만식 91 생리활성을 가진 C ― P 화합물의 화학 김용준 강익중 92 생물유기화학 서정헌 93 조직배양 김승업 94 유기전이금속화합물 조남숙 외 95 실내환경 과학 김윤신 96 유한요소법 정상권 97 대수적 위상수학 우무하 • 김재룡 98 파인만 적분론 장건수 99 응용 미생물학 박무영 100 리보플라빈 이상선 101 노화 김숙희 • 김화영 102 매트릭스 격리분광학 정기호 103 신경계 조직배양 김승업
