
김승업 서울대학교 의과대학 졸업 일본 쿄토(京都) 대학 대학원 의학박사 미국 컬럼비아 대학 펠로우 미국 펜실바 니아 대학 신경내과 부교수와 교수 독일 하이델베르크 대학, 일본 도쿄(東京) 대학, 오사카(大阪) 대학 , 미국 NIH 초빙교수 현재 캐나다 브리티시 컬럼비아 대학 신경내과 마리안코너 석좌교수 350 여 편의 저서, 논문, 초록 출간
신경계 조직배양
 신경계 조직배양
신경계 조직배양
책 머리에 동물의 뇌는 억 개가 넘는 신경세포와 그 10 배가 넘는 신경교 세포로서 구성되는 거대한 정보처리 기관이며, 이들 신경세포간 의 긴밀한 연결로 이루어진 복잡한 네트워크에 의하여 감각, 운 동 기능을 비롯하여 인지, 사고, 감정, 의지 등의 신경기능과 정 신기능을 조절, 지배하고 있다. 이러한 뇌신경계의 구조와 생리를 해명하고자 하는 생명과학의 한 분야인 신경과학은 지난 20 년 동안 세포생물학, 면역학, 유전 학, 분자생물학 등의 생명과학 관련 분야의 지식과 실험 기법의 발전에 힘 입어서 획기적인 진전을 가전 바 있다. 그러나 신경과 학은 아직도 해명되지 못한 많은 문제를 가지고 있어서 생명과학 의 각 분야에서 가장 뒤떨어전 미개척 분야로 알려져 있다. 이러한 사실을 인식하고 알츠하이머병, 파킨슨병 등의 신경변 성 질환의 원인과 치료법이 전혀 알려져 있지 않은 실정을 우려 하여 미국에서는 부시 전대통령이 20 세기의 마지막 10 년인 1990 년대를 〈 뇌신경 연구의 10 년 The Decade of Bra i n 〉이라 설정하고 신경과학의 진홍과 발전에 전력을 다하고자 호소한 바 있다. 신경조직 배양은 이러한 신경과학에 속하는 한 분야로서 지난 20 년 동안에 신경과학의 발전에 크게 기여하였다. 조직배양은 20 세기 초에 해리슨 교수가 개구리 림프액 응고 중 에 올챙이 척수의 작은 조직편을 심고 그 조직편의 신경세포에서
신경섬유가 새로 성장하는 것을 관찰한 것이 그 시초가 된다. 이 실용적이고 간편한 실험 기법이 개발된 지 90 년이 지나면서 조직 배양은 생명과학과 그 관련 분야에서 없어서는 안되는 중요한 기 술로서 그 위치가 확고하게 되었다. 1980 년대에 들어서면서 신경 과학에서도 이 조직배양 기법을 활용하여 신경분자생물학과 신경 계 바이오 데크놀러지의 분야에서 새롭고 중요한 업적이 계속해 서 나오고 있다. 미국과 유럽에서는 그 동안 신경조직 배양법의 기본 교과서가 출판되 어 수많은 연구자들의 연구에 큰 도움이 되 어 왔으나 우리 나라에서는 이러한 신경 조직배양의 이론과 실제를 서술한 기본 지침서가 없어서 대학원 학생, 초중급 연구자, 기술원 둘이 크게 곤란을 당하고 있음을 알 수가 있다. 이 책은 그러한 공백을 메우고 신경조직 배양의 이론과 실제 기술을 상세히 서술하여 새로이 신경조직 배양을 시작하고자 하 는 연구자에게 도움이 되도록 마련된 입문서이다. 이 책은 2 년전 대우학술총서의 한 권으로 출판된 『 조직배양 』 의 자매서로서 일부 중복된 부분이 있으나 이들 두 권의 책이 조직 배양 및 신경조직 배양의 이해와 학습에 도움이 되도록 기대하는 바이다. 끝으로 이 책을 마련하도록 도와주신 대우재단에 감사드리며, 이 책을 나의 양친 김정순, 임신애 두 분에게 바친다. 1996 년 1 월 김승업
신경계 조직배양
차례 책 머리에 • 5 제 1 부 신경계 조직배양의 기초지식 제 1 장 조직배양과 신경조직배양의 역사 …………………… 15 제 2 장 실험실 설비와 기구 …………………………………… 23 2. 1 멸균시설 • 24 2. 2 증류수 장치 (이온교환 컬럼) • 26 2. 3 원심분리기 • 26 2. 4 인큐베이터 • 28 2. 5 무균 벤치 • 29 2. 6 현미경 • 30 제 3 장 배양기구 ………··:……………………………………… 32 제 4 장 기구세척과 멸균 ………………………………………… 37 4. 1 세척 • 37 4. 2 멸균 • 38 제 5 장 배양액 …………………………………………………… 41 5. 1 천연 배양액 • 42 5. 2 염류용액 • 425. 3 합성 배양액 • 46
5. 4 무균검사 • 51 5. 5 항생물질 • 51 제 6 장 세포 기질과 부착인자 ………………………………… 53 6. 1 콜라겐 • 54 6. 2 라미닌 • 58 6. 3 피브로넥틴 • 59 6. 4 비트로넥틴 • 59 6. 5 젤라틴 • 60 6. 6 폴리라이신 • 60 제 7 장 무혈청 배양액의 개발 ………………………………… 62 제 8 장 배양법의 분류 …………………………………………… 68 8. 1 정치배양법 • 68 8. 2 회전배양법 • 71 8. 3 선회배양법 • 71 8. 4 부유배양법 • 71 제 9 장 일반 배양기술 …………………………………………… 73 9. 1 무균조작 • 73 9. 2 배양액 교환 • 74 9. 3 계대배양 • 75 9. 4 세포 수 계산법 • 76 9. 5 세포동결보존법 • 78 9. 6 세포 운반 • 80제 10 장 신경조직 세포 분리 기술 ……………………………81
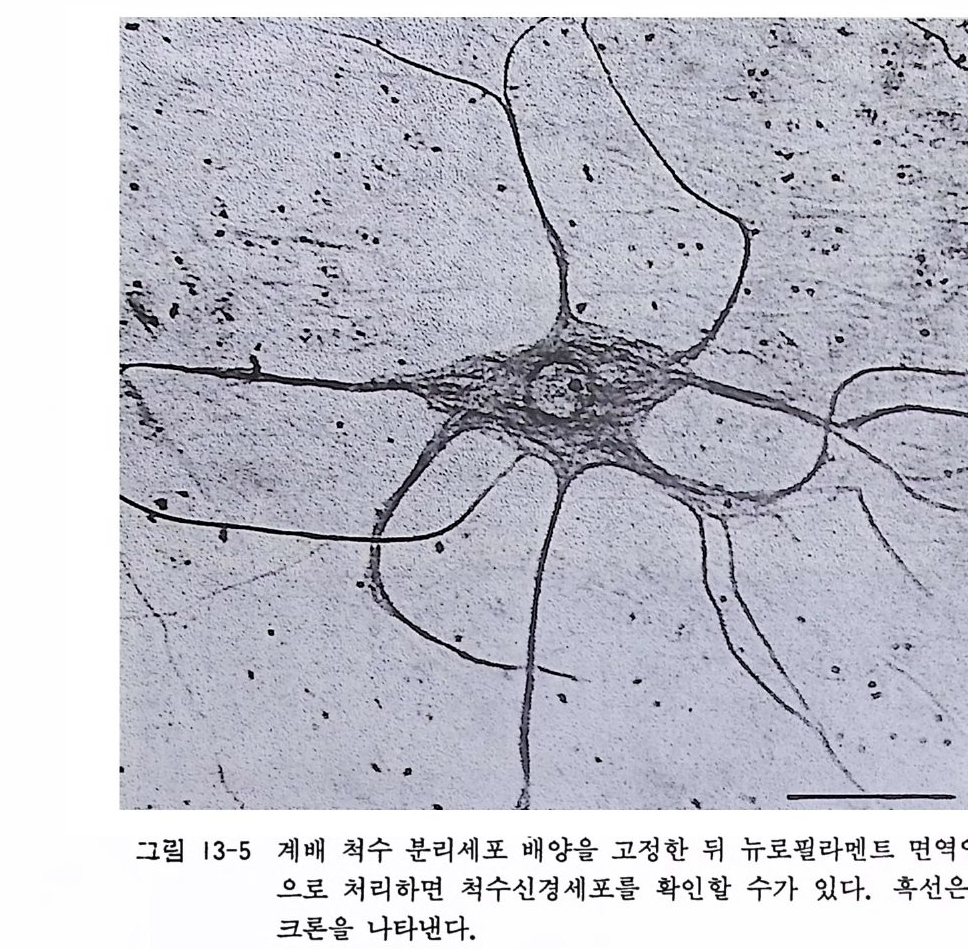
10. 1 트립신 • 82 10. 2 프로나제 • 83 10. 3 콜라게나제 • 83 10. 4 디스파제 • 84 10. 5 파파인 • 84 10. 6 금속제거제 • 85 10. 7 기계적 세포분리 • 85 제 2 부 신경계 조직과 세포의 초대 배양법 제 11 장 계배 척수후근신경절과 교감신경절의 배양 ……… 91 11. 1 계배의 적출 • 92 11. 2 계배 척수후근신경절 분리세포 배양 • 94 11. 3 맥시모우 슬라이드에 의한 계배 척수후근신경절 이식편 배양 • 100 11. 4 페트리 접시에 의한 계배 척수후근신경절 이식편 배양 • 104 11. 5 계배 교감신경절 분리세포 배양 • 106 제 12 장 계배 모양체신경절의 배양 ………………………… 109 12. 1 계배 모양체신경절 분리세포 배양 • 110 12. 2 모양체 신경영양인자의 준비 • 111 제 13 장 계배 중추신경계 조직과 세포의 배양 …………… 113 13. 1 계배 척수이식편 롤러튜브 배양법 • 114 13. 2 계배 척수신경세포 분리세포 배양 • 11913. 3 계배 망막신경세포 분리세포 배양 • 123
제 14 장 태생 래트와 마우스 척수후근신경절과 교감신경절의 배양 …………………………………·……………… 125 14. 1 태생 래트(마우스) 척수후근신경절의 분리세포 배양 • 126 14. 2 페트리 접시에 의한 태생 래트(마우스) 척수후근신경절의 이식편 배양 • 131 14. 3 신생 래트와 마우스 교감신경절의 이식편 배양 • 133 14. 4 신생 래트와 마우스 삼차신경절의 이식편 배양 • 135 제 15 장 태생 및 신생아 래트와 마우스 중추신경계 조직 • 세 포의 배양 ……………………………………………… 137 15. 1 신생 래트(마우스) 소뇌 이식편 롤러튜브 배양법 • 138 15. 2 신생 래트(마우스) 대뇌와 선조체 이식편 멀티웰-커버글 라스 배양법 • 144 15. 3 태생 래트(마우스) 척수 이식편 멀티웰-커버글라스 배 양 • 146 15. 4 태생 래트(마우스) 척수 분리세포 배양 • 150 15. 5 태생 래트(마우스) 혹질 분리세포 배양 • 152 15. 6 태생 래트(마우스)의 해마 분리세포 배양 • 156 제 16 장 성숙 포유동물 척수후근신경절과 교감신경절의 분리 세포 배양 ……………………………………………… 161 16. 1 성숙 래트(마우스) 척수후근신경절의 분리세포 배양 • 162 16. 2 퍼컬 밀도구배 컬럼에 의한 신경세포 순수분리 • 165 16. 3 성숙 래트 교감신경절 분리세포 배양 • 168제 17 장 성숙 포유동물 중추신경계조직 • 세포의 배양 …… 173
17. 1 성숙 래트(마우스) 망막 이식편 배양 • 174 17. 2 성숙 래트(마우스) 망막 분리세포 배양 • 175 제 18 장 신경세포의 순수배양 ………………………………… 180 18. 1 태생 래트(마우스) 척수후근신경절 신경세포의 순수배 양 • 181 18. 2 태생 래트(마우스) 교감신경절 신경세포의 순수배양 • 187 18. 3 태생 래트(마우스) 대뇌 신경세포의 순수배양 • 189 18. 4 항체의존 세포 분리 기술(패닝)에 의한 신생래트 망막 신 경세포 순수배양 • 194 18. 5 패닝 접시 제작법 • 198 제 19 장 슈완세포의 순수배양 ………………………………… 199 19. 1 태생 래트(마우스) 척수후근신경절 이식편 배양에 의한 슈완세포 배양• 200 19. 2 태생 래트(마우스) 척수후근신경철 분리세포 배양에 의한 슈완세포 배양 • 202 19. 3 신생 래트(마우스) 좌골신경 분리세포 배양에 의한 슈완 세포 배양 • 205 제 20 장 울리고덴드로사이트의 순수배양 …………………… 209 20. 1 퍼컬 밀도구배 컬럼에 의한 올리고덴드로사이트 순수분리 와 배양 • 210 20. 2 신생 래트(마우스) 올리고덴드로사이트의 순수분리와 배 양 • 218 20. 3 나일론 멧슈를 부착한 뷔크너 깔때기 제작법 • 224제 21 장 아스트로사이트의 순수배양 ………………………… 225
21. 1 신생 래트(마우스) 아스트로사이트의 순수분리와 배양 • 226 21. 2 무혈청 배양액을 사용한 신생 마우스 대뇌 유래의 아스트 로사이트의 순수배양 • 229 제 22 장 미크로글리아의 순수배양 …………………………… 232 22. 1 성숙 포유동물 뇌에서의 미크로글리아 분리와 배양 • 233 22. 2 신생 래트(마우스) 뇌에서의 미크로글리아 분리와 배양 • 236 제 23 장 뇌혈관 내피세포의 순수배양 ……………………… 238 23. 1 성숙 래트(마우스) 뇌혈관 내피세포의 기계적 분리 배양 법 • 239 23. 2 퍼콜 구배 컬럼에 의한 래트 뇌혈관 내피세포의 순수배 양 • 242 제 3 부 특수한 신경조직 및 비신경조직의 배양법 제 24 장 골격근 조직의 배양 ………………………………… 249 24. 1 계배 골격근의 분리세포 배양 • 250 24. 2 태생 래트(마우스) 골격근의 분리세포 배양 • 252 24. 3 젤라틴 코팅 • 254 24. 4 나일론 멧슈 봉지 제작법 • 255 제 25 장 뇌하수체 세포의 조직배양 ………………………… 256 25. 1 신생 래트나 마우스 뇌하수체 분리세포 배양 • 25725. 2 성인 뇌하수체 분리세포 배양 • 259
제 26 장 부신수질 크로마핀 세포의 배양 ••…………………· 264 26. 1 신생 래트(마우스) 부신수질 크로마핀 세포의 분리세포 배양 • 265 제 27 장 양생류와 어류 신경조직의 배양 …………………••• 269 27. 1 올챙이 척수이식편 배양 • 270 27. 2 올챙이 척수 분리세포 배양 • 271 27. 3 금붕어 망막이식편 배양 • 273 27. 4 금붕어 망막분리세포 배양 • 275 제 28 장 무척추동물 신경조직배양 …………………………… 276 28. 1 초파리 신경조직배양 • 276 28. 2 바퀴벌레 신경조직배양 • 279 제 4 부 신경계 수립세포주 제 29 장 뇌종양의 초대 배양법 ……………………………… 283 29. 1 조직소편 배양법 • 284 29. 2 분리세포 배양법 • 286 제 30 장 잡종세포 제작법 ……………………………………… 290 30. 1 폴리에틸렌글리콜 세포융합법 • 292 30. 2 전기적 세포융합법 • 295제 31 장 유전자 아입에 의한 신경계 세포주의 수립 ……… 297
제 32 장 기성 신경계 조직 수립세포주의 배양 …………… 301 제 33 장 부신갈색종 세포배양 ………………………………… 306 제 34 장 기형종 세포배양 …………………………………… 310 제 5 부 특수실험법 제 35 장 신경계 조직에서의 세포타입 특이 마카 ………… 317 35. 1 세포타입 특이 마카 • 318 35. 2 세포골격단백 • 320 35. 3 미엘린 단백 • 321 35. 4 신경전달물질과 관련 효소 • 322 제 36 장 면역세포화학 ………………………………………… 324 36. 1 형광 항체 염색법 • 326 36. 2 효소 항체 면역 염색법 • 330 제 37 장 신경계 조직 배양세포 증식의 판정 ……………… 338 37. 1 오토라디오그라피 • 339 37. 2 신틸레이션 카운터 • 340 37. 3 브로모디옥시유리딘 항체에 의한 형광염색법 • 342 제 38 장 세포 활성의 측정 …………………………………… 346 38.1 크리스탈 바이올렛 검사법 • 34638. 2 MTT 검사법 • 348
제 39 장 세포염색법 …………………………………………••• 351 39. 1 김사 염색법 • 352 39. 2 헤마독실린 염색법 • 352 39. 3 메틸그린-피로닌 염색법 • 354 39. 4 생체염색 • 355 39. 5 보디안 은영색법 • 355 39. 6 수단블랙 미엘린 염색법 • 357 39. 7 닛슬 신경세포 염색법 • 359 제 40 장 세포화학적 염색법 …………………………………… 362 40. 1 아세틸콜린 에스테라제 염색법 • 363 40. 2 카테콜아민 신경 형광영색법 • 365 40. 3 NADPH 데트라졸륨 환원효소 염색법 • 366 40. 4 모노아민 산화효소 염색법 • 368 제 41 장 전자현미경 기술 ……………………………………… 369 41. 1 투과전자현미경 • 370 41. 2 주사전자현미경 • 374 제 6 부 신경조직배양의 진보와 그 전망 제 42 장 신경조직배양의 진보와 그 전망 …………………… 379 42. 1 탈수질환 • 380 42. 2 신경변성질환 • 381 42. 3 바이러스 감염증 • 38542. 4 결어 • 386
참고문헌 • 387 용어해설 • 421 찾아보기 • 429제 1 부 신경계 조직배양의 기초지식 
제 1 장 조직배양과 신경조직배양의 역사 생물의 생존과 활동에는 그 내적 환경이 중요하다는 것을 프랑 스의 베르나르 Bernard 가 1878 년에 지적한 바 있다. 그는 이러한 내적 환경은 생물의 신전대사에 의한 결과인 동시에 피드백의 작 용으로 생물의 활동을 조절한다고 생각하였다. 또한 그는 생물 세포가 내적 환경에 미치는 영향 혹은 내적 환경으로부터 세포가 받는 영향을 연구하는 데에는 세포를 인공적인 조건 아래에서 발 육시키고 세포가 생물 전체의 영향으로부터 완전히 분리되고 차 단되어야 한다고 생각하였다. I) 이러한 학설에 의거한 실험은 프 랑스의 루 Roux 가 1885 년에 계배 신경관을 생리식염수 속에서 발육시 킨 것 이 가장 최초의 것 이 다. 1898 년에는 룽그렌 Lju ng- gr en 이 사람의 피부 조직을 암환자의 복수 속에서 일주일간 정치 한 뒤 동물에 이식하는 실험을 하였고, 1902 년에는 러브 Loeb 가 기니피그의 피부 조직을 혈장을 포함한 한천 중에서 배양하고 이 를 다른 기니피그의 피부에 이식하는데 성공하였다. 1903 년에는 졸리 J oll y가 개구리 백혈구 배양을 하였고, 1906 년에는 비비
Beebe 와 유잉 Ew i n g이 개에서 림프선종 조직을 분리하고 배양하 였다. 이와 갇이 조직배양의 발상은 19 세기 말에 시작되었지만 조직배양법이 자연과학의 기술로 정착하게 된 것은 20 세기 초 미 국의 해리슨의 공적에 의한 것이다. 미국 예일 대학의 해리슨 Harris o n 이 1906 년에 올챙이 척수조 직을 응고된 개구리 림프액 속에서 배양하여 신경섬유가 새로 성 장하는 것을 관찰하였다(그림 1-1).Z) 해리슨은 이렇게 간편하고 실용적인 배양기술을 고안함으로써 조직배양법의 시조라 불리게 되었다. 해리슨의 이와 같은 연구결과는 히스 H i s 와 라몬 이 카
 10.50 곡군`` 7 f 1111..2218 ``` 소、\4`
10.50 곡군`` 7 f 1111..2218 ``` 소、\4`
할 Ramon y Caja l 이 주장하고 있던 뉴론학설, 즉 신경계조직은 하나하나의 신경세포가 단위가 되어 구성되는 것이며 신경섬유는 신경세포체에서 나와서 다음의 신경세포체와 접촉을 한다는 학설 을 실증한 것이었다. 당시 헬트 Held 와 골지 Gol gi가 주장하던 망 사학설, 즉 신경계조직은 신경세포의 원형질의 연장에 의해서 망 사와 같은 상호연락으로 구성되었다는 학설을 실험적으로 부정한 것이다. 해리슨의 논문이 발표된 이후로 세계 각국의 다수의 학 자들이 오랫동안 노력해서 여러 가지 개선과 발전을 가져온 결과 오늘날의 신경조직배양의 터전이 이루어전 것이다 .3) 해리슨의 제자 바로우 Burrow 는 해리슨의 냉혈동물에서의 실 험기술을 온혈동물에서도 가능하도록 개량하였고, 특히 혈장이 배양성분으로써 우수하다는 것을 발견하였으며,° 바로우는 카렐 Carrel 과의 공동연구에서 각종 동물조직의 조직배양을 성공시킨 바 있다 .5) 해리슨의 초보적인 배양법을 개량하여 과학적으로 인 정받을 수 있는 실험 기술로 향상시킨 데에는 카렐의 공적이 가 장 크다. 카렐은 의과 의사로서의 오랜 경험에서 조직배양을 성 공시키려면 무균조작이 가장 중요하다고 생각하고 철저한 무균 실, 무균기구, 그리고 무균배양액을 사용함으로써 장기적인 조직 배양을 확립한 바 있다. 카렐은 또한 배양액의 구성에 대해서도 연구를 하여 조직 추출액이 각종 세포의 성장과 증식에 좋은 효 과를 가지고 있다는 연구결과에서 적절한 배양액의 구성이 조직 배양의 기초임을 보여주었다 .6) 카렐의 연구실에서는 이러한 무 균조작의 철저한 시행과 적절한 배양액 구성의 확립으로서 계배 에서 분리한 섬유아세포를 34 년간 연속 배양하는 데 성공하였다. 카렐은 이러한 성과에서, 세포가 영구적으로 생존할 수 있다고 제창하였다.” 그러나 정상 세포를 계대 배양하면 최대로 50 대 이상을 배양할 수 없다는 최근의 성과에서 보면 이러한 학설은
신빙성이 전혀 없다. 사실 카렐의 연구실에서는 20 년 이상을 계 대하고 있던 계배세포 플라스크에 카렐의 연구원이 신선한 세포 를 넣어주고 있었다는 사실이 판명되어서 하나의 스캔들로 지목 되고 있다. 아랫사람의 이러한 불미스러운 처사로 인해서 카렐의 이름에 상처가 가게 된 것이다. 1910 년에서 1950 년대까지는 카렐의 연구 이외에 앞서 말한 바 로우, 루이스 Lewis , 마리네스코 Marin e sco, 맥시모우 Maxim ow, 레비 Levi, 스트랜지웨이 Str a ng e way 등의 학자들에 의해서 조직 배양과 기관배양에서의 새로운 여턱 가지 개량과 발전을 가져온 바 있다. 이들 학자둘은 각종 동물의 신경세포와 신경교세포의 형태에 대해서 기술하였으며 이들의 조직배양은 보통 1 주일 혹은 2 주일이 한도가 되었으며 그 이상의 배양은 극히 드물었다. 재료 로서는 계배 신경조직아 많았으며 그 가운데서도 계배 척수후근 신경절세포가 많이 사용되었고 레비 Lev i와 와이스 We i ss 는 신경 섬유의 성장과 신경세포의 성숙에 대하여 상세한 기록을 남긴 바 있다 .8,9) 배양방법으로서는 올챙이 척수조직의 소편을 응고림프 액에 의해서 커버글라스 위에 고정하고 이것을 특수한 유리 슬라 이드나 페트리 접시 속에서 배양하는 방법을 해리슨이 시작하였 는데, 응고 림프액 대신에 응고 혈장(풀라스마)을 써서 배양하는 방법이 오랫동안 사용되었다. 이 배양 방법은 오늘날에도 많이 쓰이고 있다. 조직배양법에서 가장 문제가 되었던 것은 세균과 곰팡이에 의 한 감염이었다. 수혈과 혈관 봉합의 연구를 비롯한 의과학의 발 전에 기여한 공로로 노벨상을 수상받은 카렐은 철저한 무균기술 울 도입하여 세균 감영을 예방하고 조직배양의 성공에 큰 공헌을 하였지만 그 반면에 너무나 비용이 드는 무균실, 무균기구의 사 용 등 너무 장황한 준비가 필요하게 되어 많은 연구자들이 조칙
배양을 시작하기도 전에 이를 멀리한 경향이 있게 된 것도 사실 이다. 그밖에도 여러가지 제한 조건, 예컨대 특수한 배양액의 필 요나 동물 신경조직의 부위, 동물의 연령에 따라서 배양 성공율 이 저하하는 등 여러가지 부정적인 조건 등으로 조직배양 특히 신경조직배양은 최근까지 마술사의 요술인양 신비화되고 경원되 어 온 것도 사실이다. 조직배양이 커다란 진보를 가져오게 된 것은 1945 년 이후 제 2 차 세계대전이 끝난 뒤이고, 페니실린이나 스트랩토마이신 등의 항생물질을 배양액 속에 첨가함으로써 무균조작이 가능하게 된 이후의 일이다. 1945 년 이후로 신경조직배양의 역사에도 새로운 페이지가 가하게 되었고 1947 년에는 호우그 Ho gu e 가 사람 태생 신경조직을 롤러 튜브법으로 배양하고 위상차현미경 아래에서 신 경세포의 동정에 성공하였다 .IO) 1950 년대에 둘어서서 컬럼비아 대학의 머레이 Murra y와 텍사스 대학의 포메라트 Pomora t와 힐 트Hi ld 의 연구실에서 수많은 연구 업적이 나왔으며 신생과 태생 고양이, 마우스, 래트의 소뇌, 대뇌, 척수 등 중추신경조직의 장 기간의 배양이 가능해지고 신경세포와 각종 신경교세포의 동정과 형태 변화의 각종 연구가 이루어진 바 있다. 이 가운데서 가장 특기할 연구로서 조직배양하에서의 수초형성이 있다. 1955 년 피 터슨 Pe t erson 과 머레이가 계배 척수후근신경절 배양에서 처음으 로 수초형성울 관찰하였고, 11) 이어서 1957 년에 힐트가 신생 고양 이 소뇌배양에서, 12> 1958 년에는 본스틴 Borns t e i n 과 머레이가 신 생 고양이와 래트 소뇌배양에서 중추신경계 수초형성을 관찰한 바 있다 .13) 1920 년 이후 카렐의 영양 요소에 대한 연구에 이어서 루이스 Lew i s 가 동물세포의 생존과 성장의 기본 요소를 배양조건하에서 연구하였다 .14) 이러한 연구는 파커 Parker, 모간 Mor g an, 웨이모
스 Wayr no uth , 이 글 Eag le , 행 Ham 등의 후배 학자들에 의 해 서 계속되었으며, 이들은 필수아미노산, 비타민, 희금속, 염류, 뉴 클레오티드를 포함한 각종 합성배양액을 만들었으며 이러한 합성 배양액 예컨대 199, 이글 MEM, 행 F12 등의 배양액은 신경조 직배양에서 기초배양액으로서 사용되어 현재에 이르고 있다. 15) 벙기 Bun g e 는 신경조직배양의 역사를 음악의 역사와 비교하여 해리슨의 올챙이 척수조직배양의 논문이 발표된 1906 년부터 피터 슨과 머레이에 의한 수초형성의 논문이 발표된 1955 년까지 를 고 전시대, 1956 년부터 1968 년까지를 바로크 시대, 1969 년 이후를 큐비즘 시대라 구분하고 있다 .16) 고전시대에는 앞서 말한 바와 같이 해리슨, 레비, 와이스 등의 학자들이 단기간의 신경조직배 양에서 신경세포, 신경섭유의 성장을 기록하고 신경조직배양을 성공적으로 보전하는데 필수적인 기본조건에 대해서 연구하였다. 바로크 시대에는 컬럼비아 대학의 머레이를 중심으로 하여 피터 슨, 본스틴, 벙기, 크레인 Crain 등의 학자들과 텍사스 대학의 포메라트롤 중심으로 한 힐트, 럼스덴 Lumsden, 나카이 Nakai, 오카모토 Okamo t o 등의 연구자들이 은염색에 의한 우아하고 아 름다운 신경세포의 형태와 타임랍스ti me laps e 영화 촬영에 의한 역동적인 신경세포의 움직임을 기록하였다. 또한 활동전위을 전 기생리학적으로 기록하고 전자현미경적으로 시냅스 형성이 가능 함을 전시함으로서 배양신경세포가 생체 안에서의 신경세포와 다 름없이 정상적인 형태와 기능을 가지고 있음을 보고하였다. 신경 조직배양의 신빙성과 실용성이 이렇게 실증되어 새로운 전기를 가지게 되었다. 배양신경세포에 있어서의 활동전위의 기록은 크 레인이 말초신경계인 계배척수후근신경절배양에서 ,17) 그리고 중 추신경계인 고양이 소뇌배양에서 힐트와 타사키 Tasak i가 보고한 바 있다 .18) 중추신경계 조직배양에서의 시냅스 형성은 은염색 표
본에서 김 ,19 ) 피터슨 20) 에 의해서 관찰되었고, 전자현미경에서는 칼라스 Callas 와 힐트 21) 그리고 벙기 22 ) 에 의해서 보고되었다. 이 러한 신경조직배양의 초기 연구에 대해서는 1956 년과 1971 년에 발표된 머레이의 총설에 상세히 소개되어 있다 .23,24) 저자는 1966 년 머레이 교수가 있던 뉴욕의 컬럼비아 대학에 유학하여 3 년간 그곳에서 신경조직배양의 황금시대를 직접 보고 듣는 귀중한 경 험을 가진 바 있다. 벙기는 1969 년을 경계로 하여 그 이후를 큐 비즘 시대라 하였는데 그 이유는 1969 년에 신경조직의 분리세포 배양법에 대한 두 개의 논문이 발표되었기 때문이라 하였다. 이 것은 벙기가 잘못 알고 있는 것이고 이미 1956 년에 나카이가 계 배 척수후근신경절을 트립신 처리하고 신경세포를 분리하여 장기 배양에 성공한 바 있다 .25) 누가 먼저 분리세포배양을 시작하였는 지는 차치하고, 1970 년대에 들어서서 신경조직배양의 분야에는 새로운 파도가 밀려온 것이라 하겠다. 해리슨이 울챙이 척수소편 의 배양에 성공한 1906 년부터 1970 년 초기까지 이식편배양 exp la nt cultu r e 이 신경조직배양의 유일한 기술이었다. 이식편배 양의 장점은 신경조직의 분화와 그 기능의 재현을 충실하게 관찰 할 수 있다는 점이다• 또한 분리세포배양 d i sso ci a t ed cell cultu r e 에 바해서 원조직의 세포 구축을 장기간 보존할 수 있고, 시냅스 형성과 수초 형성을 쉽게 관찰할 수 있다는 장점이 있다. 그러나 현미경 아래에서 하나하나의 신경세포를 관찰할 수 없고 세포의 동정이 극히 곤란하고 전기생리학적 연구는 불가능에 가깝다• 1970 년대의 신경조직배양에서는 두 가지 새로운 기술이 도입되었 는데 그 하나가 분리세포배양이며 또 하나가 신경조직 수립세포 주 es t ab li shed cell li ne 의 도입이다. 수립세포주는 사람 혹은 동물 (주로 마우스와 래트)의 뇌종양에서 분리된 세포가 무한증식성을 획득하고 계대배양이 가능하게 된 세포 클론을 말한다. 니렌버그
Ni re nberg, 넬슨 Nelson, 슈버트 Schubert , 파이퍼 Pfe i f fer , 아마 노 Amano, 그린 Green 등에 의해서 뇌종양 유래의 신경아종세포 (뉴로불라스토마 neuroblasto m a) , 신경교종세 포 (글리오블라스토마 gliob lasto m a) , 부신갈색 종세 포 ph eochromacy tom a 의 새 로운 세 포 주가 분리되었고 이들이 정상세포가 가전 생리학적, 생화학적 성 질의 일부를 유지하고 있음이 보고되면서 그 사용이 널리 퍼지게 되었다 .26,27) 1970 년대의 뉴웨이브 new wave 시대의 연구에 관해 서는 넬슨의 총설 28) 과 사토 Sa t o 의 책자 29) 가 유용하다. 1950 년대에 처음으로 발견된 신경성장인자 (nerve grow t h fac to r , NGF) 는 1980 년대 에 들어 와서 크게 각광을 받았으며 1990 년대에 들어와서 신경성장인자와 그에 유사한 각종 신경영양 인자 neuro t ro p h i c fa c t or 가 속속히 발견되고 있는데, 이러한 신경 영양인자의 발견과 그 구조와 작용의 해명에는 신경조직배양의 공헌 이 가장 크다. 레 비 -몬탈치 니 Levi- M onta l cin i 와 코엔 Cohen 은 마우스 육종조직, 뱀독 그리고 마우스 악하선의 추출액을 계 배 후근신경절과 교감신경절의 조직배양에 작용시키면 그 신경섭 유가 경이적인 속도로 성장한다는 관찰을 하고 신경성장인자의 개념을 새롭게 창시한 바 있다 .30,31) 이 업적으로 레비-몬탈치니 와 코엔은 1986 년에 노벨상을 받은 바 있다. 신경성장인자를 비 롯하여 여러 신경영양인자는 신경세포의 형태와 기능을 유지하 고, . 이들에게 새로운 형질p heno typ e 을 부여할 수 있는데 현재 알려져 있는 것이 신경성장인자를 비롯하여 12 가지 이상 있으며 앞으로도 신경조직배양을 활용하여 발견되는 신경영양인자의 수 가 백 가지 이상 될 수 있다고 생각된다• 신경조직배양을 이용한 신경영양인자의 연구, 분자생물학적인 분석 그리고 신경질환의 조직배양 모델 등, 1990 년에 들어서서 크게 전전을 보이고 있는 연구 분야와 그 전망에 대하여는 제 42 장에서 논하기로 한다.
제 2 장 실험실 설비와 기구 제 2 차 세계대전 이전 항생물질이 나오기 전에는 조직배양 실험 실에서 무균조작을 하기 위해서 마치 큰 병원의 중앙수술실과 같 은 규모와 준비가 필요하였다. 최근에는 공기여과장비 filter ed a i r 를 써서 이러한 장황한 실험실이 필요하지 않고 무균 벤치 혹 은 무균공기상류벤치 lami na r flow hood 가 있으면 문제는 해결된 다. 조직배양에는 배양 준비와 무균 조직배양의 두 가지 임무가 있 게 되므로, 첫째로 수도와 배수구가 있는 곳을 중심으로 하여 실 험실의 반을 배양 준비 구역으로 하고 나머지 반을 무균 조직배 양구역으로 정한다. 배양 준비 구역에 최소한으로 필요한 기구로서 고압중기멸균장 치 (오토클레이브 au t oclave) 와 건열멸균기 (오븐&y heat oven), 그 리고 증류수 장치 wate r dis t il ling un it가 있다. 증류수 장치는 이 온 교환컬 럼 deio n iz i n g column 으로 대 신할 수 있는데 , 이 것 이 비 용으로 보나 생산량으로 보나 훨씬 효율적이다.
무균 조직배양 구역에는 앞서 말한 무균밴치와 정온 이산화탄 소 인큐베이터 i ncuba t or 를 두고, 이밖에 배양세포를 관찰 기록할 현미경 m i crosco p e 이 필요하다. 실험실의 사방 벽에는 배양을 위 한 유리 기구나 풀리스틱 기구를 저장할 캐비넷이나 선반이 설치 되어야 한다. 2.1 멸균시설 조직배양 실험실에서 중요한 것이 배양기구와 배양액을 준비할 때 사용되는 멸균시설이다. 이에는 전열멸균기 (오븐) (그립 2- 1 ), 고압증기 멸균기 (오토클레 이브) (그립 2-2 ) 가 있으며 , 셋째로 소량 의 배양액 혹은 열에 약한 시약을 멸균하기 위한 여과멸균기가
 ti
ti
 그림 2-2 증기멸균기 (오토콜레이브)
그림 2-2 증기멸균기 (오토콜레이브)
필요하다. 최근에는 여과멸균기로서 간편한 디스포사블 (1 회용)의 여과 필터가 나와 있어 편리하게 되어 있다(그림 2-3). 필터 구 멍 크기가 0.22 미크론과 0.45 미크론의 두 가지가 있다. 미국의 밀 리 포어 Mi llipo re 나 날전 Nalge ne 에 서 여 러 용량의 것 을 시 판하 고 있다. 2.2 증류수 장치 (이온교환 컬럼) 조직 배 양액을 만드는 물의 순도가 극히 중요하다. 미 량의 금속 에 의해서도 세포배양에 차질이 있게 되므로 고순도의 물이 필요 하다. 과거 유리 증류수 장치로 세 번 증류한 증류수를 사용하였 는데, 최근에는 여과방식 이온교환 컬럼으로 고순도의 물을 얻을 수 있게 되었다. 우리 연구실에서는 밀리포어의 밀리큐 M illi~Q 이온교환 컬럼을 사용하고 있다. 2.3 원심분리기 세포배양액에서 세포를 분리하기 위해 500-2000r p m 의 탁상 저 속원심분리기 tab le-to p cen t n ifug e 가 필요하다(그림 2-4). 조직배 양을 하는 횟수가 증가하게 되면 성능이 좋은 최고로 3000 rp m 까 지 올라가는 중성능 원심분리기가 있으면 좋겠다(그립 2-5). 원 심듀브는 멸균 풀라스틱 듀브가 여러 회사에서 생산 시판되고 있 어 편리하다. 때로는 농도구배 원심분리법 densit y grad ie n t cen t r ifug a ti on 에 의해 종류가 다른 세포를 분리 하게 되는데 , 이런 때에는 10,000-30,000 rp m의 고속원심분리기가 필요하다.
 락상완人j분리 7\
락상완人j분리 7\
2.4 인큐베이터 변온동물인 개구리나 올챙이 세포를 배양하기 위해서는 필요치 않으나 항온동물의 세포를 기르기 위해서는 인큐베이터 (lnc uba- to r 항온기)가 필요하다. 밀폐된 실험계에서라면 보통 인큐베이터 를 36- 37 °C 의 온도에 서 사용할 수 있으나 페 트리 접 시 pe tr i dis h 나 플라스크fl ask 와 같은 개방계를 쓰게 되면 이산화탄소 인큐베 이터가 절대적으로 필요하다. 이러한 이산화탄소 인큐베이터의 가스 농도는 보통 이산화탄소를 5-1 0 %를 유지하고, 인큐베이터 바닥에 물을 넣 어 서 습도를 100 %로 유지 한다 (그림 2-6 ) .
 그림 2-6 싱글도어 이산화탄소 (CO2) 인큐베이터
그림 2-6 싱글도어 이산화탄소 (CO2) 인큐베이터
2.5 무균 벤치 무균 벤치 Lami na r flow hood 는 보통 스데 인 리 스 강으로 만들 어진 폭 1-2m, 깊이 lm, 높이 2m 정도의 상자 같은 실험대로 서 천장이나 후방 부분에 있는 공기여과 장치를 통하여 무균공기 가 그 내부에 들어오게 되어 있다. 캐비넷 안에는 가스 라인과
 그림 2-7 탁상형 클린벤치
그림 2-7 탁상형 클린벤치
전공 라인이 들어 있어서 실험의 편리를 도모한다(그림 2-7, 2-8). 실험하는 사람은 캐비넷 앞에 앉아 두 손을 캐비넷 안에 넣고 실험을 한다. 최근에 국산품이 제조되고 있는데 성능이 대 단히 좋다. 2.6 현미경 조직을 배양하기 위해서는 특수한 조직 부분을 보존하고 불필 요한 조직 부분을 제거하여야 한다. 이때 꼭 필요한 기구로서 절 제용 혹은 해부용 입체 현미경 ste r eo mi cr oscop e 이 있어야 한다. 현미경 m ic rosco p e 과 라이트(광원)가 세트되는 수가 많다. 대안렌 즈가 중 zoom 이 되어 있어서 0.7 에서 3.0 의 몇 단계 배율을 선 택하게 되며, 대안렌즈가 10 배이면 전체 배율이 7 에서 30 배가 된 다(그립 2-9).
 그림 2-9 해부용 실체현미경과 광원
그림 2-9 해부용 실체현미경과 광원
살아 있는 배양세포의 형태를 매일 관찰하며, 나아가서는 배양 세포의 증식발육의 상태를 관찰하기 위해 위상차 도립현미경 inv erte d ph ase contr as t mi cro scop e 이 필수적 이다. 일반으로 대물 렌즈 4 배, 10 배, 20 배의 콤비네이션이 많이 쓰인다. 일반적인 명 시 야현 미 경 brig h t fiel d mic ro scop e 으로는 배 양세 포를 관찰할 수 없으므로 반드시 위상차현미경이라야 한다. 관찰을 기록하기 위 해 현미경 사진촬영장치 혹은 비디오카메라가 또한 필요하게 된 다 (그립 2-10) . 차이 스 Zeis s , 라이 츠 Leit z, 울림 포스 Olym pu s, 그 리고 니콘 N i kon 과 같은 회사가 고성능의 조직배양용 현미경을 시판하고 있다.
 그림 2-10 도립위상차현미경. 사전 촬영용 카메라가 부착되어 있다.
그림 2-10 도립위상차현미경. 사전 촬영용 카메라가 부착되어 있다.
제 3 장 배양기구 필자가 조직배양을 배우기 시작한 1960 년에는 조직배양에 쓰는 기구가 모두 유리로 되어 있었으며, 보통 유리로는 안 되고 미국 에서나 유럽에서 수입하는 파이렉스 P yr ex 라야 한다는 불문율이 있었다. 내가 대학원 공부를 하던 교토 대학 시철에는 일본 제품 이 신통치 않아 미국제만을 쓰던 때가 있었다. 그러나 유리를 재 사용하려면 그것을 특수한 세제로 씻고 닦은 다음에 멸균하는 번 잡하고 시간이 걸리는 조작이 필요하여 최근에 와서는 많은 연구 실에서 될 수 있는 대로 디스포사블 풀라스틱 배양기구를 쓰는 경향이 있다. 조직배양에서 사용하는 유리기구로서는 각종 배양용기인 크기 가 다론 페트리 접 시 pet r i dis h , 풀라스크 flas k, 시 험 관, 배 양액 병, 피펫 pipet, 원심 튜브 등이 있다(그립 3-1, 3-2, 3-3, 3-4, 3-5) . 조직 배 양에 또한 중요한 것 이 커 버 글라스 cover gla ss 이 다. 세포를 그 위에서 배양한 다음 여러 가지 조직염색이나 전자현미 경 관찰을 하려면 특수한 커버글라스가 필요하다.
 그림 3 기 각종 크기의 에렌마이어 플라스크
그림 3 기 각종 크기의 에렌마이어 플라스크
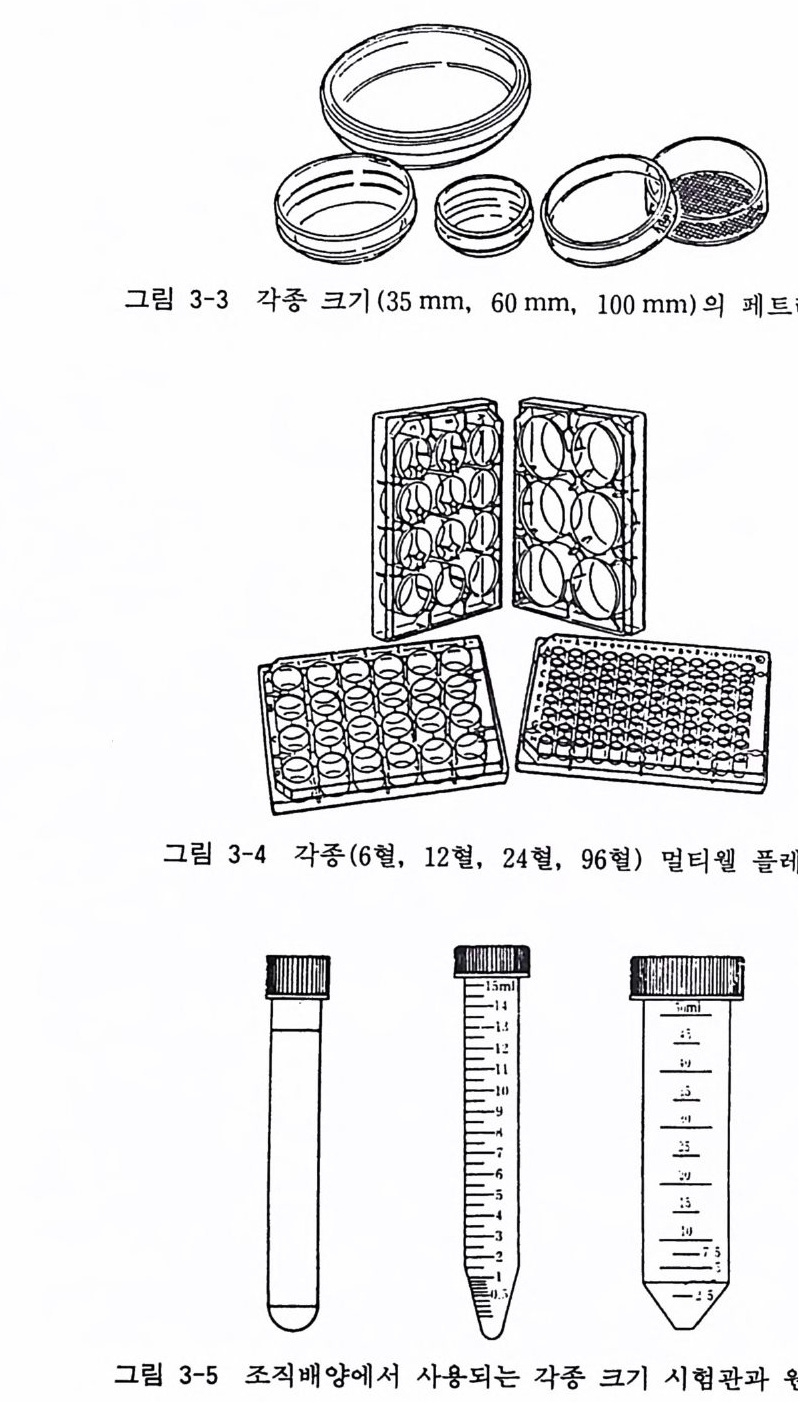 그립 3-3 각종 크기 (35 mm, 60 mm, 100mm) 의 페트리 접시
그립 3-3 각종 크기 (35 mm, 60 mm, 100mm) 의 페트리 접시
 • )
• )
1970 년에 들어서면서 풀라스틱 제품이 완전히 유리 제품을 대 체하게 되었는데, 페트리 접시나 풀라스크에는 특수한 재료를 써 서 배양세포의 접착성이 높아지게 가공되어 매우 편리하다. 그밖 에도 피펫, 원심튜브가 모두 디스포사블 풀라스틱으로 무균화되 어 있어서 편리하고 인전비를 절약하는 이점이 있다. 최근에는 챔버가 여러 개 있는 96 혈, 24 혈, 12 혈, 6 혈의 멀티웰 챔버 multiw ell chamber 가 나와 있 어 세 포 클로닝 clonin g , 독성 검 사 등에 그 위력을 보이고 있다. 조직배양에서 사용되는 각종 플라 스크, 페트리 접시, 멀티웰, 시험관, 원심튜브 등은 그립 3-1 에 서 그림 3-5 까지에서 설명하였다. 그밖에 조직배양에 필요한 기구로서는 실리콘 고무로 된 병마 개 , 러 버 풀리 스맨 rubber po li ce man, 가스 버 너 ga s burner, 피 펫 에이드pip e t aid 등이 있으며 가위, 핀셋, 시계용 핀셋, 수술칼 (메스) 등 조직분리를 위한 수술 기구가 중요하다(그립 3-6).
제 4 장 기구 세척과 멸균 4.1 세척 앞서 말한 바와 같이 최근의 조직배양 추세로는 플라스틱 기구 의 사용으로 세척 멸균의 문제가 많이 없어졌으나 배양액의 저장 울 위한 유리병, 세포를 육성시킬 커버글라스 등은 반드시 세제 로 씻은 다음 멸균해야 한다. 세제로는 알칼리성과 중성 세제가 있는데, 현재 미국, 캐나다, 일본 각국에서 가장 흔히 쓰이는 세제는 중성 세제인 세븐엑스 Seven X 이며 풀로우 Flow 사가 판매하고 있다. 유리병, 기타 유 리 기구를 세븐엑스에 하롯밤 담가 두었다가 수돗물로 세 번 가 량 씻어내고 다시 증류수(탈이온수)로 두 번 씻은 다음 건조시키 면 된다. 알칼리 세제 (라보크란 헤모졸, 엑스트란 기타)는 세척력 은 강력하나 기구 표면에서 떨어지지 않아서 몇번씩 온수로 씻어 내야 한다. 이에 비해서 세븐 엑스, 라이폰 F, 비스타, 콘트라 드 70 등의 중성 세제는 세척력은 떨어지나 씻어내기가 쉽다.
강한 산을 써서 세척할 수도 있는데, 다소 위험이 수반되므로 조심하여야 한다. 가장 보편적인 것에 50 % 황산이 있고, 그밖 에 황산과 염산을 섞어서 쓰는 실험실도 있다. 크롬산이 들어 있 는 세척액이 시판되어 이것을 써도 된다. 커버글라스는 반드시 산으로 처리할 필요가 있다. 산에 하롯밤 담근 다음 수돗물 세 번, 증류수 두 번의 세척을 거쳐서 건조시킨다. 최근에는 초음파 세척기를 써서 유리 기구를 세척할 수 있다. 세제가 들어 있는 온수 속에서 10 분 가량 초음파를 걸면 유리 기구나 커버슬립이 깨끗이 씻어전다. 4.2 멸균 세포배양액에는 많은 종류의 영양소가 있으므로 이 속에 미생 물이 있으면 이들이 급속히 증식하여 양분을 소모하고 유독 물질 울 내어서 배양세포를 죽여버린다. 따라서 멸균 방법이 극히 중 요하며, 기구, 배양액, 혈청을 멸균함에 있어 그 방법이 다르게 되어 있다(표 4-1). 고압증기멸균법은 고압증기멸균기(오토클레이브)를 써서 2 기압 120 도의 온도에서 15-20 분간을 유지하면 유리기구, 수술도구, 그 리고 배양액 중에서 단순한 염류용액 등을 쉽게 멸균할 수 있다 . 더 복잡한 배양액, 예컨대 이글용액 Eag le 's MEM 같은 배양액 가운데에는 고압증기멸균을 할 수 있도록 되어 있는 상품이 시판 되고 있다. 오토클레이브가 찰 되었는지를 알기 위해서 오토클레 이브 테이프를 유리 기구 등에 1-2cm 길이로 찰라서 붙인다• 120 도에서 15 분 증기에 접하면 테이프에 글자가 나와서 멸균이 되었음을 알 수 있다.
 표 4 기 배양 기구의 멸균법
표 4 기 배양 기구의 멸균법
유리병, 유리 페트리 접시, 피펫, 커버슬립 등은 전열 오본에 서 전열 멸균을 하는데, 그 조건은 160 도에서 90 분, 180 도에서 45 분이다. 밀리포어 필터 Mi llipo re filter 같은 필터를 써서 하는 여과멸균 법은 열에 불완전한 영양분, 예컨대 글루타민산, 포도당, 그밖에 도 호르몬, 성장인자 등을 멸균하는 데 사용된다. 일반적으로 필 터 구멍 크기가 0.22 미크론이나 0.45 미크론의 것을 사용한다. 그밖의 멸균 방법으로서는 수술기구인 가위, 메스 등을 70 % 알코올 속에서 소독하든가, 95% 알코올에 담근 다음 가스 버너 에서 불꽃멸균을 하든가 혹은 풀라스틱 기구를 에틸렌디옥사이드 로 가스 멸균하는 방법이 있다. 시판되는 풀라스틱 기구는 감마 선 멸균이 되어 있는데 이 감마선 멸균법은 고가의 기계가 필요 하므로 일반 연구실에서는 적당치가 않다. 유리 기구나 미량의
배양액, 시약 등은 자의선 아래서 20 분 가량 조사시키는 수도 있 으나, 자의선 멸균은 실패하는 수가 있어 주의가 필요하다. 여러 가지 멸균법에 대한 장점과 단접을 간략히 정리하면 표 4-2 와 갇 다.
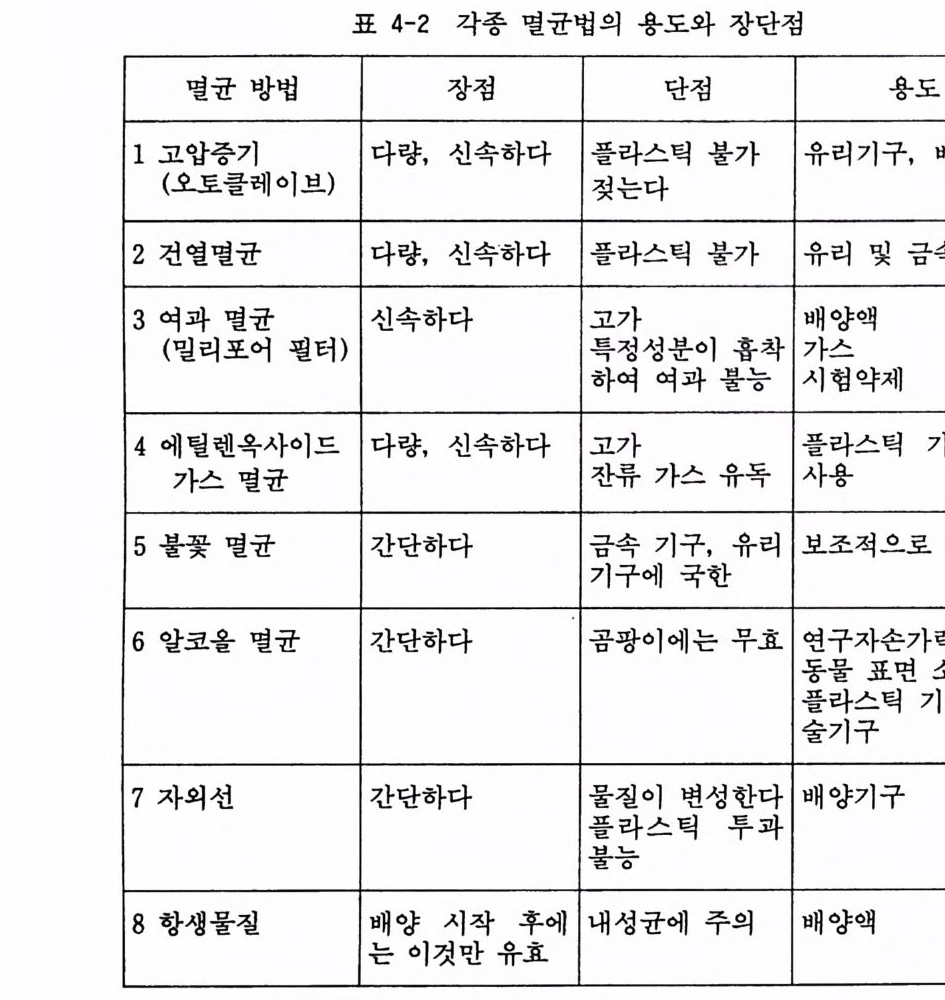 표 4-2 각종 멸균법의 용도와 장단점
표 4-2 각종 멸균법의 용도와 장단점
제 5 장 배양액 배양액은 조직배양에서 가장 중요한 요소가 된다. 이상적인 배 양액은 일정량의 기지 화학물질을 섞어서 만든 인공합성 배양액 이겠으나, 이것은 아직 실현되지 못하고 있고, 혈장p lasma, 혈 청 serum, 조직추출액 tiss ue extr a ct 이 배양에서 필요불가결의 요 소가 되어 있다. 배양세포의 증식과 생존에 필요한 인자의 해명 이 아직도 불충분하여 인공의 배양액에, 앞서 말한 혈청과 같은 천연물질을 첨가하여야 한다. 최근에는 무혈청 배양액의 개발이 많이 진전되어서 주세포나 하이브리도마 세포를 위한 무혈청 인 공합성 배양액이 각 회사에서 시판되고 있으며 초대배양에서도 단기간이면 무혈청 배양액을 사용할 수가 있다(제 7 장 참고). 배양목적, 배양세포의 종류에 알맞는 여러 가지 인공배양액이 시판되고 있는데, 여기에 일부분의 혈청을 가하면 배양세포의 충 분한 생존과 증식을 볼 수 있다. 배양액에 사용되는 증류수는 과거에는 세 번 증류한 물을 사용 하였는데, 현재로는 이온 교환 컬럼을 통과시킨 탈이온수로 충분
하며, 일반으로 시판되는 액체 배양액은 모두 이온 교환 컬럼으 로 얻은 물을 쓰고 있다. 배양세포의 생존, 증식에 문제가 있는 경우 제일 먼저 물의 순도를 검사하도록 할 것이다. 5.1 천연 배양액 천 연 배 양액 natu r al media 에 속하는 것 으로는 혈 청 serum, 혈 장p lasma, 조직추출액 tiss ue extr a ct, 락트알부민 가수분해물 lacta l bumi n hy d rolys a te , 효모추출물 ye ast extr a ct, 세 균펩 톤 bacto p e p ton e, 스킴 밀크 skim mi lk 등이 있으며 , 그밖에 도 집 착 인 자로서 콜라겐 collage n, 피 브로넥 틴 fibr onecti n, 라미 닌 lami ni n 등이 있다. 혈청이 배양액에서는 가장 중요한 천연 배양액 부분이 되겠는 데, 보편적으로 쓰이는 것이 우태아 혈청 fetal bov ine serum 과 마혈청 horse serum 이다. 그밖에 성인 혈청 human serum, 태반혈 청 human pla centa l cord serum 등이 사용될 수가 있다. 5.2 염류용액 배양액에 있어서 이온의 공급, p H 의 조정, 삼두압의 조절, 세 포 세척, 각종 시약의 용액 등에 사용하기 위하여 각종 염류용액 balanced salt soluti on 이 사용되 게 되 었다. 이 들의 대 표적 인 것은 다음과 갇다(표 5-1). 일반적으로 10 배 농축액을 만들어서 사용 시에 무균수로 희석하여 쓰게 된다. 다음에 가장 보편적으로 사용되는 얼 Earle 과 핸크스 Hanks 의
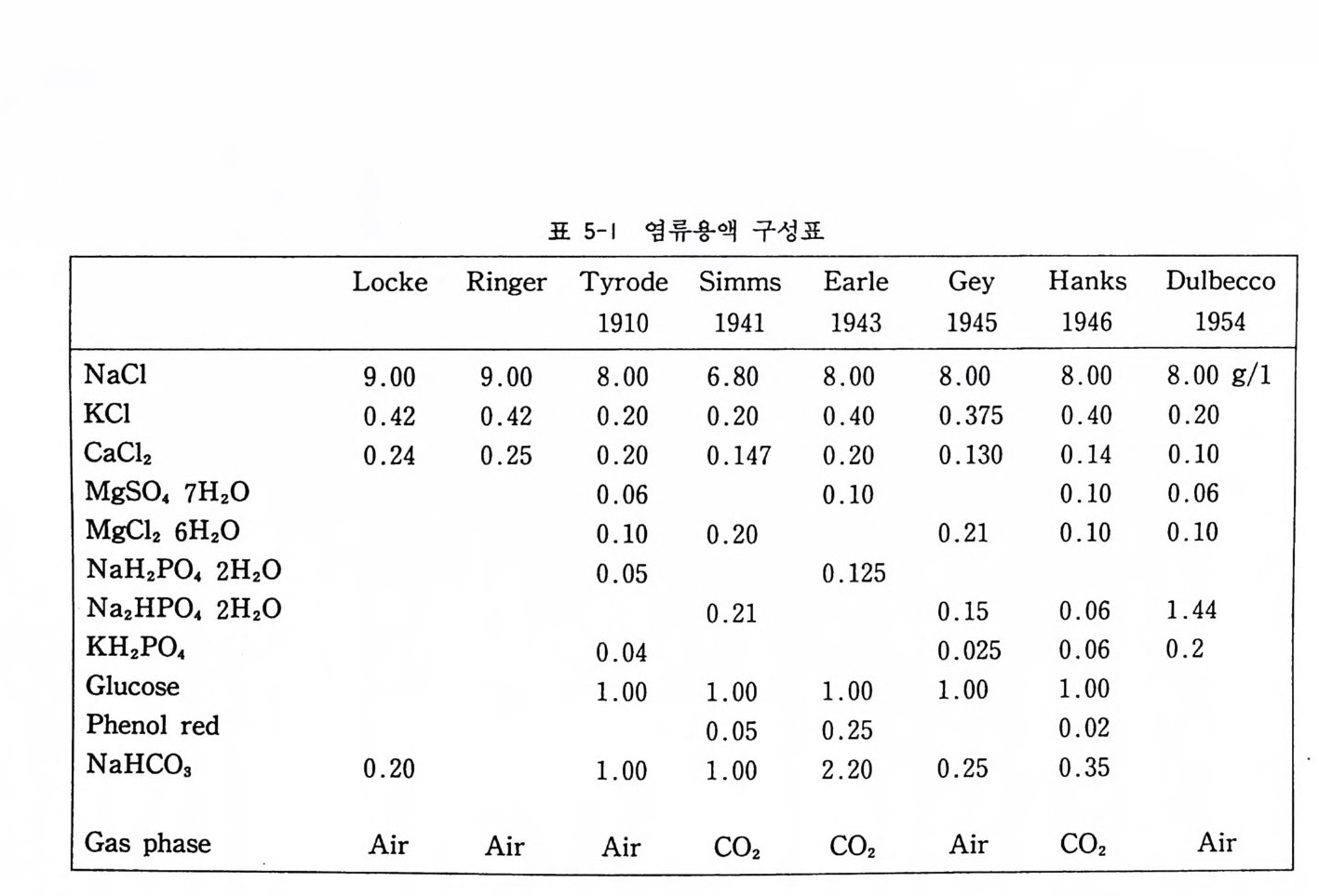 표al erE941 3 00.840 .0.200 .1005. 120 0 0.1 .2 50 .2 02 CO 2
표al erE941 3 00.840 .0.200 .1005. 120 0 0.1 .2 50 .2 02 CO 2
영류용액 제작법을 소개한다. 5.2.l 얼 염류용액과 핸크스 염류용액 〈 재료와 기구 〉 1) I 리터 유리 비커 1 2) 200ml 유리 비커 3 3) 증류수 혹은 탈이온수 4) 500 ml 유리병 2
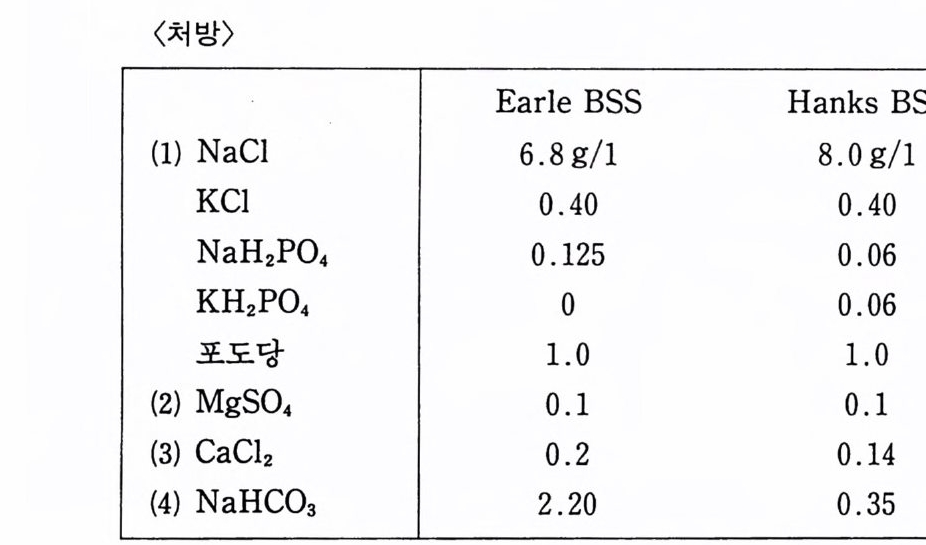 〈처방 〉
〈처방 〉
〈방법〉 1) 중류수(탈이온수) 750ml 를 1 리터 비커에 넣어 처방 (1) 을 차례로 녹인다. 처방 (2) 와 (3) 을 작은 비커에 넣어서 100ml 증류수에 녹인다. (1) 이 녹아있는 비커에 (2) 와 (3) 의 용액을 천천히 가해서 합친다. 여기에 증류수를 가해서 1 리터를 정
확히 만들고, 2 개의 500ml 유리병에 갈라서 넣고 오토클레 이브 멸균한다. 2) 멸균된 염류용액 500ml 에 0.2 % 페놀레드 멸균액 2.5 m l 울 가해서 pH 표지를 한다. 3) ( 4 ) 는 따로 준비 하되 7 . 5 % 나트륨 탄산액 (NaHCOa) 을 여 과 멸균한다. 영류 용액에 7.5 % 나트륨 탄산액을 첨가한 다. 얼액에서는 14.5 ml/500 ml, 핸크스액에서는 2.35 ml / 500 ml 을 첨 가한다. 5.2.2 칼슘-마그네슘 제거 인산염 완충용액 (CMF-PBS) 〈 재료와 기구 〉 1) 1 리터 유리 비커 1 2) 증류수 3) 500 ml 유리병 2 〈 처방 〉 NaCl 8.0 g/1 KCl 0. 2 Na2HPO ◄ 1.15 KH2 P0 4 0.2 〈 방법 〉 1) 1 리터 유리 비커에 중류수 800ml 를 넣고 위의 처방울 하 나씩 차례로 녹인다. 증류수를 더 가해서 1 리터를 만든 다 음 500ml 유리병에 갈라 넣고 오토클레이브 멸균한다. 페놀 레드나 나트륨 탄산액은 필요치 않다. 이 CMF-PBS 는 세포
를 씻어낼 때 혹은 트립신울 녹여서 조직 분리할 때 흔히 쓰 인다. 2) 멸균된 CMF-PBS 는 4°C 냉 장고에서 저 장한다. 5.2.3 나트륨 탄산(중조) 용액 나트륨 탄산 (NaHCOa) 15 g 을 200 ml 의 증류수에 녹인 다음 (7.5% 농도) 필터 여과 멸균을 하고 4•c 냉장고에 보존한다. 5.2.4 페놀레드 용액 페놀레드p henol red 1 g 을 0.1 N NaOH 40 ml 에 녹인 다음 증류수를 가해서 500ml 을 만든다. 이를 필터여과 멸균한다. 보 통 4•c 냉장고에 보존하되, 실온에서 보존할 때에는 클로로포름 2ml 롤 첨가한다. 5.3 합성 배양액 합성 배양액 syn the ti c me di um 은 인공 배양액의 기초가 되는 것으로서 그 처방은 염류액을 중심으로 하여 여기에 비타민, 아 미노산, 희금속, 당류(주로 포도당을 사용한다), 핵산, 염기, 그 밖의 대사중간 물질을 영양 성분으로서 포함하고 있다. 1950 년대 부터 여러 곳에서 합성 배양액의 처방이 발표되었는데, 그 중에 서 많이 쓰이는 것에 다음과 같은 것이 있다. 이들 합성 배양액 의 특칭은 간략하게 표 5-2 에 기술하였다.
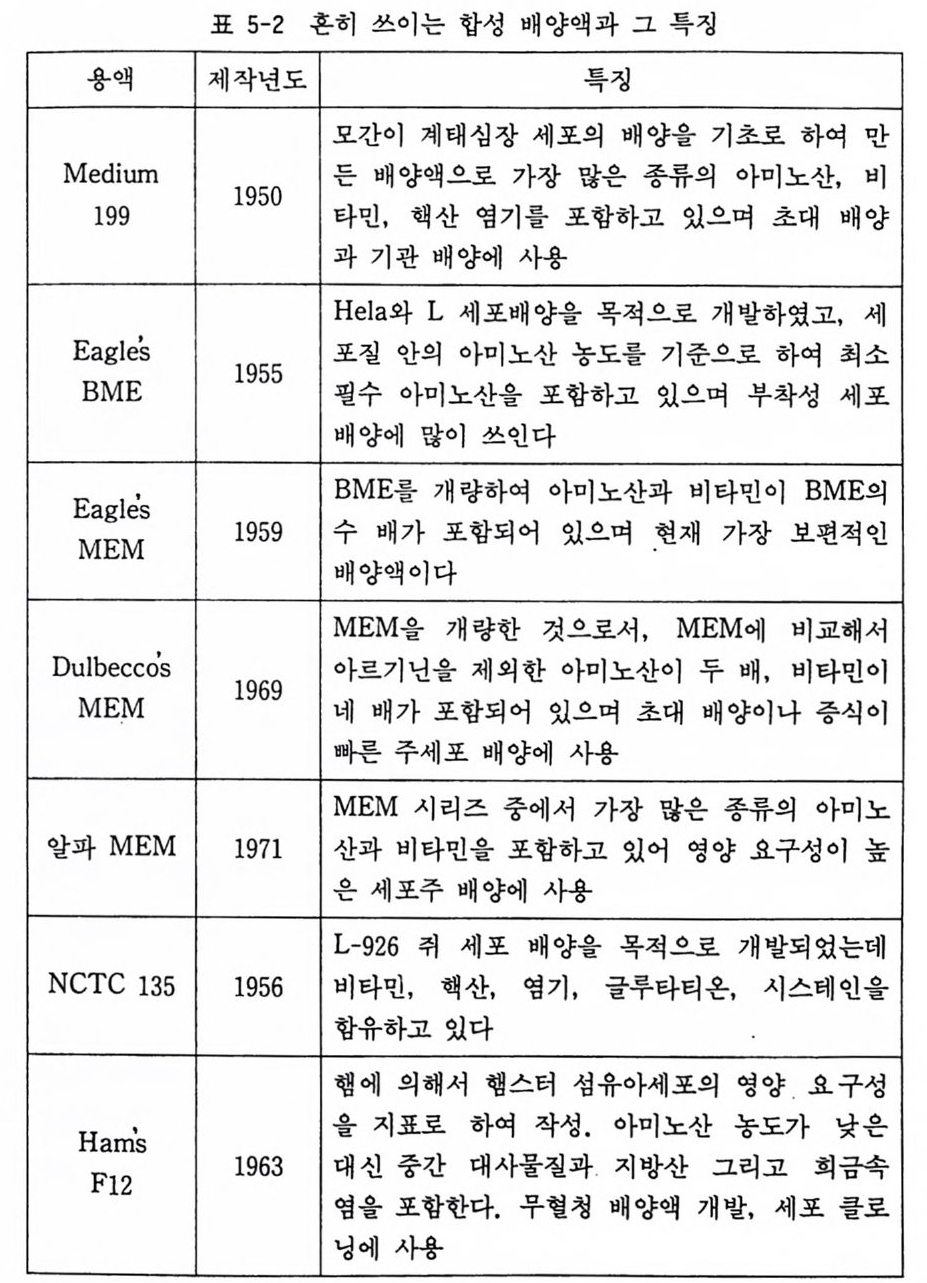 표 5-2 흔히 쓰이는 합성 배양액과 그 특칭
표 5-2 흔히 쓰이는 합성 배양액과 그 특칭
CD 199 배양액 1950 년 모간 Morga n 에 의 해 서 발표된 합성 배 양액 으로서 계 배 근육조직의 생존 일수를 지표로 하고 아미노산, 비타민, 콜레 스 데롤과 그밖에 핵산 염기 등 여러 가지 영양물질을 추가하여 영 양분이 풍부한 배양액이다. 동물세포 배양에 적합하며 백신 제조 등의 대량 배양이나 조직, 기관 배양에 많이 쓰인다. I) ® 이글 BME 배양액 1955 년 이 글 Eag le 에 의 해 서 포유동물 세 포배 양에 적 합하도록 발표된 배양액으로 합성 배양액 중에서 가장 간단한 처방을 갖고 있다. 아미노산과 비타민의 적합 농도 를 포함하고 있으며 사람 이배체세포, 사람 섬유아세포 배양에 사용된다 . 2 ) ® 이글 MEM 배양액 1955 년 이글에 의해서 발표된 이래 가장 널리 사용되는 합성 배양액이다. 배양액 중의 아미노산은 세포내 유리 아미노산의 농 도와 극히 가까운 농도를 갖도록 고안되어 있다. BME 와 비교하 여 글루타민 이의 아미노산은 두 배(아르기닌은 6 배, 히스티딘은 4 배)이며 비타민도 농도를 높이고 있다 .3) 적당한 농도의 혈청을 첨가하면 대다수의 세포배양이 가능하다. @ 그밖의 MEM 수정 합성 배양액 이글의 MEM 을 기초로 하여 각종의 MEM 수정 합성 배양액 이 발표되었는데 알파 MEM 은 스태너스 S t anners 가 마우스와 행 스터의 잡종세포 연구를 위하여 만들었으며 아미노산 8 종, 비타 민 4 종 그리고 피루브산을 첨가하여 만들었다. 달베코 Dulbecco MEM 은 최근에 많이 사용되는 배양액으로서 이글의 MEM 에 비
하여 아미노산이 2 배, 비타민은 4 배가 들어 있다. 조크리크 Joc kli ch MEM 은 NaCl 양이 감소되 어 있으며 포도당이 2 배 로 되어 있다. 부유배양용 MEM 은 부유배양에 맞도록 이글의 MEM 에 비하여 인산나트륨이 10 배로 되어 있다. @ NCTC 135 배양액 1964 년에 에반스 Evans 에 의해서 발표되었고 혈청이나 단백질 을 첨가하지 않은 조건에서 포유동물 세포를 장기간 배양하도록 고안되었다. NCTC 109 배양액은 NCTC 135 배양액에 시스데 인이 첨가되어 있다 .3) @ F-10 과 F-12 배양액 F - 10 은 1963 년 행 Ham 에 의해서 행스터 세포배양을 위하여 작성되었는데 낮은 농도의 단백질의 존재하에서 콜로니 형성이 가능하도록 되 어 있다. 4) F_l2 는 1965 년에 행 이 F-10 을 개 량하여 단백질이 없는 조건에서 세포가 생존하도록 여러 가지 아미노산 의 농도를 높이고 있다. 5) (J) L-15 배양액 1963 년 라이보비츠 Leib o v itz 에 의해서 발표된 배양액으로서 탄 산가스가 없는 인큐베이터에서 사용할 수 있고 pH 조정이 가능 하도록 유리 염기성 아미노산 (L- 아르기닌)을 사용하고 탄산나트 륨이 제거되어 있다• 글루코오스(포도당) 대신에 D- 갈락토오스가 들어있다. 아미노산 양이 많고 정상세포와 암세포가 모두 이 배 양액에서 잘 발육한다 .6)
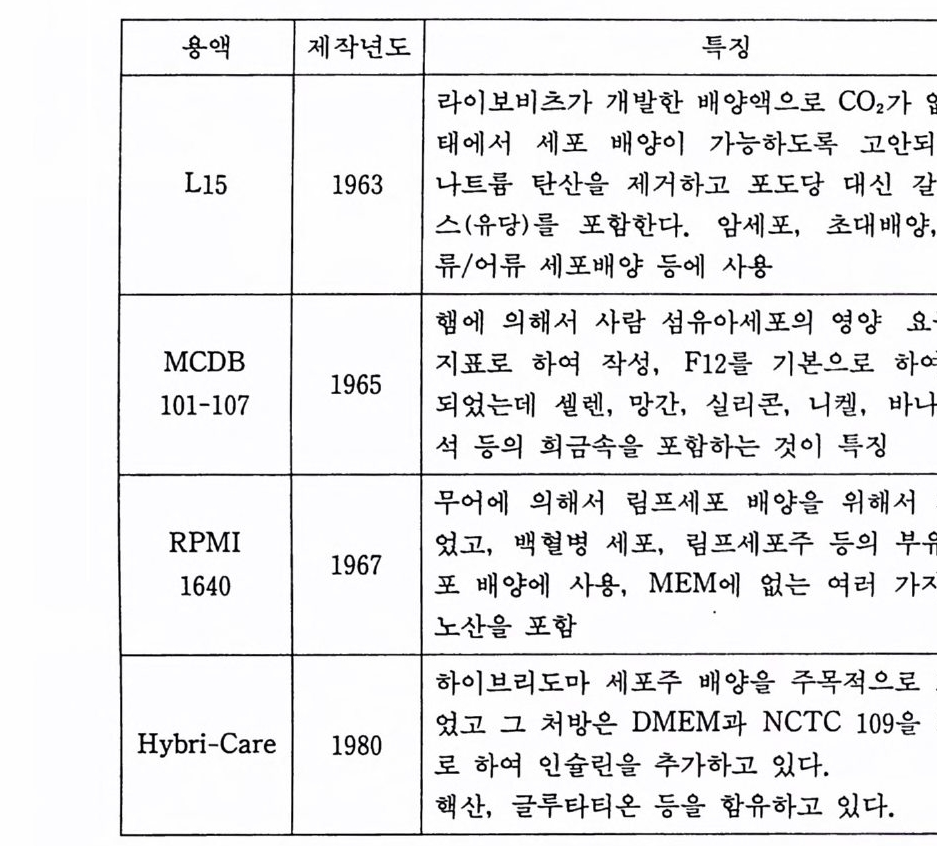 용액 제작년도 특징
용액 제작년도 특징
@ RPMI 1640 배양액 1967 년 무어 Moore 에 의해서 정상 인간 혈액세포 배양을 위하 여 작성되었고 현재 백혈구, 림프세포, 골수종 세포 등의 배양액 으로서 많이 사용되고 있다. 1> RPMI 는 로즈웰파크 연구소 Rosewell Park Memoria l Ins tit u t e 의 약자이 다.
5.4 무균검사 사용하는 배양액이 무균이 아니면 모든 배양실험이 한꺼번에 전멸하게 되므로 가장 신중을 요하는 것이 무균검사이다. 가장 간단하고 편리한 무균검사법은 사용 전에 배양액을 37 도 인큐베 이터에 며칠간 두어서, 배양액이 뽀얗게 탁하게 되면 세균이 들 어간 것이므로 이를 버린다. 티오글리콜레이트t h y o g l y cola t e 배 양액에 시험 배양액을 소량 첨가하고 37 도 인큐베이터에서 며칠 두면서 무균검사를 할 수도 있다. 시험액이 탁하게 되면 이것은 감염을 의미한다. 5.5 항생물질 조직배양을 시작하여 세포를 기르기 시작하면서부터는 무균조 작이 항생물질의 사용에 의존하게 된다(표 5-3). 가장 일반적인 항생물질로서는 페니실린-스트렙토마이신 Penic i l lin- St re p tom y cin 이 100 U/ml 과 100 µg /ml 의 농도로 사용된다. 배 양액 교환이 자 주 있는 경우에는(예컨대 1 주일에 두 번) 페니실린-스트렙토마이 신이 신뢰되나 교환이 1 주일에 한 번이라면 그 효과가 장기 지속 성인 겐타마이신 gen ta m i cin (최종 농도 25µ g /ml) 을 사용하는 것 이 바람직하다. 그밖에도 카나마이신 kanamy cin, 테트라사이클린 tet r a cy cl in e , 에리트로마이신 eryt hrom yci n 을 사용할 수도 있다. 곰팡이나 효모를 위하여 암포테리신 B amp h ote r ic in B (환기존) 나 마이코스타틴 m y cos t a ti n 을 사용하는데, 여기서 조심할 것은 동물에 따라서, 조직에 따라서 암포테리신의 독성으로 사멸하는 수가 있다는 점이다.
수술실에서 분리하여 온 인간 조직이나 도살장에서 사온 소, 돼지 조직을 사용하는 경우 염기성 세균이 혼입되어 있어서 세포 분리와 조직배양에는 성공하였으나 며칠 뒤에 세균감염으로 실험 울 포기하게 되는 수가 있다• 이러한 경우에는 날리딕스산 nali di x i c acid 을 50 µg /ml 의 최 종 농도로 사용하거 나, 설 퍼 메 톡 사졸 sulfa m eth o xazole 을 10 µg /ml 의 농도로 배 양액 에 첨 가하여 사용하면 잡균 혼입을 방지할 수 있다. 가장 이상적인 항생물질 은 광범 위 약제 broad spe c tr u m 이 어 야 하며 , 동시 에 배 양세포에 대하여 무독이어야 한다.
 표 5-3 항생물질 사용법
표 5-3 항생물질 사용법
제 6 장 세포 기질과 부착인자 세포는 다른 세포와 그 표면에 있는 분자를 통해서 접촉을 하 고 여러 가지 접합 형식울 가지게 된다. 그 예로서 갭정크션g a p jun cti on , 타이 트정 크션 tigh t jun cti on , 시 냅 스정 크션 syn a p tic jun cti on 등이 있다. 세포가 세포의 환경과 만나게 되면 그곳에는 세포의 기질 extr a cellular matr i x 이 있다. 이 세포의 기질은 초기 발생에 특히 중요한 역할을 갖게 되는데 세포와 세포의 기질의 상호작용을 통해 서 조직 구축 tiss ue archit ec tu r e 이 나 형 태 발현 mor phog enes i s 이 성취되는 것이다. 1980 년대에 세포의 기질의 대 요가 알려지게 되었는데 세포의 기질 중 · 중요한 것으로는 콜라겐 collage n 과 라미닌 lami ni n , 피브로넥틴 fibr onecti n 등의 글리코프 로데인 g l y co p ro t e i ns 이 있다. 이들의 세포의 기질은 각종 세포들 에 의해서 생성되고 세포밖으로 분비되어 축적이 된다. 이 밖에 도 콘드로넥 틴 chondronecti n, 트롬보스폰딘 throm bospo ndin 등의 세포의 기질이 있으나 그 구조나 상태가 분명치 않다. 일반으로 세포주 세포는 부유배양을 할 수 있는데 비해 초대배
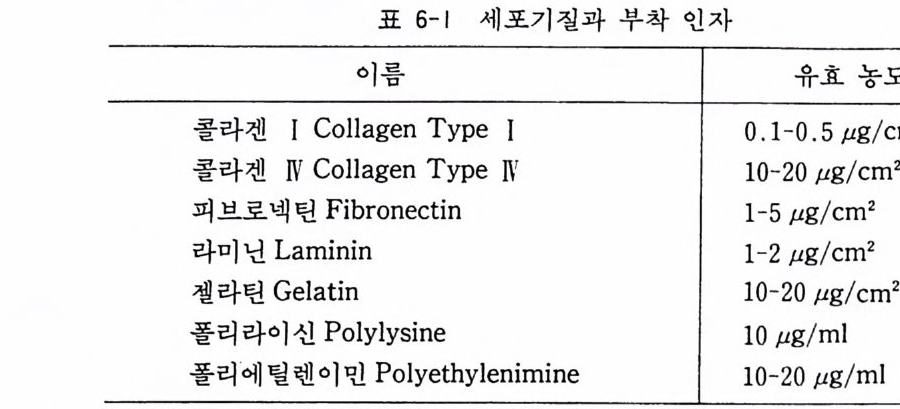 표 6 기 세포기질과 부착 인자
표 6 기 세포기질과 부착 인자
양 세포들은 표면을 가진 곳에서 성장을 잘 하게 마련이다. 그 때문에 풀라스크, 페트리 접시, 커버글라스를 사용하는데, 이때 이러한 배양기구 표면을 가공하여 세포가 잘 부착하도록 하는 것 이 중요하다. 시판되고 있는 풀라스틱 플라스크, 페트리 접시에 는 가공이 되어 세포가 부착하기 쉽게 되어 있는데, 제작회사들 이 기업비밀이라 하여 그 정체를 말하지 않아 확실치는 않다. 신경조직배양에서 현재 흔히 쓰이고 있는 세포 기질과 부착인 자의 이름과 그 유효 농도는 표 6 - 1 에서 볼 수 있다. 6.1 콜라겐 콜라겐은 섬유아세포가 만드는 단백질로서 세 개의 끈이 나선 형으로 서로 꼬아져서 섬유형으로 혹은 시트형으로 되어 있어서 세포 밖에서 축적이 된다. 섬유성으로 구성된 콜라겐은 세포 밖 에 있으면서 조직구성의 근간이 된다. 콜라겐은 부착작용을 가지 고 있어서 이것이 세포 성장, 분화, 형태 변화에 크게 영향을 준 다.
6.1.1 콜라겐 I ( I 형 콜라겐) 피부, 뼈, 근육 그 밖의 결체 조직에 많이 있고 조직배양에서 가장 많이 쓰이는 세포 기질이다. 근육세포, 신경세포, 간세포, 심장세포, 췌장세포 등 여러 조직과 세포에 적절한 기질이다 .I) 조직배양에서 쓰이는 콜라겐 1 은 래트 꼬리에서 분리하여 0.1 % 빙초산액에 녹여서 사용한다. 6.1.2 콜라겐 W( W 형 콜라겐) 모든 조직 의 기 저 막 basement membrane 의 중요한 구성 성분의 단백질이며 세포의 기질에 물리적인 안정성을 갖게 하는 것이 콜 라겐 W 라 생각되고 있다. 피부 각질세포, 간세포, 혈관내피 세 포, 암세포에서 콜라겐 W 와의 접착성이 높다. 콜라겐 W 의 생물 적 역할은 아직도 분명치 않은 · 점이 많다. 마우스 육종 Eng el breth - Holm-Swarm Sarcoma 에서 분리되는 제품이 풀로우 Flow, 깁 코 GIBCO, 시그마 Sig ma 등의 회 사에서 판매 되고 있 다. 〈방법〉 1) 콜라겐 W 분말을 0 . 25 % 초산 수용액 에 10 µg /ml 의 농도 로녹인다. 2) 배양 페트리 접시나 커버글라스 표면을 콜라겐 W 액 5-10 µg /cm2 의 농도로 덮고 실온에서 3 시간, 4 도에서 하롯밤 방 치한다. 3) 여분의 콜라겐을 제거하고 실온에서 건조시킨다.
4) 콜라겐 액의 무균화가 안된 경우에는 건조시킨 뒤 70 % 에 틸알코올에 잠시 담가서 살균하고 건조시킨다. 6.1. 3 I 형 콜라겐 분리법 1 형 콜라겐은 래트 꼬리에서 분리한다. 그 분리법은 다음과 갇다. 〈재료〉 1) 성장 래트 꼬리 2) 70% 알코올 3) 0 .1 % 빙초산액 (오토클레 이브로 무균화한다) 4) 가위, 연골도, 핀셋 5) 시험관, 원심분리 듀브, 페트리 접시 〈방법〉 1) 쥐꼬리를 비누로 간단히 씻는다. 2) 70% 알코올에서 5 분간 소독한다. 이후부터 무균조작이 된 다. 3) 꼬리를 15 cm 페트리 접시에 놓은 다음 연골도를 써서 꼬 리 끝에서부터 1 cm 간격으로 피부를 절개한다. 상방으로 꼬리 끝을 끌어올리면 은빛으로 된 콜라겐 섬유가 같이 따라 울라온다. 이를 절단하여 다른 접시에 들어 있는 0.1 % 빙 초산액에 담근다. 4) 계속하여 꼬리를 절단하고 콜라겐 섬유를 모은다. 5) 한 마리의 꼬리에서 나온 콜라겐 (10 g 정도)을 100ml 빙초 산액이 든 병에 옮긴 다음 콜드룸 (4 도)에서 마그네틱울 써서 24 시간 동안 돌린다•
6) 원심분리 듀브에 걸쭉하고 점도가 높은 액을 넣어 2000 r p m 으로 20 분간 분리 한다. 7) 상청액을 다른 병에 옮기고 4 도에서 저장한다. 약 4 개월 가 량 유용하다. 6.1. 4 건조 콜라겐 코팅 콜라겐 코팅을 두 가지 방법으로 만들 수 있다. 하나는 콜라겐 원액을 10 배로 무균증류수에 희석하고 접시나 커버글라스에 울려 놓고 여분을 빨아울린다. 이것을 하루나 이틀 실온에서 건조시킨 다. 이를 건조 콜라겐이라 한다. 이 건조 콜라겐 위에 세포부유 액을 올려 놓으면 세포배양이 시작된다. 6.1.5 습성 콜라겐 코팅 둘째번 방법은 다음과 같이 하는데, 이를 습성 콜라겐 Wet collag e n coa ti n g이 라 한다. 〈방법 〉 1) 콜라겐 원액을 커버글라스나 페트리 접시 표면을 덮울 수 있도록 코팅을 한다. 2) 다른 페트리 접시 뚜껑에 여과지 (필터페이퍼)를 부착 테이 프로 부착시키고 그 여과지에 1-2ml 의 암모니아수로 적시고 콜라겐이 코팅된 커버글라스나 페트리 접시를 덮는다. 3) 실온에서 10 분간 정치한 뒤 암모니아 노출을 중지하고 콜라 겐 도포 접시나 커버글라스를 무균 증류수로 세 번 5 분씩 씻
는다. 4) 콜라겐 코팅이 끝난 페트리 접시나 커버글라스 위에 、 · 부유 세포나 이식편을 올려 놓고 배양액을 가하면 배양이 시작된 다. 6.2 라미닌 라미닌 lam i n i n 은 모든 조직의 기저막에 특이하게 존재하는 분 자량 90 만의 당단백질이다. IV 형 콜라겐, 헤파린유산과 결합하여 기저막을 구성하고 있다. 조직배양에서는 상피세포, 간세포 등에 서 접착 작용이 있고, 신경세포의 성장 촉진의 작용도 가지고 있 다 .2) 라미닌은 암세포의 전이 능력과 관련이 있으며 전이성이 높은 악성 암세포는 라미닌 수용체를 다량 가지고 있다. 풀로우, 깁 코, 시그마 각 회사에서 무균화한 제품을 살 수가 있다. 〈방법〉 1) 라미닌을 10µ g /ml 의 농도로 무균 증류수에 녹인다. 2) 직경 3.5 c m 페트리 접시 하나에 0.5ml 의 라미닌울 분주 하고 실온에서 한 시간 방치하고 건조시킨다. 3) 커버글라스도 위의 요령으로 라미닌을 바르고 건조시킨다. 냉장고에 저장하면 한달 가량 사용할 수 있다.
6.3 피브로넥틴 피브로넥 틴 fi bronec ti n 은 악성 화세포 등에 서 분비 되 는 당단백 질 로, 분자량 25 만의 폴리펩티드 이량체이다. 세포 접착에 큰 역할 울 하므로 정상 세포 배양에 사용된다. 일반으로 정상인의 혈장 에서 분리되는데 조직배양에서는 섬유아세포, 간세포, 신경세포 등의 접착을 가져온다 . 생체 안에서는 세포접착, 세포이동, 식작 용의 촉진을 가져오며 조직 손상부에서 그 수복을 돕는 것으로 알려져 있다 .3,4) 피브로넥틴은 시판되고 있어 플로우, 깁코, 시 그마 등의 회사에서 살 수 있다. 피브로넥틴은 2-5 µg /cm2 의 세 포배양 기구 표면을 덮으면 유효하다. 〈 방법 〉 1) 피브로넥틴(시판의 것은 이미 무균화되어 있음)을 10 µg /ml 의 농도로 무균 증류수에 녹인다. 2) 직경 3.5 cm 페트리 집시 하나에 0.5 ml 의 피브로넥틴액을 분주하고 36 도에서 1 시간 인큐베이트한다. 3) 무균 증류수로 두 번 씻은 다움 다시 말린다. 4) 피브로넥틴 코팅된 접시는 실온에서 한달 가량 저장할 수 있다. 커버글라스는 위의 요령으로 하며 비슷한 방법으로 건 조시킨다. 6.4 비트로넥틴 사람의 혈장과 혈청 의 당단백 질인 비트로넥 틴 vitro necti n 은 분 자량이 75,000 이며 각종 세포가 부착, 이동, 신장, 증식의 각 작
용을 하도록 돕는다. 피브로넥틴과 유사한 작용을 가지고 있으나 화학적으로는 이 두 가지 분자는 완전히 독립되어 있다 .5) 최근까지 혈청신장인자 serum spr e adin g fac to r 라 불렸으며 헤 파란 글리코사미노글리칸g l y cosam i no g l y can, 트롬빈-안티트롬빈 m 콤플렉스 등에 부착하는 성질이 있다. 6.5 젤라틴 젤라틴g ela ti n 은 콜라겐을 변성 처리하여 수용성으로 만든 단 백질로서 조직 배양 용기의 표면에 바르면 근육세포, 혈관, 내피 세포, 데라토카시노마 세포 배양에 좋은 성적을 가진다 .5) 소나 돼지 피부에서 분리한 겔라틴이 쓰이는데 보통 2 % 수용액을 만 들어서 오토클레이브하여 무균화한다. 〈방법〉 1) 냉장고에 저장된 2 % 젤라틴 액을 온수에서 37 도까지 가열 해서 녹인다. 2) 배양 페트리 접시나 커버글라스를 가볍게 덮어서 실온에서 30 분 방치한다. 3) 여분의 첼라틴을 피펫으로 제거하고 실온에서 건조시킨다. 6.6 폴리라이신 양성 전기하전을 가전 아미노산 폴리머인 폴리라이신 Pol y -D -l y s i ne 이 최근에 많이 사용되어 풀라스틱이나 유리 풀라스크, 페
트리 접시, 멀티웰, 커버글라스를 코팅하여 사용한다 .6 ) 폴리 라이 신 po ly- D -lys i n e bromi de salt ( 분자량 30 , 000 ~ 70 , 000) (시그마에서 시판)을 0.1 m g/ml 의 농도로 증류수에 용해시킨 뒤 밀리포어 필터로 무균화한다. 사용 직전에 무균 증류슈로 10 배로 희석하고 페트리 접시, 커버글라스 등에 충분량을 가하고 20 분에 서 1 시간 동안 정치한다. 폴리라이신을 완전히 제거하고 두 번 무균증류수로 씻은 뒤에 실온에서 말린다. 폴리라이신액은 두 번 더 쓸 수 있다. 최근에는 고가인 폴리라이신에 대신하여서 값이 훨씬 낮은 폴 리에틸렌이민 po lye th y le nim i ne (시그마)을 사용하는 연구실이 있 다.” 폴리에틸렌이민은 50% 수용액으로 공급되고 있으므로 아 것을 증류수나 탈이온수에 1000 배로 희석하고 디스포사불 필터로 무균화한 뒤 사용한다. 그 사용하는 요령은 폴리라이신과 같다.
제 7 장 무혈청 배양액의 개발 조직 배양법이 시작된 초기에는 세포의 생존과 증식에 필수적 인 배 양액 성 분으로서 영 류용액 balanced salt soluti on , 배 태 추출 액 embryo extr a ct, 혈장 pla sma, 혈청 serum 등의 세포 환경 성 분을 그대로 사용하였으나 이러한 자연배양액의 아미노산, 비타 민, 당분, 금속 성분의 화학 분석이 알려지게 되자 필수영양성분 울 포함한 합성 배 양액 syn the ti c mediu m 이 개 발되 었다. 합성 배 양액 199 나 이 글의 배 양액 Eag le 's mi ni m um essenti al mediu m 등 이 그 대표적인 예가 되겠다. 그러나 조직배양 조건하에서 세포 가 생장하고 증식하려면 이러한 단순한 합성 배양액만으로는 부 족하고 일반적으로 마혈청, 우태혈청 등의 동물 혈청이 소량이나 마 반드시 첨가되어야 한다. 혈청이 조직배양액에서 가지는 기능은 다음과 같다. ® 성분이 알려지지 아니한 영양 성분의 공급 ® 영양 성분, 금속 성분의 운반자 역할 carr i er
® 세포구조에 이용되는 고분자의 직접 공급 ® 여 러 가지 의 완충 작용 buf fer i ng ® 대 사산물의 해 독 작용 deto x ic a ti on ® 단백질 분해효소 등의 증식 저해물질을 중화 ® 세포 접착 인자의 공급 위에서 성분이 알려지지 아니한 영양 성분의 공급이라 함은 세 포성장에 필수적인 성장인자gr ow t h fac t or 가 혈청 속에 포함되고 있음을 의미하는데, 그 성장인자가 무엇인가를 확인하고 그 구조 와 기능을 결정하는 것이 현재 세포생물학과 분자생물학의 가장 시급한 과제가 되 었다. 1976 년에 사토 Sato 의 연구실에서 쥐 뇌 하수체 종양 주세포 (GH3) 를 배양함에 있어서, 혈청 농도를 최소 한으로 하고 그 대신 여러 가지 호르몬과 성장인자를 추가하는 방법으로 배양을 계속한 결과 갑상선 자극 호르몬 분비 호르몬 (TRH), 소마토메딘 C, 트란스페린, 부감상선 호르몬 (PTH), 트리요도티로닌의 5 가지 호르몬의 배합으로 무혈청 배양액을 개 발하고 장기간 쥐 뇌하수체 종양 주세포를 배양하였다 .I) 우리 교실에서는 소마토메딘 C 가 인슐린과 그 약리작용이 비슷하며 수용체를 같이하는 인슐린 유사 성장인자i nsu li n like grow th fac to r I 라는 점에 유의하여 그 순수 분리가 극히 곤란한 소마토 메딘 C 대신에 인슐린을 사용하여 신경세포 배양을 위한 무혈청 배양액을 개발하였다 .2 , 3) 이 연구에서 우리가 분명히 한 것은 10 µg/m l 농도의 인슐린을 Fl2, 이글 MEM 같은 인공 배양액에 첨가하면 신경세포를 비롯하여 각종 섬유아세포, 암세포 등이 일 주일이나 이 주일의 단기간은 생존할 수가 있다는 사실이다 .3) 우리 교실에서 고안한 무혈청 배양액은 인슐린, 트란스페린, 히 드로코티존, 트리이오도티로닌과 회금속인 셀렌을 포함하고 있
다. 이렇게 무혈청 배양액에서 각종 세포가 생존하고 증식할 수 가 있다는 것은 〈 모든 세포배양에서 적절한 영양 성분, 호르몬, 세포 성장인자의 배합으로 무혈청배양이 가능하다 〉 는 것을 의미 하는 것이다. 이렇게 무혈청 배양법의 개발은 지금까지 가지고 있던 호르몬의 개념을 크게 확대시키는 것이 되었다. 호르몬의 정의는 〈 특정 내분비 기관에서 합성되어서 혈류를 통하여 다른 특정 조칙이나 기관에 작용하는 물질 〉 인데, 앞서 말한 세포 성장 인자도 호르몬이나 마찬가지로 세포 기능을 제어 조절하는 작용 울 하기 때문에 그 기능은 호르몬과 다를 것이 없다. 무혈청 배양액을 개발하려면 먼저 기초배양액을 무엇으로 하는 가를 결정한다. 가장 흔히 쓰이는 것으로서는 달베코 MEM 과 F12 가 섞 인 DME/F12 가 있고 그 밖에 이 스코베 Iscoves MEM, MCDB 105, MCDB 201, MCDB 302, NCTC 135, LIS 등 각 종 기초 배양액이 있다. 기초 배양액에 혈청을 첨가하여 세포 배 양을 하고, 점차적으로 혈청의 함유량을 감소시켜서 세포 발육이 정지되는 점을 찾는다. 이 때에 기초 배양액에 각종 영양성분과 세포 성장인자를 첨가하여 세포 발육이 다시 시작하면 각 성분의 성장 유효 농도를 측정한다 .4) 다음에 각종 영양 인자에 대한 여러 가지 고려점을 열거한다. 세포 성장 인자 각종 세포 성장 인자의 기능과 작용 양식에 대 하여서는 아직 분명치 않은 점이 많으나, 대부분의 배양세포에는 하나 또는 그 이상의 성장 인자가 필수적이다. 예컨대 쥐 태아 척수후근신경절 신경세포 배양에서 신경성장인자는 10-100ng / ml 의 농도가 필수적이며 이것이 결핍되면 신경세포가 단시간에 사멸한다. 현재 사용되고 있는 각종 세포 성장인자의 이름과 유 효 농도 그리고 유효 조직 세포에 대하여서는 표 7-1 을 참고하기
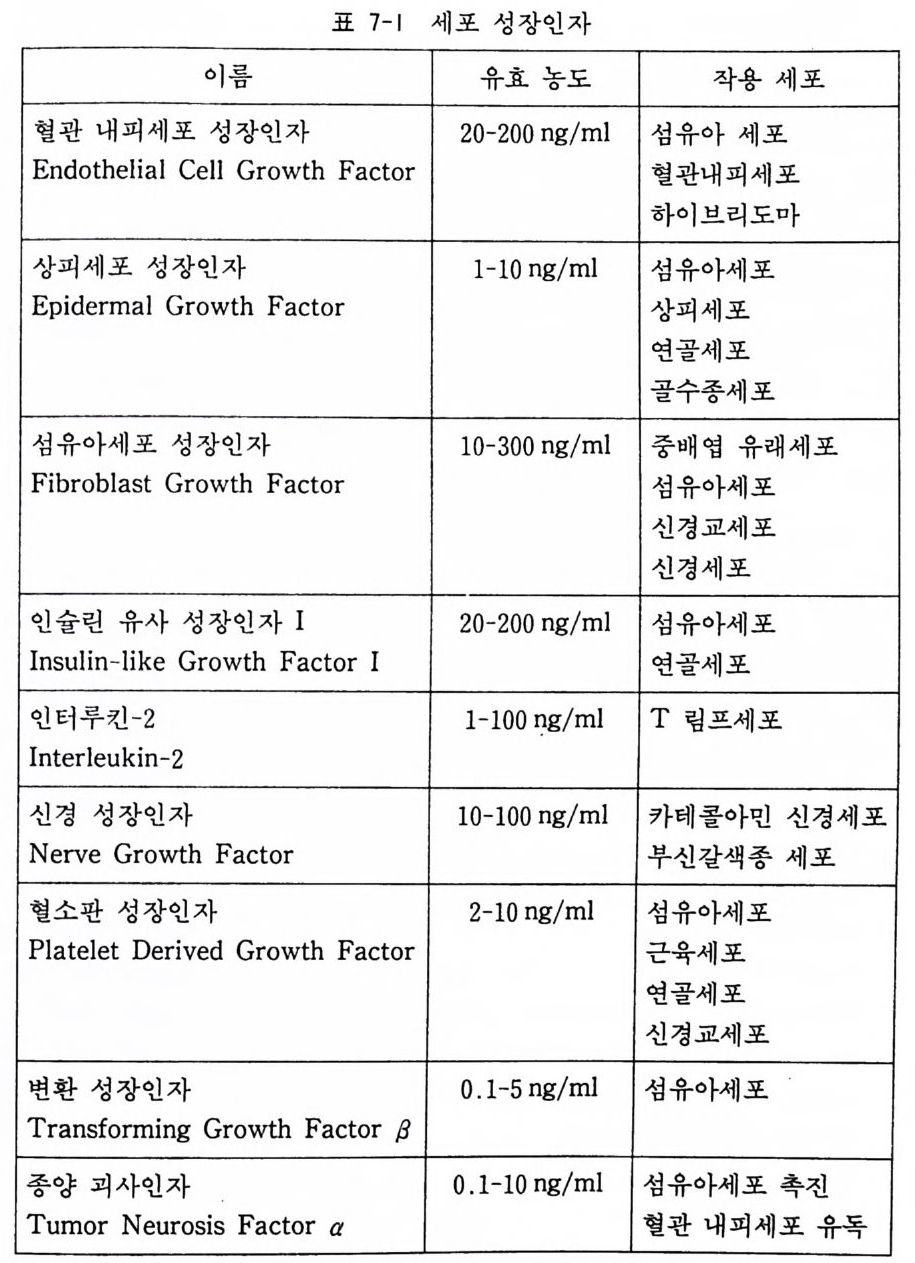 표 7-1 세포 성장인자
표 7-1 세포 성장인자
l:lt란다. 호르몬 호르몬이 각종 세포에 주는 영향은 아직 분명치 않은 점이 많으므로, 그 종류와 농도의 선택은 실험적으로 결정하는 수밖에 없다. 인슐린은 필수적이며 이것이 없으면 세포 발육이 없다. 그 농도가 생리적 혈중 농도의 1000 배로 사용되는 이유는 인슐린이 37 도의 온도에서 극히 불안정하며 기초 배양액 중의 시 스데인에 의해서 속히 분해되기 때문이다. 스테로이드 호르몬은 각종 세포의 증식과 분화를 촉진하는 것으로 알려져 있으며, 갑 상선 호르몬 역시 세포 분화와 성장에 효과가 있다고 보고되고 있다. 무혈청 배양액에 쓰이는 호르몬의 리스트는 표 7 - 2 를 참고
 표 7-2 무혈청 배양액에 쓰이는 호르몬
표 7-2 무혈청 배양액에 쓰이는 호르몬
하기 바란다. 세포기질과 세포 부착 인자 대부분의 배양세포들은 플라스크나 접시 표면에 접착한 뒤 증식을 시작한다. 기질로서 가장 혼히 쓰 이는 것이 폴리라이신이고, 콜라겐과 젤라틴도 역시 잘 쓰이는 기질이다• 부착 인자는 일반으로 배양액 속에 포함된 혈청에 의 해서 공급되고 있으므로, 무혈청 배양의 경우에는 의부에서 이를 공급하여야 한다. 접착 인자로서는 피브로넥틴과 라미닌이 무혈 청 배양액에 첨가되는 수가 있다. 부착 인자에 관하여서는 제 6 장에서 상세히 설명하였다. 무혈청 배양액 저자의 연구실에서 신경세포나 글리아세포의 배 양에 사용하고 있는 무혈청 배양액의 조성은 다음과 같다. 무혈 청 배양액은 만든 뒤에 4 도에서 2 주일, ― 20 도에서 2 개월 보존할 수 있다. 이 글 MEM, 달베 코 MEM 혹은 행 F12 100 ml 에 다음의 성분 을 첨가한다. 포도당 5mg /m l 겐타마이신 20 µg/m l 인슐린 10 µg/m l 트란스페린 10 µg/m l 히드로코티존 50nM 트리이오도티로닌 30nM 셀렌 30nM HEPES lOmM
제 8 장 배양법의 분류 일반적으로 조직배양이라고 하는 경우, 이것은 배양세포를 풀 라스틱 배양 페트리 접시나 배양 플라스크에 넣어서 이것을 이산 화탄소 인큐베이터에서 장기간 유지하는 것으로 이해하게 되는 데, 이것은 조직배양법의 한 방법에 불과하며 정치배양법 sta t i on ary cul t ure 이라 한다. 배양법은 여러 가지 콤비네이션으로 사용할 수가 있는데, 여기에서는 그 대략을 정치배양, 회전배양, 선회배양, 그리고 부유배양으로 구분하여 설명하기로 한다. 8.1 정치배양법 정 치 배 양법 sta t i on ary cu lt ure 은 세 포를 페 트리 접 시 , T- 풀라스 크, 그리고 멀티웰 챔버에 나누어 넣고 배양액을 첨가한 뒤에 인 큐베이터에 넣어서 배양하는 것을 의미하는데, 페트리 접시나 멀 티웰이 모두 밀폐할 수 없는 개방성인 용기이므로 이산화탄소를
5 %나 10 %로, 그리고 습도를 포화상태로 유지하여야 한다. 이 를 위해서 자동조절기가 있는 이산화탄소 인큐베이터가 시판되고 있다. 이산화탄소의 농도는 배양액에 들어 있는 중탄산나트륨의 농도 와 서로 평형을 이루면서 안정된 p H 를 유지하는 것이므로, 배양 액의 중탄산나트륨 농도에 따라서 이산화탄소를 5 %나 10 %로 조절하게 된다. 정치배양에서도 앞서 말한 개방계 정치배양 이의에도 폐쇄계 정치배양이 있다. 이 경우에는 일반 인큐베이터룰 사용할 수가 있다. 풀라스크나 시험관 안에 세포를 가하고 병마개로 밀폐를 하면 폐쇄계 배양이 된다. 병마개로 밀폐하기 전에 소량의 이산 화탄소를 플라스크나 시험관에 불어 넣어서 배양 성과를 개선시 킨다는 보고도 있다. 정치 배양에서 사용되는 배양기구 중에서는 여러 조직배양 학자들이 고안한 폐쇄계 배양기구가 있다. 맥시모 우 Maxim ow, 로 즈 Rose, 사 이 크 스-무 어 Sy k es-Moore, 리 턴 Leig h to n 등의 여러 기구가 판매되고 있다(그림 8-1). 이밖에도 개방계인 페트리 접시를 데시케이터 dess ic a t or 에 넣 은 다음 데시케이터를 밀폐하여 일반 인큐베이터에 정치하는 방 법을 쓸 수도 있다. 이렇게 함으로써 비용이 싸게 드는 간편한 배양시설을 할 수가 있다. 배양액의 증발을 방지하기 위하여 데 시케이터 바닥에 증류수를 넣어야 한다. 이 경우 곰팡이나 잡균 이 번식하는 것을 방지하기 위하여 소독비누(수술실에서 사용하는 종류)를 1 %의 농도로 첨가한다.
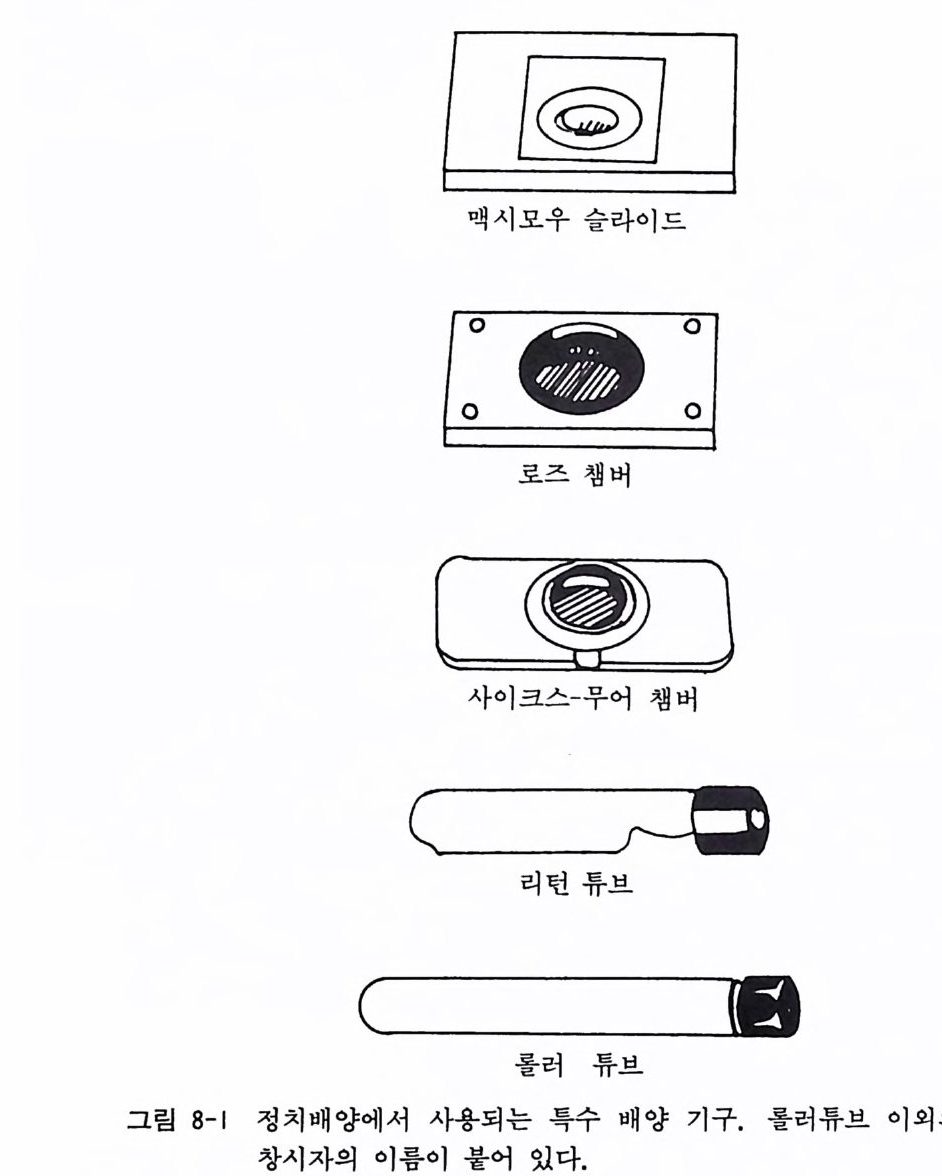 三
三
8.2 회전배양법 회전배양 roller drum cul t ure 은 세포를 시험관이나 배양병에 넣 은 다음 이들을 회전 드럼 속에 넣어서 이 드럼이 1 분 5 회전의 저속에서 1 분 50 회전의 고속 회전을 한다. 이렇게 함으로써 세포 는 액체와 가스에 차례로 노출됨으로써 증식을 빨리 하게 된다. 대량의 세포를 필요로 하는 경우, 예컨대 유전자를 도입한 세포 를 배양하는 경우 회전배양법이 쓰이는 수가 많다. 8.3 선회배양법 회전배양법에서는 배양병을 수직으로 회전시키는 데 비해 선회 배양법 rota t i on cul t ure 에서는 풀라스크(엘렌마이어 풀라스크 Ehlen meye r fl ask) 를 수평으로 선회시키는 방법을 사용한다. 일반으로 30ml 의 엘렌마이어 플라스크에 5-7ml 의 세포부유액을 넣고 1 분 에 60-80 회전을 시키면 플라스크 바닥의 중심부에 세포들이 덩어 리 를 만드는데 , 이 를 리 아그리 게 이트 reag greg a te 혹은 집 합체 배 양법이라 한다. 8.4 부유배양법 스피 너 병 spi nn er bott le 에 대 량의 세포를 배 양하는 것 이 부유배 양법 suspe nsio n cultu r e 의 가장 일반적 인 배 양법 인데 , 이를 위 해 서는 마그네트(자석)가 들어 있는 스피너병이나 회전을 시키게 하는 특수한 스터러 sti rr er 등이 필요하여 비용이 많이 드는 결접
이 있다. 우리 연구실에서는 이것을 간략하고 염가로 하기 위해 500ml 의 보통 배 양액 병 에 200-250 ml 의 세포부유액 을 가한 다음 병 바닥에 2 cm 크기 의 스터 링 마그넷 sti rr in g magn e t 을 넣고 스터 러를 1 분에 30-60 회전 시킴으로써 배양을 하고 있다. 대량의 배양을 하기 위해서 최근에 와서 여러 가지 방법이 고 안되고 있는데 , 그 중의 하나로서 마이크로캐 리 어 mi cr ocarrie r 법 이 있다. 이것은 플라스틱이나 그밖의 유기 물질로 구형 캐리어 를 만들어 그 표면에 세포를 부착시켜 부유배양법으로 세포 증식 을 하게 한다. 그 장점은 국한된 공간에서 넓은 증식 면적을 갖 게 되는 데 있다. 유전공학의 방법으로 유전자를 도입하고 생체 활성물질 bio a cti ve subs t nace 를 대량으로 분리하고자 할 때에 쓰 이는 방법이다.
제 9 장 일반 배양기술 9.1 무균조작 조직배양에서는 미생물의 오염이 없는 무균적 상태에서 장기간 세포를 유지하여야 한다. 조직 배양에서 사용되는 배양액에는 영 양가가 높은 여러 가지 화학물질이나 유기물이 들어 있으므로 세 균에게도 역시 적합한 영양액이 된다. 또한 각종 세포가 갖가지 바이러스의 숙주가 되기도 한다. 이들 미생물은 증식도가 극히 높은 까닭에 조직배양에서는 무 균조작이 가장 중요한 조건이 된다. 공기중의 먼지, 물방울, 실 험대, 벤치 그리고 실험하는 사람의 손가락 등 여러 곳에 미생물 이 존재하므로 세균이 실험조직 재료에 떨어져서 같이 붙어오지 않도록 실험실에서는 사람들의 출입을 가능한 한도에서 제한할 필요가 있다. 큰소리로 이야기 한다든가, 기침을 하는 것도 삼간 다. 벤치나 실험대 위는 실험을 시작하기 전에 70 % 알코올이나 크레졸액을 스프레이어를 써서 소독하도록 한다. 실험 시작 전에 소독비누액이나 0.3% 히비텐액으로 손을 소독하도록 한다. 실
험시에는 마스크를 하고 사람들이 주위에서 너무 움직이지 않도 록 주의하여야 한다. 배양액이 들어있는 병, 배양 풀라스크, 파스출 피펫은 사용시 화염 소독하도록 하며 핀셋, 가위 기타 수술기구는 70 % 알코올 에 담가서 소독하든가, 95 % 알코올에 잠시 담갔다가 화염 소독 한다. 실험이 끝난 뒤에 사용이 끝난 피펫, 배양병 등을 무균 벤 치에서 제거하고 무균 벤치안은 70% 알코올이나 크레졸액으로 소독해 둔다. 일반적으로 실험이 끝난 뒤에는 실험실 천장이나 무균벤치 안 에 설치된 자의선 살균등을 켜서 다음날까지 두는 것이 보통이나 요즈음은 인체에 대한 안전관리를 위해서 자의선 살균등의 사용 울 금지하는 수가 있다. 예컨대 1980 년 이래 미국의 국립의학연 구소 NIH 에서는 자의선 살균등 사용이 금지되고 있다. 9.2 배양액 교환 세포를 배양하고 있는 배양액 교환을 시작하기 전에 풀라스크 나 페트리 접시에 이상이 없는가 검사한다. 잡균이 들어가서 배 양액이 뿌영게 되었거나 노랗게 되어 있으면 다른 배양에 번지지 않도록 이를 먼저 버리고 다른 배양의 배양액 교환을 시작한다. 〈방법〉 1) T 풀라스크의 경우에는 마개를 연 다음 폴라스크입을 가스 버너 불꽃에 노출하고 불꽃 소독한다. 2) 플라스크를 세워서 배양액을 세포가 자라지 않는 쪽으로 기 울인다.
3) 파스출 피펫(혼히 진공흡입 장치에 접속한다)으로 배양액을 제거한다. 4) 새 배양액을 세포가 자라지 않는 표면을 향하여 첨가한다. 이때 피펫 에이드를 사용하면 정확한 양의 세포액을 쉽게 첨 가할 수 있다. 5) 풀라스크 입을 화염 소독하고 병마개를 한다 6) 피펫이 다른 곳에 닿은 듯하면 죽시 이것을 버리고 새 피펫 울 사용한다. 7) 다른 종류의 세포를 취급할 때에는 홉임용이나 도임용으로 새 피펫을 사용한다. 8) 일반적으로 묵은 배양액은 조금 남겨서 80 % 정도만 버린 다. 이것은 묵은 배양액이 조건 배양액이 되어 세포 증식이 나 생존에 유익하다고 생각되기 때문이다. 페트리 접시에서 배양한 경우에는 화염 소독을 못하게 되나 그 액의 교환 요 령이 플라스크나 마찬가지이다. 9.3 계대배양 세포가 증식하여 배양 풀라스크나 딧슈 안에서 빈름없이 빽빽 하게 세포충이 밀집하게 되면 이를 희석해서 새로운 플라스크에 옮기 게 된다. 이 를 계 대 배 양 subcultu r e, pa ssag e 이 라 한다. 세포 가 증식하고 있는 배양기로부터 고르게 세포부유액을 만들어서 새 배양기로 옮기게 되는데, 트립신 처리를 하거나 러버 폴리스 맨 rubber p o li ceman 을 써서 기 계 적으로 세포를 긁어내는 수가 있 다. 하이브리도마인 경우에는 간단한 피페팅으로 풀라스크 바닥 에 부착한 세포를 분리시켜서 단리세포 부유액이 될 수 있다.
〈 방법 〉 1) 동결된 2.5 % 트립신 스토크를 온수에서 녹인다. 2) 묵은 배양액을 피펫으로 홉인하여 버린다. 3) 칼슘, 마그네슘 제 거 PBS (CMF-PBS) 로 플라스크 를 한 번 씻는다. 4) 트립신 희석액 (보통 PBS 에 0. 1 % 로 희석한다)을 풀 라 스 크에 넣고 마개를 하여 인큐베이터에 10 분간 정치한다. 5) 풀라스크의 측벽을 서너 번 때려서 세포충이 풀 라스크 바닥 에서 미끌어져 분리되게 한다. 여기에 혈청 첨가 배양액 5 ml 롤 가한다. 6) 피펫으로 몇 번씩 상하로 흡입한다. 이때 세포가 분리되는 데, 거품이 안 나도록 조심한다. 거품이 나면 세포 표면에 상처가 나서 세포가 죽는다. 세포부유액을 원심 튜브로 옮긴 다. 7) 원심 (800- 1 000 rp m ) 5 분간 8) 상충액을 버리고 여기에 새 배양액 5ml 을 가한다. 9) 혈구계산판으로 세포 수를 계산한다. 10) 적당한 세포 밀도로 희석하고 새 플라스크에 도입한다. 1l) 풀라스크를 인큐베이터에 옮긴다. 이때 너무 움직이면 세포 가 풀라스크 중앙에 모이게 된다. 12) 현미경에 의한 검사는 30 분 뒤에 행한다. 9.4 세포 수 계산법 세포배양에 있어서 도입 pla ti ng 세포 수나 세포 증식도 측정 등 세포 수를 계산하는 것이 배양법의 기초 기술의 하나가 된다.
일반적으로 혈구계산판 hemoc yt ome t er 을 사용하여 세포 수를 계 산하는데 , 이 에 더 하여 트리 판블루 염 색 try p a n blue sta i n 을 하여 살아 있는 세포를 구별하는 방법을 병용하기도 한다. 트리판블루 는 죽은 세포를 파랗게 염색하나, 살아 있는 세포는 염색하지 못 하므로 트리판블루에 염색이 안 되는 세포 수를 혈구계산판에서 계산하면 살아있는 세포 수를 알 수가 있다. 이를 색소배제법이 라 한다. 혈구계산판은 2 개의 챔버로 되어 있고 한 챔버는 9 개의 1.0 mm2 의 소구역으로 나누어져 있다. 그 위에 커버글라스가 덮여 있고, 글라스와 계산판 사이가 0.1 m m 공간이 되어 있으므로 한 소구역의 용적은 0.1mm 까 된다. 여기에 10,000 배를 하면 1ml 안의 세포 수를 계산할 수 있다. 혈구계산판 계산에는 다음과 같은 오차가 있으므로 주의하는 것이 좋다. 첫째, 고르지 못한 세포 분포, 둘째, 덩어리로 되어 있는 세포를 계산, 셋째, 경계선상의 세포 수를 두 번씩 계산, 따라서 오차가 없는 측정을 위하여 최소한 세 번 세포 수를 계산 한다. 〈 재료 〉 1) 혈구계산판과 커버글라스 2) 파스출 피펫 3) 영류용액 (핸크스나 얼 영류 용액) 4) 0.4 % 트리판블루(영류 용액에 회석) 스토크액 5) 시험관 (12X75mm) 6) 카운터 7) 현미경 8) 세포부유액
〈 방법 〉 1) 트리판블루액 0.2 ml 를 영류용액 0.8 ml 에 희석 2) 혈구계산판 위에 커버글라스를 올려 놓는다. 3) 시험관에 세포부유액 0 . 5ml 와 트리판블루액 0 . 5ml 를 섞는 다. 4) 피펫으로 세포부유액, 트리판블루액을 섞은 다음 혈구계산 판의 두 챔버를 채운다. 5) 현미 경 에 서 100 배 배 율로서 10 배 대 안렌즈와 10 배 대 물렌즈 를 써서 가운데 1 개 구역과 구석의 4 개 소구역 (1 mm 3 ) 을 세 고 5 로 나눈다. 먼저 전세포 수를 카운트하고 그 다음에 트 리판블루에 염색된 죽은 세포를 센다. 이 때 소구역 하변과 우측변 선위에 울라 있는 세포는 제외한다. 6) 세포 수를 계산한다. 소구역 평균치 X10000 (1 ml 의 세포수) 1ml 의 세포 수 X 2 X 배양액 총 수一배양세포 총 수 7) 계산을 되풀이하여 재현율 오차가 15 % 이내로 한다. 9.5 세포동결보존법 배양세포를 계대유지함에 있어 여러 가지 문제점을 갖게 되는 데, 그 중 몇 가지를 열거하면 첫째, 주세포를 계대배양하는 중 에 그 형상과 성질이 변화한다. 둘째, 계대배양 중 마이코풀라스 마 등의 감염이 있다. 셋째, 유한증식성 세포주에서는 계대배양 울 오래 계속하지 못한다. 이러한 문제점을 해결해 주는 것이 세포동결보존법 cell fr eez i n g이다. 세포배양 중에 일정량의 세포를 앰플에 나누어서
동결보존하고 필요시에 하나씩 꺼내어서 실험에 사용한다. 이에 따라 매주 계대배양하는 시간과 노력을 절약할 수가 있다. 세포의 동결, 융해 후의 생존율은 동결시의 냉각속도, 보존온 도, 융해속도, 동결 보호제의 종류와 농도 등의 여러 가지 조건 이 고려되어야 한다. 다음과 같은 조건이 가장 이상적이라 생각 된다 . 동결 속도 (1 분에 1 도씩 하강), 동결보존온도(一 180 도), 급 속 융해(1분 이내), 동결보호제 사용(1 0% DMSO 나 10% 글리세 린) 〈 재료 〉 1) 세포동결액 -20% 우태아 혈청 +70% 이글 MEM 액 + 10 % 디 메 틸 설 폭사이 드 (dim eth y ls ulfo x id e , DMSO) 2) 피펫과 시험관 3) 크라이 오튜브 cryo tu be 4) 초저온 냉장고섭씨 -80 도 5) 액체질소 탱크 〈 방법 〉 1) 증식 중에 있는 세포를 분리하여 세포 수 계산에 따라 2X 106/rnl 의 세 포부유액 을 2 ml 준비 한다. 2) 세포를 1000 회전 5 분간 원심분리한다. 3) 세포 페렛에 세포동결액 4ml 를 가하여 세포부유액을 만들 고 이를 크라이오튜브에 1ml 씩 분주하여 4 튜브를 만든다. 4) 크라이오튜브 위에 마카펜으로 세포명, 날짜, 연구자 이름 울 표기한다. 5) 스티로폼 상자 속에 크라이오튜브롤 넣고 이를 -80 도 초저 온 냉 장고에 하롯밤 보존한다.
6) 동결되어 있는 크라이오튜브를 액체질소 탱크 속에 옮간다. 7) 동결보존된 세포를 융해하고 재배양하는 경우 크라이오듀브 를 45-50 도 가량의 온수에 넣 어 서 1 분 안에 녹도록 한다. 알 코올을 적신 탈지면으로 크라이오튜브 표면을 소독한다. 튜 브 내부의 세포부유액을 원심관에 옮기고 여기에 혈청 첨가 배양액 10ml 를 첨가하고 1000 회전으로 5 분간 원심분리한 다. 이것을 다시 한번 되풀이하고 T 플라스크나 페트리 접 시에 세포배양을 시작한다. 9.6 세포 운반 배양세포를 다른 대학이나 연구소 연구실에 양도하거나 받아 오는 수가 많은데, 이런 경우 다음과 같은 처리를 한다. 첫째, 위에서 기술된 대로 세포를 동결한 다음 드라이아이스가 들어 있는 컨데이너에 넣어서 밀봉한 다음 자동차, 비행기로 목 적지까지 운반한다. 목적지에 닿으면 즉시 해동하고 배양을 시작 한다. 세포를 도착 죽시 사용치 않을 경우에는 액체질소 탱크 속 에 저장한다. 둘째, 살아 있는 세포를 보내는 경우에는 T 플라스크에 세포 롤 배양하고 운반 이동 직전에 풀라스크 가득히 배양액(항생물질 과 HEPES 완충액을 첨가)을 채우고 밀봉한다. 파라핀 필름으로 병마개 주위를 든든하게 밀폐한다. 운반 중에는 대기 온도가 된 다. 목적지에 닿으면 풀라스크입을 알코올 소독하고 새 배양액으 로 갈아 넣은 다음 인큐베이터에서 정치 배양한다.
제 10 장 신경조직세포 분리 기술 신경조직을 구성하는 각종 세포와 페트리 접서나 풀라스크 표 면에서 자라는 신경세포는 콜라겐, 뮤코프로테인, 세포 접착인자 등에 의해서 상호간에 혹은 기질에 단단히 부착하고 있으며 이러 한 부착성은 칼슘과 마그네슘에 의해서 좌우되고 있다. 그러므로 이들 세포집단을 하나씩 따로 분리하려면 여러 가지 단백질 분해 효소에 의해서 소화하며 칼슘과 마그네슘을 제거하는 금속제거 제 chela t or 를 사용하여야 한다. 단백질 분해효소에는 여러 종류 가 있는데 트립신, 프로나제, 콜라게나제, 디스파제, 파파인, 판 크레아틴, 에스테라제가 여기 속한다. 신경조직의 세포 분리에 쓰이는 단백질 분해효소와 기계적 세포분리 기술을 다음에 소개 한다.
l0.1 트립신 과거에는 소나 돼지 췌장의 아세톤 분말을 사용하여 조직을 분 해하였는데, 이러한 제품은 트립신 try p s in 1 : 250 이라 불렸고 여 기 에 는 트립 신 의 에 도 키모트립 신 chy m otr y p s in 과 에 라스타제 elas t ase 를 포함하고 있어서 조직 분해에 극히 효과가 있었다. 따 라서 결정화한 순수 트립신은 이러한 비정제 트립신 1 : 250 에 비 해서 효과가 낮게 마련이다. 장시간에 걸친 트립신 처리 는 세포 를 상하게 하므로 적당한 시간이 중요하다. 트립신 처리 뒤에는 트립신 억제제 try p s in i nh i b it or 를 쓰는 것이 중요하다 . 트립신 처 리 칙후에 혈청이나 혈청이 들어 있는 배양액으로 트립신 활성을 억제하는 것은, 혈청 속에 천연 억제작용이 있기 때문이다. 최근 에는 각 회사가 2.5 % 트립신을 무균화하여 판매하고 있으므로 이것을 CMF-PBS 나 CMF- 핸크스 BSS 에 10 배로 희석해서 조직 분해, 세포 단리에 사용한다. 풀라스크나 접시에 부착한 세포 를 분리하고자 할 때에는 0.1 %에서 0.05 % 농도를 CMF - PBS 에 녹여서 사용한다. 트립신에 의한 세포 장해가 커지면 조직이 풀과 같이 점착성이 증가하여 세포 분리가 불가능함으로 이러한 경우에는 DNA 분해 효소 deoxy nu clease, DNase 를 25-100 µg /ml 의 농도로 트립 신 액 에 첨가하면 세포 분리가 용이하다. 이것은 트립신이 세포 독성 을 보여서 죽은 세포에서 DNA 가 분리되어 이것이 점착성을 증 가시키는 것이다.
10.2 프로나제 프로나제 pro nase 는 Str e p tom y ce s griseu s 균에 서 분리 한 단백 질 분해효소이며 학자에 따라서는 트립신보다 독성이 낮다고 그 사용을 권하는 수가 있다. 역시 CMF-PBS 에 0.25 %나 0.1 % 농도로 녹여서 여과 살균하여 사용한다. 10.3 콜라게나제 결체조직을 많이 포함한 종양조직이나 간, 폐 등의 조직에서 단리 세 포 를 구하고자 할 때 에 는 콜라게 나제 colla g enase 가 효력 이 있다. 트립신과 달리 세포 독성이 낮고 3 시간이 넘는 처리에 도 세포가 장해를 입지 않아서 많은 학자가 추천하고 있다. 저자 의 연구실에서 1978 년에 세계에서 처음으로 성숙한 사람의 신경 세포를 죽은 지 10 시간이 되는 사람의 신경조직에서 분리하고 두 달 넘게 배양을 하였는데, 이것은 트립신을 쓰지 않고 콜라게나 제를 사용하였기 때문이다(제 16 장 참고). 워싱턴 Wort hing ton Chemi ca l 이 나 시 그마 Sig ma 에서 판매 하고 있는 콜라게나제는 정 제되어 있지 않아서 트립신, 키모트립신 등을 소량 포함하고 오 히려 조직 분해에 효과적이다. 박테리아에서 분리한 콜라게나제 는 혈청에 의해서 억제되지 않기 때문에 콜라게나제는 생리적인 조건하에 혈청 포함 배양액에 0.1-0.25 % 농도로 쓸 수가 있다. 이것은 100-250un it /ml 의 농도가 된다. 콜라게나제를 억제하려 면 세 번 이상 원심과 부유를 되풀이하여 배양액에서 씻어내면 된다.
10.4 디스파제 디 스파제 dis p a se 는 Bacil lu s Polym y x a 균에 서 분리 한 중성 단 백질 분해효소로서 혈청이나 칼슘을 포함한 배양액 속에서도 효 력이 있으며 트립신과 달리 세포에 대한 독성이 적고 세포 분산 에 적 합하다. 1000 un it /ml 의 농도로 사용할 수 있다. 산코 Sanko 와 베 링 거 -만하임 Baerin g e r-M annheim 에 서 디 스파제 라 해 서 발매 하고 시 그마에 서 는 프로테 아제 pro te a se 라 해 서 판매 하고 있다. 10.5 파파인 파파인 pa p a i n 은 파파야 과일에서 분리되는 식물성 단백질 분 해효소로서, 이것 역시 트립신에 비교하여 독성이 적어서 트립신 울 대신하는 세포 단리에 적합한 효소이다. 파파인을 활성화하려 면 아미노산인 시스데 인 cys t e i n e 이 존재 하여 야 하며 파파인 8- 10 un it /ml 의 농도로 이글 MEM 이나 핸크스 BSS 에 녹이고 여기에 시스테인을 4m g /ml 의 농도로 첨가한 뒤 여과 멸균하여 사용한 다. 우리들은 다른 방법으로는 대단히 곤란하던 성숙 사람 망막 신 경세포의 분리 배양을 파파인을 사용함으로써 성공시켰으므로 그 사용을 추천하고자 한다(제 17 장 참고). 워 싱 턴 Worth ing ton Chem ic al 에서는 무균화하여 조직 배양용으로 제품화한 파파인울 판매하고 있다.
10.6 금속 제거제 금속 제 거 제 chelato r 인 EDTA (dia m i no eth a ne tet r a aceti c ac id sodiu m sal t)는 금속인 칼슘이나 마그네슘을 배양 환경에서 제거 함으로써 세포분리를 돕는다. EDTA 는 일반으로 칼슘-마그네슘 제 거 PBS (CMF- P BS) 에 2 % 액 으로 스토크를 만들고 사용시 에 100 배로 희석하여 0. 02 % 액으로 사용한다. EDTA 는 트립신과 함께 쓰는 것이 보통이다. 10.7 기계적 세포분리 그림 10-1 에서 보는 바와 같이 스테인레스 스틸 멧슈를 조직채 s i eve 로 만들어서, 아주 작게 자른 조직편을 그 위에 울려 놓고 여과시키는 기계적 세포분리가 있다. 멧슈 사이즈는 40(0.38 mm), 50(0.28 mm), 60(0.23 mm), 80(0. 19 mm), 100(0.14 mm) , 150 (0 .10 mm) , 200 (0 . 074 mm) 이 있으며 여 러 가지 멧 슈 의 콤비네이션으로 조직을 작게 분리할 수 있으며, 일반 신경조 직 배양의 경우에는 40, 100 멧슈를 차례로 사용하면 된다. 이러 한 조직채는 시그마에서 살 수가 있다(그림 10-1). 나일론 멧츄\ 로 봉지를 만들고 그 속에 작게 자른 조직편을 넣고 PBS 속에 서 유리봉으로 조칙을 밀어내면 세포 분리가 가능하다. 나일론 멧슈는 20, 40, 100, 200 미크론 구멍 크기가 있으므로 100 미크론 과 20 미크론의 멧슈 봉지롤 차례로 사용하는 것이 좋다(그립 10-2). 나일론 멧슈 봉지 제작법은 24.4 철을 참고할 것. 배양세포를 페트리 접시나 풀라스크에서 분리하여 계대배양을 한다든가, 생화학적인 혹은 약리 독성학적인 실험을 위하여 신속
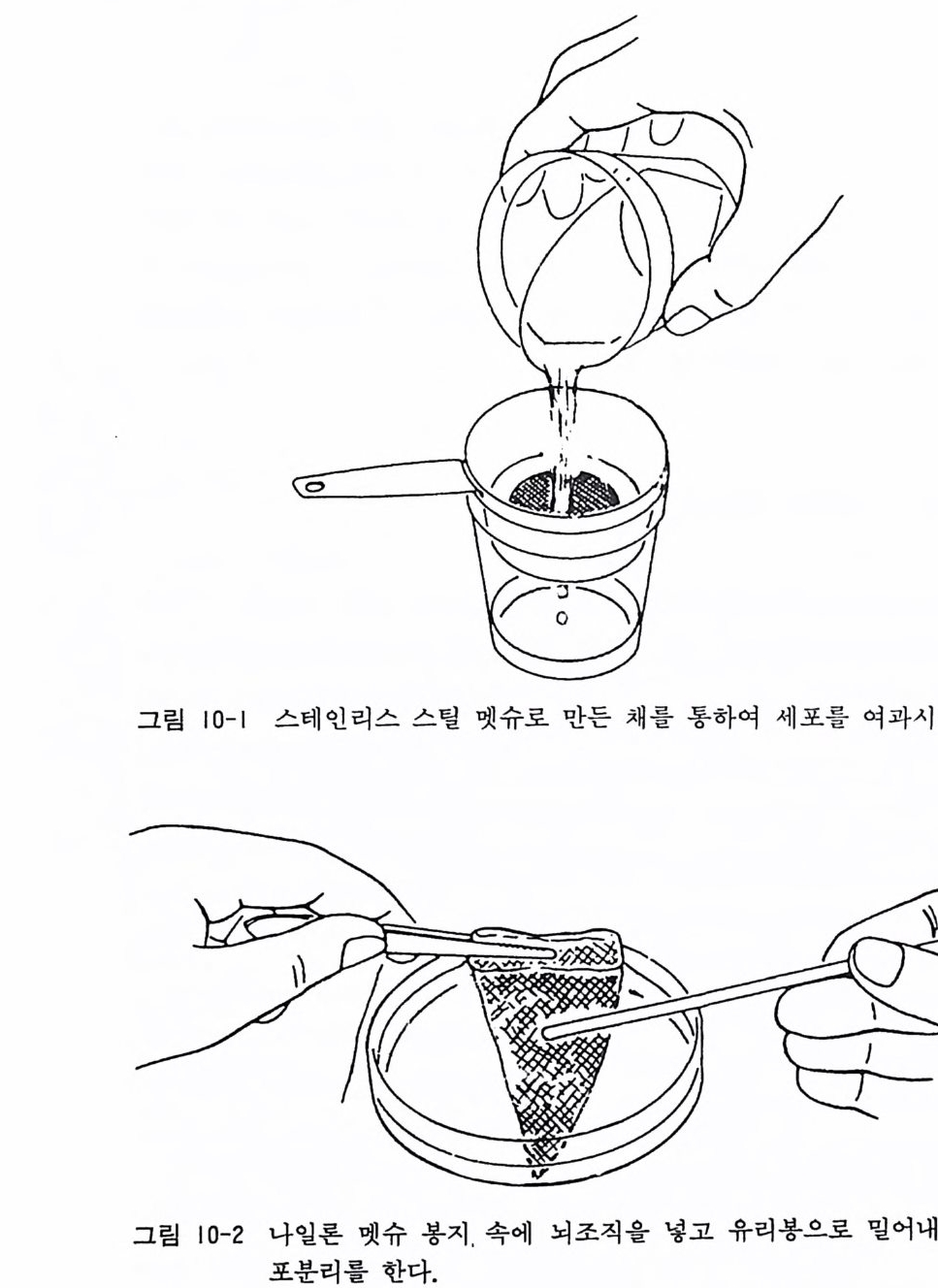 그림 |0-1 스데인리스 스틸 멧슈로 만든 채 를 통하여 세포 를 여과시킨다.
그림 |0-1 스데인리스 스틸 멧슈로 만든 채 를 통하여 세포 를 여과시킨다.
하게 세포를 분리하고자 할 때에는 러버풀리스맨 rubber po lic e -man 이 나 스크레 이 퍼 scrap e r 를 사용하여 문자 그대 로 막처 럼 퍼 져있는 세포 집단을 긁어 낼 수가 있다. 각 조직배양기구 회사에 서 여러 가지 모양의 스크레이퍼를 판매하고 있어서 편리하다.
제 2 부 신경계 조직과 세포의 초대 배양법 
제 |I 장 계배 척수후근신경절과 교감신경절의 배양 배양 기술의 향상에 따라서 각종 동물의 조직과 세포의 배양이 가능하게 되었는데, 한 가지 특별한 배양 방법으로 모든 조직과 세포 를 배양할 수 있는 것은 아니고, 각 조직과 세포에 적합한 배양법이 오랜 시간이 지나는 동안에 시행착오의 과정을 거쳐서 개발되고 개량되어 오늘에 이른 것이다. 최근까지 태생아나 신생 아 동물의 조직은 배양이 가능하나 성숙 동물의 조직은 배양이 불가능하다고 믿어져 왔다 .1) 배양 기술과 배양액 조성의 새로운 개발과 계속된 개량으로 인해서 특수 기능을 가전 간, 췌장, 혈 관, 뇌하수체, 그리고 근육조직을 성숙 동물로부터 분리하고 배 양할 수가 있게 되었다. 그러나 성숙 동물의 신경계 조직의 배양 은 극소수의 예의를 제의하고는 아직도 어려운 것이 현실이다. 성숙 포유동물의 말초신경계 신경세포의 배양은 1977 년 이후로 스코트 Sco tt 그리고 저자의 연구실에 의해서 가능하게 되었으 며 ,2 , 3) 성숙 포유동물의 중추신경계 신경세포는 지금까지 저자의 연구실에서 발표한 바 있는 망막신경세포의 배양만이 가능하다 .4)
여기에서는 신경조직의 초대배양에서 가장 초보적이면서 널리 이 용되고 있는 계배 척수후근신경절과 교감신경절의 배양을 소개하 기로 한다. 계배를 신경조직배양에 사용할 때 고려하여야 하는 것이 계배의 나이이다. 계배 척수후근신경절의 신경세포는 계배 의 나이가 많을수록 그 크기도 증가한다. 신경세포의 직경은 부 란 6 일 계배에서 8 µm 이고 10 일 계배에서는 12 µm 이며 17 일 계 배에서는 20µm 으로 증대한다. 자기 자신의 연구 과제와 그 목 적에 따라서 계배 나이를 결정하여야 한다. 일반적으로, 척수후 근신경절 spi na l dorsal root g an gli on 의 경우에는 8 일에서 12 일 계 배 에 서 , 척 수교감신 경 철 sym p a th i c ga ng lion chain 은 10 일 에 서 14 일 계배에서, 그리고 부교감신경절인 모양체신경절cili ar y g a ngli on 은 8 일에서 12 일 계배에서 분리하는 것이 적당하다. 중 추신경계에서는 척수는 6 일에서 10 일의 계배에서, 그리고 대뇌 cerebrum, 시상 op tic tec tu m , 소뇌 cerebellum 는 8 일에서 12 일의 계배에서 분리하는 것이 좋다. 11 .1 계배의 적출 척수후근신경절의 배양에는 8 일에서 12 일의 계배가 그리고 교 감신경절의 배양에는 10 일에서 14 일까지의 계배가 가장 적합하 다. 수정란을 부화장에서 사와서 서서히 회전하는 부란기에서 위 에서 말한 기간을 인큐베이트한다. 계배의 적출은 다음과 갇이 한다(그립 11-1). 〈준비〉 1) 70 % 알코올
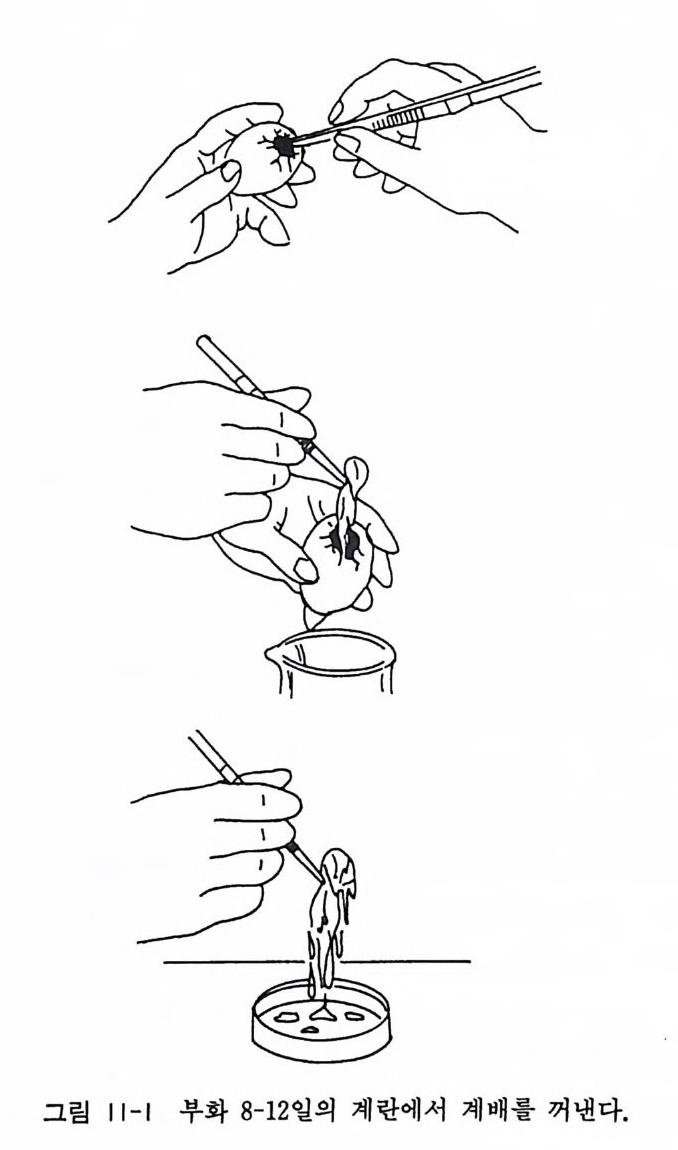 그림 | 1-1 부화 8-12 일의 계란에서 계배를 꺼낸다.
그림 | 1-1 부화 8-12 일의 계란에서 계배를 꺼낸다.
2) 10 cm 직경 페트리 접시 3) 비커 4) 칼슘, 마그네슘 제거 핸크스 영류 용액 5) 수술기구 -10cm 외과용 핀셋(곡선) 〈방법〉 1) 부란 7 일 -12 일이 되는 계란의 둔한 쪽을 위로 하여서 상부 를 70 % 알코올로 적신 티슈나 가제로 소독한다. 2) 핀셋을 사용하여 계란 상부에 구멍을 내고 원형으로 껍질을 벗긴다. 3) 끝이 곡선으로 된 의과용 핀셋으로 계배를 꺼낸다. 이때에 계배의 목을 핀셋으로 신중히 걷어서 꺼내어 핸크스액이 들 어있는 10cm 페트리 접시로 옮긴다. 11 .2 계배 척수후근신경질 분리세포 배양 〈준비〉 1) 입체 해부현미경과 조명 2) 수술기구 -10 cm 의과용 가위 (직선과 곡선) 2 개 10cm 의과용 핀셋 2 개 시계 핀셋 (No. 4) 2 개 수술용 칼 (No. 11) 2 개 3) 시험관, 페트리 접시, 원심 듀브 4) 혈구계산판 5) 칼슘, 마그네슘 제거 핸크스 염류 용액 6) 2.5% 트립신액
7) 0.2 % DNase 효소액 8) 이글 MEM 합성 배양액 9) 마혈청 10) 50 % 포도당액 11) 2 mg /m l 겐타마이신액 12) 10 µg/m l NGF( 신경성장인자) 13) 5 mg /m l 아스코르브산(비타민 C) 〈 방법 〉 1) 계배 6 마리를 부란 8 일에서 12 일되는 계란에서 분리하고 10 ml 의 핸크스액이 들어있는 10cm 직경 페트리 접시에 넣는 다. 계배 1 마리를 소량의 핸크스액이 들어있는 다른 페트리 접시에 옮기고 계배의 등을 아래로 하여 눕힌다. 여기서부터 입체 해부현미경을 사용하여 후근신경절을 적출한다. 가위로 머리를 절단하고 가슴과 배 부분을 절개한 뒤 그 속에 들어 있는 장기 (심장, 간, 위장 기타)를 깨끗이 들어낸다. 2) 계배 중앙부에서 척주 칼럼이 보이는데 요수 부근에서는 그 양측으로 신경섬유속이 꽁지처럼 노출되어서 보인다. 이들 신경섬유속을 잡아당기면 신경절 팽대부분이 보이므로 이를 핀셋으로 분리한다(그립 U-2). 후근신경절을 다수 분리하고 자 할 때에는 계배를 아래로 눕히고 먼저 몸 뒤의 피부를 제 거한다. 두명한 연골을 통하여 척수가 보이는데 시계수선용 핀셋 2 개를 써서 연골을 제거하고 다음 척수조직을 잘라낸 다. 경수에서 선골부까지 30 쌍의 후근신경절이 양측에 위치 한다. 이들 신경질을 상하지 않도록 신중히 제거하고 핸크스 용액이 들어있는 페트리 접시에 모은다. 3) 계배 6 마리에서 200 개 정도의 후근신경철이 분리되면 이들
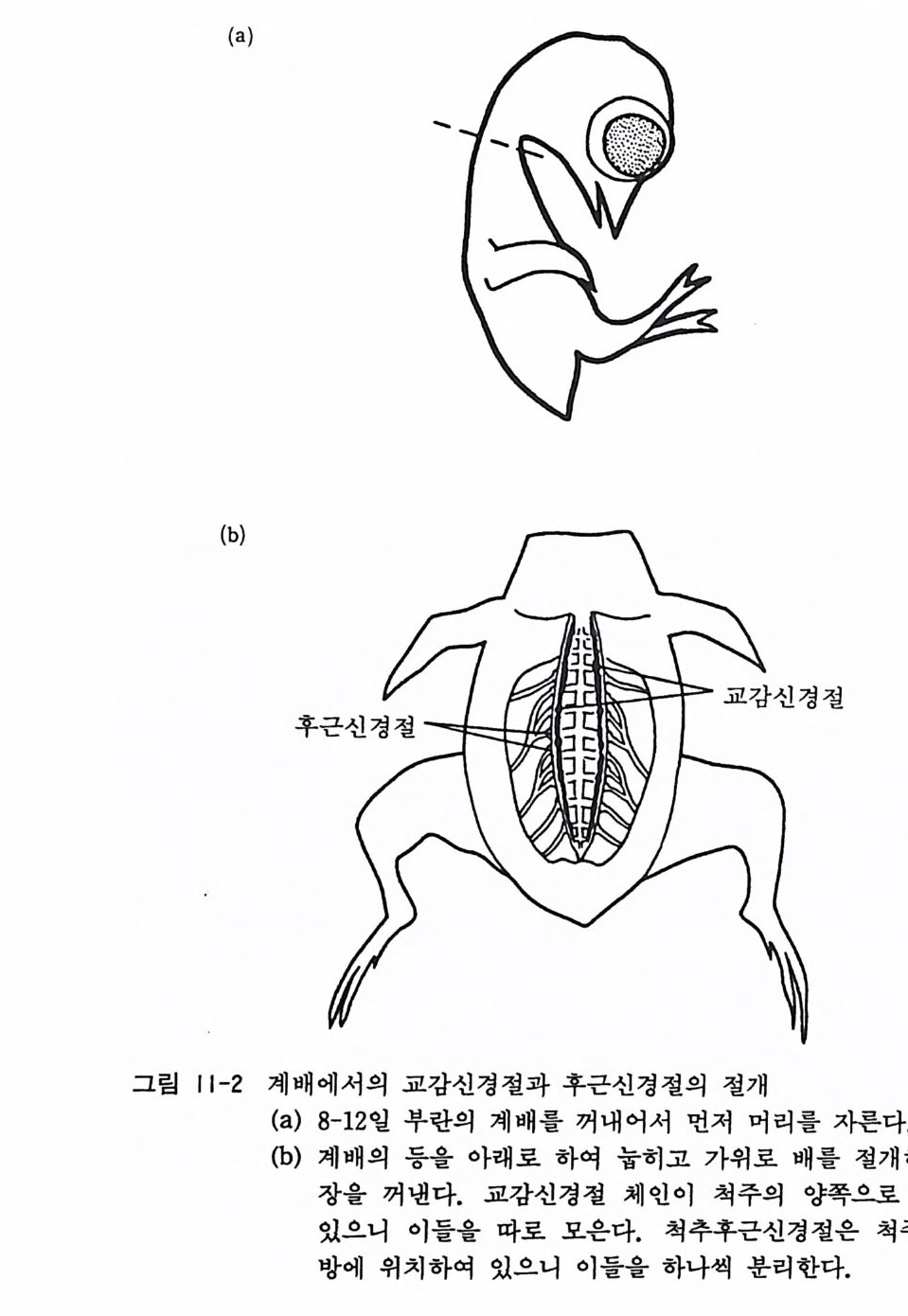 (a)
(a)
을 핸크스액과 갇이 시험관에 옮긴다. 핸크스액을 제거한 뒤 0.25% 트립신액 5ml 과 0.2 % DNase 0.2ml 이 들어있는 핸크스액을 가하고 37 도 인큐베이터에서 30 분간 인큐베이트 한다. 5 분에 한번씩 시험관을 혼들어서 조직과 효소액이 잘 혼합되도록 한다. 4) 후근신경절이 시험관 바닥에 오도록 정치한 뒤, 트립신액을 제거하고 10 % 마혈청이 들어있는 배양액 3ml 과 0.2 % DNase 0.1 ml 을 가한다. 파스출피펫으로 20 번 정도 상하로 조용히 피페팅한다. 이때 거품이 안 나도록 하는 것이 중요 하다. 파스출피펫 끝에 멸균된 미크로피펫팁울 삽입한 뒤 이 것으로 피페팅을 20 번 정도 되풀이한다. 파스출피펫 구멍의 직경은 2mm 내의이고 미크로피펫립은 0.5 m m 정도이므로 이렇게 기계적인 피페팅을 되풀이함으로써 트립신에 의해서 소화가 된 조직에서 단일세포를 분리할 수가 있다. 피페팅을 너무 강하게 한다든가 피페팅 횟수가 너무 많으면 세포막 표 면에 상처가 생겨서 세포 생존율이 떨어지게 된다. 5) 조직편이 아직도 남아 있다면, 3 분 가량 시험관을 정치한 뒤에 상청액을 다른 시험관에 옮긴다. 남은 조칙편에 4) 에서 와 마찬가지로 배양액 2ml 을 가하고 피페팅을 20 번 되풀이 한다. 6) 두 번의 세포 부유액을 합하고 이를 1200r p m 으로 8 분간 원심분리한다. 7) 상청액을 버리고 페렛에 마혈청이 들어있는 배양액 3ml 을 가하고 피페팅을 10 번 가량 하여서 세포 부유액을 만든다. 세포 부유액 1 방울을 혈구계산판에 떨어뜨려서 세포 수를 계 산하고 배양액 1ml 에 105-106 세포 수가 되도록 조정한다. 8) 폴리라이신 코팅된 커버글라스 위에 한 방울의 세포 부유액
울 올려 놓는다. 5 % CO2 인큐베이터에서 37 도로 보온한 뒤 다음날 3ml 의 배양액을 가한다. 커버글라스는 9mm 직 경이나 12mm 직경의 No.1 두께의 유리가 적당하다. 12 mm 커버글라스 8 개를 6cm 페트리 접시에 간격을 두고 위 치시킨다. 1990 년에 들어와서 미국이나 케나다에서 공급되고 있 는 유 리에는 불순물이 섞여 있어서 배양신경세포가 1 주일 안에 죽 는다는 예가 많아서 유럽에서 특별히 주문한다는 소식이 있 다. 우리 연구실에서는 미국의 알라이드 - 시그널 All i ed -Sig na l 회 사가 공급하는 아클라 Aclar 플라스틱 시 트 를 입 수 하고 업자에게 부탁하여 9mm, 12mm 그리고 22mm 직경 의 원형 커버슬립을 만들어서 이들을 사용하고 있다. 폴리라 이신 코팅에 대하여서는 제 6 장을 참고로 하기 바란다. 9) 배양액 조성은 다음과 같다. 이글 MEM 86ml 마혈청 10ml 50% 포도당액 1ml 2 mg /m l 겐타마이 신 1 ml lOµg /m l NGF( 신경성장인자) 1 ml 5 mg /m l 아스코르브산 (비 타민 C) 1 ml 〈해설 〉 1956 년에 나카이 Naka i가 계배 척수후근신경절울 트립신으로 처리하여 분리세포배양을 처음으로 보고하였는데 5) 그 이후로 이 조직배양 시스템은 오랫동안 신경과학자들에게 애용되는 실험모 델이 되었다. 이 시스템을 사용하여서 신경성장인자 NGF 의 작용 기작, 신경섬유 성장단gr o wt h cone 성장의 조절, 신경독의 병리
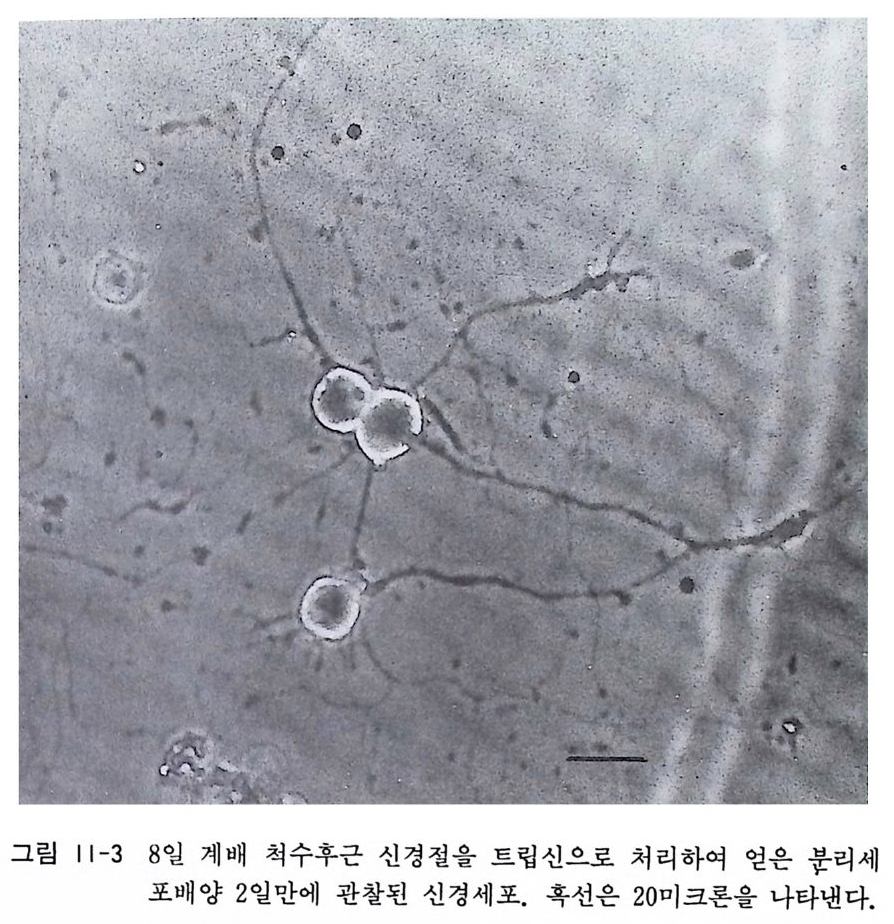 그림 | 1-3 8 일 계배 척수후근 신경절울 트립신으로 처리하여 얻은 분리세
그림 | 1-3 8 일 계배 척수후근 신경절울 트립신으로 처리하여 얻은 분리세
기전 등 신경과학에 밀접한 과제가 연구되어 왔다. 그림 11-3 에서는 8 일 계배 척수후근신경질 분리세포배양 2 일 만에 관찰된 신경세포의 위상차현미경상이다.
11 .3 맥시모우 슬라이드에 의한 계배 척수후근신경절 이식편 배양 맥시모우 슬라이드를 사용하여 계배 척수후근신경절을 장기간 배 양하고 수초 형 성 my el i n for mati on 을 조직 배 양에 서 처 음으로 보고한 것 이 1957 년 피 터 슨 Pete r son 과 머 레 이 Murray 의 업 적 인 데 이 배양법은 1970 년 초까지 신경조직배양의 주류 를 이루고 있 었다 .6) 맥시모우 이중 커버슬립 배양법 Maxim ow double cover-slip me t hod 이라 불리우는 이 배양법은 맥시모우 슬라이드와 대 형 커버글라스의 비용이 비싸고 배양 조작이 번잡하고 시간이 걷 린다는 결점이 있다. 최근에는 페트리 접시법이나 풀라스틱 미니 딧슈p las tic m i n idi sh 법을 사용하는 연구실이 증가하고 있다. 〈준비〉 11.2 에 기술한 척수후근신경절 분리세포 배양과 갇다. 다음의 것이 여분으로 필요하다. 1) 직경 15cm 대형 유리 페트리 접시 2) 맥시모우 글라스 슬라이드 3) 4cmX4cm 유리 커버슬라이드 (No.2 두께) 4) 맥시모우 술라이드 래크 rack 5) 파라핀-바셀린 (2 : 1) 혼합액 〈방법〉 1) 직경 15cm 의 대형 유리 페트리 접시 바닥에 꼭 맞도록 흰 필터페이퍼를 놓고 그 위에 12cmX12cm 의 검은 종이를 덮 는다. 페트리 접시는 알루미늄 포일에 싸고 오토클레이브롤 해서 멸균을 해둔다.
2) 페트리 접시의 검은 종이 위에 전열 멸균한 4cm X 4cm 의 정방형 유리 커버슬라이드 (No.2 두께) 2 장을 간격을 두고 놓는다. 이 커버글라스 위에 콜라겐울 코팅한 22mm 직경의 원형 커버글라스 (No . I 두께)를 놓는다. 3) 콜라겐 커버글라스 위에 계배 척수후근신경절 2 개를 5mm 의 거리를 두고 올려 놓는다 (1l . 2 철에서 기술한 바와 같이 계 배 척 수 후 근신경절을 분리한다). 4) 피펫을 사용하여 신경절 위에 배양액 소량 (0.05ml) 을 떨어 뜨린다. 5) 중앙부에 2.5cm 직경의 웅덩이가 파인 맥시모우 슬라이드 (5cm x 8cm) 의 네 구석에 멸균 바셀린을 소량 바르고 4cm X4 cm 커버글라스 위에 울려 놓는다. 이때에 맥시모우 슬 라이드 중앙부 웅덩이가 원형 커버글라스 위에 정확하게 위 치되도록 주의한다. 6) 4cmx4cm 커버글라스와 맥시모우 슬라이드 경계부를 60 도로 가온한 파라핀-바셀린 (2 : 1) 혼합액으로 코팅을 한다. 이것은 배양액의 증발을 방지하기 위한 것이다. 7) 나무로 된 특수 래크에 넣고서 36 도 인큐베이터에서 보온한 다. 8) 일주일에 두 번(월요일과 금요일) 배양액을 교환한다. 9) 배양액 교환은 다음과 같이 한다. 파라핀-바셀린으로 된 씰 울 벗긴 뒤, 수술용 칼을 왼손에, 시계 핀셋을 오른손에 잡 고 배양조직이 있는 작은 커버글라스를 신중히 들어올리고 핸크스액이 들어있는 페트리 접시로 옮긴다. 위의 방법 1) 에 서 7) 까지를 되풀이한다. 맥시모우 이중 커버슬립 배양법의 실례는 그립 11-4 에서 볼 수 있다.
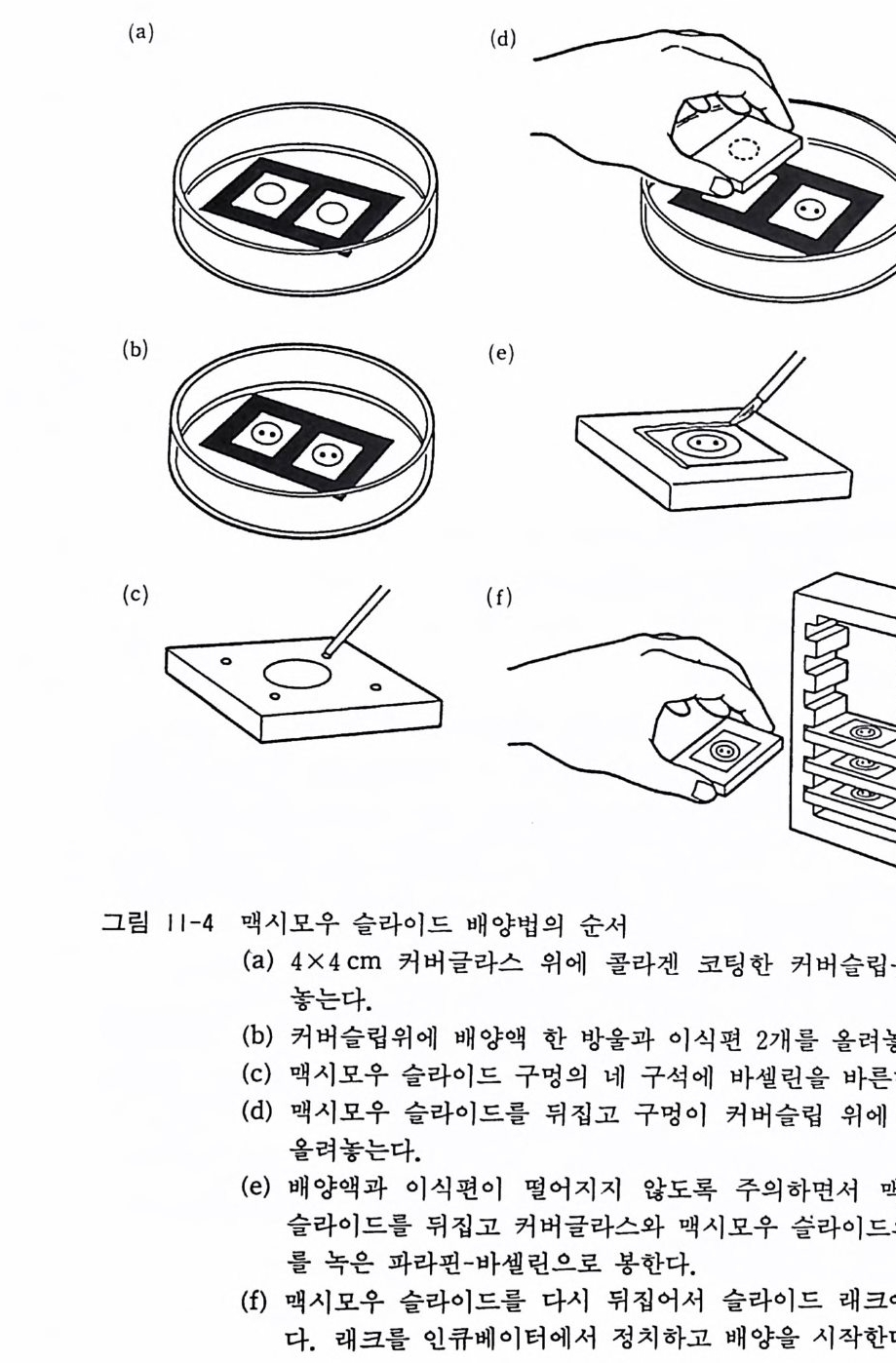 (a) (d)
(a) (d)
〈 해설 〉 신경절의 이식편 배양에서는 배양 4-6 시간 이내에 신경섬유의 성장과 슈완 schwann 세포나 섬유아세포의 유출을 볼 수 있다. 배양 후 3 일에서 8 일 정도가 되면 신경섬유가 신경절 이식편 주 위로 방사형으로 성장하여 나온다. 방추형인 슈완세포가 역시 성 장부 outg ro wt h 에 서 많이 보이 고, 때 로는 슈완세포가 신경 섬 유와 밀착되어서 접촉하고 있음을 볼 수가 있다. 배양 2 주가 넘으면 이식편 중앙부에서 신경세포의 집단을 보이게 되며 (그림 11-5), 미엘린 형성 m y e li na ti on 도 시작된다. 배양 4 주가 되면 이식편 중 앙부와 주변부에 기차 선로처럼 평행선으로 보이는 미엘린이 수 없이 보인다(그림 11-6).
 그림 11-5 계배 척수후근신경절 이식편 배양 2 주가 넘으면 이식편 중앙부
그림 11-5 계배 척수후근신경절 이식편 배양 2 주가 넘으면 이식편 중앙부
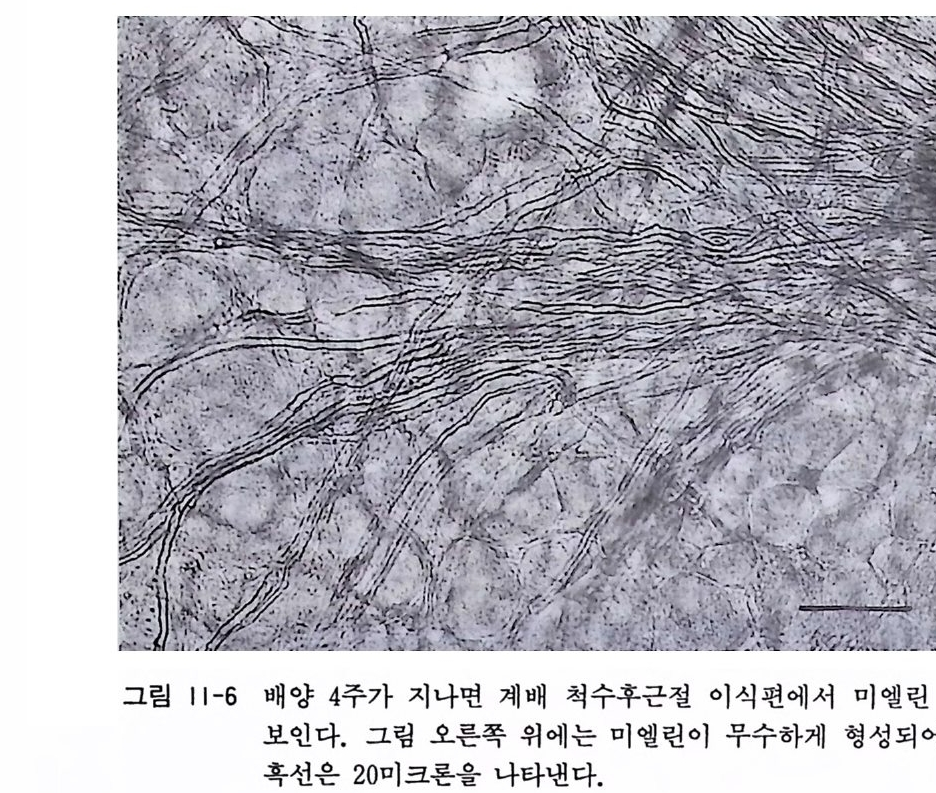 그림 11-6 배양 4 주가 지나면 계배 척수후근절 이식편에서 미엘린 형성이
그림 11-6 배양 4 주가 지나면 계배 척수후근절 이식편에서 미엘린 형성이
11 .4 페트리 접시에 의한 계배 척수후근신경절 이식 편 배양 칙경 3.5cm 의 플라스틱 페트리 접시를 맥시모우 슬라이드 대 신 사용하여 후근신경절 이식편을 배양할 수 있다. 여기서 이 페 트리 접시법을 소개한다(그립 11-7). 〈방법〉 1) 직경 3.5 cm 의 페트리 접시를 사용하되, 그 뚜껑 (커버)을
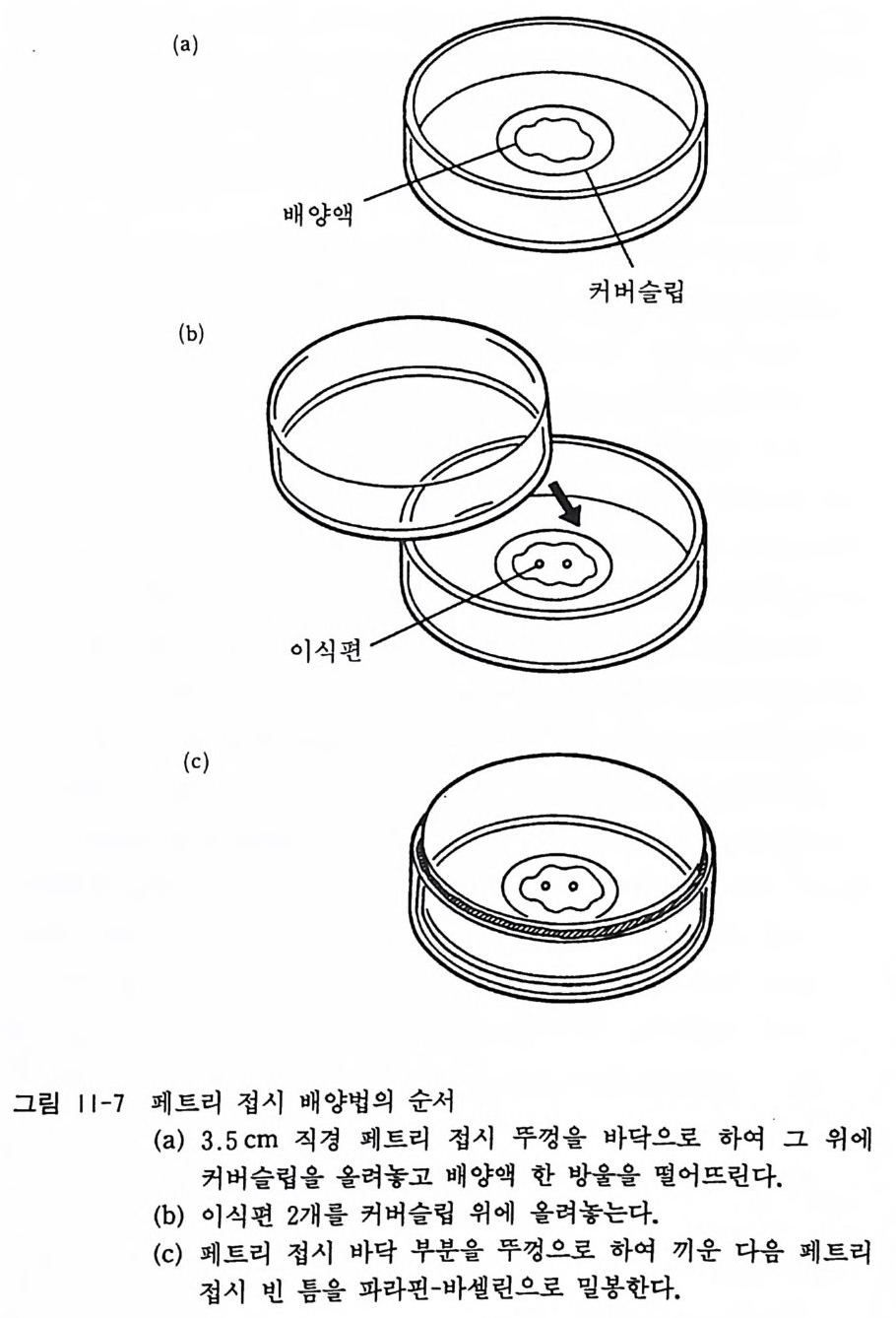 (a)
(a)
뒤집어 놓는다. 이 페트리 접시 커버 중앙에 콜라겐울 발라 놓은 22mm 직경의 원형 커버글라스를 위치시키고 배양액 한 방울 (0.05ml) 을 떨어뜨린다. 2) 계배 척수후근신경절 2 개를 5mm 의 간격을 두고 커버글라 스 위에 올려놓는다. 3) 페트리 접시의 바닥 부분을 뒤집어서 뚜껑으로 덮는다. 4) 밀폐계를 만들기 위하여 페트리 접시 위와 아랫부분의 경계 부를 60 도의 온수에서 데워서 녹인 파라핀-바셀린 혼합액 (2 : 1 비율)으로 밀폐한다. 학생들이 서예시간에 쓰는 봇을 써서 파라핀-바셀린 혼합액을 적용시키면 좋다. 파라필름이 나 테이프를 사용하여 밀봉할 수도 있다. 5) 위와 같은 페트리 접시는 한 실험에서 10 개에서 20 개를 사 용하는데 이들을 운반하기에 편리하도록 25cmX35cm 크기 의 풀라스틱 쟁반에 놓고 37° 인큐베이터에서 보온한다. 6) 일주일에 두 번(월요일과 금요일) 배양액을 교환한다. 7) 이상의 페트리 접시법은 폐쇄계 closed sys t e m 이지만 이와 반대로 개 방계 op en sys t e m 로서 배 양할 수도 있다. 우리 연 구실에서는 팔콘사 Falcon 나 코닝사 Corn i n g에서 공급하는 6 혈 멀티웰 챔버를 사용하고 있다. 멀티웰 6 개 속에 커버글라 스를 위치시키고 이식편과 배양액을 올려놓고 이식편이 콜라 겐에 밀착하도록 24 시간 기다린다. 24 시간 뒤 각 웰 속에 1 ml 의 배양액을 추가하고 배양을 시작한다. 11 .5 계배 교감신경절 분리세포 배양 계배의 교감신경절은 부란 10 일에서 12 일의 계배에서 분리하는
것이 좋다. 그 이유는 복부 교감신경절 체인이 10 일 이전의 계배 에서는 현미경 밑에서도 구별하기가 어렵고 14 일 이후에는 결체 조직이 중식하여 그 분리가 곤란하기 때문이다. 재료와 준비는 11.2 에서 기술한 척수후근신경절 배양과 같다. 〈 방법 〉 1) 계배를 소량의 핸크스액이 들어있는 페트리 접시에 등을 아 래로 하여 눕힌다. 2) 가위로 머리를 절단하고 가슴과 배 부분을 절개한 뒤 그 속 에 들어있는 장기(심장, 간, 위장 기타)를 들어낸다. 3) 척주 컬럼이 보이는데, 흉수와 요수에 걸쳐서 척주의 경계 부에 연하여서 좌우에 섬세한 교감신경절 체인이 보인다. 이 들을 시계용 핀셋으로 신중하게 들어낸다. 길이가 1.5 -2cm 정도의 체인을 척주 양측에서 분리할 수 있다(그림 11-2). 4) 교감신경절 체인을 계배 10 마리에서 분리한 뒤 11.2 에서 기 술한 척수후근절 분리세포 배양과 같은 방법으로 트립신 처 리하고 분리세포 배양을 시작한다. 교감신경절세포에서도 후 근신경절세포에서나 마찬가지로 신경성장인자 (nerve grow th fac to r , NGF) 가 그 생존과 발달에 필수영 양 요소로 되 어 있 다. 〈해설〉 계배 교감신경절과 앞에서 설명한 계배 후근신경절은 신경성장 인자 NGF 와 이와같은 그룹에 속하는 뉴로트로핀 계열, 죽 뇌유 래 신경 영 양인자 (bra i n deriv e d neurotr o p hic factor , BDNF), 뉴 로트로핀 -3 (neurotr o p h in - 3, NT-3) , 뉴로트로핀 -4 (neurotr o p hin -4, NT-4), 뉴로트로핀 -5(neuro t ro p h i n-5, NT-5) 와 갇온 신경영
양인자 (neuro t ro p h ic fac to r s, NTFs) 의 발견, 확인 그리고 생물 활성의 측정 등에 널리 사용되어 온 바 있다 .7 , 8) 계배 교감신경 철은 카데콜아민성 신경세포로 구성되어 있기 때문에 카데콜아민 성 신경세포의 신경화학과 신경약리학 분야에서 광범하게 사용되 는 실험모델이다.
제 12 장 계배 모양체신경절의 배양 계 배 의 부교감신 경 계 pa rasym p a th e ti c ga ng lion 의 하나인 모양 체신경절 ciliar y g an g l i on 은 노르아드레나린 noradrena li n 을 신경전 달 물 질 neurotr a nsmi tter 로 사 용 하 는 교 감 신 경 절 sym spa th e tic g an g li on 과는 대조적으로 아세틸콜린 ace ty lcho li ne 을 신경전달물 질로 사용하는 것이 특징이다. 모양체신경절 배양 1) 은 1984 년에 순수분리된 신경영양인자의 하나인 모양체 신경 영 양인자(cili a ry neurotr o p h ic fac to r , CNTF) 에 의해서 그 생존과 분화가 촉진된다고 보고되고 있다 .2,3) 모양 체신경절의 배양은 척수후근신경절과 마찬가지로 계배 8 일에서 12 일에 분리하는 것이 현명하다. 재료와 준비는 11.2 에서 기술한 척수후근신경절 배양과 같다.
12.1 계배 모양체신경절 분리세포 배양 〈 방법 〉 1) 계배를 소량의 핸크스액이 들어있는 페트리 접시에 배를 아 래로 하여 눕힌다. 2) 시계용 핀셋 2 개를 사용하여 안구 e y eball 주위의 피부와 근 육조직을 제거하고 안구를 신중하게 꺼낸다. 안구 를 밖으로 꺼낼 때 따라서 나오는 시신경 op tic nerve 과 바로 그 옆에 위치하고 있는 모양체신경절이 뒤에 남지 않도록 핀셋으로 시신경 후방을 절단한다(그림 12-1).
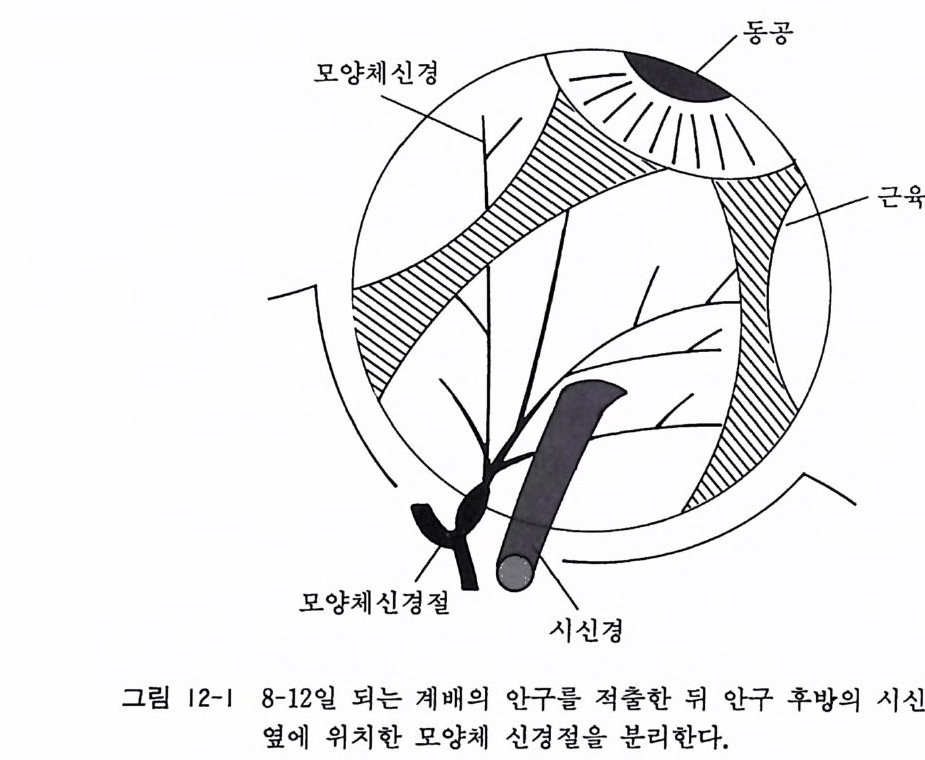 도0 고0
도0 고0
3) 모양체신경절 20 개를 계배 10 마리에서 분리한 뒤 1 1. 2 에서 기술한 척수후근신경절 분리세포 배양과 꼭같은 방법으로 트 립신 처리를 하고 세포 배양을 시작한다. 척수후근신경절 배 양의 배양액 중에 포함된 신경성장인자와 아스코르브산은 모 양체신경절 배양에는 필수적이 아니다. 4) 배양액 조성은 다음과 갇다. 이글 MEM 88ml 마혈청 10 ml 50 % 포도당액 1ml 2mg /m l 겐타마이신액 1ml 12.2 모양체 신경영양인자의 준비 모양체 신경영양인자는 최근 유전공학적인 방법으로 순수 정제 되어서 암젠 Am g en 과 리제네론 Re g eneron 과 같은 미국 제약회사 가 척수신경질환인 근위축성 척수측색경화증 am y o t ro ph ic late r al sclerosis 환자에 서 임 상실 험 중이 라 한다. 조직배양을 위해서는 시판되어 있으나 너무 고가이므로 계배 모양체신경절 분리가 끝난 안구에서 모양체 신경영양인자를 포함 한 안구 추출액을 다음과 같이 준비할 수 있다. 〈방법〉 1) 부란 8-12 일의 계배 안구를 사용한다. 2) 핸크스 용액에서 한번 씻어낸다. 3) 5ml 용량의 플라스틱 주사기 통 속에 계배 안구를 넣고 이 들을 원심시험관 속으로 밀어내어서 안구 호모지네이트롤 만
든다. 4) 계배 안구 호모지네이트와 같은 양의 핸크스액을 가하고 시 험관을 -20°C 에서 동결한다. 5) 다음날 호모지 네 이 트를 녹이 고 800 rem 회 전으로 10 분간 원 심 침전한다. 6) 상충액을 걷어내어 소량씩 분주하여서 _20°C 에서 동결하고 보존한다. 7) 계배 모양체신경절 배양액에 5 %의 비율로 가하여 조직배 양에서 사용한다. 안구 추출액을 포함한 배양액 속에서 배양 된 모양체신경절에서는 그 신경세포의 생존 수가 2-3 배가 된 다 .3)
제 13 장 계배 중추신경계 조직과 세포의 배양 제 11 장에서는 계배 척수후근신경절, 교감산경절 체인 그리고 제 12 장에서는 모양체신경절의 이식편 배양과 분리세포 배양에 대하여 상세히 기술하였고 여기에서는 계배 중추신경계 조직의 배양에 대하여 기록하기로 한다. 계배 중추신경계 조직배양의 경우 척수 spi nal cord 는 6 일에서 10 일의 계배에서, 그리고 대뇌 cerebrum, 시상 o ptic tec tu m , 소 뇌 cerebellum, 망막 reti na 에서는 8 일과 12 일 사이의 계배에서 분 리하는 것이 좋다 . 계배 척수후근신경절 이식편 배양에서는 맥시모우 슬라이드법 과 페트리 접시법을 소개하였는데 중추신경계 조직 이식편 배양 에서는 배양액 교환이 간편하고 대량 배양이 가능하며, 장기 배 양에 적 합한 롤러 튜브 배 양법 flying coverslip - ro ller tub e meth o d 룰 소개한다 .1 , 2)
13.l 계배 척수이식편 롤러 튜브 배양법 재료와 준비는 1 1. 2 에서 기술한 척수후근신경절 배양과 같으며 다음에 열거한 것이 여분으로 필요하다. 1) 16Xl25mm 시험관(구경 16mm, 길이 125mm) 2) 롤러 드럼 (브른스윅 과학 Brunswi ck Sc ien ti fic) 3) 시 험 관 래 크 tes t tub e rack 4) 22 X ll mm 장방형 커 버 글라스 (콜라겐을 발라둔다) 〈 방법 〉 1) 계배 8-12 일의 척수를 분리하고 핸크스액이 들어있는 10 cm 직경 페트리 집시로 옮긴다. 계배에서 척수를 분리하는 데 가장 편리한 방법은 먼저 계배의 배를 아래로 하여 위치 시킨 뒤 머리와 목의 경계부를 절단한다. 그 다음에 2 개의 핀셋을 써서 꼬리 부분에서 척수를 교대로 압축하면서 머리 쪽으로 밀어올린다. 이것은 마치 치약 튜브에서 치약을 짜내 는 동작과 같다. 목의 절단부에서 척수가 치약같이 나오게 된다 (그립 13-1 ) . 계 배 의 요수 lumbar cord 나 천수 sacral cord 부분에는 글리코겐체 gly c og en bod y가 있어서 배양에 방 해가 되므로 사용하지 아니한다. 2) 수술칼 (No . 11) 2 개를 사용하여 척수를 0.5-1 mm 두께의 원반으로 절단한다. 3) 칙경 15 cm 의 대형 유리 페트리 집시 (바닥에는 흰 필터페이 퍼와 검은 종이를 깔고 오토클레이브 멸균을 해둔다)의 검은 종 이 위에 맥시모우 슬라이드 2 장을 간격을 두고 놓는다. (맥 시모우 슬라이드가 없는 경우에는 직경 10cm 페트리 접시 바닥 에 콜라겐 코트한 커버글라스롤 놓을 수도 있다.)
4) 맥시모우 슬라이드 위에 미리 준비해 두었던 콜라겐울 바론 22mmXllmm 장방형 커버글라스를 10 장 간격을 두고 위치 시킨다. 배양액 한 방울을 그 위에 떨어뜨려서 건조되는 것 을 방지한다. 배양액이 고르게 퍼지도록 멸균된 유리봉으로 배양액을 퍼뜨린다. 5) 척수 소편 2 개나 3 개를 3mm 간격을 두고 커버글라스 위에 울려놓는다. 6) 대형 유리 페트리 접시의 뚜껑을 덮고 30 분간 인큐베이터에 서 정치한다. 7) 파스출 피펫으로 커버글라스 위의 여분의 배양액을 제거한 뒤 커버글라스를 16mm X 125mm 의 시험관에 집어 넣는다. 이때 주의할 것은 이식편이 커버글라스에서 미끄러져서 떨어 지지 않도록 조용히 옮겨야 한다. 이식편이 위를 향하도록 하고 시험관을 시험관 래크 위에 놓는다. 8) 시험관 속에 배양액 0.7ml 을 서서히 주입한다. 이식편이 떨어지는 경우에는 파스출 피펫으로 이것을 집어내고 커버슬 립 위에 주의 깊게 다시 올려놓는다. 이것은 어려운 것 같으 나 여러 번 연습을 하면 쉽게 할 수가 있다. 9) 스크류캡으로 시험관 입을 꼭 참근 다음, 시험관을 시험관 래크에 나란히 위치시키고 37 도 인큐베이터에 넣는다. 24 시 간 정치한 뒤에 이들 시험관을 롤러드럼에 삽입한다. 롤러드 럼은 37 도 인큐베이터 속에 장치하는데 그 속도를 1 시간에 12 번 회전 죽 5 분에 1 번 회전하도록 한다. 10) 일주일에 한번 배양액 교환을 한다. 스크류캡울 연 다음 배 양액을 파스출 피펫으로 뽑아내고, 새 배양액 0.7ml 를 가한 다. 롤러듀브 배양법의 실제는 그립 13-1 에 도시하였다. 11) 배양액 조성은 다음과 갇다.
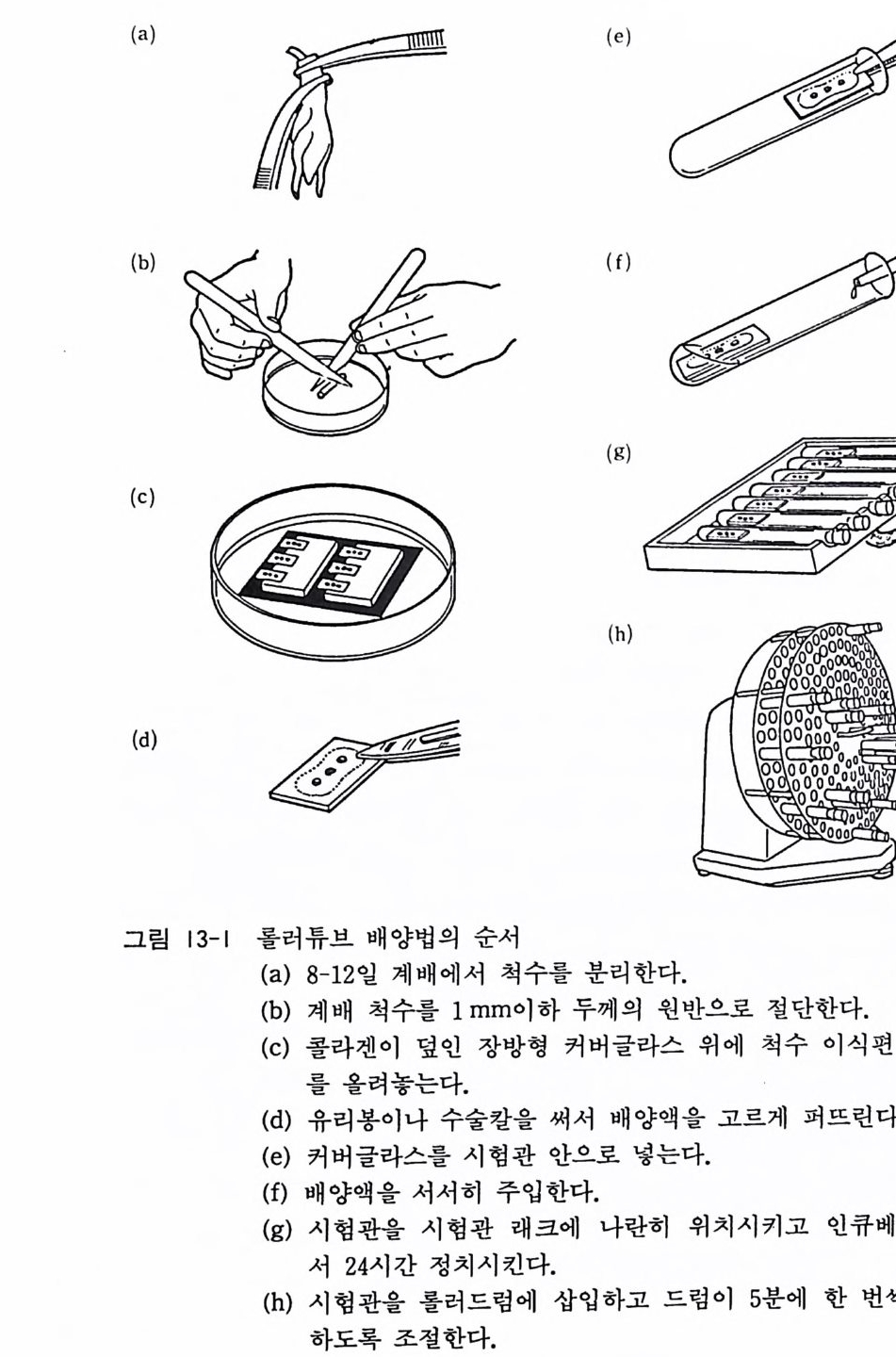 (a) 一 (e)
(a) 一 (e)
이 글 MEM 핸크스액 포함 78 ml 마혈청 20ml 50% 포도당액 1ml 2mg /m l 겐타마이 신액 1 ml 〈 해설 〉 1970 년대 초까지에는 계배 추출액을 5-10 %의 비율로 배양액 속에 가하였는데 최근에는 위에서 보인 바와 같이 마혈청이나 소 태아혈청만으로 장기 배양이 가능하다. 롤러드럼 배양법으로 저 자의 연구실에서는 마우스 태아 척수이식편 배양을 일년 이상 장 기배양한 기록이 있다. 연구실에 따라서는 1960 년대에 포메라트 Pomera t나 힐트Hi ld 가 사용하였던 것처럼 대형의 50mmXllmm 크기의 유리 커버 글라스를 사용하고 ,3) 콜라겐 기질을 쓰지 않고 풀라스마 혈장 응고 속에 이식편을 파묻어서 배양하는 수가 있으나 4) 이것은 너 무 구식인 듯하다. 자기 자신이 플라스마를 준비하기가 힘들고, 회사들이 판매하고 있는 플라스마의 품질이 좋지 않아서 트롬빈 을 가해서도 응고가 일어나지 않는 결점이 있다. 플라스마 응고 가 잘 되더라도 배양이 시작되어서 1 주일에 한 번씩 융해되어 버 리는 플라스마를 수선해야 된다는 번잡함이 큰 결점이다. 배양을 시작한 다음날부터 이식편 ' 주위에 세포유출부 ou t gro wt h 가 나오고 여기에는 주로 아스트로사이트나 섬유아세포가 있다. 이러한 유출세포 사이사이에 신경섬유가 신생하여 방사형 으로 주위에 퍼져나간다. 이식편을 만들 때에 뇌막을 완전히 제 거해 두면 배양 후에 섬유아세포의 발생이 적고 신경세포와 글리 아세포의 발육이 양호하다. 롤러튜브 배양법에서는 맥시모우 슬라이드법이나 페트리 접시
법에 비하여 이식편의 평탄화가 가속되어서 1 주일에서 2 주일 이 내에 4-8 충 정도의 세포충으로 구성된 이상적인 배양을 얻을 수 가 있다. 맥시모우 슬라이드법이나 페트리 접시법에서는 2-3 주일 뒤에 이식편 중앙에 검게 변성한 부분이 나오게 되는데 이것은 배양 개시 때에 이식편 크기를 조절하여 방지하도록 한다. 계배 척수 배양에서 보이는 산경세포의 위상차현미경과 은염색 의 사전은 그림 13-2, 13-3 에 보이고 있다. 계배의 중추신경계 조직에서 척수 이의에 대뇌 cerebrum,5) 시 구 op tic tec tu m , 소뇌 cerebellum 이 식 편 배 양과 분리 세 포 배 양은 척수 배양법에 따라서 하면 가능하다.
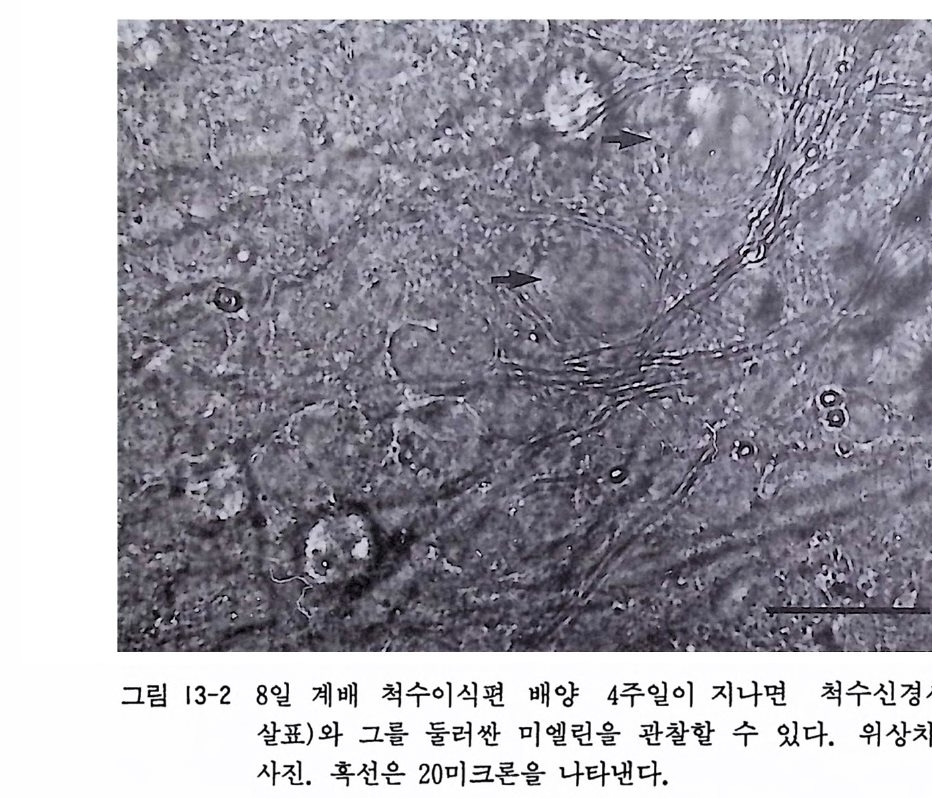 그림 13-2 8 일 계배 척수이식편 배양 4 주일이 지나면 척수신경세포(화
그림 13-2 8 일 계배 척수이식편 배양 4 주일이 지나면 척수신경세포(화
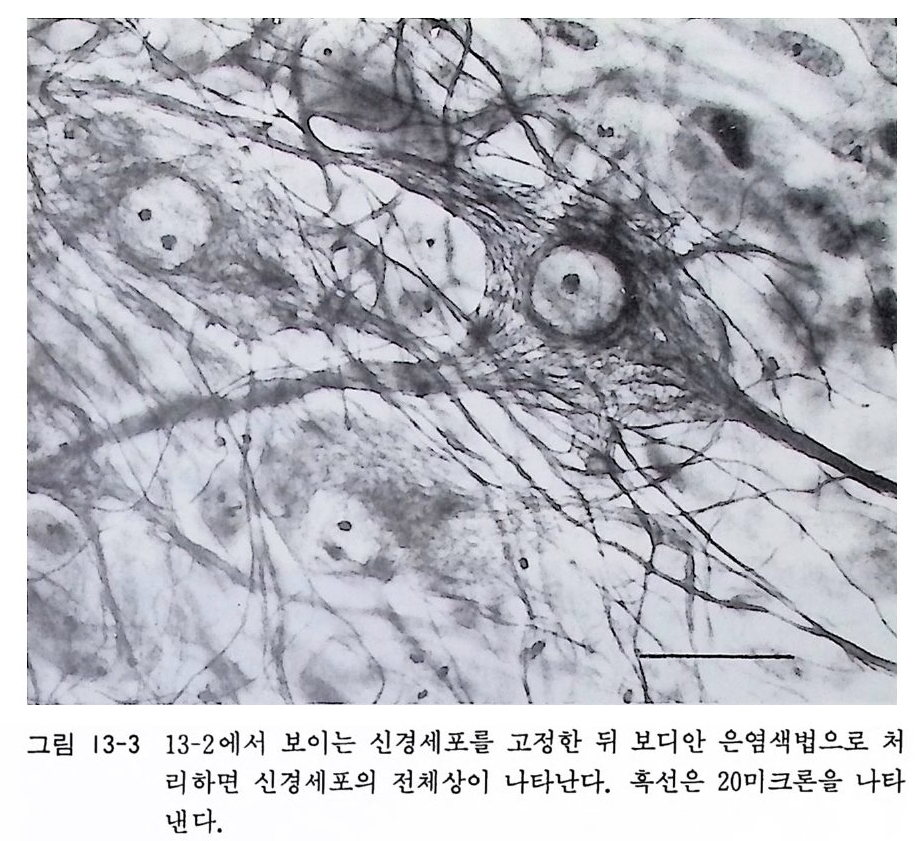 그림 |3-3 13 - 2 에서 보이는 신경세포를 고정한 뒤 보디안 은영색법으로 처
그림 |3-3 13 - 2 에서 보이는 신경세포를 고정한 뒤 보디안 은영색법으로 처
13.2 계배 척수신경세포 분리세포 배양 재료와 준비는 1 1. 2 에서 기술한 계배 척수후근신경절 배양과 같다. 〈 방법 〉 1) 12.1 에서 기술한 바와 갑이 계배에서 척수를 분리한다. 2) 수술칼 2 개로 척수를 소편으로 자르고 이롤 원심튜브에 핸
크스액과 같이 옮긴다. 3) 핸크스액을 제거하고 4. 3 ml 핸크스액, 0.5 ml 2. 5 % 트 립신액, 0.2 ml_. 0 .°'2 % DNase 액을 차례로 첨가하고 37 도 인 큐베이터에서 30 분간 정치한다. 4) 트립신액을 제거하고 DNase 를 포함한 배양액 5ml 를 가한 뒤 20 번 피페팅을 한다. 파스출 피펫 끝에 미크로피펫팁을 삽입하고 20 번 정도 피페팅을 되풀이한다. 조직편이 가라앉 은 뒤 상청액을 새 원심분리 튜브에 옮긴다. 5) 배양액을 남은 조직편에 가한 뒤 재차 20 번의 피페팅을 한 다. 상청액을 먼저 번 상청액과 합한 뒤 1000rpm 속도로 8 분간 원심분리한다. 6) 페렛에 배양액 5ml 를 가하고 세포를 부유시킨 뒤 두 번 더 씻고, 원심분리를 되풀이한다 . 7) 페랫을 배양액에 재부유시킨 뒤 혈구계산판으로 세포 수 계 산을 하고 1 X 106/ml 로 희석한다. 세포부유액은 폴리라이신 코팅된 12mm 커버글라스 (6cm 직경 페트리 집시에 8 개 를 나 란히 놓는다)에 플레이팅하고 하룻동안 37 도 인큐베이터에서 정치시킨다. 8) 다음날 배양액 3ml 를 추가한다. 배양액 교환은 일주일에 2 번 하도록 한다. 배양액 조성은 다음과 갇이 한다. 이글 MEM 88ml 마혈청 10ml 50% 포도당액 1 ml 2mg /m l 겐타마이 신액 1ml 〈해설〉 분리세포 배양에서는 각종 세포가 서로 간격을 멀리 하여 떨어
져 있으므로 이식편 배양시보다 현미경 관찰을 이상적으로 할 수 가 있다. 배양 개시 2 시간 이내에 세포가 커버글라스나 페트리 접시 표면에 부착하여 여러 가지 형태를 보이기 시작한다. 그림 13-4 와 13 - 5 에서는 4 주일 이상 분리세포 배양한 계배 척수운동신 경세포의 위상사현미경상과 뉴로필라멘트 면역 염색의 사전을 보 이고 있다. 위상차현미경 아래에서는 대형의 평탄한 형태를 가전 섬유아세포나 아스트로사이트 그리고 돌기를 가진 다각형의 신경 세포 를 대충 구별할 수 있으나 최종적인 세포동정 cellular ide nti fica ti on 은 면 역 세포화학에 의 한 특수염 색 법 으로만 가능하 다. 면역세포화학 염색법과 신경세포 타입 특이 마카 cell- typ e
 `. \ §;、`::...._0;: 초 ·· 3 을 `〉 '. ' ' .~도 . - ~- ..... , :. ,,.、`
`. \ §;、`::...._0;: 초 ·· 3 을 `〉 '. ' ' .~도 . - ~- ..... , :. ,,.、`
 .\ ...f....、...‘ t.. `.` .. . . ` `. . ...、.` ..` . . .}.Jy} . . .. ...`. 7 f > .\ i:. -t ...\J .`/\..> ' 』.. ` fJ • {....... ..\` ‘.55 `、 `` . `. : \.、...\ .` .; i ` ‘ ·、、 {` A'It. l`.\、〉 ` . | , '·` ```` ``i... . .`·`.•..`-. . •:. ’ . 2 、4 . ` :•.J ·4 ..\ \。? ~ . \.2 ’. •.`* . b:0 \, i\ .. ...\•44`、 · •·. ,5 i-‘ ` i5 t . r. ' 9 i»` ·' .? .
.\ ...f....、...‘ t.. `.` .. . . ` `. . ...、.` ..` . . .}.Jy} . . .. ...`. 7 f > .\ i:. -t ...\J .`/\..> ' 』.. ` fJ • {....... ..\` ‘.55 `、 `` . `. : \.、...\ .` .; i ` ‘ ·、、 {` A'It. l`.\、〉 ` . | , '·` ```` ``i... . .`·`.•..`-. . •:. ’ . 2 、4 . ` :•.J ·4 ..\ \。? ~ . \.2 ’. •.`* . b:0 \, i\ .. ...\•44`、 · •·. ,5 i-‘ ` i5 t . r. ' 9 i»` ·' .? .
spe ci fic markers 에 대 해 서 는 제 35 장을 참고할 것 이 다. 계배 척수신경세포 분리세포 배양에서 신경세포 상호간에, 혹 은 신경세포와 신경교세포 간에 긴밀한 상호작용이 있는데 이것 은 시냅스 형성과 미엘린 형성으로서 표현이 된다. 1972 년 저자 가 최초로 이러한 현상을 보고한 바 있다 .6) 1970 년대 이후로는 신경세포 분리배양을 사용하여 패치클램핑 pa tc h clamp ing , 홀셀클램 핑 whole cell clamp ing 등의 신경생리학
적 방법으로 신경세포의 이온 채널이나 수용체의 연구가 대단히 성행하고 있다. 형광색소인 Fura-2 나 그밖의 약품을 사용하여 분리신경세포의 세포 내의 칼슘 농도 측정도 가능하게 되었다. 앞으로 새로운 연구기술이 개발되어 분리신경세포 배양의 용도가 크게 증대할 것으로 예상된다. 13.3 계배 망막신경세포 분리세포 배양 〈 준비 〉 준비는 11.2 에서 기술한 계배 척수후근신경절 배양과 같다. 〈 방법 〉 1) 발생 8-12 일의 계배에서 좌우 양쪽의 안구를 적출하고 핸크 스액이 들어있는 10cm 직경 페트리 접시에 옮긴다. 2) 렌즈가 위에 오도록 왼쪽 손에 든 핀셋으로 안구를 누르고 오른손에 잡은 가위로 그 적도부(안구의 허리가 되는 부분)를 절단한다. 3) 핀셋으로 안구의 내부를 열고 렌즈, 유리체, 색소상피, 망 막을 핸크스액 중에 유출시킨다. 발생 8-12 일의 계배에서는 색소상피와 망막이 쉽게 분리된다. 망막은 반투명한 막으로 유리된다. 4) 수술칼을 사용해서 망막을 2mmX2mm 정도의 크기로 자 르고 이를 원심튜브에 옮긴뒤 트립신 처리를 시작한다. 트립 신 처리법은 13.2 를 참고하면 된다. 〈해설〉
망막에는 신경세포로서 강그리온 신경세포g an g li on cell, 아마 크린 신 경 세 포 amacrin e cell, 평 행 신 경 세 포 horiz o nta l cell, 쌍돌 기 신경 세 포 bip o lar cell, 그리 고 망막감각 수용세 포 ph oto r ecep- to r cell 가 있으며 글리 아세 포로서 는 뮬러 세 포 MUi ler cell 가 있 다. 래트에서는 위에 말한 각종 신경세포에 특이한 단클론 항체 가 있어서 면역세포 염색으로 구별이 가능하나 계배 망막에서 는 이것이 곤란하다. 다만 신경세포 전반을 구별하는 보디안 은 염색이나 8) 뉴로필라멘트, MAP2 등의 면역세포 염색이 가능하 다.
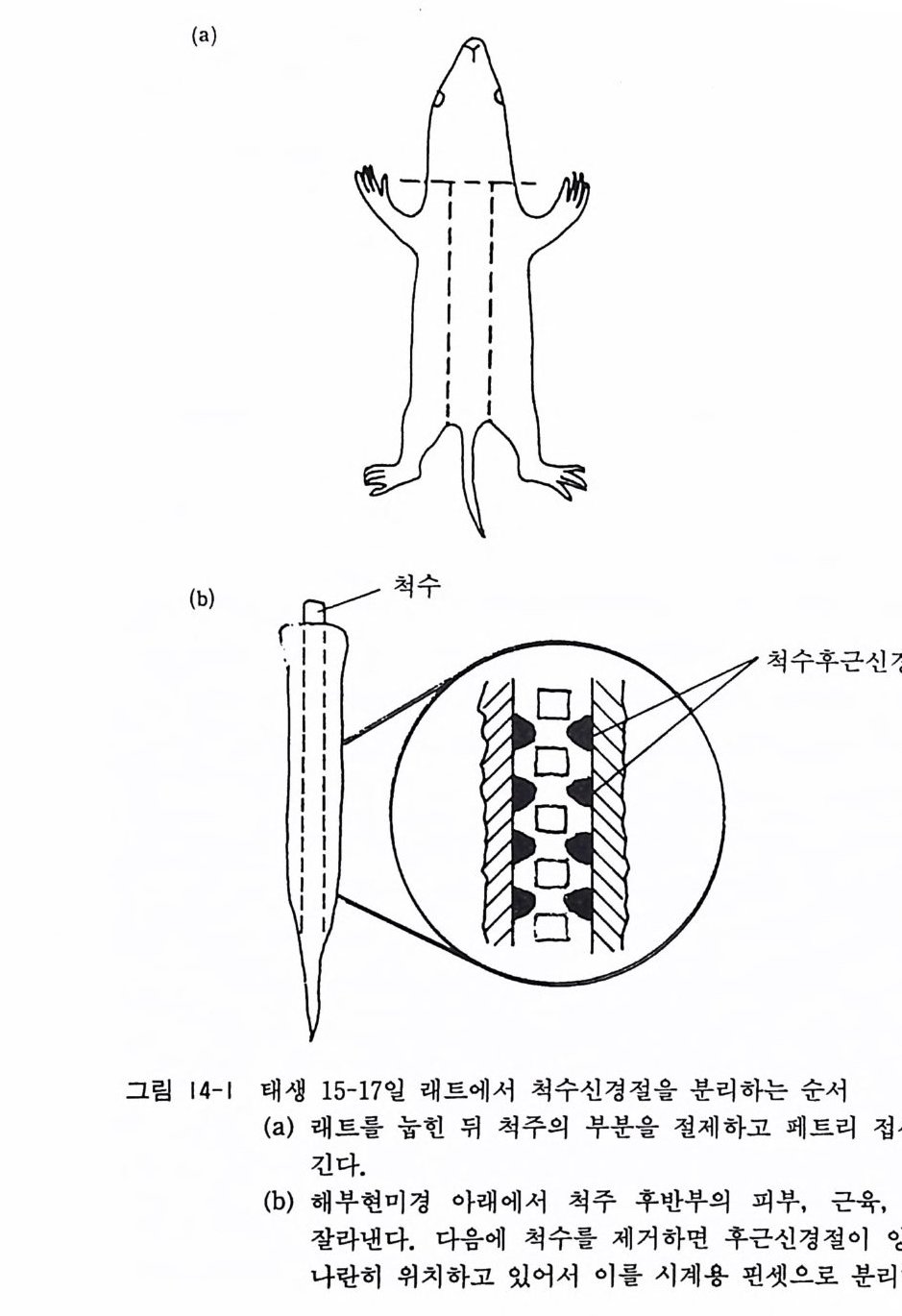
제 14 장 태생 래트와 마우스 척수후근신경절과 교감신경절의 배양 말초신경계의 조직배양에서 가장 흔히 사용되는 재료가 척수후 근신경절이다. 1957 년대에 피터슨 Pe t erson 과 더레이 Murra y가 계배 척수후근신경절 배양에서 수초 형성을 보았고 1) 래트의 척 수후근신경절 배양은 10 년이 지나서 1967 년 벙기 Bun g e 에 의해서 보고되었다 .2) 그 이후로 래트와 마우스의 후근신경절 배양은 여 러 학자들에 의해서 사용되었다. 래트나 마우스의 척수후근신경 절의 배양을 할 때 태생 중기가 좋은지 혹은 태생 후기가 좋은지 연구실에 따라 그 의견이 다르다. 후근신경절 이식배양의 경우에 는 척수배양에 가장 적합하다고 하는 태생 14-16 일이 이상적이 다. 이 시기에는 척수를 분리할 때에 요수와 천수 부분에 후근신 경철이 부착되어 있으므로 척수와 후근신경절의 이식편을 함께 배양할 수 있다. 한편 분리세포 배양의 경우에는 후근신경절을 하나씩 분리하기 쉬운 태생 16-18 일이 이상적이다. 분리세포 배 양의 경우에는 임신 동물의 임신 여부, 임신 일자가 확실치 않은 수가 많으므로 확실히 그 나이를 알 수 있는 신생아를 사용하여
도 좋은 결과를 얻을 수 있으므로 신생아 사용도 가능하다. 14.1 태생 래트(마우스) 척수후근신경절의 분리세포 배양 〈 준비 〉 1) 입체해부현미경과 조명 2) 수술기구 10cm 의과용 가위(직선과 곡선) 2 개 10cm 의과용 핀셋 2 개 시계용 핀셋 (No. 4) 2 개 수술용 칼 (No.11) 2 개 3) 시험관 , 페트리 접시, 원심튜브 4) 혈구계산판 5) 칼슘, 마그네슘 제거 핸크스 영류 용액 6) 2.5 % 트립신 7) 0.2 % DNase 효소액 8) 이글 MEM 합성 배양액 9) 마혈청 10) 50% 포도당액 11) 2 mg /m l 겐타마이신액 12) 10 µg/ ml NGF (신경 성 장인자) 13) 5 mg /m l 아스코르브산 (비 타민 C) 〈방법〉 1) 16 일 -18 일 래트와 마우스를 에텔자 속에 넣고 심마취에 의 해서 마취사시킨다.
2) 래트의 등을 아래로 하여 눕히고 배 부분을 70 % 알코올로 소독한다. 3) 배의 피부를 의과용 핀셋으로 들어울리고 배의 정중선을 따 라서 하복부를 가위로 크게 절개한다. 4) 자궁이 노출되면, 자궁의 경관부와 난관부를 절단하고 태아 가 들어있는 기다란 색 형상의 자궁을 들어내서 핸크스액 이 들어있는 10cm 페트리 접시로 옮긴다. 5) 자궁근막을 절개하고 6-12 마리의 태아를 분리한다. 6) 의과용 가위로 태아의 머리를 자르고, 등부분의 피부를 제 거한다. 척주와 척수를 포함한 태아의 후배조직을 크게 절단 하고 새 10cm 페트리 접시에 옮기고 척주 전면에 붙은 내 장을 제 거 한다 (그림 14-1) . 7) 등을 위로 하여 입체해부현미경 아래 놓는다. 10cm 곡선 의과용 가위를 사용하여 척주후배부의 근육과 척추골을 잘라 내고 척수를 노출시킨다. 척수 조직을 제거하면 경수에서 천 수에 이르는 사이에 30 쌍의 척수후근신경절이 양측에 나란히 놓여 있다. (그립 14-1) 8) 시계용 핀셋을 사용하여 조심스럽게 후근신경절을 하나씩 분리하고 CMF- 핸크스액이 들어있는 6cm 페트리 접시에 모은다. 10 마리 태아에서 400 개 이상의 신경절을 분리할 수 있다. 신경절을 모두 분리하는 동안 아직 처리하지 아니한 태아나 이미 분리된 신경절이 들어있는 페트리 집시를 얼음 위에 보존한다. 9) 모은 신경절을 소량의 핸크스액과 같이 15ml 원심튜브에 옮긴 뒤 여분의 핸크스액을 버린다. 듀브에 4.3ml CMF 핸 크스액, 0.5 ml 2.5 % 트립신액, 0.2 ml 의 0.2 % DNase 액 울 차례로 가하고 37 도에서 30 분간 인큐베이트한다. 5 분마다
 (a)
(a)
듀브를 꺼내어서 혼합이 잘 되도록 혼든다. 10) 신경절을 튜브 바닥에 남긴 채 트립신액을 뽑아버린다. 10% 마혈청이 들어있는 배양액 4ml 를 가하고 파스출 피펫 으로 20 번 상하운동에 의한 피페팅을 한다. 다음에 피펫 끝 에 멸균된 미크로피펫팁을 끼우고 20 번 피페팅을 되풀이한 다. 이 시점에서 세포의 분리가 잘 되어있을 것이나, 만일 조직편이 남아있으면 듀브를 3 분간 정치하고 세포가 부유하 고 있는 상청액울 새 튜브에 옮기고 남은 조직에 배양액 2 ml 를 가한 뒤 위에 적은 바와 같이 피페팅을 되풀이한다. 11) 세포부유액을 한 듀브에 모으고 1000r p m 에서 8 분간 원심 분리한다. 12) 상청액을 버리고 페렛에 배양액 3ml 를 가하고 1000r p m 에 서 8 분간 원심분리한다. 13) 상청액을 버리고 페렛에 배양액 5ml 를 가하여 세포를 부유 시킨 뒤 미리 준비해 두었던 폴리라이신 코팅한 커버글라스 위에 플레이팅한다(커버글라스는 6cm 페트리 접시에 놓는다). 400 개의 신경절에서 4Xl07 세포가 분리된다. 배양액의 조성 은 다음과 같다. 이글 MEM 86ml 마혈청 (혹은 우태아혈청) 10 ml 50 % 포도당액 1ml 2 mg /m l 겐타마이신액 1 ml 10 µg/m l NGF( 신경성장인자) 1 ml 5 mg /m l 아즈쿠다 산 1 ml 14) 다음날 3ml 의 배양액을 추가하여 페트리 접시를 채운다. 일주일에 두 번 배양액을 교환한다.
〈 해설 〉 계배 척수후근신경절 배양과 마찬가지로 신경조직 배양에서 가 장 널리 사용되는 배양 모델이 태생 래트와 마우스 척수후근신경 절 배양이다. 최근에는 이식편 배양보다 분리세포 배양을 사용하 는 연구자가 증가하고 있다. 계배 신경절 배양에서와 같이 신경 영양인자 연구에 커다란 역할을 하고 있다. 그림 14-2 와 14 - 3 에 서는 분리세포 배양 2 주일이 된 태생마우스 척수후근신경절 신경 세포를 보이고 있다.
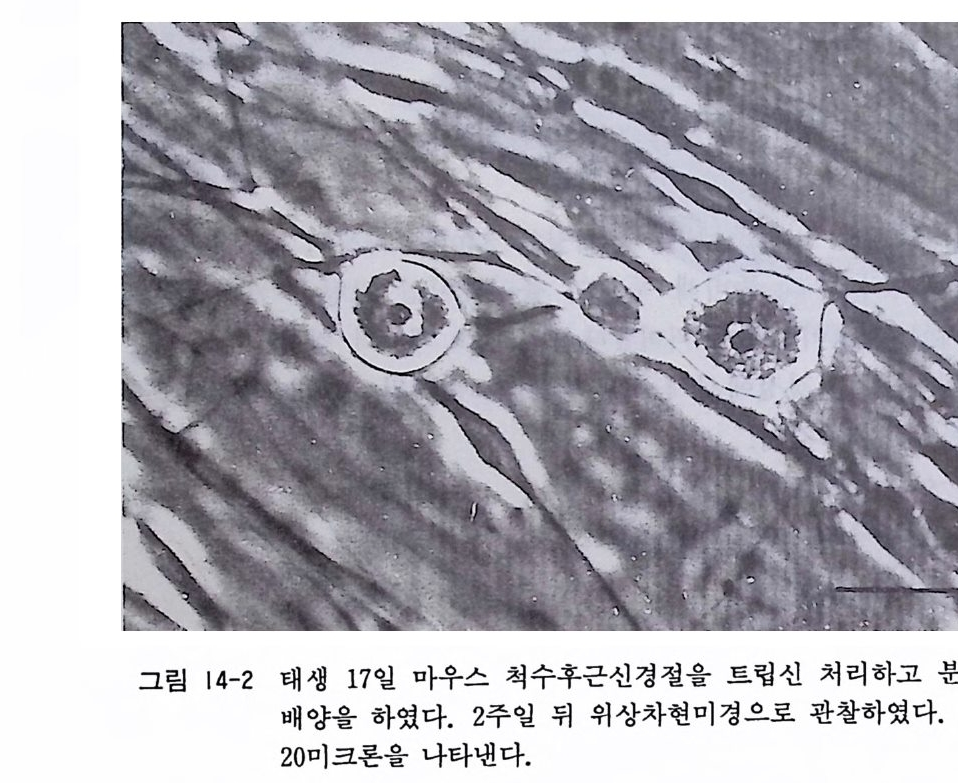 그림 14-2 태생 17 일 마우스 척수후근신경절을 트립신 처리하고 분리세포
그림 14-2 태생 17 일 마우스 척수후근신경절을 트립신 처리하고 분리세포
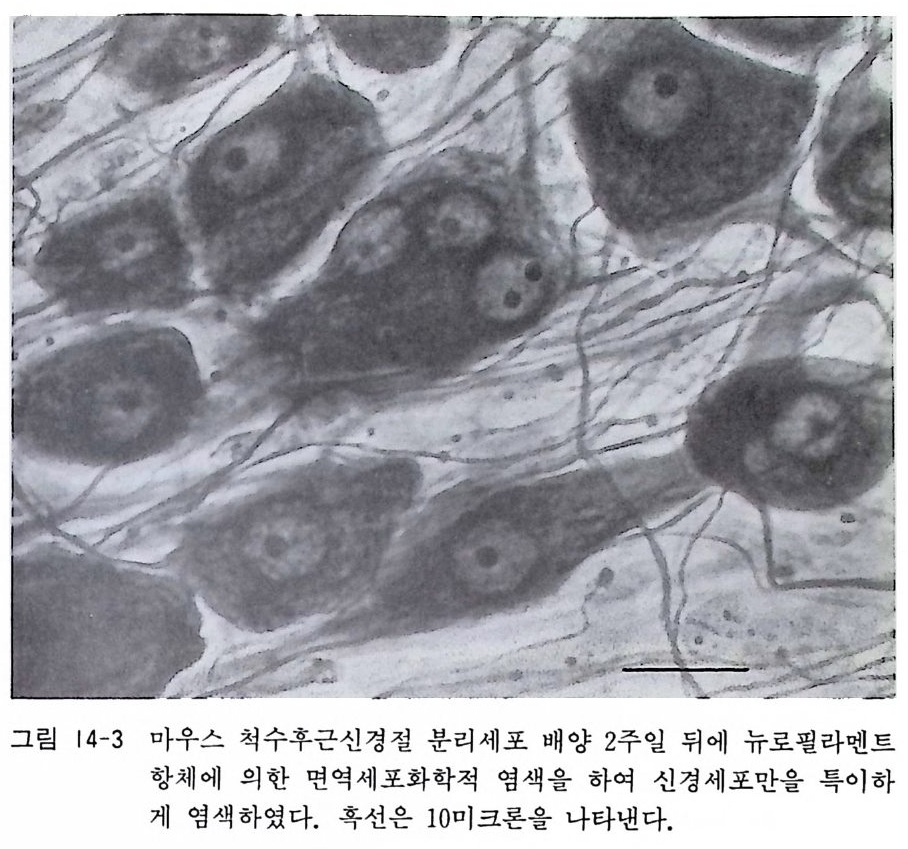 그립 14- 3 마우 스 척 수 후근 신경 절 분 리세포 배양 2 주 일 뒤에 뉴로필라멘트
그립 14- 3 마우 스 척 수 후근 신경 절 분 리세포 배양 2 주 일 뒤에 뉴로필라멘트
14.2 페트리 접시에 의한 태생 래트(마우스) 척수후 근신경절의 이식편 배양 〈 준닌 1 〉 14.1 과 준비는 같다. 〈 방법 〉 1) 14.1 순서 1)-8) 에 따라서 후근신경절을 16-18 일 래트나 마
우스 태아에서 분리한다. 2) 직경 3.5cm 의 페트리 접시를 사용하되, 그 뚜껑을 바닥으 로 그리고 접시 바닥을 뚜껑으로 뒤집어서 쓴다. 페트리 접 시 중앙에 콜라겐을 발라놓은 22mm 직경의 원형 커버글라 스를 위치시키고 배양액 한 방울(0 .05ml) 을 떨어뜨린다. 3) 래트 태아 척수후근신경절 2 개를 5mm 의 간격을 두고 커버 글라스 위에 올려놓는다. 파라필름이나 데이프를 사용하여 밀 봉할수도 있다. 4) 페트리 접시의 바닥을 뒤집어서 뚜껑으로 해서 덮는다. 5) 밀폐계를 만들기 위해서 페트리 접시 뚜껑과 바닥 사이를 60 도 온수로 덥혀 녹인 파라핀 - 바셀린 혼합액 (2 : 1) 으로 밀 폐한다. 서예용 봇을 사용한다. 6) 한 실험에 10 개에서 20 개의 페트리 접시 실험계를 만드는데 이들을 운반하기에 편리하도록 25cmX35cm 크기의 플라스 틱 쟁반에 놓고 섭시 37 도 인큐베이터에서 정치한다. 7) 일주일에 2 번 배양액을 교환한다. 배양액은 다음과 갇다. 이글 MEM • 76ml 마혈청 (혹은 우태아혈청) 20ml 10 µg/m l NGF( 신경성장인자) 1 ml 5mg /m l 아人구타산 1ml 50% 포도당액 1ml 2 mg /m l 겐타마이신액 1 ml 〈해설〉 말초신경계에서의 미엘린 형성을 위해서는 이 배양시스템이 가 장 이상적이다 .2) 다수의 연구가 1970 년과 80 년대에 나왔는데 최 근에는 분리세포 배양에 밀려서 그림자가 흐려진 감이 있다• 최
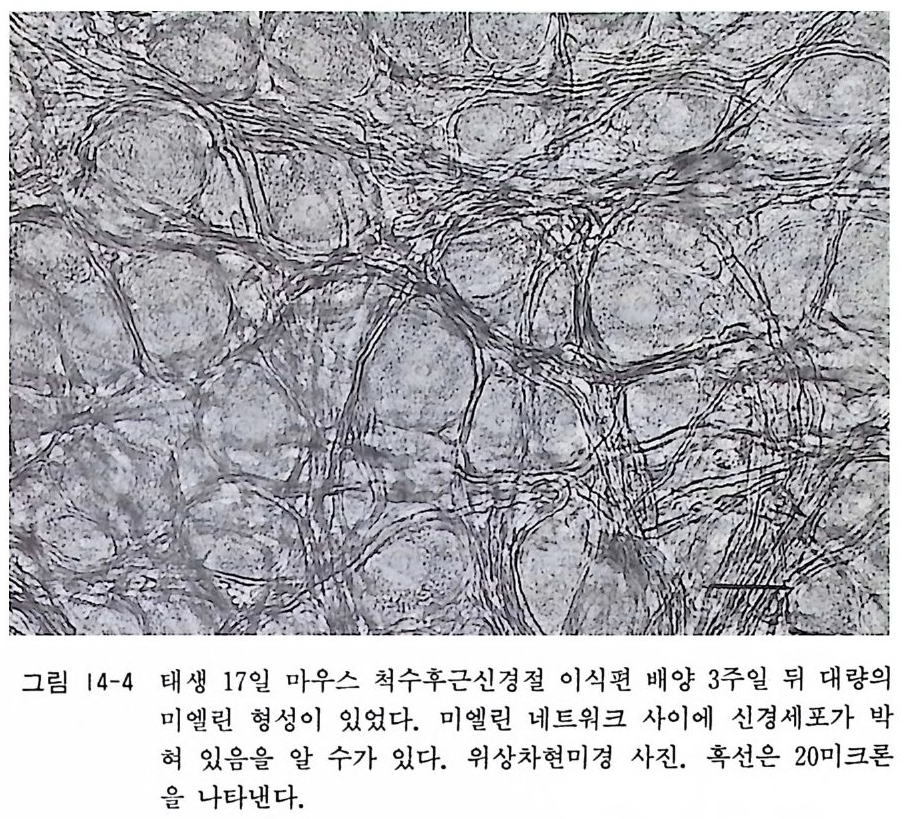 그림 14-4 태생 17 일 마우스 척수후근신경절 이식편 배양 3 주일 뒤 대량의
그림 14-4 태생 17 일 마우스 척수후근신경절 이식편 배양 3 주일 뒤 대량의
근에는 신경영양인자 연구에 널리 사용되고 있다. 그림 14-4 에서 는 태생 래트 척수신경절 이식편 배양 3 주에서 보는 신경세포와 미엘린 형성을 보이고 있다.
14.3 신생 래트와 마우스 교감신경절의 이식편 배양 〈 준비 〉 14.1 태생 래트 후근신경절 분리세포 배양과 같다. 〈 방법 〉 1) 신생 래트나 마우스를 에텔자에 넣어서 심마취로 마취사시 킨다. 혹은 -20 도 냉장고에서 10 분간 냉온마취 를 한다. 2) 상경교감신경절 sup e rio r cerv ica l g an gli on 은 반월교감신경절 semi lun ar ga ng lion 이라 불리기도 한다. 좌우 한쌍이 있어서 목의 경동맥 근처에 위치한다. 신생 래트를 코르크판이나 스 티로폼 판 위에 눕혀서 손발을 핀으로 고정한다. 3) 신생 래트 머리에서 가슴까지 70 % 알코올로 소독한 뒤 목 부분 피부를 제거한다. 목 중앙에 기관이 보이고 그 좌우로 턱에서 흉쇄골에 걸쳐서 비스듬히 흉쇄근이 있다. 이 흉쇄근 을 찰라내면 그 밀에 총경동맥 caroti d ar t er y이 있다. 총경동 맥의 주행을 위로 거슬러올라가면 총경동맥 분기부가 있다. 이 분기부 바로 아래에 상경교감신경절이 위치하므로 이를 시계용 핀셋으로 분리한다(그립 14-5). 신생아 10 마리에서 20 개의 교감신경절을 분리한다. 4) 복부교감신경질은 상경교감신경절과 마찬가지로 동물의 등 을 아래로 눕히고 적출한다. 신생 래트의 피부와 근육을 제 거한 뒤 내장도 모두 제거한다. 중앙부에 대동맥과 대정맥이 보이는데 이 두 혈관을 제거하면 그 아래에 좌우로 두 줄의 교감신경절 체인이 보인다. 이들 교감신경질 체인을 분리하 여 핸크스액이 들어있는 페트리 접시에 놓는다. 5) 14.2 순서 2)-7) 에 따라서 페트리 접시 이식편 배양을 시작
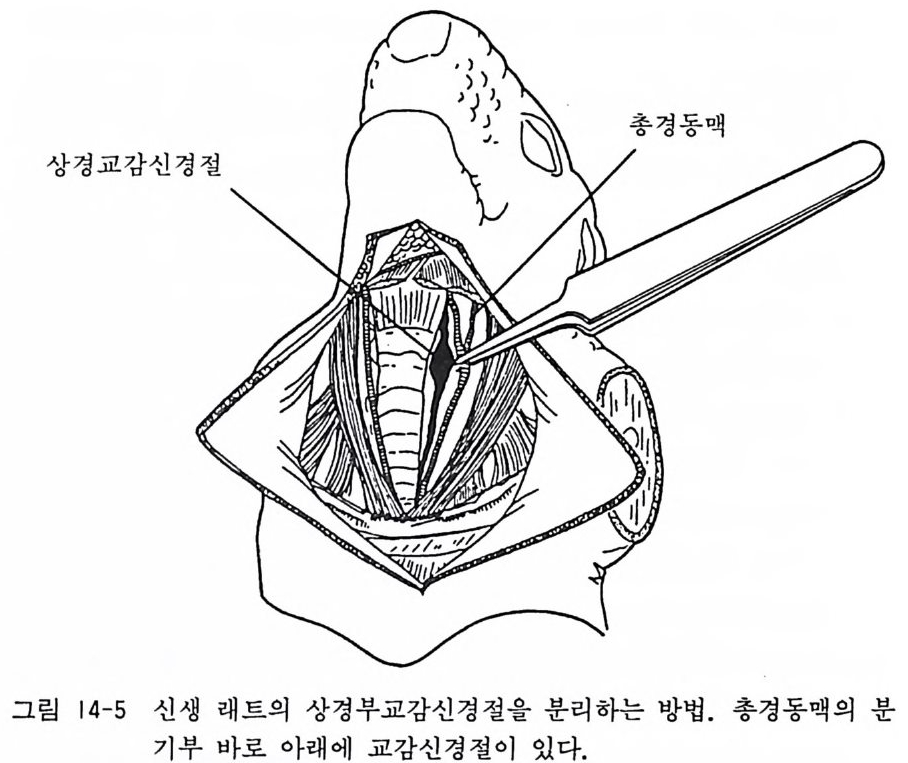 그립 14-5 신생 래트의 상경부교감신경절을 분리하는 방법. 총경동맥의 분
그립 14-5 신생 래트의 상경부교감신경절을 분리하는 방법. 총경동맥의 분
한다. 〈 해설 〉 카데콜아민 신경세포의 신경생리학, 생화학, 약리학 분야에서 널리 사용되는 실험모델이다 .3 , 4) 신경영양인자 연구에서도 최근 많이 사용되고 있다.
14.4 신생 래트와 마우스 삼차신경절의 이식편 배양 〈 준비 〉 14.1 태생 래트 후근신경절 분리세포 배양과 같다. 〈방법〉 1) 신생 래트를 심마취로 마취사시키고 비커에 담긴 70 % 알 코올에 잠시 담가서 소독하고 10cm 페트리 접시에 옮긴다. 2) 머리의 피부를 제거한 뒤 두 안구 사이에 가위 를 넣어서 좌 우로 전개하듯이 두개골을 제거한다. 후뇌에서 뇌간까지의 뇌를 끄집어낸다. 3) 두개 중앙부에 위치한 뇌하수체 좌전방과 우전방에 삼차신 경 신경절 tri g e mi na l ga ng li o n 이 보인다. 삼차신경의 팽대 부에 3 개의 신경절이 있으므로 그 앞뒤를 가위로 절단하고 신경절을 분리한다. 4) 14.2 의 순서 2)-7) 을 따라서 페트리 접시 이식편 배양을 시 작한다. 5) 페트리 접시 배양 대신에 6 혈 멀티웰 챔버에서 배양할 수도 있다. 멀티웰 6 개 속에 커버글라스를 위치시키고 이식편과 소량의 배양액을 올려놓고 이식편이 콜라겐에 밀착하도록 24 시간 기다린다. 24 시간 뒤 각 웰 속에 1ml 의 배양액을 추가 하고 배양을 시작한다. 일주일에 2 번 배양액을 교환한다.
제 |5 장 태생 및 신생아 래트와 마우스 중추신경계 조직 • 세포의 배양 1950 년대에 들어서면서 머레이 Murray, 포메라트 Pornera t, 럼 스덴 Lumsden, 힐트 Hi ld, 본스틴 Bornste i n 등의 여 러 학자들에 의하여 태생 및 신생아 포유동물의 중추신경 조직의 이식편 배양 이 다수 보고되었다. 1957 년에 힐트가 신생아 고양이 소뇌 ,1) 1958 년에는 본스틴과 머레이가 신생아 고양이와 래트 소뇌 배양 에서 처음으로 미엘린(수초) 형성을 관찰하였는데 이후로부터 포 유동물 소뇌가 배양 재료로서 많이 쓰이게 되었다 .2> 1960 년대와 70 년대에는 마우스와 래트 소뇌 이식편 배양에서 수초 배양과 신 경세포의 발육과 분화를 광학현미경과 전자현미경으로 관찰하여 신경조직배양의 초기 황금시대를 이루었다. 이때 활약한 학자들 로서는 김 Ki rn , 울프 Wol f, 알레랑 Allerand, 사일 Seil , 헨델만 Hendelrnan, 프리 바 Priv a t 등이 있다. 3-8) 척수 이식편 배양은 소뇌 배양보다 늦게 시작되어서 1965 년에 벙기 Bun g e 가 태생 래트 척수배양을 그리고 소브코비츠 Sobk ow ic z 가 1968 년에 태생 마우스 척수배양을 하여 척수신경세포의
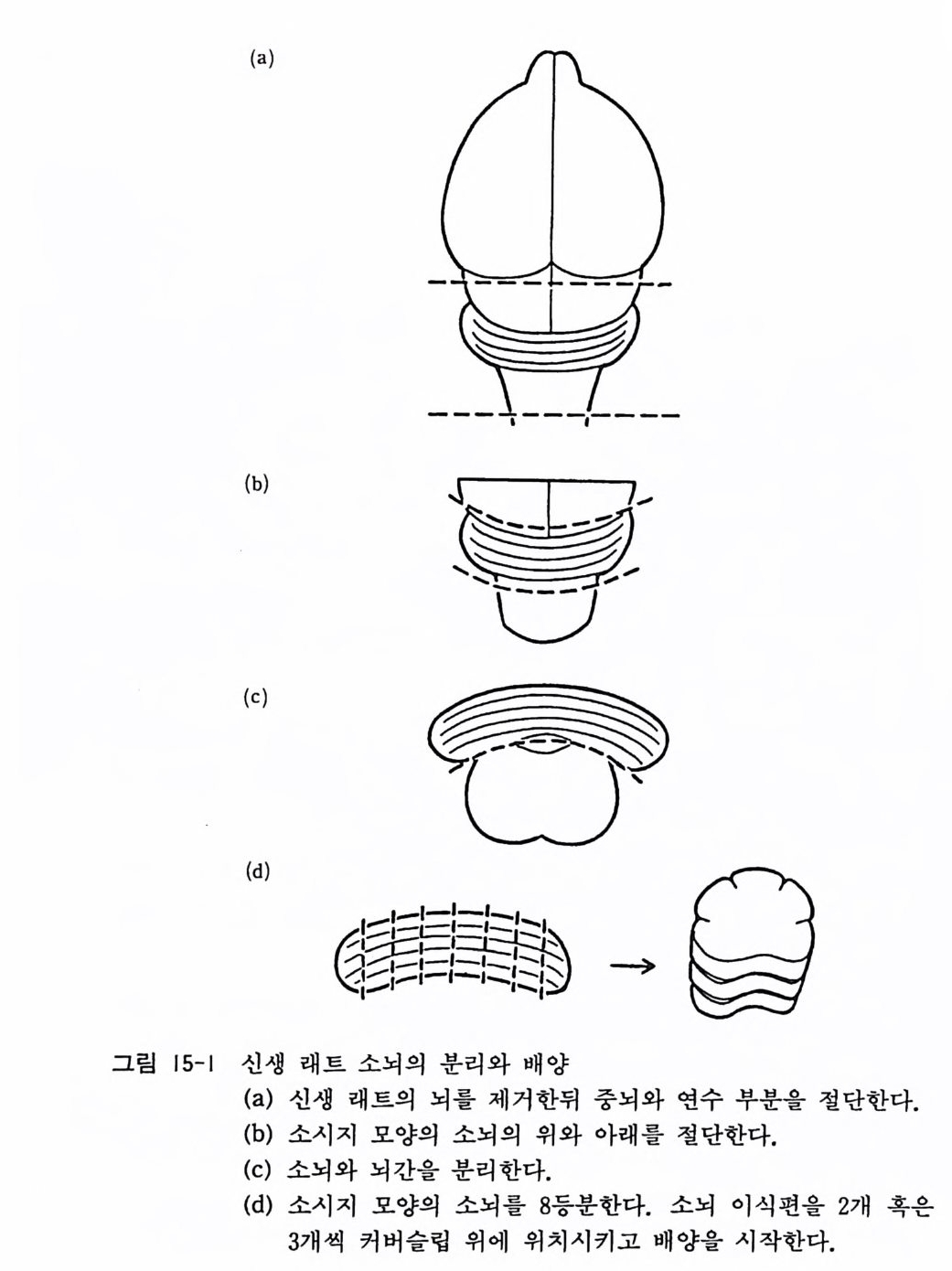
발달을 광학 및 전자 현미경으로 관찰한 바 있다 .9,IO) 1970 년대에 들어서서는 소뇌와 척수 이의에도 대뇌피질, 해마, 후구 olfa c - to ry bulb, 시 상하부 hy po th a lamus 동 중추신경 조직 의 각 부위 도 이식편 배양에 사용되었다. 1980 년대에 들어서서는 중추신경조직 이식편 배양은 분리세포 배양이 성행함에 따라서 별로 환영을 받 지 못하게 되었는데 이것은 기술적으로 간단하고 실험 결과의 분 석과 해석이 단순명쾌하게 나오는 분리세포 배양에 연구자들이 한층 더 매력을 느꼈기 때문이다. 여기에서는 먼저 신생 래트와 마우스 소뇌의 이식편 배양, 이어서 신생 래트 대뇌의 이식편 배 양과 태생 래트 척수 이식편 배양에 대하여 기술하겠다. 계속해 서 태생 래트 척수 분리세포 배양, 태생 마우스 혹질 분리세포 배양, 태생 래트 해마 분리세포 배양에 대한 소개를 하겠다. 15.1 신생 래트(마우스) 소뇌 이식편 롤러튜브 배양법 〈준비〉 1) 입체해부현미경과 조명 2) 수술기구 -10 cm 의과용 가위 (직선과 곡선) 2 개 10 cm 의과용 핀셋 2 개 시계용 핀셋 2 개 수술용 칼 (No. 11) 2 개 3) 페트리 집시, 원심튜브 4) 16mmx125mm 시험관(스크류캡이 부착되어 있음) 5) 롤러드럼 6) 시험관 래크 7) 22mmxnmm 장방형 커버글라스
8) 이글 MEM 합성 배양액 9) 마혈청 10) 50 % 포도당액 11) 2mg /m l 겐타마이신액 〈 방법 〉 1) 신생 래트 혹은 마우스를 에텔자에 넣어서 심마취로 마취사 시킨다. 혹은 ― 20 도 냉장고에서 10 분간 냉온마취를 한다. 2) 비커에 담긴 70 % 알코올에 신생 래트를 잠시 담가서 소독 시키고 10cm 페트리 접시로 옮긴다. 3) 머리의 피부를 제거한 뒤 두 안구 사이에 가위를 넣어서 좌 우로 전개하듯이 두개골을 제거한다. 후뇌에서 척수 상부까 지의 뇌를 꺼낸다. 이때 연수 위에 위치한 소시지 모양의 소 뇌를 상하지 않도록 조심해서 뇌를 제거하고 핸크스액이 든 페트리 접시에 옮긴다. 4) 현미경 아래에서 수술칼 (No . 11) 2 개의 칼날을 교차시키는 조작으로 대뇌와 중뇌를 절단한다. 소뇌가 아래로 오도록 중 뇌를 놓고 시계용 핀셋으로 뇌막을 제거한다. 5) 소시지 모양의 소뇌를 뇌간으로부터 분리하고 하나의 소뇌 를 8 등분으로 해서 슬라이스를 만든다(소뇌의 분리는 그립 15-1 참고). 6) 직경 10cm 의 페트리 접시 바닥에 미리 콜라겐울 발라둔 22mmXllmm 장방형 커버글라스를 10 장을 간격을 두고 위 치시킨다. 배양액 한 방울을 그 위에 떨어뜨려서 건조하게 되는 것을 막는다. 멸균된 유리봉으로 배양액이 고르게 퍼지 도록한다. 7) 소뇌 소편을 2 개나 3 개를 3mm 간격을 두고 커버글라스 위
 (a)
(a)
에 울려놓는다. 8) 페트리 접시 뚜껑을 덮고 30 분간 인큐베이터에서 정치한다. 9) 파스출 피펫으로 커버글라스 위의 여분의 배양액을 제거한 뒤 커버글라스를 16mm x 125mm 의 시험관에 넣는다. 이때 주의할 것은 이식편이 커버글라스에서 미끌어져서 떨어지지 않도록 주의하여 옮겨야 한다. 이식편이 위를 향하도록 하고 시험관을 시험관 래크 위에 놓는다. 10) 시험관 속에 배양액 0.7ml 를 서서히 주입한다. 이식편이 떨어지는 경우에는 파스출 피펫으로 이것을 집어내고 커버슬 립 위에 주의깊게 다시 울려놓는다. 11) 스크류캡으로 시험관 입을 꼭 잠근 다음, 시험관을 시험관 래크에 나란히 위치시키고 37 도 인큐베이터에서 24 시간 정치 시킨다. 다음날 시험관을 롤러드럼에 삽입하고 1 시간에 12 번 회전을 시킨다. 롤러드럼은 37 도 인큐베이터에 장치한다. 12) 일주일에 한번 배양액 교환을 한다. 스크류캡을 연 다음 배 양액을 파스출 피펫으로 제거하고, 새 배양액 0.7ml 를 첨가 한다. 롤러튜브 배양법의 실제는 그림 14-1 을 참고할 것. 배양액 조성은 다음과 같다. 이 글 MEM (핸크스 용액 포함) 78 ml 마혈청 20ml 50% 포도당액 1ml 2mg /m l 겐타마이신액 1ml 〈 해설 〉 소뇌 이식편 배양의 상세에 대하여서는 문헌 1-8 을 참고하기 바란다. 래트 소뇌 이식편 배양을 시작하여 3-4 일이 지나면 이식 편 주위에 방사적으로 아스트로사이트와 섬유아세포가 밖으로 퍼
져나온다. 이들 아스트로사이트나 섬유아세포 시트 위를 소뇌의 소형 신경세포인 과립세포g ranule cell 가 다수 유출해 나온다. 배 양 2 주일부터 미엘린(수초) 형성을 보게 되는데 탈수질환인 다발 성 경 화증 multip le sclerosis 이 나 실 험 적 알레 르기 성 뇌 척 수염 exp e rim enta l allergi c encep h aliti s 혈 청 을 이 러 한 배 양에 첨 가하면 전형적인 탈수병변을 볼 수 있다. 신경세포의 확인을 위해서는 칼빈딘 calbin d in 이나 파발부민 pa rvalbumi n 과 같은 칼슘 결합성 단백질에 대한 항체를 사용하여 푸르키니에 세포 Purk i n j e cell 확 인을 할 수가 있으며 신경세포 특이항원인 뉴로필라멘트 neu ro fil amen t나 MAP2(mi cr otu b ule assoc iat e d pro te i n - 2 ) 에 대 한 항
 그림 |5-2 신생 마우스 소뇌 이식편 배양 3 주일. 신경세포를 둘러싸고 미
그림 |5-2 신생 마우스 소뇌 이식편 배양 3 주일. 신경세포를 둘러싸고 미
체를 사용하여 면역세포화학적으로 신경세포 전반을 동정할 수가 있다. 고전적인 은염색인 보디안 Bod i an 법이나 홈즈 Holms 법으로 역시 푸르키니에 세포나 소뇌핵신경세포가 확인될 수 있다. 그림 15-2 에서는 위상차 현미경 아래에서 신경세포와 미엘린 형성을 보이고 있다. 그립 15-3 에서는 보디안 은영색에 의한 소뇌신경세 포의 확인을 보이고 있다.
 그림 15-3 신생 마우스 소뇌 이식편 배양 3 주일. 보디안 은염색법에 의해
그림 15-3 신생 마우스 소뇌 이식편 배양 3 주일. 보디안 은염색법에 의해
15.2 신생 래트(마우스) 대뇌와 선조체 이식편 멀티 웰-커버글라스 배양법 〈 준비 〉 15.1 과 같다. 〈 방법 〉 1) 15.1 에서 기술한 바와 같이 순서 1)-4) 를 되 풀 이한다. 2) 현미경 아래에서 수술칼 (No . 11) 2 개의 칼날 을 교차시키는 조작으로 대뇌의 전뇌부fr on t al lobe 를 2mm 의 두께로 절단 한다. 그림 15-4 에서와 같이 절단한 대뇌조직을 옆으로 눕히 고 대뇌피질을 부채 모양으로 조그마하게 절단한다. 이식편 두께는 1mm 와 2mm 의 중간으로 한다. 대뇌피질 내측에는 선조체 str i a t u m 가 있으므로 이 것 역 시 1-2 mm 두께 의 이 식 편으로 철단한다. 3) 웰이 6 개 있는 멀티웰 바닥에 미리 콜라겐을 발라둔 직경 22mm 의 원형 커버글라스를 위치시키고 배양액 한 방울을 그 위에 떨어뜨린다. 멸균된 유리봉으로 배양액이 고르게 퍼 지도록 한다. 4) 대뇌 이식편 혹은 선조체 이식편 2 개를 3mm 간격을 두고 커버글라스 위에 울려놓는다. 5) 멀티웰 커버를 덮고 5 % CO2 인큐베이터에서 2 시간 정치 한다. 이 기간에 이식편이 콜라겐에 부착한다. 6) 배양액 1ml 를 서서히 멀티웰에 첨가하고 5 % CO2 인큐베 이터에서 배양한다. 일주일에 한 번 혹은 두 번 배양액 교환 울한다. 배양액 조성은 다음과 갇다.
 (a) 五/또 三 E 또군군
(a) 五/또 三 E 또군군
이글 MEM (얼용액 포함) 78 ml 마혈청 20ml 50% 포도당액 1ml 2 mg /m l 겐타마이신액 1 ml 〈해설〉 신생 래트나 마우스 대뇌피질의 배양을 개시하는 시접에서는 아직도 신경 분화가 미숙해서 시냅스 형성도 없으며 미엘린 형성 도 시작되고 있지 않다. 배양을 시작한 후 2-3 주일이면 시냅스와 미엘린 형성이 진척되어 있어서 조칙 배양중에서 신경세포가 분 화를 계속하고 뉴론-글리아 상호작용도 계속하고 있음을 알 수가 있다. 신생 래트나 마우스 대뇌피질 신경세포의 형태학적인 혹은 전 기생리학적 발달에 대하여서는 크레인과 본스틴 Cra i n & Bornste i n , 김 Ki m , 사일 Se il이 이 미 보고한 바 있다. 11-13) 15.3 태생 래트(마우스) 척수 이식편 멀티웰-커버글 라스배양 〈준비〉 14 . 1 에서 기술한 태생 래트 척수후근신경절 배양과 갇다. 〈방법〉 1) 14.1 에서 기술한 바와 같이 순서 1)-7) 에 따라서 척수를 16-18 일 래트나 마우스 태아에서 분리한다(그립 14-1 참고). 연구자에 따라서는 13-14 일 태아에서 척수분리를 하는 경우
가있다. 2) 현미경 아래에서 수술칼 (No.11) 2 개의 칼날을 교차시키면 서 척수를 1mm 두께의 원반형 이식편으로 자론다. 3) 15.2 에서와 마찬가지로 6 웰 멀티웰 바닥에 22mm 원형 커 버글라스(미리 콜라겐을 발라둔다)를 놓고 그 위에 배양액 한 방울을 떨어뜨린다. 커버글라스 위에 척수 이식편 2 개를 3 mm 간격을 두고 올려놓는다. 4) 멀티웰 커버를 덮고 2 시간 정치한다. 이 기간에 이식편이 콜라겐에 부착한다. 5) 배양액 1 ml 를 멀티웰에 첨가하고 5 % CO2 인큐베이터에 서 배양한다. 일주일에 한 번 혹은 두 번 배양액 교환을 한 다. 배양액 조성은 다음과 같다. 이글 MEM( 얼용액 포함) 78ml 마혈청 20ml 50 % 포도당액 1ml 2mg /m l 겐타마이신액 1ml 〈해설〉 저자의 연구실에서는 맥시모우 슬라이드법이나 롤러튜브법으로 태생 래트나 마우스 척수 이식편 배양을 하여 일년 가까이 장기 배양을 한 바 있으며 광학현미경이나 전자현미경 아래에서 척수 신경세포의 형태적 발달을 보고한 논문에는 벙기 Bung e, 소브코 비 츠 Sobkowi cz, 김 Ki m 등이 있다. 9,10,U) 또한 크레 인 Cra i n 은 배 양 척수 신경세포의 전기생리학적 특칭울 보고하였다 .15) 태생 마 우스 척수 이식편 배양에서 본 광학현미경, 그리고 전자현미경에 의한 관찰은 그립 15-5, 15-6, 15-7 에서 보이고 있다.
 그림 15-5 태생 16 일 마우스 척수 이식편 맥시모우 슬 라이드 배양 4 주 일.
그림 15-5 태생 16 일 마우스 척수 이식편 맥시모우 슬 라이드 배양 4 주 일.
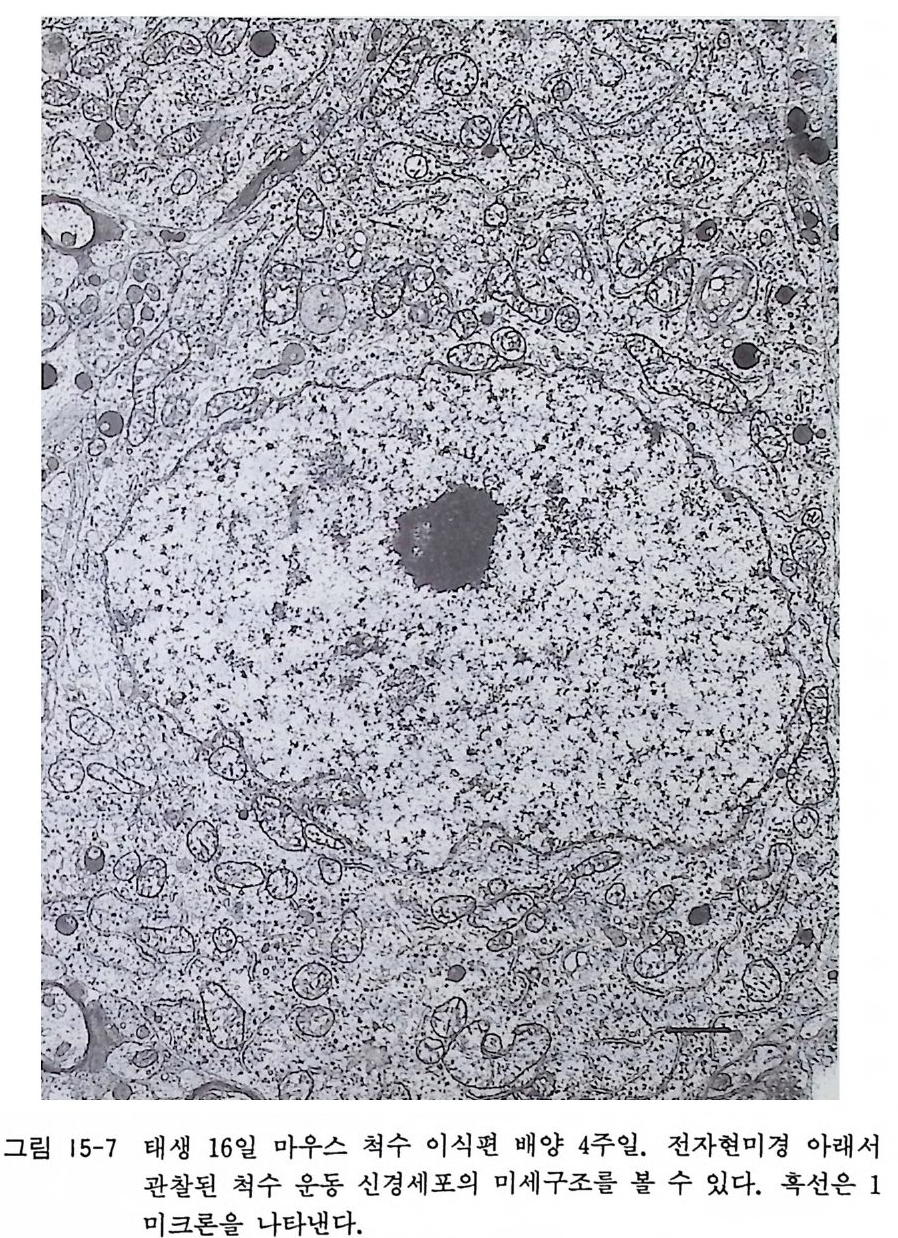 그림 15-7 태생 16 일 마우스 척수 이식편 배양 4 주일. 전자현미경 아래서
그림 15-7 태생 16 일 마우스 척수 이식편 배양 4 주일. 전자현미경 아래서
15.4 태생 래트(마우스) 척수 분리세포 배양 〈준비〉 14.1 의 태생 래트 척수후근신경절 배양과 갇다. 〈방법〉 1) 14.1 에서와 같이 순서 1)-7) 에 따라서 척수를 13 - 14 일 래트 나 마우스 태아 혹은 16-18 일 태아 6-10 마리에서 분리한다 (그림 14-1 참고) . 2) 현미경 아래에서 수술칼 (No.11) 2 개의 칼날을 교차시키면 서 척수를 1mm 두께의 조칙 소편으로 자른다. 3) 소량의 핸크스액과 같이 15ml 원심튜브에 옮간 뒤 여분의 핸크스액을 버린다. 듀브에 4.3 ml CMF- 핸크스액, 0.5 ml 의 2.5 % 트립신액, 0.2 ml 의 0.2 % DNase 액을 차례로 가하고 37 도에서 30 분간 인큐베이트한다. 5 분마다 튜브를 꺼 내어서 혼합이 잘 되도록 흔든다. 4) 척수 조직을 튜브 바닥에 남긴 채 트립신을 뽑아내어 버리 고 10% 마혈청이 들어 있는 배양액 4ml 를 가하고 파스출 피펫으로 20 번 상하운동에 의한 피페팅을 한다. 다음에 피펫 끝에 미크로피펫팁을 끼우고 피펫팅을 20 번 되풀이한다. 이 .시점에서 세포의 분리가 찰 되어있을 것이나 만일 조직편이 남아 있으면 튜브를 3 분간 정치하고 세포가 부유하고 있는 상청액을 새 튜브에 옮기고 남은 조직에 배양액을 2ml 를 가 한 뒤 위와 같이 피페팅을 되풀이한다. 5) 세포부유액을 한 튜브에 모으고 1000 r p m 에서 8 분간 원심 분리한다. 6) 상청액을 버리고 페렛에 배양액 5ml 를 가하여 세포를 부유
시킨 뒤 미리 준비해 두었던 폴리라이신 코팅한 커버글라스 위에 풀레이팅하고 5% CO2 인큐베이터에서 배양한다. 커 버글라스는 6cm 페트리 접시 바닥에 간격을 두고 위치시킨 다. 10 마리 태아에서 2X107 세포가 분리된다. 배양액의 조 성은 다음과 같다. 이글 MEM 88ml 마혈청 (혹은 우태아혈청) 20 ml 50% 포도당액 1ml 2 mg / ml 겐타마이 신액 1 ml 7) 다음날 3ml 의 배양액을 추가하여 페트리 접시를 채운다. 일주일에 두 번 배양액을 교환한다. 〈 해설 〉 근위 축성 척 색 경 화증 amy o tr o p h ic late r al sclerosis 은 신경 변성 질환의 하나로서 현재까지 불치의 병으로 알려져 있다. 이러한 척수운동신경 질환의 모델로서 널리 사용되고 있는 것이 태생 래 트나 마우스의 척수 배양이다. 병의 원인으로서는 여러 가지가 거론되고 있는데 아직도 결정적인 것이 없다. 저자의 연구실에서 는 산소 라디칼에 의한 독성으로 척수운동신경이 사멸한다는 실 험을 하였고 이에 대하여서는 수퍼옥사이드 디스뮤타제 SOD 나 카탈라제 ca t alase 와 같은 항산화효소가 이러한 신경세포의 사멸 울 방지한다는 보고를 한 바 있다 .16) 척수신경세포 배양은 또한 척수운동 신경세포 성장과 생존에 특이하게 작용하는 신경영양인 자 연구에 역시 널리 옹용되고 있다. 17-20)
15.5 태생 래트(마우스) 혹질 분리세포 배양 〈 준비〉 14.1 에서 기술한 태생 래트 척수후근신경절 배양과 갇다. 〈 방법 〉 1) 14.1 에서와 같이 순서 1)-5) 에 따라서 13-14 일 래트나 마우 스 태아 6-10 마리를 분리한다. 2) 현미경 아래에서 시계 수선용 핀셋을 사용하여 태아 머리부 분에서 먼저 피부를 벗기고 다음에 막갇이 엷은 두개골을 제 거한다. 3) 그립 15-8 에서 보이듯이 중뇌가 노출되면 그 윗부분 반을
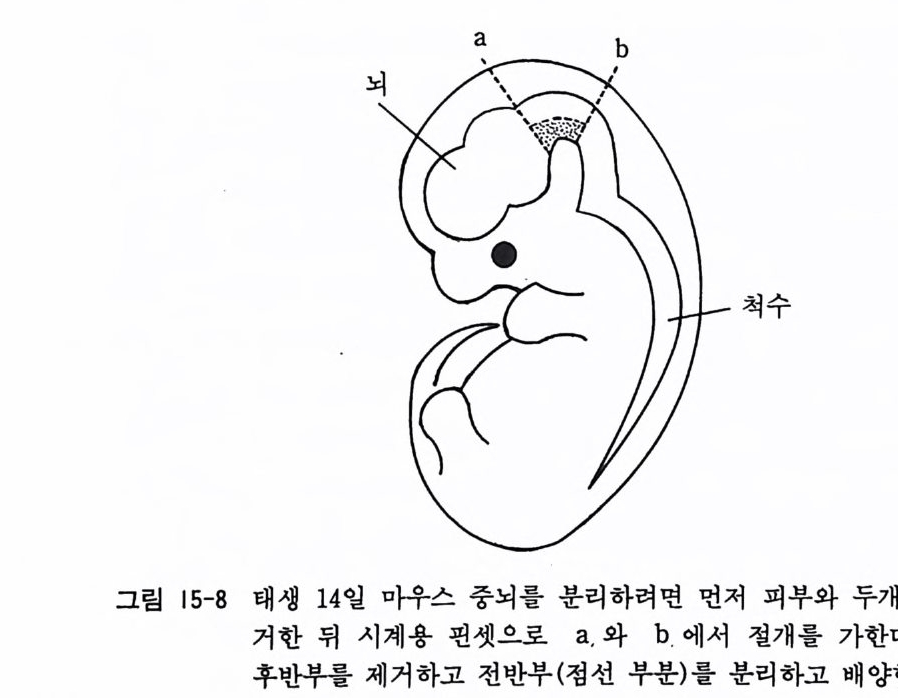 a
a
핀셋으로 제거한 다음 그 아랫부분 조직을 분리하고 CMF -핸크스액이 들어있는 페트리 접시에 옮긴다. 13-14 일 태아 에서는 뇌가 연약하여 예리한 핀셋으로 필요한 뇌조직을 절 단할 수 있다. 4) 소량의 핸크스액과 같이 15ml 원심튜브에 옮간 뒤 여분의 핸크스액을 버린다. 튜브에 2.6ml CMF- 핸크스액, 0.3ml 의 2.5 % 트립신액, 0.1 ml 의 0.2 % DNase 액을 가하고 37 도에서 30 분간 인큐베이트한다. 5) 혹질 조직을 튜브 바닥에 남긴 채 트립신액을 뽑아버리고 10% 마혈청이 들어 있는 배양액 3ml 를 가하고 파스출 피 펫으로 20 번, 피펫 끝에 미크로피펫립을 끼우고 20 번 피페팅 울한다. 6) 세포부유액을 1000r p m 에서 8 분간 원심분리한다. 7) 상청액을 버리고 배양액 3ml 를 가하고 다시 원심분리한다. 8) 상청액을 버리고 배양액 3ml 를 가하여 세포부유액을 만들 고 미리 준비해 두었던 폴리라이신 코팅한 커버글라스에 풀 레이팅하고 5% CO2 인큐베이터에서 배양을 한다. 배양액 조성은 다음과 같다. 이글 MEM 88ml 마혈청 (혹은 우태아혈청) 10 ml 50 % 포도당액 1ml 2mg /m l 겐타마이신액 1ml 9) 다음날 3ml 의 배양액을 추가하여 페트리 접시를 채운다. 일주일에 두 번 배양액을 교환한다. 〈해설〉 파킨슨 Park i nson 병은 신경변성 질환의 하나로서 현재로는 그
치료가 대단히 어렵다. 혹질의 도파민성 신경세포의 변성사가 원 인이 되어 일어나는 병이라 알려져 있다. 도파만성 신경세포의 변성사에 대하여서는 여러 가지 원인이 거론되고 있는데 그중에 는 MPTP 와 같은 신경독이 특이하게 혹질 신경세포를 공격한다 는 것이 가장 유력하다. 이러한 병인에 대한 실험연구 그리고 혹 질 도파민성 신경세포의 생리학, 생화학, 약리학을 검색하기 위 하여 래트(마우스) 혹질 신경세포 배양이 크게 활약을 하고 있 다. 21-23) 저자의 연구실에서는 태생 마우스 혹질 신경세포 배양을 사용 하여 흥분성 아미노산에 의한 혹질 신경세포의 변성 그리고 각종
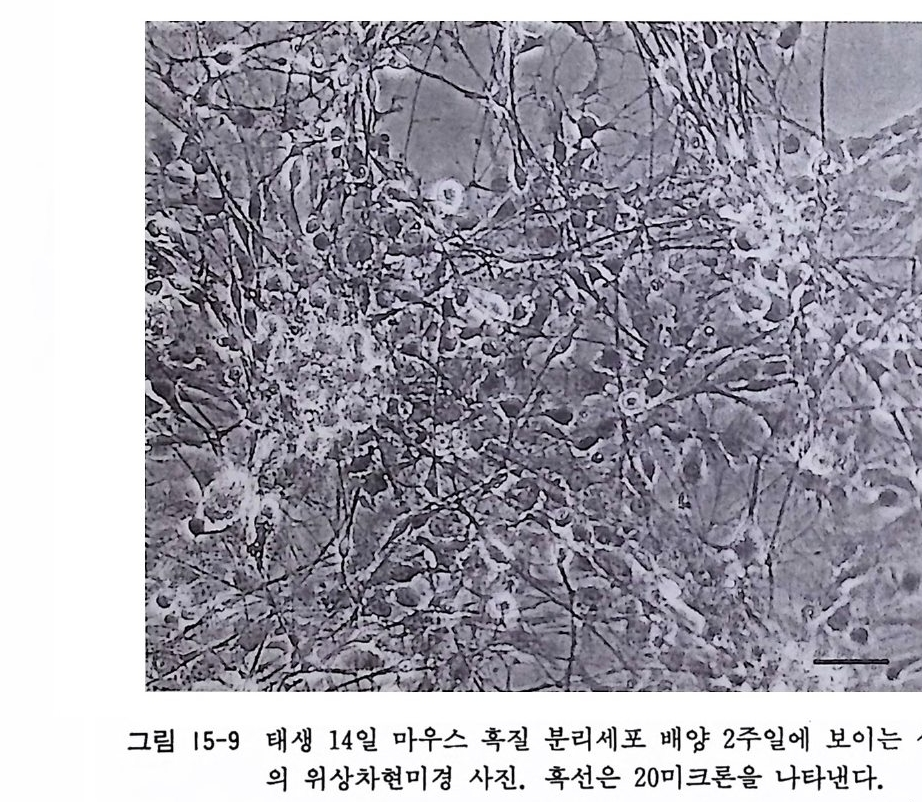 그림 15-9 태생 14 일 마우스 혹질 분리세포 배양 2 주일에 보이는 신경세포
그림 15-9 태생 14 일 마우스 혹질 분리세포 배양 2 주일에 보이는 신경세포
 `: 、 lE·
`: 、 lE·
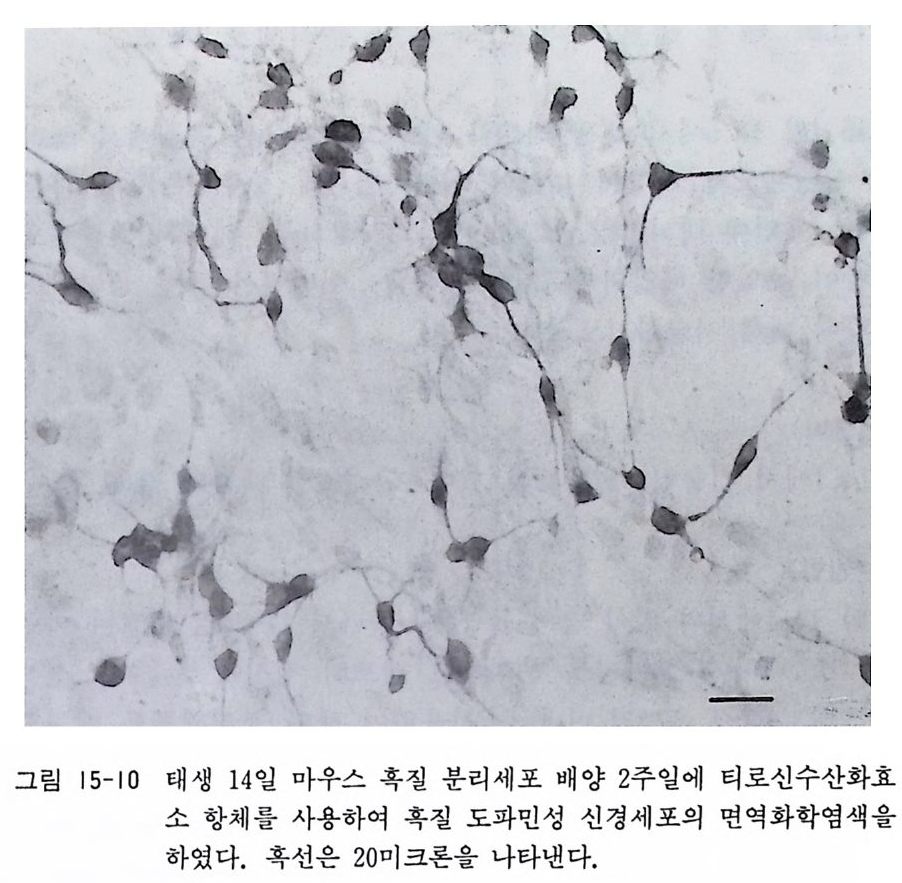
약제에 의한 방지 ,24) 그리고 혹질 신경세포에 특이하게 작용하는 새로운 신경영양인자의 연구 등이 계속되고 있다 .23) 그림 15-9 과 15-10 에서 태생 마우스 혹질 신경세포 배양의 실제를 보이고 있 다.
15.6 태생 래트(마우스)의 해마 분리세포 배양 15 .4와 15.5 에서 태생 래트와 마우스의 척수와 혹질에서 분리 한 신경세포의 배양에 대하여 서술하였는데 중추신경계의 다른 부위, 예컨대 대뇌피질, 소뇌, 시상, 시상하부, 해마의 신경세포 배양이 동일한 배양기술로서 가능하다. 여기에서 해마 분리세포 배양에 대해 서술하기로 한다. 〈준 H| 〉 14.1 에서 기술한 태생 래트 척수후근신경절 배양과 같다. 〈방법〉 1) 14.1 에서와 같이 순서 1)-5) 에 따라서 17 - 20 일 래트나 마우 스 태아 6-10 마리를 분리하고 페트리 접시에 놓는다. 신생 래트(출생 후 3 일 이내)를 사용하여 해마 분리세포를 배양할 수도 있다. 2) 태아 머리의 피부를 제거한 뒤 두 안구 사이에 가위를 넣어 서 좌우로 전개하듯이 두개골을 제거한다. 후뇌에서 연수까 지의 뇌를 꺼내어서 핸크스액이 들어있는 페트리 접시로 옮 긴다. 3) 현미경 아래에서 시계수선용 핀셋을 써서 해마를 분리한다. 먼저 대뇌와 중뇌의 경계부를 수술칼로 절단한다. 대뇌를 뒤 집은 다음 좌우 대뇌반구와 간뇌의 경계부를 수술칼로 절단 한다. 4) 대뇌반구를 뒤집으면 내측으로 해마가 바나나 모양으로 보 인다. 주위에 부착한 뇌막을 제거한 뒤 해마 조직을 분리한 다(그립 15-11 참고) . 해 마 조직을 CMF- 핸크스액 이 들어 있
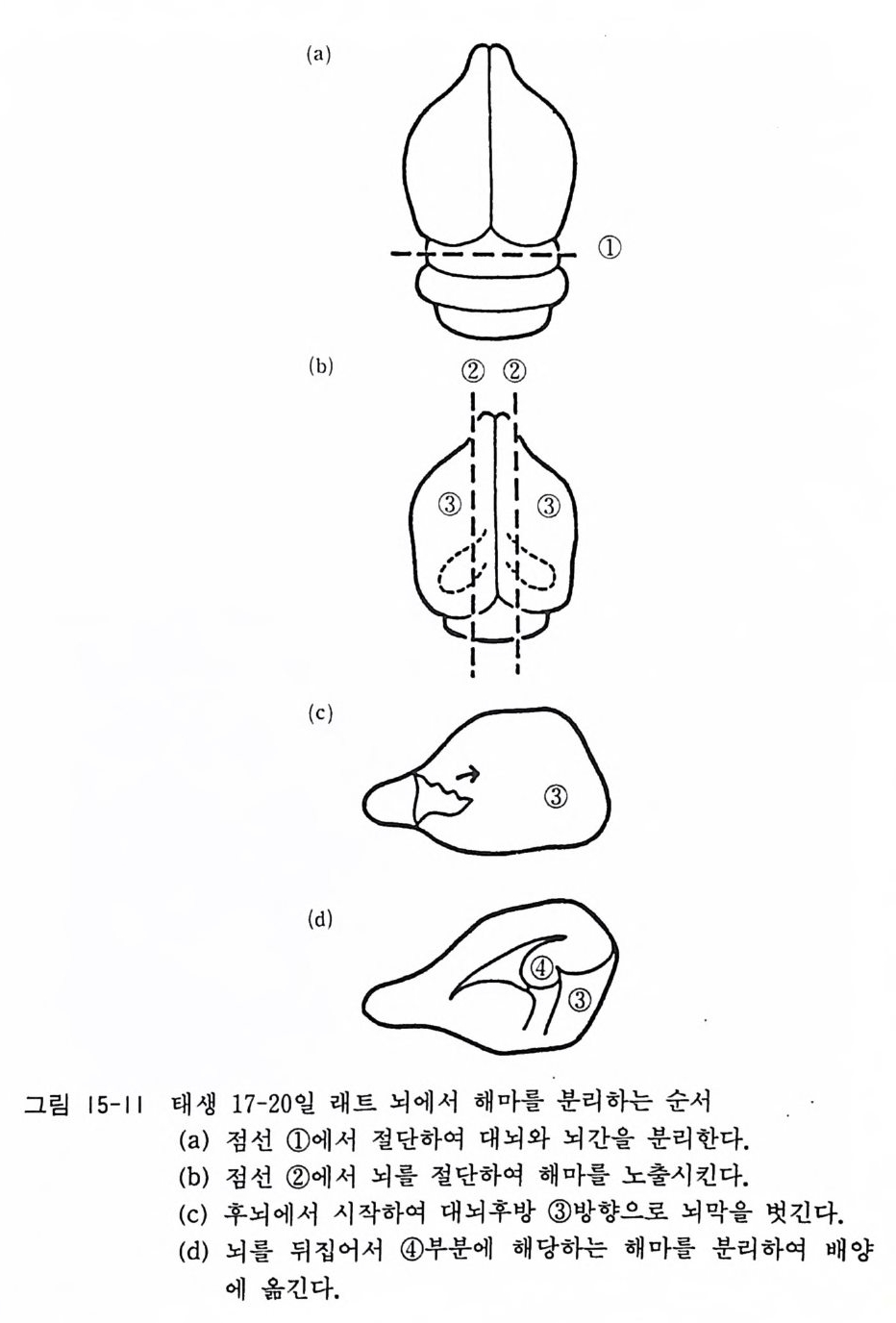 (a)
(a)
는 페트리 접시에 옮긴다. 5) 소량의 핸크스액과 같이 15 ml 원심튜브에 옮긴 뒤 여분의 핸크스액을 버린다. 튜브에 2.6ml CMF - 핸크스액, 0.3ml 2.5 % 트립신액, 0.1 ml O.2 % DNase 액을 가하고 37 도에 서 30 분간 인큐베이트한다. 6) 해마 조직을 튜브 바닥에 남긴 채 트립신액을 버리고 10 % 마혈청이 들어있는 배양액 3ml 를 가하고 파스출 피펫으로 20 번, 피펫 끝에 미크로팁을 끼우고 20 번 피페팅을 한다. 7) 세포부유액을 1000r p m 에서 8 분간 원심분리한다. 8) 상청액을 버리고 배양액 3ml 를 가하고 피페팅을 한 뒤 다 시 원심분리한다. 9) 상청액을 버리고 배양액 3ml 를 가하여 세포부유액을 만들 고 폴리라이신 코팅한 커버글라스에 풀레이팅한다(커버글라 스는 6cm 페트리 접시에 놓는다). 세포 밀도가 1 ml 당 2X 1 06 이 되도록 조절한다. 배양액 조성은 다음과 갇다. 이글 MEM 88ml 마혈청 (혹은 우태아혈청) 10 ml 50 % 포도당액 1ml 2 mg /m l 겐타마이 신액 1 ml 10) 다음날 3ml 의 배양액을 추가하여 페트리 접시를 채운다. 일주일에 두 번 배양액을 교환한다. 〈해설〉 알츠하이머 Alzhe i mer 병은 신경변성 질환의 하나로서 그 원인 과 치료가 아직도 알려지지 않은 불치의 병의 하나이다. 병변의 부위가 대뇌피질과 해마에 국한되어서 신경세포의 변성, 노인반
의 출현, 신경원섬유 변화 등의 특이한 병리상을 가지고 있다. 이러한 알츠하이머병의 원인을 규명하고 그 치료법을 개발하는 데에는 동물 실험이 필수적인데, 태생 래트나 마우스 해마 신경 세포 배양은 이러한 실험 연구에 적합한 모델이라고 할 수가 있 다 . 25 - 2 7) 아밀로이드의 신경독성, 알루미늄의 신경독성, 아포 E 의 신경독성 등 알츠하이머병의 원인이라 생각되는 여러 가지 인자의 검색에 많이 사용되고 있다. 그림 15-12, 15-13 에서는 태 생 래트 해마신경세포 배양의 실제를 보이고 있다. 해마신경세포 배양 이외에도 알츠마이머 병변의 원인 부위인 전뇌 기저핵이나 대뇌피질 신경세포배양도 알츠하이머병 연구에서 사용할 수가 있 다 . 28 )
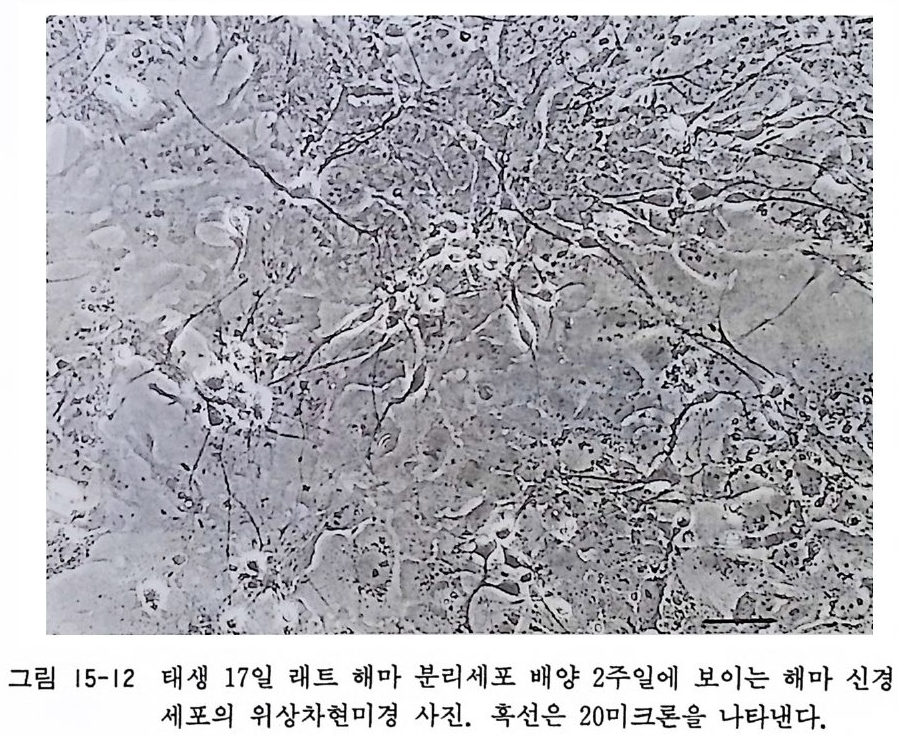 그림 15-12 태생 17 일 래트 해마 분리세포 배양 2 주일에 보이는 해마 신경
그림 15-12 태생 17 일 래트 해마 분리세포 배양 2 주일에 보이는 해마 신경
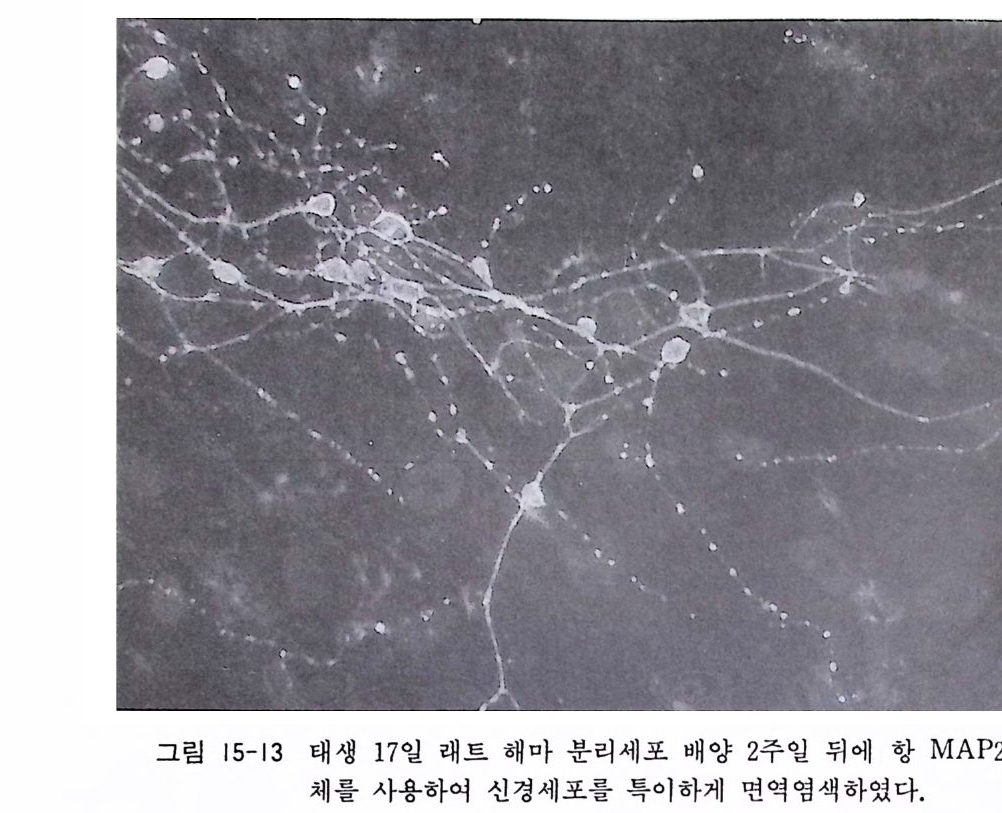 그림 15-13 태생 17 일 래트 해마 분리세포 배양 2 주일 뒤에 항 MAP2 항
그림 15-13 태생 17 일 래트 해마 분리세포 배양 2 주일 뒤에 항 MAP2 항
제 16 장 성숙 포유동물 척수후근신경절과 교감신경절의 분리세포 배양 근대 신경조직 배양에서 가장 큰 공헌을 하였던 머레이 Murra y가 1971 년에 쓴 신경조직 배양의 리뷰 논문에서는 1970 년 대 이전에 이루어진 신경조직 배양의 갖가지 연구에 대해서 언급 한 다음 신경계 조직배양에서 가장 치명적인 결점이 성숙 포유동 물의 신경세포 배양이 불가능한 것이라고 지적한 바 있다.1) 신 경세포를 성숙 포유동물에서 분리하고 배양하려는 시도는 그 전 에 여러번 있었으나 모두 실패하였다. 1950 년대에는 코스테로 Coste r o, 호우그 Hog ue , 가이거 Geig e r 등이 성인의 뇌에서부터 분리한 신경세포의 장기 배양에 성공하였다고 보고하였는데 2-4) 이들은 모두 섬유아세포나 성상교세포(아스트로사이트)를 신경세 포로 잘못 보았으며 특이 마카에 의한 신경세포의 확인이 없었기 때문에 그 결과를 믿을 수가 없다. 스코트 Sco tt가 1977 년에 성숙 마우스 척수후근신경절을 콜라 게나제로 처리하여 신경세포를 분리하고 장기 배양에 성공하였는 데 이것이 성숙 포유동물 신경세포 배양의 첫 케이스가 된다 .5)
그 다음해 저자의 연구실에서 같은 방법을 사용하여 사후 10 시간 이상되는 성인 교감신경절과 삼차신경절에서 신경세포와 슈완세 포를 분리하고 두 달 이상 배양하였다 .6) 이들 성인 삼차신경절 이나 교감신경절에서 분리한 신경세포는 전자현미경으로 관찰하 면 신경세포의 특이한 미세구조는 물론 리포푸신 lipo fu s ci n 과립 을 다수 가지고 있는 것을 볼 수 있다. 전기생리학적인 방법으로 자발성 활동전위 acti on po te n ti al 를 기록할 수가 있어서 이들 신 경세포가 조직배양 조건하에서 장기간 생존할 수 있음을 알 수가 있었다. 최근에는 성숙 래트, 마우스, 토끼 등의 말초지각신경절 과 교감신경절로부터 신경세포를 분리하고 배양하여 그 전기생리 적인 성질을 비롯하여 성장 안자에 대한 감수성, 각종 약제에 대 한 반응, 바이러스 감영 등 여러 가지 실험에서 모델시스템으로 서 활용되고 있다 .8,9) 16.l 성숙 래트(마우스) 척수후근신경절 분리세포 배양 여기에서는 성숙 래트(마우스) 척수후근신경절 신경세포의 분리 세포 배양에 대하여 소개한다. 〈준비〉 1) 입체해부현미경과 조명 2) 수술기구 -10cm 의과용 가위(직선과 곡선) 2 개 15cm 의과용 가위 2 개 10 cm 의과용 핀셋 2 개 시계수선용 핀셋 (No. 4) 2 개 수술용 칼 (No.11) 2 개
3) 시험관, 페트리 접시, 원심튜브 4) 혈구계산판 5) 핸크스 염류용액 6) 0.2 5 % 콜라게나제액 (핸크스 용액에 희석) 7) 이글 MEM 합성 배양액 8) 마혈청 9) 50 % 포도당액 10) 2 mg /m l 겐타마이신액 〈 방법 〉 1) 성숙 래트 혹은 마우스(생후 2 달 이후 2 년 이내)를 에텔자 속에서 마취사시킨다. 2) 래트롤 종이타월 위에 배를 아래로 하여 눕힌다. 70% 알 코올로 등 부분을 소독한다. 3) 15 cm 외과용 가위로 목의 연수와 척수 경계부를 절단한 다. 목에서 꼬리 부분까지의 피부를 깨끗이 박리한다. 4) 의과용 가위로 척수를 포함한 척주, 근육이 있는 부분을 목 에서 꼬리 상부까지 절단하고 페트리 접시로 옮긴다. 이때 다른 장기 가 상하지 않게 주의한다. 5) 먼저 척주부에서 근육을 제거한 뒤, 후추궁절제술의 방법으 로 척수의 후부에 덮여 있는 척추를 10 cm 곡선 가위로 절 제한다. 이때에 두 개의 평행선으로 철제를 가한다. 척추를 들어내면 척수가 보이는데 이것을 제거한다. 좌우 양측으로 척수후근신경절이 나란히 위치하고 있는데 이들을 시계수선 용 핀셋으로 하나씩 분리한다. 신경질에는 후근신경이 부착 되어 있어서 마치 올챙이같이 보인다. 이들을 핸크스 용액에 넣는다. 2 마리 동물에서 60 개 정도의 신경철을 분리할 수 있
다. 6) 소량의 핸크스액과 갇이 신경절을 3ml 의 0.25% 콜라게나 제와 0.2 % DNase 0.2 ml 가 들어있는 원심튜브로 옮기고 37 도 인큐베이터나 워터바스 속에서 3 시간 처리한다. 7) 원심튜브 바닥에 있는 신경절을 건드리지 않고 조용히 콜라 게나제액을 제거하고 3ml 의 배양액을 첨가한다. 파스출 피 펫으로 20 번 상하로 조용히 피페팅을 한다. 큰 덩어리가 남 아 있으면 3 분간 정치한 다음 상청액을 새 듀브로 옮기고 나 머지에 2ml 의 배양액을 첨가하고 피페팅을 되풀이한다. 파 스출 피펫 끝에 멸균된 미크로피펫팁을 끼우고 피페팅을 하 면 세포 분리가 쉽게 된다. 세포부유액을 하나의 시험관에 모으고 이를 1200r p m 으로 8 분간 원심분리한다. 8) 상청액을 버리고 페렛에 마혈청이 들어있는 배양액 3ml 를 가하고 피페팅을 5 번 가량 하여 세포부유액을 만든다. 세포 부유액 한 방울을 혈구계산판에 떨어뜨려서 세포 수 계산을 하고 배양액 1 ml 에 106 세포 수가 되도록 조정한다. 폴리라이신 코팅이 된 커버글라스 위에 한 방울의 세포부 유액을 올려놓는다. 5% CO2 인큐베이터에서 37 도로 보온 한 뒤 다음날 3ml 의 배양액을 첨가한다. 배양액 교환은 일 주일에 두 번 행한다. 12mm 의 커버글라스 8 개를 6cm 직 경 페트리 접시 속에 간격을 두고 나란히 위치시킨다. 커버 글라스 준비는 미리 해서 실온에서 보존하면 된다. 배양액 조성은 다음과 갇다. 이글 MEM 88ml 마혈청 10ml 50 % 포도당액 1ml 2mg /m l 겐타마이신액 1ml
〈 해설 〉 분리 직후의 배양 커버글라스 위에는 신경세포 이의에 미엘린 등의 조직 잔재가 많이 보인다. 배양 2-3 일부터 새로 나온 신경 섬유가 신경세포체에서 방사적으로 재생하는 것이 보인다. 태생 이나 신생 동물 척수후근신경절 신경세포 배양에서 필수적인 NGF 는 성숙 동물 신경절 신경세포의 생존에는 필요치 않다는 게 상식으로 되어 있다. 16.2 퍼컬 밀도구배 컬럼에 의한 신경세포 순수분리 미엘린 등의 조직잔재, 슈완세포와 섬유아세포를 제거하고 신 경세포롤 90% 정도의 순도로 배양하고자 할 때에는 다음과 같 이 퍼 컬 밀도구배 컬 럼 분리 per coll densit y grad ie n t iso lati on 를 하면 가능하다. 〈 준비 〉 1) 퍼 컬 Percoll (파마시 아사 Pharmacia ) 멸균된 상태에서 입수할 수 있다. 2) 10 배 농축 핸크스 용액, 핸크스 용액 3) 마혈청 포함 배양액 (16.1 참고) 〈 방법 〉 1) 16 . 1 에서 분리된 성숙 래트나 마우스 척수신경절 분리세포 롤 3ml 의 핸크스 용액에 부유시킨다. 2) 시험관 속에 0.9ml 퍼컬액, 0.1 ml 10 배 농축 핸크스 용 액, 2ml 핸크스 용액의 혼합액을 만든다. 이것이 30% 퍼
컬 용액이다. 3) 원심튜브에 먼저 2 ml 의 30 % 퍼컬액을 넣고 그 위에 3 ml 의 신경절 분리세포 부유액을 주의 깊게 울려놓는다. 4) 1800 r p m 으로 10 분간 원심분리를 한다. 5) 퍼컬과 핸크스 세포부유액 경계부에 미엘린과 조직 잔재가 모여 있고 그 아래의 퍼컬액 중에 그리고 튜브 바닥에 순수 분리된 신경세포가 침전된다. 6) 상청액과 미엘린을 버리고 퍼컬액과 페렛에 6ml 의 핸크스
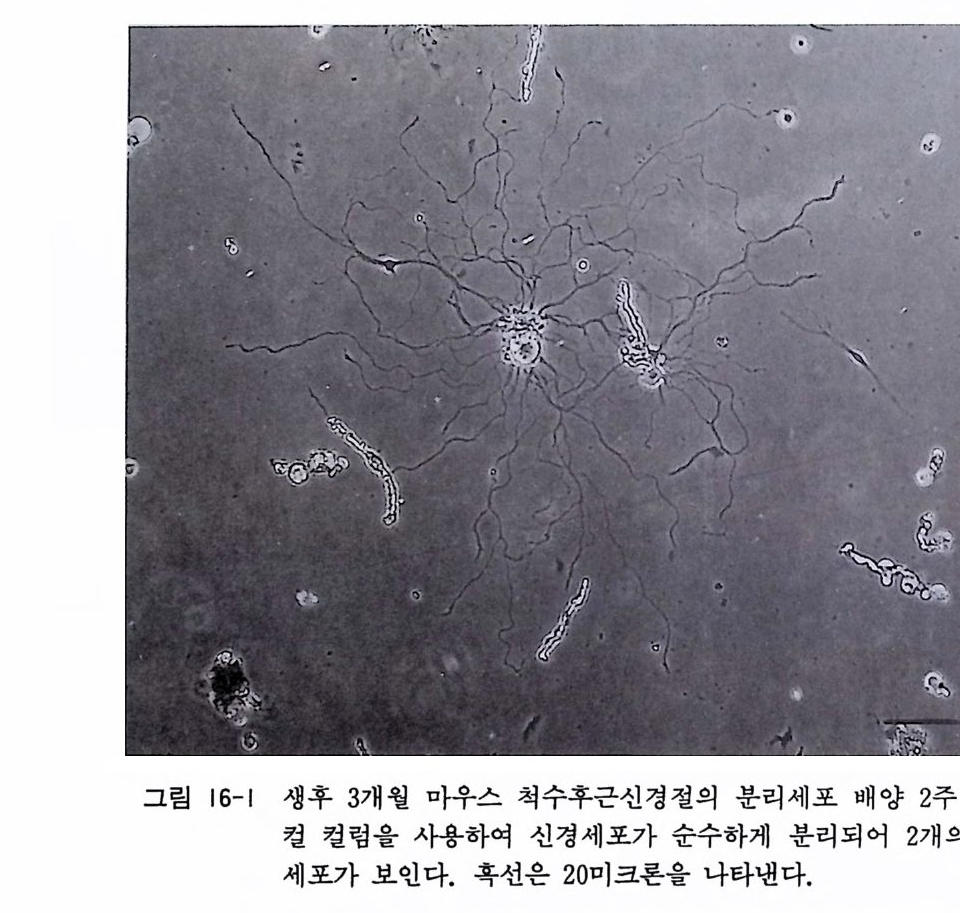 그림 16-1 생후 3 개월 마우스 척수후근신경절의 분리세포 배양 2 주일. 퍼
그림 16-1 생후 3 개월 마우스 척수후근신경절의 분리세포 배양 2 주일. 퍼
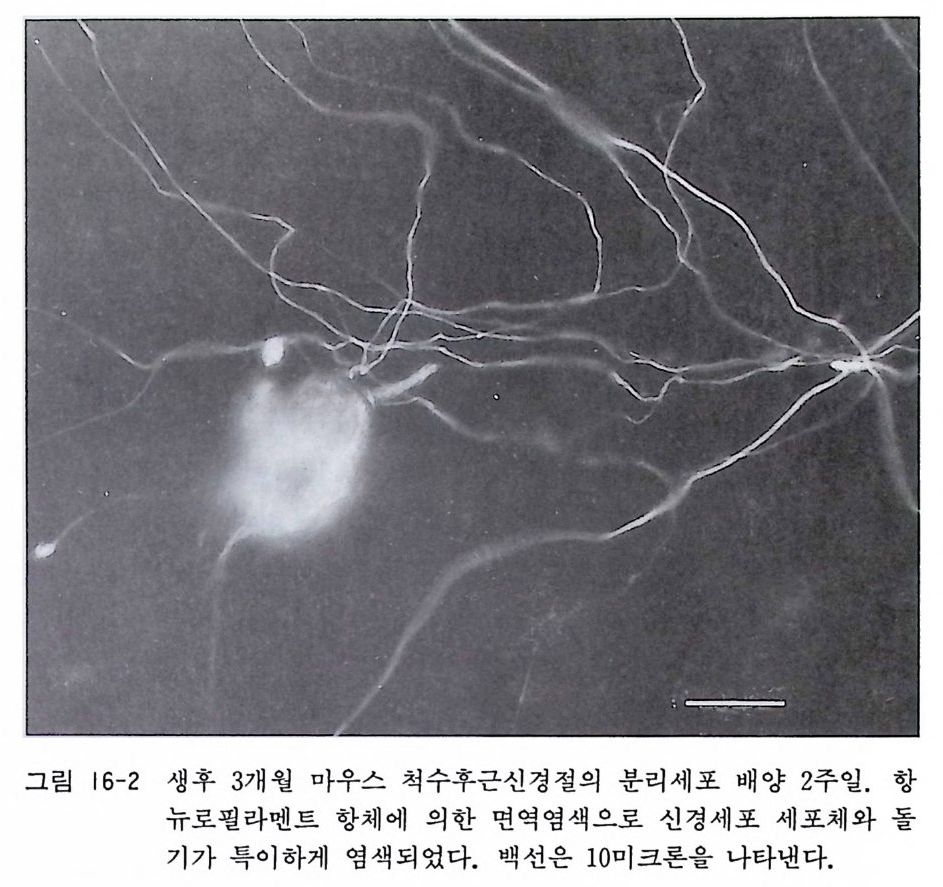 그림 |6-2 생후 3 개월 마우스 척수후근신경절의 분리세포 배양 2 주일. 항
그림 |6-2 생후 3 개월 마우스 척수후근신경절의 분리세포 배양 2 주일. 항
액을 가한 다음 1500rpm 8 분의 원심분리를 한다. 7) 상청액을 버리고 페렛을 2ml 배양액에 부유시킨 다음 커버 글라스 위에 플레이팅을 한다. 〈 해설 〉 성숙 마우스 척수후근신경절 세포배양의 실제를 그림 16-1 과 16 - 2 에서 보이고 있다. 신경세포는 분리 직후에는 효소 처리와 기계적 조작에 의해서 신경섬유가 절단되고 신경세포의 세포체만
남아서 야구공처럼 구형이 되어있다. 배양 후 2-3 일이 되면 신경 섬유가 방사형으로 다수 세포체에서 발육하게 되고 계속해서 배 양을 하면 신경성유가 하나 혹은 두 개만 남고 나머지는 퇴화를 한다. 16.3 성숙 래트 교감신경절 분리세포 배양 생후 2 달 이후 2 년이 되는 성숙 래트나 마우스를 사용하여 척 수후근신경절의 분리세포 배양에 대해서 16.1 에서 소개하였는데 교감신경절도 역시 갇은 연령의 동물에서 분리하여 배양에 사용 할 수가 있다. 교감신경철이나 후근신경절 분리 배양을 하면 신 경세포는 물론이거니와 말초신경계의 신경교세포gli al cell 인 슈 완세포도 역시 대량으로 얻을 수가 있다. 재료와 준비는 16.1 에서 기술한 척수후근신경절 배양과 갇다. 〈방법〉 1) 성숙 래트(혹은 마우스) 생후 2 달 이후 2 년 이내를 에텔자 속에서 마취사시킨다. 2) 래트를 15cmX20cm 의 카드보드 위에 등을 아래로 하여 눕히고 손발을 데이프으로 고정한다. 목과 가슴 부분을 70 % 알코올로 소독한다. 3) 머리를 좌측 혹은 우측으로 향하게 하고 핀으로 고정한다. 4) 목의 위 아래에 걸쳐서 비스듬하게 주행하는 흉쇄유양근 ste r nocleid o masto i d muscle 을 절제 하면 총경 동맥 common caroti d art er y 이 보인다. 이 총경동맥 의 주행 을 상방으로 거 슬러올라가면 경동맥 분기부가 나오는데 그 바로 아래에 상
경 교감신 경 절 sup er io r cervic a l ga ng li o n 아 위 치 한다. 이 산 경 절을 시계수선용 핀셋을 사용하여 분리하고 핸크스용액이나 PBS 가 들어있는 페트리 접시에 모은다. 상경교감신경절의 분리에 관하여서는 14.3 신생 래트(마우스) 교감신경절 배양 의 기술과 그립 14 - 5 를 참고로 할 것. 5) 래트 6 마리에서 좌우 양측으로 부터 신경절 12 개가 분리되 면 0.25 % 콜라게나제 2 ml( 핸크스 용액이나 이글 MEM 속에 녹여서 필터 여과멸균을 하고 2 ml 씩 분주한 뒤 -20 도에서 얼려 서 보관한다)와 0.2 % DNase 0.2ml 가 들어있는 원심튜브에 넣어서 37 도에서 3 시간 인큐베이트한다. 6) 바닥에 가라앉은 신경절을 건드리지 않고 콜라게나제액을 피펫으로 제거한다. 7) 마혈청이 들어있는 배양액 3ml 률 가한 뒤 파스출 피펫으로 20 번 피페팅을 한다. 이 조작으로 세포가 분리되는데 조그만 덩어리가 아직 남아있으면 듀브를 3 분간 정치한 뒤 상청액을 걷어서 새 튜브에 옮기고 나머지 2ml 의 배양액을 가하고 피 페팅을 되풀이한다. 파스출 피펫 끝에 멸균된 미크로피펫팁 을 끼우고 피페팅을 하면 세포 분리가 쉽게 된다. 8) 세포부유액울 하나의 시험관에 모으고 이를 1200r p m 으로 8 분간 원심분리한다. 9) 상청액을 버리고 페렛에 마혈청이 들어있는 배양액 1.5 ml 롤 가하고 피페팅을 10 번 가량 하여 세포부유액을 만든다. 세포부유액 한 방울을 혈구계산판에 떨어뜨려서 세포 수 계 산울 하고 배양액 1ml 에 105 세포가 되도록 조정한다. 10) 폴리라이신 코팅 된 커버글라스 위에 한 방울의 세포부유액 을 올려놓는다. 5 % CO2 인큐베이터에서 37 도 보온한 뒤 다음날 3ml 의 배양액을 첨가한다. 배양액 교환은 일주일에
두 번한다. 커버슬립 준비는 배양 시작 전에 미리 준비해 두 어야 하겠다. 12mm 직경 커버글라스 8 개를 6cm 직경 페 트리 접시 속에 간격을 두고 나란히 위치시킨다. 배양액 조성은 다음과 같다. 이글 MEM 88ml 마혈청 10ml 50% 포도당액 1ml 2 mg /m l 겐타마이 신액 1 ml 〈 해설 〉 성숙 래트나 마우스 교감신경절 배양의 방법은 다른 동물에서
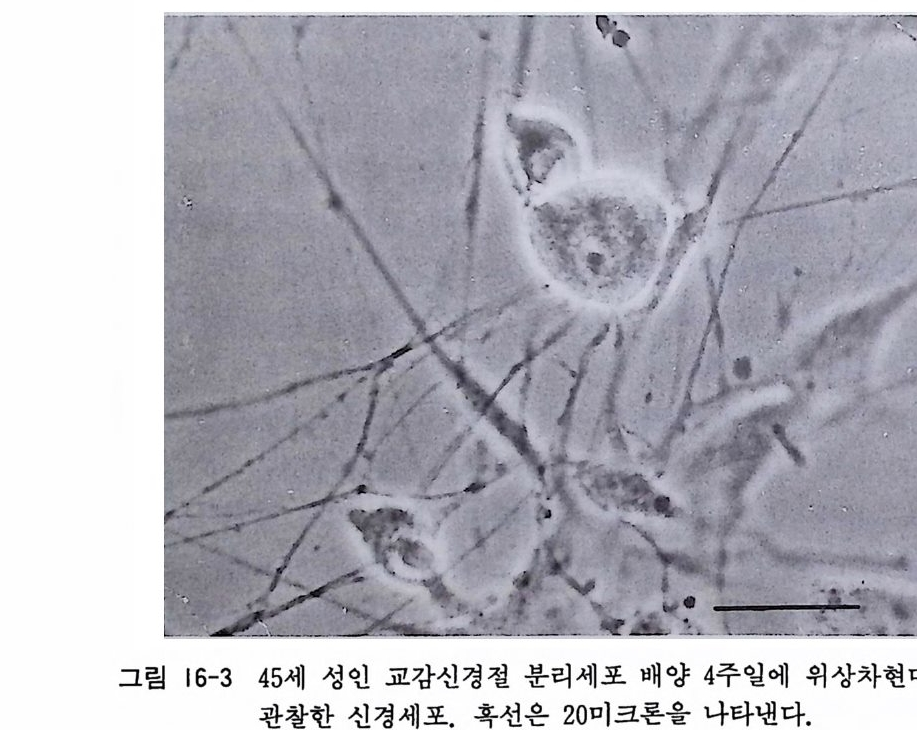 ``
``
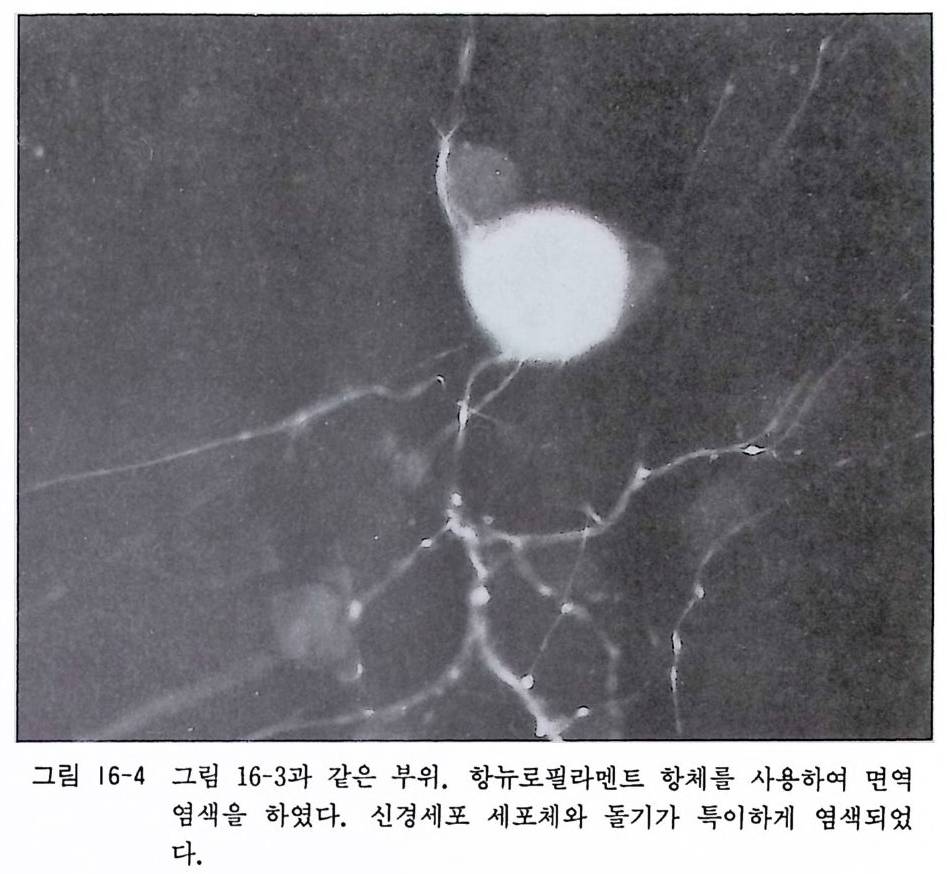 그림 16-4 그립 16-3 과 같은 부위. 항뉴로필라멘트 항체를 사용하여 면역
그림 16-4 그립 16-3 과 같은 부위. 항뉴로필라멘트 항체를 사용하여 면역
도 적용할 수가 있어서 저자의 연구실에서는 1979 년에 사후 15 시 간 이내의 성인 교감신경절 배양에서 신경세포의 생존을 확인한 바 있다. 7) 그림 16-3 과 16-4 에서는 45 세 성인 교감신경절 배양 에서 관찰된 신경세포의 위상차 현미경과 뉴로필라멘트 면역염색 의 사진을 보이고 있다. ’ 그림 16-5 에서 보는 바와 같이 교감신경 세포의 미세구조가 정상으로 보존되고 있으며 70 세의 노인에서 분리된 신경세포이기 때문에 노화과립인 리포푸신이 세포질 속에 대량으로 들어있다.
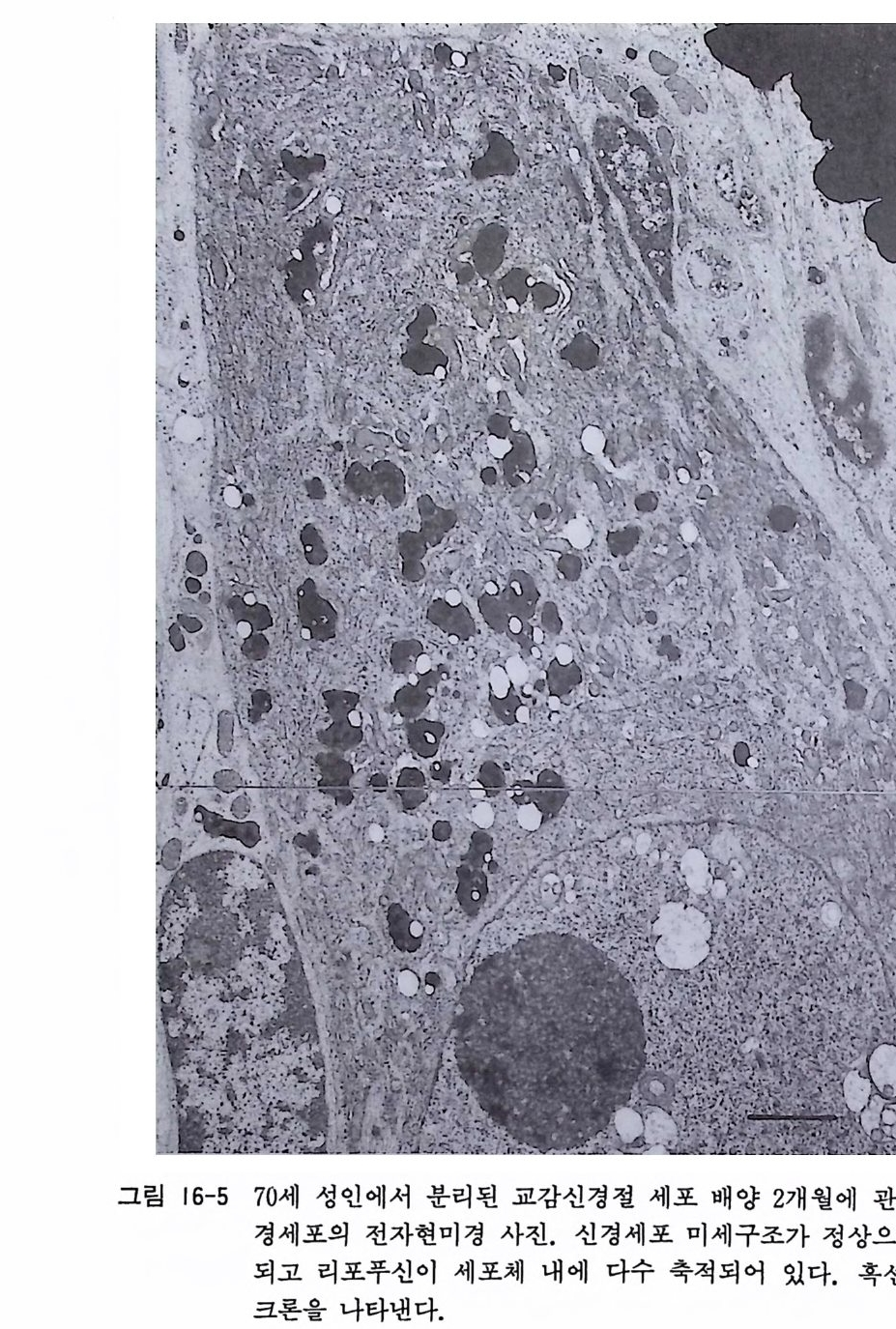 그림 16-5 70 세 성인에서 분리된 교감신경철 세포 배양 2 개월에 관찰된 신
그림 16-5 70 세 성인에서 분리된 교감신경철 세포 배양 2 개월에 관찰된 신
제 17 장 성숙 포유동물 중추신경계 조직 • 세포의 배양 성숙 포유동물의 중추신경계 조직의 배양은 제 16 장에서 서술 한 바와 같이 1950 년대 성인 뇌에서 신경세포를 분리하여 장기배 양하였다는 보고가 있었다. 이들 보고가 전혀 신용할 수 없다는 것을 알게 되자, 최근까지 여러 학자들이 성숙동물의 신경세포 배양이 불가능하다고 인식하고 전혀 그 분야의 실험에 손을 대지 않게 되었다. 이러한 상황을 보고 저자는 성숙동물 중추신경계 신경세포 배양에 도전하여서 1978 년 이후로 여러 가지 방법으로 실험한 바 있다. 그리하여 1979 년에는 저자의 연구실에서 처음으 로 성숙 포유동물 중추신경계 신경세포의 배양에 성공하였다. 성 숙한 개의 안구에서 망막 re ti na 울 분리하고 이식편 배양을 만들 어서 한 달 이상 장기배양에 성공하였던 것이다 .I) 1988 년에는 안구은행에서 보내온 60 세에서 70 세의 사람의 안구에서 망막을 분리하고 이식편 배양을 하여 역시 망막신경세포의 장기배양에 성공한 바 있다 .2) 중추신경계에서는 오직 망막신경세포의 배양 이 가능하며 대뇌피질, 해마, 소뇌, 척수 등 다른 중추신경계의
중요 부위의 신경세포 배양은 아직 그 보고가 없다. 장차 배양기술의 개선으로 성숙동물의 망막 이의의 중추신경계 신경세포 배양이 가능할 것으로 기대된다. 17.1 성숙 래트(마우스) 망막 이식편 배양 〈준비 〉 1) 해부입체현미경과 조명 2) 수술기구 -10 cm 의과용 가위 (곡선) 1 개 시계수선용 핀셋 (No. 4) 2 개 수술용 칼 (No. 11) 2 개 3) 페트리 접시, 원심튜브 4) 핸크스 영류 용액 5) IOunit s/m l 파파인 (이글 MEM 에 희석) (워싱턴 Worth i n g ton Bio c hemi ca l) 시 스테 인 (cys t e i n , 시 그마) 6) 이글 MEM 합성 배양액 7) 마혈청 8) 50 % 포도당액 9) 2 mg /m l 겐타마이신액 〈방법〉 1) 성숙 래트나 마우스 (생후 2 달 이후 2 년 이내)를 에텔자 속 에서 마취사시킨다. 2) 래트롤 배를 아래로 하여 눕힌다. 70 % 알코올로 눈 주위 를 중심으로 하여 소독한다.
3) 좌우 양측의 안구를 핀셋과 10cm 가위를 사용하여 도려낸 다. 핸크스용액이 둘어있는 페트리 접시에 안구를 모은다. 4) 왼손에 잡은 핀셋으로 안구를 고정하고 가위로 안구의 중앙 부를 절단하고 안구 내부조직을 유리시킨다. 5) 핀셋으로 절단된 안구를 펼치면 반투명한 망막이 다른 안구 조직에서 유리되고 핸크스액에서 부유한다. 6) 핸크스용액이 들어있는 다른 페트리 접시에 망막을 옮기고 두 개의 수술용 칼을 사용하여 2x 2 x2mm 크기의 소편으로 자론다. 7) 이들 망막조직 소편을 두개씩 콜라겐 코트된 22mm 직경 둥근 커버글라스나 11 X 22mm 의 사각형 커버슬립에 울려놓 는다. 8) 한 방울의 마혈청을 포함한 배양액을 커버글라스 위에 떨어 뜨리고 2 시간 동안 37 도 인큐베이터에서 정치시킨다. 9) 맥시모우 슬라이드, 페트리 접시 혹은 롤러튜브법에 의해서 배양을 시작한다 (11.3, 11. 4, 13.1 참고). 10) 배양액 조성은 다음과 같다. 이글 MEM 78ml 마혈청 (우태아 혈청) 10 ml 50 % 포도당액 1ml 2 mg /m l 겐타마이신액 1 ml 17.2 성숙 래트(마우스) 망막 분리세포 배양 1979 년 저자의 연구실에서 성숙 개 망막의 이식편 배양을 하여 성숙 신경세포의 장기배양을 처음으로 성공한 후 1988 년에는 성
인 망막 신경세포 배양에 성공한 바 있다 .2) 그 이후로는 이식편 배양 대신에 분리세포 배양기술을 사용하여 포유동물 망막 신경 세포의 배양을 여러 가지로 시도하였으나 만족스런 결과를 얻지 못하였다. 세포 분리방법으로 트립신, 콜라게나제, 디스파제, 기 계적 분리법을 사용하여서 좋은 결과를 얻지 못하였는데 파파인 울 사용한 뒤로는 생존율이 좋은 성숙 신경세포의 배양이 가능하 게 되었다. 여기에서는 파파인에 의한 분리세포 배양법을 소개하 기로한다. 〈준비〉 준비는 17.1 에서와 같다. 〈방법 〉 1) 17.1 에서 서술한 방법으로 성숙 래트나 마우스의 안구에서 망막을 분리한다. 2) 수술칼 2 개를 사용하여 분리한 망막을 소편으로 자른다. 3) 파파인 용액 2ml 가 들어있는 15ml 원심튜브에 망막 소편 을 옮기고 37 도에서 40 분간 인큐베이트한다. 파파인은 워싱 턴사나 시그마사에서 구입할 수가 있는데 100unit /m l 농도 로 이글 MEM 에 녹여서 0.2ml 씩 분주하고 一 20 도에서 보 관한다. 사용시 이글 MEM 로 10 배로 희석하고 여기에 4 m g /ml 의 시스테인을 가하고 여과멸균한다. 4) 망막 소편과 파파인이 들어있는 원심튜브에 0.1ml 의 0.2 % DNase 를 첨가한 3ml 의 배양액 (마혈청 포함)을 가하 고 20 번 피페팅을 한다. 파스출 피펫 끝에 미크로피펫립을 삽입하고 20 번 재차 피페팅을 되풀이한다. 5) 1000r p m 의 속도로 8 분간 원심분리한다.
6) 페렛에 배양액 3ml 를 가하고 세포를 부유시킨 뒤 한번 더 1000 r p m 에서 8 분간 원심분리한다. 7) 상청액을 버리고 페렛에 2ml 의 배양액을 가한 뒤 폴리라이 신 코팅된 12mm 커버슬립에 플레이팅하고 하롯동안 37 도 인큐베이터에서 정치시킨다. 8) 다음날 배양액을 추가한다. 배양액 교환은 일주일에 두 번 (월요일과 금요일) 하도록 한다. 배양액 조성은 다음과 같다. 이글 MEM · 88ml 마혈청 (우태아 혈청) 10 ml 50 % 포도당액 1ml 2 mg / ml 겐타마이신액 1 ml 〈 해설 〉 래트와 마우스와 같은 소동물의 망막 배양에서 사용하는 방법 을 그대로 응용하여 사후 5-20 시간 이내의 사람의 망막 배양이 가능하다. 그립 17-1 과 17-2 에서는 사람 망막의 이식편 배양과 분리세포 배양의 실제를 보이고 있는데 광수용체 세포p ho t orece p tor , 소형신경세포, 뮬러신경교세포 등의 형태를 알 수가 있다. 이러한 망막 각 구성세포의 확인은 세포형 특이 마커를 사용하여 면역세포화학염색을 한다(제 36 장 참고). 그림 17-3 에서는 성인 망막신경 세포롤 뉴로필라멘트 항체를 사용하여 면역염색한 것을 보이고 있다.
 그림 17-1 72 세 노인의 사후 12 시간만에 망막 을 분리하고 이식편 배양을 4
그림 17-1 72 세 노인의 사후 12 시간만에 망막 을 분리하고 이식편 배양을 4
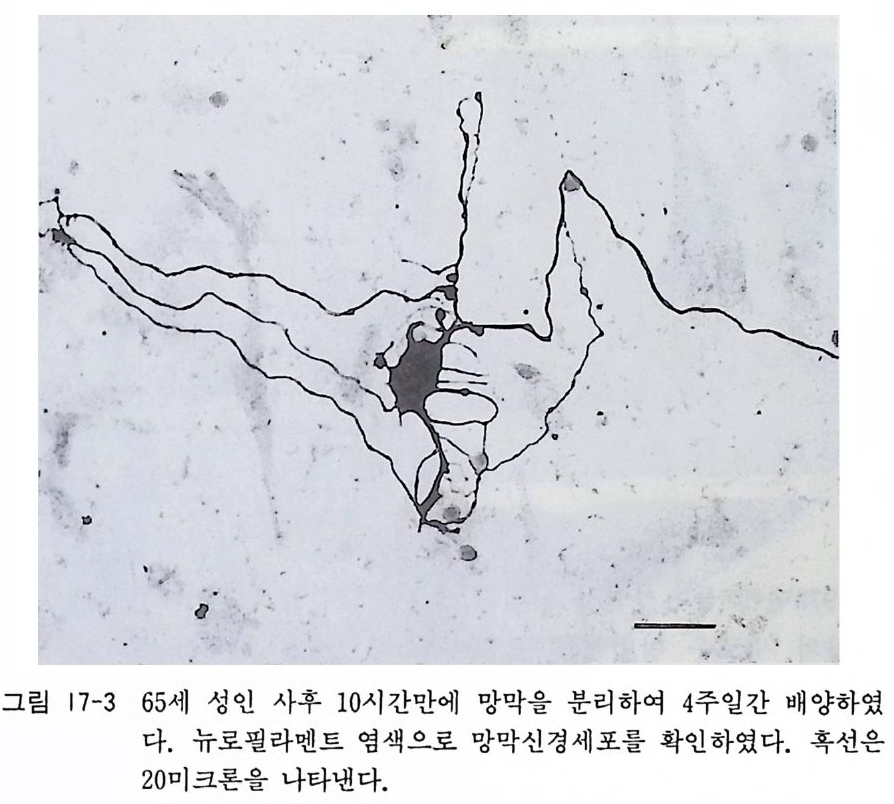 ‘-‘
‘-‘
제 18 장 신경세포의 순수배양 1970 년에 들어서면서 이식편 조칙배양법을 떠나서 신경계 조직 배양의 새로운 방법론인 분리세포 배양법을 사용하는 연구자의 수가 증가하게 되었다. 그러나 분리세포 배양에서도 각종 신경세 포와 신경교세포가 혼재하고 있는 시스템이기 때문에 이식편 조 직배양에 비해서 이렇다 할 장점이 없고 새로운 지견을 창출하지 못하여 불만이 있었다. 한편으로는 단일세포 타입이 순수한 형태 로 대량 얻을 수가 있다는 이점을 가진 수립 세포라인 esta b li sh ed celll li ne 을 실험모델로 사용하는 연구실이 증가하였다. 그러나 수립 세포라인은 뇌종양 유래 혹은 악성 형질전환 세포t rans fo rm ed cell 유래이기 때문에 정상적인 형질발현 ph enoty pic exp re - ss i on 에 문제가 있고 어느 정도까지 정상 신경세포나 신경교세포 의 형질과 기능을 충실하게 반영하고 있는가라는 커다란 의문이 있었다. 이러한 문제점을 극복하기 위하여 태생이나 신생아 정상 신경 조직에서부터 신경세포와 신경교세포를 순수하게 분리하고 이룰
조직배양 시스템에서 배양하려는 시도가 1970 년 후반에 시작되었 다. 신경조직 단일 세포 타입의 분리는 신경화학의 분야에서 1960 년대부터 활용되어 온 기술로서 신경세포, 성상교세포(아스 트로사이트), 울리고덴드로사이트, 뇌혈관, 미엘린 등이 대량으 로 정제되고 신경조직의 화학적 구성, 효소 활성, 대사반응에 크 게 활용되어 왔다. 신경화학에서 쓰이는 분리 기술은 주로 세포 타입의 비중과 크기의 차이를 이용한 것이어서 수크로오스 sucrose, 퍼 콜 per coll, 메 트리 자마이 드 metr i z a mi de , 혈 청 알부민 serum albumi n 으로 된 비 중 구배 컬 럼 densit y grad ie n t column 에 분리세포를 넣고 원심분리 뒤에 생긴 세포 밴드를 분리정제한다 는 것이다. 이 기술은 현재 신경세포와 울리고덴드로사이트 그리 고 혈관 내피세포 분리에 사용되고 있다. 신경세포를 순수배양하는 방법으로는 DNA 합성저지 약제를 사용하여 태생 래트 척수후근신경절과 교감신경절에서 90% 이 상의 순도의 신경세포를 배양하는 방법, 1,2) 신생아나 성숙 래트 척수후근신경절을 콜라게나제 처리하여 세포를 분리한 다음 이들 울 퍼콜 비중 구배컬럼에 넣어서 신경세포를 분리하는 방법, 그 리고 신경세포가 가전 특이 표면항원 spe c if ic surfa c e an tig en 을 이용하여 면역학적 수법으로 선택적으로 계배 교감신경절과 신생 래트 망막에서 신경세포를 순수분리하는 방법 3) 등이 있다. 18.l 태생 래트(마우스) 척수후근신경절 신경세포의 순 수배양 래트의 말초신경계 신경세포의 순수배양을 작성함에 있어서 가 장 재현성이 높고 기술적으로 어렵지 않은 배양 시스템은 배양액
중에 플로로디옥시유리 딘 fluo rodeoxy u rid i n e 이 나 시 토신아라비 노 시 드 cy tos in e arabin o sid e 와 갇은 DNA 합성 저 해 제 를 배 양액 중 에 첨가하여서 증식 능력을 가전 슈완세포나 섬유아세포를 제거 하는방법이다. 여기에서는우드 Wood 가개발한방법을소개한다 .I) 〈 준비 〉 1) 해부입체현미경과 조명 2) 수술기구 -10cm 의과용 가위(직선과 곡선) 2 개 10cm 의과용 핀셋 2 개 시계수선용 핀셋 (No. 4) 2 개 수술용 칼 (No. 11) 2 개 3) 시험관, 페트리 접시, 원심튜브 4) 12mm 직경 원형 커버글라스 5) 칼슘, 마그네슘 제거 핸크스 염류용액 6) 2.5 % 트립신액 7) 0.2 % DNase 효소액 8) 이글 MEM 합성 배양액 9) 마혈청 10) 50 % 포도당액 11) 2 mg /m l 겐타마이신액 12) 10 µg/m l NGF( 신경성장인자) 13) DNA 합성저해제 (풀로로디옥시유리딘과 유리딘 혹은 시토신 아라비노시드) 〈방법〉 1) 16 일 -20 일 임신 래트와 마우스를 에텔자 속에 넣고 에텔심 마취에 의해서 마취사시킨다.
2) 래트의 등을 아래로 하여 종이타월 위에 눕히고 배 부분을 70 % 알코올로 소독한다. 3) 배의 피부를 의과용 핀셋으로 둘어올리고 배의 정중선을 따 라서 하복부를 가위로 크게 절개한다. 그 아래 근육충도 역 시 정중선을 따라서 절개한다. 4) 자궁이 노출되면, 자궁의 경관부와 난관부를 절단하고 태아 가 둘어있는 기다란 색 형상의 자궁울 핸크스액이 들어있는 10cm 페트리 접시로 옮긴다. 5) 자궁근막을 절개하고 6-12 마리의 태아를 분리한다. 6) 의과용 가위로 태아의 머리를 자르고 등 부분의 피부를 제 거한다. 척주와 척수를 포함한 후배조직을 크게 절단하고 새 10cm 페트리 접시에 옮기고 척주 전면에 붙은 내장과 꼬리 를 제거한다. 7) 등을 위로 하여 입체해부현미경 아래 놓는다. 10cm 곡선 의과용 가위를 사용해 척주 후배부의 근육과 척추골을 찰라 내고 척수를 노출시킨다. 척수조직을 제거하면 경수에서 천수 에 이르는 30 쌍의 척수후근신경절이 양측에 나란히 놓여있다. 8) 시계수선용 핀셋을 사용하여 조심스럽게 후근신경절을 하나 씩 분리하고 CMF- 핸크스액이 들어있는 6cm 페트리 접시 에 모은다. 태아 한 마리에서 40-50 개의 신경절이 나오는데 10 마리 태아에서 400 개의 신경절을 · 분리할 수 있다. 신경절 의 분리는 능숙한 사람은 한 시간 정도 걸리고 초심자는 2 시 간에서 3 시간 정도 걸린다. 그동안 태아나 분리한 신경철이 든 페트리 접시를 얼음 위에 정치하도록 한다. 9) 모은 신경절을 핸크스액과 같이 15ml 원심튜브에 옮긴 뒤 여분의 핸크스액을 버린다. 원심튜브에 4.3ml CMF- 핸크스 액, 0.5 ml 2.5 % 트립신액, 0.2 ml 의 0.2 % DNase 액을
차례로 가하고 37 도에서 30 분간 인큐베이트한다. 5 분마다 튜 브를 꺼내서 혼합이 잘 되도록 흔든다. 10) 신경철을 튜브 바닥에 남간 채 트립신액을 뽑아내어 버린 다. 10% 마혈청이 들어있는 배양액 3ml 를 가하고 파스출 피펫으로 20 번 상하운동에 의한 피페팅을 한다. 다음에 파 스출 피펫 끝에 멸균된 미크로피펫립을 끼우고 20 번 피페팅 을 한다. 이 시접에서 세포의 분리가 잘 되어있을 것이나 만일 조칙 잔재가 많이 남아있으면 듀브를 3 분간 정치한 뒤 그 세포가 부유하고 있는 상청액을 새 원심튜브에 옮기고 남은 조직에 배양액 2ml 를 가한 뒤 위에 적은 바와 같이 피페팅을 되풀아한다. 11) 세포부유액을 한 튜브에 모으고 1000r p m 으로 8 분의 원심 분리를 한다. 12) 상청 액을 버 리고 페 렛에 배 양액 3 ml 를 가하고 1000 rp m 으로 8 분간 원심분리한다. 13) 상청액을 버리고 페렛에 배양액 5ml 를 가하여 세포를 부 유시킨 뒤 미리 준비해 두었던 폴리라이신 코팅한 커버글라 스 위에 플레이팅을 한다. 400 개의 신경절에서 4 X 107 개의 세포가 분리된다. 배양액의 조성은 다음과 같다. 이글 MEM 82ml 마혈청(우태아혈청) 10ml 계배 추출액 2ml 10 µg/m l NGF 1 ml 1. 5 M 영 화칼륨 (KCl) 1ml lmM 풀로로디옥시유리딘 1ml lmM 유리딘 1ml
50% 포도당액 1 ml 2 mg /m l 겐타마이신액 1 ml 14) 다음날 위에 적은 배양액 3ml 를 커버글라스가 들어있는 6cm 페트리 접시에 추가한다. 일주일에 두 번 배양액을 교환한다. 15) 2 주일 뒤부터는 90-95 % 순도의 신경세포 순수배양을 얻 을 수 있다. 순도가 높은 신경세포를 얻은 시접부터 풀로로 디옥시유리딘과 유리딘을 배양액에서 제거하여 사용한다.
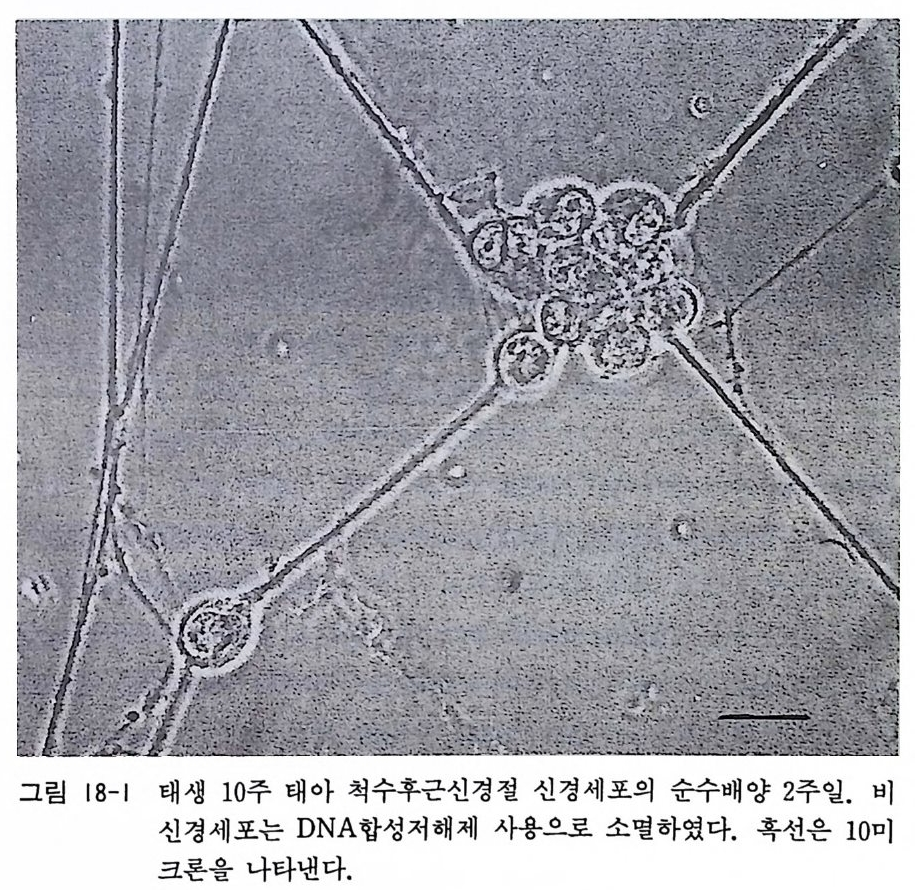 그림 18-1 태생 10 주 태아 척수후근신경절 신경세포의 순수배양 2 주일. 비
그림 18-1 태생 10 주 태아 척수후근신경절 신경세포의 순수배양 2 주일. 비
 그림 18-2 그립 18 - 1 에 보이는 신경세포를 뉴로필라멘트 항체 를 사용하여
그림 18-2 그립 18 - 1 에 보이는 신경세포를 뉴로필라멘트 항체 를 사용하여
〈해설〉 태생 래트와 마우스 후근신경절 신경세포 순수배양의 방법은 다른 동물에서도 응용할 수 있다. 그림 18-1 과 18-2 에서는 사람 10 주 태아에서 분리한 척수후근신경절 신경세포의 순수배양의 실 제를 보이고 있다. NGF 와 DNA 합성저해제를 배양액에 첨가함 으로써 신경세포만을 선택적으로 유지할 수 있다. 태생 래트와 마우스 후근신경절 신경세포 배양은 신경영양인자 에 관한 연구에서 유력한 실험모델이 되고 있다.
18.2 신생래트(마우스)교감신경절신경세포의순수배양 신생 래트 상경교감신경절의 분리세포를 혈청을 포함한 L-15 배양액에서 배양하여 신경세포의 순수배양에 성공하였는다는 보 고가 있다 .2) 그 분리방법은 기계적 분리법 즉 핀셋을 써서 교감 신경절을 조그맣게 잘라내고 피하주사침을 이용하여 세포를 분리 하고 마지막에는 보텍스믹사로 세포를 분리한다는 것이다. 저자 의 연구실에서 실제로 추시해 보니 신경세포의 대부분이 상하여 서 사멸하였다. 여기에서는 이러한 기계적 분리법보다 온화하고 신중한 트립신에 의한 신경세포 분리법에 대하여 소개한다. 〈 준바 〉 18 . 1 의 태생 래트 후근신경절 분리세포 배양과 갇다. 〈 방법 〉 1) 신생 래트 혹은 마우스를 에텔자에 넣어서 심마취로 마취사 시킨다. 혹은 신생 래트롤 조그만 상자에 넣어서 -20 도 냉 장고에서 10 분간 냉온마취를 한다. 2) 14. 3 신생 래트 교감신경절 배양의 섹션에서 기재한 대로 10 마리의 신생 래트에서 상경교감신경절 sup e rio r cerv ica l g an gli on 을 분리 한다. 3) 20 개의 교감신경절을 15ml 원심튜브로 옮긴다. 4) CMF 핸크스용액 2.6 ml, 2.5 % 트립신액 0.3 ml, 0.2 % DNase 액 0.1ml 를 교감신경절이 들은 원심튜브에 가하고 37 도에서 30 분간 인큐베이트한다. 5) 신경절을 튜브 바닥에 남긴 채 트립신액을 뽑아내어 버린 다. 10% 마혈청이 들어있는 배양액 2ml 를 가하고 파스출
피펫으로 20 번 상하운동에 의한 피페팅을 한다. 다음에 파스 출 피펫 끝에 미크로피펫립을 끼우고 20 번 피페팅을 한다. 이 시접에서 세포의 분리가 잘 되어 있을 것이나 만일 조직 잔재가 남아있으면 튜브를 3 분간 정치한 뒤 세포가 부유하고 있는 상청액을 새 원심튜브에 옮기고 남은 잔재에 배양액 1ml 를 가한 뒤 위에 적은 바와 같이 피페팅을 되풀이한다. 6) 세포부유액을 한 튜브에 모으고 1000r p m 으로 8 분의 원심 분리롤 한다. 7) 상청액을 버리고 배양액 2 ml 를 가하고 1000 r p m 으로 8 분 의 원심분리를 한다. 8) 페렛에 배양액 1ml 를 가하여 세포를 부유시킨 뒤 미리 준 비해 두었던 폴리라이신 코팅한 커버글라스 위에 플레이팅한 다. 배양액 조성은 다음과 같다. 이글 MEM 86ml 마혈청 우태아혈청 10ml 10 µg/m l NGF 1 ml 1 mM 시토신아라비노시드 1 ml 50% 포도당액 1ml 2mg /m l 겐타마이신액 1ml 9) 다음날 3ml 의 배양액을 커버글라스가 들어있는 6cm 페트 리 접시에 추가한다. 3 일 뒤에 위에 적은 배양액에서 시토신 아라비노시드를 제거한 배양액으로 교환한다. 10) 2 주일 뒤에 시토신아라비노시드 처리를 되풀이한다. 2 일간 처리한 뒤에 정상 배양액과 교환한다. 〈해설〉
앞의 18.1 에서는 척수후근신경절에서 순수한 신경세포를 얻기 위해 DNA 합성저해제인 풀로로디옥시유리딘의 첨가에 의해서, 여기에서는 교감신경절 신경세포 배양을 위해서 역시 DNA 합성 저해제인 시토신아라비노시드 사용으로 바신경세포인 슈완세포와 섬유아세포의 증식을 억제하고 배양신경세포의 순수도를 증가시 키고 있다. 이와 갇은 방법으로 중추신경계에서도 신경세포의 순 수배양이 가능하다. 이밖에도 방사선 조사에 의해서 증식세포의 증가 를 방지하고 신경세포의 순수배양이 가능한데 세슘이나 코발 트 방사능에 의해서 5000rad 의 감마선 조사를 하면 된다. 18.3 태생 래트(마우스) 대뇌 신경세포의 · 순수배양 태생 13-14 일의 래트나 마우스의 대뇌에서는 태생 7 일부터 시 작한 신경세포의 증식 , 이동이 계속되어 대뇌의 대부분의 구성 세포가 신경세포이고 가장 중요한 신경교세포인 성상교세포(아스 트로사이트)가 겨우 발생을 시작한 시기이다. 따라서 태생 13-14 일의 래트 대뇌의 분리세포 배양에서 신경세포 순수배양이 가능 하다. 세 가지의 기본적인 방법이 있는데 그 하나는 25cm2 플라 스크나 75C 而 풀라스크에 대량으로 분리세포 배양을 하고 7 일에 서 10 일 사이에 셰이킹을 하여 부착하였던 신경세포를 부유시키 고 이것을 모아서 배양하는 것이고 두번째 방법은 배양 초기에 DNA 합성저해제인 시토신아라비노시드나 풀로로디옥시유리딘 처리를 하여 섬유아세포나 성상교세포의 증식을 초기에 정지시키 는 것이고 세번째 방법은 무혈청 배양액에서 배양하여 증식세포 의 증가를 방지하는 방법이다.
〈 준비 〉 18.1 의 태생 래트 후근신경절 배양과 같다. 〈 방법 〉 1) 임신 13-14 일 래트를 에텔자 속에 넣고 에텔 심마취에 의해 서 마취사시킨다. 2) 18.1 태생 래트 후근신경절 세포 섹션에서 기술한 대로 태 생 래트롤 분리한다. 태아는 핸크스 용액이 들어있는 10cm 페트리 접시에 넣는다. 태생 래트 8-12 마리를 사용한다. 3) 태생 래트 머리피부를 제거한 뒤 두 안구 사이에 10cm 가 위로 절개를 가한다. 좌우로 전개하는 요령으로 두개골을 둥 글게 절개해서 제거한다• 후뇌 olfa c to r y bulb 에서 연수 medulla oblong a ta 에 이르는 뇌부분을 꺼내어서 핸크스 용액 이 들어있는 페트리 접시에 옮긴다. 4) 입체해부현미경 아래에서 종이를 벗기듯이 뇌막을 벗긴다. 다음에 수술용 칼 2 개를 사용하여 대뇌와 중뇌 사이를 절단 한다. 대뇌를 조그만 조직편으로 자른다. 5) 대뇌조직편을 50ml 원심튜브에 옮기고 여분의 핸크스 용액 울 제거한다. 튜브에 CMF 핸크스 용액 18ml, 2.5% 트립 신 2 ml, 0 . 2 % DNase 액 0 . 4 ml 를 가하고 37 도에 서 30 분간 인큐베이트한다. 6) 대뇌조직편을 바닥에 남긴 채 트립신액을 뽑아 버린다. 10 % 마혈청이 둘어있는 배양액 10 ml 와 0.2 % DNase 0.2 ml 를 가하고 파스출 피펫으로 20 번 피페팅을 한다. 다음에 파스출 피펫 끝에 미크로피펫립을 삽입하고 20 번 피페팅을 한다. 이 시점에서 조직편이 남아있으면 튜브를 3 분간 정치 한 뒤 세포가 부유하고 있는 상청액을 새 튜브에 옮기고 남
은 조직편에 배양액 4ml 를 가한 뒤 위와 같이 피페팅을 되 풀이한다. 7) 세포부유액을 한 튜브에 모으고 1000r p m 에서 8 분간 원심 분리한다. 8) 상청액을 버리고 배양액 4ml 를 가하고 1000r p m 에서 8 분 간 윈심분리한다. 9) 페렛에 배양액 10ml 를 가하고 세포를 부유시킨 뒤 폴리라 이신 코팅한 풀라스크에 플레이팅을 한다. 세포 수 계산울 하여 플라스크 하나에 5X106 세포가 되도록 한다. 배양액 조성은 다음과 같다. 이글 MEM 88ml 마혈청(우태아혈청) 10ml 50% 포도당액 1 ml 2 mg /m l 겐타마이 신액 1 ml 10) 7 일 뒤에 배양액울 교환하여 10ml 의 새 배양액을 넣는다. 플라스크를 셰 이 커 orbit al shaker 위 에 테 이 프로 단단하게 고정한다. 1 분에 250 회전으로 1 시간 셰이킹을 하고 주로 미 크로글리아를 포함한 배양액을 페트리 접시에 옮기고 새 배 양액 15ml 를 가한다. 플라스크롤 두번째 셰이커에 고정하 고 1 분에 250 회전의 속도로 18-20 시간 동안 셰이킹을 한다. 11) 풀라스크 셰이킹에서 모은 배양액 속의 세포는 주로 신경 세 포인데 이들을 원심튜브에 모아서 1000 rp m 에서 8 분간 원심분리한다. 12) 상청액을 버리고 페렛에 배양액 5ml 를 가하여 세포를 부 유시키고 미리 준비해 두었던 폴리라이신 코팅한 커버글라 스나 6cm 페트리 접시에 플레이팅한다. 배양액 조성은 다음과 같다.
이글 MEM 88 ml 마혈청(우태아혈청) 10 ml 50% 포도당액 1 ml 2 mg /m l 겐타마이신액 1 ml 13) 다음날 3ml 의 배양액을 커버글라스가 들어있는 6cm 페 트리 접시에 추가한다. 3 일 뒤에 위에 적은 배양액에 시토 신아라비노시드를 5µM 농도로 가해 주고 2 일간 처리해 주 면 성상교세포나 섬유아세포가 자라는 것을 방지할 수 있 다. 14) 배양 시작 3-4 일 뒤에 무혈청 배양액 DM4 로 배양액 교환 을 하면 역시 비신경세포가 자라는 것을 최소한으로 방지할 수가있다. 무혈청 배양액 (DM4) 의 조성은 다음과 같다. 이 글 MEM (혹은 F12) 97 ml 50% 포도당액 1ml 2 mg /m l 겐타마이 신 액 1 ml 영양인자-호르몬 칵테일 1ml 영양인자-호르몬 칵테일액은 각종 호르몬의 1000 배 스토 크 0.1ml 씩과 이글 MEM 0.5ml 를 혼합하여서 1 ml 의 분 주 스토크를 만들고 -20 도에서 얼려서 보관한다. 스토크의 조성은 다음과 같고 1000 배 스토크액은 멤브렌 여과 멸균한 다. 인슐린 ins uli n 10mg /m l 트란스페 린 tra nsfe r rin 10 mg /m l 셀 렌나트륨 sod ium selenit e 44 mg /m l, 3 X 10-5M 트리 요도티 로닌 tri io d oth yron ine 0 . 2 mg /m l, 3 x 10-1M 히 드로코티 존 hy dr ocort iso ne 18 mg /m l, 5 X 10-5M
〈 해설 〉 위에서 기술한 방법에 따라서 신경세포를 완전히 제거하면 플 라스크에 남아있는 세포는 주로 성상교세포(아스트로사이트)로서 풀라스크의 바닥을 덮고 있으며 그 순도가 95-99 %에 이른다. 이같이 신경세포 순수배양을 하면 동시에 성상교세포 순수배양을 얻을 수가 있다. 저자의 연구실에서도 이 방법으로 태생 인간 신 경세포의 순수배양을 하고 있는데 이러한 신경세포 배양에서 신 경 세 포가 어 떠 한 신 경 영 양인 자 neurotr o p h ic fac to r 를 생 산하고 있 는가란 과제를 면역염색, 웨스턴 불러트, 분자생물학 방법에 의 한 PCR 분석을 하고 있다. 최근 저자의 교실에서 발표한 논문 에 의 하면 태 생 인 간 신 경 세 포가 NGF, BDNF (brain deriv e d neurotr o p h ic fac to r ) , NT-3 (neurotr o p h in - 3) 그리 고 미 드카인
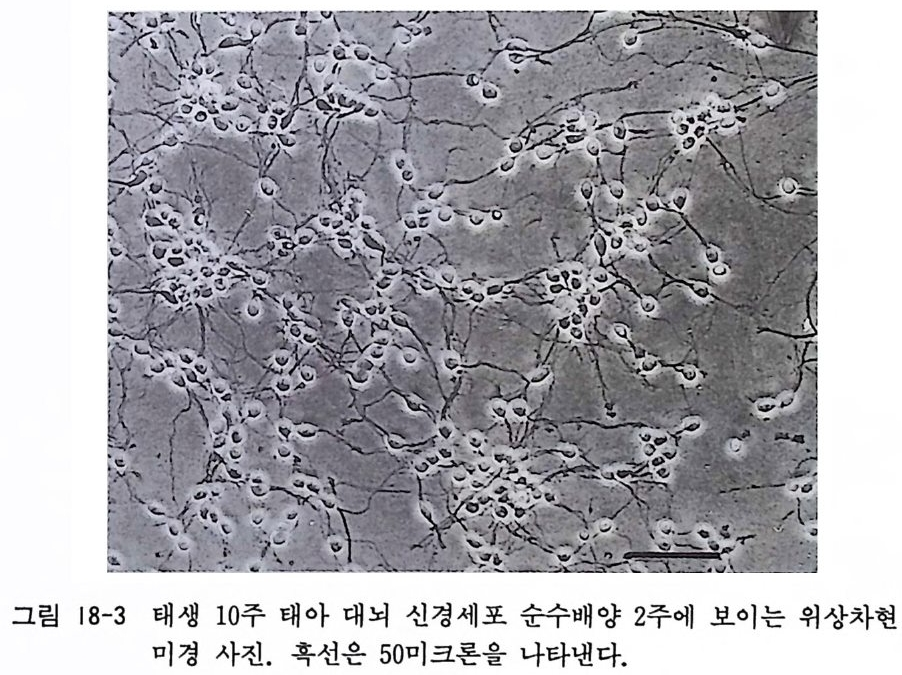 그림 18-3 태생 10 주 태아 대뇌 신경세포 순수배양 2 주에 보이는 위상차현
그림 18-3 태생 10 주 태아 대뇌 신경세포 순수배양 2 주에 보이는 위상차현
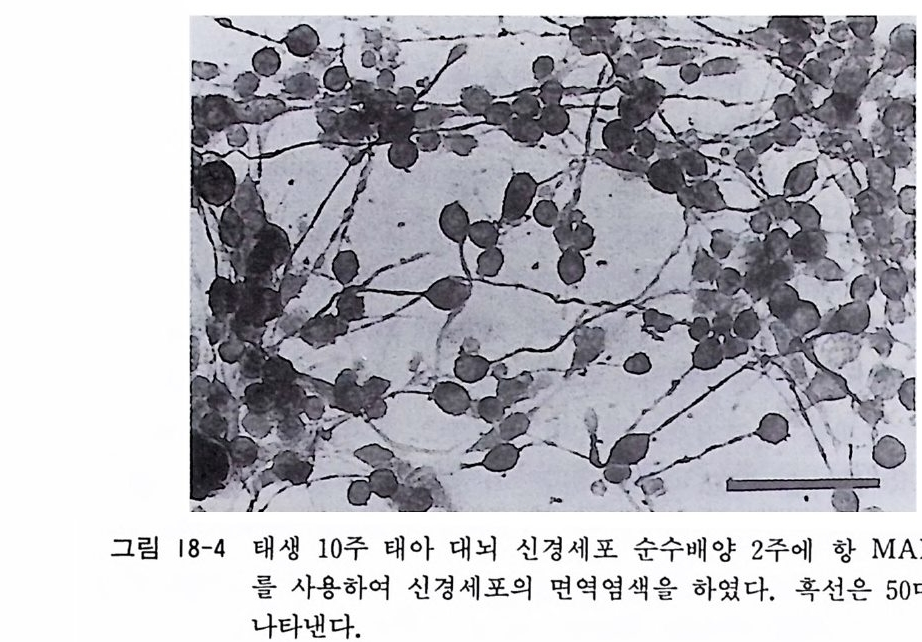 었4
었4
m i dk i ne 을 생산하고 있음을 실증하였다 .3,4) 그림 18 - 3 과 18-4 는 태생 인간 대뇌 신경세포 순수배양을 보이고 있다. 18.4 항체 의존 세포 분리 기술(패닝)에 의한 신생 래 트(마우스) 망막 신경세포 순수배양 면역조직에서 각종 면역세포, 특히 T 임파구와 B 임파구를 특 이하게 분리하는 방법으로서 개발된 항체 의존 세포분리 기술 anti bo dy media t e d cell sepa rati on 이 면역학 분야에서 크게 공헌 울 한 바 있다 .5) 이렇게 면역학적 기술을 응용한 신경세포의 순 수분리도 역시 가능하다. 흉선세포의 특이항원인 Th y -1 이 래트 에서는 T 세포 이외에도 망막의 신경세포의 특이세포 마카라 알
려져 있는데 이 Thy -1 항체를 이용한 항체 의존 세포분리 기술 을 여기에서 소개한다 .6) 이 세포 기술은 사금을 채취할 때에 쓰 는 기술과 유사하다 하여 패닝 p ann i n g이라 한다. 〈 준비 〉 1) 입체해부현미경과 조명 2) 수술기구 -10 cm 의과용 가위 (직선과 곡선) 2 개 10 cm 의과용 핀셋 1 개 시계수선용 핀셋 (No. 4) 2 개 수술용 칼 (No. 11) 2 개 3) 페트리 접시, 원심튜브 4) 핸크스 영류용액 5) 파파인 (이글 MEM 에 희석) 시 스데 인 (cy st e i n , 시 그마) 6) 이글 MEM 합성 배양액 7) 마혈청 8) 50 % 포도당액 9) 2 mg /m l 겐타마이신액 〈 방법 〉 1) 신생 래트 혹은 마우스 (생후 1 일에서 5 일)를 에텔자에 넣어 서 마취사시키든가 -20 도 냉장고에 10 분 가량 방치하여 냉 마취를 한다. 2) 래트를 종이타월 위에 눕히고 눈 주위를 70 % 알코올로 소 독한다. 3) 좌우 양측의 안구를 핀셋과 10 cm 가위를 사용하여 도려내 고 핸크스 용액이 들어있는 페트리 집시에 모은다.
4) 왼손에 잡은 핀셋으로 안구를 고정하고 가위로 안구를 절단 하고 안구 내부조직을 유리시킨다. 5) 반두명한 망막이 다른 안구조칙에서 유리되면 이들을 모은 다. 6) 파파인 용액 (10 unit s/m l) 2 ml 이 들어 있는 15 ml 원심튜브 에 망막을 옮기고 37 도에서 40 분간 인큐베이트한다. 파파인 용액의 준비는 17.2 의 성숙 래트 망막세포 배양의 섹션을 참 고할것. 7) 파파인 용액을 버리고 혈청이 들어있는 배양액 2ml 와 0.2 % DNase 용액 0.1 ml 를 가한 뒤 파스출 피펫으로 20 번 피페팅을 한다. 계속하여 파스출 피펫 끝에 미크로피펫팁을 삽입하고 20 번 피페팅을 한다. 8) 1000r p m 의 속도로 8 분간 원심분리한다. 9) 페 렛에 배 양액 2 ml 를 가하고 1000 r p m 의 속도로 8 분간 원 심분리한다. 10) 페렛에 4ml 의 이글 MEM 액을 가하여 부유액을 만들고 6 cm 직경 페트리 집시에 분주하고 1 시간 실온에서 인큐베이 트한다. 이 과정에서 섬유아세포, 미크로글리아가 접시 표 면에 부착한다. 11) 페트리 집시에 부착하지 많은 세포를 이글 MEM 액과 같 이 꺼내서 나일론 멧슈로 여과한 뒤 18 . 5 에 적은 패닝 접시 에 넣는다 (18 . 5 참고). 실온에서 1 시간 인큐베이트 한 뒤 매번 3ml PBS 를 써서 4 번 페트리 집시롤 씻어내고 부착 하지 않은 세포를 제거한다. 12) CMF- 핸주스액에 희석한 0.1 % 트립신 3ml 과 0.2 % DNase 액 0.1ml 를 패닝 집시에 가하고 실온에서 15 분간 방치한다. 마혈청 0.5ml 를 패닝 접시에 가하여서 트립신을
비활성화시킨 뒤 피페팅을 하여 접시 표면에 부착한 망막 신경세포를 원심듀브에 모으고 1000r p m 으로 8 분간 원심분 리한다. 13) 페렛에 배양액을 가하고 1000r p m 으로 8 분간 원심분리롤 되풀이한다. 14) 페렛에 혈청을 포함한 배양액 2ml 를 가하여 세포부유액을 만들어 폴리라이신 코팅한 커버글라스에 플레이팅을 하고 5 % CO2 인큐베이터에 넣어서 배양을 시작한다. 배양액 조성은 다음과 같으며 일주일에 두 번 배양액 교환 울한다. 이글 MEM 88ml 마혈청 (우태아혈청) 10 ml 50% 포도당액 1ml 2 mg / ml 겐타마이신액 1 ml 〈 해설 〉 파파인 대신 트립신을 사용하여도 되는데 파파인 효소 처리가 신경세포 분리에는 상해가 덜 하여서 건강한 신경세포를 분리할 수가 있다. 위의 방법에 의해서 95% 이상 순도의 래트 망막 신 경세포를 분리할 수가 있다. 위에 적은 Thy -1 대신에 각종 신경 세포의 표면항원인 GQ 강그리오시드에 대한 마우스 단클론 항체 A2B5 를 사용하여 신경세포의 순수분리를 할 수가 있다 .7)
18.5 패닝 접시 제작법 〈방법〉 1) 6cm 직경 페트리 접시 2 개에 O.lmg /m l 농도의 항 마우 스 IgG 혹은 항 마우스 I g M 을 녹인 PBS 2 ml 를 가하고 4 도 냉장고에서 18-24 시간 인큐베이트한다. 2) 항 마우스 I g G 나 IgM 액을 피펫으로 꺼내어서 듀브에 넣 는다. 이 I g G 액이나 I g M 액은 10 번까지 더 쓸 수 있으므로 튜브에 몇 번 사용하였나 기록해 둔다. 페트리 접시는 PBS 3ml 로 매 번 3 번 씻는다. 3) 마우스 항 Thy -1 단클론 항체 (I gG나 l g M) 를 0.1 mg / ml 농도로 PBS 에 희석하고 페트리 접시에 가한 뒤 1 시간 실온 에서 인큐베이트한다. 마우스 항 A2B5(1 g M) 로 대신 인큐베 이트할 수도 있다. 4) 항체를 제거하고 (2 번 더 쓸 수가 있다) 3 ml 의 PBS 로 페트 리 접시를 씻는다(접시는 2 주일간 유효하다). 5) 비특이반응울 방지하기 위하여 0. 2 % 우혈청 알부민이 들 어있는 PBS 2ml 를 페트리 집시에 가하고 실온에서 20 분간 방치한다. 패닝 칙전에 혈청 알부민을 제거한다. 페트리 접 시는 씻지 않고 패닝 프로세스에 들어간다.
제 19 장 슈완세포의 순수배양 슈완세포 Schwann cell 는 말초신경계에서 수초의 형성과 그 구 조와 기능을 유지하는 신경교세포로서 말초신경계 질환은 많은 경우 슈완세포의 대사이상이나 면역이상이 그 원인이라 생각되고 있다. 기앙 • 바레 말초신경영 Guil lan -Barre s y ndrome 에서는 슈완 세포의 표면항원에 대한 자가면역반응이 그 원인이라는 학설이 유력하다. 신경섬유종 neuro fi broma t os i s 은 슈완세포나 그 전구세 포의 악성 변이에 의해서 일어난 병이라는 설이 역시 유력하다. 이같은 슈완세포의 이상에 원인이 있는 질환의 병리와 그 발병기 전을 이해하기 위해서는 슈완세포의 순수배양이 유력한 방법이 되겠다. 실험동물 특히 래트의 슈완세포의 순수배양을 하는 데에는 두 가지 방법이 있는데 그 하나는 태생 래트 척수후근신경절을 이식 편으로 배양하여 DNA 합성저해제로 섬유아세포를 제거하고 슈 완세포와 신경세포를 남긴다. 그 다음에 신경세포가 모여있는 배 양의 중앙부를 잘라내고 슈완세포의 증식을 기다린다는 것이다.1)
다른 하나는 신생 래트 좌골신경의 분리세포 배양을 만들어서 DNA 합성저해제로 섬유아세포를 제거하고 신경영양인자와 콜레 라톡신에 의해서 슈완세포의 증식을 도모한다는 것이다 .2) 19.1 태생 래트(마우스) 척수후근신경절 이식편 배양 에 의한 슈완세포 배양 아래에서 소개한 신경절 이식편 배양에 의한 슈완세포의 순수 배양은 시간과 노동력을 크게 필요로 하는 방법이어서 별로 환영 받지 못하고 있는 기술이다. 그러나 역사적인 의미에서 극히 의 미 깊은 기술이기에 여기에서 소개한다. I) 〈준비〉 1) 입체해부현미경과 조명 2) 수술기구 -10cm 의과용 가위(직선과 곡선) 2 개 10 cm 의과용 핀셋 2 개 시계수선용 핀셋 (No. 4) 2 개 수술용 칼 (No. 11) 2 개 3) 시험관, 페트리 접시, 원심튜브 4) 칼슘, 마그네슘 제거 핸크스 영류용액 5) 2.5 % 트립신액 6) 0.2 % DNase 효소액 7) 이글 MEM 합성 배양액 8) 마혈청 9) 50% 포도당액 10) Zmg /m l 겐타마이신액
11) DNA 합성저해제(시토신아라비노시드와 플로로디옥시유리딘) 〈 방법 〉 1) 18.3 태생 래트 후근신경절세포 배양에서 기술한 대로 래트 태아를 16-20 일 임신 래트에서 분리한다. 태아를 10cm 페 트리 접시에 배를 아래로 해서 눕힌다. 2) 의과용 가위로 태아의 머리를 자르고 등부분의 피부를 제거 한다. 척주와 척수를 포함한 후배조직을 크게 절단하고 새 10cm 페트리 접시에 옮기고 척주 전면에 붙은 내장과 꼬리 를 제거한다. 3) 입체해부용 현미경 아래에서 10cm 곡선 의과용 가위를 사 용하여 척주후배부의 근육과 척추골을 절개하고 제거한다. 다음에 척수조직을 제거하면 경수에서 천수에 이르는 사이에 30 쌍의 척수후근신경절이 양측에 나란히 놓여있다. 4) 직경 3.5cm 페트리 접시 안에 건조 콜라겐(아래를 참고할 것)을 코팅한 22mm 직경의 원형 커버글라스를 놓는다. 커 버글라스 위에 신경절을 2 개씩 올려놓고 배양액 A 를 한 방 울 떨어뜨린다. 5) 페트리 집시의 뚜껑을 덮고 파라핀 _ 바셀린으로 밀폐한다. 페트리 접시는 37 도에서 인큐베이터에 정치한다. 배양액 A 의 조성은 다음과 갇다. 이글 MEM 82ml 우태아혈청(혹은 사람 태반혈청) 10ml 계배 추출액 2ml 50 % 포도당액 1ml 10 µg/m l NGF 1 ml 2mg /m l 겐타마이신액 1ml
l mM 시토신아라비노시드 1 ml lmM 플로로디옥시유리딘 1ml lmM 유리딘 1ml 6) 배양후 3 일에 배양액에서 DNA 합성저해제를 빼고 만든 배 양액 B 로 교환한다. 배양액 B 의 조성은 다음과 같은데 1 주 일에 2 번 교환한다. 이글 MEM 62ml 우태아혈청 (혹은 사람 태반혈청) 25ml 계배 추출액 10ml 50% 포도당액 1ml 10 µg/m l NGF 1 ml 2 mg /m l 겐타마이신액 1 ml 7) 1 주일에서 10 일 뒤에 신경세포가 밀집해 있는 이식편 주위 룰 잘라낸다. 잘라낸 신경세포 이식편은 먼저 자리에서 멀리 떨어진 슈완세포 위에 이식한다. 슈완세포는 신경섬유에 의 해서 그 증식이 수 배로 증가한다고 보고되어 있어서 신경세 포 주위에서 슈완세포 수가 크게 증가한다. 19.2 태생 래트(마우스) 척수후근신경질 분지세포 배 양에 의한 슈완세포 배양 저자의 연구실에서 슈완세포 순수배양에 사용하고 있는 배양기 술을 여기에서 소개한다. 이 방법은 19.3 에서 소개하는 좌골신경 배양과 그 기술이 비슷하다. 신경영양인자인 NGF 를 배양액 에서 제의하면 신경세포가 2 주일 뒤에 소멸하게 되어 슈완세포와 섬유아세포만 남게 되는데 섬유아세포 역시 DNA 합성저해제인
시토신아라비노시드와 풀로로디옥시유리딘을 병용하여서 사멸시 킨다. 그 다음에 슈완세포의 증식인자인 글리아 성장인자(gli a grow t h fac to r, GGF) 와 아데 닐시 클라제 효소 adeny l cyc lase 의 활 성 인자인 콜레 라톡신 cholera tox in 을 배 양액 에 넣 어 서 슈완세 포 를 최대한으로 증식시킨다. 〈 준비 〉 19 . 1 과 같다. 〈 방법 〉 1) 19.1 에서와 마찬가지로 16-20 일 래트(혹은 마우스) 태아를 사용한다. 2) 시계수선용 핀셋을 사용하여 신중하게 후근산경절을 하나씩 분리하고 CMF- 핸크스액이 들은 6cm 페트리 접시에 모은 다. 태아 한 마리에서 40-50 개의 신경절이 분리되어서 10 마 리 태아에서 400 개의 신경절을 모을 수가 있다. 10 마리의 태 아에서 신경절을 분리하려면 1-2 시간이 걸리므로 신경절이 들어있는 페트리 접시를 얼음 위에 정치한다. 3) 신경절을 핸크스액과 갇이 15ml 원심튜브에 옮긴 뒤 여분 의 핸크스액을 버린다. 원심튜브에 4.3ml 의 CMF- 핸크스 액, 0.5ml 의 2.5 % 트립신액, 0.2ml 의 0.2 % DNase 액을 차례로 넣고 37 도에서 30 분간 인큐베이트한다. 4) 신경절 조직을 바닥에 남긴 채 트립신액을 버리고, 마혈청 이 들어 있는 배 양액 3 ml 와 0 . 2 % DNase 0 . 2 ml 를 가한 뒤 20 번 피페팅을 한다. 다음에 파스출 피펫 끝에 미크로피펫팁 을 삽입하고 20 번 피페팅을 되풀이한다. 만일 조직 잔재가 남아 있으면 듀브롤 3 분간 정치한 뒤 세포부유액을 새 튜브
에 옮기고 남은 잔재에 배양액 2ml 롤 가한 뒤 위와 같이 피 페팅을 되풀이한다. 5) 세포부유액을 한 튜브에 모으고 1000r p m 으로 8 분의 원심 분리를 한다. 6) 상청액을 버리고 페렛에 배양액 3ml 를 가하고 1000r p m 에 서 8 분간 원심분리한다. 7) 상청액을 버리고 페렛에 배양액 5ml 를 가하여 세포를 부유 시킨 뒤 미리 준비해 두었던 폴리라이신 코팅한 커버글라스 나 페트리 접시에 플레이팅한다. 배양액 조성은 다음과 갇 다• 이글 MEM 81ml 우태아혈청 (혹은 사람 태반혈청) 10ml 계배 추출액 5ml 50% 포도당액 1ml 2mg /m l 겐타마이신액 1ml 10 µg/m l NGF 1 ml 0.5mM 시토신아라비노시드 1ml 8) 2 일 뒤에 NGF 와 DNA 합성저해제인 시토신아라비노시드 룰 제의한 배양액으로 교환하고 계속하여 3 일에 한번씩 배양 액 교환울 한다. 9) 2 주일 뒤에 섬유아세포가 계속해 있으면 위에 적은 배양액 에 최종 농도 5 µM 의 시토신아라비노시드를 첨가하고 2 일간 처리한다. 10) 장기간 배양하여 4 주일에서 8 주일이면 대량의 슈완세포를 분리할 수가 있다.
19.3 선생 래트(마우스) 좌골신경 분리세포 배양에 의 한 슈완세포 배양 〈 준비 〉 19.1 과 같다. 별도로 0.2 5 % 콜라게나제를 준바할 것. 〈 방법 〉 1) 신생 래트(혹은 마우스)를 에텔자에 넣어서 마취사시키든가 ― 20 도 냉장고에서 10 분간 방치하여 냉마취를 한다. 2) 래트 를 70 % 알코올이 담긴 비커에 잠깐 담가서 멸균소독 한다. 3) 래트를 페트리 접시에 배를 아래로 하여 눕힌다. 4) 허리에서 허벅다리에 걸쳐서 피부를 제거한다. 시계수선용 핀셋을 사용하여 정중선에서 의하방으로 근육충을 갈라가며 좌골신경을 찾아낸다. 약 0.8cm 정도의 좌골신경을 양측에 서 분리한다. 신생 래트 10 마리에서 좌골신경을 모은다• 5) 좌골신경을 모은 15 ml 원심튜브에 0.2 5 % 콜라게나제 1 ml 와 0.2 % DNase 0.1 m l 를 가하고 37 도에서 1 시간 인큐 ’ 베이트한다. 6) 조직을 바닥에 남긴 채 효소액을 버린 뒤에 배양액 3ml 와 0.2 % DNase 0.1ml 를 가하고 피페팅을 20 회 한다. 파스출 피펫 끝에 미크로피펫팁울 삽입하고 피페팅을 20 회 되풀이한 다. 7) 1000r p m 의 속도로 8 분간 원심분리한다. 8) 페 렛에 3 ml 의 배 양액을 가하고 lOOO rp m 의 속도로 8 분간 원심분리한다. 이 과정을 한번 더 되풀이한다. 9) 페렛에 배양액 A 를 9ml 가하여 세포부유액을 만들고 폴리
라이신 코팅한 6cm 페트리 접시 3 개에 3ml 씩 분주한다. 신생 래트 10 마리의 좌골신경에서 5X 1 06 세포가 분리될 수 있다. 배양액 A 의 조성은 다음과 같다. 이글 MEM 88 ml 우태아혈청(마혈청) 10 ml 50% 포도당액 1 ml 2 mg /m l 겐타마이 신액 1 ml 10) 배양 3 일째에 위의 배양액에 DNA 합성저해제인 시토신아 라비노시드 5µM 을 가한 배양액 B 와 교환하여 2 일간 처리 한다. 이 뒤에 다시 배양액 A 를 사용한다. 11) 배양 10 일째에는 배양액을 제거하고 0.02 % EDTA 를 포 함한 PBS 로 2 번 씻고 0.05 % 트립신이 들은 PBS 3 ml 를 가하고 37 도에서 15 분간 처리한다. 12) 파스출 피펫으로 조용하게 피페팅을 하면 슈완세포가 시트 와 같이 접시에서 벗겨져 나온다. 이들을 원심튜브에 모아 서 1000 rp m 으로 8 분간 원심분리한다. 13) 페렛에 배양액 A 6ml 를 가하고 다시 원심분리한다. 14) 페렛에 배양액 A 9ml 를 가하고 여기에 소 뇌하수체에서 분리 한 GGF 5 µg /ml 와 콜레 라독신 1 µg /ml 를 첨 가하여 서 세포부유액을 만든다. 6cm 페트리 접시 3 개에 3ml 씩 분 주하여 37 도에서 배양을 계속한다. 배양액 교환은 1 주일에 2 번 한다. 〈해설〉 신생 래트 좌골신경으로부터 슈완세포를 순수배양하는 방법은 브로카스 Brockes 에 의해서 시작되었는데 2) 그 뒤 이 방법을 사용
 그림 19-1 죽은 지 6 시간 되는 39 세 성인 삼차신경절에서 분리한 슈완세
그림 19-1 죽은 지 6 시간 되는 39 세 성인 삼차신경절에서 분리한 슈완세
하여 마우스, 고양이, 사람에서 슈완세포의 대량 배양을 하였다 는 보고가 있다 .3-5) 그림 19-1 과 19-2 에서는 사망 후 6 시간이 된 39 세 사람의 삼차신경에서 분리된 슈완세포의 배양을 보이고 있 다.
제 20 장 올리고덴드로사이트의 순수배양 울리고덴드로사이트(희돌기교세포, o lig odendroc yt e) 는 아스트로 사이트(성상교세포, astr o cy te) 그리고 미크로글리아(소교세포, m ic ro gli a) 와 함께 중추신경조직에서 신경교세포 neuro gli a 중의 하나이며 신경세포를 보조하는 역할을 가지고 있다. 올리고덴드 로사이트는 신경섬유를 여러 충으로 둘러싸서 수초(미엘란 m y e li n) 을 형성하고 또한 이들울 유지하는 기능을 가지고 있다. 수초는 신경의 흥분 전도에 중요한 역할을 가지고 있어서 급속한 도약 전도를 가져오게 한다. 다발성 경화증 mul tip le sclerosis 등 의 사람 탈수질환 demy el i na ti ng dis e ase 에서는 울리고덴드로사이 트는 질병의 표적이 되어서 탈수 부위에서 수초와 함께 소실하여 서 중추신경계의 기능 장해를 가져온다. 1,2) 이러한 탈수질환을 연구함에 있어서 대량의 올리고덴드로사이트를 순수하게 분리하 여 그 형태, 기능 대사 등을 콘트롤된 환경에서 검색하는 것이 바람직하다. 이러한 요구에 의해서 포유동물 울리고덴드로사이트 의 순수배 양과 그 배 양이 가능하게 된 것 이 1980 년 초이 다. 3, ◄)
올리고덴드로사이트 분리와 배양 방법을 두 가지로 대별할 수가 있는데 그 하나는 성숙 포유동물의 뇌에서 효소에 의한 분리세포 배양을 하고 밀도구배컬럼으로 울리고덴드로사이트를 순수분리하 는 방법이며》 다론 하나는 신생아 포유동물 뇌에서 분리세포 배양을 하여서 배양 1 주일 뒤에 셰이킹에 의해서 물리적으로 올 리고덴드로사이트를 분리시키는 방법이다 .5) 현재까지 사람, 소, 돼지, 양, 고양이, 마우스, 래트 뇌에서 올리고덴드로사이트의 순수배양의 보고가 있다 .6-9) 20.1 퍼컬 밀도구배 컬럼에 의한 올리고덴드로사이트 순수분리와 배양 올리고덴드로사이트의 순수분리에서 조속한 시간 안에 대량의 올리고덴드로사이트를 얻을 수 있고 성숙 동물의 뇌에서도 올리 고덴드로사이트롤 분리할 수 있다는 이유에서 퍼컬 컬럼이 가장 유용한 방법이라 하겠다. 저자의 연구실에서는 이 방법을 사용하 여 성숙 소, 돼지, 양 그리고 사람 뇌(사후 20 시간 이내 부검에서 얻은 샘플)에서 울리고덴드로사이트를 대량으로 분리하고 배양하 고 있다 (그립 20-1) . 〈준비〉 1) 수술기구 -15 cm 의과용 가위 1 개 10cm 의과용 핀셋 2 개 수술용 칼 (No. 11) 2 개 2) 시험관, 페트리 접시, 원심튜브 3) 오크릿지 원십튜브 Oakrid ge tub e
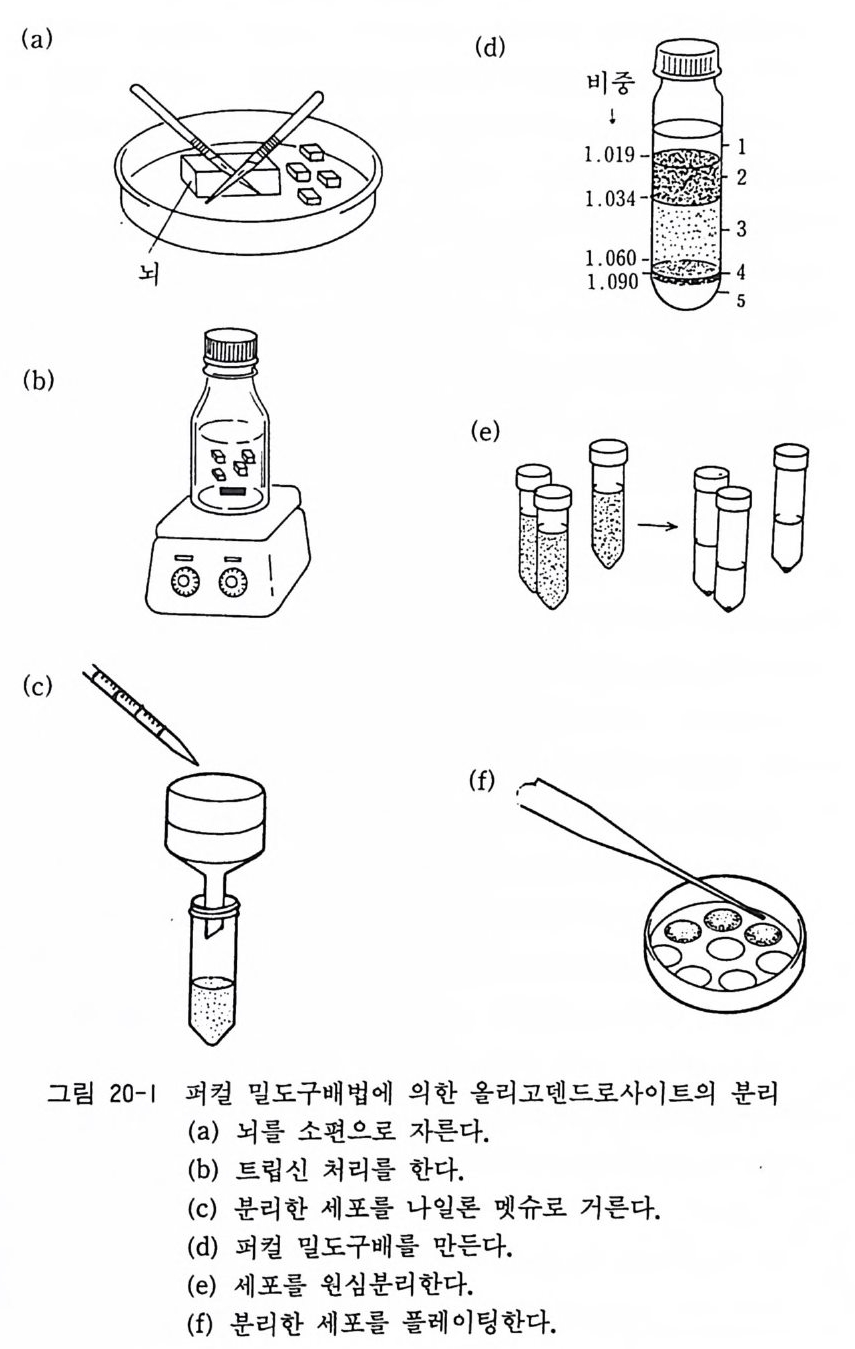 (a) (d)
(a) (d)
4) 뷔 크너 깔때 기 Bil ch ner fun nel lOOµm 나일론 멧슈를 부착시킨다. 5) 퍼콜 용액 Percoll( 파마시아 Pharmaci a) 6) PBS 7) 핸크스 영류용액 -10 배 농축 핸크스영류용액 8) 2.5% 트립신액 9) 0.2 % DNase 액 10) 이글 MEM 합성 배양액 11) 마혈청 12) 50 % 포도당액 13) 2 mg / ml 겐타마이 신액 〈방법〉 1) 도살장에서 사후 1 시간 이내의 성숙한 소, 돼지, 양의 뇌를 구매하고 오토클레이브 멸균한 1 리터 비커에 넣어서 연구실 로 운반한다(뚜껑으로는 오토클레이브 멸균한 알루미늄 포일을 사용한다). 비커에는 미리 항생물질 (lOOOun it /ml 페니실린과 1000 µg/m l 스트렙토마이신) 이 첨가된 PBS 200 ml 를 넣어둔 다. 스티로폼 보온박스에 얼음을 넣고 그 속에 비커를 보관 하면 뇌조직의 변성을 방지할 수 있다. 2) 15cm 직경의 페트리 접시에 뇌를 옮기고 항생물질이 들어 있는 PBS 로 다시 한번 씻는다. 3) 뇌를 PBS 가 담긴 새 15cm 유리 페트리 집시에 옮기고 뇌 막, 뇌혈관을 가능한 대로 제거한다. 먼저 의과용 가위를 사 용하여, 이어서 2 개의 수술칼을 사용하여 뇌를 작게 자른다. 3X3X3mm 사이즈의 뇌 조직편이 적당한 사이즈가 된다. 뇌백질을 사용하는 것이 상책이나 백질 분리에 시간이 너무
걷리게 되므로 백질과 회백질을 가리지 않고 사용한다. 4) 흡입 펌프나 전공라인에 접속된 파스출 피펫으로 PBS 를 제거하고 신선한 PBS 로 뇌조직편을 씻는다. 5) 뇌조직편을 PBS 에 희석한 0.25 % 트립신 250ml 가 들어있 는 500ml 용량 배양 유리병에 옮긴다. 이때 10ml 풀라스틱 와이드마우스 w i de mouth 피펫와 피펫에이드를 사용해서 뇌 조직편을 , 옮긴다. 트립신액은 PBS 220 ml 에 2.5 % 트립신 25 ml 과 0 . 2 % DNase 5 ml 를 첨 가하여 서 만든다. 6) 배 양병 바닥에 스터 링 마그넷 sti rr in g magn e t 을 넣 어 서 37 도 에서 1 시간 스터러 위에서 뇌조직과 트립신이 혼합되도록 한 다. 7) 피펫에이드에 접속한 10ml 와이드마우스 피펫으로 30 번 피 페팅을 한다. 다음 10ml 혈액검사용 피펫으로 30 번 피페팅 을 한다. 이 시점에서 세포 분리가 많이 진전이 되어있다. 5 분간 조직편이 가라앉는 것을 기다린다. 8) 세포부유 상청액을 뷔크너 깔때기에 설치한 나일론 멧슈(그 림 20-2) 를 통하여 여과시킨다. 여과된 세포는 3 ml 의 마혈 청이 둘어있는 50ml 풀라스틱 원심튜브에 모은다. 9) 남아 있는 뇌조직편과 세포덩어리에 PBS 100ml 를 가하고 다시 위에서와 마찬가지로 피페팅을 되풀이한다. 나일론 멧 슈를 통하여 세포를 여과시킨다. 피페팅과 여과 과정을 한번 더 되풀이한다. 10) 원심튜브에 세포부유액이 모이면(이 시접에서 50ml 원십튜 브 10 개 정도가 모인다) 1500r pm 속도로 10 분간 원심분리한 다. 11) 상청액을 버리고 페렛을 20ml 핸크스용액에 부유시킨다. 12) 40 ml 오크릿지 튜브에 퍼콜액 9 ml, 10 배 농축 핸크스액
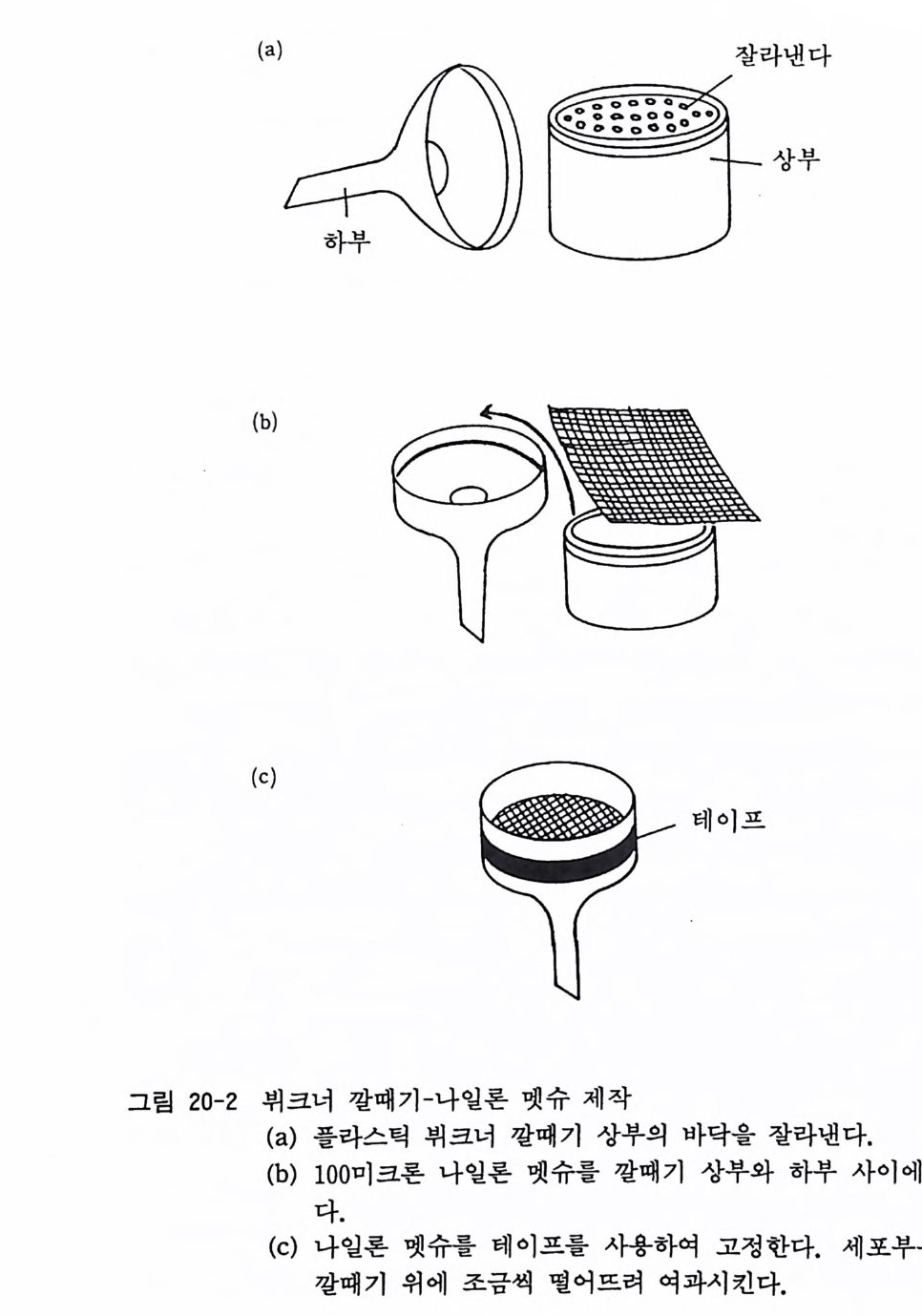 (a) 잘라낸다
(a) 잘라낸다
1ml 를 가하고 여기에 20ml 세포부유 핸크스액을 가한다. 결과적으로 30% 퍼콜액이 된다. 13) 고속원심기 Beckman 에서 퍼콜-세포혼합액을 15, 000 r p m 으 로 30 분간 원심분리한다. 14) 퍼콜 밀도구배 컬럼이 생성되어서 5 층의 구배 (그래디언트) 가 보인다. 위로부터 순서로 제 1 층에 핸크스액, 제 2 층에 수초와 데브리층(비중 1.02 5), 제 3 층에 울리고와 미크로글 리아가 부유하는 반두명충(비중 1.0 50), 제 4 층에 적혈구가 있는 충(비중 1.0 90), 제 5 층에 세포가 없는 두명충이 보인 다. 위에서 부터 제 1,2 층을 차례로 제거한다• 15) 제 3 층의 세포부유총(하나의 튜브에서 약 15ml 내의)을 새 50 ml 원심 튜브에 옮기고 30 ml 의 PBS 를 가한다. 1800 r p m 으로 15 분간 원심분리를 하여 퍼컬을 제거한다. 16) 상청액을 버리고 페렛에 5ml 의 PBS 를 가하고 1200rpm 으로 8 분의 원심분리를 한다. 17) 페렛에 4ml 의 마혈청이 들어있는 배양액울 가하여 세포부 유액을 만들고 10 개 튜브를 2 개의 듀브에 모은다. 18) 1200rpm 8 분의 원심분리를 한다. 상청액을 버리고 페렛 에 40 ml 의 배양액을 가한다. 19) 혈구계산판에 세포부유액 한 방울을 떨어트리고 현미경 아 래에서 세포 수 계산을 한다. 10o ·g 의 뇌에서 5Xl08 세포 가 분리된다. 10 개의 페트리 접시에 세포부유액을 . 8 rri l 씩 분주하고 그 위에 4ml 배양액을 추가하고 5% CO2 인큐베 이터에서 배양한다. 20) 48 시간 뒤에는 미크로글리아가 페트리 접시에 부착하고 올 리고는 세포덩어리를 형성하여 부유하고 있다. 부유액을 듀 브에 모으고 1200rpm 8 분의 원심분리를 한다.
21) 페렛에 배양액을 가하고 적당한 세포 농도로 폴리라이신 코팅한 커버글라스에 플레이팅하고(커버글라스는 6cm 페트 리 접시 바닥에 깔아 둔다), 5 % CO2 인큐베이터에서 배양 한다. 22) 24 시간 뒤에 3ml 의 배양액을 보충한다. 배양액 교환은 l 주일에 두 번 행한다. 23) 배양액 조성은 다음과 같다. 이글 MEM 92ml 마혈청 5ml 50% 포도당액 1ml 2 mg /m l 겐타마이신액 1 ml 0.25 mg /m l 암포테리신 B 1 ml 〈해설〉 분리 직후의 관찰에 의하면 올리고덴드로사이트의 숫자는 80-90 %의 순수도를 보이며 미크로글리아의 콘타미네이션이 있 기 때문에 24 시간에서 48 시간의 배양을 하게 되면 미크로글리아 가 페트리 접시 바닥에 부착하여 올리고의 순도를 98 %에까지 울릴 수가 있다. 이렇게 세포부착성의 차이를 이용하여 세포 순 도를 증가시키는 것을 부착성 의존분리법 dif fere nti al adhesio n 이 라.한다. 배양 2-3 일 안으로 울리고덴드로사이트 세포 특이 마카인 항갈 락토세 레 브로시 드 ga lacto c erebrosid e 항체 를 사용하여 면 역 염 색 을 하면 95-98 %의 세포가 염색에 양성인데 2 주일 이상 배양하고 나면 아스트로사이트, 혈관내피세포 등이 증식하여서 85 % 정도 로 그 양성도가 떨어진다. 사람 울리고덴드로사이트의 배양과 그 생물학적, 면역학적 특
성에 대하여서는 저자의 연구실에서 발표한 연구논문을 참고하기 바란다 .4,10) 그림 20-3 과 20-4 에서 사람 울리고 배양의 실제를 보이고 있 다.
 그림 20-3 72 세 성인 사후 6 시간된 뇌에서 분리된 올리고덴드로사이트 배
그림 20-3 72 세 성인 사후 6 시간된 뇌에서 분리된 올리고덴드로사이트 배
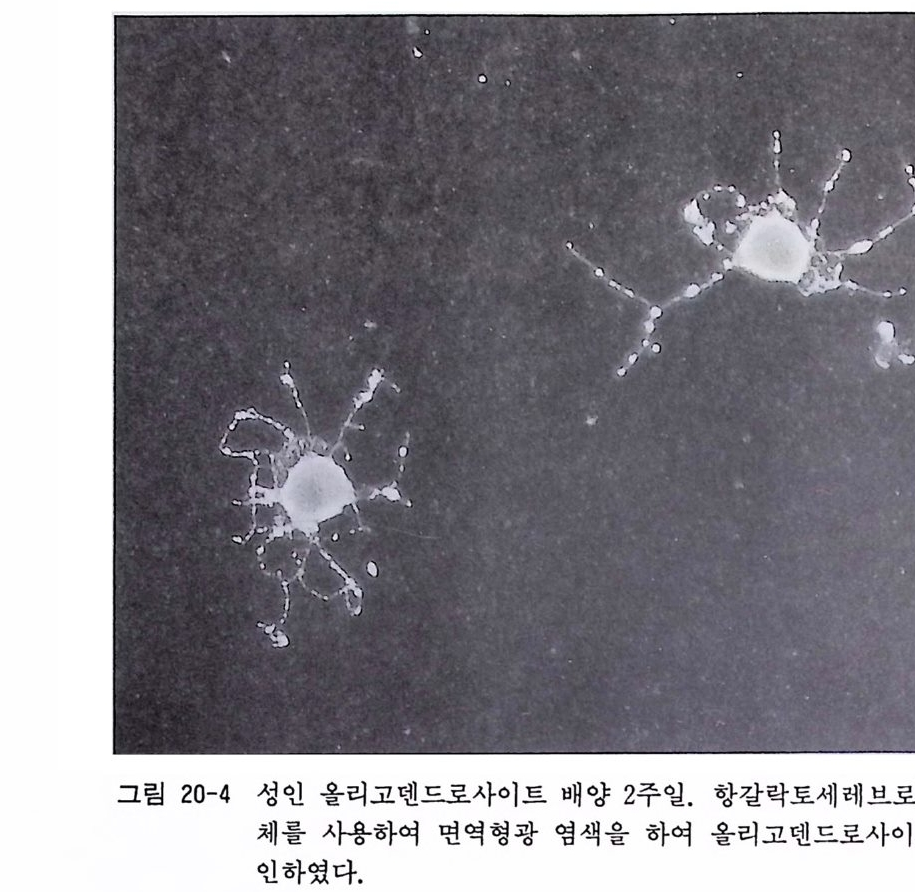 그림 20-4 성인 울리고덴드로사이트 배양 2 주일. 항갈락토세레브로시드 항
그림 20-4 성인 울리고덴드로사이트 배양 2 주일. 항갈락토세레브로시드 항
20.2 신생 래트(마우스) 올리고덴드로사이트의 순수분 리와배양 20 . 1 에서는 성숙 포유동물 뇌에서 퍼콜 밀도구배 컬럼에 의해 서 울리고덴드로사이트를 대량으로 분리하는 방법에 대하여 기술 하였는데 이 퍼컬법은 성숙 래트나 마우스 그리고 생후 2 주일에 서 4 주일의 래트나 마우스에서도 응용할 수가 있다. 그러나 신생
아 래트나 마우스에서는 적당치가 않다. 그 때문에 여기에서는 신생아 래트에서 올리고덴드로사이트를 분리하는 방법에 대하여 소개한다 .5) 원래의 방법에 의하면 기계적 세포분리법을 사용하 게 되는데 이것은 처음으로 조직배양을 시작하는 사람에게는 어 려운 방법이어서 여기에서는 트립신 처리에 의한 변법을 소개한 다. 폴 라스크 셰이킹에 의해서 울리고덴드로사이트를 분리하는 방법은 18 . 3 에서 기술한 태생 래트 대뇌신경세포 순수배양과 동 일하다. 〈 준비 〉 1) 입체해부현미경과 조명 2) 수술기구 - 10 cm 의과용 가위 (직선과 곡선) 2 개 10 cm 의과용 핀셋 2 개 시계수선용 핀셋 (No. 4) 2 개 수술용 칼 (No. 11) 2 개 3) 시험관, 페트리 접시, 원심튜브, 75cm 플라스크 4) 12 mm 직경 원형 커버글라스 5) 칼슘, 마그네슘 제거 핸크스 영류용액 6) 2.5 % 트립신액 7) 0.2 % DNase 효소액 8) 이글 MEM 합성 배양액 9) 마혈청 10) 50 % 포도당액 11) 2 mg / ml 겐타마이 신액 〈방법〉 1) 신생 래트를 에텔자에 넣어서 마취사시킨다. 혹은 신생 래
트를 -20 도 냉동실에서 10 분 방치하여 냉온마취한다. 신생 래트 10 마리를 사용한다. 2) 신생 래트 머리 피부를 제거한 뒤 두 안구 사이에 10cm 가위로 절개를 가한다. 좌우로 전개하는 요령으로 두개골을 둥글게 절개하고 제거한다. 후뇌에서 연수에 이르는 뇌부분 울 절제하여서 핸크스 용액이 들어있는 페트리 접시에 옮긴 다. 3) 입체해부현미경 아래서 종이를 벗기듯이 뇌막을 벗긴다. 다 음에 수술칼 2 개를 사용하여 대뇌와 중뇌 경계부를 절단한 다. 대뇌를 조그만 조직편으로 자른다. 4) 대뇌조직편을 50ml 원심듀브에 옮기고 여분의 핸크스용액 울 제거한다. 튜브에 CMF 핸크스 용액 18ml, 2.5% 트립 신 2 ml, 0.2 % DNase 액 0.4 ml 를 가하고 37 도에서 30 분 간 인큐베이트한다. 5) 대뇌조직편을 바닥에 남긴 채 트립신액을 뽑아서 버린다. 10 % 마혈청이 들어있는 배양액 15ml 과 0.2 % DNase 액 0.2ml 를 가하고 파스출 피펫으로 20 번 피페팅을 한다. 다음 에 파스출 피펫 끝에 미크로피펫립을 삽입하고 20 번 피페팅 울 되풀이한다. 이 시점에서 조직편이 남아있으면 튜브를 3 분간 정치한 뒤 세포가 부유하고 있는 상청액을 새 튜브에 옮기고 남은 조직편에 배양액 4ml 를 가한 뒤 위와 같이 피 페팅을 되풀이한다. 6) 세포부유액을 한 튜브에 모으고 1200r p m 으로 8 분간 원심 분리한다. 7) 상청액을 버리고 페렛에 배양액 8ml 를 가하고 1200r p m 으 로 8 분간 원심분리를 한다. 8) 페렛에 배양액 30ml 롤 가하고 세포를 부유시킨 뒤 폴리라
이신 코팅한 75 cm 플라스크 2 개에 세포부유액 15 cm 씩을 가한다. 세포 수 계산울 하여 풀라스크 하나에 5X l 06 세포 가 되도록 한다. 배양액 조성은 다음과 같다. 이글 MEM 88 ml 마혈청 (우태아혈청) 10ml 50% 포도당액 1 ml 2 mg /m l 겐타마이신액 1 ml 9) 2 일 뒤 플라스크의 배양액을 교환한다. 새 배양액 15ml 룰 가한다 . 10) 7 일 뒤 풀라스크의 배양액을 교환한 뒤에 풀라스크의 스크 류캡을 단단히 끼우고 셰이커 (Orbit al shaker, 라브라인사) 위에 데이프로 고정한다. 1 분에 250 회전으로 1 시간 셰이킹 을 하고 주로 미크로글리아를 포함한 배양액을 그대로 새 페트리 접시에 옮기고 새 배양액 15ml 를 가한다. 플라스크 를 셰이커에 고정하고 1 분 250 회전의 속도로 18-20 시간 셰 이킹을 한다. 11) 풀라스크 셰이킹에서 모은 세포부유액 속의 세포는 주로 울리고덴드로사이트인데 이들을 원심듀브에 모아서 1200 rp m 8 분의 원심분리를 한다. 12) 페렛에 배양액 5ml 를 가하여 세포를 부유시키고 미리 준 비해 두었던 폴리라이신 코팅한 커버글라스나 6cm 페트리 접시에 플레이팅한다. 13) 울리고덴드로사이트 부유액을 분리한 플라스크에 배양액 15ml 롤 가하고 셰이커에 고정한 뒤 1 분 250 회전 18-20 시간 의 셰이킹을 되풀이한다. 두번째로 울리고덴드로사이트 부 유액을 수집하고 폴리라이신 코팅한 커버글라스나 페트리 집시에 풀레이팅한다. 배양액 교환은 1 주일에 2 번 행한다.
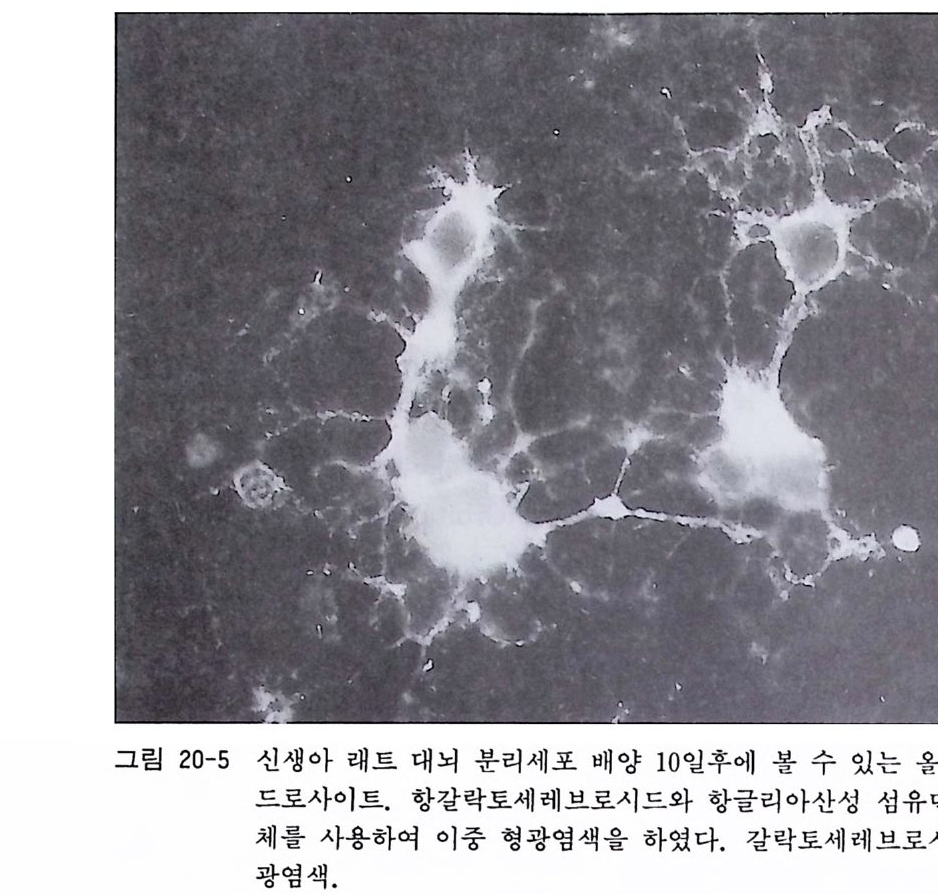 그림 20-5 신생아 래트 대뇌 분리세포 배양 10 일후에 볼 수 있는 올 리 고 덴
그림 20-5 신생아 래트 대뇌 분리세포 배양 10 일후에 볼 수 있는 올 리 고 덴
14) 플라스크 바닥에 남아있는 세포는 95 % 이상의 순도 를 가 진 성상교세포(아스트로사이트)이므로 배양액 15ml 를 가하 여 배양을 계속한다. 이를 다른 실험계로서 사용할 수가 있 다 (21.2 참고). 〈해설〉 위와 같이 신생 래트 뇌에서 분리한 올리고덴드로사이트는 이 셰이킹법으로 60-80% 의 갈락토세레브로시드 면역염색 양성이
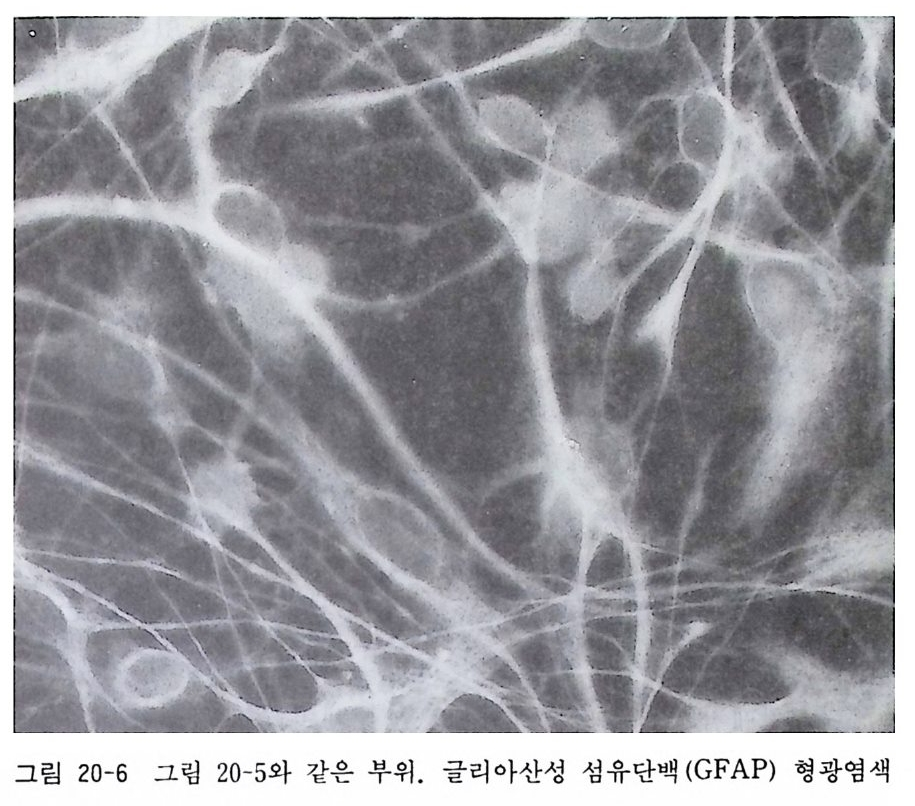 그림 20- 6 그립 2 0 - 5 와 같 은 부위. 글리아산성 섬유단백 (GFAP) 형광염색
그림 20- 6 그립 2 0 - 5 와 같 은 부위. 글리아산성 섬유단백 (GFAP) 형광염색
보통인데, 이 방법을 처음으로 고안한 UCLA 연구실에서는 98~ 99 %의 순수한 래트 울리고덴드로사이트롤 얻을 수 있다고 보고하고 있다. 그러나 이것은 너무 과장된 주장으로 생각된다. 신생 래트 뇌에서 분리한 울리고프레파레이션에는 0-2A 전구 세 포 oli go dendrocy te- ty pe 2 astr o cy te pro g en it or 가 다수 포함되 어 있어서 올리고의 발생과 분화에 대한 연구에서 귀중한 재료를 제 공하고 있다. 11, 12 ) 퍼컬 방법에서 기술한 방법에 따라서 그림 20-5 와 20-6 에서 래트 올리고덴드로사이트 배양의 실제를 보이고 있다. 플라스크에서 셰이킹을 해서 얻은 울리고덴드로사이트롤 포함한 부유세포를 24 시간 페트리 접시에서 배양하여 미크로글리
아 기타 세포를 페트리 접시에 부착시키고 부유하고 있는 올리고 덴드로사이트를 수집하면 더욱 높은 순도의 올리고덴드로사이트 를 얻을 수가 있다. 20.3 나일론 멧슈를 부착한 뷔크너 깔때기 제작법 1) 직경 10cm 의 플라스틱 뷔크너 깔때기는 미국의 날전사 Nalg en 에서 구매할 수가 있다. 2) 뷔크너 깔때기는 상부, 하부 두 부분으로 되어있다. 상부의 바닥에는 작은 구멍이 30 개 정도 있는데 가능한 대로 크게 이 바닥 부분을 찰라낸다(그림 20-2). 3) 찰라낸 상부의 바닥 부분에 100 미크론의 구멍을 가진 나일 론 멧슈를 덮은 뒤 깔때기의 하부를 이에 맞추어서 꼭 끼운 다. 깔때기 상부와 하부의 접속부를 데이프로 고정한다. 4) 알루미늄 포일로 깔때기를 싸고 이를 오토콜레이브에서 멸 균한다. 5) 사용이 끝난 깔때기는 중성세제로 씻어서 말린 뒤에 알루미 늄 포일에 싸서 다시 오토클레이브 멸균을 한다. 나일론 멧 슈가 못쓰게 될 때까지 재사용이 가능하다. 소량의 재료를 사용할 때에는 24.4 에서 소개한 나일론 봉지를 사용할 수가 있다. 나일론 멧슈는 20, 40, 100, 150, 200 미크론 구멍 크 기가 있으며 폭 1 미터, 길이 1-2 미터 크기의 것을 나이텍스 사 N yt ex 에서 살 수가 있다.
제 21 장 아스트로사이트의 순수배양 아스트로사이트(성상교세포, as t roc yt e) 는 중추신경계에서 울리 고덴드로사이트와 함께 신경세포를 보조하는 역할을 갖고 있으 며 , 혈류와 뇌 사이 에 위 치 한 관문 즉 혈류 - 뇌 관문 blood-brain -barr i er 의 유지라는 중요한 생리작용도 가지고 있다. 중추신경계 의 초기 발생 과정과 뇌 의상과 같은 조건 아래에서 아스트로사 이트가 활발히 증식하는 것은 잘 알려진 사실이나 생체 뇌안에서 이들의 증식을 조절하고 있는 기작과 그 화학물질은 아직도 명백 하지 않다. 조직배양한 아스트로사이트를 실험계로 하여 각종 성 장인자가 그 증식 능력에 어떠한 영향을 주는지 여러 연구실에서 검색을 하였는데 아스트로사이트의 증식을 촉진하는 성장인자로 서 FGF (fibr oblast grow th fac to r ) , IGF (ins uli n- lik e grow t h fac to r ) , EGF (ep ide rmal grow th fac to r ) , PDGF (pla te l et deriv e d grow t h fac to r ) , GGF (glia grow th fac to r ) 1-si 등이 알려 져 있다. 이와 반대로 아스트로사이트의 증식을 억제하는 물질이 뉴로블라 스토마 배양액과 신경세포막 성분에서 발견되었다는 보고가 있
다 .6,7) 태생 래트나 마우스 중추신경 조직에서 분리된 신경세포 가 아스트로사이트 위에서 그 발육이 활발해진다는 관찰에서 시 작되어서 아스트로사이트가 어떠한 성장인자 혹은 영양인자 neurotr o p h ic fa c t or 를 생 산하는가가 최 근에 주목을 받고 있다. 8,9) 여기에서는 아스트로사이트의 대량 순수분리와 배양에 대하여 기술한다. 21 .1 신생 래트(마우스) 아스트로사이트의 순수분리와 배양 20.2 에서 신생 래트 대뇌 유래의 울리고덴드로사이트의 순수분 리와 배양을 기술하였는데, 신생 래트 대뇌 분리세포를 풀라스크 에서 1 주일 이상 배양하고 고속도의 셰이킹을 하면 울리고덴드로 사이트가 대량으로 분리된다고 기술한 바 있다. 이렇게 올리고덴 드로사이트가 대량으로 분리된 다음에 풀라스크의 바닥에는 95-98% 순도의 아스트로사이트가 빽빽하게 시트와 같이 발육하 고 있다. 이들 순도가 높은 아스트로사이트를 트립신 처리로 얻 을 수가 있다. 〈방법〉 1) 20 . 2 에 서 기 술한 순서 1)-4) 를 충실히 따른다. 풀라스크의 배양액을 제거하고 PBS 로 한 번 씻는다. 흡인 펌프나 진공 라인에 접속된 파스출 피펫으로 배양액을 홉인하고 제거한 다. 2) 0.05 % 트립신과 0.02 % EDTA 를 포함한 PBS (PBS 100 ml 에 2.5 % 트립신 2 ml 과 2 % EDTA 1 ml 룰 첨가한다) 10 ml
를 플라스크에 가하고 10 분간 37 도 인큐베이터에 정치한다. 3) 풀라스크롤 혼들면 세포가 종이 시트와 같이 벗겨져 나온 다. 마혈청 2ml 이 들어있는 원심듀브에 세포부유액을 모은 다. 4) 풀라스크에 PBS 10ml 를 가하고 남아 있는 세포를 씻어낸 다. 5) 세포부유액을 모아서 1000 rpm 속도로 8 분간 원심분리한 다. 6) 페렛에 배양액 15ml 를 가하여서 세포를 부유시키고 폴리라 이신 코팅된 커버글라스나 페트리 접시에 플레이팅하고 5% CO2 인큐베이터에서 배양한다. 배양액 조성은 다음과 같으 며 배양액 교환은 1 주일에 두 번 시행한다. 이글 MEM 93ml 마혈청 5ml 50% 포도당액 1ml 2 mg / ml 겐타마이 신액 1 ml 〈 해설 〉 위에 기술한 방법으로 분리한 래트 아스트로사이트 순수배양의 실제를 그림 21-1 과 21 - 2 에서 보이고 있다. 강그리오시드 항체 인 A2B5 와 아스트로사이트 세포타입 특이마카인 글리아섬유성 산 성 단백 질 (glial fibr ill ar y aci di c Gp ro te i n , GFAP) 을 사용하여 이 중 염색을 하였다. 두 가지 항원이 같은 아스트로사이트에 특이하게 존재하고 있음을 알 수 있다.
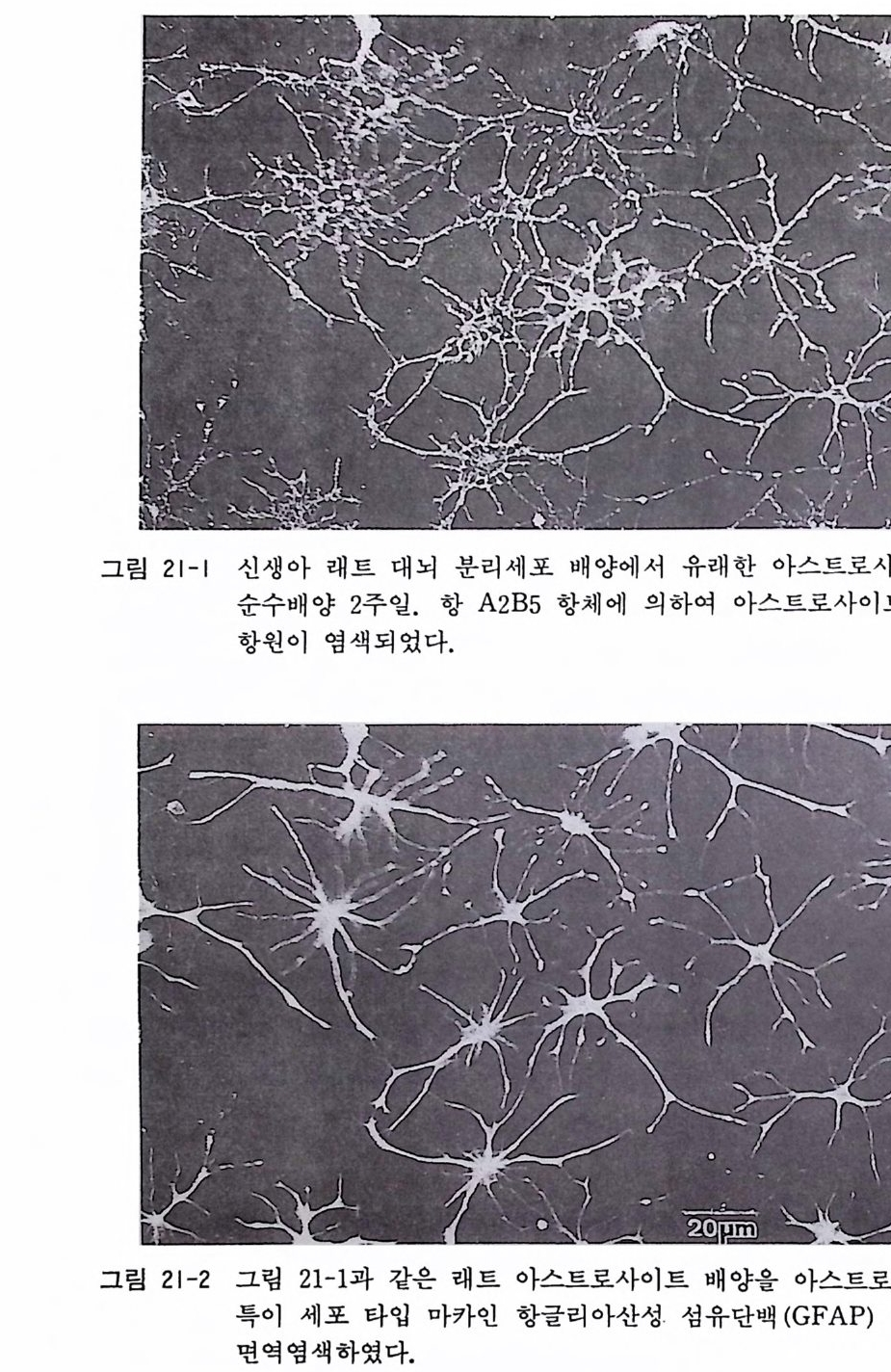 그림 21-1 신생아 래트 대뇌 분리세포 배양에서 유래한 아스트로사이트의
그림 21-1 신생아 래트 대뇌 분리세포 배양에서 유래한 아스트로사이트의
21 .2 무혈청 배양액을 사용한 신생 마우스 대뇌 유래 의 아스트로사이트 순수배양 〈 준비 〉 20.2 와 같다. 〈 방법 〉 1) 20 . 2 에서 기술한 순서 1)-6) 까지를 충실하게 따른다. 2) 상청액을 버리고 페랫에 무혈청 배양액 DM4 15ml 를 가한 다. 세포부유액을 만들고 폴리라이신 코팅한 12 혈 멀티웰 챔 버에 배양액 1ml 에 1X105 세포의 농도로 플레이팅을 한다. 뒤에 1ml 무혈청 배양액을 추가하고 37 도 인큐베이터에 정 치한다. 3) 일주일에 2 번 (월요일과 금요일) 배 양액 을 교환한다. 4) 무혈청 배양액 DM4 의 조성은 다음과 같다. 이글 MEM 혹은 Fl2 97ml 50 % 포도당액 1ml 2 mg /m l 겐타마이신액 1 ml 영양인자_호르몬 칵테일 1ml 영양인자-호르몬 칵테일액은 1000 배 스토크 0.1ml 씩과 이글 MEM 0.5ml 을 혼합하여서 lml 의 스토크롤 만들고 나누어서 ― 20 도에서 얼려서 보관한다. 영양인자-호르몬 스토크의 조성은 다음과 같고 1000 배 스토크액은 멤브레인 여과멸균한다. 인슐린 ins uli n 10mg /m l 트란스페 린 tra nsfe r rin lO mg/m l
셀 렌나트륨 sodiu m selenit e 3 X 10-5M, 44 mg / m l 트리 요도티 로닌 tri io d oty ron in e 3X 10-1M, 0. 2 mg / m l 히 드로코티 존 hy d rocorti so ne 5X 10-5M, 18 mg /m l 〈 해설 〉 무혈청 배양액에서 신생 래트 대뇌분리세포를 2 주일 이상 배양 하면 섬유아세포의 발육과 증식이 억제되고, 대다수의 신경세포 와 올리고덴드로사이트도 필수영양인자의 결핍으로 사멸하여 주 로 아스트로사이트만 남게 되므로 90-95 % 순도의 래트 아스트 로사이트 배양을 얻을 수가 있다.
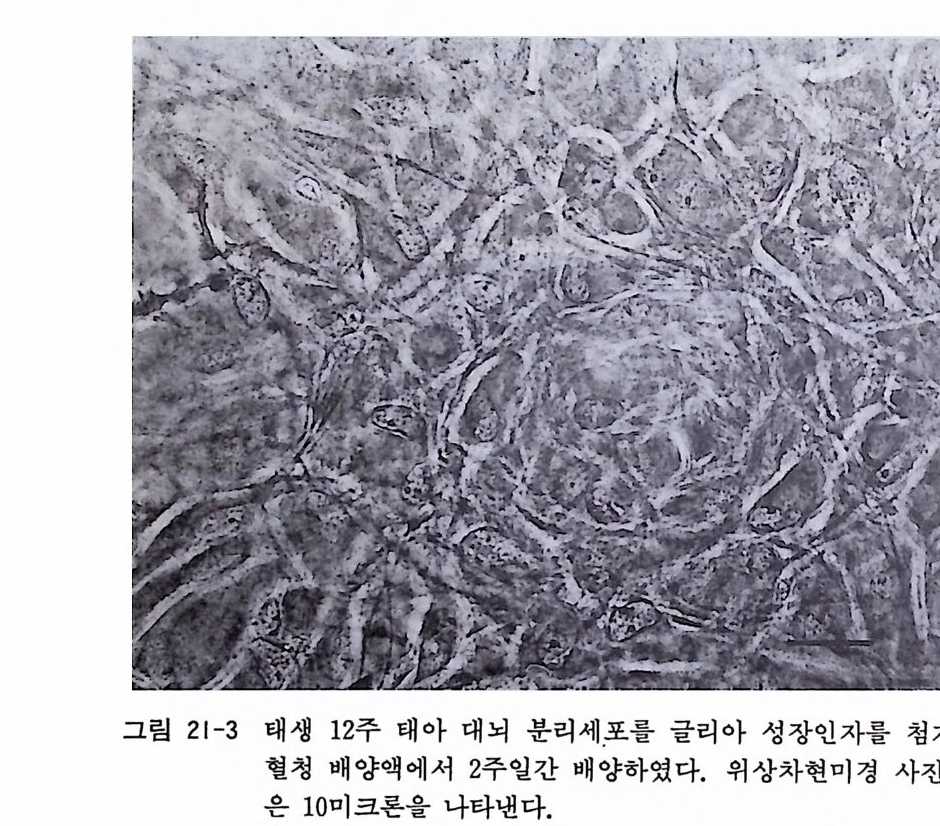 ` .’ . 글
` .’ . 글
아스트로사이트의 동정은 항글리아섬유성 산성단백질 항체를 사용하여 면역염색을 하여 GFAP- 염색 양성세포를 현미경 아래 서 판정한다. 태생 12 주 태아 대뇌 분산세포를 10 µg/ ml 글리아 성장인자 (glia gro wt h fac to r , GGF) 를 첨가한 무혈청 배양액에서 2 주일간 배양한 뒤 GFAP 면역염색을 한 실제를 그림 21-3 과 21-4 에서 보여준다.
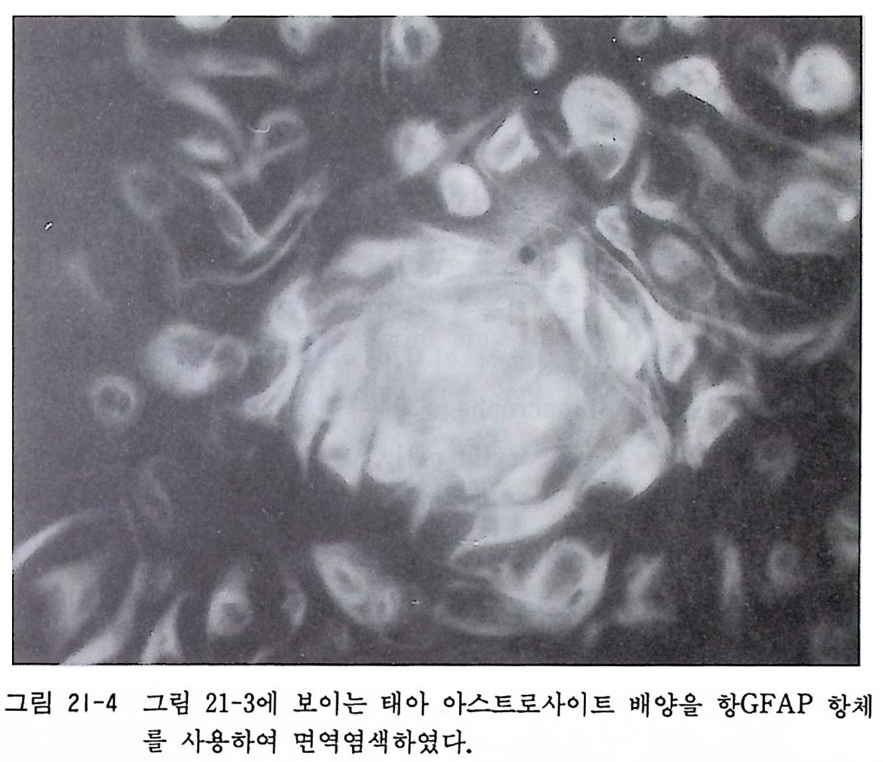 그림 21-4 그림 21-3 에 보이는 태아 아스트로사이트 배양을 항 GFAP 항체
그림 21-4 그림 21-3 에 보이는 태아 아스트로사이트 배양을 항 GFAP 항체
제 22 장 미크로글리아의 순수배양 미크로글리아 m ic ro gli a 는 아스트로사이트와 울리고덴드로사이 트와 함께 중추신경계 조직을 구성하는 신경교세포의 한 멤버로 서 신경세포를 보조하는 기능을 가지고 있다. 미크로글리아는 신경계조칙 이의의 다론 조직 특히 면역계에서 발견되는 매크로파지 macro p ha g e 와 유사한 형태와 형질을 가지 고 있어 서 탐식 작용 ph ag o cy tic acti vi t y, 항원제 시 anti ge n pre sen- tat i on , 단백 질 분해 효소 분비 작용 pro te a se secreti on 등의 특수한 기능을 가지고 있다. 최근에는 미크로글리아를 중추신경계의 탐 식세포라 규정하고, 매크로파지 세포 특이 마카를 사용하여 동정 할 수가 있게 되었다 .1) 중추신경계 조직에서 미크로글리아를 순수하게 배양하려는 시 도가 1980 년 후반에 시작이 되어서 여러 연구실에서 그 분리방법 에 대한 보고를 하였다. 또한 미크로글리아가 조직배양의 조건아 래에서 인터루킨 -1(IL-1), 조직괴사인자(ti ssue necrosis fac to r , TNF) 와 같은 사이토카인 c yt ok i ne 을 생산함으로써 신경세포나
다른 신경교세포의 증식과 분화에 영향을 준다고 보고되었다 .2-4) 22.l 성숙 포유동물 뇌에서의 미크로글리아 분리와 배양 20.1 에서 기술한 올리고덴드로사이트 순수배양의 과정에서 부 산물로 대량으로 미크로글리아를 얻을 수가 있다• 퍼컬 밀도구배 컬럼에서 울리고덴드로사이트가 모이는 제 3 층(비중 1.035 에서 1.0 6 0 사이)에는 올리고덴드로사이트와 갇이 미크로글리아가 모 이게 되므로 울리고덴드로사이트롤 순수하게 분리하려면, 이 두 가지 세포형을 분리하여야 한다. 미크로글리아는 신경세포나 올 리고덴드로사이트에 비하여 풀라스틱이나 유리 표면에 쉽게 접착 하는 성질이 있기 때문에 이러한 미크로글리아의 성질을 이용하 여 부착성 의존분리법 dif fer enti al adhes i on 의 방법으로 미크로글 리아와 울리고덴드로사이트를 순수분리하여 배양할 수가 있다. 〈 준비〉 20.1 과 같다. 성숙 소, 사람 뇌조직을 사용한다. 〈방법〉 1) 20.1 의 섹션에서 기술한 순서 1) 에서 18) 까지 충실하게 따 른다. 2) 10 개의 10cm 직경 페트리 접시에 12ml 씩 세포부유액을 분주하고 48 시간 동안 37 도 인큐베이트에 정치한다. 3) 덩어리가 되어서 부유하는 올리고덴드로사이트가 들어있는 배양액을 원심튜브에 모두 모으고 1200r p m 의 속도로 8 분간
원심분리한다. 페렛에 배양액을 첨가하고 따로 올리고덴드로 사이트 순수배양을 시작한다. 4) 페트리 접시에는 미크로글리아가 부착하고 있는데 여기에 12ml 의 배양액을 가하여서 배양을 계속한다. 배양액은 일주 일에 한 번 교환한다. 배양액의 조성은 다음과 같다. 이글 MEM 93 ml 마혈청 5ml 50% 포도당액 1 ml 2 mg /m l 겐타마이신액 1 ml 5) 면역염색이나 전자현미경으로 미크로글리아 검색을 하려면 미크로글리아가 들어있는 페트리 접시에서 커버슬립으로 리 플레이팅 re p Ia ti n g을 하여야 한다. 먼저 배양액을 제거한 뒤 CMF- 핸크스액이나 PBS 로 두 번 페트리 접시를 씻는다. 6) CMF- 핸크스액 혹은 PBS 7.6ml 과 2.5 % 트립신액 0.4 ml 를 페트리 접시에 첨가하고 10 분간 37 도에서 인큐베이트 한다. 7) 마혈청 2ml 를 페트리 접시에 가한 뒤 피페팅을 서서히 하 여서 페트리 접시에서 세포를 떨어뜨리고 부유시킨다. 8) 부유세포를 모두 원심듀브에 모으고 1200rpm 속도로 8 분 간 원심분리한다. 9) 페렛에 4) 에서 적은 배양액을 가하고 적당한 세포 밀도로 폴리라이신 코팅한 커버글라스에 플레이팅한다. 〈해설〉 미크로글리아의 세포형 확정을 위해서는 바이오틴 표지 리신렉 틴에 의한 면역 염색을 사용하면 된다. 바이오틴 표지 리신 렉틴 bio t i ny la te d Ric in communis lecti n, 시그마 에서 30 분간 인큐베 이
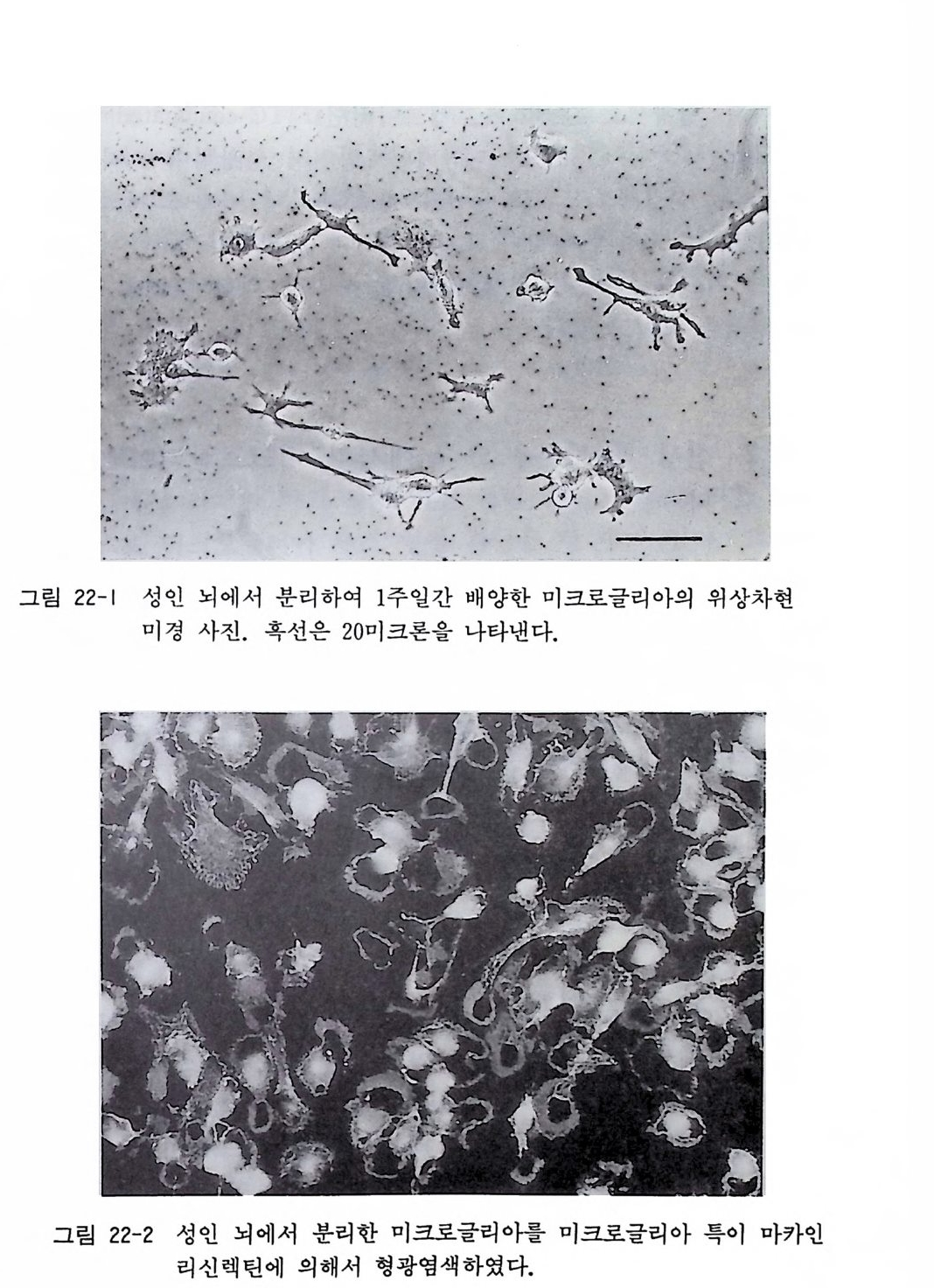 그림 22-1b ..... ...t.. .• .:f• .. .. ,i.• . tiL . . • .` . . 성미 ...:.. ...?.김〉 경인〉.홉5 . u뇌사\ '진에•서\ 흑.분선 :'~\’리 \ 은 하 〈: 여2 0 미一 1 주크`.二? .曲. ..... 일론今Jt'h .?%q 〔\. 을간. ..\ `L.’배나.f'\ . ,타 양.t.... 낸f. ... ..한』.. ...\ .... .. 다 ' g .. ?`미 一 .청’.(.?\쇼 ' , 크.. .로 \\글· 리f\.. 아 의―一 r `〈 위i상 :차 현
그림 22-1b ..... ...t.. .• .:f• .. .. ,i.• . tiL . . • .` . . 성미 ...:.. ...?.김〉 경인〉.홉5 . u뇌사\ '진에•서\ 흑.분선 :'~\’리 \ 은 하 〈: 여2 0 미一 1 주크`.二? .曲. ..... 일론今Jt'h .?%q 〔\. 을간. ..\ `L.’배나.f'\ . ,타 양.t.... 낸f. ... ..한』.. ...\ .... .. 다 ' g .. ?`미 一 .청’.(.?\쇼 ' , 크.. .로 \\글· 리f\.. 아 의―一 r `〈 위i상 :차 현
트한 뒤 형광색소 표지 스트렙트아비딘 (FITC-conj uga t ed str e - ptav i di n , 시그마)에 30 분간 인큐베이트하여서 형광현미경 하에서 관찰한다. 신경계조직 배양에서의 세포형 특이 마카에 대하여서 는 30 .1 을 참고하기 바란다. 그림 22-1 과 22 - 2 에 서 성 인 뇌 에 서 분리한 미크로글리아의 위상차현미경 사진과 리신 렉틴에 의한 형광염색을 보이고 있다. 22.2 신생 래트(마우스) 뇌에서의 미크로글리아 분리 와배양 22.1 에서는 성숙 포유동물 뇌에서 울리고덴드로사이트를 분리 하는 단계에서 미크로글리아를 대량 분리하는 방법을 소개하였는 데 여기에서도 역시 신생 래트(마우스) 뇌에서 올리고덴드로사이 트와 아스트로사이트를 순수분리하는 과정에서 그 부산물로서 미 크로글리아를 대량분리하는 방법을 소개한다. 〈 준비 〉 20.2 과 같다. 〈방법〉 1) 20.2 에서 기술한 순서 1) 에서 10) 까지를 충실하게 따른다. 2) 신생 래트 대뇌 분리세포 배양이 들어있는 풀라스크를 셰이 커에 고정하고 1 분에 250 회전으로 1 시간 셰이킹을 한다. 미 크로글리아가 부유하고 있는 배양액을 플라스크에서 모은 다 음 10cm 직경 페트리 집시나 폴리라이신 코팅한 커버글라 스 위에 플레이팅을 한다. 배양액을 교환한 뒤에 셰이킹을
하였으므로 새 배양액을 첨가할 필요는 없다. 3) 배양액 조성은 다음과 같다. 일주일에 한 번 혹은 두 번 세 포 밀도에 따라서 배 양액 교환을 한다. 이글 MEM 93ml 마혈청 5ml 50% 포도당액 1ml 2mg /m l 겐타마이신액 1ml 〈 해설 〉 22.1 에서 기재한 바와 같이 리신 렉틴 ric in lecti n 에 의한 면역 염색으로 미크로글리아 세포 형질 확인을 할 수가 있다. 그밖에 도 특이한 미크로글리아 세포 마카에 대하여서는 30.1 면역염색 법을 참고하기 바란다.
제 23 장 뇌혈관 내피세포의 순수배양 뇌혈관 cerebral mi cro vessel 을 순수하게 분리한 후 신경화학방 법을 사용하여 글루코오스, 지방산, 아미노산, 각종 이온의 혈관 투과성과 대사를 검색하는 연구가 이루어진 것은 1974 년 이후의 일이다 .1) 이러한 뇌혈관 분리 표본이 뇌혈관 관문 blood-bra i n -barr i er 의 형태학적 그리고 생화학적 레벨에서도 실험 모델로서 쓰이게 되었다. 그러나 이러한 뇌혈관 분리 표본은 여러 가지 문 제점을 가지고 있어서 장기간에 걸친 대사실험, 성장능력, 증식 능력의 측정 등의 실험은 할 수가 없었다. 이러한 뇌혈관 분리표 본의 결점을 보완하고자 만들어진 것이 뇌혈관과 뇌혈관 내피세 포의 순수배양이다. 뇌혈관 내피세포 endo the li al cell 배양은 1980 년에 들어서서 시 작이 되 었고 데볼트 DeBaul t는 마우스, 2) 보우만 Bowman 은 래트 3) 에 뇌혈관 내피세포 배양의 방법을 보고한 바 있다. 저자의 연구 실에서도 성숙 소의 뇌조직에서 분리한 미소혈관을 아스트로사이
트의 단총배양 위에서 장기간 배양하여 전자현미경으로 관찰하였 는데 생체에서 보이는 정상 뇌혈관의 미세구조가 충실하게 보존 되어 있음을 알 수가 있었다 .4) 여기에서는 포유동물 뇌혈관 내피세포 배양의 가장 일반적인 방법으로서 기계적 분리법과 퍼콜 밀도구배 분리법의 두 가지를 소개한다. 23.1 성숙 래트(마우스) 뇌혈관 내피세포의 기계적 분 리 배양법 이 방법은 뇌를 호모지나이즈한 뒤 스텐인리스 네트롤 통해서 거르면 그 위에 뇌 모세혈관이 모이게 되므로 이를 조직배양하여 서 내피세포를 분리하는 배양법이다. 장기간 배양에서 페리사이 트p er icyt e 와 섬유아세포의 콘타미네이션이 문제가 되므로 23.2 에서 소개하는 퍼콜 밀도구배 분리법을 병용하는 것이 좋겠다. 〈 준비 〉 1) 수술기 구 - 10 cm 외 과용 가위 1 개 10cm 의과용 핀셋 1 개 시계수선용 핀셋 (No. 4) 2 개 수술용 칼 (No. 11) 2 개 2) 유리 -데 프론 호모지 나이 저 homog e ni ze r 40 ml 용량 3) 세포여과기 (시그마) 4) 시험관, 페트리 집시, 원심튜브 5) 핸크스 영류 용액 6) 이글 MEM 합성 배양액
7) 마혈청 8) 50 % 포도당액 9) 2mg /m l 겐타마이신액 〈방법〉 1) 4-6 주의 래트(마우스) 2 마리를 에텔자에 넣어서 심마취로 마 취사시킨다. 2) 머리 부분을 70 % 알코올로 소독하고 뇌를 적출한다. 뇌는 핸크스 용액이 들어있는 페트리 접시에 놓는다. 3) 시계수선용 핀셋을 사용하여 뇌막과 혈관을 가능한 대로 제 거한다. 가위와 수술칼을 사용하여 뇌를 작은 조직편으로 자 른다. 4) 유리-데프론 호모지나이저에 뇌 조직편과 핸크스액 3ml 를 넣고 10 번의 상하 스트로크로 뇌를 호모지나이즈한다. 5) 뇌 호모지네이트를 튜브에 옮기고 1000rpm 속도로 6 분의 원심분리를 한다. 6) 페렛을 10ml 핸크스액에 부유시키고 0.1mm 구멍을 가진 세포여과기로 통과시킨다. 스데인리스 네트 위에 무수한 뇌 모세혈관이 걸러져서 남아 있는데 이들을 핸크스액이 들어있 는 페트리 접시에서 씻어서 분리한다. 7) 뇌 모세혈관을 핸크스액과 함께 튜브로 옮긴다. 1000rpm 속도로 6 분의 원심분리를 한다. 8) 페렛울 10ml 의 배양액에 부유시키고 콜라겐 코팅된 3.5 cm 직경 페트리 접시 5 개에 2ml 씩 분주한다. 2.2cm 칙경 원형 커버글라스에 콜라겐 코팅하여 그 위에 풀레이팅울 할 수도있다. 9) 페트리 접시를 CO2 인큐베이터에 넣어서 37 도에서 배양한
다. 배양액 교환은 1 주일에 2 번(월요일과 금요일)하며 배양액 의 조성은 다음과 갇다. 이글 MEM 86ml 마혈청 10ml 50 % 포도당액 1ml 2 mg /m l 겐타마이신액 1 ml 2 mg /m l 내피세포 성장인자 1 ml 10 mg /m l 헤 파린 1 ml 〈 해설 〉 내 피 세 포 성 장인 자 endoth e li al cell gro w th sup ple ment 와 헤 파린 he p ar i n 은 뇌혈관 내피세포 성장을 촉진하여 그 순수배양을 용이 하게 한다. 배양 개시 1-2 일 안으로 대부분 뇌 모세혈관의 소편이 콜라겐 기질에 부착하고 배양 3-4 일에는 혈관 내피세포가 소편에서 성장 해서 나온다. 배양 1 주일이면 섬유아세포와 형태가 비슷한 내피 세포가 시트와 같이 성장해서 나온다. 배양 3-4 주일이 되면 핸크 스액에 희석한 0.1 % 콜라게나제로 1 시간 처리하여 내피세포를 페트리 집시에서 유리하고 계대배양한다. 혈관 내피세포의 확인 은 제 8 인자fa c t or VllI 의 항체를 이용하여서 면역염색으로 한다. 혈관 내피세포가 자라고 있는 커버글라스를 다음과 갇이 면역영 색한다. 1) 커버글라스를 참시 PBS 에서 씻는다• 2) 영하 20 도의 냉동실에서 메틸알코올로 10 분간 고정한다. 3) 커버글라스를 공기중에서 건조시킨다. 4) 토끼 항 제 8 인자(시그마)를 PBS 로 1 : 50 으로 회석하고 실 온에서 1 시간 반웅시킨다.
5) PBS 에서 3 번 씻는다. 6) 형 광색 소 표지 -산양 항토끼 면 역 글로불린 (go at anti -r abbit im munog lo buli n- FITC, 시 그마) 을 PBS 로 1 : 50 으로 회 석 하 고 실온에서 30 분간 반응시킨다. 7) PBS 에서 3 번 씻는다. 8) 글리세롤 -PBS 에 마운트하고 형광현미경 아래서 검사한다. 23.2 퍼콜 구배 컬럼에 의한 래트 뇌혈관 내피세포의 순수배양 23.1 에서 기술한 방법에 의해서 혈관 내피세포를 배양하면 뇌 모세혈관을 구성하고 있는 내피세포 이의에도 페리사이트와 섬유 아세포가 같이 성장해 나와서 콘타미네이션이 일어나기 때문에 퍼콜 컬럼을 사용하여 내피세포만을 순수분리하여 배양하자는 것 이 이 방법의 실험목적이다 .4) 〈준비〉 23.1 과 같다. 〈방법〉 1) 4-6 주일의 래트를 23.1 의 마우스에서와 마찬가지로 마취사 시킨다. 23.1 의 조작에서 순서 1)-7) 까지 충실하게 따른다. 2) 뇌혈관 페렛을 3ml 의 0.25 % 콜라게나제액에 넣어서 37 도 에서 3 시간 처리한다. 효소 처리에 의해서 뇌혈관의 기저막 과 페리사이트가 제거된다. 3) 튜브를 1000 rp m 의 속도 10 분간 원심분리한 뒤 페렛을 10
ml 핸크스액에 부유시킨다. 4) 퍼콜 9 ml 와 10 배 농축 핸크스액 1 ml 를 혼합하고 여기에 핸크스 - 혈관부유액 10ml 를 가하면 50% 퍼콜액이 구성된 다. 오크릿지 원심듀브에 옮겨서 15000rpm 속도로 30 분간 원심분리한다. 5) 퍼콜 밀도구배 컬럼이 형성되어서 3 개의 밴드가 보인다. 제 1 층의 밴드(비중 1. 018) 에는 세포 잔재가 모이고 제 2 층 밴 드(비중 1 . 0 5 5) 에 혈관 내피세포가 모이며 제 3 층 밴드(비중 1.075) 에 적혈구가 모인다. 제 2 층 밴드를 다른 튜브에 모아
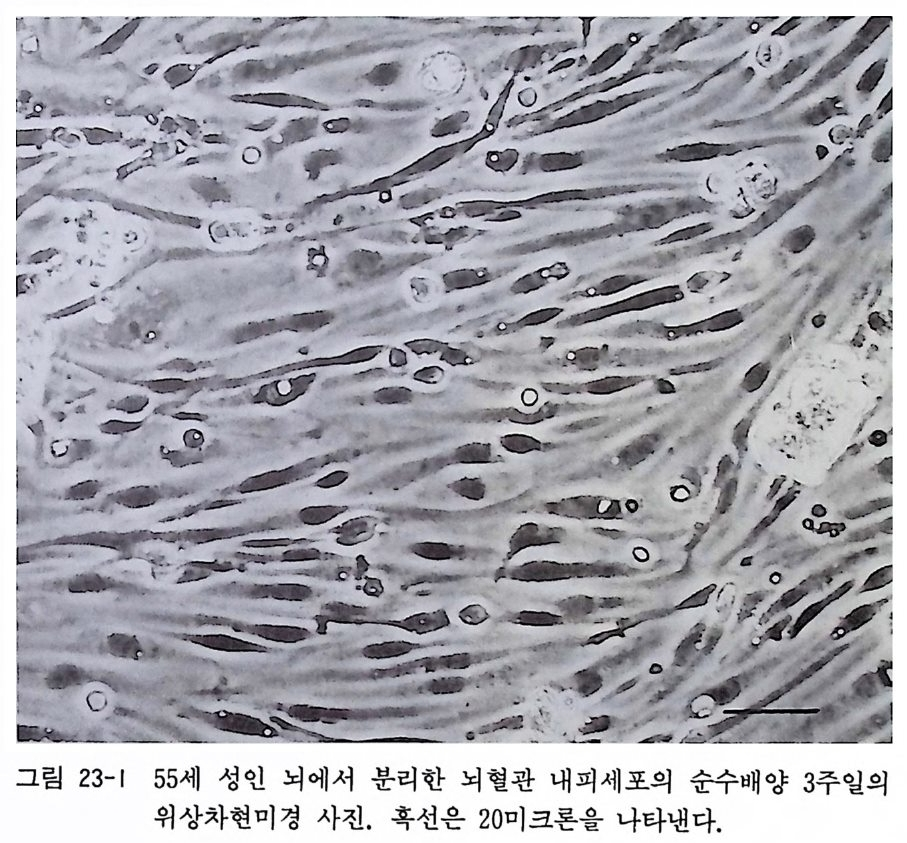 그림 23-1 55 세 성인 뇌에서 분리한 뇌혈관 내피세포의 순수배양 3 주일의
그림 23-1 55 세 성인 뇌에서 분리한 뇌혈관 내피세포의 순수배양 3 주일의
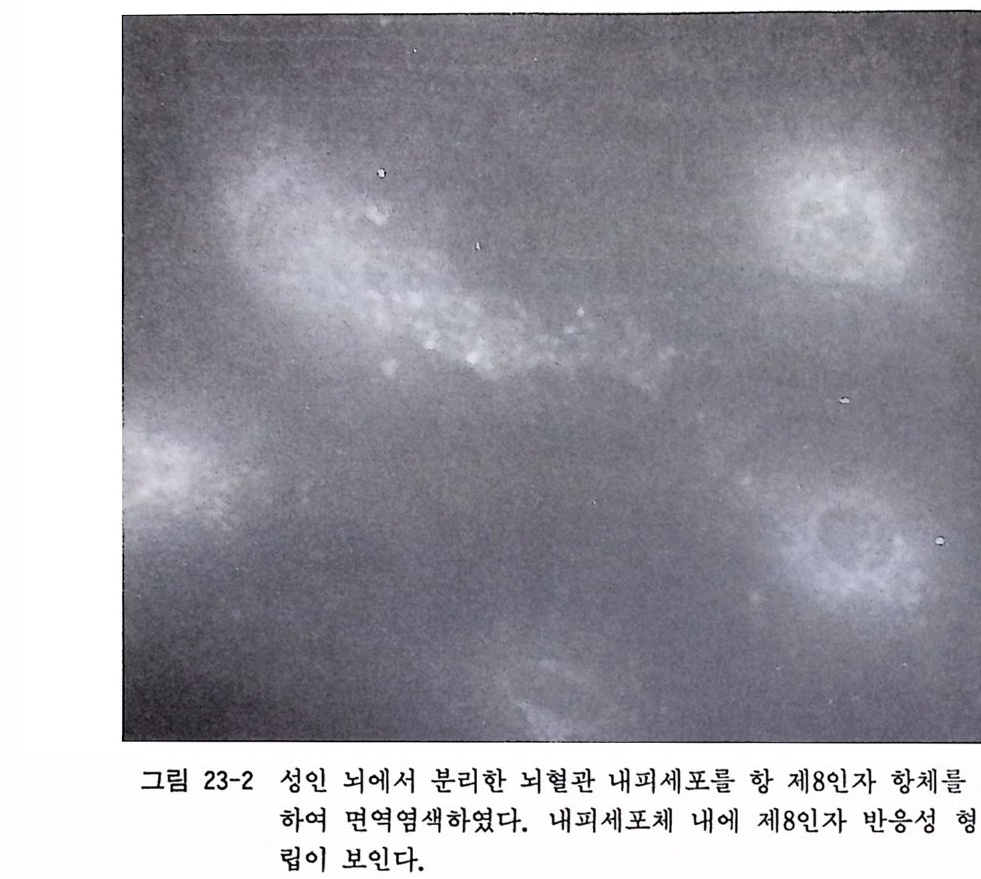 그림 23 군 성인 뇌에서 분리한 뇌혈관 내피세포 를 항 제 8 인자 항체 를 사용
그림 23 군 성인 뇌에서 분리한 뇌혈관 내피세포 를 항 제 8 인자 항체 를 사용
서 10ml 의 핸크스액을 가하고 1800r p m 으로 10 분간 원심분 리한다. 6) 페 렛울 3 ml 핸크스액 에 부유시 킨 뒤 1000 r p m 으로 6 분간 원심분리한다. 이 과정을 한번 더 되풀이한다. 7) 마지막 페렛에 배양액 3ml 를 가하고 피브로넥틴 코팅한 페 트리 접시나 커버글라스에 플레이팅하고 5% CO2 인큐베이 터에서 배양한다. 혈구계산판으로 세포 계수를 하여 1Xl05 세포 /ml 로 조정한다. 배양액 조성은 다음과 같다. 1 주일에
2 번 배양액 교환을 한다. 이글 MEM 86ml 마혈청 10ml 50% 포도당액 1ml 2 mg / ml 겐타마이 신액 1 ml 2 mg /m l 내피세포 성장인자 1 ml 10 mg /m l 헤 파린 1 ml 〈 해설 〉 보우만에 의하면 위의 방법을 사용하여서 래트와 소의 뇌조직 에서 혈관 내피세포를 순수배양할 수가 있었으며 5) 최근에는 지 스 Z i s 가 이 방법을 사용하여 사람 뇌에서 내피세포배양을 하였 다는 보고가 있다 .6) 그립 23-1 과 23 - 2 에서 내피세포 배양의 실제를 보이고 있다.
제 3 부 특수한 신경조직 및 비신경조직의 배양법 
제 24 장 골격근 조직의 배양 근육에 는 골격 근 skeleta l muscle, 심 근 cardi ac muscle, 평 활근 smooth muscle 의 세 가지 종류가 있는데 신경계 조직과 밀접한 상관관계를 가지고 있는 것이 골격근이기 때문에 여기에서는 골 격근 배양에 대하여 기술하겠으며 심근과 평활근의 배양에 관하 여서는 심장순환계의 기초 연구에 관한 덱스트를 참고하기 바란 다. 골격근 조직의 배양은 1963 년에 코니스버그 Kon igs ber g가 계배 에서 분리한 골격근 근아세포 m y oblas t의 단일세포를 배양하여 이것이 근관 m y o t ube 으로 분화하는 것을 관찰한 보고에서 시작하 였다.1) 코니스버그 이후로 계배 근육을 사용하는 근육배양계가 가장 많이 사용되어 왔으나, 최근에는 래트와 마우스의 근육배양 도 증가하고 있다. 또한 사람의 근육 질환 연구를 위하여 생검 b i o p s y에서 분리된 근육 샘플을 배양하는 데 성공하였다는 보고 가 있다 .2,3) 골격근 배양법으로서는 동물이나 사람의 골격근에서 분리한 근
아세포를 배양하는 근육세포 초대배양법과 G8 마우스나 L6 래트 근아세포주와 같은 근아세포 수립라인 4) 을 사용하는 두 가지 방 법이 있다. 근아세포 수립주라인에 대하여서는 제 32 장을 참고하 기 바란다. 24.1 계배 골격근의 분리세포 배양 〈 준비 〉 1) 입체해부현미경과 조명 2) 수술기구 -10cm 의과용 가위(직선과 곡선) 2 개 시계수선용 핀셋 (No. 4) 2 개 수술용 칼 (No. 11) 2 개 3) 시험관, 페트리 접시, 원심튜브 4) 혈구계산판 5) 칼슘, 마그네슘 제거 핸크스 영류 용액 6) 2.5 % 트립신액 7) 0.2 % DNase 효소액 8) 이글 MEM 합성 배양액 9) 마혈청 10) 50 % 포도당액 11) 2 mg /m l 겐타마이신액 〈방법〉 1) 계배 3 마리를 부란 8 일에서 12 일 되는 계란에서 분리하고 (11 . 1 을 참고할 것) 핸크스액이 들어있는 10cm 직경 페트리 집시에 넣는다.
2) 입체현미경 아래에서 계배의 대퇴근을 분리하고 새 페트리 접시에 옮긴 다음 수술용 칼 2 개를 써서 조그마한 조직편으 로 절단한다. 3) 소량의 핸크스액과 함께 근육조직편을 15ml 원심듀브에 옮 기고 핸크스액을 버린다. 핸크스액 3.5ml, 2.5 % 트립신액 0.4 ml, 0.2 % DNase 액 0.1 ml 를 첨가하고 37 도에서 30 분 간 인큐베이트한다. 인큐베이션 중에는 5 분에 한번씩 시험관 을 혼들어서 조직과 효소액이 잘 혼합되도록 한다. 4) 근육조직을 듀브 바닥에 남겨두고 트립신액을 제거한 뒤, 10% 마혈청이 들어있는 배양액 3ml 를 가한다. 파스출 피 펫으로 20 번 상하로 조용히 피페팅을 한다. 다음 파스출 피 펫 끝에 멸균된 미크로피펫립을 삽입하고 피페팅을 20 번 되풀이한다. 5) 조직편이 아직 남아있으면 3 분 가량 튜브를 정치한 뒤 상청 액을 다른 시험관에 옮긴다. 4) 에서와 마찬가지로 배양액 2 ml 를 가하고 미크로피펫팁을 삽입한 피펫으로 20 번 피페팅 을 되풀이한다. 6) 두 번의 피페팅으로 나온 세포부유액을 합하고 1000r pm 속도로 8 분간 원심분리한다. 7) 상청액을 버리고 페렛에 배양액 6ml 를 가하고 피페팅을 한 다. 깔때기형으로 만든 나일론 멧슈 (20 미크론 멧슈)를 새 원 심튜브에 세우고 이를 통해서 세포부유액을 여과한다. 나일 론 멧슈는 오토클레이브에서 미리 멸균해 둔다. 8) 세포부유액을 1000 rpm 속도로 8 분간 원심분리한다. 9) 페렛에 배양액 8ml 를 가한 뒤 6cm 페트리 접시 2 개에 4 ml 씩 분주하고 37 도에서 1 시간 인큐베이트한다. 그동안에 섬유아세포의 대부분이 페트리 접시 바닥에 우선적으로 부착
하고 근아세포 m y oblas t는 액중에서 부유하게 된다. 10) 부유세포를 모으고 콜라겐이나 젤라틴 코팅한 커버글라스 위에 플레이팅을 하고 5% CO2 인큐베이터에서 배양한다. 젤라틴 코팅의 방법은 24.3 을 참조한다. 혈구계산판으로 세 포수 계산을 하고 1X 1 06 세포 /ml 의 세포 농도로 희석한 다. 배양액 교환은 일주일에 두 번 한다. 배양액 조성은 다 음과같다. 이글 MEM 합성 배양액 83ml 마혈청 10ml 50% 포도당액 1ml 2 mg /m l 겐타마이신액 1 ml 계배 조직 추출액 5ml 24.2 태생 래트(마우스) 골격근의 분리세포 배양 〈준비 〉 24.1 의 계배근육 배양과 같다 . 〈방법〉 1) 14.1 에서 기술한 대로 16-18 일 임신 래트에서 태아를 분리 한다. 2) 10cm 의과용 가위를 사용하여 래트 태아에서 대퇴근을 분 리하고 핸크스액이 들어있는 페트리 접시에 옮긴다. 3) 24.1 의 계배 근육배양에서 기술한 순서 3)-10) 를 따라서 래 트 근육 분리세포배양을 시작한다.
〈 해설 〉 위에 소개한 근조직 배양법은 가장 일반적인 기술인데 이밖에 도 여러 가지 다른 방법이 있다. 예컨대 트립신 대신에 파파인, 디스파제, 콜라게나제 등의 효소를 사용하는 방법이 있다. 배양 2 일째부터 근아세포가 융합을 시작하여 배양 5-6 일째에는 가느다 란 근관 m y o t ube 의 형성을 볼 수가 있다. 배양 2 주일부터는 근관 이 더욱 성숙해지고 골격근에서 특징적으로 볼 수 있는 횡문 cross s t r i a ti on 이 나오기 시작한다. 그림 24 - 1 과 24-2 에서는 태생 래트 골격근 배양의 실제를 보이고 있다.
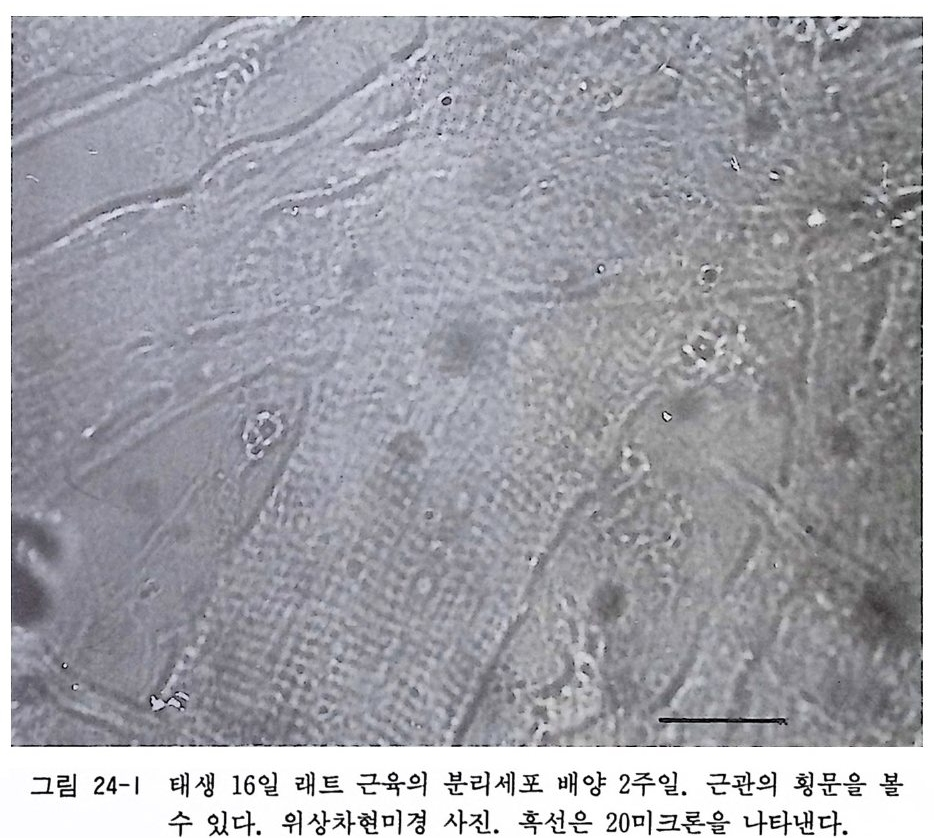 그립 24-1 태생 16 일 래트 근육의 분리세포 배양 2 주일. 근관의 횡문을 볼
그립 24-1 태생 16 일 래트 근육의 분리세포 배양 2 주일. 근관의 횡문을 볼
 그립 24-2 태생 16 일 래트 근육의 분리세포 배양 2 주일• 배양을 고정하고
그립 24-2 태생 16 일 래트 근육의 분리세포 배양 2 주일• 배양을 고정하고
24.3 젤라틴 코팅 증류수 100ml 에 백색 겔라틴 2 g을 가하고 워터바스 위에서 용해시킨다. 다음에 오토클레이브에서 멸균한 다음 냉장고에 보 관한다. 사용시에는 젤라틴액을 온수에서 따뜻하게 한 다음 충분 량의 젤라틴액으로 페트리 접시나 커버글라스 표면을 덮고 10 분 뒤에 여분의 젤라틴을 제거하고 무균 벤치 속에서 건조시킨다. 젤라틴 코팅한 접시는 3 주일 가량 유효하게 사용할 수가 있다.
24.4 나일론 멧슈 봉지 제작법 나일론 멧슈는 그 구멍 크기가 20, 40, 100, 200 미크론의 여 러 가지가 있으며 폭 l 미터, 길이 1_2 미터 크기의 것을 살 수 가 있다. 올리고덴드로사이트 배양 (20.1) 에서는 100 미크론 나일 론 멧슈를 사용하는 데 반하여 태생 래트 중추산경 분리세포 배 양, 계배 근육분리세포 배양, 태생 래트 근육분리세포 배양에서 는 20 미크론 나일론 멧슈를 사용한다. 나일론 멧슈를 4X 4 cm 크기로 절단하고 이룰 접은 뒤 두 장 의 슬라이드글라스 사아에 끼운다. 나일론 멧슈 조각이 슬라이드 글라스 사이로 2mm 정도 나오도록 하고 이 부분을 가스나 알코 올 램프 화염에 노출하면 융합이 되어서 삼각형의 ·봉지가 만들어 진다. 최근에는 플라스틱 실러 pla stic sealer 가 시판되고 있어서 이것을 사용하면 융합이 깨끗하게 된다. 나일론 봉지를 20 개 이 상 만들어 유리 페트리 접시에 넣고 오토클레이브에 의해서 멸균 해 둔다(그림 10-2 참고). 이돌 나일론 봉지는 사용한 뒤에 중성 세제로 씻은 다음 말려서 다시 쑬 수가 있다.
제 25 장 뇌하수체 세포의 조직배양 1970 년대 초기 부터 각종 동물의 뇌 하수체 전 영 ante r io r pitui - t ar y의 이식편 배양과 분리세포의 보고가 나오기 시작하였다 .1-3) 이들의 조직배양의 재료로서는 주로 래트의 뇌하수체가 사용되었 으며 뇌하수체 전영에 존재하는 성장호르몬 분비세포 somato t r o p h , 프롤락틴 분비세포 mammo t ro p h, 부신피질 자국호 르몬 (ACTH) 분비세포 cort ico tr o p h , 성선자국호르몬 (LH 와 FSH) 분비 세 포 go nadotr o p h , 갑상선 자국호르몬 분비 세 포 th y ro tr o p h 의 5 가지 종류의 세포를 순수 분리하려는 여러 가지 방법이 시도된 바 있다. 이는 뇌하수체를 콜라게나제, 디스파제, 트립신 등의 효소로 분리세포를 만든 다음에 각종 세포 타입의 비중 밀도의 차이를 이용하여 혈청 알부민, 퍼콜, 메트리자마이드 등에 의해 서 농도 구배컬럼을 만들어서 각종 세포를 분리하는 방법이다. 이론적으로는 좋아보이나 실제로는 원하지 않는 다른 종류의 세 포가 혼재하는 등 불편한 점이 적지 않다. 뇌하수체 세포 배양에 대 하여 서 는 틱 시 에 -비 달 Ti x ie r -Vi da l 과 팔쿠하 Farqu har 의 전문
서를 참고하기 바란다 .4) 여기에서는 신생 래트 뇌하수체 배양과 성인 뇌하수체 배양 5 ) 을 다루기로 한다. 25.1 신생 래트나 마우스 뇌하수체 분리세포 배양 〈 준비 〉 1) 입체해부현미경과 조명 2) 수술기 구 -10 cm 의 과용 가위 (직 선과 곡선) 2 개 10cm 의과용 핀셋 2 개 시계수선용 핀셋 (No. 4) 2 개 수술용 칼 (No. 11) · 2 개 3) 페트리 접시, 원심듀브, 피펫 4) 0. 25 % 콜라게나제 (이글 MEM 에 녹이고 필터여과 멸균) 5) 이글 MEM 합성 배양액 6) 마혈청 7) 50 % 포도당액 8) 2 mg /m l 겐타마이신액 〈 방법 〉 1) 신생 래트(마우스) 5-10 마리를 에텔자에 넣어서 심마취로 마취사시킨다. 2) 비커에 담긴 70% 알코올에 래트롤 잠깐 담그고 소독시킨 뒤 10 cm 페트리 접시로 옮긴다 . 3) 머리의 피부를 제거한 뒤 두 안구 사이에 가위를 넣어서 좌 우로 전개하듯이 두개골을 제거한다. 핀셋을 써서 뇌를 꺼낸 다. 뇌저 중앙에 직경 3X2mm 사이즈의 뇌하수체가 보이는
데 이를 핀셋으로 분리하고 핸크스액이 담긴 페트리 접시로 옮긴다. 4) 수술칼을 사용하여 뇌하수체를 3 등분한다. 2ml 콜라게나제 가 들어있는 원심튜브에 넣어서 2 시간 37 도에서 인큐베이트 한다. 5) 뇌하수체 조직을 튜브 바닥에 남긴 채 콜라게나제액을 제거 하고 10% 마혈청이 들어있는 배양액 2ml 를 가하고 20 번 피페팅을 한다. 파스출 피펫 끝에 미크로피펫립을 끼우고 20 번 피페팅을 되풀이한다. 6) 세포부유액을 1000rpm 속도로 8 분간 원심분리한다. 7) 상청액을 버리고 페렛에 배양액 2ml 를 가하고 1000rpm 8 분의 원심분리를 한다. 8) 상청액을 버리고 페렛에 배양액 2ml 를 가하고 미리 준비해 두었던 폴리라이신 코팅한 커버글라스 위에 플레이팅하고 5 % CO2 인큐베이터에서 배양한다. 배양액 조성은 다음과 같다. 이글 MEM 합성 배양액 88ml 마혈청 (혹은 우태아혈청) 10 ml 50% 포도당액 1ml 2 mg /m l 겐타마이신액 1 ml 9) 다음날 3ml 의 배양액을 커버글라스가 들어있는 6cm 페트 리 접시에 추가한다. 〈해설〉 배양 24-48 시간 안에 8-10 µm 직경의 원형 혹은 타원형의 뇌 하수체 유래 호르몬 분비세포를 커버글라스 위에서 볼 수가 있 다. 각종 호르몬 분비세포의 확정은 면역조직학 염색법에 의해서
행한다(제 36 장 참고) 25.2 성인 뇌하수체 분리세포 배양 저자의 연구실에서는 죽은 지 10 시간 이내인 사람의 병리해부 시 분리한 뇌하수체 전염의 조직배양을 하여 여러 가지 호르몬의 분비 를 확인한 바 있다. 여기에 그 방법을 기술한다. 〈 재료 〉 25.1 과 같다. 〈 방법 〉 1) 병리 해부에서 얻은 뇌하수체는 항생물질을 포함한 CMF -PBS 가 소량 들은 10cm 유리 페트리 집시에 옮기고 수술 칼 두 개를 교차시키는 상태로 작게 잘라낸다. 압박하지 말 고 예리하게 조직을 자른다. 2) 조직편을 원심튜브로 옮긴다. 3) 여분의 CMF-PBS 를 버리고 이글 MEM 에 희석한 0.25 % 콜라게나제액 3ml 를 가한 뒤 듀브를 인큐베이터에 옮기고 37 도에서 3 시간 정치한다. 15 분에 한 번씩 튜브를 혼든다. 4) 3 시간 뒤 튜브 바닥에 있는 조직편을 둔 채 콜라게나제액을 걷어서 버린다. 5) 10 % 마혈청을 포함한 배양액 5ml 룰 튜브에 첨가하고 20 번 피페팅을 한다. 조직편이 가라앉는 것을 기다려서 상청액 울 걷어서 새 원심튜브에 옮긴다. 6) 배양액 3ml 를 남아 있는 조직편에 첨가한 뒤 피펫 끝에 미
크로피펫립을 끼우고 피페팅을 20 번하여 세포분리를 한다. 7) 세포부유액을 합쳐서 1200rpm 8 분간 원심분리한다. 페렛 울 10 ml 핸크스액 에 부유한다. 8) 40 ml 오크릿지 원심튜브에 퍼콜액 9 ml, 핸크스액 10 배 액 1ml 를 가하고 여기에 핸크스액 10ml 와 위의 세포부유액 10 ml 모두 합하여 30 ml 의 용액을 혼합한다. 9) 고속원심분리기에서 15, 000 rpm 속도로 25 분간 원심분리한 다. 튜브에 5 층의 농도 구배 densit y grad ie n t 가 보인 다. 위 에 서부터 제 1 층은 투명한 핸크스액 , 제 2 층은 죽은 세포와 세 포 잔재가 모인 우유색 충, 제 3 층은 반두명한 세포를 포함 한 층이고, 제 4 층은 한 줄로 보이는 적혈구 충, 제 5 층은 바닥이며 세포가 없는 충이다. 제1, 2 층을 버리고 제 3 층을 튜브에 모은 뒤(약 15ml 정도) 여기에 30ml 의 핸크스액을 넣어서 희석하고 1800r p m 으로 10 분간 원심분리한다. 10) 상청액을 버리고 페렛에 10 % 마혈청을 포함한 배양액 3 ml 를 가하고 1500r p m 으로 8 분간 원심분리한다. 11) 페렛에 배양액을 8ml 가하고 피페팅을 한 뒤 60mm 패트 리 접시 2 개에 분주하고 5 % CO2 인큐베이터에서 배양한 다. 폴리라이신 코팅한 12 mm 풀라스틱 커버글라스에 세포 롤 플레이팅하면 세포화학영색이나 전자현미경 검색에 편리 하다. 배양액은 일주일에 두 번 교환한다. 배양액 조성은 다음과같다. 이글 MEM 합성 배양액 88ml 마혈청 10ml 2 mg /m l 겐타마이신액 1 ml 0.25mg /m l 암포테리신액 1ml
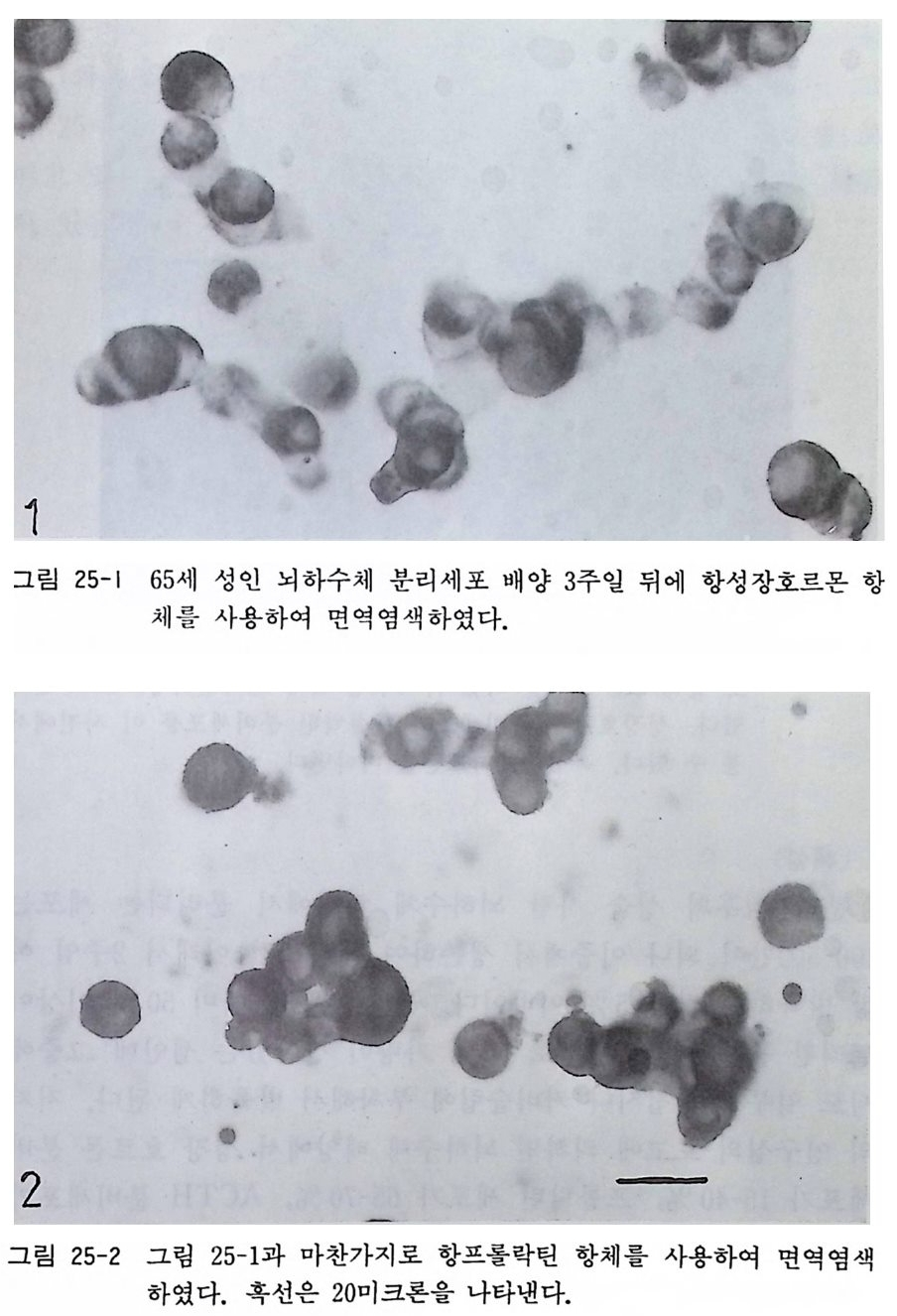 1
1
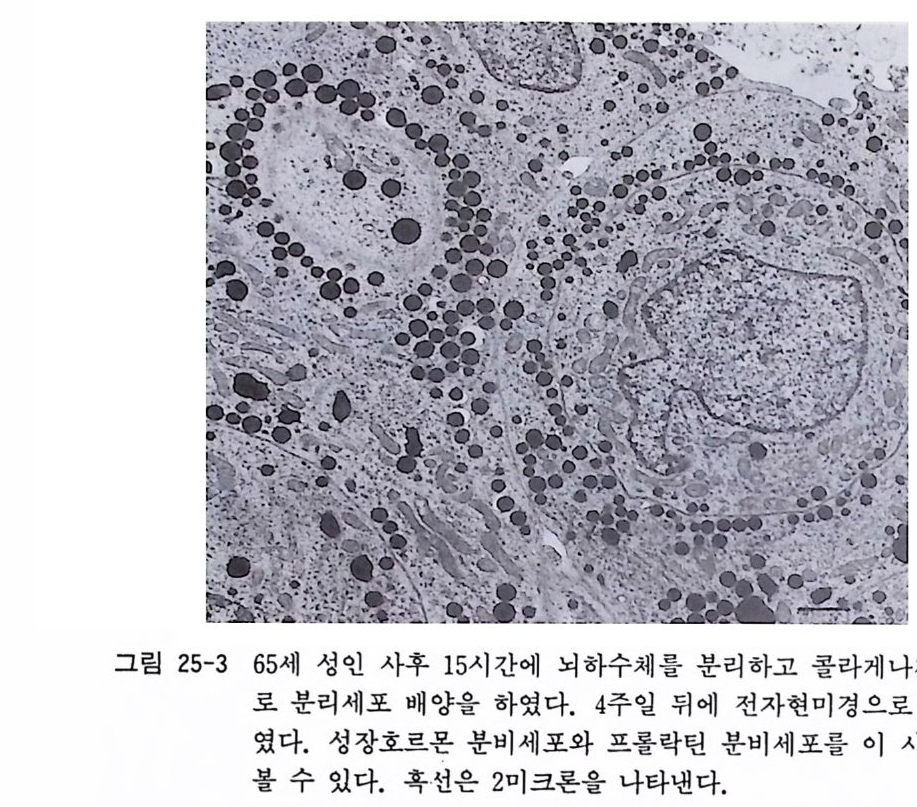 그림 25-3 65 세 성인 사후 15 시간에 뇌하수체 를 분리하고 콜라게나제 처리
그림 25-3 65 세 성인 사후 15 시간에 뇌하수체 를 분리하고 콜라게나제 처리
〈해설〉 분리 직후의 성숙 사람 뇌하수체 하나에서 분리되는 세포는 100-500 만이 되나 이중에서 생존하여 배양조건 아래서 2 주일 이 상 발육하는 것이 5 % 이하이다. 분리 직후에 이미 50 % 이상이 트리판 블루에 염색됨으로 50 % 가량이 살아있는 셈인데 그중에 서도 일부만이 접시나 커버슬립에 부착해서 발육하게 된다. 저자 의 연구실의 보고에 의하면 뇌하수체 배양에서 성장 호르몬 분비 세포가 15-40 %, 프롤락틴 세포가 65-70 %, ACTH 분바세포가 20 %, LH/FSH 세포가 10 %, TSH 분바세포가 10 %의 비율을 보이고 있었다 .5,6) 전자현미경 아래에서도 각종 호르몬 분비세포
의 분비과립의 크기로서 각 종류의 확정이 가능하였다. 그림 25-1 과 25-2 에서 성인 뇌하수체 배양의 실제를 보이고 있다. 그 립 25-3 에서는 성인 뇌하수체 배양세포의 전자현미경 사전을 보 이고 있는데 크기가 다른 호르몬 분비 과립이 세포체 안에 가득 차 있음을 볼 수 있다.
제 26 장 부신수질 크로마핀 세포의 배양 신경융 neural cres t은 태생기의 신경발생기에 신경세포와 내분 비세포로 분화하는 독특한 성격을 가지고 있다. 부신수질 adrenal medulla 크로마핀 세 포 chromaff in cell 는 이 신 경 융 유래 세 포의 분화에 있어서 각종 약물의 영향을 검색하는 데 유력한 모델이 될 수 있다. 태생이나 신생 래트의 크로마핀 세포를 NGF 존재 하에서 배양하면신경세포로변환하여서 신경섬유가신생하게 되고, I) 임신 래트에게 NGF 를 대량으로 두여하면 태어난 신생 래트의 크로마핀 세포가 교감신경세포로 형질변환이 될 수 있다 .2) 크로 마핀 세포의 신경세포로의 형질 변환은 신생 동물에만 한하지 않 고 래트, 기니피그, 사람에서도 성숙 동물의 크로마핀 세포를 NGF 존재하에 배양하면 가능하다는 것이 알려져 있다 .3) 최근 크로마핀 세포의 배양이 주목을 받고 있는데 그 이유는 중추신경 계 변성질환인 파킨슨병 Parkin s on's dis e ase 환자에서 부신수질 조직의 뇌 자가이식으로 임상증세가 개선되었다는 보고가 있었기 때문이다 .4) 현재는 부신수질 뇌내 자가이식은 효과가 없다고 알
려져 있으나 사람 태아 크로마핀 세포를 배양한 뒤 이를 파킨슨 병 환자의 뇌에 이식하는 것은 이론적으로 가능성이 있다 .5) 크 로마핀 세포의 조직배양을 이용하여 카데콜아민 분비 신경세포의 성질과 파킨슨병의 병인과 치료를 연구하는 것은 앞으로도 계속 될 것이다. 26.1 신생 래트(마우스) 부신수질 크로마핀 세포의 분 리세포 배양 〈 준비 〉 1) 입체해부현미경과 조명 2) 수술기구 -10 cm 의과용 가위 (직선과 곡선) 2 개 10 cm 의 과용 핀셋 (곡선) 2 개 시 계수선용 핀셋 (No. 4) 2 개 수술용 칼 (No. 11) 2 개 3) 페트리 접시, 원심튜브, 피펫 4) 0.25 % 콜라게나제 (이글 MEM 에 녹이고 필터여과 멸균) 5) 이글 MEM 합성 배양액 6) 마혈청 7) 50 % 포도당액 8) 2mg /m l 겐타마이신액 〈방법〉 1) 신생 래트나 마우스 5-10 마리를 에텔자에 넣어서 심마취로 서 마취사시킨다. 2) 비커에 담긴 70 % 알코올에 래트를 잠시 담그고 소독시킨
뒤 10cm 페트리 접시에 옮긴다. 3) 래트를 눕히고 가위로 배의 피부의 정중선을 자른다. 그L°.= 도 역시 절개한 뒤 신장을 찾아내고 그 상부에 부착되어 있 는 부신을 분리하고 핸크스 용액이 들어있는 10cm 페트리 집시에 옮긴다. 현미경 아래에서 부신을 절개하고 부신수질 을 분리한다. 4) 수술칼 2 개를 교차하면서 부신수질을 소편으로 절단한다. 5) 조직편을 원심듀브에 옮기고 여분의 핸크스액을 제거한 뒤 0.25% 콜라게나제액 2ml 를 가하고 37 도에서 2 시간 인큐베 이트한다. 6) 듀브 바닥에 있는 조직을 남겨두고 콜라게나제액을 제거한 다. 7) 10 % 마혈청을 포함한 배양액 3ml 를 튜브에 가하고 20 번 피페팅을 한다. 계속하여 피펫 끝에 미크로피펫팁을 끼우고 20 번 피페팅을 되풀이한다. 8) 세포부유액을 1000rpm 속도로 8 분간 원심분리한다. 페렛 에 배양액 3ml 를 가한 뒤 원심 분리를 한번 더 되풀이한다. 9) 페렛에 배양액 2ml 을 가하고 5Xl06 세포가 되도록 조절한 뒤 폴리라이신 코팅한 커버글라스에 플레이팅하고 5% CO2 인큐베이터에서 배양한다. 배양액 조성은 다음과 갇다. 일주 일에 두 번 배양액 교환을 한다. 이글 MEM 합성 배양액 87ml 마혈청 (혹은 우태아혈청) 10ml 10 µg/m l NGF 1 ml 50% 포도당액 1 ml ` 2mg /m l 겐타마이신액 1ml
〈 해설 〉 위의 방법은 신생 래트나 마우스의 부신 크로마핀 세포의 배양 에서 적합한 방법으로서 여러 연구자가 이 방법을 쓰고 있다. 저 자의 연구실에서는 10-12 주 사람 태아 · 부신의 분리세포 배양을 하였는데 이때에는 콜라게나제 대신 0.25 % 트립신액을 사용하 여 분리세포를 만들었다. 그림 26-1 과 26-2 에서 사람 태아 부신 크로마핀 세포 배양의 실제를 보이고 있다. 대량으로 크로마핀 세포를 분리하고자 할 때에는 소의 부신 조직을 사용하는데, 콜 라게나제액에 의한 부신조직의 관류와 밀도구배 컬럼을 사용하여 그 순도가 높아지게 된다 .6)
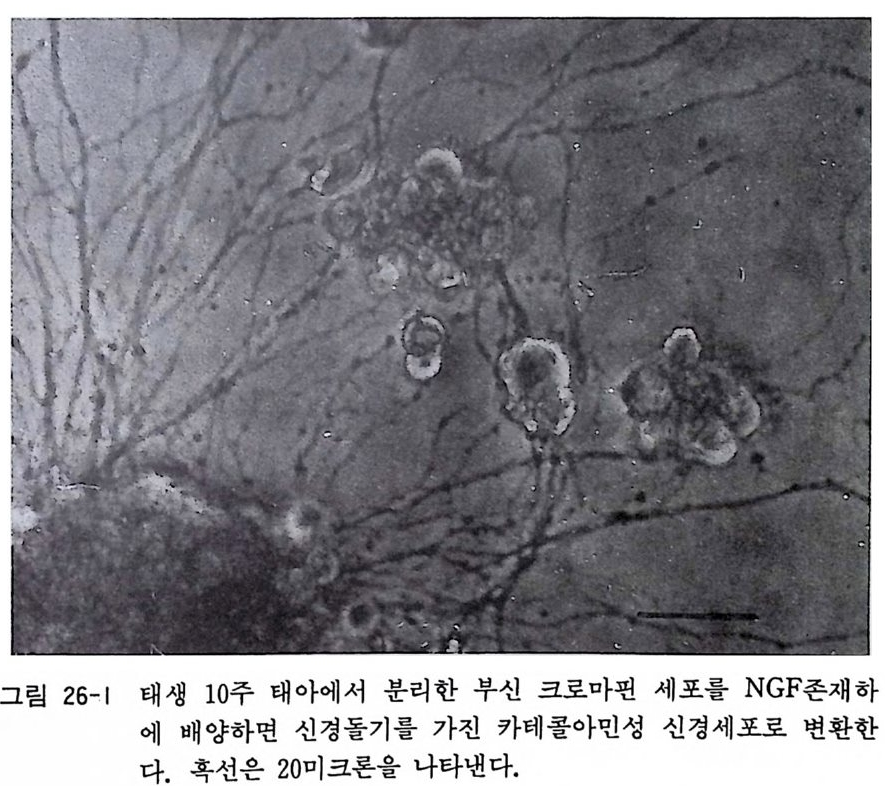 그립 26 기 태생 10 주 태아에서 분리한 부신 크로마핀 세포를 NGF 존재하
그립 26 기 태생 10 주 태아에서 분리한 부신 크로마핀 세포를 NGF 존재하
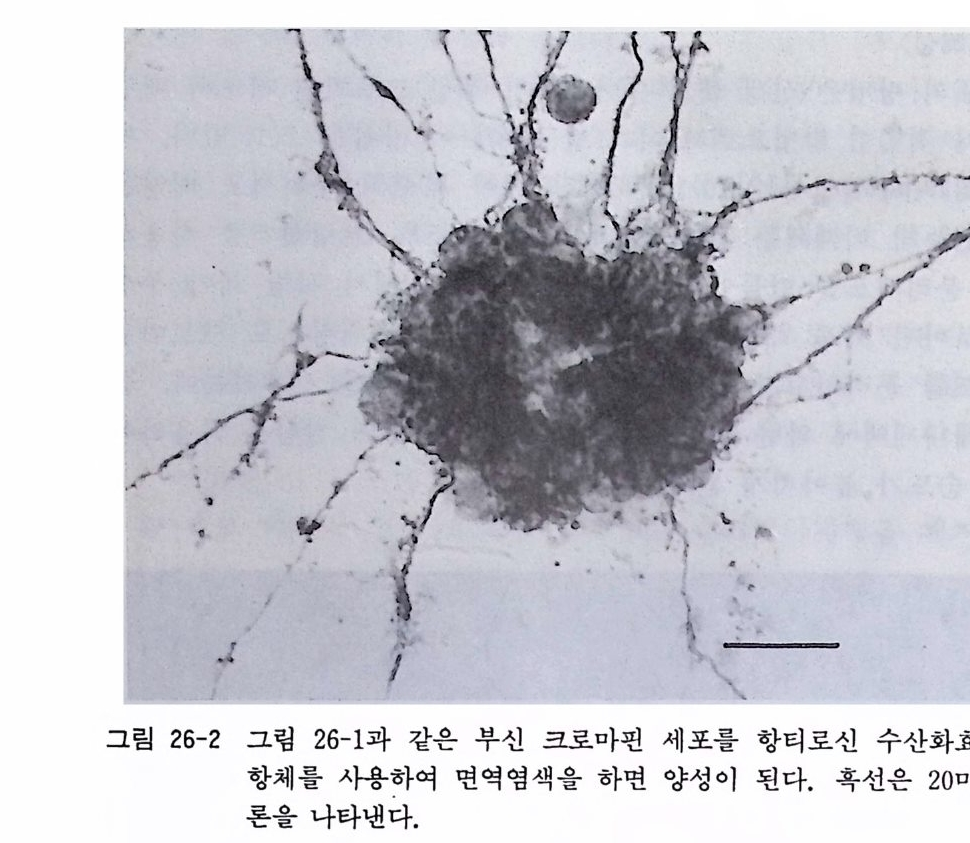 T
T
제 27 장 양생류와 어류 신경조직의 배양 실험생물학의 분야에서는 오랫동안 개구리와 갇은 양생류를 실 험계로서 사용하였다. 예컨대 조직배양의 아버지라 불리는 해리 슨 Harr i son 은 양생류인 올챙이 척수의 이식편 배양에서 신경섬 유가 새로 발육해 나오는 것을 관찰하였다. I) 그러나 최근의 포 유류와 조류의 조직을 사용한 신경조직 배양에 비하면 양생류와 어류의 신경조직을 재료로 하는 조직배양의 보고는 그 수가 극히 한정 되 어 있다. 크루즈 Kruse 와 패 터슨 Patt er son 이 저술한 조직 배양 매뉴얼에서도 양생류와 어류의 비신경계 조직의 배양에 대 해서는 여러 가지 서술이 있는데 비하여 신경조직의 배양에 대해 서는 한마디도 언급하지 않고 있다. 2) 양생류와 어류의 신경계 발달에 관해서는 이미 기초지식이 많 이 축적되어 있고 재료를 쉽게 얻을 수 있으며 포유류의 신경조 칙에 비해서 그 재생 능력이 대단히 높다는 등 양생류와 어류 신 경조직의 실험 모델로서의 가능성을 높게 평가할 필요가 있다. 여기에서는 양생류와 어류 신경조직을 사용한 조직배양 모델에
대해서 소개한다. 27.1 올챙이 척수이식편 배양 〈준비〉 1) 수술기구 -10 cm 의과용 가위 1 개 10cm 의과용 핀셋 2 개 시계수선용 핀셋 (No. 4) 2 개 수술용 칼 (No. 11) 2 개 2) 페트리 접시, 커버글라스, 피펫 3) PBS 4) 라이보비츠 L-15 합성 배 양액 5) 우태아혈청 6) 2mg /m l 겐타마이신액 7) 1 M HEPES 완충액 〈방법〉 1) 개 구리 Rana pipien s 의 태 생 기 인 올챙 이 의 발달단계 (스테 이 지)를 테일러 Ta y lor 와 콜러스 Kollros 의 아틀라스에 의해서 확정한다 .3) 비커에 들어 있는 70% 알코올 속에 올챙이를 참깐 담가서 소독한 다음 PBS 가 들어있는 페트리 접시로 옮긴다. 2) 의과용 가위를 써서 올챙이 등의 피부, 근육, 척추연골을 제거한 다음 시계용 핀셋 2 개를 써서 척수를 분리한다. 3) 척수를 10cm 직경 유리 페트리 접시로 옮기고 수술칼 2 개 를 써서 0.5-1mm 두께의 디스크로 절단한다.
4) 미리 콜라겐을 코팅한 22mm 직경 원형 커버글라스나 llX 22mm 장방형 커버글라스를 3.5cm 직경 페트리 접시의 중 앙에 놓고 한 방울의 배양액을 떨어뜨린다• 5) 척수 소편 2 개를 5mm 간격을 두고 커버슬립 위에 올려놓 는다. 6) 페트리 접시를 데시케이터 안에 놓고 실온(1 8-20 도)에서 배 양을 개시한다. 높은 습도를 유지하기 위하여 데시케이터 바 닥에 증류수를 넣는다. 7) 배양액의 조성은 다음과 같다. L-15 배 양액 78 ml 우태아혈청 20ml 2 mg / ml 겐타마이신액 1 ml 1 M HEPES 완충액 1 ml 27.2 올챙이 척수 분리세포 배양 〈준비 〉 27.1 의 올챙이 척수 이식편 배양과 같으나 다음을 따로 준비한 다. 1) 파파인 (워싱턴 혹은 시그마) 100 unit /m l 2) 시스테 인 (cy st e i n , 시그마) 〈방법〉 1) 27.1 순서 l) 에서 3) 까지를 되풀이한다. 2) L-15 합성 배 양액 1. 8 ml 에 파파인 (100 un it/m l) O. 2 ml, 그 리고 시스테인 8m g을 가하고 여과멸균한다. 활성화한 파파
인액은 10unit /m l 최종 농도를 가진다. 3) 활성화한 파파인액이 들어있는 원심튜브 속에 척수조직을 넣고 실온에서 30 분간 인큐베이트한다. 4) 파스출 피펫으로 20 번 피페팅을 한다. 계속하여서 피펫 끝 에 미크로피펫립을 끼우고 20 번 피페팅을 되풀이한다. 5) 원심듀브롤 1000rpm 속도로 8 분간 원심분리한다. 6) 페렛에 배양액 2ml 를 가하고 원심분리를 되풀이한다. 7) 페렛에 배양액 2ml 를 가하여 세포부유액을 만들고 폴리라 이신 코팅한(혹은 라미닌 코팅한) 12 mm 직경 원형 글라스 (혹은 아클라플라스틱) 커버슬립에 50, 000-100, 000 세포를 플 레이팅한다. 8) 배양액 구성은 27.1 과 같다. 플레이팅한 다음날 배양액을 추가하여 페트리 접시를 채운다. 배양액 교환은 일주일에 두 번 행한다. 〈해설〉 일반으로 포유동물이나 조류 조직배양을 위한 배양액이나 염류 용액은 삼투압이 높기 때문에 L-15 나 이글 MEM 에서 중탄산나 트륨을 제거하여 양생류나 어류 조직에 맞는 삼투압으로 조정한 다. 다른 연구실에서는 파파인을 사용하지 않고 척수조적을 수술 용 칼로 섬세하게 자론 다음 PBS 속에서 피페팅을 되풀이함으로 써 분리세포를 얻을 수 있다고 한다 .4) 분리세포 배양의 경우에 는 혈청 농도를 20 %에서 10 %로 줄이는 것이 좋다. 연구실에 따라서는 우태아혈청이나 마혈청 대신 개구리 혈청을 사용하여 3-4 개월간의 장기 배양이 가능하다고 한다.” 양생류나 어류에서 는 포유류나 조류와 달라서 혈당량이 낮기 때문에 포도당액을 첨 가할 필요가 없다. 올챙이 척수 이의에도 척수후근절 신경세포
배양이 가능하며 NGF 에 의해서 신경섬유 신장 효과를 볼 수가 있다고 한다. 또한 올챙이 척수-근육 공동배양이 가능하며 신경 -근육 접합부의 전기생리학적 연구에 사용되고 있다 .6,7) 27.3 금붕어 망막이식편 배양 어류의 신경조직 배양으로서 현재까지 보고된 것은 금붕어 망 막의 이식편 배양과 분리세포 배양이 8 , 10) 있다. 〈 준비 〉 27.1 과 같다. 〈 방법 〉 1) 배양 개시 2 주일 전에 금붕어 시신경을 절단한다. 이것은 망막 신경절세포g an gli on cell 의 재생 능력을 최고로 가져가 기 위함이다• 2) 금붕어를 비커 속에 들어있는 70% 알코올 속에 참깐 담가 서 소독한 다음 10 cm 페트리 집시로 옮긴다. 3) 안구를 분리한 뒤 안구를 철단하고 각막, 초자체, 색소상피 를 차례로 제거한다. 4) 망막을 2x2mm 크기로 절단한 뒤, 콜라겐 코팅한 커버글 라스 위에 플레이팅한다. 커버글라스는 페트리 접시에 넣고, 페트리 접시는 고습도인 데시케이터에서 배양을 한다. 배양액 조성은 다음과 같다. L-15 배양액 78 ml 우태아혈청 20ml
2 mg / ml 겐타마이 신액 1 ml 1 M HEPES 완충액 1 ml 〈 해설 〉 그림 27-1 에서는 금붕어 망막이식편 배양에서 신경섬유와 신경 세포, 글리아세포가 이식편 주위로 유출해 나오는 것을 보이고 있다.
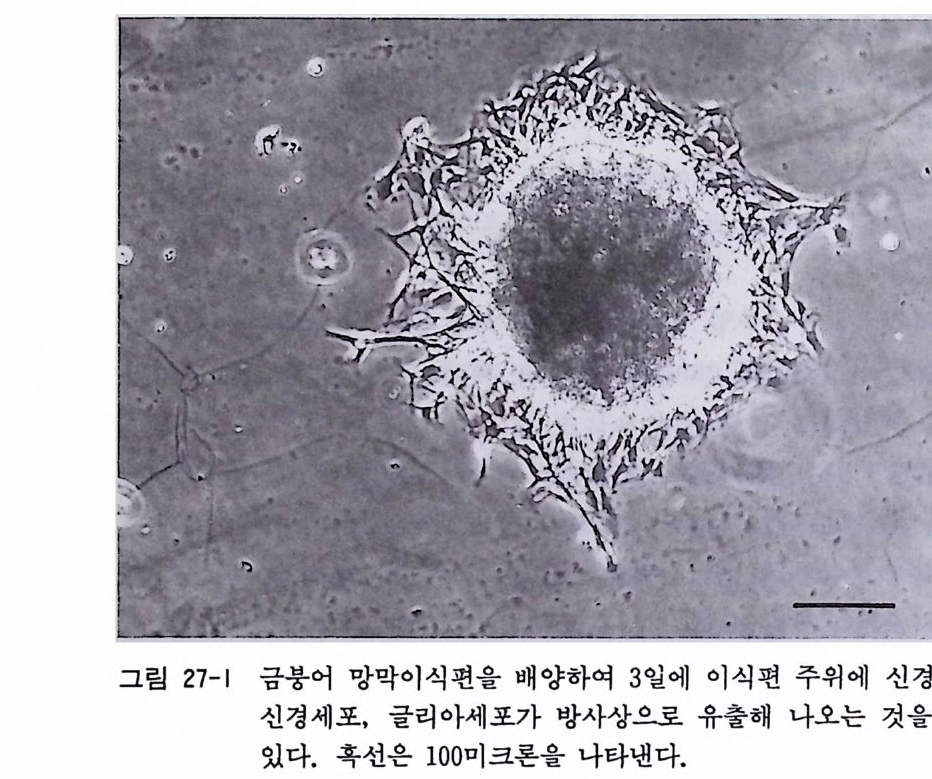 -‘ ..
-‘ ..
27.4 금붕어 망막분리세포 배양 〈 준비 〉 27.1 과 같다. 파파인액을 27 . 2 에 기술한 대로 준비한다. 〈 방법 〉 1) 활성화한 파파인액 (10 unit /m l 파파인과 4mg /m l 시스테인이 들 어있는 L-15 배양액)이 들어있는 원심듀브에 망막조직을 넣 고 실온에서 30 분간 인큐베이트한다. 2) 파스출 피펫으로 20 번, 미크로피펫팁을 끼운 피펫으로 20 번 피페팅을 한다. 3) 원심튜브를 1000rpm 속도로 8 분간 원심분리한다. 4) 페렛에 배양액 2ml 를 가하고 원심분리를 되풀이한다. 5) 페렛에 배양액 2ml 를 가하여 세포부유액을 만들고 폴리라 이신 코팅한 커버슬립에 50, 000-100, 000 세포 농도로 플레이 팅을한다. 6) 배양액 구성은 27 . 1 의 올챙이 척수배양과 같다.
제 28 장 무척추동물 신경조직배양 무척수동물의 조직배양은 유전학, 생리학, 병리학의 분야에 중 요한 공헌을 하여 지금에 이르고 있으며,I) 특히 곤충조직 배양 온 곤충이 식물과 동물의 질병을 매개하는 벡터가 되기 때문에 그 본태를 해명하고자 곤충조직 배양이 성행하였다 .2) 무척수동 물의 신경계조직배양은 최근까지 별로 주목을 받지 못한 분야인 데 그간 초파리 ,3,4) 바퀴벌레 ,5,6) 거머리 7,8) 달팽이 9,10) 아프리지 아 11,12) 등의 연구가 있다. 28.1 초파리 신경조직배양 초파리 Drosop hi l a melang a.st er 의 신경조직배양은 20 년 전에 시 작되었는데, 발생 초기의 유충 전체를 배양하기 때문에 이러한 배양에는 신경조직 이의의 다론 조직이 많이 혼재하고 있었다 .3) 최근 부화 96 시간인 제 3 기 유충의 뇌반구와 신경절을 분리세포
배양하였다는 보고가 있다 .4) 이 보고에 의하면 세 종류의 신경 세포가 그 크기에 따라서 분류되며, 활동 전위의 발생, 나트륨 채널의 발현 등 전기생리학적인 연구가 가능하였다고 한다. 〈 준비 〉 1) 수술기구-시계수선용 핀셋 (No. 4) 2 개 수술용 칼 (No. 11) 2 개 2) 페트리 접시, 원심듀브, 피펫 3) 슈나이 더 Schneid e r 곤충용 영 류용액 4) L-15 합성 배 양액 5) 마혈청 (혹은 우태아현청) 6) 2 mg / ml 겐타마이신액 7) 10 unit /m l 파파인 (4 mg /m l 시스테인 첨가) 〈 방법 〉 1) 제 3 기의 초파리 유충(부화 후 96 시간)을 70 % 알코올 속에 담가서 소독한 뒤 증류수에서 2 번 씻는다. 2) 슈나이더액이 들어있는 10cm 페트리 접시 속에서 해부용 입체현미경 아래서 뇌와 신경절을 분리한다(그림 28-1). 유 충의 머리와 꼬리의 구별이 곤란한데, 두 개의 안데나 같이 생긴 기관이 보이면 이것이 꼬리가 된다. 3) 수술용 칼로 뇌와 신경절을 작게 철단한다. 4) 파파인액이 들어있는 원심튜브에 뇌조직을 넣고 실온에서 30 분간 인큐베이트한다. 5) 파파인액을 버린 뒤 슈나이더액 2ml 를 가하고 20 번 피페팅 을 한다. 피펫 끝에 미크로피펫립을 끼우고 20 번 피페팅을 되풀이한다.
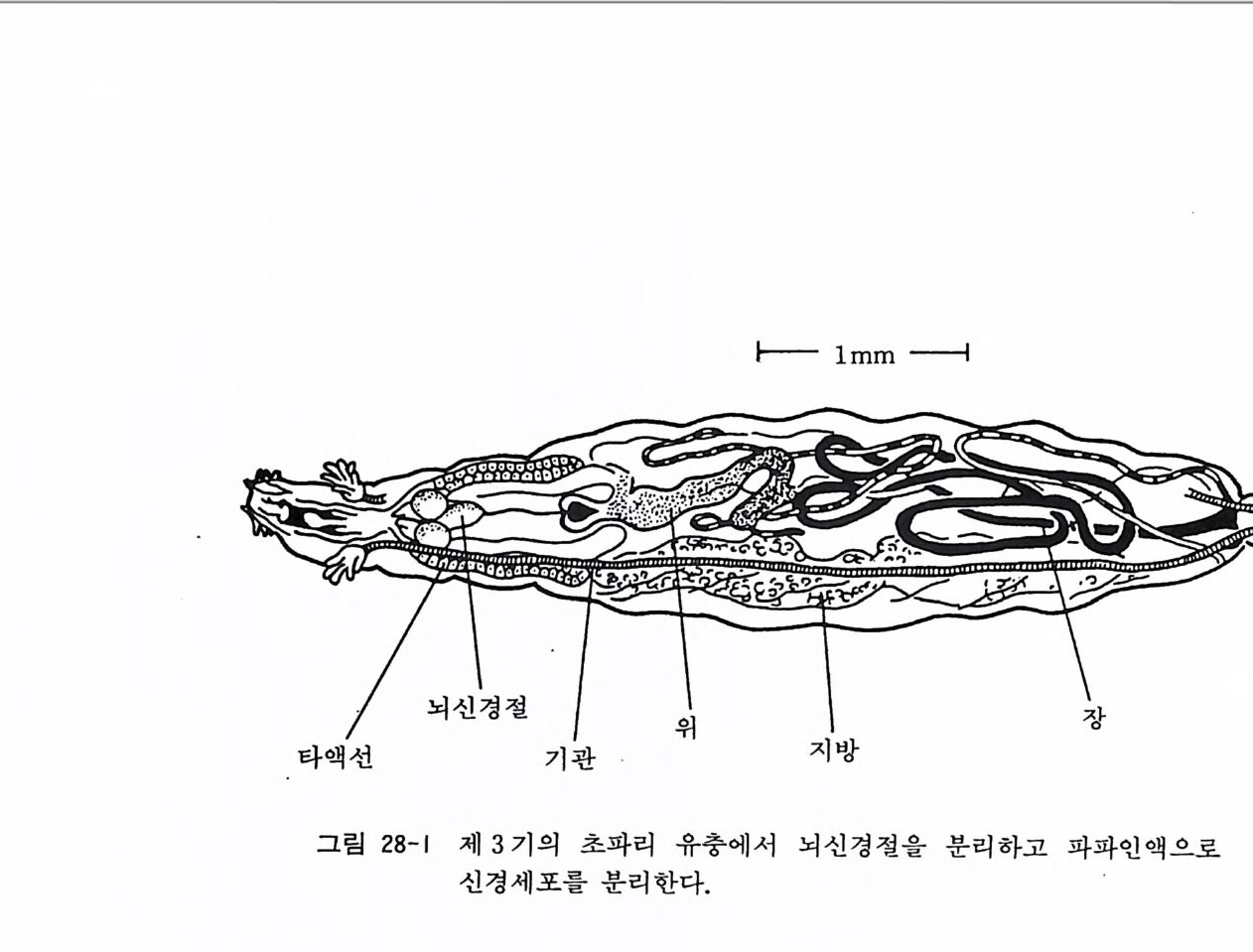 하
하
6) 1000 rpm 속도로 8 분간 원심분리한다. 7) 페렛에 배양액 2ml 를 가하고 원심분리를 되풀이한다. 8) 페렛에 배양액 2ml 를 가하여 세포를 부유시키고 세포부유 액 (1X 1 06 세포 /ml) 을 조정하고 폴리라이신 코팅한 커버글라 스에 플레이팅한다. 9) 배양액 구성은 다음과 같다. 슈나이더액 45 ml L-15 배양액 44ml 마혈청 20 ml 2 mg / ml 겐타마이신액 1 ml 10) 24 시간 뒤 3ml 의 배양액을 추가하여 페트리 접시를 채운 다. 페트리 접시는 바닥에 증류수를 넣은 데시케이터에 넣 어서 밀봉하고 실온에서 배양한다. 배양액 교환은 일주일에 두번한다. 〈 해설 〉 곤충 조직배양에서 사용되는 각종 염류 용액은 시그마, 깁코 등에서 살 수가 있다• 그레이스 Grace 영류용액에서는 염화나트 륨을 완전히 제거하였고 슈나이더액에서는 염화나트륨이 3 분의 1 이하가 되었으며, 염화칼륨과 염화마그네슘의 농도가 증가되어 있다. 28.2 바퀴벌레 신경조직배양 바퀴벌레 유충 Pe rip lane ta ame ri cana 에서 분리한 신경절은 콘 웰 Cornwell 의 아틀라스에 의 해서 그 발달 단계를 결정 한다. 13)
바퀴벌레 유충 신경절 배양의 상세는 레비-몬탈치니 Levi- Mon t alc i n i의 논문을 참고하기 바란다. 5,6) 〈 준비〉 28 . 1 과 같다. 〈방법〉 1) 16-18 일째의 바퀴벌레 알 케이스 e gg case 를 70 % 알코올에 참깐 담가서 소독한 뒤 증류수에서 두 번 씻는다. 2) 슈나이더액이 들어있는 페트리 접시 속에서 알 케이스를 깨 고 유충을 분리한다. 시계수선용 핀셋을 써서 뇌, 식도하신 경절, 흉부신경절, 복부신경철을 분리한다. 3) 미리 콜라겐을 코팅한 22mm 직경 원형 커버글라스를 3.5 cm 칙경 페트리 접시의 중앙에 놓고 한 방울의 배양액을 떨 어트린다. 4) 분리한 신경절을 2 개씩 커버글라스 위에 올려 놓는다. 5) 페트리 접시를 데시케이터 안에 놓고 29 도 인큐베이터에서 배양을 시작한다. 높은 습도를 유지하기 위하여 데시케이터 바닥에 증류수를 채운다. 6) 배양액 조성은 다음과 같다. 슈나이더 용액 45ml L-15 배양액 44ml 마혈청 20ml 2mg /m l 겐타마이신액 1ml
제 4 부 신경계 수립세포주 
제 29 장 뇌종양의 초대배양법 뇌종양 brain tum or 에서 는 악성세포가 무한중식을 계속하고 있 기 때문에 뇌종양에서 세포를 분리하고 이를 배양하여 주세포를 수립하는 것이 쉬울 것이라 생각되나 실제로는 이것이 대단히 어 려워서 뇌종양 배양에 적합한 기술과 수많은 시행착오가 필요하 다. 정상 조직에서 분리된 세포가 계대배양되는 경우에 이들이 무 한히 증식하는 것이 아니고 인간태아 폐 섬유아세포에서 보더라 도 50 대 정도에서 사멸하게 된다. 마우스나 행스터 세포에서는 이보다도 짧아서 20 대 전후의 계대에서 끊어지게 마련이다. 뇌종 양에서 분리된 세포는 정상 신경조직에서 분리된 아스트로사이트 나 혈관 상피세포와 같은 유한 증식성을 가진 세포에 비하여 무 한 증식성을 가지므로 이것이 이점이 될 수가 있겠다. 사람의 뇌종양 조직은 수술실에서 수술 샘플로서 얻어오는 것 이므로 어느 정도 무균상태를 유지할 수 있다. 한편 수술실에는 여러 가지 잡균의 감염이 있다는 가능성을 생각하여서 뇌종양 조
직은 항생물질을 고농도로 포함한 핸크스 용액이나 PBS 가 들은 비커나 샘플 컨데이너에 넣은 뒤 조직배양실로 가져오도록 한다. 뇌종양의 초대 배양법에는 두 가지 기본 방법이 있는데 그것은 조직소편 배양법과 분리세포 배양법이다. 29.1 조직소편 배양법 조직소편 배양법 exp la nt cul t ure 은 이식편 배양법이라 불리기도 하는데 극히 간편하고 손쉽게 조직배양을 시작할 수 있는 장점과 또한 극히 제한된 소량의 조직으로도 시작할 수 있는 장점이 있 다. 조직을 소편으로 자를 때에는 예리한 수술칼 sca p e! 이나 안과 용 가위를 사용하여 조직편을 예리하게 만들 것이며 조직소편 속 으로 가스나 영양물질이 충분히 침투할 수 있도록 그 크기가 1-lOmm 까 넘어서는 안된다. 한편 조직편이 너무 작으면 그 속 에 포함되는 세포 수가 너무 적어서 발육이 안되는 수가 있으니 조심하여야 한다. 약 10 년 전까지는 응고혈장p lasma clo t을 써서 조직소편을 커버슬립이나 페트리 집시 바닥에 부착하도록 하였는 데 최근에는 좋은 혈장의 입수가 힘들어서 사용되지 않는다. 신 경조직 배양에서 최근 많이 쓰이는 세포기질로서는 콜라겐이 있 다. 1 형 콜라겐을 래트 꼬리에서 분리하고 커버글라스나 페트리 접시에 코트하는 방법은 6.1 섹션에서 소개하였으니 이를 참고하 기 바란다• 사람이나 동물의 뇌종양 조직의 초대배양을 시작하는 데 흔히 쓰이는 방법은 샌드위 치 sandw ich 법 과 에 칭 etc h in g 법 의 두 가지가 있어서 여기에 소개한다(그립 29-1 과 29- 2) . 샌드위치법은 조직소편을 35mm 페트리 접시 바닥에 위치하고 그 위에 22X22mm 유리 커버슬립을 울려 놓는다. 커버슬립의
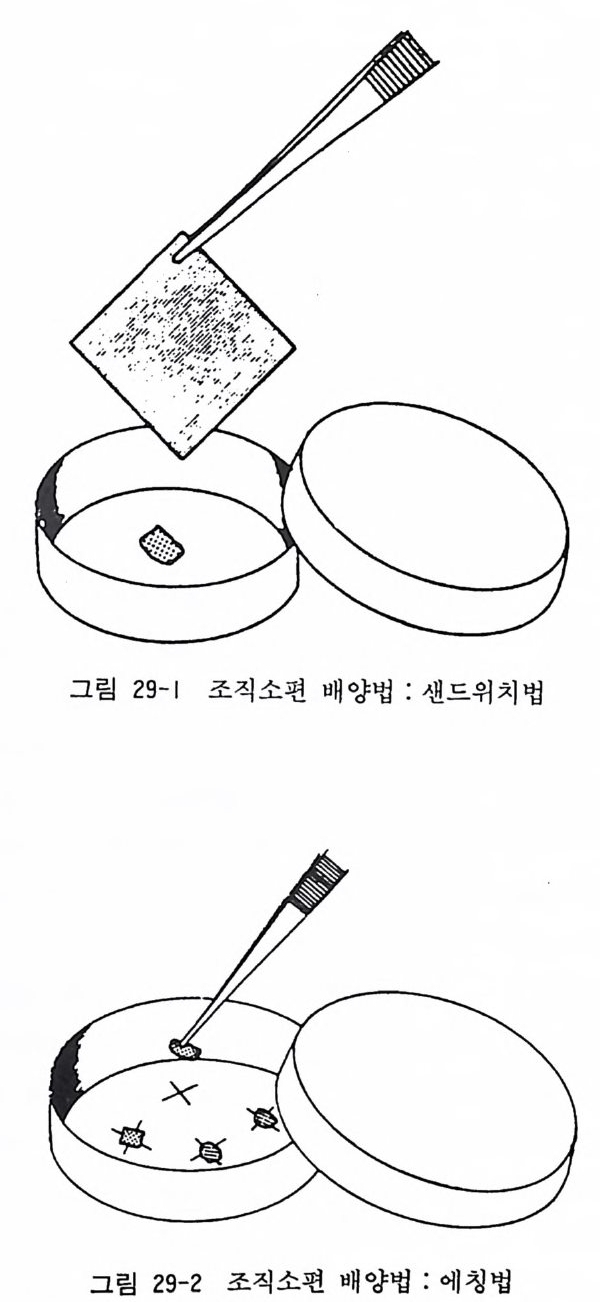 그림 29-1 조직소편 배양법 : 샌드위치법
그림 29-1 조직소편 배양법 : 샌드위치법
압력으로 조직소편이 유리되는 것을 방지하게 된다. 접시에 2 ml 의 배양액을 주입하고 배양을 시작한다(그립 29-1). 에칭법은 35mm 페트리 집시 바닥에 3-4 개의 X 를 수술칼을 써서 그어 놓는다. 조직소편을 X 위에 울려놓고 약간의 압력을 가하면 접시 상처 위에 소편이 부착하게 된다. 30 분 가량 기다린 뒤에 2ml 의 배양액을 가하고 배양을 시작한다(그립 29-2). 일반 적으로 2-3 일 안으로 세포가 조직편 주위에서 이동해 나오는 것 이 보이나 예의적으로 1-2 주일이 걸리는 수도 있다. 29.2 분리세포 배양법 뇌종양 조직을 단일 세포로서 분리하고자 할 때에 효소 처리에 의한 분리와 기계적인 처리에 의한 세포 분리의 두 가지 방법이 있다. 효소를 쓰는 경우에는 트립신, 프로테이네즈, 파파인 혹은 콜라게나제 등의 효소를 하나 선택해서 사용한다. 효소액 속에서 소화가 된 조직은 염류용액이나 효소억제액을 써서 효소를 씻어 내고 피페팅을 되풀이하여 단독 세포로 분리한다. 세포덩어리가 아직도 남아 있는 경우에는 20-100 미크론 구멍 크기를 가전 나 일론 멧슈를 써서 걸러낸다• 나일론 멧슈의 입수가 힘든 경우에 는 여성 나일론 양말을 잘라내어서 오토클레이브 멸균을 하여 사 용토록 한다• 조직이나 세포 잔재가 많이 있는 경우에는 1 분에 500 회전 (50 g) 정도의 원심 침전을 시키고 씻어낼 수 있다. 세포 분리법에는 세 가지 주의가 필요한데 그것은 다음과 같다. 첫째, 조직을 소편으로 조그마하게 자르는데 이것은 조직 표면 이 효소에 닿는 부분을 . 많이 하기 위한 것이다. 너무 작게 조직 울 자르면 세포들이 파괴되고 너무 크면 효소 작용이 낮아서 세
포 분리가 곤란하다. 따라서 1mm3 정도 혹은 그보다 약간 큰 정도가 이상적이다. 둘째, 자기 연구 목적에 이상적인 효소와 그 사용 농도를 결정 하여야 한다. 참고문헌에 사용 농도가 지시되어 있으므로 이것을 참고로 하여 조직과 사용목적에 따라서 경험적으로 결정하도록 할 것이다. 셋째, 효소액 속에서의 인큐베이션 시간이 역시 중요하다. 너 무 오래 효소액에서 소화하면 조직과 세포가 파괴되고, 너무 짧 은 시간이면 조직에서 세포가 분리되지 않는다. 일반적으로 0.2 5 % 트립신에서는 20 분에서 30 분인데, 이것도 트립신의 종류 와 제조자에 따라서 품질이 천차만별로 다르기 때문에 경험적으 로 자기가 결정하여야 한다. 일반적으로 트립산울 사용하여 조직 에서 세포를 분리하는 경우에 사용하는 방법은 다음과 같다• 1) 적당한 크기의 종양조직을 유리 페트리 접시나 맥시모우 슬 라이드에 옮기고 그 속에서 예리한 안과 수술용 가위나 수술 칼 2 개를 써서 l mm3 정도 크기로 찰라낸다. 2) 조그맣게 자른 조직편을 50ml 원심듀브나 100ml 배양액 병에 옮기고 전자에는 20ml 후자에는 30ml 의 염류용액이나 PBS 를 추가한다. 조직편을 옮기는 것은 10ml 피펫을 사용 한다. 3) 원심튜브를 37 도 온도를 유지하고 있는 워터바스 wa t er ba t h 에 옮겨서 5 분 정도 보온한다. 4) 조직 위에 있는 상청액을 버리고 10-20ml 의 0.25 % 트립 신액을 추가하여 37 도 워터바스에서 25-30 분간 보온한다. 적 당한 속도로 진동할 수 있는 셰이킹 워터바스가 있으면 이상 적이다. 5) 원심튜브롤 꺼내어 상청액을 버린다. 조직편이 끈끈하게 되
어서 액을 버릴 수가 없는 경우에는 0.2 % DNA 효소 (DNase, 시그마) 를 4_5 방울 첨 가하고 2-3 분 기 다리 면 된 다. 이러한 젤이 나오는 것은 파괴된 세포에서 DNA 가 분리 되어서 서로 엉키기 때문이다. 6) 혈청을 포함한 배양액 10ml 를 추가하고 10ml 피펫으로 20 번 가량 위 아래로 피페팅을 한다. 이 기계적인 조작으로 세 포가 조직에서 분리된다. 이때에 거품이 나올 정도로 강하게 피페팅을 하면 세포 표면이 상해서 사멸함으로 조심스럽게 피페팅을 한다. 7) 2-3 분 기다려서 세포덩어리가 가라앉는 것을 기다리고 상청 액을 다른 50ml 원심튜브에 옮긴다. 남아 있는 세포덩어리 에 10ml 의 배양액을 새로 가하여 피페팅을 계속하고 세포 분리를 한다. 8) 분리된 세포를 포함한 부유액을 모아서 1000 회전 8 분의 저 속 원심을 한다. 상청액을 버리고 가라앉은 세포에 신선한 배양액을 가하고 저속 원심을 한다. 이 조작을 한번 더 되풀 이하고 마지막으로 세포부유액을 폴리라이신 코팅한 커버글 라스나 페트리 접시에 플레이팅하여 배양을 시작한다. 9) 일주일에서 열홀이 지나서 상피상 세포가 증식하게 되면 일 주일에 두 번 배양액을 교환한다. 열흘이 지나면 섬유아세포 가 우월해지는데, 뇌종양 세포는 모자이크처럼 자라나는데 비해서 섬유아세포는 방추형태로 넓게 퍼져 나오므로 그 형 태 구별이 간단하다. 현미경을 보면서 섬유아세포를 러버풀 리스맨으로 긁어내어서 뇌종양 세포 콜로니를 남긴다. 10) 콜로니를 분리하여 뇌종양세포의 순수 배양이 가능하다. 도립위상차 현미경 아래서 콜로니 colon y를 선택하고 페트 리 접시 밀을 마카펜으로 표시를 한다. 배양액을 버리고 콜
로니가 있는 부분을 가운데로 하고 실리콘 그리스를 바른 유리 실린더 (직경 0.8cm, 높이 0.8cm) 를 세운다. 0.05 % 트립신과 0.02 % EDTA 가 들어있는 CMF-PBS 를 0.3 m l 실린더 안에 주입하고 5 - 10 분 뒤에 분리되는 세포를 모아서 3.5cm 직경 페트리 접시에서 배양한댜 배양액은 앞서 말 한 10 % 우태아혈청을 포함한 달베코 MEM 이다(배양을 계 속 하면 뇌종양세포가 증식하여 순수배양이 가능하게 된다) (그 립 29- 3) . 유리 실린더는 클로닝 실린더 clonin g cyl ind er 라 하며 미리 오토클레이브 멸균을 해둔다.
 그림 29- 3 콜로니 형성을 위한 클로닝실린더법
그림 29- 3 콜로니 형성을 위한 클로닝실린더법
제 30 장 잡종세포 제작법 생체내에서나 배양 조건하에서나 세포는 서로 경계를 가지고 있어서 세포가 서로 접촉을 가졌을 경우 서로 융합하는 수가 없 다. 그러나 세포에 특수한 처리를 하여 2 개 이상의 세포가 경계 면이 없는 하나의 세포막 안에 둘러싸인 융합체를 만들어 내는 경우 이룰 세포융합 cell fus io n 이라 한다. 형성된 융합세포가 더 나아가서 핵의 융합을 가져올 수가 있는데, 이 경우 유전적 조성 이 다른 핵이 융합하여 두 가지 다른 세포의 유전자의 발현이 있 울 수 있다. 이를 잡종세포 hy br i d cell 라 한다 .1-3) 이 러 한 세포 융합을 일으킴 에는 폴리 에 틸 렌글리콜 po lye th y le ne g l y col 을 써서 화학적으로 융합시키는 방법, 바이러스를 써서 생 물학적으로 융합시 키는 방법 , 그리고 강력 한 전장 electr o p h oresis 를 써서 물리적으로 융합시키는 방법이 있다. 후자는 전장융합법 elect ro p o rati on 이 라 하며 이 를 위 한 기 구 electr o p o rato r 가 시 판되 고있다. 세포 융합을 시킨 뒤에 잡종세포를 선택적으로 찾아내는 것을
잡종형성법 hy b rid i z a ti on 이라 하며, 이룰 사용하여 체세포유전학 somati c cell ge neti cs 이 최근 크게 전보한 바 있다. 이 선택법에 서 흔히 쓰이는 것에 히폭산틴 구아닌 포스포라이보즈트란스페라 제 (HGPRT) 결손 세 포주와 티 미 딘키 나제 (TK) 결손 세 포주를 쓰 는 방법이다. 그 배양액 속에 뉴클레오티드의 신생 합성을 방지 하는 아미노프테 린 ami no p ter in e , 그리고 뉴클레오티 드의 전구체 인 티 미 딘 thy m i di n e 과 히 폭산틴 hyp o xanth i n e 을 포함한 HAT 액 울 선택배양액으로 사용한다. 이 조건하에서는 잡종세포만이 살 아 남게 된다. 1970 년 초에 들어와서 마우스 신경아세포종(뉴로불라스토마 neuroblasto m a) 주세 포와 신경 세 포, 아스트로사이 트 등의 각종 신경계 세포 사이에서 잡종세포를 수립하고 이들을 사용하여 신 경 기능의 조절 기능 연구를 하는 것이 유행하였다. 미국의 니렌 버 그 Ni re nberg 와 그 공동연구자인 미 나 Mi na , 아마노 &nano, 넬슨 Nelson 등이 이들 잡종세포를 사용하여 신경섬유의 성장, 아세틸콜린 감수성, 활동전위의 발생, 신경세포 특이 효소의 발 현 등 신경과학의 기초연구를 하였다 .4-7) 마우스 뉴로불라스토마와 글리오블라스토마 사이에 생긴 잡종 세포에서는 부모세포에서 볼 수 없는 콜린아세틸트란스페라제 choli ne acety ltra nsfe r ase 활성 과 시 냅 스 syn a ps e 형 성 등의 뉴론 에 특이한 형질이 나와 있어서 전기생리학적 연구에서 많이 쓰이 고 있다 .4,6) 마우스 뉴로블라스토마와 마우스 신경세포 사이의 잡종세포에서는 역시 콜린아세틸트란스페라제 활성이 특히 높아 서 이들 잡종세포주를 이용하여 신경전달물질의 유전자 발현에 대한 조절 기능 연구를 여러 연구실에서 계속하고 있다 .7,8) 또한 마우스 뉴로불라스토마와 사람 태생 척수후근신경절 세포 사이의 이종간 잡종세포에서 뉴로필라멘트, A2B5 항원 등 뉴론 특유의
형질 발현이 보고되어 있다 .9) 위와 같이 증식성이 낮거나 증식 성이 정지된 신경세포나 올리고덴드로사이트와 뉴로블라스토마나 글리오불라스토마 사이에서 생긴 잡종세포는 신경세포나 올리고 덴드로사이트의 형질을 유지하면서 증식을 계속할 수 있기 때문 에 신경생물학 연구에 수많은 가능성을 가져다 줄 수가 있다고 하겠다. 30.1 폴리에틸렌글리콜 세포융합법 〈준비〉 1) 세포 (1) 마우스 뉴로블라스토마 주세포 N18TG2 HGPRT 효소 결손주로서 미국 배양세포은행 (ATCC) 에 서 구할 수 있다. (2) 태생 마우스 척수 후근신경절 분리세포 배양 2) 시험관, 원심튜브, 페트리 접시, 멀티웰 3) 스톱워치 4) 10 % 우태아혈청 /90 % 이글 MEM 5) HAT 100 배액 (히폭산틴 5X10-3 M, 아미노프테린 5X10-4M, 티미딘 1x10-3M) 6) 50 % PEG( 분자량 1000 의 폴리에틸렌글리콜 10 g을 50ml 병에 넣어서 오토클레이브 멸균하고, 식은 다음에 합성 배양액 20ml 를 가해서 혼합한다.) 〈방법〉 1) 두 세포군을 혼합하여 1200r p m 으로 5 분간 원심분리한다.
2) 상청액을 버리고 페렛에 PEG 액 0.5ml 를 넣어서 세포를 부유시킨다. 3) PEG 를 가한 1 분 뒤에 이글 MEM 10 ml 롤 천천히 첨가하 고 1200r p m 으로 5 분간 원심분리한다. 4) 이글 MEM 으로 세 번 세척한다(상청액을 버리고 페렛에 MEM 을 넣어서 피페팅을 한다). 이 단계에서 PEG 이 제거된 다. 5) 세포 수를 계산하고 우태아혈청 첨가 배양액에 희석한 다음 페트리 접시나 멀티웰에 플레이팅을 한다. 6) 하루 뒤에 HAT 배양액 (우태아혈청 배양액에 HAT 100 배액
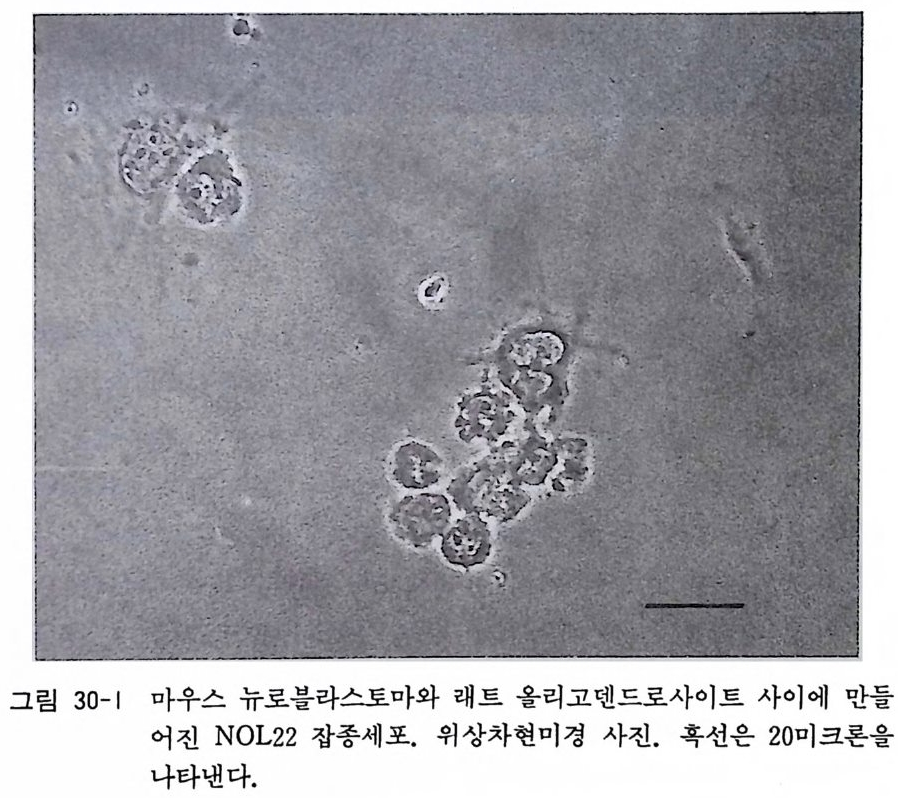 그림 30-1 마우스 뉴로불라스토마와 래트 울리고덴드로사이트 사이에 만들
그림 30-1 마우스 뉴로불라스토마와 래트 울리고덴드로사이트 사이에 만들
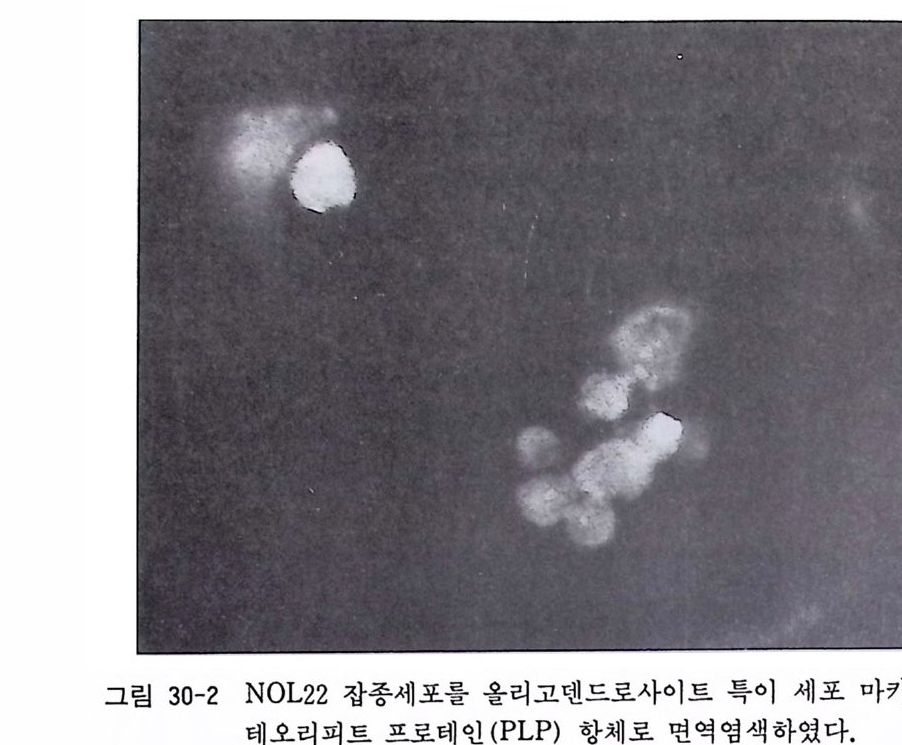 그림 30-2 NOL22 잡종세포 를 을리고덴드로사이트 특이 세포 마카인 프로
그림 30-2 NOL22 잡종세포 를 을리고덴드로사이트 특이 세포 마카인 프로
을 1 %로 첨가한다)으로 액교환울 한다. 7) 1 주일에서 2 주일 사이에 콜로니가 발생하면 이를 분리 배양 한다. 8) 모노레이어로 배양되고 있는 세포를 사용하는 경우에는 두 종류 세포를 6cm 직경 페트리 접시에 고밀도로 가득하게 배양하고 다음날 배양액을 버린 뒤 PEG 액을 첨가한다. 여 기서 접시를 흔들어 액이 혼합되도록 한다. 인큐베이터에 접 시를 정치하고 2 분에서 5 분 뒤에 PEG 액을 제거한다. 이글 MEM 으로 세 번 세척을 하고 마지막에 10% 우태아혈청을 포함한 배 양액을 가한다.
〈 해설 〉 저자의 연구실에서 최근 만들어 낸 마우스 뉴로블라스토마 N18TG2 와 래트 올리고덴드로사이트 간의 잡종세포 NOL14 와 NOL22 에서는 올리고덴드로사이트 특이 단백 질인 MBP (my el i n basic pro te i n ) 와 PLP (pr ote o lip id pro te i n ) 그리 고 신 경 세 포의 특 이 마카인 A2B5 항원을 가지고 있는데 이들은 뉴로블라스토마 에서 볼 수가 없는 형질이다 .10) (그림 30-1, 30-2) 30.2 전기적 세포융합법 최근에는 각 바이오텍 기구회사가 판매하고 있는 전자세포 융 합장치 elec t ro p ora t or 를 사용하여 잡종세포를 만들 수가 있게 되 었다. 직류 전기펄스로 이루어지는 세포 융합은 PEG 법에서 보 는 세포 독성이 없기 때문에 높은 확률로 잡종세포를 만들 수 있 다. 다만 전자세포 융합장치가 고가인 것이 단점이라 하겠다. 〈 준비 〉 30 .1 의 PEG 법 과 같다. 〈 방법〉 1) 직경 3.5cm 의 페트리 접시(미리 폴리라이신 코팅해 둔다) 중앙에 1Xl06 세포 농도가 되는 태생 마우스 척수후근신경 절 신경세포(슈완세포가 일부 있음)를 플레이팅한다. 2) 24 시간 뒤에 마우스 후근신경절세포 위에 1Xl06 세포농도 가 되는 마우스 N18TG2 세포를 플레이팅한다. 3) 배양액을 제거하고 2ml 의 전기융합용액 (lOmM 인산완충액
에 280 mM 마니 톨 manit ol, 1 mM Mg C l2, O. 1 mM CaCl2, 10 mM HEPES 를 첨가)을 가한다. 4) 0.5cm 간격으로 2 개의 스테인리스 전극을 위치시키고 0.6-1. 2 kV/cm, 50-100 µsec 의 직류 펄스를 1 초 간격을 두 고 5 번 가한다. 5) 전기융합 용액을 제거하고 우태아혈청이 들어있는 배양액을 7} 한다. 6) 24 시간 뒤에 HAT 가 들어있는 선택배양액과 교환한다. 7) 일주일에 2 번 배양액 교환을 하고 잡종세포 콜로니가 나오 는 것을 기다린다.
제 31 장 유전자 이입에 의한 신경계 세포주의 수립 최근 암유전자의 검색과 유전자의 발현 조절을 연구하는 방법 으로서 유전자 이입 gen e tra nsfe r 이 성행하고 있다. 이 기술은 각종 세포 DNA 를 배양세포에 이입 t rans f ec ti on 하고서 유전자 활 성을 나타내게 하는 기술이다. 저자의 연구실에서는 초대신경조 직 배양계에서 이 기술을 이용하여 슈완세포의 주세포라인 MSl 을 수립하였다 .I) 이러한 유전자 이입에 의해서 정상 신경계세포 의 형질을 유지한 채로 무한중식성을 가전 주세포라인이 수립되 고 무수한 세포를 증식시켜서 각종 생화학적인 혹은 분자생물학 적인 실험에 사용할 수가 있다. 유전자 이입 기술에는 여러 방법이 있어서 인산칼슘침전법 2,3) 과 리포솜도입법 4) 의 두 방법이 사용되고 있다. 인산칼슘침전법 은 독성이 있어서 최근에는 리포솜도입법이 많이 사용되고 있다. 〈준1:1 1 〉 1) 6cm 페트리 접시에 아스트로사이트, 슈완세포 등 중추신
경계나 말초신경계의 신경교세포의 순수배양(제 20, 21, 22 장 참고)을 해둔다. 2) 페트리 집시, 원심튜브 3) 유전자 DNA( p SVneo3) 를 0.4 mg DNA/ml 로 조절해 둔 다. 4) 리 포펙 틴 용액 (Lip o fe c ti n, GIBCO-BRL 사) 0 . 8 mg /m l 5) 이글 MEM 합성배양액 6) 우태아혈청 7) 50% 포도당액 8) 2 mg / ml 겐타마이신액 〈방법〉 1) 6 cm 직 경 페트리 접 시 하나에 유전자 DNA 10-20 µg과 리포펙틴액 30-50 µg이 들어가게 DNA - 리포펙틴액을 만든 다. 5 개의 페트리 접시롤 실험에 사용한다면 DNA 0.4 m g / ml 와 리포펙틴 0.8m g /ml 를 각각 0.25ml 준비해 둔다(증류 수에 희석한다). 2) DNA 0.25ml 과 리포펙틴액 0.25ml 롤 서서히 혼합하고 실 온에서 15 분간 정치한다. 3) 기다리는 동안에 페트리 접시의 세포를 이글 MEM 으로 두 번 씻어낸다. 페트리 접시에 2.5ml 의 이글 MEM 을 첨가한 다. 4) 페트리 접시 하나에 DNA- 리포펙틴액 0.1ml 를 첨가하고 페트리 접시를 서서히 혼들어서 액이 균일하게 퍼지도록 한 다. 5) 페트리 집시를 37 도 인큐베이트에서 정치하고, 24 시간 뒤에 10% 우태아혈청을 포함한 배양액과 교환한다. 배양액 조성
 그림 31-1 유전자 이입으로 제작된 MSl 마우스 슈완세포주 라인. 정상슈
그림 31-1 유전자 이입으로 제작된 MSl 마우스 슈완세포주 라인. 정상슈
은 다음과 같다. 이글 MEM 88ml 우태아혈청 10ml 50% 포도당액 1 ml 2 mg /m l 겐타마이신액 1 ml 6) 3 일 뒤에 위의 배양액에 G418( 네오마이신, 시그마) 200 µg/ ml 를 첨가한 선택 배양액으로 액교환한다. 7) 일주일에 두 번 선택 배양액으로 배양액 교환을 한다. G418( 네오마이신)에 저항성이 있는 세포가 자라나서 콜로니 형성을 하기 시작하면 제한희석법에 의해서 세포클로닝을 한 다. 이 전과정이 일반으로 3-4 주일 걸린다. 〈해설〉 저 자의 연구실에서 사용한 pS Vneo3 풀라스미드 pla smi d 는 폴 리오마 바이러스의 하나인 SV40 의 T 항원 유전자와 네오마이신 저항 유전자를 가지고 있다. 유전자 이입의 결과는 사용하는 세 포의 상태에 따라서 크게 영향을 받는다. 나의 교실에서 얻은 마 우스 슈완세포주라인 MSI 의 경우에는 정상 슈완세포가 가전 형 질인 Po 단백질, 라미닌, s100 단백질 발현을 볼 수가 있으며 배가시간 double tim e 이 28 시간이다 .1) (그림 31-1)
제 32 장 기성 신경계조직 수립세포주의 배양 신경계의 발생과 분화를 연구함에 있어서 가장 곤란한 것은 여 러 가지 종류로 나누어전 세포들이 복잡하게 서로 작용하고 있어 서 실험 결과의 분석이 어렵다는 것이다. 같은 종류의 신경조칙 세포를 대량으로 얻을 수가 있었으면 하는 것이 신경과학 연구자 들의 오랜 꿈이었다. 이것을 해결하는 데에는 두 가지 방책이 있 는데 그 하나는 신경세포나 신경교세포를 순수분리하는 방법이고 다른 하나는 신경세포나 신경교세포의 주세포라인의 배양이다. 제 19 장에서 제 23 장까지 소개한 바 있는 신경세포, 슈완세포, 울리고덴드로사이트, 아스트로사이트, 뇌혈관 내피세포의 순수배 양은 전자를 대표하는 것이고 이들 순수배양한 신경세포나 신경 교세포를 사용하여 여러가지 홍미 있는 연구가 수행되어 왔다. 그러나 세포의 순수도가 90 % 정도로 머물러서 다른 종류의 세 포가 섞여있다는 점과 신경세포의 경우에는 전혀 증식하지 않아 서 대량의 세포를 구하기가 곤란하다는 점이 결접으로 남아 있게 되었다• 신경계세포의 주세포라인의 배양은 이러한 결점을 극복
하는 좋은 모델 시스템이 될 수가 있다. 신경계 세포의 주세포라인의 배양에는 뉴로블라스토마(신경아 세포종 neuroblasto m a) , 글리 오블라스토마 (신경 교아세 포종 gliob las- tom a) , 올리고덴드로사이토마(희돌기교세포종 olig o dendrocy tom a) , 슈와노마 Schwannoma, 부신갈색종 ph eochromocy tom a 등이 있으 며 이들 주세포들은 사람이나 실험동물의 뇌종양에서 분리된 세 포를 조직배양에 옮겨서 무한증식성을 갖게 한 것이다. 1- 6 ) 뇌종 양의 세포를 분리하고 배양에 옮기는 기술은 제 29 장 뇌종양의 초대배양법에서 소개한 바 있다. 1970 년대에 신경과학에서 일어난 새로운 경향의 하나는 又0 1 A0 1 - 신경조직의 분리세포배양과 신경계조칙 유래의 수립세포주 es t ab -
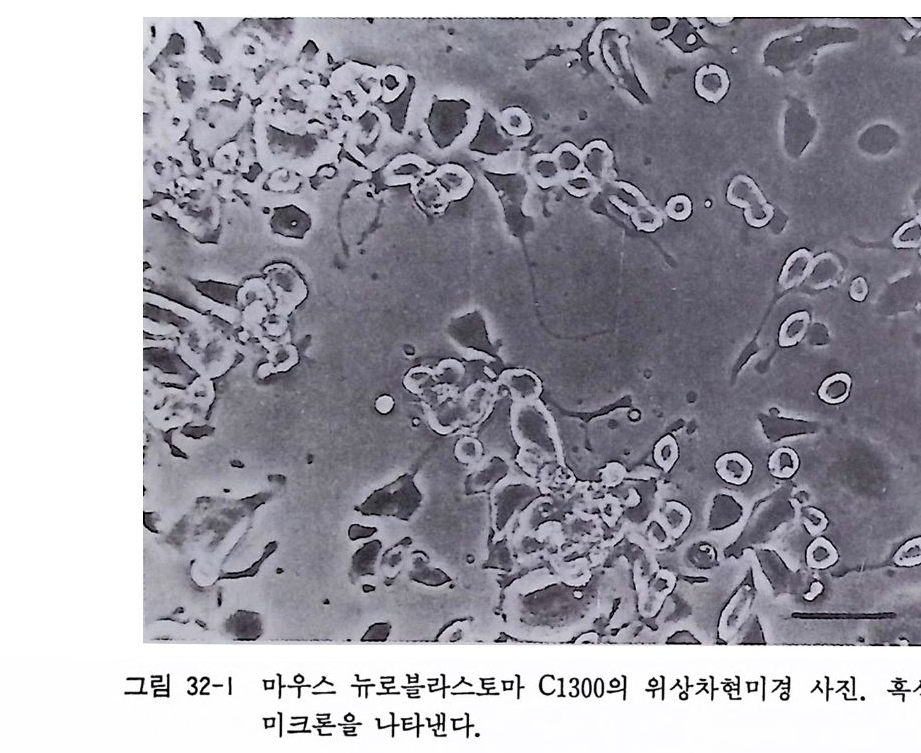 之`.
之`.
lish ed cell li ne 의 배양이 널리 연구모델로서 사용하는 것이었다. 마우스 뉴로블라스토마 C1300 은 AJ 계통 마우스 복강내에서 발 견된 교감신경절 원발의 종양으로서 지금까지 50 년 이상이나 동 물 이식을 계속하여 보존이 되어온 역사를 가지고 있으며 사토 Sato 와 슈버트 Schubert 의 연구실에서 1969 년에 조직배양계로 옮 기는데 성공하였다. 3,4) (그립 32-1). 그 뒤 N4, N18 등의 C1300 세포의 클론이 니렌버그 N i renber g 연구실에서 클론화되어 각 국 연구실에서 사용되고 있다. 특히 HGPRT 효소 결손주인 N18TG2 는 신경계 혹은 비신경계 각종 세포 사이에 잡종세포를 작성하는데 애용되는 세포주이다(제 30 장 참고). 래트 글리오마 C6 는 사토 Sa t o 의 연구실에서 발암물질인 메틸니트로소우레아를 임신 위스타 계통 래트에 두여한 뒤 그 태아의 뇌속에 생긴 뇌종
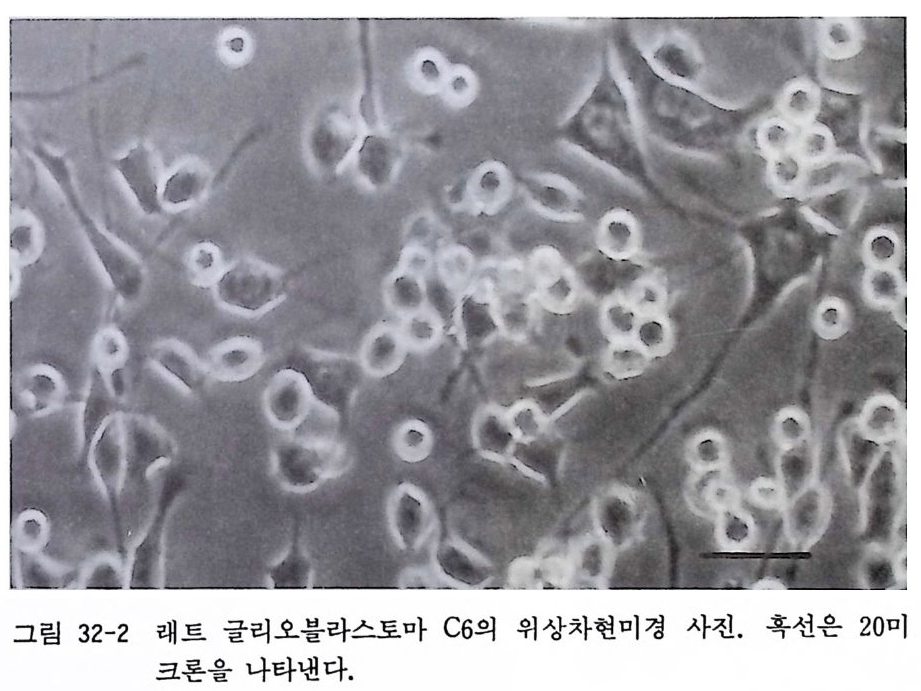 그림 32 군 래트 글리오블라스토마 C6 의 위상차현미경 사진. 혹선은 20 미
그림 32 군 래트 글리오블라스토마 C6 의 위상차현미경 사진. 혹선은 20 미
양을 조직배양계로 옮겨서 만든 것이다. C6 세포는 아스트로사 이트의 마카인 s100 단백질을 많이 가지고 있으며 그 일부의 세 포는 글리 아산성 단백 질 GFAP 을 표현한다 5) (그립 32-2) . 1970 년 이후로 여러 연구실에서 새로운 뇌종양 주세포가 사람과 실험동 물에서 분리되었다. 신경계 수립세포주 라인이 신경계조직 초대배양에 비하여서 여 러 가지 장점이 있는데 그것은 첫째, 유전적인 그리고 기능적인 균일성을 가지고 있다. 둘째, 발육능력이 엄청나게 크다. 셋째, 대량의 세포를 단시간에 수집할 수 있다. 넷째, 실험의 재현성이 높다. 다섯째, 연구자 사이에서 쉽게 주고받고 할 수가 있다. 또 한 각국에 존재하는 세포은행을 통하여 연구자가 원하는 수립세 포주 라인을 입수할 수가 있다. 여섯째, 분자생물학적인 유전자 조작이 가능하다. 일곱째, 신경계에서 특이한 항원과 신경영양인 자의 수집이 가능하다. 표 32-1 에서는 실험적으로 널리 사용되고 있는 신경계조직 세 포주와 유래와 특칭을 열거하였는데 이들은 미국 세포은행 (Americ a n Ty pe Cultu r e Collecti on , ATCC) 에 서 구할 수가 있 다. 풀라스크에 살아있는 세포 혹은 바이알에 동결되어 있는 세 포의 두 가지 방법으로 구입할 수가 있다. ATCC 의 주소, 전화번호, 팩시밀리 번호는 다음과 갇다. Americ a n Ty pe Cultu r e Collecti on 12301 Parklawn Dr ive Rockvil le, MD 29852 USA Tel (301) 881-2600 FAX (301) 816-4361
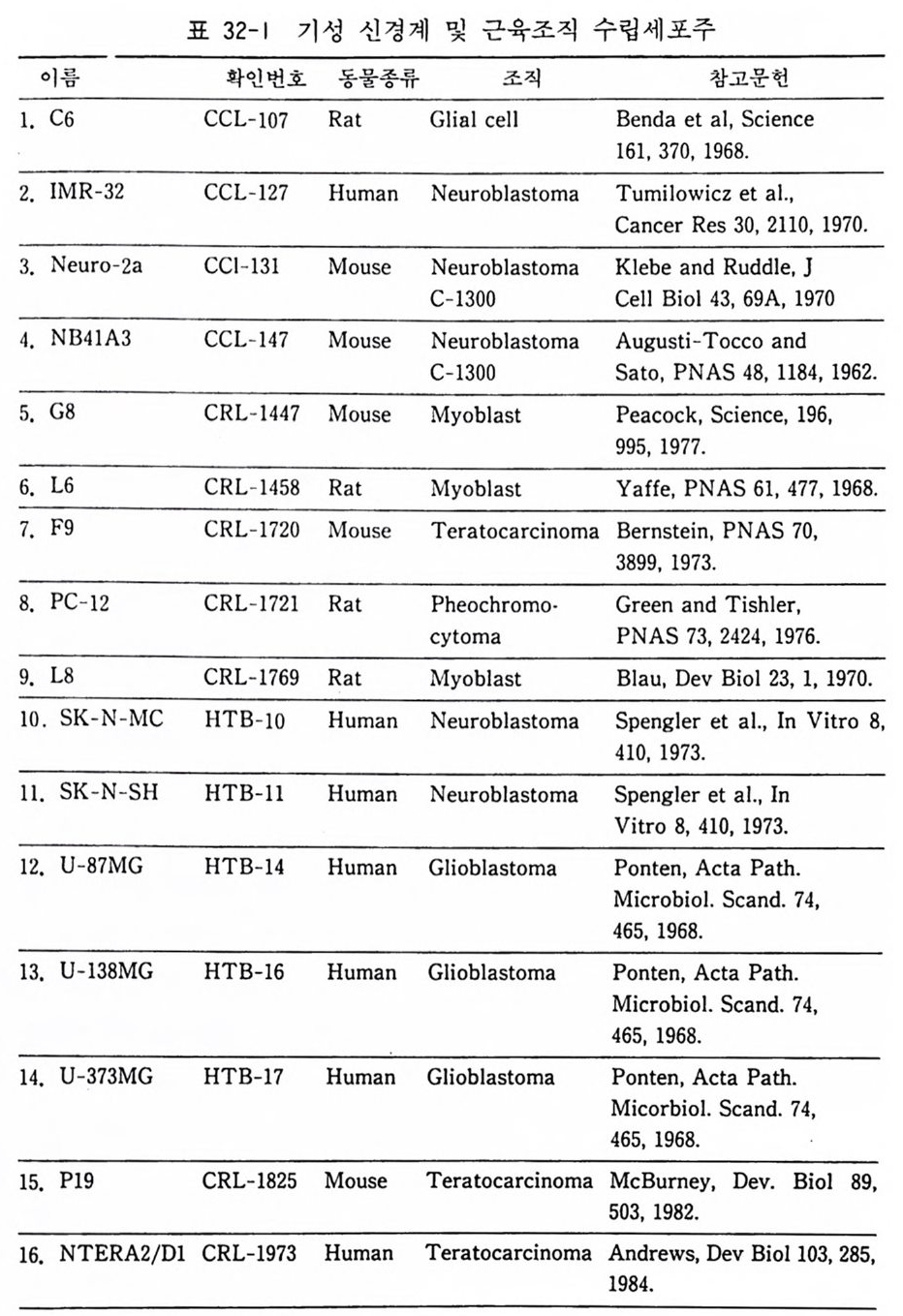 표 32-1 기성 신경계 및 근육조직 수립세포주
표 32-1 기성 신경계 및 근육조직 수립세포주
제 33 장 부신갈색종 세포배양 부신갈색종p henochromoc yt oma 은 부신수질 크로마핀 세포가 악성화하여 생기는 종양이다. PC12 세포는 1976 년 그린 Greene 과 티슐러 T i schler 가 래트 부신갈색종을 조직배양계로 옮겨서 배 양을 시작하였는데 20 년 동안에 세계 각국에서 세포생물학은 물 론 분자생물학의 분야에서 널리 사용되는 배양계가 되었다 .I) PC12 세포의 특칭은 신경성장인자 NGF 의 존재하에서는 신경섭 유가 성장하고 활동전위롤 내는 교감신경세포로 분화한다는 것이 다. 현재 사용되고 있는 PC12 세포주는 대부분이 200 대 넘어 계 대배양되어 온 것으로서 그 일부의 세포주는 여러 가지 형질의 변화가 있다고 지적되고 있다. PC12 세포를 분양받은 경우에는 빠른 시기에 동결하여 보존해 두는 것이 좋겠다. 〈준비〉 1) 페트리 접시, 풀라스크, 원심튜브, 피펫 2) 달베코 MEM 합성 배양액
3) 우태아혈청과 마혈청 4) 2 mg /m l 겐타마이신액 〈 방법 〉 1) 6 cm 페트리 접시나 T25 플라스크에 10 µg/m l 폴리라이신 액 2 ml 를 가하고 30 분간 정치한 뒤 아롤 제거하고 증류수로 한번 씻는다. 2) 폴리라이신 코팅한 6cm 페트리 접시나 T25 풀라스크에 5 X1 04 세포 /ml 의 농도로 PC12 세포를 넣고 배양을 시작한 다. 6cm 페트리 접시에는 3ml 그리고 T25 풀라스크에는 5 ml 의 세포배양액을 넣는다. 3) 배양액 교환은 3 일에 한 번씩 하며 1 주일이나 10 일마다 계 대 배 양 subcul t ure 을 한다. 4) 계대배양은 다음과 같이 행한다. 플라스크의 마개를 단단히 잠근 뒤, 플라스크 양쪽 측면을 손바닥으로 10 번 가량 강타 한다. 배양중인 세포가 시트같이 떨어져 나오는데 이것을 피 페팅을 10 번 가량함으로서 세포를 분리할 수 있다. 페트리 집시의 경우에는 배양액 을을 빨아올린 뒤 세포 시트 위에 강하 게 불어내리면 세포가 분리되어 나온다. 5) 세포 분리는 0.05 % 트립신과 0.02 % EDTA 를 포함한 CMF-PBS 에 10 분 처리한 뒤 분리할 수 있다. 세포를 모아 서 1000 rpm 8 분간 원심침전한다. 6) 페렛에 20ml 의 배양액을 가하고 4 개의 T25 풀라스크에 분 활한다. 7) PC12 세포의 신경세포 분화롤 보고자 할 때에는 배양액 속 에 10 µg/m l NGF( 시그마)를 200 대 1 로 회석하여 50 ng / ml 농도로 첨가한다.
배양액 조성은 다음과 같다. 달베코 MEM(DMEM) 89 ml 우태아혈청 5ml 마혈청 5ml 2 mg /m l 겐타마이신액 1ml 〈 해설 〉 신경계의 발생과 분화 그리고 신경세포의 생존 을 조절 하 는 물 질로서 신 경 영 양인 자 neurotr o p h ic fac to r 의 중요성이 최근 크게
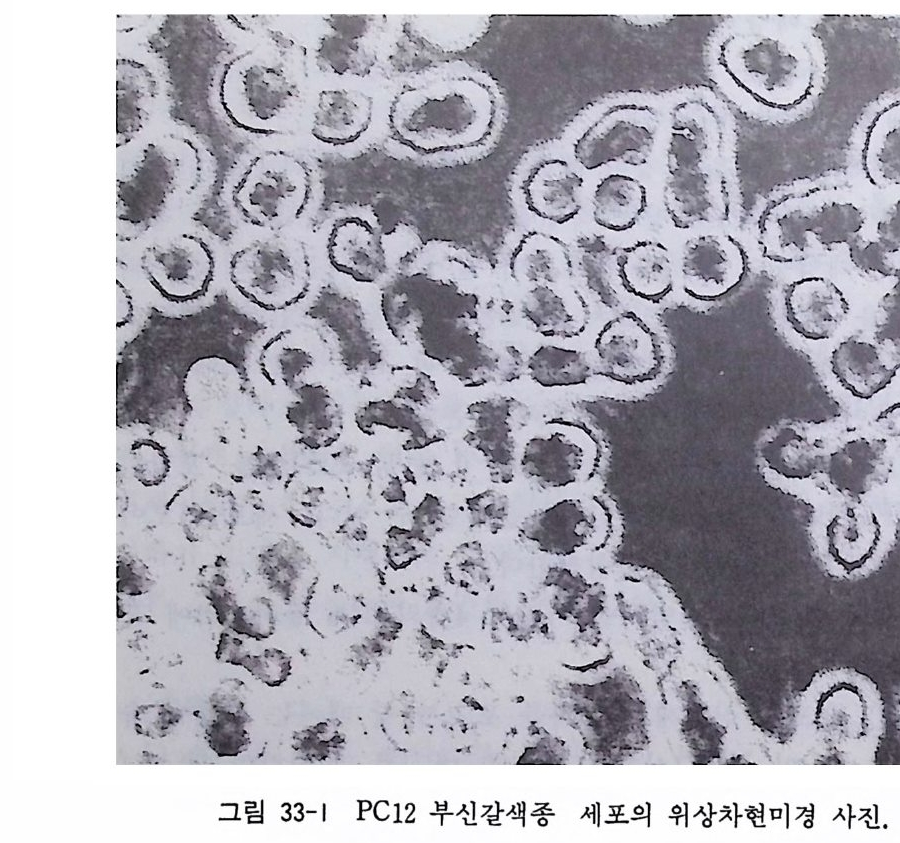 \L ..-B`. a · `f5
\L ..-B`. a · `f5
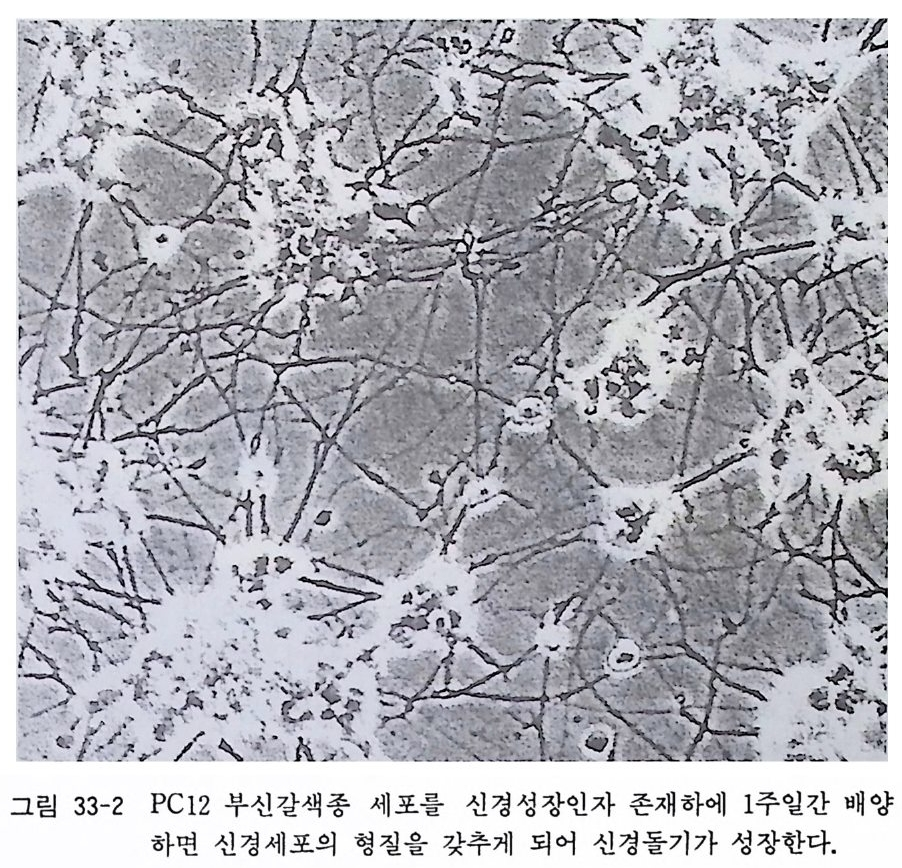 그림 33-2 PC12 부신갈색종 세포를 신경성장인자 존재하에 1 주일간 배양
그림 33-2 PC12 부신갈색종 세포를 신경성장인자 존재하에 1 주일간 배양
강조되고 있다. 이들 신경영양인자 중에서도 1950 년대에 처음느 로 발견된 신경성장인자 (NGF) 의 존재하에서 종양세포인 PC12 세 포가 교감절신경세포로 분화한다는 것이 알려진 이후에 PC12 세 포가 여러 가지 다론 신경영양인자 (NTF) 죽 영기성 섬유아세포 증식 인자 (bas ic fibr oblast grow th fac to r, bFGF) , 인터루킨 -6, 상 피 성 장인 자 (ep ide rmal grow th fac to r, EGF) , 뉴로트로핀 -3 (neur-otr o p in- 3, NT-3) 에 의해서도 그 분화가 유도된다고 보고되고 있 다. 2,3) 그립 33-1 과 33-2 에서는 PC 12 세포의 NGF 아래서 신경 세포로 분화되는 것을 보이고 있다.
제 34 장 기형종 세포배양 기형종(데라토카시노마t era t ocar ci noma) 은 사람이나 마우스 생 식기관에 발생하는 악성 종양이다. 이 종양의 근간세포는 배아성 암종세포 embryo nal carin o ma cell 라 불리는 미분화 세포이다. 이 EC 세포는 무한증식성과 종양형성능을 가전 악성 암세포인데 여 러 가지 종류 세포로 분화할 수 있는 다분화 능력을 가전 점에서 초기배에서 보는 미분화 세포와 극히 닮은 성질을 가전다. EC 세포는 대량 배양할 수가 있고 배양조건을 바꿈으로서 초기 배아 발생처럼 여러 가지 방향으로 분화시킬 수가 있다는 특칭이 있 다. EC 세포의 주화세포중에는 분화능이 저하되어서 보통의 배 양 조건 아래 에 서는 분화하지 않고, 레 티노산 reti no ic ac id 이 나 디 메 틸설폭시 미 드 (dim eth yls ulfo x im i de , DMSO) 같은 화학물질에 의해서 비교적 짧은 기간에 분화를 유도할 수가 있어서 신경세포 나 근육세포로 변환한다 .1-4)
〈 준비 〉 1) P19 세포주 : 미국 세포은행 Americ a n Ty pe Cultu re Collec- ti on 에 서 살 수가 있다. 2) 레티노산(시그마) : 10-3M 농도로 디메틸설폭시드에 녹이고 3 주일 안에 사용한다. 3) 디 부티 릴 사이 클릭 AMP (Dibu ty ryl cyc l i c AMP, 시 그마) : 10- 4 M 농도로 PBS 에 녹이고 여과멸균한 뒤 4 도에서 보존 4) 이소부티릴 메틸 크산틴 (Isobu tyry l meth y l xanth i n e , 시그 마) : 10- 2 M 농도로 PBS 에 녹이고 여과 멸균한 뒤 4 도에서 보존 5) 페트리 접시, 원심튜브, 피펫 〈 방법 〉 1) P19 는 0.1 % 겔라틴을 바론 풀라스틱 접시에서 배양하며 2 일에 한 번 배양액울 교환한다. 배양액은 10 % 우태아혈청 을 포함한 달베코 MEM 액이다. 2) 계대배양은 4 일에 한 번씩 트립신에 의해서 한다(그립 34- 1) . 계대배양은 배양액을 버린 뒤 0.05 % 트립신과 0 . 02 % EDTA 를 포함한 CMF-PBS 를 3 ml 가해서 37 도 인 큐베아터에서 보온한다. 트립신 -EDTA-PBS 액을 조심스럽 게 빨아서 버린 뒤, 신선한 배양액을 5ml 가하고 피페팅을 하여 세포를 분리한다. 6cm 직경 페트리 접시에 5Xl05 세 포의 농도로 분주한다. 3) P19 세포 5Xl05 세포 농도로 페트리 접시에 분주하고 10 % 우태아혈청을 포함한 달베코 MEM 배양액에 1X10-6 M 레티노산 , 10-6M 디부티릴사이클릭 AMP, 10- ◄ M IBM 중 한 가지를 첨가한다. 배양액은 2 일에 한 번씩 교환하는
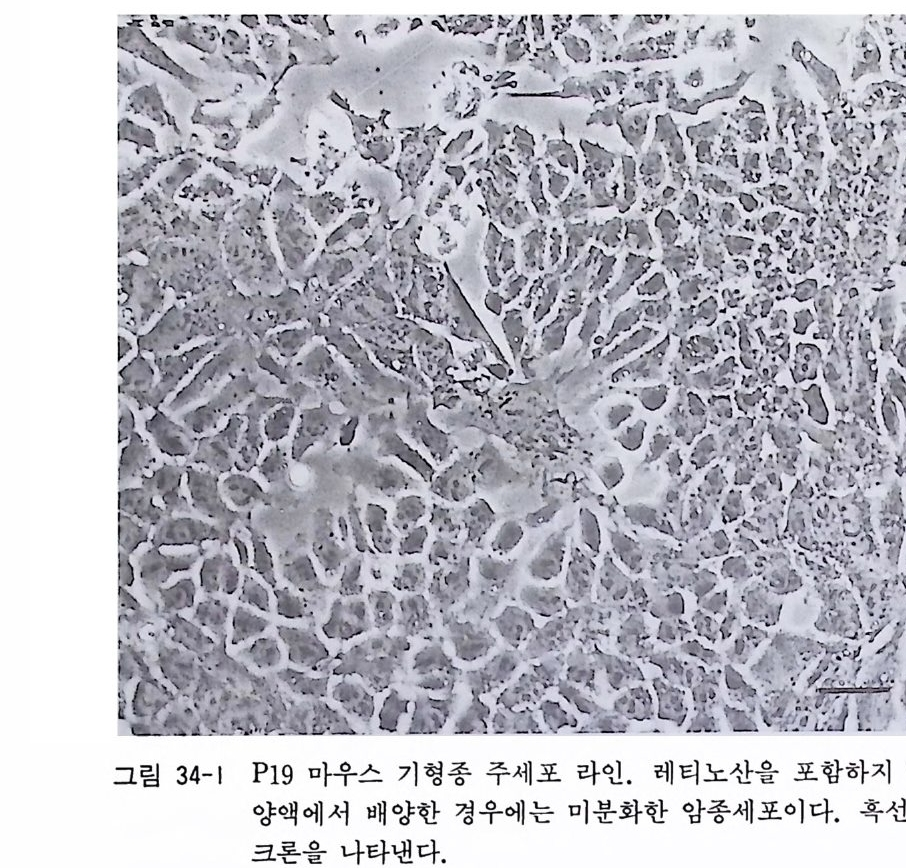 그림 34-1 P19 마우스 기형종 주세포 라인. 레티노산 을 포함하지 않은 배
그림 34-1 P19 마우스 기형종 주세포 라인. 레티노산 을 포함하지 않은 배
데, 1 주일 뒤에는 Pl9 세포가 모두 내배영 세포로 분화된 다. 레티노산에서 배양한 세포는 그 일부분이 신경세포로 분 화한다(그립 34-2). 디부티릴사이클릭 AMP 처리 후에는 근 육세포로 분화하며 IBM 처리 후에 피부 각질세포로 분화한 다. 4) 계대 배양할 때에 젤라틴을 바른 접시 대신에 박테리아용 접시에 세포를 분주하면 세포가 30-100 개 가량 들어있는 세 포 크럼프(덩어리)를 만든다. 이 세포 크럼프는 마우스 정상
 그림 34-2 Pl9 마우 스 기형종 주 세포 를 레티노산 존재하에 배양하면 1 주
그림 34-2 Pl9 마우 스 기형종 주 세포 를 레티노산 존재하에 배양하면 1 주
배 발생에 있어서 수정 후 4-5 일에 해당되는 배아가 된다. 〈 해설 〉 그림 34-1 에서는 레티노산을 첨가하기 전에 Pl9 기형종 세포가 분화하기 전의 상태를 보이고 있으며 그림 34-2 에서는 레티노산 첨가 후 10 일뒤에 기형종 세포가 신경세포로 분화하여 신경섬유 가 방사형으로 발육하고 있음을 볼 수 있다. 신경세포에 대한 세 포타입 특이 마카, 예컨대 MAP2, 뉴로필라멘트에 대한 항체를
사용하여 면역염색으로 신경세포로 분화한 P19 기형종 세포를 특 정할 수가 있다.
제 35 장 신경계조직에서의 세포타입 특이 마카 사람의 뇌 전체의 신경세포 수는 100 억에서 200 억 개 사이라 하며 그 가운데서 대형의 신경세포는 10 만 아상의 시냅스를 만들 고 있어서, 신경세포 간의 결합에 의해서 구성되는 신경계의 네 트워크는 우리들의 상상을 초월하여 극히 복잡한 양상을 보인다. 이러한 신경계의 발생은 신경관에서의 신경상피세포의 증식, 신 경아세포의 이동, 신경세포돌기의 신장, 시냅스의 형성, 글리아 세포의 증식과 이동, 수초(미엘린) 형성, 그리고 혈관설 1 관문 형성 등의 각 프로세스가 질서정연하게 차례로 이루어짐으로서 완성이 된다. 이러한 신경계의 초기 발생과 성장에 관한 기본적 인 문제를 연구하는 방법으로서 신경조직배양법은 가장 유력한 실험기술로서 사용이 된다. 일반으로 신경조직배양의 재료로 쓰 이는 조직은 태생 혹은 신생 동물에서 유래함으로 신경조직배양 법을 사용하여 행하는 실험은 필연적으로 신경계 발생의 각 현상 과 그 해명을 대상으로 한다. I)
35.1 세포타입 특이 마카 뇌신경계는 근육, 간, 심장 등의 다른 조직에 비해서 다양한 세포타입으로 구성되며 이러한 갖가지 세포타입이 어떠한 구조와 기능을 가지는가를 해명함에 있어서 가장 문제가 되는 것이 어떻 게 이들 세포타입을 구별하는가 하는 것이다. 1970 년대 후반까지 는 고전적인 은염색, 닛슬염색, 카테콜아민 형광염색 등에 의해 서 신경세포를 식별하는 것만이 유일한 어프로치였다. 1970 년에 들어서서 면역학의 분야에서 세포 표면 항원에 대한 특이항체를 사용하여 각종 면역세포를 염색 구별하는 방법이 도입된 이후로 이 면역세포 화학영색법이 그대로 신경조직배양에서도 적용이 되 었다. 2-4) 아스트로사이트에 특이한 글리아섬유성 산성단백질 (GFAP), 울리고덴드로사이트의 표면에 특이한 갈락토세레브로시드 ga lacto c erebrosid e , 그리고 신경세포 표면에 특이한 A2B5 와 파 상풍독 tet a n us tox in 에 대 한 특이 항체 를 사용해 서 각각 아스트 로사이트, 올리고덴드로사이트 그리고 신경세포의 확정이 가능하 게 되었다 .5-7) 1975 년에 단클론 항체 제작법이 소개되어서 각종 의 특이 항체가 제작되었고 이들이 면역염색법에서 응용되고 있 다. 신경조직에는 다른 조직에서 볼 수 없는 특칭적인 단백질이 있어 서 이 에 는 세 포골격 단백 cy tos keleta l pro te i n s , 수용체 단백 recep tor pro te i n s , 이 온채 널 ion channel pro te i n s , 미 엘 린 단백 my el in pro te i n s 등이 있어서 각각 신경조직에 있어서 특이한 기 능을 가지고 있다. 이밖에도 신경전달물질 관련효소 neuro t rans mi tter assoc iat e d enzym es, 신 경 세 포 접 착 인 자 cell adhesio n molecules 등 신경조직에만 특이하게 존재하는 단백질이 있다. 위에서 말한 바와 같이 신경조직에 단백을 항원으로서 제작된 폴
리클로날 혹은 단 클로날 항체는 세포타입 특이 마카로서 쓰인 다. 이들 세포타입 특이 마카는 표 35-1 에서 보여준다. 표 35-1 신경조직배양에 있어서의 세포타입 득이 마카 1. 신경세포 Neurofi lam ent MAP2 Neuron spe ci fic enolase A2B5 Teta n us tox in Neurotr a nsmi tter s (acety lc holi ne , dop a mi ne , seroto n in , GABA, glu ta m i c aci d) Neurop e pt ide s (substa n ce P, somato s ta t i n, VIP) Neurotr a nsmi tter assoc iat e d enzy m es (choli ne acety lt ra nsfe r ase, acety lc holin e ste r ase, glu ta m i c ac id decarboxy la se, tyr osin e hy d roxy la se) 2. 아스트로사이트 Gli al fibr ill ar y acid i c pro te i n (GFAP) Gluta m i ne syn the ta s e s100 pro te i n i 올리고덴드로사이트 Galacto c erebrosid e Sulfa tide My el i n basic pro te i n (MBP) My e li n pro te o lip id pr ote i n (PLP) My el i n assoc iat e d gly c o p ro te i n (MAG) Cy cl ic nucleoti de ph osph ohy d rolase (CNP) Carbonic anhy d rase C 4. 슈완세포 Lami ni n NGF recep tor (low aff ini t y) Galacto c erebrosid e s100 pro te i n
5. 섬유아세포 Fib r onecti n Thy -1 6. 미크로글리아 MAC-I Labelled late x beads Ric inu s communis lecti n Non-spe c i fic este r ase 7. 뇌혈관 내피세포 Facto r \께 35.2 세포골격단백 신경조칙에는 특칭적인 세포골격단백의 존재가 알려져 있는데 이들은 세포 미 세구조상의 크기 에 의 해 서 미 소관 (mi cro tu b ules, 직경 24run), 중간직경 필라멘트(i n t ermed i a t ed filam ents , 직경 10 run), 미소 필라멘트 (m ic ro fi lamen t s, 직경 5nm) 의 세 종류로 구 별된다. 이중 미소관은 신경세포의 세포체와 돌기에 존재하고 있 어 축색 수송에서 중요한 역할을 가지고 있다. 미소관은 알파와 베타의 두 종류의 튜브린 서브유니트로서 구성되고 이밖에도 MAPl (mi cro tu b ule-assoc iat e d pro te i n 1) , MAP2, 타우 단백 tau pro te i n 등의 결합단백이 튜브린과 결합하여 미소관을 구성한다 .8) 이 중염 색 법 double sta i n i n g 에 의 해 서 연구된 바에 의 하면 배 양 신경세포의 수상돌기와 축색돌기가 각각 MAP2 와 뉴로필라멘트 neurofi lam ents , 아스트로사이트에서는 글리 알 필라멘트gli al filam ents 가 특칭 적 이 다. 7,9,10) 이 밖에 도 데 스민 desmi n, 비 멘틴 vim enti n 그리고 케 라틴 kerati n 이 신경조직 에 존재 한다. 데스민
은 배양 아스트로사이트에서 관찰된다는 보고가 있으나 이에 반 대하는 논문이 있어서 앞으로 그 검토가 필요하다. 비멘틴은 아 스트로사이트의 글리알 필라멘트에서 GFAP 와 공존해 있으나 성 숙함에 따라 비멘틴이 소멸한다. 또한 미분화 산경세포에서 비멘 틴과 뉴로필라멘트의 공존이 보고되고 있으나 신경세포가 성숙함 에 따라 비멘틴이 소멸한다. 한편 울리고덴드로사이트에서는 중 간 직경 필라멘트가 존재하지 아니한다. 신경 상피세포에서 분화 해 오는 맥 락총 상피 세 포 choroid ple xus ep ithe li al cells 에 서 는 케 라틴의 존재가 특징적이다. 뉴로필라멘트와 GFAP 는 신경조직배 양에 있어서 신경세포와 아스트로사이트롤 확정함에 있어서 가장 전형적인 세포타입 특이 마카이다. 35.3 미엘린 단백 중추신경계와 말초신경계의 미엘린은 전자현마경 하에서 그 구 조가 극히 유사하나 생화학적으로는 단백질 조성상에 많은 차이 가 있다. 중추신경 미엘린의 중요한 것으로는 미엘린 영기성 단 백 my e lin basic pro te i n , MBP , 프로데 어 리 피 드 단백 pro te o li pid pro te i n , PLP , CNP 2, 3-cyc l ic nucleoti de -3-ph osph ohy d rolase 가 있으며 이 밖에 도 미 엘 린 관련 단백 my el in - assoc iat e d gly c o - pro te i n , MAG) 이 있다. 11,12) 말초신경 미 엘린에서는 중추신경 미 엘린의 MBP 에 대응하는 P2 단백이 있고 그밖에도 PO 단백과 MAG 가 그 구조 조성에 참가한다 .12) 중추신경계에서는 올리고덴 드로사이트가, 말초신경계에서는 슈완세포가 미엘린 형성에서 중 요한 역할을 하고 있어, 이들 세포는 상기한 미엘린 단백을 합성 하고 있다. 이러한 이유로 미엘린 단백인 MBP, PLP, CNP 가
올리고덴드로사이트의 특이 마카가 되고 P2 와 PO 단백이 슈완세 포의 특이 마카가 된다. 미엘린의 가장 특징적안 당지질인 갈락 토세레브로시드g alac t ocerebros i de 는 올리고덴드로사이트의 표면 항원으로서 면역염색의 감수성이 높고 배양계에 있어서 울리고덴 드로사이트 확정에 가장 널리 사용된다. 신경계에서는 이밖에도 강글리오시드 ga ng lios id e 등의 각종 당지 질의 존재 가 알려 져 있 다. 13) 그 가운데서 홍미가 있는 것은 GT 강글리오시드에 특이한 A2B5 항체 6) 인데 이 항체는 각종 신경세포를 특이적으로 염색함 과 동시에 울리고덴드로사이트 -2 형 아스트로사이트 전구세포 0 -2A Pro g en it orcells 를 인식한다고 보고되고 있다. 13) 35.4 신경전달물질과 관련 효소 신경계에 있어서의 각종 신경전달물질과 신경펩티드 neuro p e pti des 의 뇌내 분포와 동태의 연구가 최근 성행하고 있다. 이것 은 특이 항체를 쉽게 얻을 수 있다는데 원인이 있다. 지금까지 소마토스타틴, 서브스탠스 P, 서브스탠스 Y, VIP 등의 신경펩 티드와 도파민, 세로토닌, 아세틸콜린, GABA 등의 신경전달물 질에 특이한 항체가 상업적으로 판매되고 있다. 이밖에도 신경전 달물질 관련 효소에 특이한 항체가 면역세포화학에서 흔히 쓰이 고 있어서, 그 가운데서 아세틸콜린 합성효소 cho li ne acety lt ra nsfe r ase, 티 로신 수산화효소 tyros in e hy d roxy la se, 글루 타민산 탈탄산효소g lu t am ic aci d decarboxy la se 등이 각종 신경세 포 확정에 사용된다 .14) 아세틸콜린 합성효소는 콜린성 신경세포 의 마카로서 가장 · 특이적이나 그 항체가 대단히 고가이고 다른 동물에서는 교차반웅이 없다는 등의 문제가 있어서 이러한 경우
에는 아세틸콜린 에스데라제 acety lc holi ne ste r ase 효소화학법에 의 해서 콜린성 신경세포의 확정을 할 수가 있다.
제 36 장 면역세포화학 신경조직 가운데서 여러 가지 다른 종류의 세포가 어떠한 기능 을 가지고 있으며 서로 어떠한 상관관계를 갖는가를 검색함에 있 어서 제일 먼저 일어나는 의문은 어떻게 서로 다른 종류의 세포 를 인식하고 판정하는가 하는 것이다. 이 문제 때문에 면역학과 그에 관련된 분야는 오랫동안 그 연구의 전전이 지체된 바 있다. 1970 년대에 들어서서 면역계 세포인 임파구의 B 세포와 T 세포 분별을 염색법으로 확실하게 되었는데 이것은 각 세포 표면에 특 이한 항원에 대해서 특별하게 인식하는 항체를 사용하여 이루어 전 것이다. 이러한 면역 염색법으로 시작하여 지난 20 년 동안에 면역학은 급속한 전보를 가진 바 있으며 또한 1975 년 쾰러 Kohler 와 밀스틴 Mi lste i n 에 의 해 서 개 발된 단클론 항체 제 작법 의 보급으로 면 역 세 포화학 im munocy toc he-mi st r y 의 기 술은 크게 발 전한 바 있다 .1) 최근의 면역세포화학의 발달에 의해서 콜린성 신경세포는 아세 틸콜린 합성효소, 카데콜아민 신경세포에서는 티로신 수산화효
소, 그리고 GABA 신경세포에서는 글루타민산탈탄산 효소 등의 신경전달물질 관련효소를 면역조직화학적으로 증명할 수가 있다. 이에 더하여 아세틸콜린, 도파민, 에피네프린, 세로토닌, GABA, 그밖의 여러 가지 신경전달물질에 대한 특이 항체와 단 클론 항체가 만들어지고 이들 특이 항체의 사용으로 면역조직화 학은 신경과학 그리고 신경조직배양에서 중요한 부분을 차지하게 되었다. 면역염색법에는 두 가지 기본적 기술이 있는데 항체 an ti bod y를 형 광색 소 혹은 페 록시 다제 pe roxid a se 나 알칼리 포스파타제 alka- line ph osph ata s e 같은 효소와 결합시켜서 직접 조직에 있는 항원 an tig en 에 작용시켜서 관찰을 하는 직접법(그림 36 - 1) 과 항원에
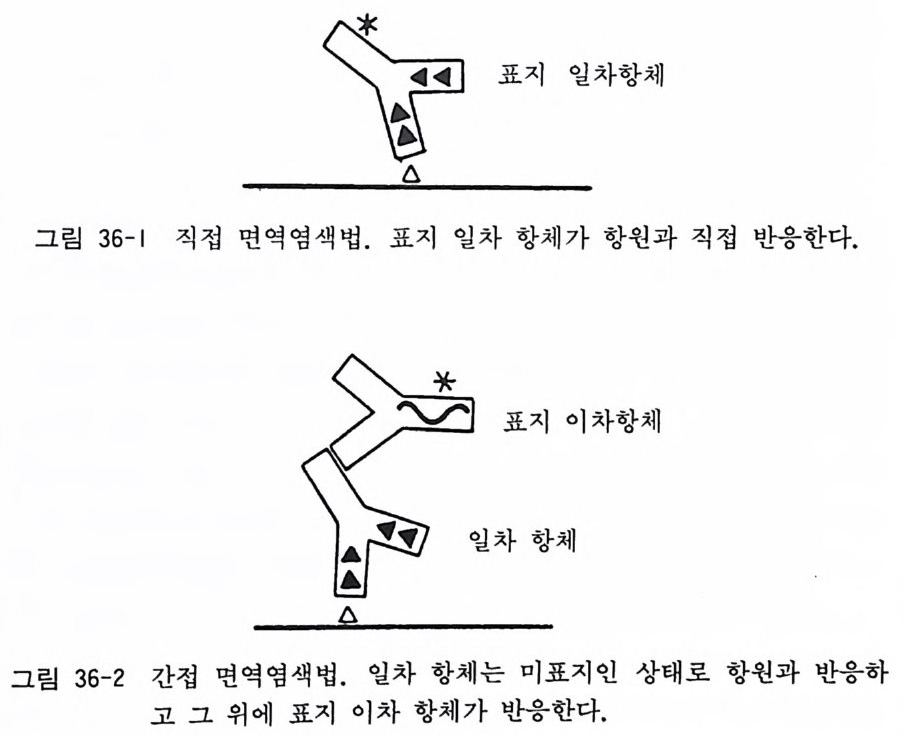 \ 표지 일차항체
\ 표지 일차항체
작용시키는 항체 (일차 항체)는 미표지로 반응을 시작하고 그 다 음 단계에서 그 항체에 대한 항체(이차 항체)에 형광색소나 효소 표지를 하여 간접적으로 항원을 검출하는 간접법(그립 36-2) 의 두 가지 가 있다. 2-5) 직접법은 비특이 반응이 적고 간접법에 비하여 특이성이 높으 나 그 감도가 많이 떨어전다• 이에 더하여 일차 항체에 일일이 형광색소나 결합효소 표지를 결합시키는 번잡한 조작을 자기 연 구실에 하여야 하는 결점이 있다. 그 반면 간접법은 표지 이차 항체가 여러 가지 결합형으로 제작되어서 저렴한 가격으로 쉽게 입수할 수 있는 이점이 있다. 최근에는 PAP 법, ABC 법 등 간접 법을 보충한 감도가 높은 기술이 고안되어 면역염색법이 널리 사 용되고 있다. 36.1 형광 항체 염색법 여기에서는 신생 래트 대뇌 분산 세포 배양을 써서 아스트로사 이트(성상교세포)의 특이 항원인 글리아산성 섬유단백질 GFAP, glial fibr ill ar y aci di c pro te i n 의 형 광 항체 염 색 의 조작을 소개 한 다. 일차 항체의 대조군으로서 정상토끼 혈청 혹은 NS-1 마우스 골수종 세포 배양액을 희석하여 사용한다. 형광 현미경은 차이스 Zeis s , 라이츠 Leit z, 올림퍼스 0lym pu s· 그리고 니콘 Ni ko n 등의 회사에서 시판하고 있다. 형광 현미경은 광원으로서 형광을 발생 시키게 하는 자의선 근원인 초고압 수은 램프, 광선 스펙트라에 서 파장을 선별하는 여기필터, 대물렌즈와 접안렌즈 사이에서 형 광 색조의 조정을 하는 흡수필터, 암시야 콘덴서, 현미경사진 촬 영을 위한 카메라와 그 조정장치의 몇 가지 콤비네이션으로 되어
있다. 형광현미경은 광원의 조사 방향에 따라서 투과형과 낙사형 두 가지가 있는데 최근에는 낙사형이 주류가 되어있다. 면역염색 표본을 관찰함에 있어서 형광을 일으키는 광선의 파장을 선택하 는 데 필터의 콤비네이션에 따라서 UV, BV, V, B, G 의 다섯 가지가 있으며 플로레신(녹색 형광)의 경우에는 B 콤비네이션, 로다민(적색형광)의 경우에는 G 콤비네이션 그리고 쿠마린(청색 형광)의 경우에는 V 콤바네이션을 사용한다. 〈 재료 〉 1) 일차 항체-토끼 항 인간 GFAP 항체 2) 이 차 항체 -플로레 신 fluo rescein 표지 산양 항 토끼 IgG 항체 3) 희 석 액 -HHH (Hanks 액 -HEPES-horse serum) 핸크스액에 10 mM HEPES 완충액과 1 % 마혈청을 첨가하 여서 항체의 희석액으로 사용한다. 4) 미크로피펫와 피펫팁 5) 미크로 튜브 6) 챔버 10cm 페트리 접시 바닥에 파라필름을 꼭 맞게 깔고, 페 트리 접시 뚜껑 내면에는 필터페이퍼를 데이프로 붙이고 물 로 적셔서 페트리 접시 내부가 마르지 않도록 한다. 〈 방법〉 1) 래트 대뇌 세포배양 커버슬립을 한번 PBS 로 씻고, 미리 차게 해두었던 메틸알코올 속에서 영하 20 도에서 10 분간 고 정한다• 2) 고정된 커버슬립을 PBS 로 두 번 씻는다. 3) 커버슬립을 챔버로 옮기고 토끼 항 GFAP 항체 (HHH 나
PBS 로 1 : 100 으로 희석)을 20 µI( 커버슬립 사이즈에 따라서 양 이 다르다) 울려서 1 시간 실온에서 반응시킨다. 4) 커버슬립을 PBS 로 3 번 씻는다. 5) 풀로레 신 표지 산양 항 토끼 IgG 항체 (HHH 나 PBS 로 1 : 50 으로 회석) 20 µI 을 을려서 1 시간 실온에서 반응시킨다. 6) 커버슬립을 PBS 로 3 번 씻는다. 7) 유리 슬라이드 위에 글리세린 -PBS9 : 1 혹은 폴리비닐알코 올-글리세린 (36 . 2 . 5 참고)으로 봉입한다. 8) 형광 현미경 밀에서 검색한다. 〈해설〉 신생 래트 대뇌 세포배양에서 아스트로사이트가 GFAP 항체에
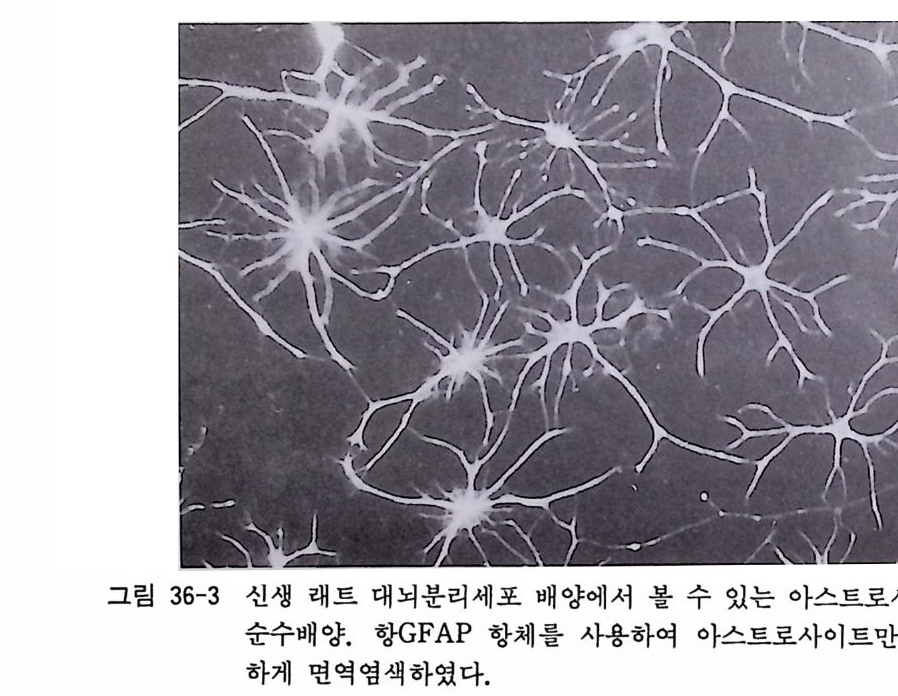 그림 36-3 신생 래트 대뇌분리세포 배양에서 볼 수 있는 아스트로사이트의
그림 36-3 신생 래트 대뇌분리세포 배양에서 볼 수 있는 아스트로사이트의
염색한 것은 그림 36 - 3 에서 보이고 있다 .6) 성인 척수후근 신경 절 dorsal root ga ng lion 세 포를 분리 하여 10 % 마혈 청 을 포함한 배양액에서 2 주일간 배양한 다음 그 신경세포를 마우스 항 신경 필라멘트 neuro fi lamen t 단클론 항체를 사용하여 면역염색한 것이 그립 36 - 4 이다. 그 염색방법은 위에서 설명한 형광항체 염색법과 같다 .7)
그림 36-4 성인 척수 후근신경절 배양 2 주일 후에 항뉴로필라멘트 항체를 사용하여 신경세포만을 특이하게 면역염색하였다.
36.2 효소 항체 면역염색법 항원의 세포 표면 혹은 세포내 존재를 증명하려면 효소단백질 을 항체와 결합시켜서 항체의 마카 marker 로서 쓸 수가 있다. 항 원과 항체를 작용시켜서 그 존재를 항체로 표지하고 있는 효소를 가시화시킴으로써 관찰한다는 원리이다. 마카로서 사용되는 효소 에는 여러 종류가 있는데 그중에서 가장 보편적인 것이 페록시다 제와 알칼리포스파타제이다. 효소 항체 면역염색법에는 효소 표 지 항체 간접 법 , 효소 표지 프로데 인 A 법 , PAP (pe roxid a se -anti pe roxid a se) 법 , ABC (avi di n - bio t i n comp le x) 법 의 네 가지 가 있다. 36.2.1 페록시다제 표지항체 염색법 이 간접법의 원리는 그림 36-2 에 설명되어 있다. 〈방법 〉 1) 배양 커버슬립을 꺼내서 세포 표면 항체의 경우에는 살아있 는 그대로, 혹은 4% 파라포름알데히드에서 5 분간 고정한 뒤, PBS 로 씻고 챔버로 옮긴다. 세포내 항원인 경우에는 산성 알코올(알코울 대 빙초산 95 : 5) 중에서 영하 20 도에서 10 분간 고정한다. 고정한 뒤 PBS 로 두 번 씻고 챔버로 옮 긴다. 2) 일차 항체를 HHH 나 PBS 로 20 배에서 100 배로 희석하고 20 짜를 커버슬립에 울려놓고 60 분간 실온에서 인큐베이트 한다. 3) 커버슬립을 PBS 로 세 번 씻는다.
4) 페록시다제 표지 이차 항체 (산양 항토끼 혹은 산양 항 마우스 l gG) 를 PBS 로 40 배에서 100 배 희석하고 커버슬립에 첨가하 여 실온에서 30 분간 반응시킨다. 5) 커버슬립을 PBS 로 세 번 씻는다. 6) DAB 액에 과산화수소를 넣어서 그 속에서 커버슬립을 5-10 분간 반응시킨다 (36.2 . 2 참고). 7) 커버슬립을 PBS 로 두 번 씻은 뒤 알코올 탈수, 크실렌 xy le ne 두철을 하고 퍼마운트p ermoun t에 봉입한다. 8) 현미경 아래서 검색한다. 36.2.2 DAB 용액 DAB 는 시그마에서 살 수가 있으며 DAB 용액은 다음과 같이 작성한다. 1) 디 아미 노 벤 지 딘 (DAB, 3, 3-dia m i no benzid i n e tet r a hy d rox- ych lorid e ) 5 m g을 0 . 05 M 트리 스 완충액 (pH 7 . 6) 10 ml 에 녹이고 필터페이퍼로 여과시킨다. 2) 30 % 과산화수소액 (H202) 0 . 01 ml 을 0 . 6 ml 증류에 희 석 하 고, 그 0.1 ml 을 10ml DAB 액에 첨가한다. 3) DAB 액 속에 염화암모늄니켈 nic k el ammoniu m sulfa te 30 mg 혹은 염 화코발트 cobalt chlorid e 40 m g을 첨 가한 뒤 에 과 산화수소액을 추가하면 페록시다제 염색이 증가하여 반응의 색조가 갈색에서 흑색으로 바뀌고 콘트라스트가 높아진다. 36.2.3 ABC 면역염색법 ABC 법의 원리는 그립 36-5 에 설명되어 있다.
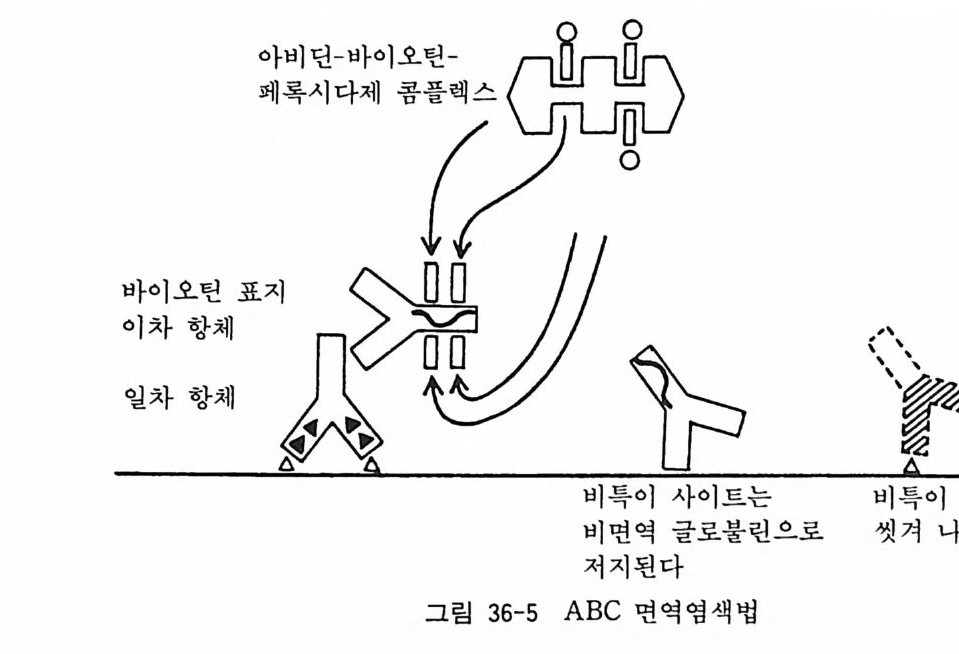 바이오틴
바이오틴
〈 방법 〉 1) 배양 커버슬립을 4 % 파라포름알데히드 중에서 5 분간 실온 에서 고정, 혹은 산성 알코올 중에서 15 분간 영하 20 도에서 고정한뒤, PBS 로 두 번 씻고 챔버로 옮긴다. 2) 비면역혈청(이차 항체와 같은 동물, 일반으로 산양이나 토끼) 의 40-100 배 희석액 (PBS) 을 커버슬립에 올려놓고 20 분간 실 온에서 반웅시킨다. 3) 커버슬립에서 비면역혈청을 씻어내지 않고 가볍게 부로팅을 하여 버리고, 일차 항체 희석액 (100-1000 배)을 울려놓고 실 온에서 1-2 시 간 반응시 킨다. 경 우에 따라서는 24, 48, 72 시 간 동안 4 도에서 반응시킨다. 4) 커버슬립을 PBS 로 세 번 5 분씩 씻는다. 5) 바이오틴 bio t i n 표지 이차 항체를 PBS 로 40-100 배 희석하
여 커버슬립에 울려놓고 30 분간 실온에서 반응시킨다. 6) 커버슬립을 PBS 로 3 번 5 분씩 씻는다. 7) 아비딘액과 페록시다제 결합 바이오틴액의 혼합액 (벡터사 Vec t or 의 ABC 액은 A 액과 B 액을 0.04 ml 씩 섞어서 이룰 PBS 나 트리스완충액 5ml 에 희석한다)을 커버슬립에 울려놓고 30 분간 실온에서 반응시킨다. 8) 커버슬립을 PBS 로 3 번 5 분씩 씻는다. 9) DAB 반응을 5 - 10 분간 한다 (21. 2.2 참고). 10) 탈수, 두철, 퍼마운트 봉입을 한 뒤 현미경 아래에서 관찰 한다. 36.2.4 PAP 면역 염색법 PAP( perox i dase-an tip erox i dase) 법의 원리는 그림 36-6 에 설명 되어 있다. 〈 방법 〉 1) 배양 커버슬립을 4% 파라포름알데히드 중에서 5 분간 실온 에서 고정, 혹은 산성 알코올 중에서 15 분간 영하 20 도에서 고정한 뒤, PBS 로 두 번 씻고 챔버로 옮긴다. 2) 비면역 혈청(이차 항체와 같은 동물, 일반으로 산양이나 토끼) 의 40-100 배 희석액 (PBS) 을 커버슬립에 울려놓고 20 분간 실 온에서 반응시킨다. 3) 커버슬립에서 비면역혈청을 씻어내지 않고 가볍게 부로팅을 하여 버리고, 일차 항체 희석액 (100 배 -1000 배)을 울려 놓고 실온에서 1-2 시간 반응시킨다. 4) 커버슬립을 PDS 로 3 번 5 분씩 씻는다.
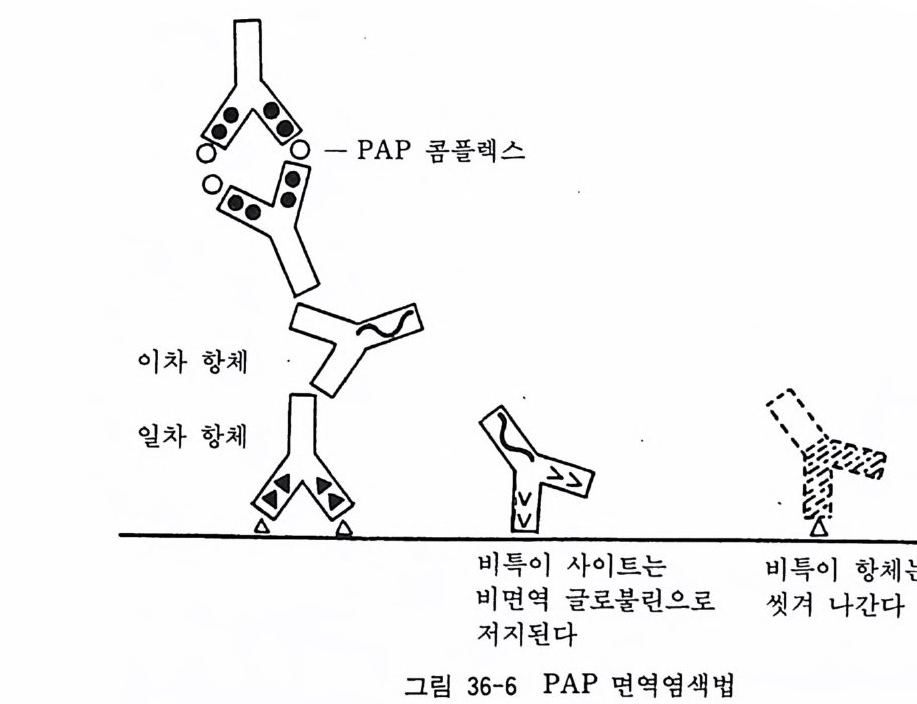 일차 항체 \ ,、 , ```”,``` 太篠.5` -· / ,4., < Ss
일차 항체 \ ,、 , ```”,``` 太篠.5` -· / ,4., < Ss
5) 미표지 이차 항체를 PBS 로 40-100 배 희석하여 커버슬립에 울려놓고 30 분간 실온에서 반응시킨다. 6) 커버슬립을 PBS 로 5 분씩 3 번 씻는다 7) 커버슬립에 PAP 희석액(일차 항체가 단클론 마우스이면 마우 스 PAP, 토끼이면 토끼 PAP 를 PBS 로 100 배 희석)을 올려놓 고 30 분간 실온에서 반응시킨다. 8) 커버슬립을 PBS 로 3 번 5 분씩 씻는다. 9) DAB 반응을 5-10 분간 한다 (21 . 2 . 2 참고) 10) 탈수, 투철, 퍼마운트 봉입을 한 뒤 현미경 아래에서 관찰 한다.
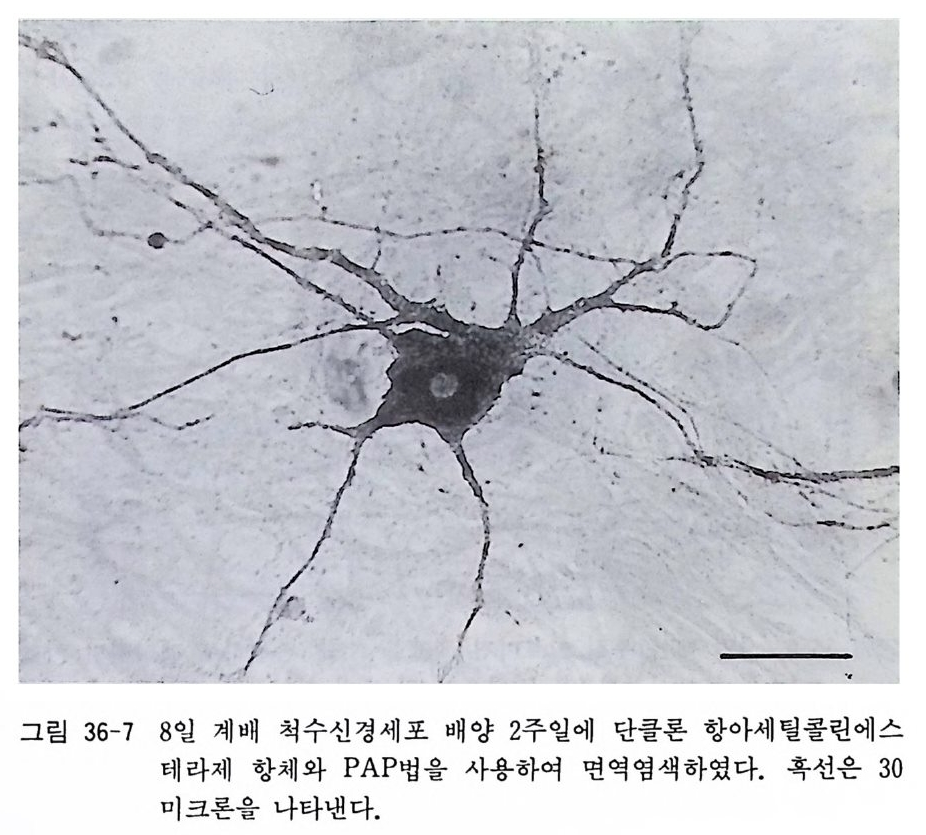 ` A ·•
` A ·•
〈 해설 〉 그립 36 - 7 에서는 계배 척수 신경세포를 단클론 항아세틸콜린에 스데라제 acety l cholin e ste r ase 항체와 PAP 법을 써서 염색하였고 그립 36-8 에서는 사람 태아 부신 크로마핀 세포를 토끼 항티로신 수산화효소 tyros in e hyd roxy la se 항체 와 ABC 법 을 써 서 염 색 하였 다.
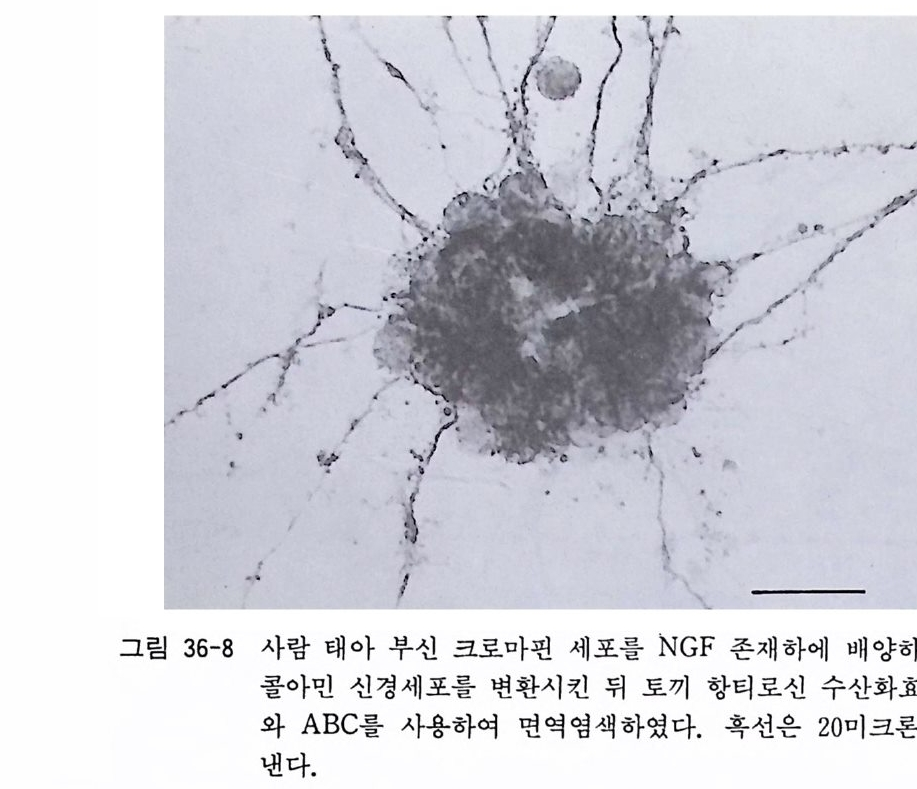 .) C ,,/;” `‘` '
.) C ,,/;” `‘` '
36.2.5 봉입제 폴리비닐알코올_글리세린 제작법 형광염색이나 면역염색을 한 뒤에 배양세포를 가전 커버글라스 를 적당한 봉임제로 슬라이드글라스에 접착시키고 현미경 아래에 서 관찰을 하게 된다. 형광염색이나 효소 세포화학염색을 한 프 레파레이션은 탈수와 투철의 과정을 거치지 않기 때문에 수용성 인 봉임제가 필요하다. 가장 일반적으로 사용되는 봉입제로서 폴 리비닐알코올-글리세린의 제작법을 여기서 소개한다.
〈 방법 〉 100ml 용량의 유리 비커에 폴리비닐알코올(시그마) 10 g을 넣 은다음 0. 2 M 트리스 • 영산완충액 (pH 8. 5) 40 ml 을 가하고 60 도 에서 가열한다. 폴리비닐알코올이 녹으면 이를 식힌 다음 글리세 린 20ml 을 가하고 잘 섞이도록 유리봉으로 짓는다. 폴리비닐알 코올-글리세린액은 3000r p m 에서 10 분 원심분리를 하고 그 상청 액을 봉임제로 사용한다. 상청액은 4 도나 영하 20 도에서 보존하면 1 년 정도 사용할 수 있다. 폴리비닐알코올 봉임제를 젤바톨 Gelva t ol 혹은 엘바톨 Elva t ol 이라 하여 시판되고 있다.
제 37 장 신경계 조직 배양세포 증식의 판정 배양세포의 증식 능력을 검사하는 데 가장 보편적으로 사용되 는 기 술은 트리 티움 표지 티 미 딘 tri t iat e d thy midi n e 을 취 득한 세 포를 신틸레 이 션 카운터 sci nt i llat i on counte r 로 그 방사능을 자동 적으로 측정하는 방법과 오토라디오그라피예 의해서 사전유제 emulsio n 위에 은입자 s il ver gr a i n 로서 검출하는 두 가지 방법이 있다. 세포가 증식하려면 DNA 합성을 하는데 이것은 세포주기 cell cyc l e 중에서 S 기에서만 가능하며 이때에 세포가 티미딘을 취득하여 DNA 로 변환시키는 것을 이용한 것이다. 최근에든 티 미 딘의 유도체 인 브로모디 옥시 유리 딘 bromodeoxy u rid i n e 에 대 한 단클론 항체를 사용하여 형광면역염색법으로 신경계 세포의 증식 능력을 검색할 수가 있게 되었다.
37.l 오토라디오그라피 배양 중에 있는 신경계세포의 증식 동태를 티미딘에 의한 오토 라디오그라피 au t orad i ogra p h y로 검색한 연구가 있어서 아스토로 사이트 ,1 , 2) 올리고덴드로사이트 ,3,4) 슈완세포 5,6) 가 신경영양인자에 의해서 증식능이 증가한다는 보고가 있다. 〈 준비 〉 1) 트리티움 표지 티미딘 2) PBS 3) 고정액 -4 % 파라포름알데히드 4) 사진 유제 -Kodak NTB- 2 5) 사전 현상액 -~odak D-19 사전 정착액 6) 밀봉상자 〈 방법 〉 1) 배양세포가 자라고 있는 커버슬립을 트리티움 표지 티미딘 (0.5 미크로큐리 /ml 최종 농도)이 들어있는 배양액 속에서 2-12 시간 인큐베이트 한다 . 2) 커버슬립을 PBS 속에서 2 번 간단히 씻는다 3) 커버슬립을 4 % 파라포름알데히드 고정액 속에서 5 분 실온 에서 고정한다. 4) 커버슬립을 PBS 속에서 2 번 씻는다 5) 커버슬립을 헤마톡시린액 속에서 2 분간 염색한다. 6) 커버슬립을 PBS 속에서 2 번 씻는다. 7) 커버슬립을 드라이어를 써서 건조시킨 다음, 유리 슬라이드
위에 배양세포가 위에 오도록 퍼마운트로 정착시킨다. 8) 여기서부터는 Kodak No.2 dark red 필터를 사용하고 암실 에서 행한다. 사진 유제를 워터바스 속에서 45 도로 가온해서 녹이고 그 5ml 를 증류수 5ml 로 희석한다. 9) 커버슬립이 올라 있는 슬라이드를 한 장씩 사진 유제 속에 2 초씩 담그고 서서히 수직으로 끌어울린 다음 세워서 10-15 분간 건조시킨다. 10) 슬라이드를 실리카겔 건조제가 들어있는 밀봉상자에 봉하 고 데이프를 써서 밀봉한다. 냉장고 속에서 4 도의 온도에서 7 일 -10 일간 노출시 킨다. 11) 현상은 실온에서 5 분간, 정착은 10 분간, 수선은 15 분간으 로 그뒤에 충분하게 건조시킨다. 12) 건조시 킨 뒤 90 % 알코올, 95 % 알코올, 100 % 알코올 2 번, 크실렌 2 번을 통하여 탈수와 두석을 한 뒤 퍼마운트로 포매한다. 13) 현미경 아래서 관찰한다. 37.2 신틸레이션 카운터 트리티움 표지 티미딘은 세포가 증식할 때에 일어나는 DNA 합성에 필수적인 뉴클레오티드인데 티미딘울 취득한 세포는 증식 사이클에 있다고 할 수 있다. 사전 감광의 원칙으로 사전 유제를 사용하여 티미딘 취득을 판정하는 오토라디오그라피는 암실이 필 요하고 그 결과를 알게 되기까지 1 주일 이상이 필요하다. 그러한 이유로 단시간 안에 결과를 알 수 있는 신틸레이션 카운터법 sci nt i llat i on conute r 이 많이 사용되 나, 다른 종류의 세 포가 전혀
들어있지 않고 순수한 세포로 구성되었다는 증거와 대량의 세포 가 필요하다는 이유로서 그 결과를 신뢰할 수 없다는 결점이 있 다. 〈 준비 〉 1) 트리티움 표지 티미딘 2) PBS 3) 1 % 나트륨아자이드 (NaN3) 4) 0 . 2 N 수산화나트륨 (NaOH) 5) 1 N 염산 6) 신틸레 이션액 (뉴잉글랜드 뉴크레아의 A q uasol 나 &nersham 의 ACSII 등이 시판되 어 있다) 〈 방법 〉 1) 3.5 cm 페트리 접시 12 장에 세포부유액 (5X104/ml) 을 2 ml 씩 주입하고 37 도에서 4 시간 배양한다. 2) 배양액을 버리고 신선한 배양액 1ml 를 주입한다. 세포 증 식 자극제 혹은 억제제를 0.01 ml 씩 추가하고 1 시간 배양한 다. 3) 각 페트리 집시에 트리티움 표지 티미딘 (0.5 미크로큐리 /ml 최종 농도)을 가하고 37 도에서 4-24 시간 배양한다• 4) 인큐베이터에서 페트리 접시를 꺼내고 아이스박스 위에서 1 % 나트륨아자이드 액을 0.01 ml 추가하여 반응을 정지시 킨다. 5) PBS 로 2 번 세포를 씻고, 0.2N 수산화나트륨 액을 0.5ml 첨가하여 세포를 용해시킨다. 6) 5 분 뒤 용해 세포액을 플라스틱 시험관에 옮기고, 0.5ml 의
0.2 N 수산화나트륨액으로 페트리 접시를 씻어내서 그 내용 을 다시 시험관에 추가한다. 7) 1 N 염산 0.2ml 로 중화시킨 다음, 50 % 트리크롤 초산 (TCA) 0.15 ml 을 첨가하여 4 도에서 30 분 정치하면 침전이 생긴다. 8) 원심침전한 뒤 상청액울 버리고 침전을 5 % 트리크롤 초산 으로 한 번 씻는다. 9) 침전을 글라스 필터 위에 옮기고 5 % 트리크롤 초산 2 번, 에틸알코올, 아세톤의 차례로 씻어낸다. 10) 글라스 필터를 바이알에 넣고 신틸레이션액 10ml 을 첨가 한 뒤 베타카운터에서 방사능을 측정한다. 37.3 브로모디옥시유리딘 항체에 의한 형광염색법 1982 년에 티미딘 유도체인 브로모디옥시유리딘에 대한 단클론 항체가 만들어지고 이것을 사용하는 면역염색법의 개발로서 증식 세포의 확인이 짧은 시간 안에 이루어지게 되었다 .7) 이에 더하 여 세포 타입 특이 마커를 응용하는 이중 면역염색법으로 증식세 포의 형질 표현도 동시에 알 수가 있다. 여기에서는 아스트로사 이트에 있어서의 GFAP 와 브로모디옥시유리딘 항체에 대한 이중 면역염색의 실제를 소개한다 .8-10) 〈준비〉 1) 브로모디옥시유리딘 BrdU( 시그마 )-lmM 농도로 증류수에 녹이고 필터여과하고 멸균해 둔다. 이것을 100 대 1 로 배양 액에 첨가한다.
2) 2 N 염산 3) 0.1 M 봉산-봉사 완충액 (pH 9.0 ) 4) 브로모디옥시유리딘 단클론 항체 (벡턴-디킨슨사 Becto n -Di ck - ins on) 〈 방법 〉 1) 17 - 18 일 래트 태아 뇌에서 분리한 아스토로사이트 배양 · (15 . 6 참고) 에 서 그 배 양액 중에 BrdU 를 10-5M 농도로 첨 가하고 12 - 24 시간 동안 37 도에서 인큐베이트한다. 배양액 중 에 10- s M 의 플로로디 옥시 유리 딘 fluo rodeoxy ur id i n e 을 첨 가 하면 세포내 티미디레이트 합성을 억제하고 BrdU 표지가 좋아진다. 2) 배양 커버슬립을 PBS 로 두 번 씻은 뒤 一 20 도의 냉동실에 서 에 틸알코올로 20 분간 고정 한다. 3) PBS 속에서 2 번 씻는다. 4) 토끼 항 GFAP 항체 (PBS 에서 1 : 100 희석) 속에서 실온에 서 30 분간 반응시킨다. 5) PBS 속에서 3 번 씻는다. 6) 풀로레신 표지 산양 항토끼 l g G(PBS 에서 1 : 50 희석) 속에 서 실온에서 30 분간 반응시킨다. 7) PBS 속에서 3 번 씻는다. 8) 2N 염산에서 20 분간 반웅시켜서 DNA 를 변성시킨다. 이어 서 봉산-봉사 완충액에서 10 분간 중화시킨다. 9) PBS 속에서 3 번 씻는다. 10) 항 BrdU 항체 (PBS 에서 1 : 20 회석) 속에서 실온에서 30 분 간 반웅시킨다. 11) PBS 속에서 3 번 씻는다.
12) 로다민 표지 산양 항마우스 I g G(PBS 에서 1 : 50 회 석 ) 속 에서 실온에서 30 분간 반응시킨다. 13) PBS 속에 서 3 번 씻 는다. 14) 젤바톨 (36.2 . 5 참고)로서 글라스 슬라이드 위에 봉입하고 형광현미경 아래서 검사한다. 〈 해설 〉 그림 37-1 과 37-2 에서는 사람 태아 아스트로사이트 배양에서
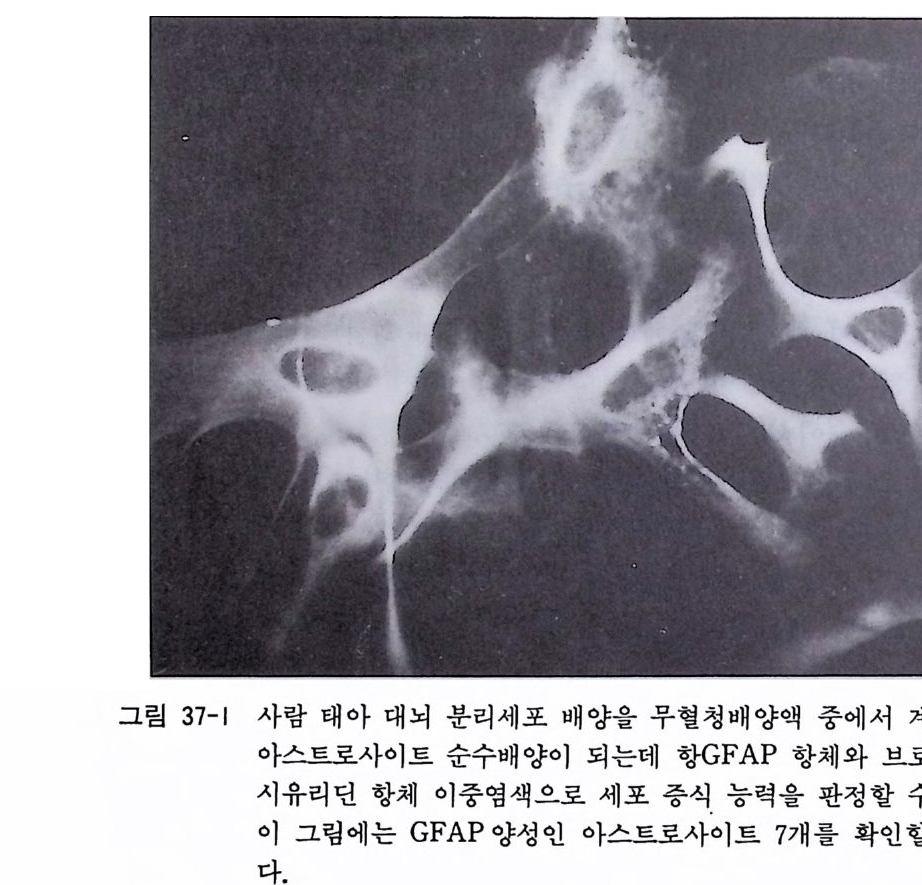 그림 37-1 사람 태아 대뇌 분리세포 배양을 무혈청배양액 중에서 계속하면
그림 37-1 사람 태아 대뇌 분리세포 배양을 무혈청배양액 중에서 계속하면
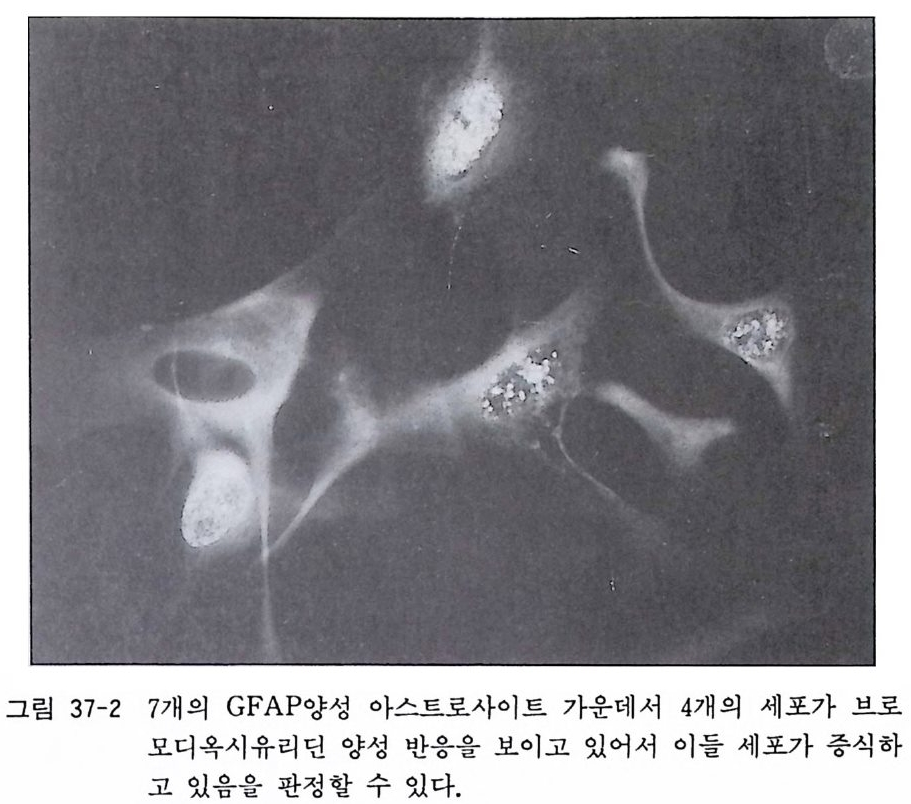 그림 37-2 7 개의 GFAP 양성 아스트로사아트 가운데서 4 개의 세포가 브로
그림 37-2 7 개의 GFAP 양성 아스트로사아트 가운데서 4 개의 세포가 브로
증식하는 세포가 확인되었는데 녹색 형광으로 아스트로사이트 마 카인 GFAP 반응이 그리고 적색 형광으로 브로모디옥시유리딘 반응이 관찰되었다.
제 38 장 세포 활성의 측정 38.l 크리스탈 바이올렛 검사법 화학물질이 배양세포에 미치는 영향을 알고자 할 때, 그 물질 이 독성을 보이는 그 농도를 알아보는 것이 중요하다. 여기에서 는 가장 보편적이고 간이한 방법으로 96 혈 멀티웰 미크로타이터 폴레이트 mul ti well mi cro ti ter p la t e 와 ELISA 비색계를 사용하는 방법을 기술한다. 신생 마우스 대뇌 배양을 사용하여 염화메틸수은 me t h y l mercu ry chlorid e 의 신경 독성을 검 사하는 실험 을 소개 하기로 한 다. 〈준비〉 1) 0.4 % 크리스탈 바이올렛 수용액 2) 메틸알코올 3) 96 혈 멀티웰 플레이트, 피펫, 시험관
4) ELISA 바색계 〈 방법 〉 1) 96 혈 멀티웰 플레이트에 신생아 마우스 소뇌 분리세포 배 양을 시작한다. 배양 7-10 일경에 실험을 시작한다. 2) 영 화메 틸수은 meth y lm ercury chlorid e 의 8 rnM 스토크 용액 울 증류수로 만든다. 96 혈 멀티웰에서 배양액을 제거한 뒤 0.1 ml 의 5 % 마혈청 포함 배양액을 각 웰에 분주한다. A 와 B 열에 160µM 염화메틸수은을 포함한 배양액 0 . 1ml 를 분주한다. 배양액과 수은액을 잘 혼합한 뒤 A 와 B 열에서 0.1ml 를 C 와 D 열에, C 와 D 열에서 E 와 F 열로 차례로 희석 을 계속한다. 그 배열은 다음과 같다. A 와 B 80µM 염화메틸수은 C 와 D 40 E 와 F 20 G 와 H 10 I 와 J 5 K 와 L 양성 대 조군 (수은액 을 첨 가하지 않는다) 3) 3 일간 배양한 뒤, 각 웰을 PBS 로 한 번 씻고 메탄올로 5 분간 고정한다. 0.4 % 크리스탈 바이올랫액으로 20 분간 염 색한 뒤 PBS 로 한 번 씻는다. 4) 염색한 표본은 ELISA 비색기에서 OD 590 으로 측정한다. 양성대조군의 결과를 100 %로 한 뒤 각 웰의 OD 결과를 % 로 표시한다. 가로축에 약품의 농도를, 세로축에 OD 결과를 표시한 뒤 50 % 저해치를 알아본다. 5) ELISA 비색계가 없는 경우에는 각 웰의 세포 염색도의 높 고 낮음을 육안으로 혹은 현미경 하에서 조사하여 세포장해
도를 0 에서 4 로서 표지한다. 0 은 양성대조군, 4 는 세포가 모 두 사멸하여 웰에서 떨어져 나간 경우, 2 는 양성대조군의 50 % 정도의 경우로 한다. 38.2 MTT 검사법 최근에는 MTT 검사법이 위에 기술한 크리스탈 바이올렛 검 사법에 대신하여 널리 사용되고 있다. 그 원리는 다음과 같 다. MTT< 3 - (4 , 5-d i m eth y lt h i a z ol-2 - yl ) -2, 5-d i p h eny l tet r a zoliu m brom i de 〉 는 수용액 속에서는 무색인데 살아있는 세포의 미토콘 드리아 안의 효소에 의해서 물에 녹지 않는 보라색 색소로 변환 한다. 색소의 강도를 ELISA 비색계에서 측정하여 살아있는 세 포의 증식, 독성검사 등에 활용할 수가 있다 .I) MTT 는 시그마 에서 살 수가 있다. MTT 검사법 이의에도 XTT, 뉴트럴레드검 사법 등이 개발되어서 세포 활성 검사에 쓰이고 있다 .2) 〈준비〉 1) 5 mg /m l MTT (시 그마) 수용액 2) 0.04N 염산을 첨가한 이소프로필알코올 3) 96 혈 멀티웰 플레이트, 피펫, 시험관 4) ELISA 비색계 〈방법〉 1) 38.1 의 방법에서 1)-2) 의 순서로 실험을 한다. 2) 3 일간 배양한 ’ 뒤 배양을 씻지 않고, 0.01 ml MTT 액 (PBS 에 5 mg /m l .£. 스토크를 만든다) 을 각 웰에 추가하고 4 시 간 37
도 인큐베이터에서 정치 배양을 계속한다. 3) 멀티웰 플레이트를 인큐베이터에서 꺼내고 각 웰에 이소프 로필 알코올 -0.04 N 영산 용액 0.1 ml 씩을 첨가한다. 4) ELISA 비 색 계 에 서 파장 570 nm 로 OD 를 측정 한다. 〈 해설 〉 그림 38-1 에서는 MTT 검사법으로 메틸염화수은 독성을 측정
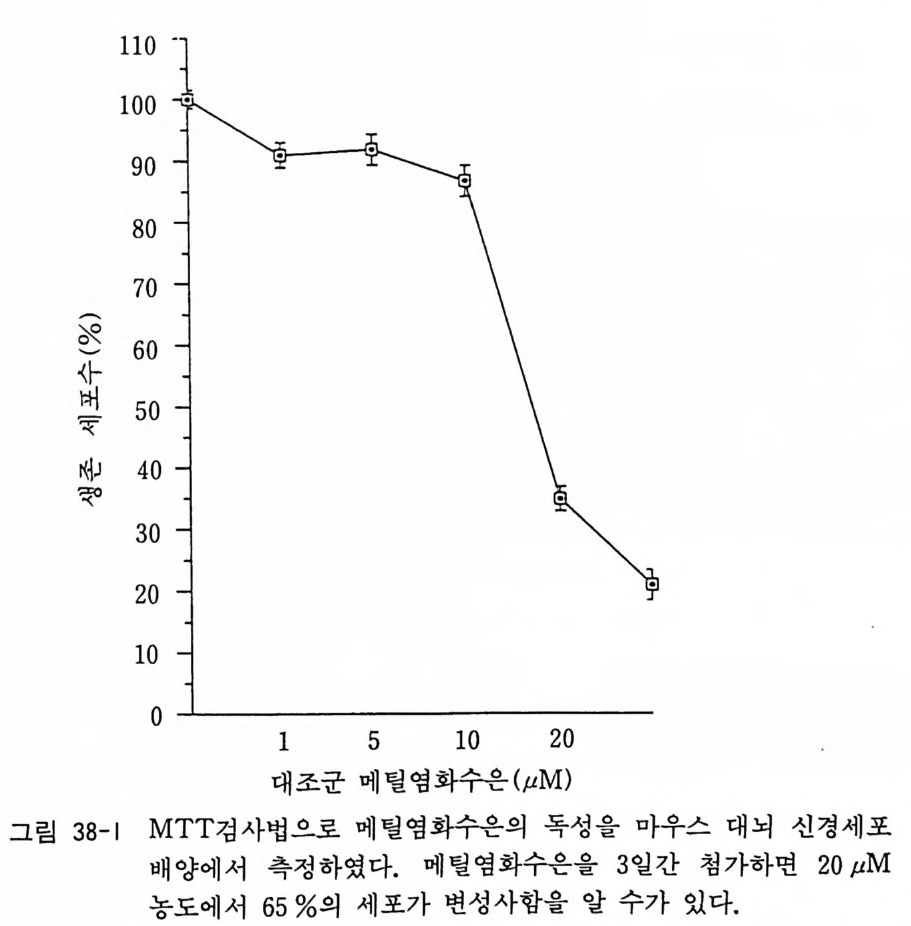 1l001'-r_ 09-I. 08070605040302010
1l001'-r_ 09-I. 08070605040302010
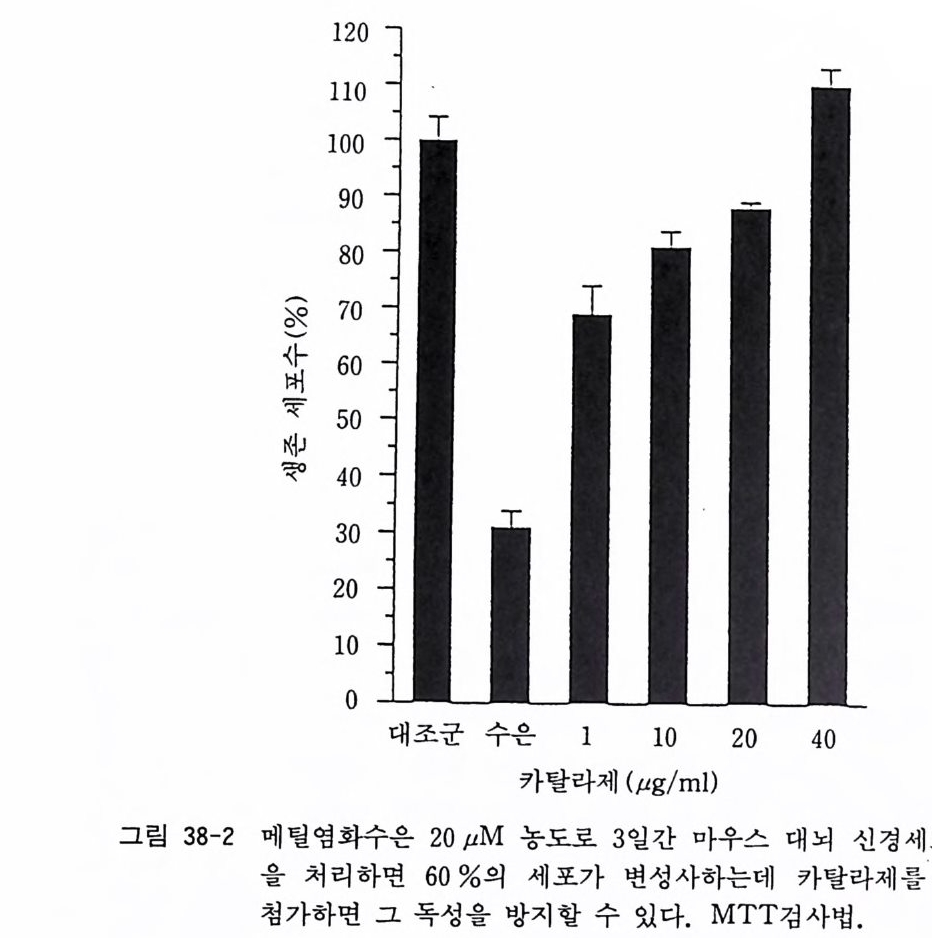 120
120
한 실례를 보인다. 메틸염화수은을 3 일간 배양액에 첨가하였던바 신생아 마우스 대뇌 신경세포 배양에서 다수의 세포가 죽었음을 알 수가 있다. 예컨대 20µM 농도에서 생존한 세포가 35% 이하 임을 알 수가 있다. 그림 38-2 에서는 역시 MTT 검사법에 의한 세포 생존도 측정 울 하였다. 카탈라제를 첨가하면 메틸염화수은의 독성을 방지할 수있다.
제 39 장 세포영색법 신경조직 배양세포는 그 유래하는 조직, 세포의 기능과 영양상 태, 그리고 배양 조건에 따라서 그 모양이 달라진다. 따라서 신 경조직 배양세포에 의한 실험을 하게 되면 먼저 그 세포의 형태 롤 여러 가지 조건 아래서 관찰하고 그 내용을 잘 알아두어야 하 겠다. 여기에서는 가장 기초적인 세포고정, 염색법으로서 김사염 색 Gi em sa s t a i n 과 헤마톡실린-에오신 염색, 그리고 메틸그린피로 닌 염색법에 대해 간략히 기술한다. 특수 신경조직 염색법으로서 신경세포 특이 염색법인 보디안 Bod i an, 홈즈 Holms, 빌쇼우스키 Bie l schowsky 은염색법이 있으며 그밖에 신경조칙 이식배양에서 보이는 미엘린울 특수 염색하는 수단블랙 염색법이 있다 .1-5)
39.1 김사 염색법 〈준비 〉 1) 김사액 2) 배양세포(커버글라스) 3) PBS 4) 산성메틸알코올(메틸알코올 : 빙초산 3 : 1) 5) 봉임제(퍼마운트, 기타) 〈방법〉 1) 커버글라스(세포가 그 위에 있음)를 PBS 에서 씻는다. 2) 산성메틸알코올에서 30 초 가량 고정한다. 3) 커버글라스를 실온에서 말린다. 4) 김사액 (1 : 10 온수로 희석한다) 속에서 30 분 동안 실온에서 염색한다 5) 증류수로 두 번 씻고 완전히 실온에서 말린다. 6) 봉임제(퍼마운트)로 슬라이드글라스 위에 봉입한다. 〈결과〉 세포질은 보라색, 호영기성 과립은 푸른색, 호산성 과립은 빨 간색이 된다. 39.2 헤마독실린 염색법 〈준비〉 1) 헤마톡실린액 Harr i s hemato x y lin
2) 배양세포 3) PBS 4) 포르말린 5) 암모니아수 (1 : 50) 6) 알코올 7) 크실렌 8) 봉임제 〈 방법 〉 1) 커버글라스를 PBS 에서 씻는다. 2) 10 % 포르말린에 실온에서 10 분간 고정. 3) PBS 에서 두 번 씻는다. 4) 헤마독실린액을 커버글라스 위에 분주한다. 실온에서 한 시 간 정치한다. 5) 증류수로 씻는다. 6) 암모니아수로 씻는다. 이때 염색이 파랗게 된다. 7) 증류수로 씻은 다음 .알코올로 탈수하고 다음에 크실렌에서 두석을 한다. 8) 퍼마운트로 슬라이드 위에 봉입한다• 〈 결과 〉 핵이 푸른색으로 염색된다. 해마톡실린 다음에 5 분 가량 에오 신액 eos i n 에 염색하면 세포체 속이 빨갛게 염색될 수 있다.
39.3 메틸그린-피로닌 염색법 〈 재료 〉 재료는 39.2 와 같다. 〈 방법 〉 1) 커버글라스를 95 % 알코올에서 10 분간 고정한 뒤 증류수에 서 씻는다. 2) 메틸그린-피로닌액으로 10 분간 실온에서 염색한 뒤 가볍게 증류수에서 씻는다. 3) 필터페이퍼로 물을 빨아들이고 100% 아세톤으로 급속히 탈수한다. 4) 아세톤-크실렌 혼합액에서 1 분, 투석을 한 다음 슬라이드 위에 퍼마운트로 봉입한다. 메틸그린-피로닌액 조성 용액 A : 5 % 피로닌 수용액 17. 5 ml 2% 메틸그린 수용액 10.0 ml 츠。 르,,소-T 250. 0 ml 용액 B : 0 . 2M 산 초산 완충액 (pH 4.8) 250 ml 사용 직전에 A 와 B 를 같은 양을 섞는다. 〈결과〉 DNA 는 녹색으로, RNA 는 적색으로 염색된다.
39.4 생체염색 세포를 고정하거나 죽이지 않고 색소로 염색하는 것을 생체염 색 vit al sta i n i n g 이 라 한다. 트리 판 블루 try p a n blue, 리 튬 카민 lithi u m carmi ne 등의 산성 색 소, 뉴트럴 레 드 neutr a l red, 메 틸 렌 블루 meth y le ne blue 등의 염 기 성 색 소, 빅 토리 아 블루 vic tor ia blue, 아이 소졸엑 트 바이 올렛 iso solecht vio l et 등의 중성 색 소를 사용할 수 있다. 원액으로서 물에 녹인 것을 쓰고 이를 PBS 에 희석해서 사용한다. 39.5 보디안 은염색법 〈 준비 〉 1) 고정 액 -불화암모늄 ammoniu m bromi de 10 g을 증류수 500 ml 에 녹이고 여기에 폴마린액 70ml 를 첨가한다. 2) 프로타르골 p ro t ar g ol- 윈트롭 wi nt h r op , 크로마 Chroma 사에 서 팔고 있는 은화합물이다. 사용 직전에 프로타르골 0.07g 을 10ml 에 녹인다. 3) 환원 액 -히 드로퀴 논 hy d roq u in o ne 1 g을 증류수 100 ml 에 녹 이고 여기에 폴마린액 5ml 를 첨가한다. 4) 1 % 염 화금 go ld chlorid e 2 % 수산 oxali c ac id 5) 정 착액 -5 % 티오황산나트륨 sodiu m thios ulfa te 〈방법〉 1) 배양 커버슬립을 PBS 로 한 번 씻은 뒤 고정액 중에서 실
온에 서 6-24 시 간 고정 한다. 2) 고정액을 버리고 PBS 로 두 번 씻은 뒤 95% 알코올을 넣 고 실온에서 5-7 일간 재고정한다. 3) 증류수에서 3 번 5 분씩 수선한다. 4) 새로 만든 0.7 % 프로타르골액 중에서 37 도에서 24 시간 인 큐베이트한다. 5) 증류수에서 3 번 5 분씩 수세한 뒤 환원액 중에서 10 분간 처 리한다. 배양신경세포가 갈색이 된다. 6) 증류수에서 2 번 5 분씩 수세한 뒤 l % 염화은액 속에서 3 분 간 처리한다. 7) 증류수에서 2 번 5 분씩 수세한 뒤 2 % 수산액 속에서 5 분간 처리한다. 배양신경세포가 보라색으로 염색이 된다. 8) 증류수에서 수세한 뒤 정착액에서 8 분간 처리한다. 9) 증류수에서 수세한 뒤 알코올, 크실렌을 통하여 탈수, 두철 한 뒤 퍼마운트에 봉입한다. 〈해설〉 배양신경세포의 세포체, 신경돌기 중의 신경원섬유가 보라색에 서 검은색으로 염색되며 신경교세포, 섬유아세포 등 비신경세포 핵도 역시 염색된다. 이 방법은 보디안 Bod i an 의 방법을 저자의 연구실에서 신생 마우스 소뇌와 태생 마우스 척수신경세포 조직 배양을 위해서 개량한 것이다 .1,2) 그림 39-1 에서 계배 척수전각 운동신경세포 배양에서 보이는 신경세포의 은염색을 보이고 있 다.
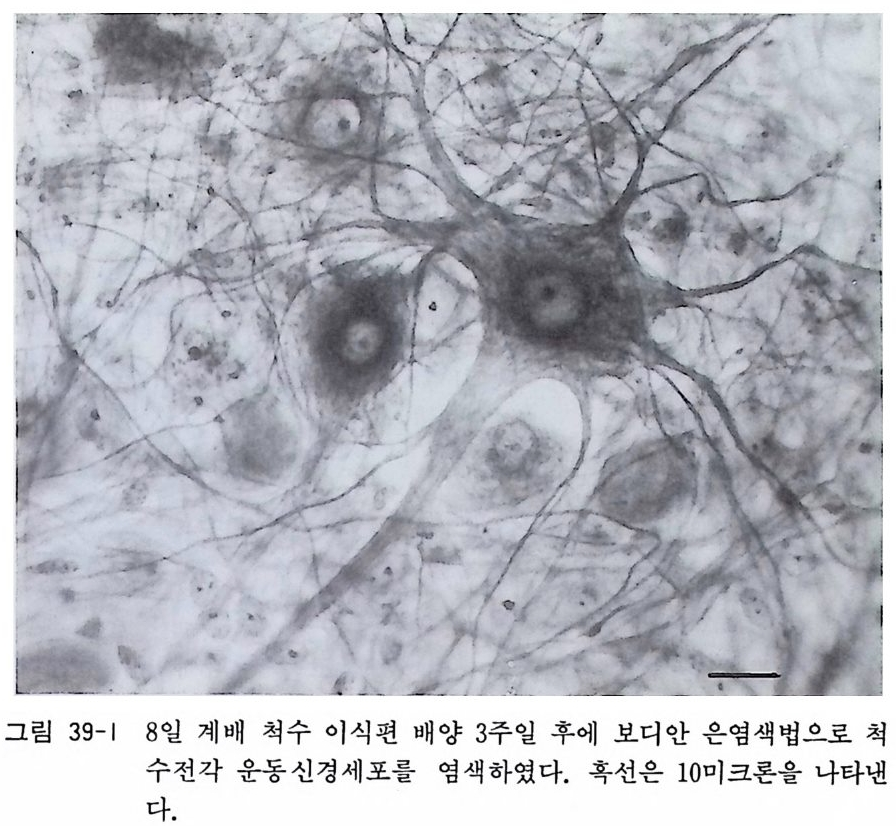 그림 39-1 8 일 계배 척수 이식편 배양 3 주일 후에 보디안 은염색법으로 척
그림 39-1 8 일 계배 척수 이식편 배양 3 주일 후에 보디안 은염색법으로 척
39.6 수단블랙 미엘린 염색법 〈 준비 〉 1) 수단블랙 sudan black B I . 5 g을 70 % 에 틸 알코올 100 ml 에 넣고 5 일간 스터러에서 혼합시킨다. 사용 직전에 쓸 만큼 필 터여과한다. 6 개월간 실온에서 안정하다. 2) 0.2 % 에리트로신 eryt hros in e 수용액 3) I % 오스뮴 osmi um tet r o xid e 수용액
〈 방법 〉 1) 배양 커버슬립을 PBS 로 한 번 씻고 10 % 폴마린액에서 2 시간 고정한다. 2) 증류수로 3 번 씻는다. 3) 70 % 알코올에 2 분간 담근다. 4) 1.5 % 수단블랙액 중에서 실온에서 30 분간 염색한다. 5) 70 % 알코올 속에서 10 초씩 담근 다음 현미경 아래에서 검 사한다. 미엘린 염색이 보이면 알코올에 의한 분별을 중지한 다. 6) 증류수로 1 번 씻는다. 7) 0.2 % 에리트로신액 중에서 15 분간 염색한다. 8) 증류수로 1 번 씻는다. 9) 1 % 오스뮴 수용액 중에서 20 분간 고정한다. 10) 증류수로 씻은 다음 폴리비닐알코올-글리세린(젤바톨)에 포매한다. 〈해설〉 태생이나 신생 래트(마우스) 중추신경조직이나 후근신경절 이 식편 배양을 2 주일 이상 계속하면 미엘린(수초) 형성이 있게 되 는데 이들을 현미경 아래에서 관찰하려면 미엘린 염색이 반드시 필요하다. 그림 39-2 에서는 마우스 척수후근신경절 이식편 배양 3 주만에 보인 수초 염색의 실제를 보인다.
 v --:.~.. . 쓰 ·i `
v --:.~.. . 쓰 ·i `
39.7 닛슬 신경세포 염색법 〈 준비 〉 1) 1 % 톨루이 딘 불루 tol uid i n e blue 혹은 티 오닌 thion in e 수용 액 2) 0.2 M 초산완충액 (pH 4.3 ) 3) 디 옥산 d i oxane 과 에 틸 알코올
〈 방법 〉 1) 배양 커버슬립을 10 % 폴마린액에서 1 시간 고정한다. 2) 증류수에서 3 번 5 분씩 수세한다. 3) 0.05 % 톨루이딘블루액 (초산완충액 9.5ml 에 1 % 톨 루이딘 블루 0.5ml 를 첨가한다)속에서 37 도에서 2 시간 동안 인큐베 이트한다. 4) 증류수에서 3 번 수세한다 . 5) 다음의 액 중에서 5 분씩 분별과 탈수를 한다. 100 % 알코올 100 % 알코올 : 디옥산 (1 : 1) 디옥산 I 디옥산 Il 6) 크실렌에서 두석을 하고 퍼마운트에 봉입한다.
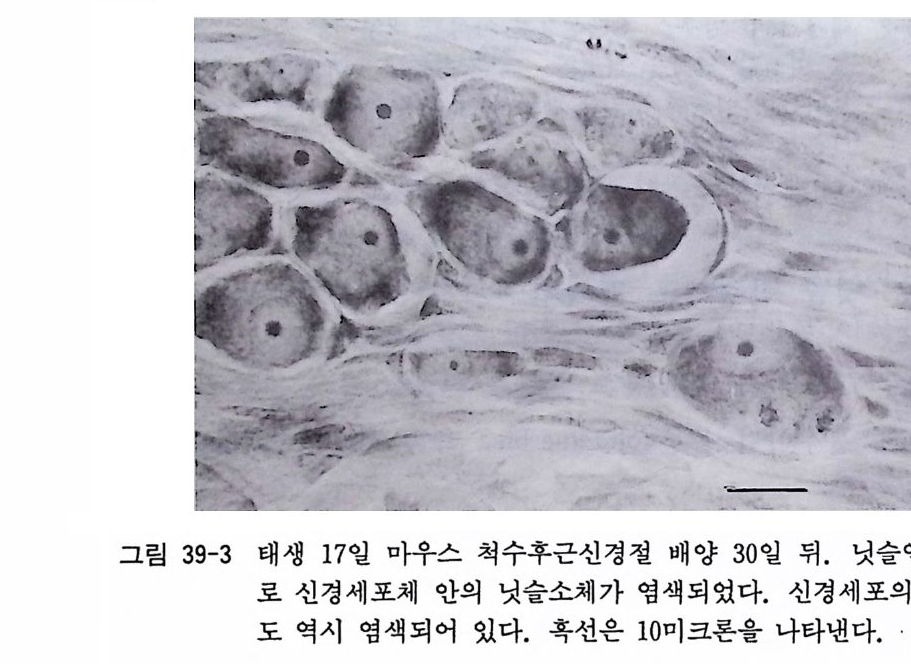 그림 39-3 태생 17 일 마우스 척수후근신경절 배양 30 일 뒤. 닛술염색법으
그림 39-3 태생 17 일 마우스 척수후근신경절 배양 30 일 뒤. 닛술염색법으
〈 해설 〉 그림 39 - 3 에서 보듯 배양신경세포의 닛슬 소체가 염색되어 있 다. 이것은 태생 마우스 척수후근신경절 신경세포배양 한달 후의 것이다.
제 40 장 세포화학적 염색법 세포의 구조와 기능을 화학적으로 검색하고 그 생물학적인 의 의를 밝히는 것이 세포화학의 목적이며, 여러 종류의 염색법을 사용하여 화학물질, 효소의 소재를 증명하는 것이 세포화학이다. 1970 년대 후반에 들어서서 신경 세포 타입 특이 마카에 의해서 면역염색법으로 신경조직의 각 세포 타입이 확정되기까지는 세포 화학적 염색법이 그 유일한 방법이었다 .1,2) 조직배양에서 존재하는 세포 타입을 확인하는 것은 그 예로서 콜린성 신경세포를 확인함에는 아세틸콜린에스테라제 효소염색 그리고 카데콜아민 신경세포를 증명함에는 포름알데히드에 의한 형광발색법이 있다. 최근에 들어와서 면역조직화학의 발달로 세 포 화학 염색법의 유용성이 크게 줄어들었으나 아직도 여러 연 구자에게 사용되고 있는 현상이다. 흥분성 아미노산에 의한 신 경독성, 저산소중에서의 배양모델에서 NADH- 테트라졸륨 환원 효소 양성의 신경세포가 선택적으로 생존한다는 보고에서 보듯이 각종 효소 염색법의 리바이벌이 있는 것이 현상이다.
40.1 아세틸콜린 에스테라제 염색법 아세틸콜린 에스데라제 ace ty lcho li ne es t erase 는 신경전달물질인 아세틸콜린을 분해하는 효소로서 아세틸콜린을 전달물질로 하는 뇌영역에서는 그 효소 활성이 높다. 여기에서는 카노프스키 Karnovsky 와 루츠 Roots 의 방법 을 소개 한다. 3) 최근에 는 이 효소 염색법의 감도를 높이기 위하여 타고 Ta g o 의 변법이 사용되기도 한다 .4) 〈 준비 〉 1) 반응액 요드아세틸콜린 5mg lOO mM 마레인산 완충액 (pH 6.0 ) 6.5 ml lOO mM 구연산나트륨액 0 . 5 ml 30 mM 유산동액 1 ml 5mM 페리시안칼륨액 1ml O. lm M 콜린에스데라제 저해제 1ml 2) 콜린에스테라제 저해제 : 이 소음파 (iso -OMPA, tet r a is o p ro p yl pyro p h osph orami de , 시 그 마)는 특이한 콜린에스데라제 저해제로써 아세틸콜린에스데 라제 활성에는 영향이 없다. 〈방법 〉 1) 배양 커버슬립을 10 % 폴마린액 중에서 4 도에서 10 분간 고 정한다. 2) 증류수에서 세 번 수세한다. 3) 효소반응액 중에서 실온에서 1-2 시간 반응시킨다.
4) 현미경 아래에서 반응을 체크하고 신경세포 염색이 충분하 면 증류수에서 한 번 수세한다. 5) 알코올에서 탈수하고 크실렌에서 투석한 뒤 퍼마운트 속에 봉입한다. 〈 해설 〉 그림 40-1 에서는 태생 마우스 척수 분리세포 배양에서 보이는 척수전각 운동신경세포가 아세틸콜린 에스데라제 염색에 강하게 양성이다.
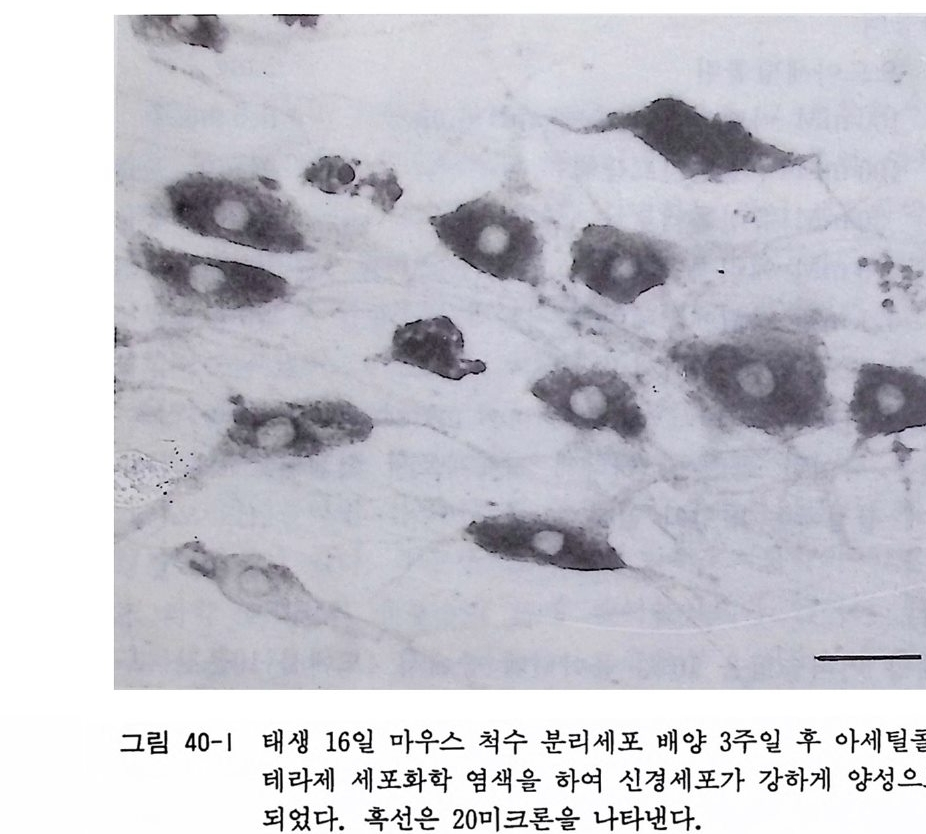 그림 40-1 태생 16 일 마우스 척수 분리세포 배양 3 주일 후 아세틸콜린에스
그림 40-1 태생 16 일 마우스 척수 분리세포 배양 3 주일 후 아세틸콜린에스
40.2 카데콜아민 신경 형광염색법 1960 년대에 카테콜아민 형광범이 소개되었을 적에는 글리옥실 산 대신에 파라포름알데히드를 사용하였고 습도 조절이 엄격하지 아니하면 형광을 볼 수가 없었으며 형광현미경이 너무 고가여서 엄두도 내지 못하던 고등기술이었다. 여기에서는 글리옥실산에 의한 카테콜아민 형광염색법을 소개한다 .5) 〈 준비 〉 1) 글리옥실산(g l y ox yli c aci d, 시그마) 2 g을 PBS 100 ml 에 녹 인다. 2) 드라이어 〈 방법 〉 1) 배양 커버슬립을 PBS 에서 2 분간 씻는다. 2) 2 % 굴리옥실산액 중 4 도에서 5-10 분간 인큐베이트한다. 3) 드라이어를 사용하여 커버슬립을 건조시킨다. 4) 오븐 속에서 90 도에서 5 분간 처리한다. 5) 폴리비닐알코올-글리세린(겔바톨)에 포매하고 형광현미경 아래서 관찰한다. 〈 해설 〉 그림 40-2 에서는 신생 래트 상경부 교감신경절의 분리세포 배 양 2 주일에 관찰된 카테콜아민 양성 신경세포를 보이고 있다.
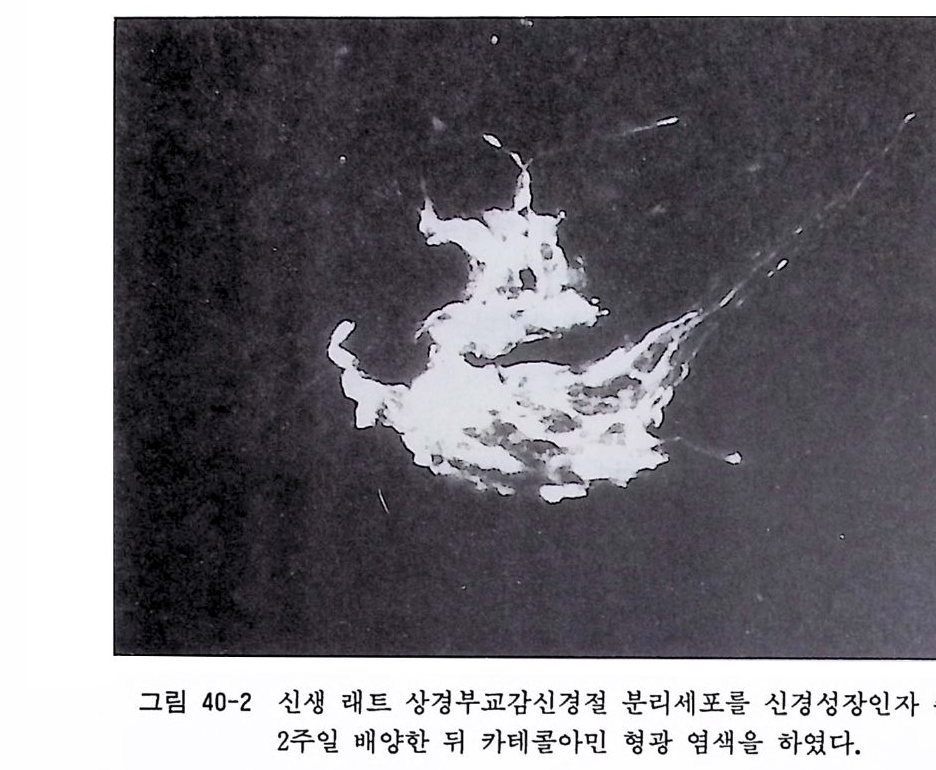 그림 40-2 신생 래트 상경부교감신경절 분리세포를 신경성장인자 존재하에
그림 40-2 신생 래트 상경부교감신경절 분리세포를 신경성장인자 존재하에
40.3 NADPH 데트라졸륨 환원효소 염색법 NADPH 테 트라졸륨 환원효소 (NADPH tet r a zoli um reducta s e) 는 세포의 호흡 체인의 초기 스탭에서 NADPH 를 시토크롬 C 로 환원하는 세포 생존에 중요한 효소이며 신경세포에서는 그 활성 이 높은 것으로 알려져 있다. 최근에는 이 효소가 산화질소 생성 효소 nit ric oxid e syn the ta s e 와 공존하고 있음이 알려 져 서 NOS 의 효소 분포의 연구에도 쓰이고 있다.
〈 준비 〉 1) 반응액 : NADPH (nic o ti na mi de adenin e din u cleoti de ph osph ate reduced for m, 시 그마) 1. 5 mg TNBT (tet r a nit ro blue tet r a zoli um , 시그마) 1.5 mg O.l M 인산완충액 (pH 7.4) 1 ml 증류수 2ml 2% 나트륨아자이드 0.1 ml 2) 고정액 : 1% 염화나트륨을 포함한 10% 중성 폴마린액 〈 방법 〉 1) 신선한 배양 커버슬립을 반응액 중에서 37 도에서 1 시간 반 응시킨다. 2) PBS 에서 두 번 씻는다. 3) 고정액 속에서 10 분간 실온에서 고정한다. 4) 15 % 알코올에서 2 분간 씻은 뒤 PBS 에서 두 번 씻고 폴리 비닐알코올-글리세린(겔바톨)에 포매한다. 〈 해설 〉 신경계 조직 배양세포 중에서도 신경세포에 NADPH 데트라졸 륨 환원 효소의 반웅이 강하고 신경세포 세포질 속에서 보랏빛 과립상의 효소반응을 볼 수가 있다.
40.4 모노아민 산화효소 염 색 법 모노아민 산화효소 (MAO) 는 미토콘드리아에 존재하고 아드레 날린, 트립타민 , 세로토닌 등의 카테콜아민을 산화하는 카데콜아 민 신경전달물질 관련 효소의 하나이다. 〈 준비 〉 1) 반웅액 : 트립 타민 영 산염 (try p tam i ne , 시 그마) 1mg 황산나트륨 0.8mg TNBT (tet r a nit ro blue tet r a zoliu r n, 시 그마) 1 mg O.l M 인산완충액 (pH 7.6) 1 ml 증류수 3ml 2) 고정액 : 1 % 염화나트륨을 포함한 10 % 중성 폴마린액 〈 방법 〉 1) 신선 배양 커버슬립을 반응액 중에서 37 도에서 1 시간 반응 시킨다. 2) PBS 에서 두 번 씻는다. 3) 고정액 속에서 10 분간 실온에서 고정한다• 4) 30 % 알코올에서 2 분간 씻은 뒤 PBS 에서 두 번 씻고 폴리 비닐알코올 - 글리세린에 포매한다. 〈해설〉 신경계 조직 배양세포 중에서 신경세포, 아스트로사이트 , 올리 고덴드로사이트에 모두 모노아민 산화효소 활성을 볼 수 있으나 섬유아세포에서는 활성이 극히 낮다.
제 41 장 전자현미경 기술 전자현미경 기술 elec t ron m i crosco py은 1960 년 이래 의학과 생 물학의 분야가 획기적인 전보를 가져온 이유 중의 하나가 되거니 와, 그 이전에는 생각지도 못하던 세포의 미세구조가 세밀하게 하나씩 가시화되었다. 전자현미경 아래에서 명확하게 보이는 각 세포 소기관이 담당하고 있는 생리기능이 무엇인가 하는 의문에 서 시작되어 생화학, 생리학, 분자생물학의 각 분야가 역시 활성 화되어 오늘날의 의생물학의 폭발적인 지식 축적의 기반이 되었 다. 전자현미경의 분야에서는 투과전자현미경과 주사전자현미경을 중심으로 하여 이에 세포화학, 면역화학, 동결절편 제작법, 원소 분석법 등의 기술을 병용함으로서 새로운 연구 분야가 크게 전개 되고 있다. 조직배양세포를 전자현미경하에서 관찰하는 것도 이 러한 새로운 전개의 하나가 된다. l) 신경계 조직 배양세포를 위상차현미경 아래에서 관찰하고 사전 촬영을 하여 세포 형태, 증식 상태 등을 기록으로 남길 수 있으
나, 이들 신경 계세포의 기능과 분화를 파악하려면 광학현미경 관찰만으로는 충분치 않고 생화학적 분석, 전기생리학적 분석, 그리고 전자현미경에 의한 세포의 미세구조 관찰이 중요한 방법 이 된다 .2) 41 .1 두과전자현미경 두과전자현미 경 tra nsmi ss io n electr o n mi cro scop y 의 기 본구조는 광학현미경과 비슷한데 광원으로서 열전자 elec t ron 를 사용하며 이것이 자계형 렌즈(전자코일을 튜브형으로 제작)을 지나면서 그 궤도가 바뀐다. 이 전자가 20-100 nm 의 두께가 있는 에폭시 시 료를 투과하여 또 한번 자계 형 렌즈를 통과하면서 영 상을 만들게 된다(그림 41-1). 두과전자현미경에서 관찰된 배양신경세포의 실 례를 그림 41_2 에서 볼 수가 있다. 다음에 적은 방법에서는 아클라 풀라스틱 (Aclar) 커버슬립이 에폰과 중합하지 않는 성질을 이용하여 세포와 커버슬립 경계면 이 깨끗하게 분리되는 경우를 설명하였다. 배양세포를 풀라스틱 페트리 접시나 플라스크에 배양한 경우에는 접시나 플라스크를 원상태로 고정, 탈수를 하고 프로필렌 옥사이드 대신에 100 % 알코올에서 알코올-에폰 혼합액에서 처리하고 그 다음에 에폰액 으로 포매하도록 한다. 풀라스틱과 에폰수지를 분리하려면 자기 가 원하는 장소 주위를 찰라내고 액체질소 속에 3 분 가량 넣었다 가 공기 중에 꺼내면 간단하게 풀라스틱이 벗겨진다. 유리 슬라 이드 위에서 배양한 경우는 에폰 포매 뒤에 유리 슬라이드를 핫 플레이트 ho t pla te 위에서 150 도의 열에서 1 분간 가열하고 유리 와 에폭시수지의 열팽창률을 이용하여 순간적으로 분리한다.
 -..―.‘: 尸,- .`、, .. ? .、.、,'. ’' . L. . . J51.- '> .·- .. m 언 7 ..2.~: · . • `
-..―.‘: 尸,- .`、, .. ? .、.、,'. ’' . L. . . J51.- '> .·- .. m 언 7 ..2.~: · . • `
〈 재료 〉 1) 신경계조직 배양세포(아클라 풀라스틱 커버슬립 위에서 배양) 2) 25 % 글루타르알데히드 3) 0.15 M 카코딜레이트 완충액 cacody la te buff er , pH 7.3 4) 2 % 오스뮴 osmi um tet r o xid e 5) 알코올 6) 프로필렌옥사이드 〈방법〉 1) 신경계조직 배양세포(아클라플라스틱 커버슬립 위에서 배양) 를 PBS 에서 잠깐 씻는다.
2) 2.5 % 글루타르알데히드 (0.15M 카코딜레이트 완충액에 희석 한다)에서 30 분간 실온에서 고정한다. 3) 커버슬립을 PBS 에서 1 분간 씻는다. 4) 2 % 오스뮴 수용액에서 30 분간 실온에서 고정한다. 값비싼 오스뮴울 절약하기 위해 커버글라스 위에 오스뮴을 소량 산 같이 덮는다. 5) 다시 PBS 에서 1 분간 두 번 씻는다. 6) 70 %, 80 %, 90 %, 95 %, 100 % 알코올로 차례로 탈수한 다. 실온에서 각각 5 분씩 시행한다. 7) 알코올, 프로필렌옥사이드(1 : 1) 에 10 분간 정치한다. 8) 프로필렌옥사이드에 10 분간 정치 9) 프로필렌옥사이드-에폰 (1 : 1) 속에서 두 시간 정치하는데, 이 과정까지는 실온에서 한다. 10) 적당한 풀라스틱 용기 바닥에 아클라 커버슬립을 놓고(배 양세포를 위로 향한다) 그 위에 에폰액을 봇는다. 에폰액 처 방은 다음과 같다. A액 : Ep o n 812 62ml DDSA 100ml B 액 : Ep o n 812 100 ml MNA 89ml A 액과 B 액을 4 : 6 의 비율로 혼합한 뒤 DMP-30 을 전체 양의 1. 5% 가 되도록 첨가하고 잘 섞이도록 나무 막대로 혼합한다. 11) 오븐 (59 도)에서 2-3 일간 경화시킨다. 12) 에폰 블록을 현미경 마이크로톰으로 미세 절편을 만들고 우라닐아세데이트 uran y l ace t a t e 와 구연산연 lead cit ra t e 액으 로 전자염색을 한 다음 현미경 아래에서 관찰한다.
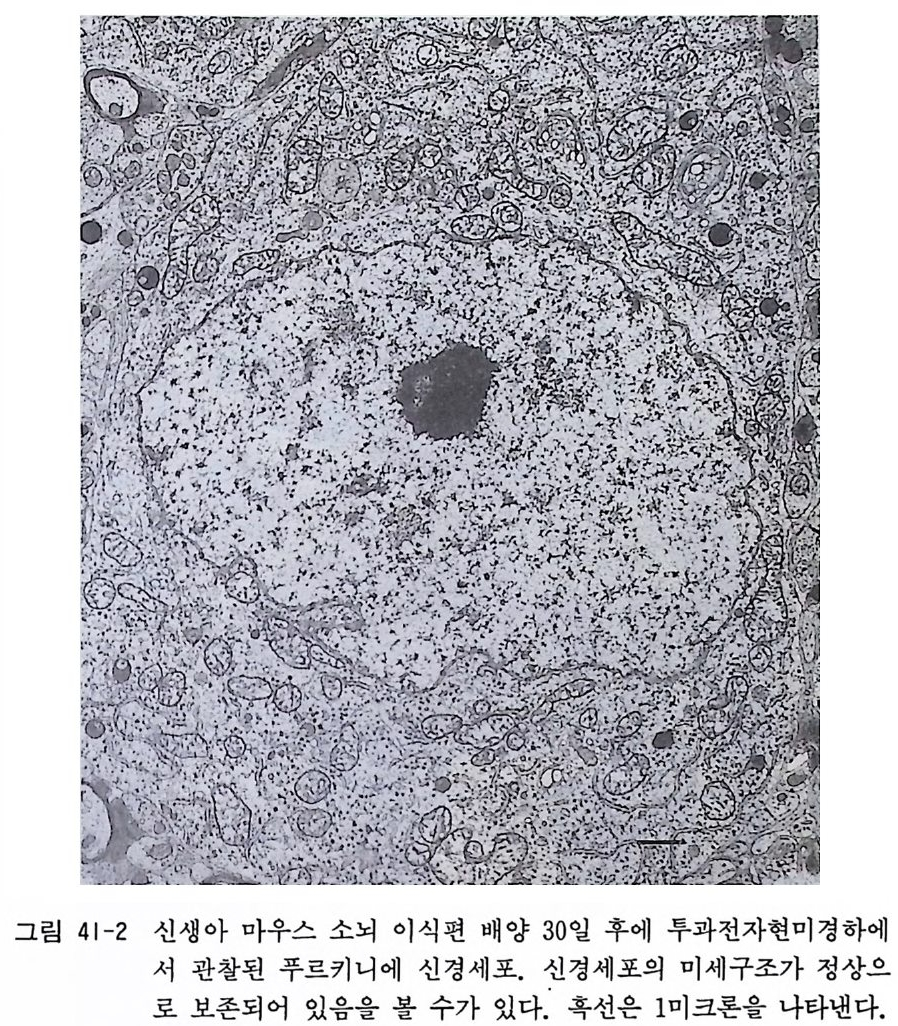 그림 41-2 신생아 마우스 소뇌 이식편 배양 30 일 후에 두과전자현미경하에
그림 41-2 신생아 마우스 소뇌 이식편 배양 30 일 후에 두과전자현미경하에
〈 해설 〉 그립 41-2 에서는 신생아 마우스 소뇌 엑스플란트 배양 4 주일에 전자현미경 관찰을 하였다. 푸르키니에 신경세포, 시냅스, 미엘 린을 관찰할 수가 있다.
41 .2 주사전자현미경 물질의 표면에 전자 electr o n 롤 쪼이 면 전자는 그 물질의 구성 원자와의 상호작용으로 산란하게 되어 점차로 그 에너지를 상실 하고 마지막에 가서는 그 물질에 흡수되어 버린다. 이 과정에서 전자 혹은 전자파가 물질 표면에서 전공 중으로 방사된다. 이렇 게 의부로 방사되는 전자와 전자파가 보이는 반응을 종합하여 영 상을 만들고 재료 형태를 관찰하거나 원소 분석을 하는 장치가 주사전자현미경 scannin g electr o n mi cr oscop y 이다(그립 41- 3) . 따 라서 주사전자현미경은 세포 표면의 형상과 변화를 입체적으로
 凌거. 걸*-?• 궁.' ·국 . 정;- 효.· _ :::, 그 1 . 』 '구。 ·' ~·-: •.
凌거. 걸*-?• 궁.' ·국 . 정;- 효.· _ :::, 그 1 . 』 '구。 ·' ~·-: •.
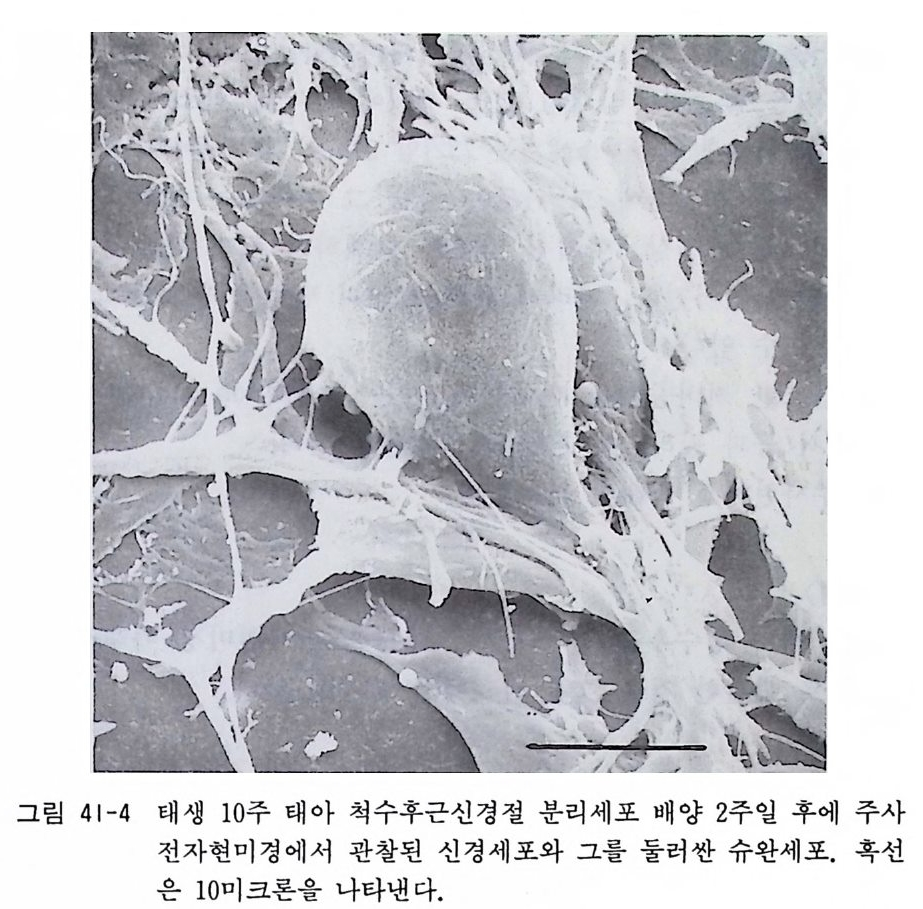 、묘 ` 조....' \ `I''/~`’
、묘 ` 조....' \ `I''/~`’
관찰하기에 적합한 기술이다. 〈 재료 〉 4 1. 1 과 같음 〈 방법 〉 1) 투과전자현미경에서와 같은 방법으로 고정, 탈수를 한다. 2) 초산 아밀에서 15 분간 2 번 실온에서 치환한다.
3) 건조용 네트 박스 내부에 필터페이퍼를 깔고 그 위에 커버 슬립을 놓는다. 임계점 건조 장치에 넣어서 건조시킨다. 임 계점 (예컨대 탄산가스에서는 73 기압 31 도가 임계점이다)을 경계 로 하여 순식간에 그 위로는 기체 그 아래로는 액체로 변환 하는 데 기체상, 액체상 양쪽이 존재하지 않기 때문에 표면 장력이 없어진다. 따라서 세포 표면의 변형이 최소한으로 방 지될 수 있다. 4) 전조한 커바슬립을 꺼내어서 이온 스퍼터 ion spu tt er 에 넣 어서 금과 팔라듐의 합금으로 코팅을 한다. 5) 주사현미경 아래서 관찰한다. 〈해설 〉 사람 태아 척수후근 신경세포의 배양을 주사현미경 아래서 입 체적으로 관찰한 것을 그립 41-4 에서 보이고 있다.
제 42 장 신경조직배양의 전보와 그 전망 신경조직배양은 동물 실험에 바하여 다음과 같은 장점이 있다. 첫째, 다수의 실험계를 단시간 안에 작성할 수 있어서 유지 비용 이 적게 들고 대단히 경제적이다. 둘째, 영양인자, 이온, 온도, 산소농도 등 화학적 혹은 물리적 환경의 조절이 쉽다. 셋째, 영 양인자, 호르몬, 대사전구물질, 독성물질 등이 뇌-혈관 관문 blood-braiu - barrie r 에 의해서 선택적으로 봉쇄되는 in v i vo 와 달 리 신속하게 뇌실질에 운반이 된다. 넷째, in v i vo 와 달리 다른 뇌신경계, 내분비계, 그리고 혈관계에 의한 통제와 수식을 받지 않는다. 다섯째, 살아 있는 신경세포를 현미경으로 직접 관찰하 고 그 형태 변화와 미묘한 운동을 계속 기록할 수 있다. 여섯째, 살아 있는 신경세포에 직접 미소전극을 삽입하고 레이저 광선의 조사, 유전자와 항체의 미량 주사 등 극히 델리케이트한 실험이 가능하다. 일곱째, 배양액을 회수하여 신경세포가 생산 분비하는 활성물질을 채집하고 분석할 수 있다. 여덟째, 실험이 끝난 뒤 단시간 안에 고정, 염색을 할 수가 있어서 광학현미경은 물론 전
자현미경 관찰에서 이상적인 표본을 얻을 수 있다. 이상과 같은 신경조직배양의 장점을 최대한으로 이용하여 1960 년 이후에 신경 질환의 조직배양 모델이 수없이 작성되었는데 이 들을 다음에서 소개 한다. 42.1 탈수질환 사람의 중추신경 의 탈수질환 demy e li na ti ng dis e ase 의 대 표적 인 것 에 다발성 경 화증 multip le sclerosis 이 있는데 이 것 은 자기 면 역 에 의한 신경질환 au t o i mmune d i sease 이라 생각되고 있다. 이 학 설은 신경조직배양에서 많은 지지롤 받고 있다. 1961 년 본스틴 Bornste i n 과 아펠 Ap pe l 이 래 트 소뇌 배 양에 EAE (exp e rim enta l allergi c enceph alomy e li tis) 동물의 혈 청 을 첨 가한바 급성 의 탈수 현상 dem y e li na ti on 을 관찰한 이후로 신경조직배양에 의한 다발성 경화증의 연구가 많이 발표되었다 .l) EAE 에서의 탈수기전으로서 첫째, 면역글로불린에서만 탈수 현상을 볼 수 있다. 둘째, 보체 comp lim ent 의 존성 이 다. 셋 째 , 뇌 백 질 whit e matt er 에 의 해 서 탈 수 활성이 흡수된다. 이 3 가지 원인으로 EAE 혈청 중에 존재하 는 항미엘린 항체가 그 원인이라 알려져 있다• 다발성 경화증 환 자의 혈청, 뇌척수액, 뇌조직 추출액을 신경조직배양에 첨가하여 탈수현상을 관찰할 수 있다 .2,3) 이들의 실험은 액성 면역 humoral i mm unity에 의하여 탈수가 일어남을 증명하고 있는데, 이와 달 리 세포성 면 역 cellular im mun ity 울 통하여 서 임 파구나 매 크로파 지 macro p ha g e 에 의한 탈수 기구가 중요하다는 것이 알려져 있 다. 이 분야에서는 올리고덴드로사이트를 순수배양하고 이를 표 적세포로 하여 임파구에 의해서 일어나는 세포성 면역의 연구가
성행하고 있다 .4 , 5) 최근에는 사이토카인 c yt ok i ne 에 의해서 MHC 항원의 표현이 변화하고 있다는 보고가 있다. 6) 말초신경계에서는 래트 척수후근신경절 배양에 EAN(exp e ri- menta l allergi c neur iti s) 동물의 혈청 혹은 기 앙-바레 Guil lan -B arre 신경영 환자의 혈청을 첨가하여 탈수를 관찰하고 이들 질 환이 자기면역에 의한 것이라는 학설이 유력하다》 이같이 신경 조직배양은 미엘린 형성의 프로세스 그리고 탈수 질환의 발병기 전 해명에 적합한 실험 모델이 되고 있다. 최근에는 울리고덴드로사이트와 그 전구세포 pre cursor 의 존재 그리 고 이 들 세 포가 신경 영 양인 자 neurotr o p h ic fac to r 에 의 하여 증식하는가 등의 문제가 중요한 연구과제로서 신경조직배양에서 다루어지고 있다 .8 , 9) 42.2 신경변성질환 • 신 경 변 성 질 환 neurodeg e nerati ve dis e ase 의 주된 것으로서 알츠 하이 머 Alzheim er 병 , 파킨슨 Parkin s on 병 , 헌팅 턴 Hunti ng ton 병 그리 고 근위 축성 축색 경 화증 (amy o tr o p h ic late r al sclerosis , ALS) 이 있다• 신경변성질환의 발병 기전에 대하여서는 수많은 학설이 나와 있는데 다음과 같은 것이 최근 주목을 받고 있다. 첫째, 홍 분성 아미 노산에 의 한 신경 독성 exc itot o x ic ami no aci ds , 둘째 , 산소 라디칼에 의한 신경독성 oxy ge n radic a ls, 셋째, 신경영양인 자의 결 핍 defi cien cy in neurotr o p h ic fac to r s 그리 고 넷 째 , 마 크로 글리 아의 활성 화 acti va ti on of mi cr og lia 등이 다. 이 상과 같은 여 러가지 신경세포에 대한 독성이 단독으로 혹은 협동적으로 작용 하여, 신경세포의 변성 나아가서는 사멸을 가져오게 된다. 따라
서 신경조칙배양에서는 위에 열거한 여러 가지 작용인자에 의한 신경독성을 검색하고 이룰 예방하는 방책을 찾아내는 데 위력을 발휘하게 된다. 알츠하이머병 환자의 뇌는 신경세포 수의 감소, 노인반 sen il e pla qu e , 그리고 신경원섬유변화 neurofi br ill ar y tan g le , NFT 의 출 현이 가장 특칭적이다. 1960 년에 시작되어서 최근까지 NFT 를 실험적으로 배양신경세포에서 만드는 실험이 성행하였는데 콜키 틴 계통의 유사분열 저해제 spi nd le i nh i b it or 나 알루미늄염을 배양 액에 첨가하고 신경세포 체내에 NFT 를 관찰하는 연구가 다수 보고되었다 .10-12) 그러나 이들 NFT 는 알츠하이머병에서 보는 이 상 필라멘트(p a i red heli ca l filam ents , PHF) 의 구조를 갖지 않고 직선형의 구조이기 때문에 최근에는 주목을 받지 못하고 있다. 알츠하이머병에서는 마이넬트 기저핵의 콜린성 신경세포의 변성 이 있어서 이것이 알츠하이머병 발병의 원인이 되고 있다고 생각 되는데 이러한 콜린성 신경세포의 생존과 기능 증가를 위하여 신 경성장인자 (NGF) 를 알츠하이머병 환자에게 투여하려는 시도가 있다 .13) 이 배경에는 래트 전뇌 콜린성 신경세포 배양에서 NGF 가 신경세포의 생존과 아세틸콜린 합성효소의 활성을 증가시킨다 는 보고가 있기 때문이다 .14,15) 알츠하이머병 환자의 뇌에서 신경 세포 수가 감소하는데 이 것을 신경 영 양인자 neurotr o p h ic fac to r 의 결핍에 원인이 있다고 하는 학설이 있다. 16) 그러나 이와 반대로 래트 대뇌 신경세포 배양에서 알츠하이머 뇌 추출물이 정상 뇌 추출물보다 더 높은 신경영양작용을 보였다는 보고가 있다 .17) 베 타 아밀로이드의 일부를 구성하는 28 잔기 풀리펩티드를 래트 해 마 신경세포 배양에 첨가하면 이들 신경세포의 생존이 월동 높아 졌다는 보고도 있다. 18) 그러나 이와 반대로 아밀로이드 전구물질 (APP) 유전자를 전입시킨 PC12 세포에서는 세포내에서 합성된
펩티드 때문에 세포가 죽었으며, 이들 세포의 조건 배양액 condit ion ed med i um 을 첨가하면 래트 해마 신경세포가 변성한다 는 보고가 있다 .19) 이같이 배양신경세포를 실험모델로 하여 알츠 하이머병의 병인을 검색하는 연구가 계속될 것이며 특히 아밀로 이드 단백질 속에 존재하는 신경영양인자 또는 신경독성물질의 탐색이 앞으로 계속될 것이다. 파킨슨병은 혹질의 도파민성 신경세포가 탈락하고 혹질-선조체 로가 변성하는 신경질환이다. MPTP 혹은 6-hy d roxy d op a mi ne 을 혹질에 주사하여 파킨슨병의 동물 모델을 작성한 뒤에 이들 동물(래트와 원숭이)의 선조체 부위에 조직 배양한 태생 동물의 혹질, 교감신경절세포, 부신 크로마핀 세포를 이식하여 임상 증 상이 개선되었다는 보고가 있다 .20,21) 이러한 동물 실험에서의 성 공에 기초를 두고, 1986 년에서 1990 년 사이에 200 케이스가 넘는 파킨슨 환자의 부신수질 조직 자가이식이 있었다. 초기에는 L - 도파 투여의 저하 등의 증세 개선으로 효력이 있다고 하였으나 지금에 와서는 수술 그 자체가 무효라는 결론이 나와 있다. 최근 에 와서는 신선하게 얻어전 혹은 배양을 한 사람 태아 (8-10 주) 중뇌 도파민 신경세포를 파킨슨병 환자의 선조체에 이식한다는 새로운 치료법이 시도되고 있다. 유럽과 미국에서만 100 케이스가 넘는 태아 신경세포 이식이 파킨슨병 환자에게 시행되어 있다 .22) 파킨슨병 환자의 뇌에서 혹질의 도파민성 신경세포가 선택적으 로 변성하는데 대하여 여러 가지 학설이 나와 있는데 그중의 하 나는 흥분성 아미노산이 글루타민산에 의한 신경독성으로 혹질세 포가 공격을 받는다는 것이다. 최근 저자의 연구실에서 태생 마 우스 혹질 조직배양에 0.2 µM 농도의 글루타민산울 10 분간 노출 하고 24 시간 뒤에 검사해 본 결과 50 % 이상의 도파민성 신경세 포가 죽었음을 알 수가 있었다. 이러한 글루타민산의 신경독성은
MK 801 을 포함한 NMDA 수용체 길항제를 투여한 배양에서는 완전히 방지되어 있었다 .23) 이갇이 흥분성 아미노산과 신경변성질환의 관련에 대하여서 조 직배양을 사용하여 연구하는 성과가 많이 나오게 되었다. 글루타 민산과 이에 유사한 아미노산은 흥분성 신경전달물질로서 정상 신경생리기능을 함과 동시에 과량에서는 각종 신경세포 를 변성시 키고 파괴하는 작용을 가지고 있는 것이다 .24,25) 이같이 흥분성 아미노산에 의해서 유발된 질환으로서는 뇌출혈, 경련, 뇌허혈, 간질 등의 급성 질환이 있으며 만성질환으로서는 파킨슨병, 헌팅 턴병, 알츠하이머병, ALS 가 이것에 해당된다고 생각된다. 구암 ALS- 파킨슨-치 매 (ALS-PD) 증후군은 ALS, 파킨슨병 그리 고 치 매 세 가지 병의 증상을 모두 가지고 있기 때문에 이러한 병명을 갖게 되는데 최근 소철나무 열매에서 나오는 흥분성 아미노산 BMAA ({J- N-meth y la mi no alanin ) 이 배 양 래 트 척 수신경 세 포를 파 괴하는 사실이 보고되어서 26) 흥분성 아미노산이 신경변성질환의 발생에 크게 관여하는 것이 아닌가 하는 가정 아래 여러 사람이 이러한 연구를 계속하고 있다. 흥분성 아미노산에 의한 배양 신 경세포의 변성과 탈락 그리고 각종 길항체에 의한 예방은 최근의 리뷰에 상세히 기록되어 있다 .25) 근위축성 측색경화증 (ALS) 환자의 혈청을 배양액 중에 첨가하 면 배양한 마우스 척수 운동신경세포가 사멸한다는 보고가 있는 대 ,27) 이에 대해서는 몇몇 연구실에 그러한 사실이 없다는 보고 가 역시 나와 있어서 ALS 환자 혈청의 신경독성에 관해 앞으로 도 연구가 계속될 것이다. ALS 환자 척수에서 운동신경세포가 선택적으로 변성하고 사 멸하는데 대해서는 그 원인이 확실치가 않다. 최근 신경변성질환 에서 산소 Oxy ge n 라디칼이 선택적으로 신경 세포의 번성을
일으킨다는 학설이 유력하게 되었다. 최근 저자의 연구실에서는 태생 마우스 척수신경세포 배양에서 활성산소의 신경독성에 대하 여 연구한 바 있다. 척수 신경세포 배양을 크산틴 산화효소 xanth in e oxid a se 와 히 폭산틴 hyp o xanth i n e 이 들어 있는 배 양액 에 서 4 시간 동안 인큐베이트하면 활성 산소가 생성되어 이에 의하 여 신경세포가 선택적으로 파괴된다. 이때에 카탈라제 cata l ase, SOD 와 같은 항산화제 an ti ox i dan t s 나 MK801 와 같은 NMDA 수 용체 길항체를 배양액에 넣어주면 활성산소의 산경독성을 완전히 차단할 수가 있었다. 28) 42.3 바이러스 감염증 신경조직배양을 사용하여 바이러스 감염의 병리학적 연구가 1970 년대부터 성행하게 되었다. 동물실험과 달라서 면역계의 통 제를 받지 않는 환경 아래 바이러스-숙주간의 상관관계를 장기간 동안 관찰할 수 있는 것이 신경조직배양의 장점이다. 항신경성 바이러스인 광견병 바이러스 ,29) 흥역 바이러스와 SSPE,3,30) 헤르 페스 바이러스 31,32) 등을 신경조직배양에 감염시켜서 각종 신경세 포와 글리아 세포의 바이러스 감수성을 광학현미경과 전자현미경 레벨에서 연구하는 것이 그 주된 것이다. 최근 주목을 받고있는 레트로바이러스 re t rov i rus 의 분야에서도 신경조직배양이 사용되 고 저자의 연구실에서는 HTLV-I 의 직접 감염을 사람 글리아세 포 배양에서 확정한 바 있다 .33> HTLV-I 감염으로 일어난 신경 병인 HAM(HTLV-I assoc iat e d my el op a th y ) 환자의 혈청이 마우 스 소뇌배양에서 탈수 반응을 일으켰다는 보고가 있다 .34) HIV 의 분야에 있어서도 배양한 사람 미크로글리아 세포에서 HIV-I 의
직접감염이 있다는 보고를 볼 수 있다 .35) 42.4 결어 신경조직배양을 사용하여 신경질환의 모델을 만들고, 이들 질 환의 발생 기 전 pa th o mechanis m 을 연구한다는 어 프로치 는 앞으로 도 계속되리라 생각된다. 여기에서는 탈수질환, 신경변성질환 그 리고 바이러스 감염증을 다루었지만 이밖에도 선천성 신경계 발 생 이 상과 관련된 마우스 신경 질병 뮤탄트 neurolog ica l muta n ts 신경조직의 신경조직배양, 중금속과 유기 용매에 의한 신경중독 증의 배양모델, 성숙 동물 신경조직의 배양에 의한 신경 노화모 델 등 신경조직 배양에서 검색의 대상이 되고 있다. 최근 분자생물학적 개념과 방법론이 신경조직 배양도 도입되어 서 참신하고 자극적인 업적이 나오기 시작하였다. 타우단백질의 안티센스 울리고뉴클레오티드를 래트 소뇌 배양에 첨가하였더니 미크로듀뷸 형성에 변화가 와서 대형 신경세포의 수지돌기 형성 에 이상이 . 있었다는 보고 ,36) 레트로 바이러스 벡터를 감염시킨 초기 신경상피세포를 조직 배양중에서 추적하여 그 발달과정을 검색한다 .37) 그리고 순수배양한 사람 태아 아스트로사이트가 NGF, BDNF, NT-3 는 생산하나 CNTF 는 생산을 못 한다는 증거를 PCR 을 써서 검색한다는 연구 38) 를 볼 때에 분자생물학적 기술의 적용에 의해서 신경조직배양의 분야에서 새로운 세계가 우리 앞에 전개되고 있음을 알 수가 있다.
참고문헌 제 1 장 1) Bernard C: Le<;ons sur les ph enomenes de la vie communs aux anim aux et aux veg e ta u x. Paris , 1878. 2) Harriso n RG: Observati on s on the livi n g develop ing nerve fibe r. Anat Rec 1, 116, 1906. 3) Harris o n RG: The grow t h of nerve fibe r as a mode of pro to p la smi c movement. J Exp Zool 9, 787, 1910. 4) Burrows MT: The cultiv a ti on of tiss ues of the chic k embry o outs i d e the body . J Amer Med Ass 55, 1379, 1910. 5) Carrel A, Burrows MT: Cultiv a ti on of adult tiss ues and orga ns outs i d e of the body. J Amer Med Ass 55, 1379, 1910. 6) Carrel A: A mt ho d for the ph y si o l og ica l stu d y of tiss ues in vit ro . J Exp Med 38, 407, 1923. 7) Carrel A: The new cy tol og y. Sc ien ce 73, 297, 1931 . 8) Levi G, Mey e r H: Reacti ve , reg res siv e and reg e nerati ve pro cesses of neurons, cultiv a te d in vit ro and inj u r ed wi th mi - cromanip u lato r . J Exp Zool 99, 141, 1945. 9) Weis s P, Hisc oe H: Exp e rim ents on the mechanis m of nerve grow th. J Exp Zool 107, 315, 1948. 10) Hog ue MJ : Human fetal brain cells in tiss ue cultu r e: Their ide nti - 11) Peftiec r as toi onn E aRn , dM muortria liyt yM. RJ :E Mxpy eZl iono l s h1e0a6t, h 8 5fo, r1 m94a7t i on in cultu r e of
av ian spi na l ga n glia. Amer J Anat 96, 319, 1995 12) Hi ld W: My el i no g e nesis in cultu r es of mammali an nervous tiss ue. Zeit Zellfo rs ch 46, 71, 1957. 13) Bornste i n MB, Murray MR : Seria l observati on s on pa tt er ns of gr ow 血 my el i n for mati on , main t e n ance and deg e nerati on in cultu r es of newborn rat and kit ten cerebellum. J Bio p h y s Bio c hem Cy tol 4, 499, 1958. 14) Lewis W: Mali gna nt cells. Harvey Lectu r e 31, 214, 1935. 15) Eagl e H: Nutr i t ion needs of mammali an cells in tiss ue cultu r e. Sc ien ce 122, 501, 1955. 16) Bunge RP: Chang ing use of tiss ue cultu r e 1950- 19 57. In: The Nervous Sy st e m , Vol. 1, Brady RO(Ed), p 31, Raven pre ss, New York, 1975. 17) Crain SM: Resti ng and acti on po te n ti al s of cultu r ed chic k embryo spi na l ga ng lion cells. J Comp Neurol 104, 285, 1956. 18) Hi ld W, Tasaki I: M orph olog ica l and ph y si o l og ica l pro p e rti es of neurons and glial c~lls in tiss ue cultu r e. J Neurop h y si o l 25, 277, 1962. 19) Ki m SU: Neurons in tiss ue culutr e . Arch Hist o l Jap 23, 401, 1963. 20) Pete r son ER , Crain SM, Murray MR . Di ffer enti at i on and pro -long e d main t e n ance of bio e lectr i c a lly acti ve spi na l cord cul- tur e. Zie t Zellfo r sch 66, 130, 1965. 21) Callas G, Hi ld W: Electr o n mi cro scop ic observati on s of syn ap tic endin g s in cult ur es of mammali an centr a l nervous tiss ue. Zeit Zellfo r sch 63, 686, 1964. 22) Bung e RP, Bung e M, Pete r son ER: An electr o n mi cro scop ic stu d y of cultu r ed spi na l cord. J Cell Bio l 24, 163, 1965 23) Murray MR: Nervous tiss ue in vit ro . In: Cells and Ti ss ues in Cultu r e, Vol. 2, Wi llm er F (Ed), p 373, Plenum Press, New York,
1965. 24) Murray MR: Nervous tiss ues iso late d in cultu r e. I n: Handbook of Neurochemi st r y , Vol. 5A, Lajt ha A(Ed), p 373, Plenum Press, New York, 1971 . 25) Nakai J: Diss oc iat e d dorsal root ga ng lia in tiss ue cultu r e. Amer J Anat 99, 81, 1956. 26) Aug u sti -T occo G, Sato G: Esta b li sh ment of fun cti on al clonal line s of neurons from mouse neuroblasto m a. Proc Nat Acad Sc i USA 64, 311, 1969. 27) Schubert D, Hump h reys S, Bato n i R, Cohen M: In vit ro dif fere nti - ati on of a mouse neuroblasto m a. Proc Nat Acad Sc i USA 64, 316, 1969. 28) Nelson P: Nerve and muscle cells in cultu r e. Phy si o l Rev 55, 1, 1974. 29) Sato G: Tis s ue Culutr e of nervous Sy st e m . Plenum pre ss, New York, 1973. 30) Levi- M onta l cin i R, Hamburge r V: •S electi ve grow th- s t i m ulati ng eff ec ts of mouse sarcoma on the sensory and sym pa th e ti c nervous sys t e m of the ch ick embryo . J Ex p Zool 116, 321, 1951 . 31) Legvrio- Mw tohn tsa tli cmi nuil a Rt,i nCg o ah ge en n St : i sIno lvaitte rdo farnodm i ns n vaivk oe evfef neuc mts . o Pf r ao cn eNrvaet Acad Sc i USA 42, 695, 1956. 제 5 장 1) Morga n J, Morto n H, Parker RC: Nutr i t ion of an im al cells in tiss ue cultu r e. Init ial stu d ies on a syn the nti c mediu m . Proc Soc Exp Bio l Med 73, 1, 1950.
2) Eag le H. Nutr i t ion needs of mammali an cells in tiss ue cultu r e. Sc ien ce 122, 501, 1955. 3) Evans VJ : Chemi ca lly defi ne d media for culti va ti on of long -ter m cell str a in s from fou r mammalia n spe ci es . Exp Cell Res 36, 439, 1964. 4) Ham RG: An im p ro ved nutr i e n t soluti on for dip lo id chin e s e ham- ste r and human cell line s. Exp Cell Res 29, 515, 1963. 5) Ham RG: Clonal grow t h of mammalia n cells in a chemi ca lly defi ne d, syn the ti c mediu m . Proc Nat Acad Sci USA 53, 288, 1965. 6) Leib o vit z A : The grow t h and main t e n ance of tiss ue/c e ll cultu r es in free ga s exchang e wi th the atm osph ere. Amer J Hy g 78, 173, 1963. 7) Moore GE, Gerner R, Frankli n H: Cultu r e of normal human leuko- cy tes . J Amer Med Ass 199, 519, 1967. 제 6 장 1) Bornste i n MB: Reconsti tut e d rat tai l collag e n as substr a te for tiss ue cultu r e on coverslip s in Maxim ow sli de s and roller tub es. Lab Invest 7, 134, 1958. 2) Manth o rp e M, Long o F, Davis G, Varon S: Lami ni n pro mote s neuriti c rege nerati on from cultu r ed pe rip h eral and centr a l neurons. J Cell Bio l 97, 1882, 1983. 3) Akers RM, Mosher D, Lil ien J: Promoti on of reti na l neurit e outg row th by substr a tw n bound fibr onecti n. Dev Bio l 86, 179, 1981 . 4) Grin n ell F: Cellular adhesiv e ness and extr a cellular substr a ta . Int
Rev Cy tol 58, 65, 1978. 5) Carbonett o S. The extr a cellular matr i x of the nervous sys te m . Trends Neurosci 7, 382, 1984. 6) Yavin E, Yavin Z: At tac hment and cultu r e of dis s oc iat e d cells from rat embryo hemi sp h ere on po ly- l ys in e coate d substr a te . J Cell Bio l 62, 540, 1974. 7) Ruegg U, Heft i F: Growt h of dis s oc iat e d neurons in cultu r e dis h es coate d wi th syn th e ti c pol ym e ric ami ne s. N eurosci Lett 49, 319, 1984. 제 7 장 1) Hay a shi I, Sato G: Rep la cement of serum by hormones pe rmi ts grow t h of cells in a defi ne d mediu m . Natu r e 259, 132, 1976. 2) Sny d er E, Ki m SU: Hormonal req u ir m ent for neuronal surviv a l in cultu r e. Neurosci Lett 13, 225, 1979. 3) Sny d er E, Ki m SU: Insulin : Is it nerve surv iva l fac to r ? Brain Res 196, 565, 1980. 4) Romi jn H, Habets A, Mud M, Wolte r s P: Nerve grow th, syn ap tog e n esis and bio e lect_ ric acti vi t y in fetal rat cerebral corte x tiss ue cultu r ed in serum-fr ee , chemi ca lly defi ne d mediu m . Dev Brain Res 2, 583, 1982. 제 11 장 1) Murray MR: Nervous tiss ue iso late d in cultu r e. In: Handbook of Neurochemi st r y , Vol SA, Lajt ha A( Ed .), Academi c pre ss, New
York, p 373, 1971 . 2) Scott B: Adult mouse dorsal root ga ng lia neurons in cell cultu r e. J Neurobio l 8, 417, 1977. 3) Ki m SU, Warren K, Kali a M: Tis s ue cultu r e of adult human neurons. Neurosci Lett er s 11, 137, 1979. 4) Ki m SU, Takahashi H: Tis s ue cultu r e stu d y of adult h uman reti na neurons. Invest Op h th a l 29, 1372, 1988. 5) Nakai J. Dis s oc iat e d dorsal root ga ng lia in tiss ue cultu r e. Amer J Anat 99, 81, 1956. 6) Pete r son ER, Murray MR : My e li n sheath for mati on in cultu r es of avia n spi na l ga ng lia. Amer J Anat 96, 319, 1955. 7) Levi- M onta l cin i R, Ang e lett i P: Essenti al role of NGF in the surviv a l and main t e n ance of dis s oc iat e d sensory and sym pa - the ti c nerve cells in vitro . Dev Bio l 7, 653, 1963. 8) Manth o rp e M, Skap e r S, Varon S: Neurotr o p h ic fac to r s and the ir anti bo die s : In vitro mi cro assay for titra ti on and screenin g . Brain Res 230, 1981 . 제 12 장 1) Manth o rp e M, Davis G, Varon S: Purif ied pro te i n s acti ng on cultu r ed chic k embryo ciliar y ga ng lion neurons. Fed Proc 44, 2253, 1985. 2) Lin L, Mi sm er D, Lil e J , Armes L, Butl e r E, Vannic e J, Coll ins F: Purif ica ti on , clonin g and exp re ssio n of ciliar y neurotr o p h ic fac to r . Sc ien ce 246, 1023, 1989. 3) Barbin G, Manth o rpe M, Varon S: Purif ica ti on of the chic k ey e cil iar y neurotr o p hic factor . J Neurochem 43, 1468, 1984.
제 13 장 1) Ki m SU, O h T, Jo h nson D:Increased acti vi t y of choli ne acety lt ra ns- fer ase in develop ing cultu r es of chic k spi na l cord. Neuro- bio l og y, 5, 119, 1975. 2) Ki m SU, Weng e r E: De novo for mati on of syn a p s e in cultu r es of chic k neural tub e. Natu r e (New Bio l ) 236, 152, 1972. 3) Pomerat C, Coste r o I: Tis s ue cult ur e of cat cerebellum. Amer J Anat 99, 211, 1956. 4) Gahwi le r B: Orga noty pic monolay e r cultu r es of nervous tiss ue. J Neurosci Meth 4, 329, 1981 . 5) Ki m SU, Tunnic l if f G: Morph olog ica l and cy toc hemi ca l develop - ment of chic k cerebrum cultu r ed in vit ro . Exp Neurol 43, 515, 1974. 6) Ki m SU: Formati on of syn ap s es and my e li n sheath s in cultu r es of dis s oc iat e d chic k embryo nic spi na l cord. Exp Cell Bio l 73, 528, 1972. 7) Akag a wa K, Barnsta b le C. Identi fica ti on and characte r i za ti on of cell type s in monolay e r cultu r es of rat reti na usin g monoclonal anti bo die s , Brain Res 383, 110, 1986. 8) Ki m SU: Neuronal type s in long -ter m cultu r e of avia n reti na , Exp e rie n ti a 27, 1319, 1971 . 제 14 장 I) Pete r son ER, Murray MR: My e li n sheath for mati on in cultu res of avia n sp ina l ga ng lia. Arner J Anat 96, 319, 1955. 2) Bung e M, Bung e RP, Pete r son ER, Murray MR . A ligh t and
electr o n mi cro scop e stu d y of long -ter m orga niz e d cultu r es of rat dorsal root ga ng lia. J Cell Bio l 32, 439, 1967. 3) Main s R, Patt er son P: Prim ary cultu r es of dis s oc iat e d sym pa th e ti c neurons. I Esta b li sh ment of long -ter m go rwt h in cultu r e and stu d ie s of dif fer enti at e d pro p e rti es . J Cell Bio l 59, 329, 1973. 4) O'Lagu e P, Pott er D, Furshap a n E. Stu d ie s on rat sym p a th e ti c neurons develop ing in cell cultu r e. Devel Bio l 67, 384, 1978. 제 15 장 1) Hi ld W: My e li no g e nesis in cultu r es of mammali an nervous tiss ue. Zeit Zellfo r sch 46, 71, 1967. 2) Bornste i n MB, Murray MR: Seria l observati on s on pa tt er rns of grow th, my e li n for mati on , main t e n ance and deg e nerati on in cultu r es of newborn rat and kit ten cerebellum. J Biop h y s Bio c hem Cy tol 4, 499, 1958. 3) Kim SU: Observati on s on cerebellar gran ule cells in vit ro . A.si l ve r and electr o n mi cro scop ic stu d y. Zeit Zellfo r sch 107, 454, 1970. 4) Wolf M: Anato m y of cultu r ed mouse cerebellum. J Comp Neural 140, 281, 1970. 5) Allerland CD: Patt er ns of neuronal dif fer enti at i on in develop ing cultu r es of neonata l mouse cerebellum. J Comp Neurol 142, 167, 1971 . 6) Seil F: Cerebellum in cult ur e. In: Revie w s in Neurosci en ce, Vol 4, Schneid e r D(Ed), p 105, Raven Press, New York, 1979. 7) Hendelman W, Marshall K, Ag ge rwal A, Wojt ow i cz J: Orga niz a - tion of pa th w ays in cultu r es of mouse cerebellum. In: Ti ss ue and Orga n Cultu r es in Neurobio l og y, Fedoroff S(E d ), p 539,
1977. 8) Priv a t A. Postn a ta l matu r ati on of rat Purkin j e cells cultiv a te d in the absence of tw o aff er ent sys t e m s. J Comp Neurol 166, 201, 1976. 9) Bung e R, Bung e M, Pete r son ER: An electr o n mi cro scop ic stu d y of cultu r ed spi na l cord. J Cell Bio l 24, 163, 1965. 10) Sobkowi cz H, Guil ler y R, Bornste i n MB . Neuronal orga niz a ti on in long ter m cultu r es of the spi na l cord of the fet a l mouse. J Comp Neurol 132, 365, 1968. 11) Crain SM, Bornste i n MB: Bio e lectr i c acti vi t y of neonata l mouse cerebral corte x durin g grow th and dif fer enti at i on in tiss ue cultu r e. Exp Neurol 10, 425, 1965. 12) Ki m SU: Ligh t and electr o n mi cro scop ic stu d y of mouse cerebral neocorte x in tiss ue cultu r e. Exp Neurol 35, 305, 1972. 13) Seil F, Kelly J, Leim an A: Anato m i ca l orga niz a ti on of cerebral neocorte x in tiss ue cultu r e. Exp Neurol 45, 435, 1974. 14) Ki m SU: Ef fec ts of choles~erol bio s yn the sis inh ib i t or AY-9944 on orga noty pic cultu r es of mouse spi na l cord. Lab Invest 32, 720, 1975. 15) Crain SM: Neurop h y si o l og ic Stu d ie s in Ti ss ue Cult ur e. Raven Press, New York. 1976. 16) Mi ch ik a wa M, Lim K, McLamon J, Ki m SU: Oxy ge n rad ica l ind uced neuroto x ic ity in spi na l cord neureon cultu r e. J Neuro-sci Res 37, 62, 1994 . . 17) Mi ch ik a wa M, Ki ku chi S, Ki m SU: Leukemi a inhibi t or y fac to r media t e d inc rease of cholin e acety lt r a nsfe r ase acti vity in mouse spi na l cord neurons i~ cult ur e. Neurosci Lett 140, 75, 1992. 18) Mi chika wa M, Ki ku chi S, Muramats u T, Ki m SU: Reti no ic ac id
repo nsiv e ge ne pro duct, mi dk in e , has neurotr o p h ic fun cti on s for mouse spi na l cord and dorsal root ga ng lion neurons in cultu r e. J Neurosci Res 35, 530, 1993. 19) Dohrmann U, Edg a r D, Sendtn e r M, Thoenen H: Muscle deriv e d fac to r s tha t sup po rt s urviv a l and pro mote fibe r outg row t h from chic k spi na l moto r neurons in cultu r e. Devel Bio l 118, 209, 1986. 20) McManaman J, Op pe nheim R, Prevett e D , Marchett i D. Rescue of moto r neurons from cell death by a pu rif ied skeleta l muscle po lyp e p tide : Ef fec ts of ChAT develop m ent fac to r , CDF. Neur- on 4, 891, 1990. 21) Mi ch el P, Heft i F: Toxic ity of 6-hy d roxy d op a mi ne and dop a mi ne for dop a mi ne rgi c neurons in cult ur e.: J Neurosci Res 26, 428, 22) De1n9i9s 0- D. . o nin i S, Glowi ns ki J, Prochia n tz A: Sp eci f ic inf l ue nce of str i a t a l tar ge t neurons on the in vit ro grow t h of mesencep h alic dop a mi ne rgi c neurti es , J Neurosci 3, 2292, 1983. 23) Ki ku ch i S, Muramats u H, Muramats u T, Ki m SU: Mi dk in e , a novel neurotr o p h ic fac to r , pro mote s surviv a l of mesenceph ali c neurons in cult ur e. Neurosci Lett 160, 9-1993. 24) Ki ku ch i S, Ki m SU: Gluta m ate neuroto x ic ity in mesenceph ali c dop a mi ne rgi c neurons in cultu r e. J Neurosci Res 36, 558, 1993. 25) Ki m SU: Morph olog ica l develop m ent of neonata l mouse hippo camp u s cult ur ed in vit ro . Exp Neurol 41, 150, 1973. 26) Banker G, Cowan WM: Rat hip po camp a l neurons in dis p e rsed cell cultu r e. Brain R es 126, 397, 1977. 27) Dott i G, Sulliv a n C, . Banker G: The esta b li sh ment of po larit y by hippo camp a l neurons in cultu r e. J Neurosci 8, 14.5 4, 1988. 28) Huett ne r J, Baug h man R. Prim a ry cult ur e of ide nti fied neurons from the vis u al corte x of po stn a ta l rats . J Neurosci 6, 3044,
1986. 제 16 장 1) Murray MR: Nervous tiss ues iso late d in cultu r e. In: Handbook of Neurochemi st r y , Vol5A, Lajt ha A(Ed), p 373, Raven pre ss,N ew York,1 9 71 . 2) Coste r o I, Pomerat C: Cultiv a ti on of neurons from the adult h uman cerebral and cerebellar cort ex . Amer J Anat 98, 405, 1958. 3) Hog u e M: A stu d y of human brain cells in tiss ue cultu r e. Amer J Anat 93, 397, 1953. 4) Geig e r R: Subcultu r e of adult mammali an brain corte x in vit ro . Exp Cell Res 14, 541, 1958. 5) Scott B: Adult mouse dorsal root ga ng lia neurons in cell cultu r e. J Neurobio l 8, 417, 1977. 6) Ki m SU: Ti ss ue cultu r e of mammali an sym p a th e ti c neurons. J Neurop a th Exp Neurol 37, 621, 1978. 7) Ki m SU, Warren K, Kali a M: Ti ss ue cultu r e of adult human neurons. Neurosci Lett 11, 137, 1979. 8) Fukuda J, Kamey a ma M: A tiss ue cultu r e of nerve cells from adult mammali an ga ng lia and some electr o p h y si o l og ica l pro p er t ies of the nerve cells. Brain Res 202, 249, 1980. 9) Ki m SU, Tomonag a M, Ghett i B: Neurofi br ill ar y dege nerati on in cultu r ed adult mouse neurons ind uced by may tan sin e . Acta Neurop a th 52, 161, 1980.
제 17 장 1) Messin g A, Ki m SU: Long -t e r m cultu r e of aduft mammalia n centr a l nervous sys t e m neurons. Exp Neurol 65, 293, 1979. 2) Ki m SU, Takahashi H: Tis s ue cultu r e stu d y of adult h uman reti na neurons. Invest Op h th a l 29, 1372, 1988. 제 18 장 1) Wood P: Sep a rati on of Schwann cells and neurons from normal pe rip h eral nervous tiss ue. Brain Res 115, 361, 1976. 2) Main s R, Patt er son P: Prim ary cultu r es of dis s oc iat e d sym p a th e ti c neurons. J Cell Bio l 59, 329, 1973. 3) Morett o G, Xu R, Walker D, Ki m SU: Co-exp r essio n of mRNA for neurotr o p h ic fac to r s in human neurons and glial cells in cultu r e. J Neurop a th Exp Neural 53, 78, 1994. 4) Sato h J, Morett o G, Muramats u T, Chang H, Cho J, Ki m SU: Mi dk in e tha t pro mote s surviv a l of fet a l human neurons is pro duced by fetal astr o cy tes in cultu r e. Devel Brain Res 75, 201, 1993. 5) W ys ocki L, Sato V: Pannin g for lym ph ocy tes : a meth o d for cell selecti on . Proc Nat Acad Sc i USA 75, 2844, 1978. 6) Barres B, Sil ve rste i n B, Corey D, Chun L: Immunolog ica l, mor- ph olog ica l and electr o p h y s io l og ica l varia t i on among reti na ga ng lion cells pu rif ied by pa nnin g . Neuron 1, 791, 1988. 7) Eis e nbart h G, Walsh F, Ni re nberg M: Monoclonal anti bo dy to a pla sma membrane anti ge n of neurons. Proc Nat Acad Sc i USA 76, 4913, 1979.
제 19 장 1) Wood P: Sep a rati on of fun cti on al Schwann cells and neurons from normal pe rip h eral nervous tiss ue. Brain Res 115, 361, 1976. 2) Brockes J, Fie l ds K, Raff M: Stu d ie s on cultu red rat Schwann cells. Esta b li sh ment of pu rif ied po p u lati on s from cultu r es of pe rip h - eral nerve. Brain Res 165, 105, 1979. 3) Morett o G, Ki m SU, Shin D, Ri zz uto N: Long -ter m cultu res of human adult Schwann cells iso late d from auto p s y mate i r a ls. Acta Neurop a th 64, 15, 1984. 4) Sobue G, Brown M, Ki m SU, Pleasure D: Axolemma is a mi tog e n for human Schwann cells. Annals Neurol 15, 449, 1984. 5) Levi A, Bung e RP, Lofg ren J, Meim a L, Heft i F, Ni ko li cs K, Sli w kowski M: The inf l ue nce of hereg ulins on human Schwann cell pro li fer ati on . J Neurosci 15, 1329, 1995. 제 20 장 1) McAlpi ne D, Lumsden C, Acheson E: Multip le Sclerosis . A Reap- pr ais a l, Churchil l-Livi n g s to n , Edin b urgh , 1972 2) McFarlin D, McFarland H: Multip le sclerosis . New Eng J Med 307, 1183, 1982. 3) Lisa k R, Pleasure D, Sil be rberg D, Mannin g M, Said a T: Lon g-ter m cultu r e of bov ine oli go dendrocy tes iso late d wi th a pe rcoll grad ie n t. Brain Res 213, 165, 1981 . 4) Ki m SU, Sato Y, Sil be rberg D, Pleasure D, Rorke L: Lon g-ter m cultu re of human oli go dendrocy tes . J Neurol Sc i 62, 295, 1983. 5) McCarth y K, DeVell is J: Prep a rati on of sep a rate astr o g lial and
oli go dendrog lial cell cultu r es from rat cerebral tiss ue. J Cell Biol 85, 890, 1980. 6) Gebic k -Harte r P, Aklth a us H, Schwartz P, Neuhoff V: Bulk sepa rati on and long -ter m cultu r e of oli go dendrocy tes from pig brain . J Neurochem 43, 357, 1981 . 7) Szuchet S, Ste f a n sson K, Wolman R, Dawson G, Amason B: Main t e n ance of iso late d olig o dendrocy tes in long -ter m cultu r e, Brain Res 200, 151, 1980. 8) Norto n W, Farroq M, Fie l ds K, Rain e C: The long -ter m cultu r e of bulkis o late d bovin e oli go dendrog li a freo m adult brain . Brain Res 270, 295, 1983. 9) Hira y a ma M, Sil be rberg D, Lis a k R, Pleasure D: Long -ter m cul- tur e of oli go dendrocy tes iso late d from rat corpu s callosum by pe rcoll densit y grad ie n t. J Neurop a th Exp Neurol 42, 16, 1983. 10) Ki m SU: Neurobio l og y of human olig o dendrocy tes in cultu r e. J Neurosci Res 27, 712, 1990. 11) Raff M, Mi ller R, Noble M: A gli a l pro g e nit or cell tha t develop s in vit ro int o an astr o cy te or an oli go dendrocy te dep e ndin g on cultu r e mediu m . Natu r e 303, 390, 1983. 12) Raff M: Gli al cell div e rsif ica ti on in the rat op tic nerve. Sc ien ce 243, 1450, 1989. 제 21 장 1) Ki m SU, Ste m J, Ki m M, Pleasure D: Cultu re of pu rifi ed rat astr o cy tes in serum-fr ee med ium sup ple mente d wi th mi tog e n. Brain Res 274, 79, 1983. 2) Sheff ield W, Kim SU: My e li n b asic pr ote i n causes pr oli fer ati on of
lym p h ocy tes and astr o cy tes in vit ro . Brain Res 132, 580, 1977. 3) Sim p so n D, Morris o n R, DeVell is J, Herschman H: EGF bin d in g and mi tog e nic acti vi t y on pu rif ied po p u lati on s of cells from the CNS. J Neurosci Res 8, 453, 1982. 4) Yong V, Ki m SU: Grow th fac to rs for fet a l and adult human astr o cy tes in cultu r e. Brain Res 444, 59, 1988. 5) Ric h ardson W, Prin g le N, Mosley M, Dubois - Dalcq M: A role for PDGF in normal gliog e nesis in the centr a l nervous sys te m . Cell 53, 309, 1988. 6) Ito J, Kato T, Hara F: Dete c ti on and pa rt ial pu rif ica ti on of glial grow th inh ib i t or y fac to r in the condit ion ed mediu m of neurob-lasto m a cells. Neurochem lnte r nat 11, 331, 1987. 7) Sobue G, Pleasure D: Astr o g lial pro li fer ati on and ph enoty pe are modulate d by neuronal pla sma membrane. Brain Res 324, 175, 1984. 8) Unsic k er K, Reic h ert H, Schmi dt R: Astr o g lia and FGF have neurotr o p h ic fun cti on s for cultu r ed pe rip h eral and centr a l nervous sys t e m neurons. Proc Nat Acad Sc i USA 84, 5459, 1987. 9) Morett o G, Xu R, Walker D, Ki m SU: Co-exp re ssio n of mRNA for neurotr o p h ic fac to rs in human neurons and glial cells in cultu r e. J Neurop a th Exp Neurol 53, 78, 1994. 제 22 장 1) Gi ul i an D, Baker T: Characte r iz a ti on of amoeboid mi cro g lia iso -late d from develop ing mammali an brain . J Neurosci 6, 2163, 1986. 2) Giu l i an D, Baker T, Sh in L, Lachman L: Inte r leukin - I of the CNS
is pro duced by amoeboid mi cro g lia. J Exp Med 164, 594, 1986. 3) Saw a da M, Kondo N, Suzumura A, Marunouchi T: Producti on of tum or necrosis fac to r -a l ph a by mi cro g lia and astr o cy tes . Brain Res 491, 394, 1989. 4) Walker D. Ki m SU, McGeer P: Comp le ment and cy tok in e ge ne exp re ssio n in cultu r ed mi cro g li a deriv e d from po st- m orte m human brain s . J Neurosci Res 40, 478, 1995. 제 23 장 1) Brendel K, Mezan E, Carlson E. Isolate d brain mi cro vessels: A pu rif ied , meta b oli ca lly acti ve pre p a rati on from bovin e cerebral corte x . Sc ien ce 185, 953, 1974. 2) De Bault L, Kahn L, Frommes S, Canc illa P: Cerebral mi cr ovessels and deriv e d cells in tiss ue cultu r e. In Vi tro 15, 473, 1979. 4) Ki m SU, Lee KH: Human and bovin e cerebral mi cro vessels in tiss ue cultu r e. Neurolog y 38, 295, 1988. 5) Bowman P, Enn is S, Rarey K, Betz A, Goldste i n G: Brain mi - crovessel endoth e li a~ cells in tiss ue cultu r e: A model for stu d y of blood-brain - barrie r pe rmeabil ity. Annals Neurol 14, 396, 1983. 6) Dorovon i-Z is K, Pramey a R, Thomp s on G: Isolati on and cultu r e of human brain mi cro vessel endoth e li al cells. J Neurop at h Exp Neurol 48, 365, 1989.
제 24 장 1) Konig s berg I: Clonal analys is of my o g e nesis . Scie n ce 140, 1273, 1963. 2) Askanas V, Gallex G: Sy n ergi st i c inf l ue nce of po lyp e p tide grow th fac to r s on cultu r ed human muscle. Arch Neural 42, 749, 1985. 3) W itko wski J: Ti ss ue cultu r e stu d ie s of muscle dis o rders. Tech-niq u es, cell gr ow th, morph olog y and cell surfa c e. Muscle nerve 9, 191, 1986. 4) Yaff e D: Rete n ti on of dif fer enti at i on po te n ti al i ties durin g pro -long e d cultiv a ti on . Proc Nat Acad Sci USA 61, 477, 1988. 제 25 장 1) Vale W, Grant G, Amos M, Guil lem i n R: Cultu r e of enzym a ti ca lly dis p e rsed ante r io r pitui t ar y cells: Functi on al vali da ti on of a meth o d. Endocrin o log y 91, 562, 1972. 2) Hop k in s C, Farqu har M: Hormone secreti on by cells dis s coia t e d from rat ante r i or pitui t ar ie s . J Cell Bio l 59, 276, 1973. 3) Hy m er W, Evans W, Kraic e r J, Mastr o A, Gris w old E: Enric h ment of cell type from the rat adrenohy po p h y si s by sedim enta t i on at unit grav it y. Endocrin o log y 92, 275, 1973. 4) Ti xi e r -Vi da l A, Farqu har M: The Ante r io r Pit ui t ar y, Academi c Press, New York, 1975. 5) Ki m SU, Shin D, Morett o G: Isolati on , cultu r e and cell type ide nti - fica ti on of adult h uman pitui t ar y cells. A cta Neurop a th 68, 205, 1985. 6) Shin D, Ki m SU: Hormonal secreti on by adult pitui t ar y cells in
cultu r e. Human Path 19, 83, 1988. 제 26 장 1) Unsic k er K, Kris c h B, Ot ten U, Thoenen H: NGF ind uced fibe r outg row th from iso late d rat adrenal chromaff in cells. Proc Nat Acad Sc i USA 75, 3498, 1978. 2) Aloe L, Levi -M onta l cin i R: NGF-in d uced tra nsfo r mati on of im - matu r e chromaff in cells in viv o int o sym pa th e ti c neurons. Proc Nat Acad Sc i USA 76, 1246, 1979. 3) Tis h cler A, DeLell is R, Bia l es B, Carabba V, Wolfe H: NGF- ind uced neurit e outg r owt h from normal human chromaff in cells. Lab Invest 43, 399, 1980. 4) Madrazo I, Drucker R, Dia z V, Marti ne z J, Torres C, Becerril J: Op e n mi cro surgi ca l auto g rap h of the adrenal medulla to the rig h t caudate nucleus in tw o pa ti en ts wi th Parkin s on's dis e ase. New Eng J Med 316, 831, 1987. 5) Kamo H, Kim SU, McGeer P, Shin D: Transpl a nta t i on of cult ur ed fet a l human adrenal chromaff in cells to rat brain . Neurosci Lett 57, 43, 1985. 6) Fenwic k E, Fajd iga P, Howe N, Liv e tt B: Functi on al and mor- ph olog ica l characte r iz a ti on of iso late d bovin e adrenal medu-llary cells. J Cell Bio l 76, 12, 1978.
제 27 장 1) Harris o n RG: The outg row th of the nerve fibe r as a mode of pro to p la smi c movement. J Exp Zool 9, 787, 1910. 2) Kruse P, Patt er son M: Ti ss ue Cultu r e: Meth o ds and Ap lica ti on s, Academi c Press, New York, 1973. 3) Tayl o r A, Kollros J: Sta g e s in the normal develop m ent of Rana pipien s larvae. Anat Rec 94, 7, 1946. 4) Evers J, Laser M, Sun Y, Xi e Z, Poo M: Stu d ie s of nerve-m uscle int e r acti on s in Xenop u s cell cultu r e. J Neurosci 9, 1523, 1989. 5) Pollak E, Koves J: In vit ro cultiv a ti on of larval frog spi na l cord. Tis s ue Cul Manual 1, 193, 1975. 6) Peng H, Chang P: Formati on of po st- s yn a p tic spe ci al i za ti on in- duced by late x beads in cultu r ed muscle cells. J Neurosci 2, 1760, 1982. 7) Xi e Z, Poo M: Init ial events in the for mati on of neuromuscular syn a ps e . Proc Nat Acad' Sc i USA 83, 7063, 1986. 8) Landreth G, Ag ran off B: Exp la nt cultu r e of adult g o ld fish reti na : A model for the stu d y of CNS rege nerati on . Brain Res 161, 39, 1979. 9) Schawarz M, Ag ran off B: Outg row th and main t e n ance of neurite s from cultu r ed go ld fish reti na l ga ng lion cells. Brain Res 206, 331, 1981 . 10) Vi el mett er J, Stu e rmer C: Gold fish reti na l axons respo nd to po sit ion spe ci fic pro p erties of tec ta l cell membranes in vitro. Neuron 2, 1331, 1989.
제 28 장 1) Vag o C: Inverte b rate Tis s ue Cultu r e, Academi c Press, New York, 1971. 2) Maramorosch K: Inverte b rate Tis s ue Cultu r e: Research Ap pli c a- tion s, Academi c Press, New York, 1976. 3) Seecof R, Tep litz R, Gerson I, Ikeda K, Donaldy ]:Di ffer enti at i on of neuromuscular jun cti on s in cultu r es of embryo nic drosop h il a cells. Proc Nat Acad Sc i USA 69, 566, 1972. 4) Wu C, Suzuki N, Poo M: Diss oc iat e d neurons from normal and muta n t drosop h il a larval CNS in cell cultu r e. J Neurosci 3, 1889, 1983. 5) Chen J, Levi- M onta l cin i R: Axonal outg row th ari d cell mi gr ati on in vitro from nervous sys t e m of cockroach embryo s. Sc ien ce 166, 631, 1969. 6) Lev i-M onta l cin i R, Seshan K: Long -ter m cultu r es of embryo nic and matu r e ins ect nervous and neuroendocri ne sys te m . In: Tis s ue Cultu r e of the Nervous Sy st e m , Sato G(Ed), p 123, Plenum Press, New York, 1973. 7) Ready F, Ni ch ols J: Identi fied neurons iso late d from leech CNS make selecti ve connecti on s in cultu r e. Natu r e 281, 67, 1979. 8) Die t z e l I, Drap e au P, Ni ch ols J: Volta g e dep e ndence of 5- . hy d roxy try p tam i ne release at syn a p s e betw een ide nti fied leech neurons in cultu r e. J Phy si o l 372, 191, 1986. 9) Barker D, Wong R, Kate r S: Sepe rate fac to r pro duced by the CNS of the snail, Heli so ma, sti m ulate neurit e o utg row th and choli ne meta b oli sm in cultu r ed neurons. J Neurosci Res 8, 419, 1982. 10) Cohan C, Hayd on P, Kato r S: Sin g le channel acti vi t y dif fer s in grow i ng and non-gr ow i ng grow th cones of iso late d neurons of
Helis o ma. J Neurosci Res 13, 285, 1985. 11) Dag o n D, Levit an I: Isolate d ide nti fied ap ly s ia neurons in cell cult ur e. J Neurosci 1, 736, 1981 . 12) Schacher S: Differ enti al syn a p se for mati on and neurit e out- grow t h at tw o branches of the gian t cells of Ap ly s i a in dis o c ia- ted cell cultu r e. J Neurosci 5, 2028, 1985. 13) Cornwell PB : The Cockroach. Hutc h ins on, London, 1968. 제 30 장 1) Sorie u l S, Ep h russi B: Karyo log ica l demonstr a ti on of hy b rid i z a - tion of mammali an cell in vit ro . Natu r e 190, 653, 1961 . 2) Ponte c orvo G: Producti on of mammali an somati c hy b rid s by means of po lye th y le ne gly c ol tre atm ent. Somat Cell Genet 1, 397, 1975. 3) Littlef i el d J: Selecti on of hy b rid s from mati ng s of fibr oblasts in vit ro and the ir pre sumed recombin a nts . Sc ien ce 145, 709, 1964. 4) Mi na J, Glazer D, Ni re nberg M: Geneti c dis s ecti on of neural pro p e rtie s usin g somati c cell hyb rid s . Natu r e new Bio l 235, 225, 1972. 5) Amano T, Hemp re cht B, Kemp e r M: _High acti vity of choli ne acety lt r a nsfe r ase ind uced in neuroblasto m a x glia hyb rid cells. Exp Cell Res 84, 391, 1974. 6) Nelson P, Chris t i an C, Ni re nberg M: Sy na ps e for mati on betw een clonal neuroblasto m a x gliom a hy b rid cell and str i a t e d muscle cells. Proc Nat Acad Sc i USA 73, 123, 1976. 7) Greene L, Shain W, Chalazonit is A , Breakfi eld X, Mi na J, Coon H, Ni re nberg M: Neuronal pro p e rt ies of hy b rid neuroblasto m a x
sym pa th e ti c ga n glion cells. Proc Nat Acad Sc i USA 72, 4923, 1971 . 8) Hammond D, Wain e r B, Tonsga rd J, Heller A: Neuronal pro p - ertie s of clonal hy b rid cell line s deriv e d from centr a l choli ne r- gic neurons. Sc ien ce 234, 1237, 1986. 9) Dic k son J, Flanig a n T, Walsh F: Exp re ssio n of human neuronal anti ge ns in a mouse neuroblasto m a x human dorsal root ga n- glion cell hy b rid . EMBO J 2, 283, 1986. 10) Sug a ma S, Wata b e K, Cashman N, Ki m SU: Exp r essio n of olig o dendrog lial anti ge ns in an mouse neuroblasto m a x oli go dendrog lia cell hy b rid . J Neurop a th Exp Neural 48, 366, 1989. 제 31 장 1) Wata b e K, Yamada -M, Kawamura T, Kim SU: Transfe c ti on and sta b le tra nsfo r mati on of adult m ouse Schwann cells wi th S V- 40 large T anti ge n ge ne. J Neurop a th Exp Neural 49, 455, 1990. 2) Graham F, Van der Eb A: A new tec hniq u e for the assay of infec ti vi t y of human adenovir u s-5 DNA . Vir o log y 52, 456, 1973. 3) Chen C, Okaya ma H: Hi gh eff icien cy tra nsfo r mati on of mam- . mali an cells by pla smi d DNA . Mol Cell Bio l 7, 2745, 1987. 4) Felgn e r P, Gadek T, Holm M, Roman R, Wenz M, Danie l sen M: Lip o fe c tin : A high ly eff icien t, lipid- med iat e d DNA tra nsfe c ti on pro cedure. Proc Nat Acad Sc i USA 84, 7413, 1987.
제 32 장 1) Pfe iffer S, Bets c hart B, Cook J, Manc ini P, Morris P: Glia l cell line s. In: Cell, Ti ss ue and Orga n Cultu r es in Neurobio l og y, Fedoroff S(Ed), p 287, Academi c Press, New York, 1977. 2) Nelson P: Neuronal celllin e s. I n: Cell, Tis s ue and Orga n Cultu r es in Neurobio l og y, Fedoroff S(ED), p 347, Academi c Press, New York, 1977. 3) Aug u sti -T occo G, Sato G: Esta b li sh ment of fun cti on in g clonal line s of neurons from mouse neuroblasto m a. Proc Nat Acad Sc i USA 64, 311, 1969. 4) Schubert D, Hump h reys S, Bato n i C, Cohen M: In vit ro dif fer enti a- tion of a mouse neuroblasto m a. Proc Nat Acad Sc i USA 64, 316, 1969. 5) Benda P, Lig h tb o dy J, Sato G, Levin e L, Swet W: Dif fer enti at e d rat glial cell str a in in tiss ue cultu r e. Sc ien ce 161, 370, 1968. 6) Lendahl U, Mckay R: The use of celllin e s in neurobio l og y. Trends Neurosci 13, 132, 1990. 제 33 장 1) Greene L, Tis c hler A: Esta b li sh ment of a noradrenergi c clonal line of rat ph eochromocy tom a cells whic h respo nd to NGF. Proc Nat Acad Sc i USA 73, 2424, 1976. 2) Greene L, Alett a J, Rukenste i n A, Greene S: PC 12 · ph eo-chromocy tim a cells: C ultu r e, NGF tre atm ent and exp erim enta l exp lo it at i on . Meth o d Enzym o l 147, 207, 1987.
3) Guroff G: PC 12 cells as a model of neuronal dif fer enti at i on . In: Cell Cultu r e in the Neurosci en ce, Bott en ste i n J and Sato G(Eds), p 245, Plenum pre ss, 1985. 제 34 장 1) McBurney M, Vi llen euve E, Edwards M, Andera P: Contr o l of muscle and neuronal dif fer enti at i on in a cultu r ed embryo nal carci no ma cell line . Natu r e 299, 165, 1982. 2) Rudnic k i M, McBurney M: Cell cultu r e meth o ds and ind ucti on of dif fer enti at i on of embryo nal carci no ma cell line s. In: Tera- toc arci no ma and Embryo nic Ste m Cells: A Practi ca l Ap pro ach, Roberts o n E(Ed), p 19, IRL Press, Oxfo r d, 1987. 3) McBurney M, Reuhl K, Ally A, Nasip u ri S, Bell J, Craig J: Differ enti at i on and matu r ati on of embryo nal carci no ma deri- ved neurons in cultu r e. J Neurosci 8, 1063, 1988. 4.) Mi ch ik a wa M, Xu, R Muramats u H, Muramats u T, Ki m SU: Mi dk in e is a media t o r of reti no ic ac id ind uced neuronal dif fer -enti at i on of embryo nal carci no ma cells. Bio c hem Bio p h y s Res Comm 192, 1312, 1993. 제 35 장 1) Ki m SU, Tomonag a M: Neural Ti ss ue Cultu r e. Fuji ta Ki ka ku, Toky o , 1989. 2) McKay R, Raff M, Reic h ardt L. Monoclonal anti bo d ies to Neural Anti ge ns. Cold Sp r in g Harbor Lab, New York, 1981 .
3) Kennedy P: Neural cell markers and the ir ap pli c a ti on to neural- og y. J Neuroim munol 2, 35, 1982. 4) Ki m SU: Anti ge n exp r essio n by glial cells grow n in cultu r e. J Neuroim munol 8, 255, 1985. 5) Mi rs ky R, Wendon L, Black P, Sto l kin C, Bray D. Teta n us tox in : A cell surfa c e marker for neurons in cultu r e. Brain Res 148, 251, 1978. 6) Eis e nbarth G, Walsh F, Ni re nberg M: Monoclonal anti bo dy to a pla sma membrane anti ge n of neurons. Proc Nat Acad Sc i USA 76, 4913, 1979. 7) Raff M, Fie l ds K, Hakomori S, Mi rs ky R, Pruss R, W int e r J: Cell type spe ci fic markers for dis t i ng u is h in g and stu d y ing neurons and glial cells in cultu r e. Brain Res 174, 283, 1979. 8) Caceres A, Banker G, Bi nd er L: Immunocy toc hemi ca l locali za ti on of tub uli n and mi cr otu b ule-assoc iat e pro te i n 2 durin g the devel-op m ent of hip po camp a l neurons in cultu r e. J Neurosci 6, 714, 1986. ~) Shaw G, Banker G, WEber K: An im munofl uo rescence stu d y of neurofi lam ent pro te i n exp re ssio n by develop ing hip po camp a l neurons in tiss ue cult ur e. Eur J Cell Bio l 39, 205, 1985. 10) Lee V, Wu L, Schlaep fer W: Monoclonal anti bo die s recog nize neurofi lam ent tri p le t pro te i n s . Proc Nat Acad Sc i USA 79, 6089, 1982. 11) Camp a g no ni A: Molecular bio l og y of my e li n pro te i n s from the centr a l nervous sys t e m . J Neurochem 51, 1-1 4 , 1988. 12) Lees MB, Brosto f f SW: Prote i n s of my e li n. In: My e li n, Second Ed, Morell P(Ed), p 197, Plenum Press, New York, 1984. 13) Ki m SU, Morett o G, Lee V, Yu R: Neuroim munolog y of ga ng - Iios id e in human neurons and gli a l cells in cultu r e. J Neurosci
Res 15, 303, 1986. 14) Cuello A: A Meth o ds in Neurosci en ce: Immunohis t o c hemi st r y . 제 36 장 1) Koehler G, Mi lste i n C: Conti nu ous cultu r e of fus ed cells secreti ng anti bo dy of pre -defi ne d spe ci ficity. Natu r e 256, 495, 1975. 2) Cuello A: A Meth o ds in Neurosci en ce: Immunohis t o c hemi st r y . Joh n Wi ley , New York, 1983. 3) Polak J, Immunocy toc hemi st r y , Practi ca l Ap plica ti on s in Path o l-og y and Bio l og y. Joh n Wrig h t, Bris t o l , 1983. 4) Joh nsto n A, Thorpe R: Immunochemi st r y in Practi ce . Blackwell, Oxfo r d, 1987. 5) Beesley JE: Immunocy toc hemi st r y : A Practi ca l Ap pro ach. IRL Press, Oxfo rd , 1993. 6) Ki m SU: Anti ge n exp re ssio n of glial cells gro wn in cultu r e. J Neuroim munol 8, 255, 1985. 7) Kim SU, Tomonag a M, Ghett i B: Neurofi br ill ar y deg e nerati on in cultu r ed adult mouse neurons ind uced by may tan sin e . Acta Neurp a th 52, 161, 1980. 제 37 장 1) Ki m SU, Ste r n J, Ki m M: Pleasure D: Cultu r e of pu rif ied astr o cy tes in serum free mediu m sup ple mente d wi th mi tog e n. Brai n Res 274, 79, 1983. 2) Pruss R, Bart let P, Gavrilo v ic J, Lis a k R, Ratt ra y S: Mi tog e ns for
glial cells. Devel Brain Res 2, 19, 1982. 3) Ecclesto n P, Sib e rberg D: Fib r oblast grow t h fac to r is a mi tog e n for olig o dendrocy tes in vit ro . Devel Brain Res 21, 315, 1985. 4) Wood P, Bung e RP: Evid e nce tha t axons are mi tog e nic for oli go dendrocy tes iso late d from adult anim als. Natu r e 320, 756, 1986. 5) Raff M, Abney E, Brockes J, Hornby -S mi th A: Schwann cell gro wt h fac to r s. Cell 15, 813, 1978. 6) Ratn e r N, Bung e RP, Glaser L. Schwann cell pro li fer ati on : Am overvie w . Ann N Y Acad Sc i 486, 170, 1986. 7) Gratz n er H. Monoclonal anti bo dy to 5-bromodeoxy ur id i n e and 5-i o d odeoxy u rid i n e : A new reag e nt for dete c ti on of DNA re- plica ti on . Scie n ce 218, 474 , 1982. 8) Yong V, Ki m SU: a new double labell ing im munofl uo rescence tec hniq u e for the dete r mi na ti on of pro li fer ati on in human astr o cy tes in cultu r e. J Neurosci Meth 21, 9, 1987. 9) Yong V, Ki m SU: Grow th fac to r s for fetal and adult human glial cells in cultu r e. Brain Res 444, 59-66, 1988. 10) Morett o G, Yoo A, Ki m SU: Human astr o cy tes and cy tok in e s. Tumor necrossis fac to r alph a and int e r fe r on ga mma do not pro mote astr o cy tic pro li fer ati on . Neurosci Lett 151, 17, 1993. 제 38 장 1) Mosman T: Rap id colorim etr ic assay for cellular grow th and surviv a l: Ap plica ti on to pro li fer ati on and cy tot o x ic ity assays . J Immnol Meth 65, 55, 1983. 2) Lip m an J, Fli nt 0: Cell cultu r e sys te m s and in vitro tox ic ity
tes ti ng . Cy tot e c hnolog y 8, 129, 1992. 제 39 장 1) Lillie R: Hi st o p a th o log ic Technic and Practi ca l Hist o c hemi st r y . Thir d Ed, MeGraw-Hi ll, New York, 1965. 2) Pearse AGE: Hi st o c hemi st r y . Theoreti ca l and Practi ca l. Thir d Ed, Churchil l-L iv i n g s to n , 1980. 3) Bodia n D: A new meth o d for sta i n i n g nerve fibe rs and nerve endin g s in pa raff in secti on s. Anat Rec 63, 89, 1936. 4) Kim SU: Observati on s on cerebellar gran ule cells in tiss ue cultu r e. A silv er and electr o n mi cro scop ic stu d y . Zeit Zellfo r sch 107, 454, 1970. 5) Toran-Allerand D: Golgi -C o x modif ica ti on s for the im p re g n ati on of whole mount pre p a rati on s of orga noty pic cultu r es of the CNS. Brain Res 118, 293, 1976. 제 40 장 1) Lil lie R: His t o p a th o g ic Technic and Practi ca l Hist o c hemi st r y . Thir d Ed, McGraw-Hi ll, New York, 1965. 2) Pearse AGE: Hi st o c hemi st r y : Theoreti ca l and Ap plied . Thir d Ed, Churchil l-Living s to n e, Edin b urgh , 1980. 3) Karnovsky M, Rp pts L: A dire ct colorin g thi o c holi ne meth o d for choli ne ste r ase. J Hi st o c hem Cy toc hem 12, 219, 1964. 4) Tago H, Kim ura H, Maeda T: Deta i l ed acety lc holi ne ste r ase fibe r and neuronal sta i n i n g in rat brain by a new and hig h ly sensit ive
his t o c hemi ca l pro cedure. J Hist o c hem Cy toc hem 34, 1431, 1986. 5) Lind vall 0, Bjo r klund A, Hokfe lt T , Lju n g d ahl A: Ap plica ti on of the gly o xy li c aci d meth o d to vib r ato m e secti on s. Hist o c hemi e 35, 31, 1973. 제 41 장 1) Wi ss e E: Electr o n Mi cro scop y and Cy toc hemi st r y . North Holland, Amste r dam, 1973. 2) Pete r s A, Palay S, Webste r H: The Fin e Str u ctu r e of the Nervous Sy st e m . Saunders, Phil ad elph ia , 1976. 제 42 장 1) Bornste i n MB, Ap pe l S: The ap plica ti on of tiss ue cultu r e to the stu d y of EAE: pa tt er n of demy e li na ti on . J Neurop a th Exp neurol 20, 141, 1961 . 2) Ki m SU, Murray MR, Tourte l lott e W, Parker J: Demonstr a ti on in tiss ue cultu r e of my e lin o to x ic ity in cerebrospi na l flui d and brain extr a cts from multip le sclerosis pa ti en ts . J Neurop a th Exp Neurol 29, 420, 1970. 3) Rain CS: Ultr a str u ctu r al app li c a ti on s of cultu r ed nervous sys t e m to neurop a th o log y. Prog r Neurop a th 2, 27, 1973. 4) Kawai K, Zweim an B: Characte r is t i cs of in vitro cy tot o x ic . eff ec ts of my e li n basic pro te i n reacti ve T cell line s on syn e rge neic olig o dendrocyt es . J Neuroim munol 26, 57, 1990. 5) Sato h J, Ki m SU, Kastr u ckoff L: Absence of natu r al kil ler cell
acti vity ag a in s t oli go dendrocy tes in multip le sclerosis . J Neuro- im munol 28, 75, 1990. 6) Sato h J, Ki m SU, Kastr u koff L, Takei F: Exp re sio n and ind ucti on of ICAMs and MHC anti ge ns on cultu r ed murin e oli go den-drocy tes and astr o cy tes . J Neurosci Res 29, 1, 1991 . 7) Hira no A, Cook S, Whit ak er J, Dowl ing P, Murray MR: Fin e str u ctu r al Aspe cts of demy e li na ti on in vit ro . J Neurop a th Exp Neurol 30, 249, 1971 . 8) Raff M: Gl ial cell div e rsif ica ti on in the rat op tic nerve. Sc ien ce 243, 1450, 1989. 9) Sato h J, Ki m SU: Proli fer ati on and dif fer enti at i on of fet a l human olig o dendrocy tes in cultu r e. J Neurosci Res 39, 260, 1994. 10) Seil F, Lamp e rt P: Neurofi br ill ar y tan g le s ind uced by vin c ris t i ne and vinv lasti ne sulfa te in centr a l and pe rip h eral neurons in vitro . Exp Neurol 21, 219, 1968. 11) Seil F, Lamp e rt P, Klatz o I: Neurofi br ill ar y sph eroid s ind uced by alumi nu m ph osph ate in dorsal root ga ng lion neurons in vit ro . J Neurop a th Exp Neurol 28, 74, 1969. 12) Kim SU, Tomonag a M, Ghett i B: Neurofi br ill ar y deg e nerati on in cultu r ed adult mouse neurons ind uced by may tan sin e . Acta Neurop a th 52, 161, 1980. 13) Phelps C, Gag e F, Growdon J. Heft i F, Harboug h R, Joh nson M: Pote n ti al use of NGF to tre at Alzheim er's dis e ase. Neurobio l Ag ing 10, 205, 1989. 14) Hartik k a J, Heft i J: Comp a ris o n of NGF's eff ec ts on develop m ent of sep tum , str i a t u m and nucleus basali s c holi ne rgi c neurons in vitro . J Neurosci Res 21, 352, 1988. 15) Hata n aka H, Ni ho nmats u I, Tsukui H: NGF pro mote s surviv i a l of cultu r ed magn o cellular choli ne rgi c neurons from nucleus
basali s o f Magn e rt in po stn a ta l rats . Neurosci Lett 90, 63, 1988. 16) Ap pe l S: A unif ying hyp o th e sis for the cause of amy o tr o p h i~ lalte r al sclerosis , Parkin s onis m and Alzheim er's dis e ase. Ann Neurol 10, 499, 1981 . 17) Uchid a Y, !hara Y, Tomonag a M: Alzheim er's dis e ase brain extr a ct sti m ulate s the surviv a l of cerebral cort ica l neurons from neonata l rats . Bio p h y s Bio c hem Res Comm 150, 1263, 1988. 18) Whit so n J, Selkoe D, Cotm an C: Amy lo id beta pro te i n enhances the surviv a l of hip po camp a l neurons in vit ro . Scie n ce 243. 1488, 1989. 19) Yanker B, Dawes L, Fis h er S, Komaroff L, Oste r -Granit e M, Neve R: Neuroto x ic ity of a frag m e nt of the amy lo id pre cursor associa t e d wi th Alzheim er's dis e ase. Sc ien ce 250, 417, 1989. 20) Kamo H, Ki m SU, McGeer P, Shin D: Functi on al recovery in a rat model of Parkin s on's dis e ase foll owi ng tra nspl a nta t i on of cul- tur ed human sym p a th e ti c neurons. Brain Res 397, 372, 1986. 21) Redmond D, Natt ol i n F, Collie r T, Leranth C, Robbin s R Roth R: Cryo p re servati on , cultu r e and tra nspl a nta t i on of human fetal mesenceph ali c tiss ue int o monkeys . Sc ien ce 242, 768, 1988. 22) Lind vall 0, Sawle G, Wi dn er H, Roth w ell J, Bjo r klund A, Brooks D, Brundin P, Marsden D, Odin P, Rehncrona S. Ev ide nce for long -ter m surviv a l and fun cti on of dop a mi ne rgi c graf t s in pro g res siv e Parkin s on's dis e ase. Ann Neurol 35, 172, 1994. 23) Ki ku chi S, Ki m SU: Gluta m ate n~uroto x ic ity in mesencep h ali c dop a mi ne rgi c neurons in cultu r e. J Neursci Res 36, 558, 1993. 24) Olney J, Sharpe L: Brain lesio n s in an inf a n t rhesus monkey tre ate d wi th monosodiu r n glu ta m ate . Sc ien ce 166, 386, 1969. 25) Choi D: Gluta m ate neuroto x ic ity and dis e ase of the nervous
sys t e m . Neuron 1, 624, 1988. 26) Sp e ncer P, Nunn P, Hug o n J, Ludolph A, Ross S, Roy D: Guam ALS-pa rkin s onis m -dementi a link ed to a pla nt exc itan t neur- oto x in . Sc ien ce 237, 517, 1987. 27) Wolfg ram F, Mey e rs L: Amy o tr o p h ic late r al sclerosis . Ef fec t of serum on ante r io r horn cells in tiss ue cultu r e. Sc ien ce 179, 559, 1973. 28) Mi ch ik a wa M, Lim K, McLarnon J, Ki m SU: Oxy ge n radic a l ind uced neuroto x ic ity in spi na l cord neuron cultu r es. J Neuro-sci Res 37, 62, 1994. 29) Mats u moto S, Sneid e r L, Kawai A, Yonezawa T: Furth e r stu d ie s on the rep lica ti on of rabie s -lik e vir u s in orga niz e d cult ur e of mammali an nervous tiss ue. J Vi ro l 14, 981, 1974. 30) Ecob-Jo h nsto n M, Bornste i n M, Rain e C: Long -ter m inf e c ti on of cultu r ed hamste r dorsal root ga ng li a wi th Halle SSPE vir u s. Neurop a th Ap pl Neurobio l 3, 267, 1977. 31) Feldman L, Shep pa rd R, Bornste i n MB . Herpe s sim p le x vir u s-host cell relati on ship in orga niz e d cultu r es of mammalia n nervous tiss ue. J Vi ro l 2, 621, 1968. 32) Piz e r L, Ki m SU, Ny st r o m P, Coate s V: Herpe s sim p le x vir u s rep lica ti on in ph ep c hromocy tom a cell line tha t respo nds to NGF. Acta Neurop a th 44, 9-14, 1978. 33) Wata b e K, Said a T, Ki m SU: Human and sim i an glial cells infec te d by HTLV- 1 i n cultu r e. J Neurop a th Exp Neurol 48, 610, 1989. 34) Iwasaki Y, Tsukamoto T, Terunuma H, Osame M: Mechanis m of neural tiss ue damag e in human retr o vir u s inf e c ti on . In: HTLV-1 and the Nervous Sy st e m , Roman G and Osame M(Eds), p 325, Lis s , New York, 1989.
35) Watk in s B, Dorn H, Kelly W, Armstr o ng R, Pott s B, Dubois - Dalcq M: Sp e c ific tro p ism of HIV-1 for mi cro g lial cells in pr im ary human brain cultu r es. Sc ien ce 249, 549, 1990. 36) Caceres A, Kosik K. Inhib i t ion of neurit e p o larity by tau anti se nse oli go nucleoti de s in pr im ary cerebellar neurons. Natu r e 343, 461, 1990. 37) Luskin L, Pearlman A, Sanes J: Cellli ne ag e in the cerebral cor tex of the mouse stu d ie d in viv o and in vit ro wi th a recombin a nt retr o vir u s. Neuron 1, 635, 1988. 38) Morett o G, Xu R, Walker D, Ki m SU: Co-e x p r essio n of mRNA for neurotr o p h ic fac to rs in human neurons and glial cells in cultu r e. J Neurop a th Exp Neurol 53, 78, 1994.
용어해섣 계대배양 Subcul t ure 세포가 증식하여 배양기 바닥에서 밀도가 높아 지면 이들을 희석하여 새 배양기에 옮기게 되는데 이것이 계 대 배 양이 다. 계 대 Passa g e 라 하기 도 한다. 글리아세포 G li al cell 신경계는 뉴론과 글리아세포의 두 가지 세포로 구성된다. 글리아세포에는 아스트로사이트, 울리고덴드로사 이트, 미크로글리아 세 가지로 구·성되며 뇌실을 싸고 있는 상의 세 포 epe d ym a l cell, 망막의 뮬러 세 포 Muller cell, 소뇌 피질의 버그만세포 Ber gm an cell 도 특이한 글리아세포의 일 종으로 간주된다. 수초를 형성하는 글리아세포로서 중추신경 계에서는 을리아덴드로사이트, 말초신경계에서 슈완세포 Schwann cell 가 있다. 기 관배 양 Or gan cultu r e 기 관 전체 혹은 그 일부를 조직 배 양 조건에 서 보존하고 조직 분화를 관찰하고자 하는 배양법. 기 질 Subs t ra t e 배 양세포가 발육할 수 있느 유리나 풀라스틱 딧슈, 풀라스크 등의 배양기 표면, 그리고 콜라겐, 젤라틴, 폴리라 이신 등 배양기 표면을 덮은 물질을 총칭하여 말한다. 내피 세포 Endo th e li al cell 혈관의 내 강을 구성 하는 세포로서 배 태 의 중배영에서 유래한다. 뇌혈관 내피세포는 다른 조직 유래 내 피세포와 생리적 성질이 다르다고 한다. 뉴로필라멘트 Neuro fil amen t 신경세포 골격 구성 성분의 하나로 세 포체에서 돌기(축색, 수상둘기)에 걸쳐 길이로 뻗어 있는 섬 유 모양의 구조물이다. 이것은 세포막 및 소포, 미토콘드리 아 등 세포내 구조물과 연관되어 있고 그들 상호간에도 연결
이 있다. 뉴론 Neuron 신경세포, 죽 신경계의 구성단위이다. 이것은 세포체와 두 개의 돌기인 수상돌기와 축색으로 이루어진다. 단충배양 Monola y er cultu r e 세포를 배양기 바닥에 부착하여 증식하 는 경우를 말하며 대다수의 세포배양이 이 형식으로 증식한 다. 동결 보존 Cr y o preserva ti on 섭 씨 영 하 100 도 이 하에 서 세 포, 조직 , 배태 (엠브리오), 종자, 정자 등을 동결하여 저장하는 기술. 모노클론 항체 Monoclonal anti bo dy B 임파세포의 클론이 생산하는 면 역항체를 말하는데 보통 비장 유래 B 세포와 골수종세포의 잡 종세포에서 생산되는 항체를 의미한다. 무균기 술 Ase ptic tec hniq u e 조직 배 양에 서 곰팡이 , 세 균, 바이 러 스, 마이코플라스마 등의 미생물이 조직배양 속에 들어가지 않도 록 예방하는 기술이다. 무한증식 Inf ini t e g row th 초대 배 양 세 포의 계 대 를 계 속하다가 증식 을 무한히 할 수 있는 능력을 가지게 된 수립세포계 es t ab li shed cell li ne 를 말한다. 무혈청배양액 Serum free mediu m 배양세포에게 영양을 공급하는 배 양액으로서 혈청을 포함하지 않고 그 구성 성분이 알려져 있 는 무기염, 비타민, 아미노산, 호르몬, 성장인자로만 되어 있는배양액 배가시 간 Doub li n g tim e 배 양 세포수가 두 배로 증가하는 데 소요되 는시간. 배양액 Med i um 무기염, 아미노산, 비타민 그 밖의 영양분을 포함한 용액으로서 그 속에서 배양세포가 최소한 24 시간 이상 생존 할 수가 있다. 부유배 양 Sus p ens i on cultu r e 세 포가 배 양기 바닥에 부착하지 않고 부 유한 상태에서 증식하는 경우이며 혈액 유래세포, 복수세포 등이 이 상태에서 발육한다.
부착인자 A tt achmen t fac to r 배양기 표면에 코팅하여 배양세포의 접 착을 크게 증가시키는 물질로서 폴리라이신, 피브로넥틴, 라 미 닌, 콜라겐 등이 있다. 분리 세포배양Di sso ci a t ed cell cultu r e 조직을 효소적 처리나 기계적 처리에 의해서 단독세포로 분리한 뒤 아를 배양하는 기술로 서, 트립신과 콜라게나제에 의한 효소분리법이 가장 많이 사 용된다. 분열지수 M it o ti c ind ex 일정 시간 안에 분열하는 세포수를 전체 세 포수에 대하여 퍼센트로 표시한 지수. 분화Diff eren ti a ti on 배양세포에서 본래의 유래세포에서 보이는 특이 한 형태와 기능이 그대로 보존되는 현상• 세포골격 C yt oskele t on 각종 세포 특히 뉴론은 여러 세포골격 단백들 의 협동으로 그 기능과 형태가 유지된다. 세 가지 종류로 대 별된다. 액틴 ac ti n 과 그 관련 단백, 미세관 m ic ro t ubule 과 그 관련 단백 그리고 뉴로필라멘트 neuro fil amen t가 그것이다. 세포계 Cell li ne 초대베양에서 시작된 세포가 계대배양에 의해서 증 식하고 수대 이상 배양이 가능한 경우. 세 포기 질 Cellular matr i x 배 양세 포가 배 양 기 구 표면에 용이 하게 부착 하도록 하는 물질로서 피브로넥틴, 라미닌, 콜라겐 등이 있 다. 부착인자라 불리기도 한다. 세포밀도 Po p ula ti on densit y 배양기의 단위면적 안에 있는 세포수. 세 포 밀 도 증식 저 지 현 상 Dens ity depe n dent grow th inhibi t ion 세 포가 증식하여 세포밀도가 높아지면 세포의 증식이 저지된다. 세포배양 Cell cultu re 분산된 세포를 시험관 속에서 in vit ro 24 시간 이상 발육시키는 방법. 세포융합 Cell fus io n 두 세포가 융합하여 하나의 세포가 되는 것이 며, 실험적으로는 바이러스, 폴리에틸렌글리콜, 전기융합에 의해서 융합조작을 한다. 수립주세포 Es t ab li shed cellli ne 초대배양에서 시작된 세포가 무한증
식성을 가진 경우이며 단일세포가 증식하여서 그 자손만으로 구성된 세포계를 수립주세포라 한다. 수초 M y e li n 축색을 여러 겹으로 둘러싸고 있는 막으로 충추신경계에 서는 올리고덴드로사이트에서 만들어지고 말초신경에서 슈완 세포가 만든다. 이는 전기적 절연체의 역할을 하여 신경전도 룰 빠르게 하는 데에 관계된다 sal t a t or y conducti on . 슈완세포 Schwann cell 축색으로 이루어전 말초 신경섬유는 슈완세포 로 둘러싸여 있다. 말초신경에는 유수섬유와 무수섬유가 있 으며 유수섬유는 한 개의 축색을 수초형성을 이루는 슈완세 포가 둘러싸고 무수섬유는 한 개 또는 그 이상의 축색둘이 슈완세포 돌기에 둘러싸여 있으나 수초 형성을 이루지 않는 다. 시냅스 S yn a p se 뉴론과 뉴론 사이의 기능적 연결부위이다. 이 연결부 위를 통하여 뉴론간의 신호전달이 이루어지며 화학적 전달물 질로 신호전달이 일어나는 것을 화학적 시 냅스 chem ic al s yn a p se 라 하고 전기적으로 신호전달이 이루어지는 것을 전 기 적 시 냅 스 elec t r ic al s y nap se 라 한다. 신경성장인자 Nerve grow th fac to r 신경의 성장 촉진을 유발하는 거 대 분자로서 독사의 뱀독과 생쥐 악하선에서 발견된 것을 시 작으로 여러 동물 조직에서 얻을 수 있다. 신경세포 이동 Neural mi grat i on 척추동물의 신경세포는 성숙한 뇌에 서 영주하는 장소와 별개의 장소에서 발생된다. 신경세포 이 동이란 세포가 기원하는 장소에서 종착지인 성숙 뇌의 영주 장소로 이동하는 현상을 말한다. 이는 축색의 성장과는 다른 것이다. 신경 전 달물질 Neuro t ransm itt er 뉴론과 뉴론 사이 또는 뉴론과 주효 장기 사이의 집합부에서 신경자극을 전달하는 화학적 물질이 다. 말초신경에서 잘 알려진 아세틸콜린, 노르에피네프린을 비롯하여 중추신경계에서 GABA, 글리신, 글루타민산 등 아
미노산과 도파민, 세로토닌, 히스타민 등의 아민류 그리고 엔돌핀, 엔케팔린, VIP, 뉴로텐신 등의 펩티드 등 약 50 종 이 거론되고 있다. 신경영양인자 Neuro t ro ph i c fac to r s 뉴론이 생체에서나 배양중에서 장기간 생존하는 데 필수적인 성장인자를 총칭하여 신경영양 인자라 하는데 신경성장인자 NGF 가 1950 년대에 처음으로 발 견된 이후로 다수의 신경영양인자가 발견되었다. NGF, BDNF, NT-3, NT- 4, CNTF, FGF, EGF, PDGF, TGF 등이 현재 알려져 있다. 알츠하이머 병, 파킨슨병, 헌팅턴 병 등 신경변성 질환은 신경세포가 신경영양인자의 결핍으로 변성 사함으로써 기인된다는 설이 유력하다. 악성 형질전환 Ma lig nan t tra nsfo r mati on 형질전환한 세포가 정상조 직을 무질서하게 침투하는 능력을 갖게 되는 현상. 영류용액 Balanced salt soluio n 배양액 중에서 나트륨, 칼륨, 칼슘, 마그네슘 등의 무기이온을 공급하고 삼투압과 p H 를 조절하 며 각종 시약의 용매로 사용하는 용액이다. 얼 Earle, 핸크스 Hanks, 가이 Ge y 등 여러 처방이 있다. 유전자형 Geno typ e 세포의 유전 형질의 특징. 유한증식 F i n it e grow t h 생물체에서 분리되어서 배양되는 세포는 대 부분이 계대를 계속하면 중도에서 증식이 중지된다. 이배체세포 D iplo i d 성영색체를 제의한 모든 염색체의 수가 이배수 있으며, 세포가 유래된 원래 동물체의 세포와 형태와 수가 동일한 경우 사람에서는 46, 마우스에서는 36, 래트에서는 40 이 정상이다. 이식편 배양 Ex p lan t cultu re lmm3 정도의 조직편을 콜라겐이나 젤라 틴과 갇은 기질 위에 올려놓고 배양하며 조직의 원래 형태나 기능을 가능한 한 유지하면서 배양하는 기술. 잡종세포 H y br i d 두 가지 다론 종류의 세포가 세포융합 기술에 의해 하나의 세포막 안에 들어가서 단일세포가 되는 것.
접촉억제 현상 Con t ac t inh ib i t ion 세포가 증식하여 배양기 바닥에 빈 틈없이 발육하고 세포가 서로 접촉하게 되면 세포증식이 저 지되는 현상이다. 조직 배 양 T i ssue cultu re 세 포, 조직 혹은 기 관을 시 험 관 속에 서 in vit ro 24 시간 이상의 기간 동안 보존 혹은 발육시키는 방법을 의미하여 세포배양 cell cultu r e, 조직배양ti ssue cultu re, 기관 배양 or g an cultu re 그리고 식물조직배양p lan t tiss ue cultu re 이 여기에 포함된다. 조직추출액 T i ssue extr a ct 천연배양액에 속하는 물질로서, 계태추 출 액이 가장 보편적으로 사용된다. 이 밖에도 근육추 출 액, 뇌 추출액에서는 특징적인 성장촉진인자가 발견되었다. 조 직추 출액을 배양액의 일부로 첨가하면 세포증식과 분화촉진의 효 과가 있다고 보고되고 있다. 증식곡선 Grow t h curve 세포를 배양하면서 정기적으로 세포수 를 계 산하여 이것이 두 배가 되는 시간을 계산한다. 착상효율 Pla ti n g eff icien cy 일정 시간 안에 도입된 세포 가운데 배양 기구 표면에 착상한 수를 퍼센트로 표시한 것. 천연배양액 Na t ural mediu m 배양세포가 생존하고 증식함에는 배양액 속에 혈장, 혈청, 조직추출액, 효모추출액, 스킴밀크 등의 천연배양액이 일부분 포함되어야 한다. 천연배양액에서 가장 흔히 쓰이는 것이 혈청이며 이에는 우태아 혈청fet al bovin e serum, 신생우혈청 ca lf serum 그리고 마혈청 horse serum 등 이 있다 . 초대배양 P ri mar y cultu re 동물조직이나 기관에서 직집적으로 분리된 세포나 조직편을 배양조건하에서 발육시키는 기술, 초대배양 이 성공적으로 계대배양되면 세포계 cell li ne 가 된다. 카테콜아민성 뉴론 Ca t hecholam i ne neuron 벤젠 링에 두 개의 -OH 기와 또 하나의 에틸아민 측쇄를 가전 화합물, 죽 도파민, 노르에피네프린, 에피네프린을 말하는데 이들 카데콜아민에
의해서 자극 전달이 아루어지는 뉴론을 말한다. 콜린성 뉴론 Chol i ner gic neuron 아세틸콜린에 의하여 자극전달이 이 루어지는 뉴론을 말하며 말초 부교감신경, 운동신경 그리고 자율신경 절전 섬 유 pr eg a ng li o n ic fi ber 가 이 것 에 속하고 중 추신경계에도 콜린성 뉴론이 존재한다. 클로닝 Clon i n g 소수의 단독세포를 페트리집시나 멀티웰 플레이트에 뿌려서 단일세포의 자손으로만 된 세포 집단을 얻는 기술. 클론 Clone 단일세포가 증식하여서 세포 집단을 만드는 것. 클론효율 Clon i n g eff icien cy 도입된 단일세포 가운데서 클론을 만드 는 세포수 를 퍼센트로 표시한 것. 탈분화 Ded iff eren ti a ti on 배 양세 포에 서 본래 의 유래 세 포에 서 보는 형 태와 기능이 상실되는 상태로서 분화의 반대. 포화밀도 Sa t ura ti on densit y 특정 배양조건하에서 배양기의 단위면적 안에서 발육할 수 있는 최고 세포수. 표현형 Pheno typ e 배양세포가 표현하는 형태적, 생화학적 그리고 생 리적인 특징. 합성 배 양액 S yn t he ti c mediu m 배 양세포가 생 존하고 증식 함에 있어서 최소한으로 필요로 하는 영양분인 무기이온, 아미노산, 비타 민, 핵산 영기, 회금속염을 포함하고 있는 배양액으로서 199 배양액, 이글 MEM, 행 F-12 등이 이에 속한다. 핵형 Kar y o typ e 염색체는 유전자를 지니는 실체이며 DNA, 단백질 그리고 소량의 RNA 를 포함하며 동물의 종류마다 고유의 형 태와 수를 가지고 있다. 이를 핵형이라 하는데 악성화한 세 포, 선천성 질환 환자의 세포, 바이러스나 방사선에 쪼인 세 포에서는 핵형에 변화가 온다. 형질유도 Induc ti on 자국에 의해서 세포형질 표현이 증가되는 현상이 며, 예컨대 호르몬 자극에 의해서 배양세포에서 효소생산이 증가하는 경우를 말한다. 형질전환 Trans fo rma ti on 바이러스, 방사선, 발암물질 등에 의해서
배양세포가 세포 형태와 세포 성질이 영구히 변화하는 것.
찾아보기
기 각종 멸균법의 장단점 40 간접면역염색법 325 갈락토세레브로시드면역염색 216 개방계배양 106 건 조 콜 라겐 57 겐타마이신 51, 52 계대배양 75 계배 골격근 배양 250 계배 교감 신경절 배양 96 계배 망막 배양 123 계배 모양체 신경절 배양 109 계배의 적출 92, 93 계배 중추신경조직 배양 113 계배 척수배양 114, 119 계배 척수후근신경절 배양 94, 96, 100, 104 고속원심분리기 26 고압 증기멸균(오토클레이브) 23 곤충 신경조직 배양 276, 279 골격근 배양 249 공기 여과장비 23 교감신경 철 배 양 96, 133 근위축성 측색경화증 (ALS) 1:;1,
384 근육(골격근) 배양 249 근육 조직 수립세포주 305 글리아 산성섬유 단백 면역 염색 223, 227, 326 글리아 성장인자 (GGF) 203, 231 굴리 오블라스토마 302 금붕어 신경조직 배양 273 금속제거제 81, 85 기계적 세포분리 85 기구 세척 37 기앙 • 바레 신경영 199, 379 기형종 세포배양 310 김사염색법 352 L 나일론 멧슈 봉지 85, 86, 255 나트륨탄산 용액 46 날리딕스산 52 내피세포 성장인자 241 노인반 380 농도 구배 원심분리법 26 뇌종양 유래 수립세포주 302 뇌종양 초대 배양법 283 뇌하수체 세포배양 256 뇌혈관 관문 379 뇌혈관 내피세포 배양 238 뉴로블라스토마 292, 302
뉴로필라멘트 면역염색 142, 179 닛슬 신경세포 염색 359 C 다발성 경화증 380 달백코 배 양액 47, 48 대뇌 배양 113, 144 데시케이터 69 디메틸설폭사이드(D MSO) 79 디스파제 84 디아미노벤지딘(D AB) 331 DNA 분해 효소 (DNase) s2 DNA 합성저해제 182 근! 라미닌 58 래트(성숙) 교감신경 철 배 양 168 래트(성숙) 뇌혈관 내피세포 배 양 239 래트(성숙) 망막 배양 174, 175 래트(성숙) 척수 후근신경절 배 양 162 래트(신생) 교감신경 철 배 양 133 래트(신생) 뇌하수체 세포 배양 254
래트(신생) 대뇌 배양 144 래트(신생) 미크로글리 아 배 양 236 래트(신생) 부신수질 크로마핀 세포 배양 265 래트(신생) 삼차신경 절 배 양 135 래트(신생) 선조체 배양 144 래트(신생) 소뇌 배양 138 래트(신생) 아스트로사이트 배양 226 래트(신생) 울리고덴드로사이트 배양 218 래트(신생) 좌골신경 슈완세포 배양 205 래트(태생) 골격근 배양 252 래트(태생) 슈완세포 배양 200, 202 래트(태생) 척수 배양 146, 150 래트(태생) 척수 후근신경절 배 양 126 래트(태생) 해마 배양 156 래트(태생) 혹질 배양 152 러버풀리스맨 75, 87 로즈 Rose 챔버 69 롤러드럼 116 롤러 듀브 배 양법 114, 116, 138 리신 렉틴 염색 234 리 톤 Le ig h t on 배 양기 69
리포펙틴 298 □ 마우스(성숙) 교감신경절 배양 168 마우스(성숙) 뇌혈관 내피세포 배양 239 마우스(성 숙) 망막 배 양 174, 175 마우스 (성숙) 척수후근신경절 배양 162 마우스(신생) 교감신경절 배양 133 마우스(신생) 뇌하수체 세포 배 양 257 마우스(신생) 대 뇌 배 양 144, 189 마우스(신생) 부신수질 크로마핀 세포 배양 265 마우스(신생) 삼차신경절 배양 135 마우스(신생) 선조체 배양 144 마우스(신생) 소뇌 배양 138. 마우스(신생) 아스트로사이트 배 양 226 마우스(신생) 올리고덴드로사이 트 배양 218 마우스(신생) 좌골신경세포 배양
205 마우스(태생) 슈완세포 배양 200. 202 마우스(태생) 척수배양 146, 150 마우스(태생) 척수 후근 신경절 배양 126 마우스(태생) 해마 배양 156 마우스(태생) 혹질 배양 152 마이코스타틴 51, 52 마이크로캐리어 72 망막 세포 배양 123, 270 망사학설 17 매크로파지 232 맥시모우슬라이드 배양법 69, 100, 102 MAP2 면 역 염 색 142, 160 멀티웰-커버글라스 배양법 144 멀티웰챔버 (플레이트) 35, 36 메틸그린-피로닌 염색 354 메틸염화수은 244 면역세포화학 324 면역염색법 324 멸균 시설 24 모노아민 산화 효소염 색법 368 모양체 신경영양인자 109, 111 모양체 신경절 배양 109 무균 검사 51 무균 배양실 23 무균 벤치 23, 29
무균조작 73 무척추동물 신경조직 배양 276 무혈청 배양액 62, 229 무혈청 배양액 조성 67, 229 미엘린단백 321 미크로글리아 순수배양 232 미크로글리아의 활성화 385 l:l 바로크 시대 20 바이러스 감염증 385 바퀴벌레 신경조직 배양 279 배양기구 32 배양기구 멸균 37, 38 배양세포의 성장과 증식 배양액 41 배양액 교환 74 배양준비실 23 변환성장인자 65 보디안 은염색법 143, 355 봉임제 336 부교감계 신경계 배양 109 부신 갈색종 세포 배양 306 부신수질 크로마핀 세포 배양 264 부유 배양법 72 부착인자 53 뷔크너 깔때기 나일론 멧슈 제작
214, 224 브로모디옥시유리딘 항체 342 비디오 카메라 31 비트로넥틴 59 人 사 람 (성 인) 교 감 신 경 절 배 양 171 사람(성인) 뇌하수체 세포 배양 259 사람(성인) 망막배양 177 사람(성인) 슈완세포 배양 207 사람(성인) 울리고덴드로사이트 배양 216 사람(태아) 대뇌 신경세포 배양 193 사람(태아) 척수 후근 신경절 배 양 186 사이 크스-무어 (Sy k es-Moore) 챔 버 69 사진촬영장치 31 산소라디 칼 381, 384 상피성장인자 (EGF) 65 샌드위치법 284 생체염색 355 생체활성물질 72 선회배양법 71 설퍼메독사졸 52
섬유아 성장 인자 (FGF) 65 성숙동물 말초신경조직 배 양 161 성숙동물 중추신경조직 배양 163 세븐엑스 37 세포골격 단백 319 세포기질 53 세포내 칼슘 농도 측정 123 세포동결보존법 78 세포 분리 기술 81 세포성 면역 380 세포성장인자 64 세포수 계산법 76 세포영색법 351 세포운반 80 세포융합 292 세포 증식의 판정 338 세포 타입 특이마카 317 세포 화학적 염색법 362 세포 활성의 측정 346 소뇌 배양 113, 138 수단블랙 미엘린 염색법 357 수립세포계 279 수술기구 35, 36 수초(미엘린) 염색 357 수초 형성 19, 100 슈나이더 영류 용액 277 슈와노마 302
슈완세포 배양 199 스크레이퍼 87 스터링 마그네트 72 스데인리스스틸 멧슈 85, 86 스트렙토마이신 51, 52 습성 콜라겐 57 시험관 32, 34 신경계 수립세포주 301, 305 신경변성질환 381 신경섬유종 199 신경성장인자 (NGF) 22, 65, 107 신경세포 순수 배양 180 신경세포의 순수 배양 180, 187 신경 아세포종 세포 302 신경 영 양인자 308, 381 신경영양인자의 결핍 381 신경원섬유 변화 382 신경전달물질 322 신경 전달물질 관련 효소 322 신경조직배양의 역사 13 신경조직배양 창시자 17 신틸레이션 카운터 340 실험실 기구 ’ 23 실험실 설비 23 。 A2B5 항체 227 ABC 염색법 331
ATCC( 미국 세포은행) 304 아밀로이드 382 아세틸콜린 에스데라제 염색법 363 아스트로사이트 순수배양 225 알츠하이머병 158 암포테리신 B( 환기존) 51, 52 액성면역 380 양생류 신경조직배양 269 어류 신경조직배양 269 에칭 배양법 283 여과 멸균기 (여과필터) 25, 26 열 영류용액 44 영류용액 42, 43 L-15 배 양액 49, 50 오본 24 오토라디오그라피 339 오토클레이브 21, 25 올리고덴드로사이트 순수배양 209 올챙이 척수배양 16, 270 원심분리기 26, 27 원실 듀브 32 위상차 도립 현미경 31 유전자 이입 297 이글 배양액 47, 48 이식편 배양 21 이온교환 컬럼 23, 26 인산염 완충용액 (버퍼) 45
인슐린 67 인슐린 유사성장인자(I G F) 65 인온 스퍼터 376 인큐베이터 24, 28 인터루킨 -2 65 일구구 199 배양액 47, 48 MTT 검사법 348 ELISA 비색계 347 F-12 배 양액 47, 49 HGPRT 결손 세포주 291 NADPH 데트라졸륨 환원 효소 염색 365 NCTC 135 배 양액 47, 49 大 자가면역반응 199 잡종세포 제작법 300 전기적 세포융합법 295 전자 현미경 369 전자현미 경 표본제 작법 371 정치배양법 68 제 8 인자 241 젤라틴 60, 254 조직소편(이식편) 배양법 284 조직 추출액 41 종양 괴사인자 65 좌골신경배양 205 주사 전자현미경 374
중성능 원심분리기 26, 27 중성세제 37 증류수 장치 23, 26 직접면역염색법 325 天 척 수 배 양 114, 119, 146, 150, 270 척수 후 근신경절 배양 96, 126, 162 천연 배양액 42 초대 배양법 89 초파리 신경조직배양 276 더 카나마이신 51, 52 카데콜아민 신경 형광영색법 365 커버글라스 32 콜라게나제 83 콜라겐 54 콜라겐 분리법 56 큐비즘 시대 20 크로마핀 세포 264 크리스탈 바이올렛 검사법 346 클로닝 실린더법 288
E 탁상 저속원심분리기 26, 27 탈수 질환 380 테라토카시노마(기형종세포) 배 양 310 테트라사이클린 51, 52 투과전자현미 경 370 트립신 82 트립신 억제제 82 티로신 수산화 효소 염색 268 티미딘 340 티오글리콜레이트 51 立 파킨슨 병 153, 264, 383 파파인 84, 176, 196, 275 패닝에 의한 세포분리 194 패닝접시 제작법 198 패치 클램핑 123 패트리집시 배양법 104, 105, 131 퍼 콜 밀도구배 컬 럼 165, 210, 242 페놀레드 용액 46 페니실린 51, 52 페록시다제 표지 항체 염색법 330
페트리집시 32, 34 폐쇄계 배양 106 폴리라이신 60 폴리비닐알코올-글리세린봉임제 336 폴리에틸렌글리콜 세포융합법 292 폴리에틸렌이민 61 프로나아제 83 풀라스크 32, 33 피브로넥틴 59 피펫 32 피펫 에이드 36 PAP 면역 염색법 333 RPMI 1640 배양액 50 o-_ 합성배양액 20, 46, 47 항생물질 51 항체 의존 세포분리 기술 194 해마 배양 156
해마목시린 염색 353 해부입체현미경 30 핸크스 영류용액 44 행 FIZ 배양액 47, 49 헤파린 241 혈관 내피세포 배양 238 혈관 내피세포 성장인자 65 혈소판 성장인자 (PDGF) 65 혈장 42 혈청 42 혈청의 영양 역할 62 형광 항체염색법 326 형광현미경 327 호르몬 66 훌샐 클램핑 123 환기존(암포테리신 B) 51, 52 회전 배양법 71 효소 표지 항체 면역 염색법 330 혹질 배양 152 흥분성 아미노산 384 HAT 배양액 292
김승업 1936 년 생. 경기고 2 년때 검정고시로 서울의대 입학 1960 서울대학교 의과대학 졸업 1965 일본 교토(京都)대학 대학원 의학박사 1966~1969 미국 컬럼비아대학 펠로우 1972~1983 미국 펜실바니아대학 신경내과 부교수와 교수 독일 하이델베르크대학, 일본 도쿄(東京)대학, 오사카(大阪)대학 초빙교수 1983~ 현재 캐나다 브리티시 컬럼비아대학 신경내과 마리안 코너 석좌교수 350 여 편의 저서, 논문, 초록 출간 신경계 조직배양 대우학술총서 • 자연과학 103 1 판 1 쇄 찍음 -1996 년 1 월 10 일 1 판 1 쇄 펴 냄 -1996 년 1 월 30 일 지은이一―金承業 펴낸이 __- 朴孟浩 펴낸곳-(주)민음사 출판등록 —19 91 . 12. 20. 제 16-490 호 서울특별시 강남구 신사동 506 대표전화 515-2000, 팩시밀리 515-2007 값 22,000 원 ©김승업, 1996 자연과학 생물학 KDC/472. 028 Prin t e d in Seoul, Korea ISBN 89-374-3593-4 94470 89-374-3000-2 (세트) 지은이와 합의하여 인지를 붙이지 않습니다.
대우학술총서 (자연과학) 1 소립자와 게이지 상호작용 김진의 52 수리분류학 고철환 2 동역학특론 이병호 53 찰스 다윈 정용재 3 질소고정 송승달 여 금속부식 박용수 4 상전이와 임계현상 검두철 55 양자광학 이상수 5 촉매작용 진종식 56 효소반응속도론 서정헌 6 뫼스바우어 분광학 옥항남 57 화성암 성인론 이민성 7 극미량원소의 영양 승정자 58 확률론 구자홍 8 수소화봉소와 유기봉소 화합물 59 분자 분광학 소현수 윤능민 60 벡터속 이론 양재현 9 항생물질의 전합성 강석구 61 곤충신경 생리학 부경생 10 국소적 형태의 Ati ya h-Sin g e r 62 에너지띠 이론 모혜정 지표이론 지동표 63 수학 기초론 김상문 11 Muco p ol y sacchar i des 의 64 신경과학 박찬웅김승업 생화학 및 생물리학 박준우 65 BCH 부호와 Reed-Solomon 부호 12 천체물리학 홍승수 이만영 13 프로스타글란딘 합성 김성각 66 양자 전기역학 김영덕 14 천연물화학연구법 우원식 67 군환론 박재걸 15 지방영양 김숙희 68 대수기하학 조영현 16 결정화유리 김병 호 69 양자 장이론 이재형 17 고분자의 화학반응 조 의 환 70 해양오염과 생태계 심 재 형 18 과학혁명 김영식 71 비기체 연소합성 ( SHS ) 여철현 19 한국지질론 장기홍 72 크로마토그래피 이대운 20 정보이론 한영열 73 곤충의 사회행동 추종길 21 원자핵반응론 정운혁 74 동위원소 지질학 김규한 22 파괴역학 김상철 75 X- 선 결정학 김양 23 분자궤도 이론 이익춘 77 통계역학 조순탁 24 반응속도론 정경훈 78 고분자의 구조와 형태학 이석현 25 미분위상수학 이현구 79 LC 에 의한 광학 이성질체의 분리 26 자기공명방법 조성호 현명호 27 플라스마물리학과 핵융합 최덕인 80 신경전달물질 서유헌 28 천문관측과 분석 이 시 우 81 발생과 유전자 발현 이양 립 29 석탄에너지 변환기술 김상돈 82 스테로이드 화학 김완주김득준 30 해양미고생물학 백광호 83 다체계론 엄정인 31 편미분방정식론 김종식 84 중핵반응론 김병택 32 대통일이론 소광섭 85 비가역 열역학 이철수 33 금속전자계의 다체이론 김덕주 86 등각장론 임채호 34 액정중합체 진정일 87 방사선생물학 남상열 35 복합재료 권숙안 88 석유지질학 이용일 36 단백질 생합성 박인원 89 베르누이 시행의 통계적 분석 37 한국의 광물종 김수진 배도선 · 김성인 38 일반상대론 이철훈 90 신경세포생리학 강만식 39 레이저 광산란 분광학 김종진 91 생리활성을 가진 C — P 화합물의 40 복소다양체론 김상문 화학 김용준 · 강익중 41 역학적 연구방법 김일순 92 생물 유기화학 서정헌 42 핵구조물리학 민동필 93 조직배양 김승업 43 후리에 해석과 의미분 작용소 94 유기 전이금속 화합물 김도한 윤석승이재근·장인순조남숙 44 한국의 고생물 이하영 95 실내환경과학 김윤신 45 질량분석학 김명수 96 유한요소법 정상권 46 급변론 박대현 97 대수적 위상수학 우무하김재룡 47 생체에너지 주충노 98 파인만 적분론 장건수 48 리이만 기하학 박을룡 99 응용미생물학 박무영 49 군표현론 박승안 100 리보플라빈 이상선 50 비선형 편미분 방정식론 하기식 101 노화 김숙희김화영 51 생체막 김형만 102 매트릭스 격리분광학 정기호