
서정헌 1948 년 출생 1971 년 서울대학교 문리과대학 화학과 졸업(이학사) 1975 년 미 국 시 카고대 학 화학과 졸업 (Ph. D ) 미국 노스웨스턴대학 연구원 1987 년 제 1 회 한국과학상 화학부문 수상 1 9<) 3 년 제 7 회 한국과학상 대상 수상 현재 서울대학교 자연과학대학 화학과 교수 100 여 편의 논문이 있음
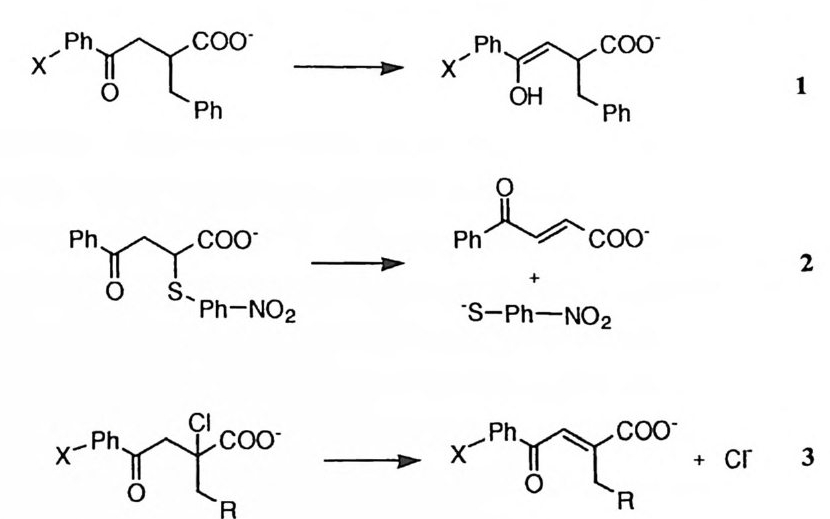
생물유기화학
 생물유기화학
생물유기화학
책머리에 앞으로 화학이 어느 방향으로 발전하겠는가? 인류가 가까운 장래에 극복해 야 할 문제점들을 몇 가지 꼽으라면 에너지, 식량, 환경, 질병 퇴치 등이 있 다. 이러한 문제점을 해결하는 데 화학이 어떻게 기여할 수 있는가를 살펴보면 화학이 발전할 방향을 짐작할 수 있을 것이다. 에너지 문제를 해결하는 방안 중에 태양 에너지를 화학 에너지로 전환하는 것이 있고, 식량 문제에는 인공적으로 광합성에 의하여 식량을 제조하는 것이 궁극적인 해결책이 될 것이다. 훼손된 환경을 복원하기 위해서는 각종 오염 물 질을 안전한 형태로 전환시키는 촉매를 고안해야 할 필요가 있고, 질병 퇴치를 위해서는 질병의 원인을 분자 단위에서 파악하고 이룰 예방 혹은 치료할 수 있 는 유기분자를 설계하는 것을 해결 방안의 하나로 꼽을 수 있다. 화학에서 새로운 발견이 이루어지지 않으면 이와 갇은 방안을 실현하는 것은 불가능하다. 그렇다면 화학에서는 어떠한 작업을 통하여 이러한 문제점을 해결 하는 실마리를 찾아갈 것인가? 앞으로 화학 분야의 학문적 혹은 산업적 활동 의 큰 줄기는 에너지, 식량, 환경, 질병 퇴치 등의 문제점을 해결할 수 있는 기능을 가진 분자를 고안하는 작업과 긴밀한 관계를 가지게 될 것이다. 이러한 기능을 가전 분자를 창안할 때 우선 생체계에서 이루어지는 화학 작 용에 관심을 가질 수밖에 없다. 아것은 아러한 기능을 가장 효율적으로 수행하 는 계가 생체이기 때문이다. 현재로서는 생체가 이러한 기능을 수행하는 원리 롤 파악하고 이를 모방하는 것이 인공적인 시스템을 고안하는 가장 손쉬운 방 법인 것이다. 생물 유기화학은 유기화학적 기법을 사용하여 생물학의 문제점을 해결하고, 생물학적 기법을 사용하여 유기화학의 문제점을 해결하고, 생체를 모방하여 인 공적인 기능성 화합물을 제작하는 학문 분야이다. 이 분야는 최근 2Qa l 년 사
이에 두드러지게 성장하였다. 에너지, 식량, 환경, 질병 퇴치 등의 문제점에 생체 모방적인 기법을 사용하 여 접근하는 작업에서 선도적인 역할을 담당하는 분야 중의 하나로 유기화학과 생물학이 접합하는 경계에 위치한 생물 유기화학을 꼽을 수 있는 것이다. 생물 유기화학이 본격적으로 발달한 연륜이 짧기 때문에 국내뿐 아니라 국의 에도 종합적인 교과서로 만족스러운 것이 드문 형편이다. 이 책은 1990 년과 1992 년에 저자가 서울대학교 화학과 대학원에서 강의한 자료를 중심으로 집필 하였다. 200 자 원고지 1,00W ol- 정도의 분량에 생물 유기화학의 주요 관십 분야를 심 도 있게 기술하는 것은 어려운 일이다. 특히 생물학과 유기화학을 두루 섭렵하 면서 현재 관심의 초점이 모아지고 있는 주제를 모두 다룬다는 것은 불가능한 일인지도 모론다. 우선, 국내의 대학원 학생을 위한 입문서 혹은 이 분야에 관 심을 가진 분들에 대한 소개서 역할이라도 이 책이 제대로 맡아준다면 성공한 것으로 칠 수 있을 것이다. 이 책은 학사과정에서 유기화학과 생화학을 이수한 학생들을 위한 교재의 수 준에서 집필하였다. 이 책이 우리나라의 생물 유기화학 분야의 연구 활동이 국 제적으로 지도적인 수준에까지 발전하는 데 믿거름이 되기를 소망한다. 미비한 부분과 찰못된 부분을 시정하고, 계속 발전해 가는 이 분야의 연구 결과를 보 충할 수 있는 기회가 언젠가는 있으리라 스스로 위로하면서, 이 책이 발간되는 직접적인 계기를 마련해 준 대우재단에게 감사한다. 1994 년 2 월 서정헌
차례
책 머리에 5 제 1 장 서론 13 제 1 절 생물 유기화학의 영역 13 제 2 절 이 책의 구성 15 제 1 부 유기화학의 기법을 이용한 생체 현상의 탐구 제 2 장 효소 작용 원리에 대한 물리 유기화학적 접근 19 제 1 절 효소 반응과 물리 유기화학 19 제 2 절 각종 촉매 작용기의 작동 원리 21 (1) 특정 산 및 일반 산 촉매작용 21 (2) 특정 염기 및 일반 염기 촉매작용 22 (3) 친핵성 촉매작용 22 (4) 정전기적 촉매작용 23 (5) 혐수성 작용 24 제 3 절 인접효과 24 제 4 절 다중 작용기 촉매효과 27 제 5 절 용매효과 27 제 6 철 금속 이온의 촉매작용 29 제 7 절 착물 형성에 의한 촉매작용 29 제 8 절 특정 효소의 반응 메커니즘 구명 31 • 참고문헌 31제 3 장 생체 분자의 유기화학 33
제 1 절 인산 유도체 33 제 2 절 아실 유도체 40 • 참고문헌 44 제 4 장 금속 이온과 유기 생체 분자 46 제 1 절 금속 이온 자체의 촉매작용 47 (1) 천전자체의 활성화 47 (2) 이탈기의 활성화 48 (3) 산의 활성화 50 • (4) 저해적인 역반응 경로의 봉쇄 51 (5) 생산적인 형태의 제공 52 제 2 절 금속에 배위된 물 분자의 촉매작용 53 (1) 친핵체로서의 공격 54 (2) 일반 산 촉매작용 53 제 3 절 금속에 배위된 수산화 이온에 의한 촉매작용 55 (1) 친핵체로서의 공격 55 (2) 일반 염기 촉매작용 56 제 4 절 이핵체 금속 이온의 촉매작용 57 제 5 절 금속 이온과 유기 작용기의 협동 58 제 6 절 복수 개의 촉매 레퍼토리의 작동 59 제 7 절 금속 이온의 성질과 리간드의 구조가 촉매 효율에 마치는 효과 60 제 8 절 전망 61 • 참고문헌 제 5 장 효소반응 메커니즘 연구의 예 : 카르복시펩티다제 A 66 제 1 절 기질의 특이성 66 제 2 절 구조 68제 3 절 Glu-270 의 촉매 역할 69
제 4 절 Tyr-248 의 촉매 역할 85 제 5 절 Zn( Il )이온의 촉매 역할 87 제 6 절 기타 작용기의 역할 88 제 7 절 모형 연구 88 제 8 절 결론 92 • 참고문헌 93 제 6 장 신의약 설계 98 제 1 절 효소 저해 98 제 2 절 수용체 인식 99 제 3 절 DNA 조절 1O1 • 참고문헌 103 제 2 부 생물학의 기법을 이용한 유기화학의 탐구 제 7 장 유기 합성과 효소 107 제 1 절 입체 선택적인 유기 합성에 대한 요소의 이용 107 (1) 효소 작용의 반응 및 기질 특이성 107 (2) 효소의 거울상 이성질 특이성의 이용 113 (3) 효소의 프로키랄적 입체 특이성의 이용 116 (4) 효소 득이성의 복합적인 이용 122 (5) 복수 효소의 이용 123 (6) 동위원소 라벨의 입체 특이적인 도입 124 제 2 절 효소의 유기 용매 127 제 3 절 효소의 가공 130 • 참고문헌 133제 8 장 항체 효소 138
제 1 절 서론 138 제 2 절 항원과 항체 139 제 3 절 과거의 관련 연구 보고 140 제 4 절 항체 효소의 개발 사례 141 (1) 전이상태와 상호 보완적인 항체 141 (2) 특정 촉매 작용기의 유도 145 (3) 유기 합성을 위한 항체 효소 147 (4) 기타 148 • 참고문헌 151 제 3 부 생체 모방 화학 제 9 장 분자 인식 153 제 1 절 분자 인식의 연구 동향 153 제 2 절 시클로덱스트린 154 제 3 절 시클로판 162 제 4 절 칼릭스아렌 167 제 5 절 크라운 에테르 171 제 6 절 기타 176 • 참고문헌 181 제 10 장 막 모방 화학 185 제 1 절 생체 막 185 제 2 절 미셀 187 제 3 절 인공 막 191 • 참고문헌 196제 11장 합성 고분자와 인공 효소 198
제 1 절 미세 환경 198 제 2 절 결합 부위 201 제 3 절 유기 촉매 작용기 204 제 4 절 인공 금속 효소 209 제 5 절 분자 형태 조정 212 • 참고문헌 214 찾아보기 217제 1 장 서론 제 1 절 생물 유기화학의 영역 자연과학을 물질과학과 생명과학으로 구분할 수 있다. 물질과학에는 비생명 체를 다루는 수학, 물리학, 화학, 지구과학 및 이들의 응용분야인 기계공학, 전자공학, 화학공학 등이 포함된다. 생명과학에는 생명체를 디루는 생물학 및 이의 응용분야인 의학(치의학, 수의학 포함), 약학, 농학 등이 포함된다. 화학 과 생물학의 경계에서 물질과학과 생명과학이 접한다고 볼 수 있다. 이미 오래 전부터 화학과 생물학의 경계에서 생화학과 분자 생물학이 형성되어 발전해 왔 다. 근래에는 생화학과 분자 생물학의 구분이 모호해져서 거의 동일시되고 있 다. 이에 따라 생화학은 화학과 생물학의 경계분이라기보다 생명과학의 한 분 야로 독자성을 가지게 되었다. 현재로는 물질화학과 생명과학의 경계에서 교량 역할을 생물 유기화학, 생물 무기화학 및 생물 물리화학이 담당한다고 보는 것이 타당하다. 여기에서는 유 기분자 혹은 무기분자의 물질적 성격을 다루는 화학의 제 분야에서 도출된 지 식을 생명 현상의 구명에 적용하고 또한 생명과학의 지식을 이용하여 분자들의 물질로서의 행동을 연구하고 있다. 모든 생명현상은 유기반응이다. 따라서 생명현상을 가장 정밀한 차원에서 본 격적으로 다루고 있는 분야는 생물 유기화학이라고 불 수 있다. 현재로는 반웅 기 차원에서 생명현상의 원리를 구명함으로써 생명현상을 원자 차원에서까지 연구할수있다. 생물 유기화학을 정의하는 방법은 여러 가지가 있겠지만, 저자는 〈유기화학
의 기법을 이용하여 생명과학의 문제점을 해결하고 , 생물학의 기법을 이용하여 유기화학의 문제점을 해결하며, 생체를 모방한 기능성 유기 화합물을 독자적인 영역으로 연구하는 학문분야 〉 라고 정의하고 있다. 좀더 구체적으로 생물 유기화학이 취급하는 영역을 파악하려면 우선 이 분야 의 국제 학술지인 Bio o rga n ic Chem istry에 두고되는 눈문들이 다루는 다음과 같은 주제들을 살펴볼 수 있다. 1) 새로운 효소반응과 대사 경로에 관한 화학 2) 효소 메커니즘과 효소 구조 3) 효소 및 대사 경로 연구에 대한 반응속도 , 동위원소, 입체 화학 등 기법의 응용 4) 효소, 펩티드 및 기타 생체 분자에 대한 화학적 모형 5) 효소와 조효소를 모방한 촉매의 개발 6) 효소를 이용한 유기 합성 반응 7) 생물학의 발견에서 도출되는 새로운 화학적 원리 8) 메커니즘에 근거하거나 위치 지향적인 효소 저해제의 개발 9) 효소 저해 작용에 대한 화학적 원리 10) 페로몬 등 화학적 정보 매개 물질의 구조와 작용 메커니즘 11) 호르몬 작용의 분자 차원 메커니즘 및 악리 작용의 분자적 근거 12) 수용체의 분자 인식에 관련된 원리 13) 핵산의 화학적 수정 및 이룰 이용한 유전 물질의 조작 또한 이 분야의 국제 학술회의로서 국제 순수 및 응용 화학 연맹 (IUPAC) 의 후원으로 1993 \1 6 월에 일본에서 열린 생물 유기 국제 학술회의에서 다루었던 주제를 살펴봉으로써 현재 생물 유기 분야에서 현재 활발히 이루어지고 있는 연구활동의 성격을 단편적으로나마 파악할 수 있다. 1) 효소의 기능과 모델링 2) 생물학적 및 인공적 수용체 3) 생합성 경로와 생리 활성 물질 4) 유전 공학의 유기화학적 측면
5) 생체 고분자 및 인공 복합분자 배열체 6) 생화학적 과정의 시뮬레이션 생물 유기화학의 연륜은 비교적 짧다 . 생명현상을 유기화학의 차원에서 본격 적으로 연구한 최초의 학자로는 미국의 시카고대학과 하버드대학에 재직한 바 있는 웨스트하이머 교수를 꼽을 수 있다. 웨스트하이머 교수가 연구생활을 시 작한 1940 년대 이후부터 생물 유기화학이 시작되었으며 물리 유기화학적인 기 법을 생체분자의 화학 현상을 구명하는 데 본격적으로 활용한 벤더, 브레슬로, 카이저 교수 등에 의하여 1960 년대 이후 확산되기 시작하여 지금은 수많은 연 구가 이루어지는 분야로 성장하게 된 것이다. 제 2 절 이 책의 구성 생물 유기화학의 전영역을 다루고 있는 만족스러운 교과서나 참고서적은 국 내의를 막론하고 전무한 형편이다. 이것은 생화학과 유기화학의 넓은 영역을 두루 섭렵하면서 생물 유기화학자들이 관심을 가지고 있는 주제를 십도 있게 다루는 것이 한 개인의 능력으로는 쉽지 않기 때문이다. 현재 의국에서 출판된 종합적인 생물 유기화학 서적으로 듀가의 Bio o rga n ic Chemi str y : A Chemi ca l Ap pro ach to Enzym e Ac ti on 이 있다. 그러나 이 책 은 부제에서 볼 수 있듯이 효소의 유기화학적 측면만을 중점적으로 다룬 한계 가 있다. 이 책이 다루는 영역에서 전개되는 연구활동은 잘 소개하고 있지만, 이 책은 구성이 매우 산만하여 소재들 간의 연결이 적절하지 못하다는 결접이 있다. 이 책에서는 제한된 지면 속에서도 학사과정 4 학년 혹은 대학원과정 학생들 이 생물 유기화학에 대한 기초적인 사항에 적절히 접할 수 있도록 세 가지 영 역으로 구분하여 생물 유기화학을 기술하고 있다. 제 1 부에서는 유기화학의 기법을 이용하여 생물학적인 문제점을 해결하는 예 를 몇 가지 다루고 있다. 우선, 효소반응 원리를 구명하는 데 사용되는 물리
유기화학적인 접근방법에 대하여 설명한다. 그리고, 각종 생체분자의 유기화학 에 대한 전형적인 예로 인산 유도체와 아실 유도체의 유기화학을 소개한다. 그 다운]는 유기 생체반응에 대한 금속 이온의 루이스산 촉매작용에 관하여 논의 한다. 유기화학적 기법으로 효소반응 메커니즘을 구명하는 예로 카르복시펩티 다제 A 의 메커니즘 연구를 설명한다. 마지막으로 신의약을 설계하는 데 동원 되는 주요 개념을 설명하고 유기화학이 기여하는 바를 검토한다. 유기화학적인 기법을 사용하여 생물학적 문제점을 해결하는 바는 이의에도 다수 있지만 이 정도로 기본적인 사항은 짚었다고 볼 수 있다. 조효소 등에 관한 유기화학이 다루어지지 않은 점은 아쉽지만, 저자가 수년 전 저술한 『 효소반응속도론 』 을 참조하면 생체분자에 대한 유기화학이 어느 정도 보완될 수 있다고 본다. 제 2 부에서는 생물학의 기법을 이용하여 유기화학의 문제점을 해결하는 예를 다루고 있다. 이 목적으로 유기 합성에 있어 효소가 응용되는 바를 다양하게 설명한다. 최근에 등장한 항체 효소에 대한 소개를 포함시켜, 유기 반응에 필 요한 촉매를 생체에게 주문하여 제작하게끔 하는 것을 다룬다. 제 3 부에서는 생물 유기화학이 가지는 독자적인 영역으로 간주할 수 있는 생 체 모방 화학에 관하여 설명하기로 한다. 생체를 모방하는 데에는 일차적으로 분자 인식이 개입하는데, 이것과 관련하여 최근에 활발하게 이루어지고 있는 각종 호스트 분자에 관한 연구 동향을 논의한다. 생체막이 가지는 독특한 기능 인 자체 배열현상을 모방하여 각종 기능성 거대 분자를 설계하려는 노력이 기 울어지고 있는 막모방 화학도 다룬다. 마지막으로 인공적으로 효소의 특성을 가전 촉매를 제작하려는 시도에 관한 내용을 인공 고분자와 인공 효소라는 제 목으로취급한다.
제 1 부  유기화학의 기법을 이용한 생체 현상의 탐구
유기화학의 기법을 이용한 생체 현상의 탐구
제 2 장 효소 작용 원리에 대한 물리 유기화학적 접근 제 1 절 효소 반응과 물리 유기화학 효소란 모든 생명현상의 원인이 되는 화학반응을 생체의 조건하에서 효과적 으로 진행되게 하는 촉매이다. 효소는 단백질로 구성되어 있으며 경우에 따라 작은 유기 화합물인 조효소나 금속 이온을 포함하기도 한다. 효소를 구성하는 단백질의 형태가 효소의 활성도에 큰 영향을 미치는데, 변성, 화학적 수정 등 으로 형태가 변호円 l 때 효소의 활성이 완전히 상실되기도 한다. 단백질은 아미노산이 펩티드 결합에 의하여 중합됨으로써 얻어진다. 이때 펩 티드 결합의 사슬은 단백질 구조의 척추에 해당한다. 각각의 구성 아미노산은 고유한 견사슬을 가지는데 겹사슬은 성격에 따라 형수성, 극성, 이온성 등으로 구분된다. 효소는 분자량이 수만, 수십만인 거대 분자이다. 그러나 대부분의 경우 기질의 분자량이 수백에 불과하여도 활성을 나타낸다. 효소라는 둥근 거 대 분자 표면에 비교적 작은 분자가 수용되는 틈이 있다. 이것을 활성자리라고 부르는데 효소의 촉매작용은 활성자리 속에서 효소의 촉매 작용기와 기질의 반 웅 부위 간에 일어나는 화학반응으로 간주할 수 있다. 효소 촉매 반응이 여타의 촉매 반응과 구분되는 특징은 다음의 세 가지 점이 다. 첫째로, 효소와 기질 간에 착화합물이 형성된다. 둘째로, 105-101 내에 달 하는 고도의 증속효과를 나타내어 생체의 조건하에서 필요한 모든 화학반응이 신속히 진행되게 한다. 셋째로, 관여하는 반응의 유형과 기질의 구조에 대하여 고도의 선택성을 보이는 특이성을 가진다. 이러한 효소 촉매 반응의 특칭은 기질이 활성자리 속에 갇히어 착화합물을
형성하기 위하여서는 효소가 제시하는 구조적인 요건을 충족시켜야 하며, 이러 한 과정으로 형성된 생산적인 착화합물에서는 효소의 촉매 작용기와 기질의 반 응부위가 효과적으로 반응을 일으킬 수 있도록 서로 인접해 있다는 것으로 설 명이 가능해진다. 이 장에서는 효소가 기질과 착화합물을 형성하는 과정과 효소-기질 착화합물 에서 촉매 작용기가 고도의 증속효과를 시현하는 과정에 개입되는 촉매 요인을 물리 유기화학적 기법으로 도출하거나 확인하는 예를 소개하고자 한다. 효소 반응은 모든 생명현성을 일으키는 화학반응에 해당되므로 그 메커니즘 울 구명하는 것은 생명현상을 작용기적 치원에서 밝히는 것을 의미한다. 그런 데 효소는 거대 분자이어서 그 반응 메커니즘을 연구하는 데 초점을 활성자리 에만 맞추어도 현대과학이 보유한 기법으로는 단지 피상적이며 단편적인 정보 만을 얻을 수 있을 뿐이다. 크기가 작은 유기분자의 반응 메커니즘은 최근 3, 40 년간 물리 유기화학적 지식이 축적되어 정교한 차원으로 이해되고 있다. 이에 반하여 개별적인 효소들의 반응 메커니즘을 유기 반응 메커니즘이 규명된 정밀도로 파악하는 것은 불가능하다. 이 생진체행내되에고서 는있으 산며화 환이원에, 개합입성되, 는분 해효,소 의재 배종치류 도등 매거우의 다모양든하 다종.류 의” 이화렇학듯반 무응 수한 효소에 대하여 반응 메커니즘을 일일이 결정하는 것은 현재로서는 불가능 한 일이다. 그러나 생명현상의 기본적인 작동 원리를 이해하려는 자연과학의 가장 큰 목적을 달성하기 위해서는 효소 반응 메커니즘의 규명을 게을리할 수 없다. 물리 유기화학적 기법을 이용한 효소 촉매 요인의 규명에서는 효소에서 작동 하리라고 추측되는 촉매 요인을 비교적 소형이며 인공적으로 합성된 유기 화합 물을 구사하여 시험하고 있다. 소형인 유기 화합물에서 작동하는 촉매 요인이 효소에서도 가능하리라고 가정하는 것은 타당성이 있다. 이러한 촉매 요인은 개별적인 효소의 메커니즘을 연구할 때 단위 과정으로 간주되며 복수의 단위 과정이 복합적으로 참여하는 것으로 고도의 촉매효과를 설명할 수 있는 것이다.
제 2 절 각종 촉매 작용기의 작동 원리 효소의 활성자리 속에 위치한 각종 아미노산의 건사슬은 효소의 촉매작용에 중요한 역할을 담당한다고 일반적으로 가정한다. 형수성 곁사슬은 형수성 환경 울 제공하여 촉매작용에 기여한다. 국성인 건시술은 일반 산, 일반 염기, 친해 체, 정전기적 작용중심 등으로효소반응에 침여한다. 각종 아미노산 곁사슬에 부착된 작용기가 효소 반응에 참여하는 방식을 평가 하기 위하여 이들 작용기들이 소형 유기 화합물의 반응에 촉매로 참여할 때의 메커니즘이 자세히 연구되어 있다. 소형 유기 화합물의 반응에서 작동하는 기 본적인 단위 촉매작용을· 유형별로 소개하면 다음과 같다. 2),3) ( I ) 특정 산 및 일반 산 촉매작용 양성자 (H + ) 는 용매화되어 용액내에 존재한다. 양성자가 반응부위로 이동할 경우 루이스산으로 작용하여 반응성을 증가시킬 수 있다. 이때 반응속도는 용 매화된 양성자의 농도에 의존하게 되는데 이를 특정 산 촉매작용이라고 한다. 특정 산에 의한 촉매작용의 예로 1 과 같은 이탈기의 이탈 능력의 제고, E} 같은 친전자체의 반응성 향상 등을 둘 수 있다. H+ R-OH ~ R-+OH2 一 R+ + H20 1 유 H+ +유 H Nu- 언 유 R-C-OR' ~ R-C-OR' ~ R-9-0R' _,._ ~ R-C-Nu + R'OH 2 ~u 특정 산의 농도는 동상적인 반응 조건하(예 : 중성 p H 인 수용액)에 매우 낮다. 이러한 경우에도 반웅 매체 내부에 존재하는 약산들이 반응에 참여하여 전이상 태를 안정화시킬 수 있다. 이는 전이상태에서 양성자가 약산의 짝염기에서 반 웅村 쪽으로 디소간 이동하여 뇨} 갇이 촉매효과를 유발하는 것으로 볼 수 있다. 이와 같은 촉매작용은 개입된 산에 대하여 특정한 구조를 요구하지 않으
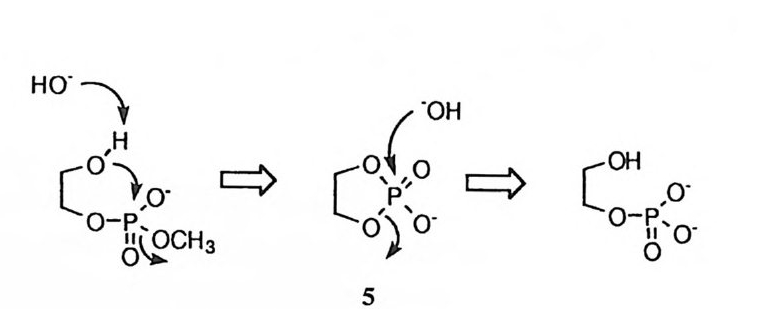
R-c유- OR' 二 [ 나° )-+ O-R-'- A]d -:t :~ ~ R-g_ N u + R'OH + HA 3 1>- Nu 므로 일반 산 촉매작용이라고 부른다. 일반 산 촉매효과는 일반 산 촉매의 산 도가강할수록크다. (2) 특정 염기 및 일반 염기 촉매작용 특정 산 및 일반 산에 대칭되는 개념으로 특정 염기 (수용액에서는 수산화 이 온)와 일반 염기를 고려할 수 있다. 특정 염기의 농도가 충분히 큰 경우 특정 염기의 참여에 의하여 반웅이 진행될 수 있다(예 : 4). 반면 특정 염기의 농도가 낮아서 이에 의한 속도가 매우 느린 경우 반응계에 존재하는 약염기가 전이상 태를 안정화시켜 반응속도를 증가시킬 수 있다(예 : 5) . R-CoII - OR' ~ R-
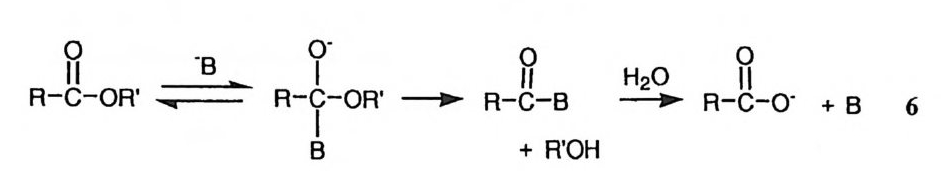 R-C유- OR' =·B= 댜o· - OR' 一 댜0 -B 목 R- 0산 -o· + B 6
R-C유- OR' =·B= 댜o· - OR' 一 댜0 -B 목 R- 0산 -o· + B 6
다 빨라야 하는 제약조건이 뒤따른다. 6) 친핵체의 반응성이 강하면 중간체의 형 성이 용이한 대신 그 파괴가 곤란해질 것임을 추측할 수 있다. 따라서 친핵성 메커니즘으로 진행하는 경우에는 중간체의 형성 및 파괴를 모두 촉진시키는 구 조적인 특징을 촉매가 지니고 있다. (4) 정전기적 촉매작용 반응속도가 증가하는 요인 중에 반응물 간의 유효농도가 증가하는 것이 포함 된다. 수용액 중에서는 이온 간의 회합이 불가능하여 반대된 전하를 띤 두 이 온 간에 유효농도가 증가하는 것이 어렵다. 그러나 유기 용매의 함량을 증가시 켜 용매의 극성을 저하시키거나 수용액내에서 협수성인 환경을 칭~옹}면 (예 : 미 셀) 정전기적인 작용에 의한 이온 간의 회합이 중전된다. 이에 따라 유효한 상 대농도가 증가되며 반응속도가 증가할 수 있다. 이러한 이온 간의 회합뿐 아니라 전이상태의 안정도를 정전기적인 효과에 의 하여 싱승시킴으로써도 반응속도를 증가시킬 수 있다. 이러한 촉매반응의 예로 아세탈 가수분해의 전이상태에서 카르보늄 양이온과 인접한 분자내 음이온 간 의 정전기적 작용에 의한 안정화 효과 {7 화군) 아미드 가수분해의 전이상태에서
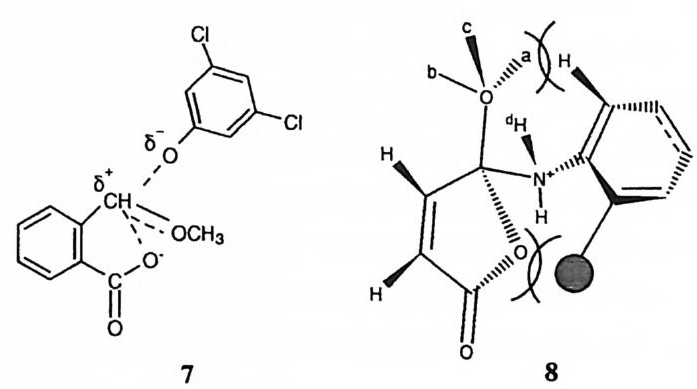 5.bCH:clc IIO c bH)H
5.bCH:clc IIO c bH)H
암모늄 양이온과 인접한 분자내 음이온(約]서 빗금천 부분계 위치한 카르복실 음이 온) 간의 정전기적 작용에 의한 가속효과 등을 둘 수 있다. 8) (5) 혐수성 작용 극성이 큰 용매 속에서는 형수성이 큰 분자들끼리 회합하는 경향이 크다. 이 러한 회합작용은 분자 간의 유효농도를 증가시킴으로써 반응속도를 제고할 수 있다. 형수성 분자의 회합은 국성 용매내에서 국지적인 협수성 환경을 조성해 준다. 반응에 따라서는 협수성 환경에서 속도가 크게 증가하기도 하므로 국지 적인 형수성 환경을 조성함으로써 촉매효과를 유발할 수 있다. 제 3 절 인접효과 효소는 반드시 기질과 착화합물을 형성한 뒤 촉매반응을- 일으킨다. 이것은 효소와 기질 간의 반응이 분자 간 반응이 아니고 분자내 반응임을 뜻한다. 분 자 간 반응을 분자내 반응으로 전환시킴으로써 두 반응부위 간에 충돌하는 빈 도를 증가시키며 따라서 반응물 간의 유효농도를 제고할 수 있다. 9)-11) 분자내 반응으로 두 반응부위 간의 충돌 빈도가 단순히 증가할 뿐이라면 반 응부위의 유효 몰농도(E M) 는 용매의 농도(물이 용매인 경우 55M) 를 초과하지 않을 것이다. 그러나 모형 연구의 결과 EM 이 1010M 을 초과하는 경우도 흔히 관찰된다. 예를 들어 표 1 에 수록한 밀레암산 유도체의 가수분해 반응에 대한 분자내 카르복실산의 유효농도는 1013M 에 달한다. 9) 화합물 9-14ol] 서 분자내 카르복실기의 EM은 분자 구조에 매우 민감하게 의 존하여 1013_l03M 의 큰 범위내에서 분포하고 있다. 이러한 EM 의 차이는 말레 암산의 올레핀 탄소에 치환된 알킬기에 의한 입체효과로 설명할 수 있다. 죽, 103 !} 1 텨 알킬기가 도입될 때 입체효과로 인하여 각도 a와 /3를 감소시키며 두 반응宇서인 CONHMe 기와 COOH 기 사이의 간격을 줄일 수 있다. 반면, 1 파 1~ 합형 혹은 다형 고리가 도입될 때에는 도리어 각도 a, fJ를 증가
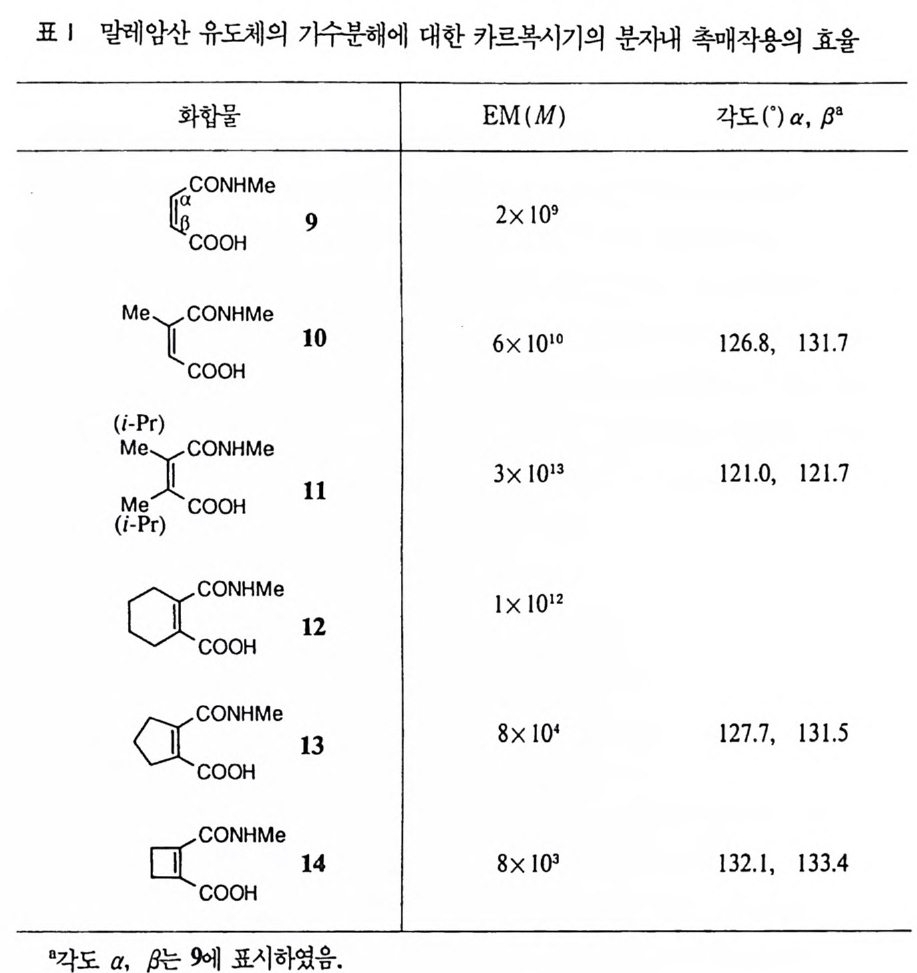 표 l 말레암산 유도체의 가수분해에 대한 카르복시기의 분자내 촉매작용의 효율
표 l 말레암산 유도체의 가수분해에 대한 카르복시기의 분자내 촉매작용의 효율
시켜 두 반응부위 간의 거리를 늘일 수 있다. 이러한 상반된 효과에 기인한 반 응부위 간의 거리 차이는 사소한 것이지만 반응속도에는 101 백의 차이를 초래 한다. 화합물 11 과 같이 두 반응부위 간의 간격이 압축된 경우에는 바닥 상태 에서 두 반응부위 주위의 수화 껍질도 이미 제거되었을 뿐 아니라 카르복실기 의 산소 원자가 아미드기의 탄소 원자에 밀착되어 이 두 원자 간의 거리는 결 합 길이에 육박하고 있을 가능성이 크다. 표 1 의 결과는 입체효과에 의하여 반 응부위 간의 인접효과가 증대되는 폭을 짐작할 수 있게 한다. 뿐만 아니라 비 교적 간단한 유기분자에서 이러한 큰 가속효과를 관찰할 수 있는 것은 효소의
경우 동일한 입체 압축효과에 의해 고도의 촉매효과를 거둘 수 있을 것임을 시 사해준다. 말레암산 유도체는 효소 - 기질 착화합물의 모형으로 간주할 수 있다. 즉 아미 드기는 가수분해되는 기질에 해당하고 카르복실기는 효소의 촉매 작용기에 해 당하는 것이다. 화합물 15-19 에 대한 모형 연구 결과 1~ 든 마이클형 첨가반응 (20) 을, 15, 17, 19 는 가수분해 반응 (21) 을, l Si곤 두 가지 반응을 동시에 일으 킴을 알 수 있었다. 12),13) 이것은 효소-기질 착화합물 속의 구조를 사소하게 변 화시켜도 효소가 관여하는 화학반응의 경로가 바뀌게 되며 따라서 효소의 특이 성이 크게 영향을 받을 수 있음을 시사한다.
 〈;겁:,
〈;겁:,
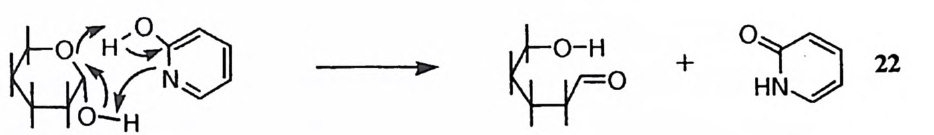
제 4 절 다중작용기 촉매효과 단일 작용기가 분자내 촉매로 작용할 때에도 인접효과에 의하여 큰 가속효과 를 시현할 수 있음을 이미 설명한 바 있다. 효~ 경우에는 활성자리 속에 여 러 개의 촉매 작용기를 보유하고 있다고 일반적으로 가정하고 있다. 여러 개의 촉매 작용기가 참여하는 경우 가속효과는 단일 작용기의 경우와 비교하여 큰 폭으로 증대될 것이 예측된다. 두 개 이상의 촉매 작용기가 단일 단계 반응에 참여할 경우 협동성의 정도에 따라 가속 정도가 각각의 촉매 작용기에 의한 가 속 정도의 곱보다 더 큰 상승효과를 기대할 수 있다. 세 개 이상의 작용기가 협동하면서 상승효과를 보인다면 효소가 성취하는 높은 가속 현상을 쉽게 설명 할수있는것이다.
 广: ::o “' 尸。 + 0H: 22
广: ::o “' 尸。 + 0H: 22
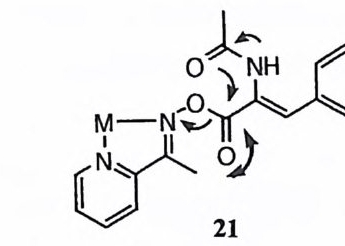
두 작용기 촉매작용은 소형의 유기분자의 반응에 대하여 상당히 연구된 바 있다. 그러나 고도의 협동성에 의한 싱승 작용은 성취된 바 없다. 헤미아세탈 류의 변광 회전에 대한 2- 히드록시피리딘의 촉매작용 (22) 은 전형적인 두 작용 기 촉매작용의 예로 흔히 인용되고 있다 .3) 2 2oj]서 2- 히드록시피리딘의 히드록 시기는 일반 산으로, 피리딘 질소는 일반 염기로서 참여하여 헤미아세탈 고리 의 파괴를 가속시키고 있다. 제 5 절 용매효과 용매의 성격이 바뀜에 따라 분자 간의 상호 작용, 반응속도 등 효소 작용에 관련된 여러 가지 요인이 영향을 받게 된다. 소형 유기분자의 반응의 속도에 미치는 용매효과의 가장 두드러진 예로 음이온 친핵체와 중성 기질 사이에 일
어나는 SN2 반응을 들 수 있다. 14) 이 경우 용매를 물에서 아세돈, 디메틸 선폭 시드 (DMSO), 핵사메틸 포스포라미드 등 극성 비양성자성 용매로 바꿀 때 반 옹 속도가 101 백 이상 증가한다. 효소의 활성자리에는 형수성 사슬, 국성 작용기 등이 위치하고 있다. 따라서 그 내부의 용매로서의 구조는 물과는 판이하며 특이한 성질을 지녔으리라고 짐 작된다. 효소가 관여하는 화학반응이 용매의 성격에 민감한 경우 활성자리 속 의 환경이 이에 적합하게끔 준비되어 있을 것임도 추측할 수 있다 . 효소 반응의 중요한 특징 중의 하나인 효소-기질 착화합물의 형성에는 효소 의 활성자리 속의 각종 작용기와 기질의 작용기 사이에 작동하는 형수성 작용 뿐 아니라 극성 작용도 개입하고 있다. 형수성 작용은· 극성이 큰 용매에 의해 쉽게 수용되며 극성 작용은 극성이 낮은 용매에서 중요해질 것이다. 이는 효소 의 활성자리 내부의 성질이 이러한 이중성을 지녀야 함을 암시한다. 효소에 의 한 극성 작용 및 형수성 작용의 동시 수용에 대한 모형으로 DMSO 속에서 음 이온성 에스테르와 양이온성 아민 간의 반응을 연구한 바 있다. 15) 속도 자료에 서 에스데르와 아민 사이에 꼬과 갇은 착화합물이 형성됨을 알 수 있었는데 끄의 형성에는 이온 간의 국성 작용뿐 아니라 아민과 에스테르의 형수성 부위 간의 상호 작용도 중요하였다. 따라서 DMSO 는 이 두 가지 상반된 작용을 모두 수 용하였다. 이러한 측면에서 효소의 활성자리 속의 성격에 대한 모형으로 DMS 예 취급할 가치가 있는 것이다.
 。
。
제 6 절 금속 이온의 촉매작용 생체내에서 금속 이온이 촉매로 작용하는 경우는 산화환원제로서 참여하는 것과 루이스산으로 참여하는 것으로 대별할 수 있다. 15) 금속이 관여하는 생체 내 산화환원 반응의 종류는 매우 다양하며 생화학 및 무기화학적 측면에서 많 은 연구가 수행되어 있다. 금속 이온이 루이스산으로 작용할 때 히드로늄 이온에 의한 촉매작용과 비교 하여 여러 가지 차이점을 보인다. 즉, 생체 조건(pH 7) 하에서 히드로늄 농도 가 매우 낮아 특정 산 효과를 기대하기 어려움에 반해 금속 이온의 경우 비교 적 높은 농도를 얻을 수 있고, 금속 이온이 지니는 전하도 흔히 +2 이상이며, 배위 결합을 통하여 반응부위의 정확한 위치에 금속이 결합할 수 있어 강력한 산 촉매로 반응에 참여할 수 있게 된다. 금속 이온의 촉매 요인에 대한 물리 유기적 접근은 루이스산으로서 유기반응 에 참여하는 경우를 중심으로- 수행되어 왔다. 지금까지 보고된 연구의 결과에 서 도출해 낸 촉매 요인은 제.Po버因 자세히 논의하게 된다. 제 7 절 착물 형성에 의한 촉매작용 효소 반응의 중요한 특칭 중의 하나가 효소-기질 착화합물의 형성임을 전술 한 바 있다. 효소-기질의 착화합물게서는 효소의 촉매 작용기와 기질의 반응부 위 간에 높은 반응속도를 실현할 수 있도록 구조적인 조건이 구비되어 있다. 이러한 구조를 지닌 착물의 형성은 엔트로피의 측면에서 불리할 것으로 예상되 지만 실제로 효소-기질 착화합물의 형성 상수는 103M4 이상으로. 열역학적인 견지에서 자발성이 높은 과정이다. 효소와 기질 간에 착물이 형성될 때 다양한 종류의 분자 간의 힘이 복합적으 로 조합되고 형성된 착물 속에서는 반응부위 간에 정교한 입체적인 배치가 달 성된다. 이러한 요인을 유기분자를 사용하여 실현해 보려는 노력이 여러 분야 에서 경주되어 왔다.
협수성 효과와 정전기적 효과로서 촉매와 기질 간의 착물 형성 상수를 제고 하고 형성된 착물 속에서 적절한 촉매 요인을 제공함으로써 증속효과를 시현하 는 것의 예로 제 11Po네 서 다루게 될 미셀과 제 11 장에서 논의할 폴리머를 들 수 있다. 폴리머의 구조를 적절히 변화시키면 폴리머가 지니는 형수성 효과 혹은 다중 전하에 의한 정전기 효과를 이용하여 기질과 비교적 강한 착물을 형성할 수 있다. 폴리머 촉매의 표면으로 선택적으로 끌려온 기질은 폴리머 표면의 미 세 환경에 의한 용매효과, 폴리머에 공유적으로 부착된 작용기의 공격 등으로 인하여 증속효과를 보이게 된다. 미셀의 경우에도 형수성 효과와 정전기적 효 과에 의하여 기질과 칙물을 형성하게 된다. 미셀 주위의 미세 환경에 의하여 빠른 속도로 기질이 반응을 일으킬 수 있다. 폴리머 유도체와 미셀의 경우, 기질과 형성한 착물에서 기질의 위치가 좁은 구역 속에 제한되어 있지 못하다. 따라서 촉매에 공유적으로 도입된 작용기와 긴밀한 인접관계를 성취하기가 거의 불가능하다. 이러한 단점을 극복하기 위하 여 비교적 작은 공간을 내부에 지니고 있는 호스트 분자와 이에 결합하는 게스 트 분자 간의 착물 형성과 이에 수반되는 촉매효과의 연구가 활발히 진행되고 있다. 제筑。데서 다루게 될 시클로덱스트린 유도체, 크라운 에테르 유도체 등 을 그 예로 들 수 있다. 이러한 호스트-게스트 촉매계의 경우에 반응부위 간에 매우 생산적인 입체 관계를 성취한 예가 보고되고 있는데 대부분의 경우 에스 데르를 기질로 사용하고 있다. 호스트 촉매에 친핵체를 부착시킴으로써 호스트 게스트 착물에서 호스트의 친핵체가 에스데르를 매우 신속하게 공격하게 한 사 례는 상당히 보고되어 있지만, 대부분의 경우에 그 결과 형성되는 아실흡누매 중간체는 빠른 속도로 파괴되지 않고 있다. 죽 촉매가 재생되어 에스테르의 가 수분해가 완결되는 효율이 낮아 진정한 촉매계로 작용하지 못하는 것이다. 효 과적인 촉매작용을 성취하기 위하여서는 중간체의 형성뿐 아니라 파괴도 증속 되어야 하는데 호스트-게스트 계에서 이 두 측면을 동시에 높은 효율로 충족시 킨 예는 혼하지 않은 것이다.
제 8 절 특정 효소의 반응 메커니즘 구명 물리 유기화학적 방법으로- 도출해 낸 촉매 요인은 효소 반응 메커니즘을 논 의하는 데에 기본적인 개념으로 사용되고 있다. 이러한 일반론적인 촉매 요인 의 규명 이의에도 물리 유기화학적 기법은 모든 효소의 메커니즘 연구에 동원 된다. 즉, 여타의 화학반응과 마찬가지로 효소 반응의 메커니즘 연구에 대한 기본적인 도구는 반응속도론이다. 그리고 이것만으로 해결할 수 없는 문제점을 풍기 위하여 여러 가지 물리적 기법을 사용하는 것이다. 제~o네서 카르복시펩티다제 A 의 촉매 반응의 메커니즘을 예로 들어 논의할 것이지만, 각종 물리 유기화학적 기법을 구사하여 특정한 효소의 반응 메커니 즘을 규명하고 있으며, 대형 분자인 효소를 사용하여 직접적으로 판단하기 어 려운 사항은 모형 분자를 이용하여 유용천· 정보를 수집하고 있다. 이상에서 물리 유기화학적 기법을 사용하여 효소의 촉매 요인을 구명하거나, 효소의 모형을 개발하거나, 효소를 사용하여 얻은 메커니즘 자료를 평가하는 예를 간략하게 소개하였다. 이러한 연구는 효소 반응 메커니즘의 연구뿐 아니 라 유기 반응과 무기 반응의 메커니즘 연구와도 밀접한 관계를 지니고 있다. 소기의 구조적인 요인을 모형분자에 도입하기 위하여서는 정교한 합성 유기적 인 기법도 필요하게 된다. 효소 반응, 유기 반응, 무기 반응, 유기 합성, 고분 자 반응 등이 서로 유기적으로 연관되어 생명현상의 기본 원리를 구명하려 시 도하는 것이 이 분야의 주제라고 할 수 있는 것이다.
참고문헌 1) Sp ec to r , L. B. Covalent Cata ly si s by Enzym es, Spr inge r-Verlag : N. Y., 1982. 2) Jen cks, W. P. Cata ly si s in Chemi stry and Enzym olo gy , McGraw-H ill :
N. Y., 1969. 3) Bender, M. L. ; Berge r on, R. J. ; Kom iya ma, M. The Bio o rg an ic CIU! m is try of Enzym a.tic Cata lysis, Wi ley : N. Y., 1984. 4) Suh, J. ; Lee, B. H. J Org. Chem., 1980, 45, 3103. 5) Suh, J. ; Lee, E. ; My un g , Y. C. ; Kim , M.; Kim , S. ]. Org. Chem., 1985, 50, 977. 6) Suh, J. ; Cheong, M. ; Han, H. Bio o rg. Chem., 1984, 12, 188. 7) Fife , T. H. ; Przy st a s , T. J. J Am. Chem., Soc, 1980, 102, 292. 8) Suh, J. ; Kim, M. J. ; Kim, C. B. J Org. Che1n., 1983, 48, 2453. 9) Kirb y, A. J. Adv. Phys . Org. Chem., 1 980, 17, 183. 10) Meng er , F. M. Acc. Chem. Res., 1985, 18, 128. 11) Czar nik, A. W. In Mecluin i s H c Pri nc i ple s ofE nzy, ne Acti vit y , Lieb man, J. F. ; Greenberg, A. Ed., VCH : New York, 1988 ; Chap ter 3. 12) Suh, J. ; Baek, D. J. Bio o rg. Chem., 1981, 10, 266. 13) Suh, J. ; Kim, M. J. ; Seong, N. J. ]. Org. Chem., 1981, 46, 4354. 14) Parker, A. J. Adv. Phys . Org . Chem., 1967, 5, 173. 15) Suh, J. ; Kim, Y. ; Lee, E. ; Chang, S. H. Bio o rg. Chem., 1986, 14, 33.
제 3 장 생체 분자의 유기화학 생체 분자는 대부분이 유기분자이어서 유기화학 분야에서 축적된 지식에 입 각햐겨 그 구조와 기능을 설명하게 된다. 생체 분자에 포함된 다양한 형태의 유기 작용기에 대하여 일일이 유기화학적인 특성을 설명하기에는 지면에 제한 이 있어서, 생체 분자에서 가장 중요한 역할을 담당하는 몇 가지에 대하여서 특징을 설명하고자 한다. 생체 고분자 중 가장 중요한 것으로 유전 정보를 저장하고 전달하는 핵산과, 세포내에서 제반 화학반응이 일어나도록 하는 효소를 둘 수 있다. 핵산은 인산 디에스테르기를, 그리고 효소는 아미드기를 연결 작용기로 하여 중합체 구조를 이루고 있다. 제筑데서는 이러한 작용기에 대한 유기화학적 측면을 취급하게 된다. 제 1 절 인산유도체 핵산인 DNA 와 RNA는 뉴클레오티드의 중합체이다. 뉴클레오티드는 질소 를 함유하는 염기 (푸린 혹은 피리미딘 유도체), 당 (D- 리보오스 혹은 D-2- 데옥시 리보오스) , 그리고 하나 또논 하나 이상의 인산기로 아루어져 있다. 핵산의 모 노머는 한 개의 인산기를 가전 뉴클레오티드인데, 이들이 중합되어 형성된 DNA 의 일부 구조를 그림 1 에 표시하였다. 정보의 보존과 전달 과정에는 핵산 이 적절한 수준의 안전성을 보유하는 것이 필요한대, 이러한 화학적 성질은 인 산 디에스데르 결합의 안정성에 기인한다.
 ~ H2bc:二 ;_?-O- H2bc:二 ;I-o~
~ H2bc:二 ;_?-O- H2bc:二 ;I-o~
핵산 이의에도 생체내에서 중요한 기능을 담당하는 인산 유도체의 예로 ATP 혹은 GTP와 같은 에너지 저장 물질과 c-AMP 혹은 c-GMP 와 같은 조절 기능 물질, NAD+ 혹은 NADP+ 같은 조효소가 있다• 이중에서 ATP, c-AMP 등과 같은 화합約 생체 기능을 발휘하는 데에는 인산 무수물 혹은 인산 에스데르 결합의 가수분해 반응이 개입하고 있다.
 H (20c H O\H쵸 O c‘Pf-oc0 H 3
H (20c H O\H쵸 O c‘Pf-oc0 H 3
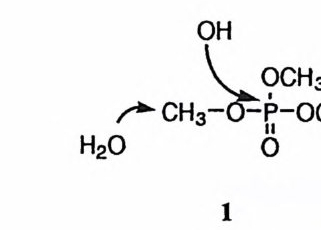
스테인르산를 (H 3형P성O4한)다 은. 삼 ”염 이기중 에산서이 기트 리때에문스에테 모르노는에 화스테학르적,으 로디 에가스장테 불르안 정및 하트며리 에가 장 쉽게 친핵체의 공격을 받게 된다. 인산의 트리알킬 에스테르의 경우에는 친 핵체가 인 혹은 탄소를 공격할 수 있다. 단단한 2) 염기인 수산화 이온은 단단한 산인 인 원자를 공격하고 무른 염기인 물 분자는 무른 염기인 탄소를 공격하여
SN2 빈념을」 일으키는 것 (1) 이 알려져 있다. 3)-5) 아릴 에스데르의 경우에 친해 체는 통상적으로 인을 공격하게 된다.
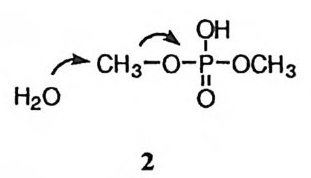 ~ CHr3-o -언r. -o cH3
~ CHr3-o -언r. -o cH3
인산 디에스데르는 일염기 산인데 PKa 가 1. 5 정도이어서 낮은 p H 에서는 중 성형으로 존재하고 중간 이상의 p H 에서는 음이온으로 존재한다. 序牛형의 고리 형 에스테르 혹은 강하게 활성화된 아릴 에스테르를 제의하고는 인산 디에스테 르의 음이온형은 정전기적 척력 때문에 수산화 이온과의 반응성이 매우 낮다. 디메틸 포스페이트의 음이온은 수산화 이온과 매우 느리게 반응하며 (반감기 : 100C 에서 약 20 년) P-0 결합의 절단이 C-0 결합의 절단보다 우세하다. 한 편하,는 데중,성 이형때은 물물 과분 자10는0° C메 에틸서 산4소 X 에1Q -S6SN-2l 의반 응속을도 일상으수켜( 반C감—기 0: 5(결),'-합] 간을) 로단 반절응하 는 경로 (2) 가 P-0 결합을 단절하는 것보다 4 배 정도 우세하게 일어난다. 6) 인산 디아릴 에스테르는 가수분해될 때 , C-0 결합은 단절되지 않고 P-0 결합만이 단절된다. 디아릴 에스데르의 아릴기에 전자를 강하게 당기는 치환 체가 존재하면 가수분해가 쉬워진다. 예를 들어 pH 4 에서 디페닐 포스페이트 의 가수분해 속도 상수는 100°C 에서 3X 1Q -9S-1( 반감기 : 약 穴i)이고 비스 (2, 4 - 디니트로페닐) 포스페이트의 가수분해 속도 상수는 74. ff C 에서 1.8X10-ss-1 이다 .7),8) 인산 모노에스데르는 이염기 산 (PK1 가 약 1, p.K:i가 약 6.5) 으로 p H 에 따라 중성형, 모노음이온, 혹은 디음이온으로 존재한다. 아주 낮은 p H 에서는 중성 형이 산 촉매에 의하여 가수분해되고 아주 높은 p H 에서는 디음이온형이 수산 화 이온에 의하여 가수분해된다. 중간 p H 에서는 모노음이온형이 Elcb 형 제거 반응과 비슷한 메커니즘 (3) 으로 가수분해되는데, 인산 모노에스데르의 모노음 이온형의 반응성은 인산 트리에스테르보다는 낮고 인산 디에스데르보다는 높
 H-o,)
H-o,)
다. 9),10) 책 메커니즘에서 인산의 수소는 이탈 알콕시기의 산소를 안정화하기 위하여 양성자를 제공하는 역할을 하고 있다. 아릴 모노에스데르의 이탈기의 이탈 능력이 좋을 경우에는 이탈기를 양성자 화할 필요가 없어져서 디음 이온형도 쉽게 가수분해 반응 (4) 을 일으킨다. 11) ,12 ) 인산 에스데르의 인 원자에 친핵체가 공격하여 치환반응을 일으킬 때, 그림 까 같이 여러 유형의 메커니즘이 가능하다. 13) 첫번째의 분리성 메커니즘은 위의 3 및 4가 속하는 메커니즘이다. 인산 모노 에스테르의 경우에 이 메커니즘에 따르면 메타포스페이트 이온 (PO i )이 중간체 로 포함되는데 이것이 중간체인지 혹은 전이상태인지에 대하여 많은 연구가 수 행된 바 있는데, 용매로부터 완전히 자유로운 형태로는 형성되기가 어렵다는 방향으로 의견이 모아지고 있다. 인산의 OH 기를 계속하여 에스테르회·하여 디에스데르나 트리에스데르를 만 들거나 옥시 음이온이 금속에 배위됨으로써 인 주위의 음전하가 작아지면 음이 온성 친핵체가 인을 공격할 수 있게 되고 이에 따라 메커니즘이 분리성에서 회 합성으로 전환된다. 회합성에 속한 두번째와 세번째의 메커니즘은 5 배위 화합 물이 중간체로 존재하는가 혹은 전이상태일 뿐인가에 따라 차이가 있는데, 반 웅의 구조와 반응조건에 따라 메커니즘이 유동적이 된다. 5 배위 중간체의 수
명이 충분히 길어져서 중간체로 존재하게 되면 친핵체가 이탈기의 반대 방향에 서 접근하는가 혹은 인접 방향에서 접근하는가에 따른 메커니즘상의 변화가 가 능하다.
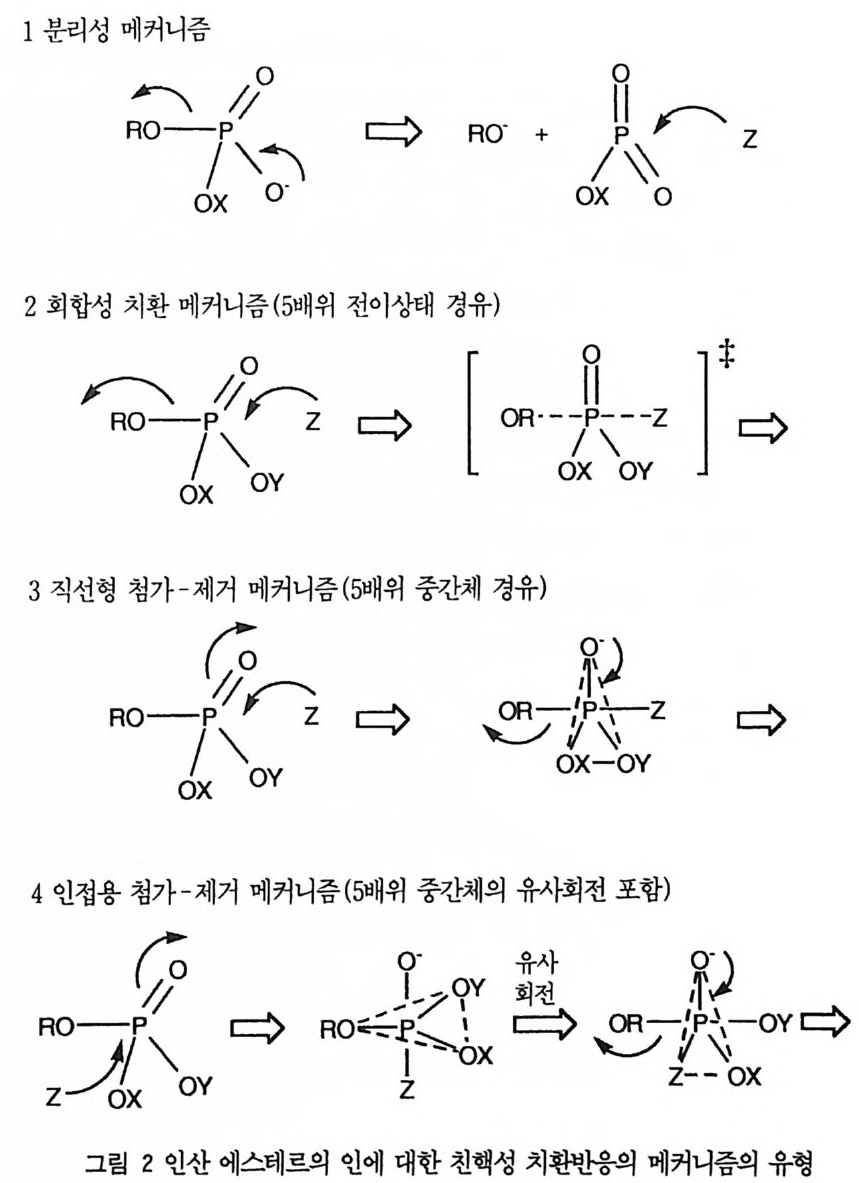 1 분리성 메커니즘
1 분리성 메커니즘
인의 5 배위 중간체는 다음과 같은 특성을 가진다. 13),14) I) 양삼각뿔 구조를 가지고 꼭대기 방향에 엄 H, 적도 방향에 37H 의 리간드가 위치한다. 2) 중심 인 원자에 공격해 들어오거나 인 원자로부터 이탈하는 원자단은 꼭 대기 방향에서 출입한다. 3) 인 원자를 포함하여 4-6 각형의 고리가 형성될 때 꼭대기 방향의 위치와 적도 방향의 위치를 각각 한 개씩 접유함으로써 고리의 왜력을 경감한다. 4) 전기음성도가 큰 리간드가 꼭대기 방향을 차지한다. 5) 리간드는 유사 회전에 의하여 위치를 바꿀 수 있다. PFs t 꼭대기 방향과 적도 방향의 두 위치를 차지하고 있는 리간드가 동일 하지 않기 때문에 두 가지 종류의 불소 리간드를 가지고 있다. 그러나 핵자기 공명 스펙트럼싱에서는 이 두 가지 불소가 동일한 것으로 나타난다. 이것은 유 사 회전에 의하여 두 리간드가 핵자기 공명의 시간 스케일 이내에 서로 자유로 이 변환하기 때문이라고 설명한다. 15) 유사 회전은 적도 위치 리간드 중 하나가 받침이 되고, 나머지 네 리간드의 위치가 이동하여 꼭대기 위치와 적도 위치의 리간드가 자리를 바꾸는 것 (그림 3) 을 일컫는 것으로 인산 에스데르의 반응 메 커니즘을 설명하는 데 도입되었다. 16)
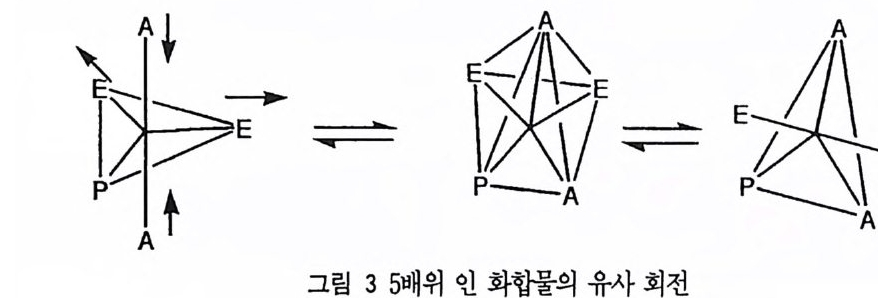 그림 3 5 배위 인 화합물의 유사 회전
그림 3 5 배위 인 화합물의 유사 회전
그림 2 에 포함된 세번째 및 네번째 메커니즘은 이탈기와 친핵체의 상대적인 위치에 차이가 있다. 네번째 메커니즘에서도 공격과 이탈은 꼭대기 위치에서 일어나고있다. 앞서 설명한 바 있듯이 디메틸 포스페이트의 가수분해 속도는 매우 느린데,
 H 。c \ H?,\c,pH I3 0。 C°°,'P)' :°0- ~ cO HP`5 o0 ,0
H 。c \ H?,\c,pH I3 0。 C°°,'P)' :°0- ~ cO HP`5 o0 ,0
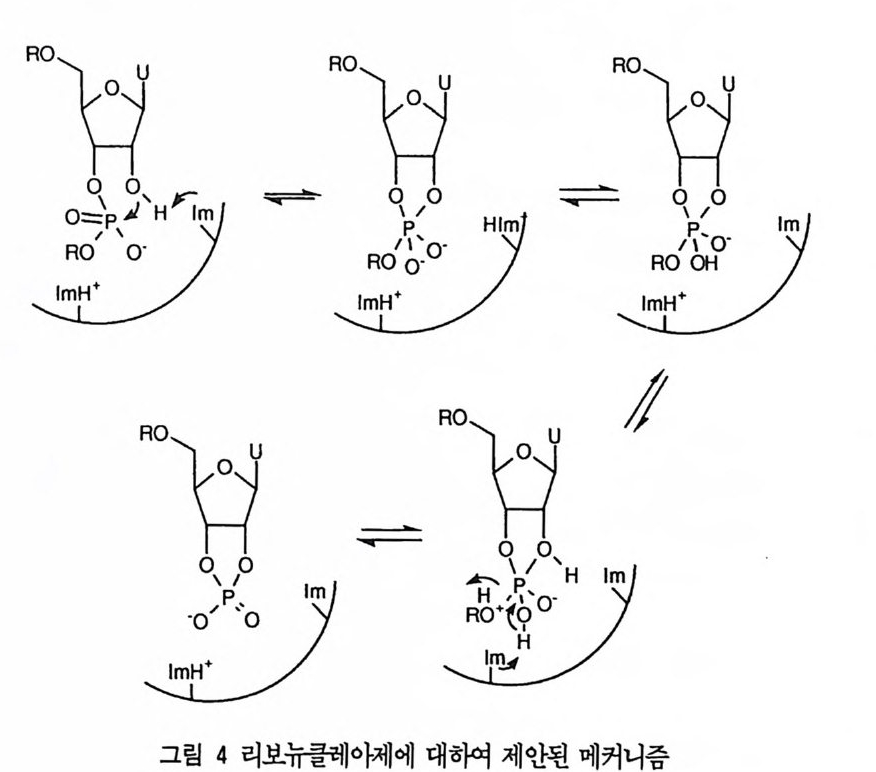
이에 비하여 2 - 히드록시에틸 메틸 포스페이트는 25'C , lN NaOH 의 조건하에 반감기가 2~] 불과하다. 이것은 인접한 히드록시기가 분자내 친핵성 공격에 의하여 합才형 중간체 를 형성하고 이 중간체 (에틸렌 포스페이트)는 분자내에 존 재하는 왜력 을 경감하기 위하여 신속하게 가수분해되기 때문 (5) 으로 볼 수 있 다 .1 4) 이 결과는 RNA 의 가수분해와 연관시킬 수 있다. DNA는 유전 정보를 보
 \odp Ro 〉R 0\0\koRO
\odp Ro 〉R 0\0\koRO
 2_[:O ? 亡 H2 。 \O(I`IP E HO— 。P `o ·
2_[:O ? 亡 H2 。 \O(I`IP E HO— 。P `o ·
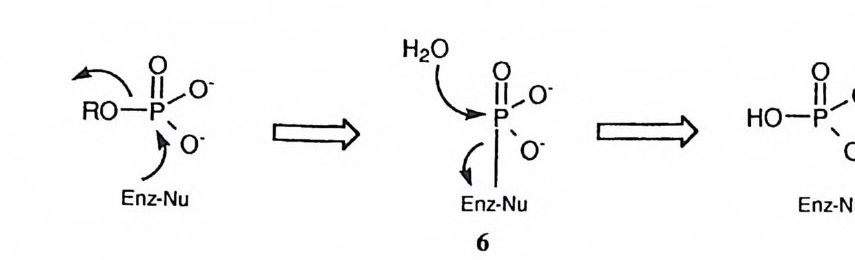
존하기 때문에 상당한 안정성을 가져야 하는 데 비하여 RNA 는 유전 정보를 전달하는 역할을 담당하기 때문에 쉽게 제거할 수 있어야 한다. 이에 따라 RNA는 DNA 보다 화학적으로 불안정하다. 이들 핵산의 구조의 차이는 리보 오스 부분에 히드록시기를 가지고 있는지의 여부에 달려 있다. RNA 는 인산 디에스테르 부위에 인접한 위치에 히드록시기를 가지고 있기 때문에 요} 유사 한 메커니즘에 의하여 쉽게 가수분해되는 것이다. 리보뉴클레아제는 RNA의 가수분해에 관여하는 효소인데, 이 효소에 관하여 브레슬로가 17) 제안한 메커니 즘(그림 4) 을 참고로 소개한다. 이 메커니즘에서 두 개의 히스티딘 잔기의 이미 다졸기가 일반 염기로도 작용하고 이탈기를 양성자화하는 데 필요한 양성자를 제공하기도한다. 인산 모노에스테르가 효소에 의하여 가수분해되는 경우에는 3 혹은 心仕근 달 리 효소의 작용기가 친핵체로 작용하여 포스포릴-효소 중간체를 형성 (6) 하게 된다. 산 포스파타제의 경우에는 이미다졸이 친핵체로 작용하고, 아연 금속 효 소인 알칼리 포스파타제의 경우에는 세린의 히드록시기가 친핵체로 작용한 다 .14) 인산 디에스데르 혹은 인산 모노에스데르의 가수분해에 관여하는 효소의 메 커니즘에서 인접형 첨가-제거 메커니즘(그림 2) 이 작동한다는 증거는 아직 얻 어진 바가없다 .l4) 제 2 절 아실유도체 생체계에 존재하는 아실 유도체의 결합 중에서 가장 양이 많은 것은 단백질 의 기본 결합인 아미드 결합이다. 이의에도 지방질의 에스테르 결합, 아세틸
CoA 의 티오에스테르 결합 등도 생체계에서 중요한 역할을 담당하는 아실 유도 체의 예로 들 수 있다. 유기 반응 메커니즘 중에서도 아실 유도체의 가수분해 가 물리 유기화학 분야에서 일찍이 많은 연구의 대상이 되었고, 효소 반응 메 커니즘 중에서도 단백질 가수분해 효소의 메커니즘이 가장 먼저 연구되었다. 이에 따라 아실 유도체의 화학예 관한 정보가 많이 축적되어 있다.
 0} --N:H 구 O} ---N,\R 2 _ -O) =맛R 2
0} --N:H 구 O} ---N,\R 2 _ -O) =맛R 2
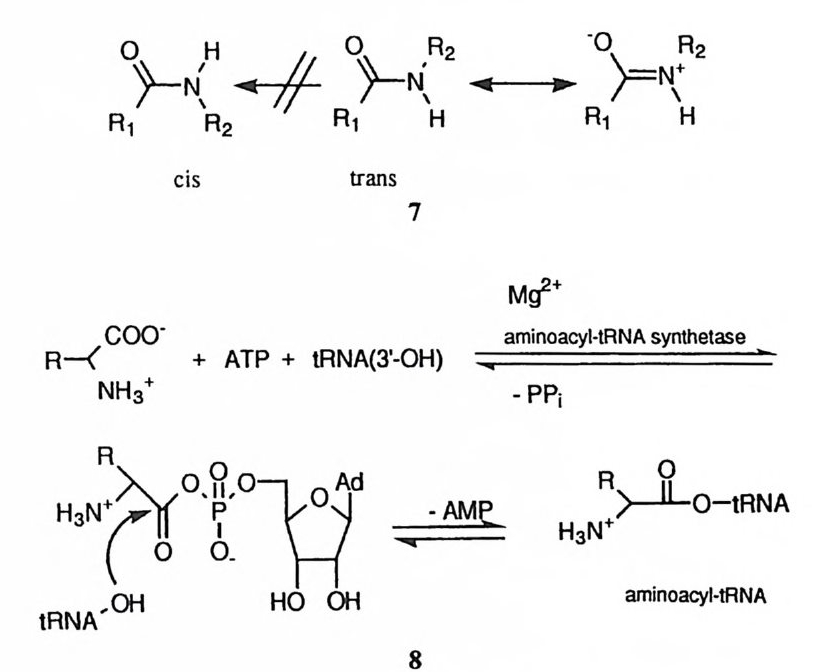
절아하미리드만 큼결 합C 은一 NC =결N합 이주중위결의합 회의전 기이여 억도제가 ( 7큰) 되 공어명 있구다.조 로l4) 설이에명 하따는라 것 C이— -적N 결합은 이중결합의 성격을 강하게 가지고 상당한 안정성을 가지고 있다. 또한, 아미드는 시스형과 트란스형이 이성질체로 존재하는데 시스형에서는 입체 장애 가 존재하여 트란스의 형태가 더 안정하다. 18) 아실 유도체는 아실기에 연결되어 있는 이탈기의 염기도가 낮을수록 불안정 햐겨 쉽게 친핵체의 공격을 받아 치환반응을 일으키게 된다. 생체내에서 단백 질이 합성되는 과정에서는 아실기가 ATP 에 의하여 활성화된 뒤에 디시
t RNA 로 이동하여 아미노아실 - t RNA의 형태로 전환 된 뒤, 아민의 공격을 받 아 아미드 결합을 형성 (8) 한다. l4) 아실 유도체의 천핵성 치환반응에 관한 메커니즘 연구 중 가장 고전적인 것 은 에스테르 가수분해에 관한 것이다. 산 촉매 반응인가 혹은 염기 촉매 반응 인가, 일분자성인가 혹은 이분자성인가, 알킬 탄소국낡: 결합이 단절되는가 혹 은 아실 탄소-산소 결합이 단절되는 것인가에 따라 메커니즘이 분류된 바 있 다 .19) 아실 유도체의 치환반응의 대부분에서 아실 탄소 - 산소가 단 절 되며, 이때 친 핵체와 아실 탄소 간의 결합 형성과 이탈기와 아실 탄소 간의 결합 단절의 시 차에 따라 회합성, 분리성, 혹은 동시성으로 메커니즘을 구분하는 경향이 최근 에 중요시되고 있다. 26)
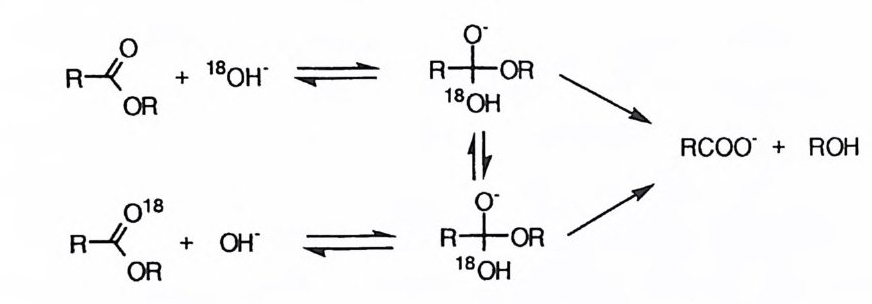 。 o-
。 o-
회합성 메커니즘에서는 친핵체가 아실기에 첨가되어 정사면체 중간체가 형성 된 뒤에 이탈기가 떨어져 나간다. 이에 대한 명확한 증거로 최초에 수집된 것 은 H2l8Q 속에서 에스테르를 알칼리 가수분해시키는 도중에 산울 가하여 반응 을 중지시킨 후, 미반응 에스테르의 아실기에 180 가 도입된 것을 발견한 실험 으로부터이다. 21) 이 결과는 정사면체 중간체가 형성되고, 이 정사면체 중간체 로부터 생성물이 생성되는 단계가 속도 결정 단계이고, 정사면체 중간체내에서 양성자 이동이 신속히 일어난다고 가정 (9) 함으로써 설명되는 것이다. 정사면체 중간체는 매우 불안정하고 반응성이 높아 그 존재를 확인하기가 어 렵다. 메틸 아세데이트와 수산화 이온이 반응하여 형성된 정사면체 중간체의 반감기가 10-1 초임이 알려져 있다. 22) 정사면체 중간체 혹은 이에 관련된 유사
 o- 。
o- 。
체를 합성 혹은 분리하려는 시도가 광범위하게 이루어진바, 그 예는 10, 23) 11, 2 ◄) 12, 2sJ 13, 26) 14, 21) 152s) 등이다. 분리성 메커니즘에서는 이탈기의 제거가 선행되어 아실리움 양이온이 형성된 뒤 친핵체가 공격하여 치환반응이 완결된다. 근자에 들어 이 메커니즘으로 진 행되는 반응이 희귀한 것은 아니라는 것이 알려졌다. 29) 아실리움 양이온은 아 실기의 点 1 치에서 전자가 제공 (16) 됨으로써 흔히 안정화된다. 30) 동시성 메커니즘은 최근에 페놀 음이온 사이에 아실기가 교환되는 반응에 대 하여 그 가능성 (17) 이 제안된 바 있고 ,31) 또한 이를 반박하는 증거도 제기된 바 있다 .32)
참고문헌 1) Bunto n , C. A. Acc. Gliem . Res., 3 , 257, 1970. 2) Pearson, R. G. ; Songs t a d , J. J Am. Chem. Soc., 8 9, 1827, 1967. 3) Barnard, P. W . C. ; Bunto n , C. A. ; Llewellyn , D. R. ; Oldham, K. G. ; Sil ve r, B. ; Vernon, C. A, Chem. Ind. (London) , 760, 1955. 4) Vernon, C. A. Chem. Soc., Spe c ia l Pub!., N o. 8, 17, 1957. 5) Barnard, P. W . C. ; Bunto n , C. A. ; Llewellyn , D. R. ; Vernon, C. A. ; Welch, V. A. J Chem. Soc., 2670, 1961. 6) Bunto n , C. A. ; Mhala, M. M. ; Oldham, K. G.; Vernon, C. A. J Chem. Soc., 3293, 1960. 7) Barnard, P. W . C. ; Bunto n , C. A . ; Kellerman, D. ; Mhala, M. M. ; Sil v er, B. ; Vernon, C. A. ; Welch, V. A. J Chem. Soc. B, 227, 1966. 8) Bunto n , C. A. ; Farber, S. J. J Org. Chem. , 3 4, 767, 1969. 9) Cox, D. J. ; Ramsay, 0. B. Chem. Rev., 6 4, 317, 1964. 10) Bru ice , T. C. ; Benkov ic, S. J. Bio o rga n ic Mechanis ms, Benja m in : New York, 1966, Chap ter s 5-7.
11) DiS a bato , G . ; Jen cks, W. P.]. Am. Chem. Soc., 83, 4400, 1961. 12) Bunto n , C. A. ; Fendler, E . J. ; Fendler, J. H. ]. Am. Chem. Soc., 89, 1221, 1967. 13) Cullis, P. M. In Enzy, ne Mechanis ms, Page , M. I. ; Wi lliam s, A. Eds., Roy al Socie ty of Chem ist ry : London, 1987, Chap ter 11. 14) Dug as , H. Bio o rga n ic Chemi stry. A Chemi ca l Ap pro ach to Enzy,n e Acti on , Sp ri n g e r -Verlag : New York, 1989, 2nd Ed., Chap ter 3. 15) Berr y, R. S. ] . Chem . Phys ., 32, 9 33, 1960. 16) Westh e im er, F. H. Acc. Chem . Res., 1, 70, 1968. 17) Breslow, R. ; Labelle, M. J Am. Chem. Soc., 108, 2655, 1986. 18) Sukumaran, D. K. ; Porok, M. ; Lawrence, D. S. J Am. Cheni. Soc., 1 991, 111, 706. 19 ) Jon es, R. A. Y. Phys ica l and Mechanis tic Orga n ic Chemi stry, Cambri dg e Univ e rsity Press : Cambrid g e , 1 983, 2nd ed., Chap ter 12. 20) Wi lliam s, A. In Enzy, ne Mechanis ms, Page , M. I. ; Wi lliam s, A. Eds., Roy al Socie ty of Chem ist r y : London, 1987, Chap ter 8. 21) Bender, M . L. ]. Am. Chem. Soc., 1951, 73, 1626. 22) Guth r ie , J . P. ]. Am. Chem. Soc., 1973, 95, 6999. 23) Goto , T. ; Kish i, S. ; Takahashi , S. ; Hira ta , Y. Tetr a hedron, 1965, 21, 2059. 24) Huber, R. ; Bode, W. Acc. Chem. Res., 1978, 11, 114. 25) Rog er s, G. A. ; Bruic e, T. C. J Am. Chem. Soc., 1973, 95, 4452 ; 1974, 96, 2473 and 2481 . 26) Gravit z, N. ; Jen cks. W . P. J Am. Chem. Soc., 1 974, 96, 489. 27) Cap on , B. ; Ghosh, A. K. ; Gri ev e, D. McL . A Ace, Chem. Res., 1981, 14, 306. 28) McClelland, R. A. ; Santr y, L. J. Acc. Chem. Res., 1 983, 16, 394. 29) Doug la s, K. T. ; Wi lliam s, A. Chem. Rev., 1975, 75, 627. 30) Cevasco, G. ; Guanti, G. ; Hop k in s, A. R. ; Thea, S. ; Wi lliam s, A. ]. Org. Chem., 1985, 50, 479. 31) Ba-Saif, S. ; Luth ra, A K. ; Wi lliam s, A. ]. Am. Chem. Soc., 1987, 109, 6362. 32) Suh, J. ; Heo, J. S. ] . Org . Chem., 1990, 55, 5531 .
제 4 장 금속 이온과 유기 생체 분자 효소 중에는 활성을 가지기 위하여 금속 이온이 필수적으로 요구되는 것이 있는데 이를 금속 효소라고 한다. 금속 효소의 활성자리에는 금속 이온이 포함 되어 여러 가지 촉매작용을 수행하고 있는 것이다. 유기 반응에서 금속 이온이 수행할 수 있는 촉매작용은 루이스산 촉매작용, 산화환원 촉매작용, 유기 금속 화합물을 통한 촉매작용 등 세 가지로 분류할 수 있다. I) 생체내에서 각종 유기 반응에 대하여 금속 이온은 주로 루이스산 촉 매 혹은 산화환원 촉매로 작용하며, 유기 금속 화합물을 통한 촉매작용은 소수 만 알려져 있다. 루이스산 촉매작용에서 금속 이온은 촉매 역할을 수행하는 동 안 산화상태가 변화하지 않는다. 금속 이온이 루이스산 촉매로 작용하는 금속 효소의 예는매우많은숫자에 달하며 ,2) 유기 반응의 거의 대부분의 유형이 이 러한 금속 효소에 의하여 촉매작용을 받고 있다. 이 장에서는 금속 이온이 루 이스산으로 생체 유기 반응에 참여하는 촉매작용에 대한 유기화학적 측면을 다 룬다. 금속 이온이 산화환원 촉매로 생체 반응에 관여하는 것은 생물 무기화학 쪽으로 치우천 분야이기 때문에 여기에서는 디루지 않기로 한다. 유기 반응에서 루이스산 촉매로 작용하는 금속 이온의 촉매 원리에 대한 지 식은 유기 반응, 무기 반응, 효소 반응 등의 메커니즘을 연구하는 데 매우 중 요한 위치를 차지하게 된다. 뿐만 아니라 이러한 금속 이온의 촉매 원리는 생 체 모방 촉매를 고안하는 데에도 중요하게 사용할 수 있다. 금속 이온은 인공 효소가 기질의 구조를 인식하여 착화합물을 형성하는 과정에 참여할 수 있을 뿐 아니라 이 착화합물내에서 일어나는 반응 촉진 단계에도 관여할 수 있기 때 문 O] 다.
금속 이온의 루이스산 촉매작용에 관하여서는 지난 20 여 년간 많은 연구가 보고된 바 있다. 3)-6) 그러나 금속 이온의 촉매작용에 대한 세부적인 사항을 이 해할 수 있으려면 여러 가지 메커니즘상의 문제점에 대한 해답을 구하여야 한 다. 금속 이온이 수행할 수 있는 촉매 역할은 무엇인가. 금속에 배위된 물 분 자와 금속에 배위된 수산화 이온이 가지고 있는 촉매 특성은 어떠한가. 금속 이온 그리고 금속에 배위된 물 분자 혹은 수산화 이온은 유기 촉매 작용기와 어떻게 협동할 수 있는가. 리간드의 구조 혹은 금속 주위의 기하학적 형태 등 에 의하여 촉 매 효율이 어떻게 영향을 받을 것인가. 이러한 사항은 금속 이온 이 유기 반응과 생물학적인 과정에서 수행하는 루이스산 촉매작용을 연구하는 데에 제기되는 질문의 예로 들 수 있다. 제 1 절 금속 이온 자체의 촉매작용 또 다른 루이스산안 히드로늄 이온과는 달리 금속 이온은 중성 혹은- 염기성 p H 에서도 높은 농도를 가질 수 있고, +2 이상의 복수 전하를 가질 수 있다. 이의에도 금속 이온은 킬레이트화를 통하여 유기분자의 특정 부위에 결합할 수 있다. 유기분자에 결합된 금속 이온은 루이스산으로 작용함으로써 다른 촉매 요인의 도움이 없이도 여러 가지 독자적인 촉매 역할을 수행할 수 있다. (I) 친전자체의 활성화 유기 천전자체가 금속 이온에 배위됨으로써 활성화되는 경우가 많다. 예를 들어 카르보닐 유도체나 아실 유도체에 친핵체가 공격할 때 카르보닐 산소에 형성되는 음전하는 금속 이온에 의하여 안정화된다. 에스데르 가수분해 , 7),8) 아 미드 가수분해 ,9) 알데히드 혹은 케돈의 수화 혹은 환원 ,10)-13) 니트릴의 수 화, 14 ), 1 5 ) 옥상가세트산 혹은 옥살프로피온산의 탈카르복실화 1 6 ) ,17) 등은 이 효과 에 의하여 금속 이온 촉매작용을 받을 수 있다. 카르보닐 산소가 금속 이온에 배위된 뒤 수산화 이온이 금속에 배위된 카르 복실기를 공격함으로써 (1) 에스테르나 아미드가 가수분해되는 예가 많으리라고
 H 입〈X 야R\/ 止 XR5
H 입〈X 야R\/ 止 XR5
여겨진다. 그러나 이 메커니즘은 반응속도론적으로 동일한 메커니즘인 금속에 배위된 수산화 이온에 의하여 카르보닐 탄소가 공격받는 경로 (2) 와 구별하기가 어려운 경우가많다 .18) 리간드의 치환속도가매우느린 Co(III) 에 의한 촉 매작 용에 대하겨서는, 180 츄척 실험에 의하여 아미노산 에스데르 혹은 아미드의 가수분해 반응이 금속에 의하여 촉매작용을 받을 때 이 두 메커니즘이 모두 작 동하고 있음을 밝힌 바 있다. 9),1 9 ),20)
 ++
++
2-시 아노 -1, 10 페난트롤린 혹은 2- 시아노피리딘의 Ni (Il) 혹은 Cu(Il)- 착 화합물게서는 금속 이온이 니트릴기와 작용하는 것이 분자 구조상 불가능하다• 그러나 금속 이온은 니트릴기의 수화 반응의 전이상태에서 질소 원자를 결합 (3) 하는 것이 가능하고 이에 따라 전이상태에서 질소 원지에 형성되는 음전하 를 안정화하여 촉매작용을 시현할 수 있다고 보고된 바 있다. 14),1 5 ) 포스포릴 유도체의 친핵성 반응에서 포스포릴 산소에 형성되는 음전하도 금 속 이온과의 작용에 의하여 안정화될 수 있다. 21),22) (2) 이탈기의 활성화 알콕시 산소 원자가 금속 이온에 결합되면, 천전자성 탄소 중심으로부터 산
소가 이탈하는 능력이 제고된다. 이에 따라 에스데르 가수분해, 23) 아세탈 가수 분해, 24) 혹은 에폭시드 가수분해 25) 등에서 금속 이온의 촉매작용이 관찰된 바 있다.
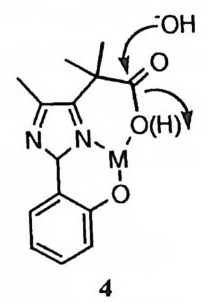 4
4
수산화 이온과 산화 이온 (02 - ) 까지도 금속에 배위됨으로써 아실 중심으로부 터 이탈하는 능력이 제고된다. 예를 들어, 금속에 배위된 카르복실 음이온의 아실 탄소에 수산화 이온이 공격 (4) 함으로써 카르복실기가 가수분해되는 반응 이 보고된 바 있다. 18) 옥심이 아탈기일 때는 옥심 질소 원자가 배위자리이기 때문에 알콕시 음이온 이 이탈기인 경우와 비교하여 금속과의 결합이 훨씬 용이하다. 이에 따라 아 실, 26)- 2 8) 포스포릴, 29) 혹은 술포닐 307] 에서 옥심이 이탈하는 능력이 금속 이 온에 의하여 제고되는 예가 보고된 바 있다.
 RCON` R 十o
RCON` R 十o
아미드기의 질소 원자가 금속 이온에 배위되면 아미드의 가수분해가 촉진될 수도 있다. /3-락탐은 고리의 왜력 때문에 질소 원자가 금속 이온에 쉽게 배위 (5) 될 만큼 염기성을 띠고 있다 .31),32) 반면에 일반적인 아미드의 경우에는 아
미드 질소의 염기도가 매우 낮아 아미드의 탄소 원자가 정사면체 중간체로 변 하겨야만 아미드의 질소 원자가 금속 이온과 결합 (6) 할 수 있을 것이다. 33) 그 런데, 정사면체 중간체에서 아민이 이탈되려면 아민이 양성자와 결합하여야 하 는데, 이 아민의 질소 원자가 금속 이온에 배위됨에 따라 질소 원자에 양성자 가 결합하는 것이 저해되어 도리어 아민의 이탈을 금속 이온이 지체시킬 수도 있다 .34) 유황, 35) 할로겐 36) 등 기타 이탈 원자가 금속 이온에 배위되어 이탈 능력이 제고되는 예도 보고되어 있다. (3) 산의 활성화 금속 이온에 배위되면 물의 산성도가 증가한다. 37),38) 또한, 알콜의 산소, 옥 심의 아민 질소, 혹은 아민의 질소가 금속 이온에 배위하면 알콜, 39) 옥심 40)-42) 혹은 아민 43),44) 의 산성도가 각기 제고된다. 이에 따라 각각의 짝염기가 중성 혹 은 산성 용액에서도 상당한 농도로 존재하게 된다. 아러한 짝염기는 금속 이온 에 배위될 때 염기성이 감소하게 될 것이다. 그러나 이 효과는 친핵성의 감소 가 염기성의 감소에 비교하여 작다든지, 짝영기의 농도 증가의 폭이 친핵성의 감소보다 더 크다든지, 혹은 금속 이온의 템플레이트 효과가 뛰어나다든지 하 는 등의 반대적인 보완 효과로 인하여 카르보닐, 울레핀, 혹은 포스포릴 등에 대한 전체적인 친핵성 반응의 가속 효과를 시현하게 된다.
 :[
:[
금속 이온아 리간드에서 비교적 먼 위치에 결합된 양성자를 활성화하는 경우 도 있다. 예를 들어 금속 이온에 배위된 아실 혹은 카르보닐 화합물의 Ca-H 결합이 활성화됨으로써 카르보 음이온 중간체 (7, 8) 를 경유하는 양성자 교환,
에피머화, 알돌 축합, 엔울화 반응 등이 촉진될 수 있다. 45 ) - 4 7) (4) 저해적인 역반응 경로의 봉쇄 3 카르복시 0~ 피린의 가수분해는 Fe(III) 혹은 Al(III) 이온에 의하여 촉매작 용을 받는다. 48 ) 이 촉매작용은 저해적인 역반응 경로를 금속 이온이 봉쇄하는 이유만으로 일어나고 있다. 기질의 카르복실 음이온이 에스데르 결합에 친핵성
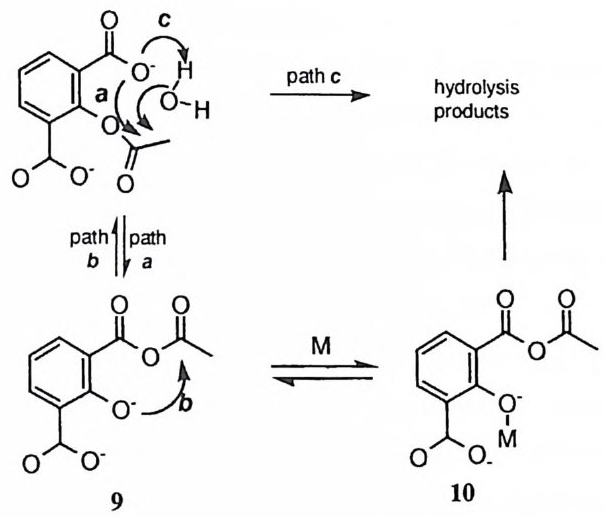 o검 o· 0 H path c hpryod drou lcy tss i s
o검 o· 0 H path c hpryod drou lcy tss i s
공격 (a 경로)을 가함으로써 얻어지는 산무수물 중간체 (9) 에서는 산무수물 결합 에 대한 페놀 음이온의 공격 (b 경로)이 매우 효과적이다. 따라서 금속 이온이 없을 경우에는 기질의 가수분해는 분자내 카르복실 음이온에 의한 일반 염기 촉매작용에 의한 경로 (c 경로)을 통하여 진행된다. Fe(III) 혹은 Al(III) 이온을 가하면 금속 이온이 산무수물 중간체의 살리실산 부분과 결합함으로써 페놀 음 이온을 봉쇄 (10) 하게 된다. 금속 이온이 존재하면 기질은 일반 염기 경로를 거 치지 않고 천핵성 경로를 거치게 되는데 이는 산무수물 중간체가 포획됨으로써 입증되었다. 많은 수의 효소 반응은 공유 중간체를 포함하게 된다. 49) 이러한 효소 반응이 치환 반응을 경유하게 되면, 효소의 촉매 작용기에 의하여 기질에서 이탈기가 절단된 이후에도 이탈기는 반옹부위 주변에 머물게 된다. 그런데 반응부위에
매우 인접한 곳에 이탈기가 자리잡으면 중간체에 대한 이탈기의 역공격도 매우 효과적이 된다. 이 역공격에 의하여 전체적인 효소 반응속도가 지연될 것이기 때문에 효소는 이탈기를 반응부위에서 신속히 분리하든지 혹은 이탈기의 반응 성을 봉쇄하여야 한다. 따라서 금속 효소에 따라서는 단순히 이탈기와 결합하 여 저해적인 역반응 경로를 봉쇄함으로써 금속 이온이 촉매작용에 참여하는 경 우도있을것이다. (5) 생산적인 형태의 제공 촉매 효율을 괄목하게 제고하려면 반응물의 〈 유효 농도 녔t 5 0) 고도로 증가시 켜야 한다. 금속 이온은 템플레이트 효과에 의하여 분자 간 반응을 분자내 반 응으로 전환시킴으로써 유효 농도를 증가시킬 수 있다. 금속 효소에서 금속 이온은 단백질의 형태 변화를 유도함으로써 촉매 작용기 둘의 위치를 매우 미세한 수준으로까지 조정할 수 있다. 이 효과는 아직까지 소형 분자를 사용하여서는 성공적으로 재현한 바가 없다. 효소 반응에서는 기질과 효소의 착화합물 형성에서 방출되는 자유 에너지가 기질의 구조에 왜력 혹은 변형을 유도하는 데 사용될 수 있으며 기질의 구조에 이러한 왜력과 변형을 도입함으로써 반응속도를 크게 증가시킬 수 있다. 51)
 Energy
Energy
2,2' -디피리딜 부분에 금속이 킬레이트화될 때의 결합력이 3 및 3' 위치에 연 결된 반응 중심의 주변에 왜력을 유도하는 데 이용되어 (11) ' 고리화, 제거 반 응, 라세미화 반응 등을 가속하는 사례가 보고된 바 있다. 52) 이와 유사하게 디 -2- 피리딜 케돈이 Co(ll )이온에 킬레이트화되면 카르보닐 기에 왜력을 유도하게 되고, 그 왜력을 경감하기 위하여 카르보닐기의 수화가 촉진되는 사례가 보고된 바 있다. 11)
 OH. M-O
OH. M-O
Co( 매) 착화합물에 킬레이트화된 인산 모노에스데르의 가수분해 과정에서 의부 수산화 이온이 친핵체로 인 원자를 공격하여 금속 이온에 킬레이트화된 5 배위 수의 포스포란 중간체를 형성할 수 있다. 이때 킬레이트화된 에스테르 의 4- 원자고리에 유도되는 왜력 (12) 은 정사면체 인 원자가 5 배위 구조로 전환 되는 과정의 전이상태에서 경감될 수 있다. 53) 여러 가지 광학 선택적인 유기 합성 반응에서 금속 이온이 루이스산 촉매로 사용된 예가 보고되어 있다. 54), 5” 이러한 입체 선택성은 금속 이온이 전이상태 에서 한 가지의 입체 이성질체의 형태를 선택적으로 안정화시킴으로써 유발되 는것이다. 제 2 절 금속에 배위된 물 분자의 촉매작용 금속 이온이 물에 용해되면, 금속에 배위된 물 분자와 금속에 배위된 수산화 이온도 동시에 얻어지고 또한 중요한 촉매 역할을 수행할 수 있다. ( I ) 친핵체로서의 공격 2- 피리딘알도옥심의 아세틸 에스데르의 가수분해에 대한 Cu (Il)이온의 촉매 작용에서 수산화 이온의 농도에 속도가 비례하는 경로와 p H에 속도가 무관한 경로 등 두 가지 반응 경로가 관찰된 바 있다. 56) 속도 자료를 세부적으로 분석 한 결과 금속에 배위된 물 분자는 금속에 결합된 에스데르의 카르보닐 탄소에 친핵체로서 공격함 (13) 을 알 수 있었다. 물 분자의 염기성과 친핵성은 물 분자 가 금속 이온에 배위될 때 크게 감소한다. 이 반응에 대하여 보여주는 Cu( Il ) 에 배위된 물 분자의 효과적인 친핵성 공격은 다른 물 분자에 의한 일반 염기
 GBH Y{ °/
GBH Y{ °/
촉매작용이 효과적으로 일어나며 금속 이온의 템플레이트 효과에 의하여 친해 체와 친전자체 간의 상호 유효 농도가 매우 높아지기 때문이라고 볼 수 있다. 금속게 배위된 카르복실산의 락본화 반응 및 금속에 배위된 아미드의 가수분 해 반응에서 p H에 속도가 무관한 반응 경로가 관찰된 바 있다. 이것을 금속에 배위된 물 분자가 카르복실기 혹은 아미드기에 친핵체로 공격하여 반응이 진행 되는 것으로 해석하여 보고한 사례가 있다. 57),58) 그러나 이 반웅 경로는 금속에 배위된 수산화 이온이 친핵체로 작용한 다음 속도 결정 단계인 히드록실기나 아민이 끊겨나가는 과정에서 특정 산인 히드로늄 이온이 참여하는 메커니즘으 로 더 잘 설명된다 .59) (2) 일반산촉매작용 금속 이온에 배위되면 물 분자는 약산으로 변하고 이에 따라 일반 산 촉매로
 X = H or CH2coo·
X = H or CH2coo·
도 작용할 수 있다. 이 촉매 역할은 최근에 5%(v/v) 의 물을 포함하는 디메틸 술폭시드에서 금속 이온과 카르복실 음이온의 공동 촉매작용에 의하여 알킬 아 미드가 가수분해되는 반응 (14) 에서 최초로 관찰되었다. 60),61) 여기에서 카르복 실 음이온이 에스데르의 카르보닐 탄소를 공격하여 형성된 알콕시 음이온은 금 속 이온에 의하여 안정화된다. 이와 같이 형성된 정사면체 중간체에서 아민이 축출되는 과정에서 금속에 배위된 물 분자가 일반 산 촉매로 작용하여 이탈기 의 질소 를 양성자화시켜 주고 있다. 제 3 절 금속에 배위된 수산화 이온에 의한 촉매작용 금속 이온의 종류에 따라 금속에 배위된 수산화 이온이 중성 혹은 심지어 산 성 용액에서도 상당한 농도로 존재한다. 37),38) ( I ) 친핵체로서의 공격 금속게 배위된 수산화 이온이 친핵체로 작용하는 것은 여러 종류의 반응에서 관찰된 바 있다. 앞서 언급한 바 있는 4 큰 금속에 배위된 수산화 이온이 금속 에 결합된 에스테르를 공격하여 형성된 것이다. 18) 금속에 배위된 물 분자가 에 스테르를 공격하는 1E] 반응에서도 금속에 배위된 수산화 이온의 공격이 동시 에 일어나고 있다. 56)
 1M5/ NR2\ :\\ [ HN }一1 6 广i 亡〉R
1M5/ NR2\ :\\ [ HN }一1 6 广i 亡〉R
아미드의 가수분해가 금속에 배위된 수산화 이온의 친핵성 공격에 의하여 진 행되는 예로 쩌t 들 수 있다. 19),57) 금속에 배위된 수산화 이온이 카르보닐 탄소 에 인접한 곳에 위치함으로써 아미드기가 효과적으로 가수분해되는 예 (15) 가
보고된 바 있다. 62),63) 아민 질소가 정사면체 중간체에서 떨어져 나울 때는 전이 상태에서 이 질소 원자에 음전하가 생기는 것을 방지해 줄 양성자 공여체가 필 요하다. 금속에 배위된 수산화 이온이 친핵체로 공격할 때 이 수산화 이온이 이탈하는 아민 질소를 양성자화해 주는 양성자의 공급자 역할도 (16) 담당할 수 있다. 금속거) 배위된 수산화 이온이 알데히드 64) 혹은 산무수물 65) 등 다른 카르보닐 중심에 친핵성 공격을 가하는 예도 카르보닉 안히드라제 혹은 카르복시펩티다 제 A 와 같은 금속 효소의 모형으로 연구된 바 있다. 66)
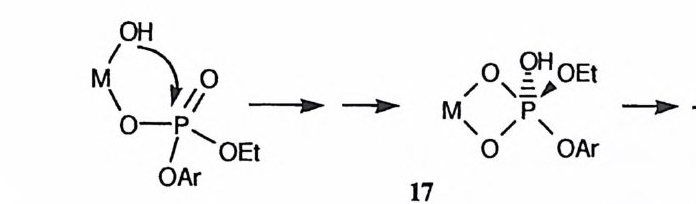 M/O\o H\—, IPIO\ _~ 一> M/\o /gP \ 0E t _► ~
M/O\o H\—, IPIO\ _~ 一> M/\o /gP \ 0E t _► ~
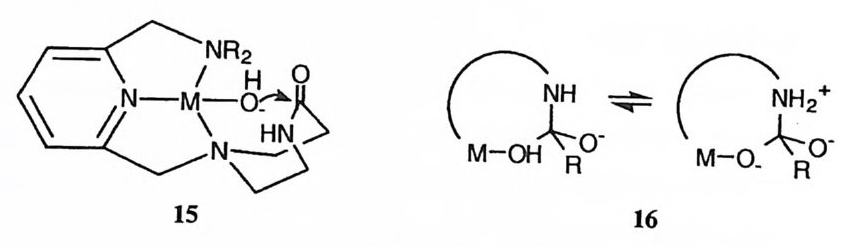
금속에 배위된 수산화 이온은 인산 에스테르에 대하여서도 천핵체로 작용할 수 있다. 예를 들어, 인산 디에스데르의 가수분해는 금속 이온의 촉매작용을 받는데, 금속에 배위된 수산화 이온이 친핵체로 작용하여 止才형 고리를 포함하 고 있는 중간체 (예 : 17) 를 경유하게 된다. 66)-68) Co( 매)에 배위된 수산화 이온이 울레핀 탄소를 친핵체로서 공격하여 이중결 합을 수화시키는 반응도 관찰된 바 있다. 69) (2) 일반 염기 촉매작용 금속에 배위된 수산화 이온은 약염기이기 때문에 일반 염기 촉매로 작용할 수 있다. 그러나 이 촉매 역할은 최근에서야 m-(2- 이미다졸릴아조)페닐 P 나尸구 엔술포네이트의 가수분해에 대한 Cu (Il)이온의 촉매작용에서 최초로 관찰되었 다. 70) 이 반요]서 첨가 중간체 사이에 양성자가 이동되는 단계 (18) 에서 히드 록 (Il ) 이온이 일반 영기로 참여하고 있다.
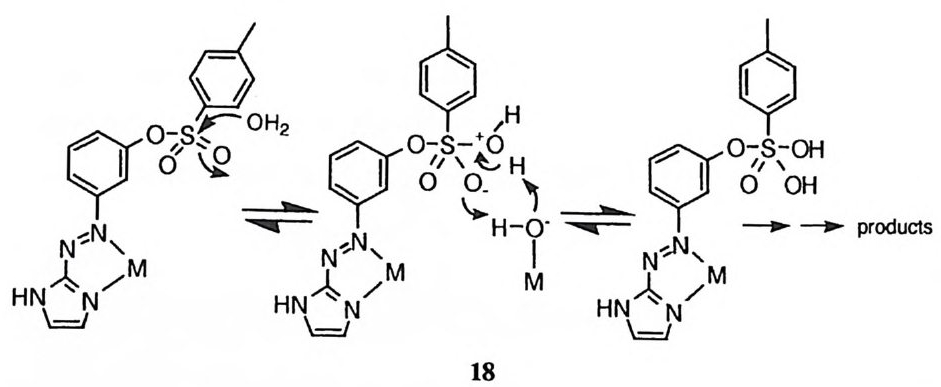 戶: -! ' 亨
戶: -! ' 亨
제 4 절 이핵체 금속 이온의 촉매작용 2- 아세틸피리딘케토옥심과 6- 카르복시 -2- 피리딘카르복스알도옥심의 가수분 해에 대한 Zn(Il) 이온의 촉매작용에서 Zn(Il) 이온 두 개가 참여하는 반응 경 로가 관찰된 바 있다.?" 상세한 메커니즘 분석 결과 이핵체 Zn (Il)이온이 촉매 단위로 참여 (19) 하고 있으며 이것과 속도론적으로 동일한 메커니즘인 두 개의 Zn (Il)이온이 분리되어 참여하는 경로 (20) 는 작동하지 않는 것이 밝혀졌다 .71)
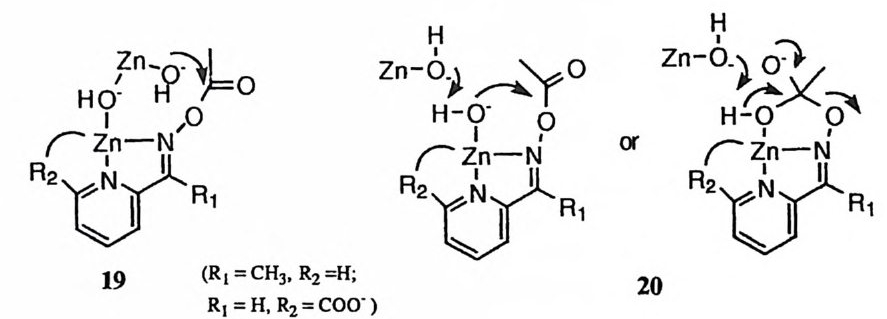 ZRn-HH% - ?一 :Y p:° or znR- 「OH.H 汶 -0건 〉 군 A1
ZRn-HH% - ?一 :Y p:° or znR- 「OH.H 汶 -0건 〉 군 A1
뿐만 아니라, 6- 카르복시 -2- 피리딘카르복스알도옥심의 가수분해의 경우에는 일핵체 Zn (H)이온이 참여하는 촉매작용은 전혀 관찰되지 않았다. 이핵체 Zn (l )이온의 평형 농도는 일핵체와 비교하여 훨씬 작을 것이다. 그럼에도 불구· 하고 이핵체 이온에 의한 촉매작용이 효과적으로- 진행되는 것은 전이상태의 구 조와 관련이 있다고 여겨진다. 전이상태에서 형성되는 고리의 안정도가 고리의
구조가 바뀜에 따라 크게 영향을 받을 것이기 때문이다. 몇 가지 다른 반옹에 서도 금속 이온의 촉매작용에 두 개의 금속 이온이 전이상태에 포함되어 있는 예가 보고되고 있다. 72),73) 정확한 메커니즘 분석은 이루어져 있지 않지만 이러 한 반舒]서도 이핵체 금속 이온이 촉매 단위로 포함되어 있을 가능성이 있다. 제 5 절 금속 이온과 유기 작용기의 협동 금속 이온의 촉매 효율은 다른 촉매 요인이 금속 이온과 협동하여 참여할 때 더욱 제고될 것이다. 유기 반응에 대한 금속 이온의 촉매작용에 일반 산이나 일반 염기가 침여하는 예로서 피루브산 엔을 음이온의 케돈화에 대한 아세트산 과 Cu(Il )이온의 공동 촉매작용이나 74) 아미드 가수분해에 대하여 Co(III) 에 결 합된 수산화 이온과 완충제의 공동 촉매작용을 75) 둘 수 있다.
금속 이온과 유기 작용기의 협동은 유기 촉매 작용기와 금속 결합 부위가 모 두 반응자리에 인접하여 있을 때 매우 효율적으로 진행될 것이다. 예를 들어, 금속 이온과 아미드 산소 원자가 매우 효율적으로 협동하여 아릴 에스데르 결 합이 파괴되는 예 (21) 가 관찰된 바 있다. 76),77) 2- 피리딘카르복스알도옥심의 Zn (Il)착화합물이 아세트알데히드의 수화에 촉매로 침여할 때, 킬레이트화된 리간드의 옥심 음이온이 일반 영기로 작용하여 Zn( Il )에 배위된 아세트알데히 드의 카르보닐기에 물 분자가 공격하는 것을 도와준다고 보고된 바 있다. 78) Zn(Il ) 금속 효소인 카르복시펩티다제 A(CPA) 에 대하여서는 제病》 ]서 다 루게 되듯이 효소 자체의 메커니즘에 대하여서도 많은 논쟁이 진행중이며 모형
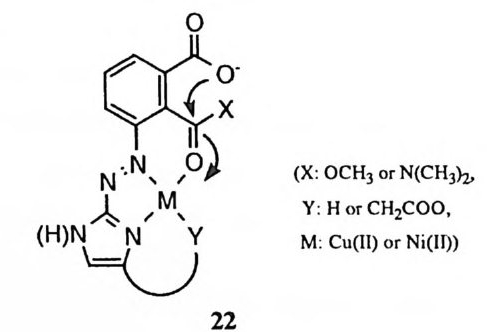 며N •. N 、 :,,占) (X: OCH3 or N(CH3 n. ,
며N •. N 、 :,,占) (X: OCH3 or N(CH3 n. ,
연 구도 상당량에 걸쳐 보고된 바 있다. CPA 의 모형으로 설계된 화합물 중에 가장 많은 수의 촉매 특성 을 성공적으로 재현한 것은 2 - 이미다졸릴아조 부분을 금속 결합자리로 포함하고 있는 화합물 (22) 이다. 60), 61 ) ,7 3 ) 이 효소 모형에서 금 속 이온, 카르복실 음이온 및 용매가 효과적으로 협동하여 알킬 아미드 혹은 알 킬 에 스 데르 결합을 절단하고 있다. 제 6 절 복수개의 촉매 레퍼토리의 작동 대부분의 유기 촉매 작용기와는 달리 금속 이온은 두 가지 이상의 촉매 역할 울 동시에 수행하는 예가 혼하다. 예를 들어 19 로 71) 표시된 에스데르 가수분해 의 촉 매작용에 관여하고 있는 금속 이온의 촉매 레퍼토리는 금속에 의한 템플 레이트 효과, 이핵체 금속 이온의 참여, 금속 이온에 의한 이탈 옥심의 활성 화, 금속 이온에 배위됨으로써 물 분자의 이온화의 제고 그리고 금속에 배위된 수산화 이온의 친핵성 공격 등이다. 14로 60),61) 표시된 아미드 가수분해에서 금속 이온은 정사면체 중간체의 옥시 음이온을 안정화시켜 주고 있을 뿐 아니라 금속에 배위된 물 분자의 산성도를 제고시키고, 고리형 전이상태의 적절한 기하학적 구조를 제공하고 있다. 뿐만 아니라, 금속에 배위된 물 분자는 일반 산으로 작용하고 있다. 포스핀산의 에스데르와 인산의 모노-, 디-, 혹은 트리에스테르의 가수분해에 대한 금속 촉매작용에서는 12 혹은 17 의 예와 같이 #R 형 구조의 중간체가 혼
히 개입된다 .53),66)-68) 이 경우에 템플레이트 효과, 포스포릴 산소 원자에 형성 된 음이온의 안정화, 기질 구조내의 왜력 유도, 금속에 배위된 물 분자의 산성 도의 제고 등이 금속 이온에 의하여 달성되고 있으며 금속에 배위된 수산화 이 온은 친핵체로 참여하고 있다. 제 7 절 금속 이온의 성질과 리간드의 구조가 촉매 효율에 미치는 효과 생물학적 반응뿐만 아니라 간단한 유기 반응에서 수행하는 금속 이온의 촉 매 작용에서도 금속 이온의 종류와 리간드의 종류 및 정확한 위치가 중대한 역할 울 담당할 것이라 함은 의문의 여지가 없다. 금속 이온의 종류가 바뀔 때 유기 반응에 대한 금속 촉매작용의 효율이 괄목 하게 바뀌며 이러한 변화를 설명할 수 없는 사례가 흔히 보고되고 있다. 72) 인산 디에스데르의 가수분해에 대한 금속 이온의 촉매작용에서 Ir( 매)은 Co( fil)보다 효율이 훨씬 떨어진다. 이 사실은 Ir( 매)이온의 반경이 더 크며 따 라서 Ir( 매)에 배위된 수산화 이온이 반응 중심인 원자를 분자내에서 공격 (17) 하여 仁淸짜리 중간체를 형성하기가 더 어렵기 때문이라고 설명된 바 있다. 68 ) 반뭉개 中斗서는 금속 이온이 바뀔 때 메커니즘이 달라질 수도 있다. 79) 여러 가지 리간드를 가진 Cu(Il) 착화합물이 에스데르 가수분해에서 반응성에 차이 가 나는 것을 리간드의 전자 공여 능력에 따라 Cu( Il )이온의 루이스 산도가 감 소하는 것과 관련지어 설명한 보고도 있다• 80) cis-디아콰데트라아자 Co (매 ) 착 화합물에 의한 인산 디에스데르 가수분해의 촉매작용에서 테트라아자 리간드의 구조가 바뀔 때 촉매 활성도가 괄목하게 바뀌는 것이 관찰된 바 있다. 66) 이러 한 사실은 ¥-l-형 중간체에서의 결합각과 관련지어 설명되었다. Zn (Il)쭙 효소에 대하여 많은 경우에 카르복실 음이온-이미다졸 -Zn(Il) 의 세 개의 작용기가 구조적으로 연계되어 촉매작용에 참여하고 있음이 보고된 바 있다 .81) 이는 Zn (Il)이온의 성질이 카르복실 음이온과 이미다졸의 상호작용 울 통하여 수정되고 있음을 암시한다. 1~ 2~ 표시된 아미드 가수분해에서
이미다졸 N — H 가 이온화해야만 금속 촉매 효과가 작동함이 관찰되었다. 60),61) 이미다졸 고리의 전자 밀도가 증가하면 중심 금속 이온의 촉매 활성도를 제고 하고 있는데, 이는 위에서 언급한 금속 효소에서 관찰되는 카르복실 음이온-이 마디좀 -Zn(Il )의 세 작용기의 공동 참여와 유사한 것이다. 그러나 왜 이미디졸 고리에서 전자 밀도가 증가하면 중심 금속 이온의 촉매 효율이 제고되는가에 관한 이유는 현재로서 알려진 바 없다. 제 8 절 전망 유기 반응에 루이스산 촉매로 사용할 수 있는 금속 원자의 종류는 란타니드 혹은 악티니드를 포함하여 매우 큰 수에 달한다. 각 금속 원자의 여러 가지 산 화상태까지 고려하면 이 숫자는 훨씬 커지게 된다. 그런데 메커니즘에 관한 자 료는 소수의 유기 반응과 소수의 금속 이온에 대하여서만 수집된 바 있다. 더 욱이 금속 이온의 반응성에 대한 리간드의 효과에 대하여서는 거의 알려진 바 가 없다. 따라서 이 분야의 연구 활동이 진행됨에 따라 유기 반응에 대한 금속 이온의 루이스산 촉매작용에 대한 새로운 사실이 속속 밝혀지게 될 것이다. 생체 모방 촉매에 의한 분자 인식 및 촉매작용은 현재 매우 활발한 연구의 대상이 되고 있다. 이에 따라 금속 이온의 촉매 레퍼토리를 다수 활용하는 효 율적인 인공 금속 효소의 설계도 광범위하게 추구될 것으로 전망된다. 82)-86)
참고문헌 1) Bender, M. L. Mechanis ms of Homoge n eous Cata ly si s from Proto n s to Prote ins , Wi ley - l nte r scie n ce : New York, 1971, p. 212. 2) Vallee, B. L. ; Wacker, W. E. C. In Handbook of Bio c hemi stry and Molecu-
lar Bio lo gy , Fasman, G. D., Ed., C RC Press : Cleveland, 1976, 3rd ed., V ol. 2, pp. 276-292. 3) Satc h ell, D. P. N. ; Satc h ell, R. S. Annu. Re p. Prog. Che m. Sec t. A , 1979, 75 , 25. 4) Hay, R. W. In Comp relze 函 ve Coordin a ti on Che mi str y, Wi lki n s on, G., E d., Perga m on : Oxfo r d, 1987, Vol. 6, pp. 412-485. 5) Suh, J. Bio o rg. Chem ., 1990, 18, 345. 6) Suh, ]. Acc. Clzem. Res., 1990, 25, 273. 7) Buckin g h a m, D. A. ; Dekkers, J. ; Sarge s on, A. M. ; Wein , M. ]. A111. Cli em . Soc, 1972, 94, 4032. 8) Hay, R. W. ; Nolan, K. B. J Clzem . Soc. Dalt on Tra ns., 1974, 2542. 9) Buckin g h a m, D. A. ; Sarge s on, A. M. ; Keene, F. R . ]. Am. Chem. Soc., 1974, 96, 4981 . 10) El-Hi laly , A. E. ; El-Ezaby , M. S. ] .lnorg. N ucl. Chem., 1976, 38, 1533. 11) Suh, M. P. ; Kwak, C.- H ., S uh, J. I norg. Chem ., 1989, 28, 50. 12) Suh, M. P. ; Kwak, C.-H ., Suh, J. J Clzem . Soc. Dalto n , 1991, 1165. 13) Creig h ti on , D. J. ; Hajd u , J. ; Sig ma n, D. S. J Am. Chem. Soc., 1976, 98 , 4619. 14) Breslow, R. ; Fair w eath e r, R. ; Keana, J. J Am. Chem. Soc, 1967, 89, 2135. 15) Breslow, R. ; Sch rnir, M. J Am. Chem. Soc., 1971, 93, 4960. 16) Covey, W. D. ; Leussin g , D. L. ]. Am. Chem. Soc., 1974, 96, 3860. 17) Kubala, G. ; Mart ell , A. E. J Am. Chem. Soc., 1 982, 104, 6602. 18) Wells, M. A. ; Bruic e, T. C. J Am. Chem. Soc., 1977, 99, 5341 . 19) Buckin g h am, D. A. ; Foste r , D. M. ; Sarge son, A. M. ]. Am. Che m. Soc., 1974, 96, 1726. 20) Sutt on , P. A . ; Buckin g h a m, D. A. Acc. Chem. Res., 1987, 20, 357. 21) Gellman, S. H. ; Pett er , R. ; Breslow, R. J Am. Chem. Soc., 1986, 108, 2388. 22) Meng er , F. M. ; Tsuno, T. ]. Am. Chem. Soc., 1989, 111, 4903. 23) Fif e, T. H. ; Przy s ta s , T. J. J Am. Chem. Soc., 1 982, 104, 2251 . 24) Przys t a s, T. J. ; Fife , T. H. ]. Am. Chem. Soc., 1980, 102, 4391 . 25) Hanzlik , R. P. ; Hamburg, A. ]. Am. Chem. Soc., 1 978, 100, 1745.
26) Suh, J. ; Lee, E. ; Jan g , E. S . Inorg. Chem. , 1981, 20, 1932. 27) Suh, J. ; Suh, M. P. ; Lee, J. D. Inorg. Che- ,n., 1985, 24, 3088. 28) Suh, J. ; Kwon, B. N. ; Lee, W. Y. ; Chang , S. H. Inorg. Che1n., 1 987, 26, 805. 29) Hsu, C.- M . ; Coop er man, B. S. J Am. Chem. Soc., 1976, 98, 5652. 30) Suh, J. ; Koh, D. ]. O rg. C he1n., 1 987, 52, 3446. 31) Gensmante l, N. P. ; Gow li ng , E. W. ; Page , M. I. J Chem. Soc. Per k in Il , 1978, 375. 32) Schwart z, M. A. Bio o rg. C he m ., 1 982, 11, 4. 33) Houg ht o n , R. P. ; Putt ne r, R. R. Che- ,n. Commun., 1970, 1270. 34) Suh, J. ; Mi n, D . W. ]. O rg. C hem., 1991, 56, 5710. 35) Satc h ell, D . P. N. Che m. Soc. Rev., 1977, 6, 345. 36) Blackburn, G. M. ; Ward, C. R . M. J Chem. Soc. Chem. Commun., 1 976, 79. 37) Basolo, F. ; Pearson, R. G. Me c/1 1.ln is ms of Inorga n ic Reactio n s, Wi le y : New York, 1968, 2nd ed., p. 32. 38) Barnum, D. W. Ino r g. C he1 n., 1983, 22, 2297. 39) Eik i , T . ; Kawada, S . ; Mats u shim a, K . ; Mori, M. ; Taga k i, W . Che- ,n. Lett. , 1980, 997. 40) Breslow , R. ; Chap m an, D. J Am. Chem. Soc., 1 965, 87, 4195. 41) Lau, H.-p. ; Guts c he, C . D . J Am. Chem. Soc., 1 978, 100, 1857. 42) Suh, J. ; Cheong, M. ; Han, H. Bio o rg. Che1n., 1984, 12, 188. 43) Harrow field , J. M. ; Jon es, D. R. ; Lin d oy, L. F. ; Sarge s on, A. M. J Am. Che- ,n. Soc., 1 980, 102, 7733. 44) Dixo n, N. E. ; Sarge s on, A. M. ]. Am. Chem. Soc., 1 982, 104, 6716. 45) Akaobri, S. ; Ota n i, T. T. ; Marshall, R . ; Wi ni t z, M. ; Greenste i n , J. P . Arch. B io c he- ,n. Bio p h y s . , 1959, 83, 1. 46) Cox, B. G. ]. Am. Che- ,n. Soc., 1 974, 96, 6823. 47) Buckin g h am, D. A. ; Ste w art , I. ; Sutt on , P. A. ]. Am. Che- ,n. Soc., 1990, 112, 845. 48) Suh, J. ; Chun, K. H. ]. Am. Clze -,n. Soc., 1 986, 108, 3057. 49) Spe c to r , L. B. Covale11t Cata lysis by Enzym e s, Spr in g e r-V erlag : New York, 1982.
50) Kir b y, A. J. Adv. Phys . Org. Che1n., 1980, 17, 183. 51) Jen cks, W. P. Cata lysis in Chemi stry and Enzym ology , McGraw-Hi ll : New York, 1969, p. 294. 52) Rebek, J., Jr. ; Coste ll o, T. ; Watt ley , R. J Am. Chem. Soc., 1 985, 107, 7487. 53) Anderson, B. ; Mi lbu rn, R. M. ; Harrow field , J. M. ; Robert so n, G. B. ; Sar- ges on, A. M. J Am. CA奭 . Soc., 1977, 99, 2652. 54) Fin n M. G. ; Sharp le ss, B. In As ymn zetr ic Syn the s is, Morriso n J. D., Ed., Academ ic Press : Orlando, 1985, Vol. 5, pp. 247- 30 8. 55) Noy o ri, R. ; Takaya , H. Acc. Chem. Res., 1990, 23, 345. 56) Suh, J. ; Cheong, M. ; Suh, M. P. J Am. Clzem. Soc., 1982, 104, 1654. 57) Boreham, C. J. ; Buckin g h a m, D. A. ; Franci s, D. J. ; Sarge s on, A. M. J Am. Chem. Soc., 1981, 103, 1975. 58) Boreham, C. J. ; Buckin g h a m, D. A. ; Keene, F. R. J Am. Chem. Soc., 1979, 101, 1409. 59) Suh, J. ; Han, H. Bio o rg. Chem., 1984, 12, 177. 60) Suh, J. ; Hwang, B. K. ; Koh, Y. H. Bio o rg. Chem. , 1 990, 18, 207. 61) Suh, J. ; Park, T. H. ; Hwang, B. K. ]. Am. Clzem. Soc., 1992, 114, 5141 . 62) Groves, J. T. ; Chambers, R. R., Jr. J Am. Chem. Soc., 1984, 106, 630. 63) Groves, J. T. ; Baron, L. A. J Am. Chem. Soc., 1989, 111, 5442. 64) Woolley, P. Natu re , 1975, 258, 677. 65) Breslow, R. ; McClure, D. E. ; Brown, R. S. ; Eis e nach, J. J Am. Chem. Soc., 1975, 97, 194. 66) Ch in, J. Acc. Chem. Res., 1991, 24, 145. 67) Ch in, J. ; Banaszezyk , M. ; Jub ia n , V. ; Zou, X. J Am. Chem. Soc., 1989, 111, 186. 68) Hendr y, P. ; Sarge s on, A. M. J Am. Chem. Soc., 1989, 111, 2521 . 69) Gahan, L. R. ; Harrow field , J. M. ; Heri t, A. J. ; Lind oy, L. F. ; Whimp , P. 0. ; Sarge s on, A. M. J Am. Chem. Soc., 1985, 107, 6231 . 70) Suh, J. ; Kim, J. ; Lee, C. S. ]. Org. Chem., 1991, 56, 4364. 71) Suh, J. ; Han, 0. ; Chang, B. J Am. Chem. Soc., 1986, 108 , 1839. 72) Suh, J. ; Chang, B. Bio o rg. Chem., 1987, 15, 167.
73) Suh, J. ; Chung , S. ; Lee, S. H. Bio o rg. C hem. , 1987, 15, 383. 74) Mi ller, B. A. ; Leussin g , D . L. J Am. Chem. Soc., 1 985, 107, 7146. 75) Schep ar t z, A. ; Breslow, R. J Am. Chem. Soc., 1987, 109, 1814. 76) Suh, J. ; Kim , S. M. Bio o rg. Chem. , 1989, 17, 169. 77) Fif e, T. H. ; Przys t a s , T. ]. ; Puja r i, M. P.] . Am. Chem. Soc., 1 988, 110, 8157. 78) Woolley, P. R. J Chem . Soc. Chem. Commun, 1975, 579. 79) Leach, B. E . ; Ang el ic i, R. J. J Am. Chem . Soc., 1 968, 90, 2504. 80) Nakon, R. ; Rechani, P . R. ; Ang el ic i, R. ]. Am. Chem. Soc., 1974, 96, 2117. 81) Chris ti a n son, D. W. ; Alexander, R. S. ]. Am. Chem. Soc., 1989, 111, 6412. 82) Rana, T. M. ; Meares, C. F. J. Am. Chem. Soc., 1990, 112, 2457. 83) Schep ar t z, A. ; Cuenod, B. J . Am. Chem. Soc., 1990, 112, 3247. 84) Hoy er , D. ; Cho, H. ; Schultz, P. G. ]. Am. Chem. Soc., 1990, 112, 3249. 85) Mack, D. P. ; Dervan, P. B. ]. Am. Chem. Soc., 1990, 112, 4604. 86) Suh, J. ; Cho, Y. ; Lee, K. J. ]. Am. Chem. Soc., 1991, 113, 4198.
제 5 장 효소 반응 메커니즘 연구의 예 : 카르복시펩티다제 A 생명체의 각종 생명현상의 원리를 가장 기본적으로 파악하기 위하여서는 생 체 반응에 촉매로 관여하는 효소의 작용 원리를 구명하여야 한다. 현대 과학의 수준게서 효소 반응의 메커니즘을 가장 정밀하게 이해하는 것은 효소의 활성자 리에 있는 각 아미노산 잔기에 포함된 작용기의 치원에서 효소의 촉매작용을 파악하는 것이다. 효소 반응의 메커니즘의 연구에는 물리 유기화학 분야에서 유기 반웅 메커니즘을 구명하는 데 사용한 각종 기법을 활용하게 된다. 이 장 에서는 물리 유기화학의 분야에서 개발된 각종 연구 방법을 응용하여 효소 반 응 메커니즘을 규명하는 작업을 설명하려 한다. 이 목적으로 카르복시펩티다제 A의 메커니즘 연구를 예로 들기로 한다. 제 1 절 기질의 특이성 카르복시펩티다제 A( 이하 CPA 로 약칭함)는 생체내에서는 단백질의 카르복시 말단에 인접한 아미드기를 가수분해시키는 데에 사용되는 단백질 가수분해 효 소이다. I) 그러나 생체 바깥에서는 아미드 이의에도 에스테르와 티울에스테르와 같은 카르복실산 유도체의 가수분해에도 활성을 나타낸다. 표 l 에 CPA 에 의 해 가수분해받는 카르복실 유도체의 구조적 특이성을 요약하였다. 카르복실 유도체의 가수분해 이의에도 1-3 과 갇은 반응들이 CPA 에 의하여 촉매작용을 받는다는 사실이 보고되어 있다. l7),8) 의 반응은 케돈의 엔올화 반응이며 29) 와 식 310) 의 반응은 f3-제거 반응
 X,,,Phr[ co o· _——> x,,.Ph:\c: 1
X,,,Phr[ co o· _——> x,,.Ph:\c: 1
 표 I CPA 에 의하여 가수분해받는 카르복실 유도체 기질의 구조적 특이성
표 I CPA 에 의하여 가수분해받는 카르복실 유도체 기질의 구조적 특이성
이다. 이 반응을 일으키는 화합물의 구조는 CPA 의 기질의 구조와 유사하여 카르복실기, a- 탄소의 형수성 결사슬, T 위치의 카르보닐기 등을 포함하고 있 다. 즉 이러한 구조적 요건을 충족시킴으로써 CPA 의 활성자리에 끼여들어 착 화합물을 형성하게 되고 , 이 착화합물내에서 효소의 촉매 작용기가 기질의 카 르복실기의 /3 위치에 있는 C-H 결합을 공격함으로써 엔올화 혹은 제거 반응 이 일어나게 되는 것이다.
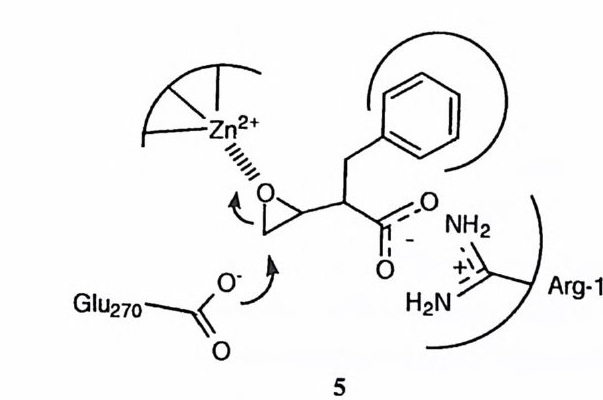
제 2 절 구조 CPA 에 대하여 프로카르복시펩티다제 A 가 선구 물질로 존재한다• 11),12) CPA 의 분리 정제 방법으로는 세 가지가 보고되어 있는데, 13)-15) 분리 방법에 따라 각기 다른 형태의 CPA 가 얻어진다. CPA 의 아미노산 순서는 화학적 방법 16) 및 X- 선 결정학 방법으로 1 ),17) 절정 되어져 있다. CPA 의 분리 방법에 따라 N- 말단 아미노산의 위치에 차이가 나 게 된다. N- 말단 아미노산에 차이가 나는 CPA 에 대하여 속도론적인 차이점 으로 두드러진 것은 아직 보고된 바가 없다. CPA 는 소의 췌장으로부터 분리 하는데, 이때 사용한 소의 종류에 따라 유전학적인 이유로 몇 군데의 아미노산 이 바뀐 두 가지 {CPAval 및 CPALeu) 의 이성질체 효소가 얻어진다. CPA 결정의 삼차원적 구조는 X- 선 회절법으로 측정되었는데, l),l7) CPA 의 활성자리에는 Zn( Il )이온이 위치해 있다. 이 Zn(Il )이온에는 Giu - 72, His-6 9 및 H i s-196 이 리간드로 배위되어 있다. 디펩티드인 Gl y -T yr은 N- 말단이 아 실화되어 있지 않아 CPA 에 의하여 극히 느린 속도로 가수분해된다. Giy - T yr을 CPA 와 결합시킨 다음, 이때 형성된 착화합물의 결정 구조를 CPA 와 비교함으로써 활성자리의 위치를 찾아내고 활성자리 내부에서의 효~ 기질 간의 구체적인 상호작용을 밝힐 수 있었다. Gl y -T yr과 CP A;가 형성한 착화합 물에 대하여 결정한 구조는 반응을 일으키지 못하는 비생산적인 형태에 관한 것이라는 제약점을 수반한다. 따라서 이 착화합물의 결정 구조로부터 도출한 효소와 기질 간의 상호작용은 실제 효소 반응의 동적인 반응 과정에서 관여하 는 것과 차이가 있다. 그러나 결정학적인 구조로부터 Zn (Il)이온 이의에도 Glu-270, Ty r-2 48 및 Ar g -14 화 촉매작용에 일차로 개입되는 주요 작용기임 을 알 수 있었다. 이에 따라 CPA 반응 메커니즘의 연구는 Zn (Il)이온, Glu- 27 뼈 카르복실기, Ty r-2 4~ 페놀기, Arg- 1 4~ 구아니디늄기의 정확한 역 할의 규명에 초점을 맞추게 되었다.
제 3 절 Glu-270 의 촉매 역할 Glu - 270 이 활성자리에 위치해 있으며 중요한 촉매 역할을 담당할 것이라는 점은 X- 선 결정학 연구결과로부터 먼저 제시되었다. 1),1 7 ) Glu-27 떠 화학적 수정은 N- 브로모아세틸 -N- 메틸페닐알라닌을 사용하여 수행 (4) 되었다. 20) 이 수정 시약은 CPA 의 기질이 갖추는 구조적인 요건들을 구비함으로써 CPA 의 활성자리에 끼여들어 착화합물을 형성하고, 이 착화합물 내에서 Glu-270 의 카르복실기와 반응하여 CPA 를 알킬화시킨다. 이 화합물은 아미드이지만 아미드 질소에 붙은 수소가 메틸기로 치환되었으므로 CPA 에 의 하여 아미드의 가수분해를 받지 않는다. 이 화학적 수정의 결과로 얻어진 효소 는 활성을 완전히 상실하였다. 이 결과로부터 Glu-27~ 카르복실기가 CPA 의 촉매작용에 필수적으로 필요하다고 할 수는 없다. 그 이유는 Glu-270 에 커 다란 분자가 공유 결합에 의해 연결되면 단순히 입체적으로. 활성자리를 봉쇄함 으로써 CPA 의 활성이 상실될 수가 있기 때문이다.
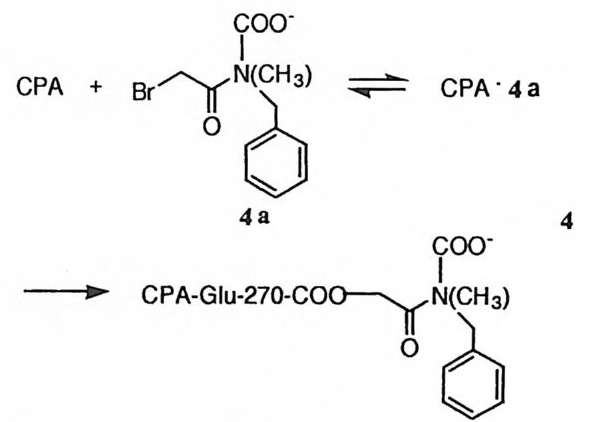 CPA + Br\90:0)· = CPA 4 a
CPA + Br\90:0)· = CPA 4 a
4oJ] 의한 Glu-27~ 화학적 수정 실험으로부터 두 가지의 중요한 정보가 제 공되었다. 첫째는 Glu-27 뼈 카르복실기의 p Ka 값을 ~ E 파 반응하는 속도 의 pH 의존도로부터 결정할 수 있었던 점이다. EI 착화합물 속에서 측정된 , Glu-27 뼈 pK a 값은 7. 邸로서, 카르복실기에 대하여 통상적으로 물속에서 관찰되는 pK a 값인 4oJ] 비하여 두드러지게 높다. 이것은 Glu-270 이 위치한
활성자리 내부의 미세환경이 매우 높은 협수성을 띠기 때문으로 해석된다. 둘 째는 Glu-27 떠 카르복실 음이온이 브롬화 알칸인 4~ 반응하는 속도가 매우 빠르다는 사실이다. 이것은 EI 착화합물 내부에서 Glu-27 個l- 4~ 반응부위 조!<>ii 상당히 효과적인 인접 효과가 존재하기 때문으로 볼 수 있다. 그리고 음 이온 친핵체에 의한 SN2 반응이 바극성 매질 속에서 증속된다는 사실과도 결부 시킬수있다.
 。 J _,___ ' Arg- 1 45
。 J _,___ ' Arg- 1 45
최근에는 에폭시드 저해제에 의하여 Glu-270 이 알킬화되는 것 (5) 이 보고되 었다 .21) 특히, 이 경우에는 Glu-27~] 카르복실기가 알킬화되었음을 X- 선 결 정학으로 입증한 바 있다. 22) CPA 는 생성물인 a- 아미노산, a- 히드록시산, 혹은 a- 메르캅토산과 착화합 물을 형성하는데, 이들 생성물은 경쟁적 저해제로 작용한다. 따라서 CPA 촉 매 반응의 속도론적 행동은 생성물에 의한 경쟁적 저해 작용 (6) 에 의하여 특칭 지어진다.
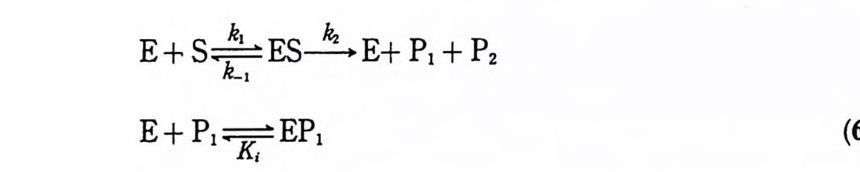 E+S~&k1 ES 一& E+P1+P2
E+S~&k1 ES 一& E+P1+P2
6g. CPA 에 대해 수집된 속도 자료를 설명할 수 있는 가장 간단한 반응식이
된다. 6oll 대해서 유도되는 ~a t과 Kma p社근 다음과 같다. k.:at = kz (7) Kmap p= Km= (k_1 + ki)/k 1 (8) 경우에 따라서는 @] 미카엘리스-멘텐 식을 조금 더 복잡하게 변형시키기도 한다. 이때 flcn t 및 Kma p呼 표현도 바뀌게 된다. 수정된 반응식과 그에 따른 변수의 표현을 예시하면 다음과 갇다. E + S 一kk_1 I ES 一& ES1 一k, E + P, + P2 E+ Pl=K=i : EPI (9) 찌대해서 ~at = fvi~/ (fvi + ~) (10) Kmap p = Km~/ (fvi + ~) (11) = ~ (k_. + !vi) Ik1 (!vi + ~) E+ S ;:k:k-it t= = ES ;:k:&_i2= = ES1 一& E+ P1 + P2 E+P1~KiE P1 (12) 1 짜 대해서 kcet = /¾~/ (/¾ + ~) (13) Kmap p = (結싸 + k-1~ + k_,k._2 ) /k1 (% + & + 回 (14)
CPA 에 의한 카르복실 유도체의 가수분해 반응에 대하여 속도론적 자료를 수집하여 분석한 후, &at, k.:a1 /Kmap p 및 Kmap Po ll 대한 pH 의존도를 작성 하면 다음과 같은 몇 가지 범주가 나타난다. 幅 /Kma p¢1 pH 의존도는 기질과 착화합물을 형성하고 있지 않은 자유로 운 상태에 있는 효소 속에서의 촉매 작용기의 pK a 값을 나타낸다. CPA 의 福 /Kma p回 pH 곡선은 기질의 종류세 따라 세 가지 유형을 가진다. 4),2 3 )- 3 1) 〈 범주 |-I> p Ka 가 6-6.5 인 작용기가 염기성형으로 존재하고 p Ka 가 약 9 인 작용기가 산성형으로 존재하여야 활성을 보이는 기질 : 대부분의 기질이 여기에 속함. 〈 범주 1-2> p Ka 가 6.5-7 인 작용기가 염기성형이고 p Ka 가 7.5-8 인 작용기 가 산성형일 때에 활성을 보이는 기질 : 0- 아세틸 -L- 만델산 혹 은 0- 니트로벤조일 -L- 만델산 등이 여기에 포함되는데, 이들은 매우 느리게 가수분해되는 에스테르 기질임 . 〈범주 1-3> p K가 약 8 인 작용기가 염기성형이고 p K가 약 9.5 인 작용기가 산성형일 때 활성을 나타내는 기질 : D- 티올에스데르인 S - 신나 모일 -D-a- 메르캄토-f3 - 페닐프로피온산이 여기에 속함. kea t의 pH 의존도는 기질의 종류에 따라 다음과 갇이 분류할 수 있 다 23),25),26),28),3 2 ),33) 〈범주 2-1> p Ka 가 약 6 인 작용기가 영기성형이기만 하면 활성을 보이는 기 질 : 울리고펩티드들이 여기에 속함. 〈범주 2-2> p Ka 가 약 7 인 작용기가 염기성형이고 p K가 약 8 인 작용기가 산성형일 때 활성을 보이는 기질 : 범주 1-2 에 속한 기질이 여기 에해당됨. 〈범주 2-3> p H 初}지는 범주 2-1 의 !?.;a t과 같은 pH 의존도를 보이지만 pH 9 인 이싱에서는 p比가 상승할 때 kca t이 급상승하는 기질 : o- 신 나모일 -L- /3페닐락트산이 여기에 속함.
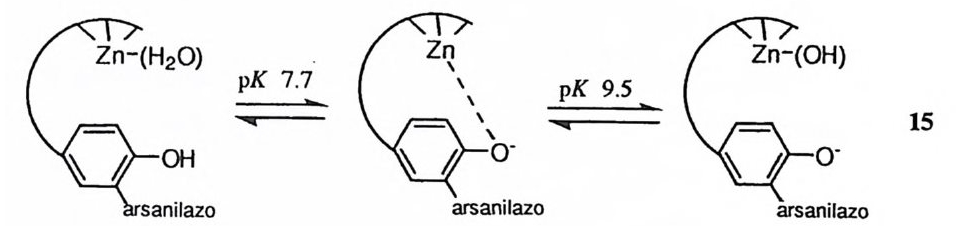
〈 범주 2-4> & m 이 p H 에 무관한 기질 : o- 히퓨릴 -L- {J-페닐락트산, 0-(a- 벤조일아미노)신나모일 -L- {J-페닐락트산, 0 - (a- 벤조일아미노)신 나모일 -L - 만델산 등이 여기에 속함. 이상에 열거한 법주들을 특정짓는 pK a 값을 모으면, 약 6.5, 약 8.0 및 약 9 인 적어도 세 가지의 촉매 작용기가 존재함을 알 수 있다. 이들이 어느 아미 노산 잔기의 결사슬인 것에 대한 주장은 여러 가지가 제시되어 있지만, 화학적 수정과 분광학적 측정 의 결과를 종합하면 디움과 같이 결론을 내릴 수 있다. 4로 표시된 화학적 수정의 결과로부터 EI 착화합물에서의 Glu-27~ p Ka 가 7.0 임이 판명되었다. EI 의 활성자리는 자유로운 CPA, 즉 E 긱 활성자리보다 다소 형수성이 높을 것을 감안하면 E1l 서의 pK a 값 6 . 허문 Glu-27Q ol] 결부시 키는것이 타당하다. CPA 의 T yr -24 짜 특이하게 아르사닐아조화시켜 얻은 아조 CPA 에 대하여 아조페놀기의 분광학적 특성을 이용하여 다음과 같은 이온화 상수를 측정한 바 있다. 30),31 )
 이 KPl一 :iazo 麟
이 KPl一 :iazo 麟
15olj 표시된 평형 상수는 통상적인 Ka 와는 다소 다른 의미를 지닌다. 그러 나 해당되는 p Ka 와 동일한 p H 에서 반응물의 절반이 생성물로 전환된다는 물 리적 의미를 지니므로 속도론적으로 도출되는 p Ka 와 동일하게 취급할 수 있 다. 통상적인 페놀의 p K가 10-11 이지만 페놀 음이온이 금속 이온에 배위됨으 로써 p Ka 가 7 . 7로 감소한다. pK a 9 . 5 인 두번째 평형 단계는 금속 이온에 대 하여 페놀 음이온과 수산화 이온이 경쟁하는 과정으로 이해할 수 있다. 아르사 닐아조기의 전자적 효과를 감안하면 속도론적으로 결정한 pK a ~ Ty r-2 78 에 , pK a ~ Zn( Il )이온에 · 배위된 물 분자에 결부시키는 것이 타당하다. 기질
에 따라서는 Zn(Il) 이온의 배위자리를 Ty r- 24~ 페 놀 응이온이나 수산화 이 온이 점령하고 있으면 CPA 와 결합하지 못하여 활성을 보이지 않는 경우도 있 다. 반면에 Ty r-2 4~ 페놀이 이온화하여 염기성이 되어야 친핵체로 작용하여 활성을 보이는 수도 있다. 이상의 pH 의존도의 분석 결과, Glu-270SI pK a 값이 약 6 . 5 이고 그 카르 복실기가 이온화되어 음이온으로 존재하여야 대다수의 기질에 대하여 CPA 가 활성을 가짐을 알 수 있다.
 znIl0lH3°
znIl0lH3°
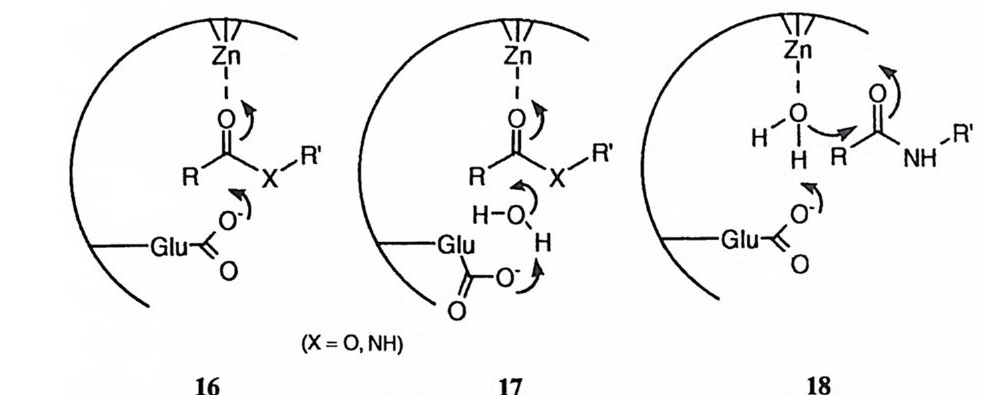
이상에서 논의한 X 선 결정 구조, 화학적 수정, 반응 속도론 등의 결과로부 터 Glu-270 이 CPA 의 반응게 필수적인 역할을 담당한다고 결론내릴 수 있다. 그 다음 단계로는 Glu-270 이 가지는 정확한 촉매 역할이 무엇인지를 결정하는 문제가 남아 있다. Glu-27~ 카르복실 음이온이 지닐 수 있는 촉매 역할로 16-1 쩌 표시한 친핵체 혹은 일반 염기가 가능하다.
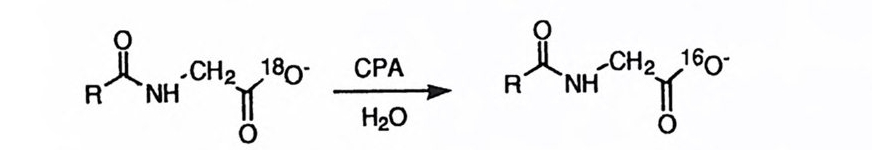 Ri NH CH2 『。 1 80- 一CHP2A0 R J。 lN H/ CH2 f。 160· 19
Ri NH CH2 『。 1 80- 一CHP2A0 R J。 lN H/ CH2 f。 160· 19
이러한 메커니즘을 구별하려는 시도가 최근 20 여 년간 계속되고 있다. 일반 염기 메커니즘을 선호하는 학자들이 발표한 논문의 수는 다수에 달하지만, CPA를 대상으로 직접 수집한 연구 결과로서 타당성을 지닌 증거로 일찍이 제
시된 것은 브레슬로 등이 보고한 180- 교환 실험의 결과를 꼽을 수 있다 .34) , 35) N- 벤조일글리신 혹은 N- 아세틸글리신의 카르복실기에 1e 0-을 표지한 뒤, 이 180 이 물의 160 과 교환되는 반응 (19) 의 속도를 CPA 존재하에서 측정하였 다. 그 결과, a- 아미노산을 반응계에 가해 주면 괄목할 만한 증속 현싱이 관 찰됨에 반하여 a- 히드록시산울 가할 때에는 속도 증가가 거의 나타나지 않았 다.
 R) 0 lNH.,. cH2l (180 · + HX-CHR-coo· ~ R) 0 lNH., .cH2llx— CHA -COO·
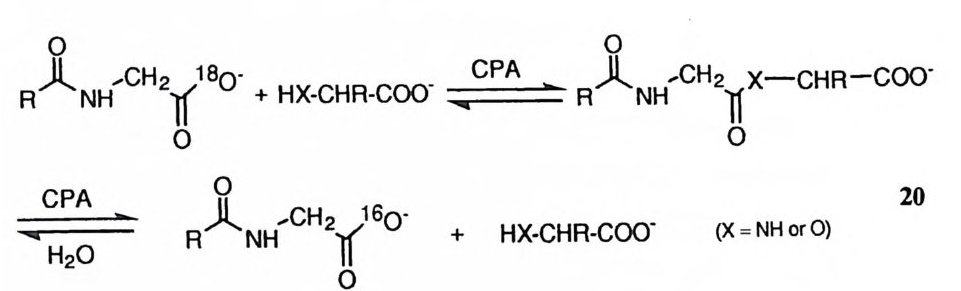
R) 0 lNH.,. cH2l (180 · + HX-CHR-coo· ~ R) 0 lNH., .cH2llx— CHA -COO·
Glu-270 이 일반 염기로 작용하는 경우 이 180- 교환 반응은 CPA 의 활성자 리 내부에서 아실-글리신과 a- 아미노산(혹은 a- 히드록시산)이 아미드(혹은 에스 데르)를 합성하였다가 다시 가수분해됨으로써 (20) 일어난다고 볼 수 있다. 에 스데르보다 아미드가 열역학적으로 더 안정하므로, 아미드의 합성 속도가 에스 데르의 합성 속도보다 더 클 것을 예측할 수 있다. 이에 따라 180 교환 속도가 a- 히드록시산이 존재할 때보다 a- 아미노산이 존재할 때 더 빠르다는 실험 결 과를 일반 염기 메커니즘으로 설명할 수 있는 것이다.
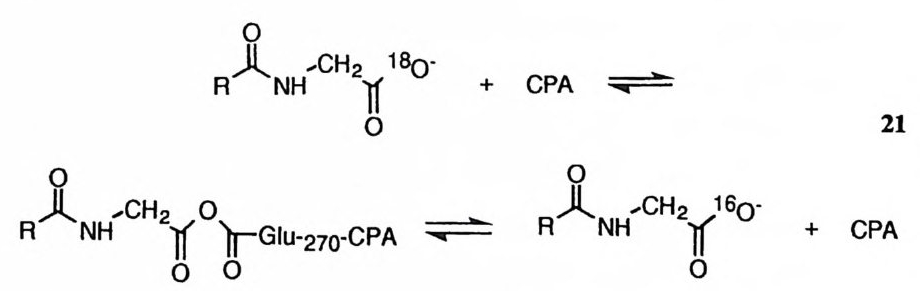 RJ 。 lN H ,,야 2I 1% + CPA ~
RJ 。 lN H ,,야 2I 1% + CPA ~
반면에 Glu-270 이 친핵체로 작용한다면 아실-글리신과 Glu-270 사이에 산 무수물 중간체가 형성되고 이 중간체가 도로 파괴됨으로써 (21) 18 Q-교환이 일
어나게 된다. 이 경우 a- 히드록시산 혹은 a- 아미노산을 가해 줄 때 두드러 진 180 - 교환 속도의 차이를 예측하기 곤란하다. 따라서 브레슬로 등은 친핵성 메커니즘을 배제하고 일반 염기 메커니즘을 선택하였다.
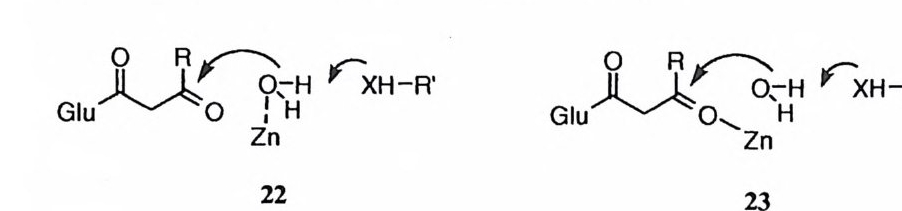 Glu U-O0 Z!n~ HH r XH-R' Glu 丈文0言\ Zn~ HH rxH-A'
Glu U-O0 Z!n~ HH r XH-R' Glu 丈文0言\ Zn~ HH rxH-A'
그러나 브레슬로 등이 발표한 논문의 각주에서 34) 언급되었듯이 친핵성 메커 니즘아 작동하더라도, 산무수물 중간체가 아실-글리신으로부터 형성되거나 파 괴되는 속도가 가해 준 a- 아미노산에 의하여 촉진된다면 180- 교환 실험의 결 과가 설명된다. 피리딘옥심의 아세틸 에스데르의 가수분해에 대한 금속 이온의 촉매작용을 소재로 한 모형 연구의 결과, 금속 이온에 배위된 물 분자가 아실 화합물에 친핵체로서 공격할 때 일반 염기에 의한 보조를 받음이 밝혀졌 다. 36),37) 아실 -CPA 중간체의 파괴가 물 분자의 공격에 의해 일어난다면 (22 혹 은 23) , a- 히드록시산보다 a- 아미노산이 훨씬 강한 염기이므로 a- 아미노산을 가해 주었을 때 아실 -CPA 중간체가 더 큰 속도로 파괴될 것이다. 따라서 아 실 -CPA 중간체의 파괴 및 그 역반응인 재합성은 a- 아미노산의 존재하에 촉진 될 수 있고, 이것으로써 관찰된 180- 교환 속도를 설명할 수 있는 것이다. 이러 한 이유로 브레슬로의 180- 교환 실험은 친핵성 메커니즘과 일반 영기 메커니즘 울 구별할 수 없는 것이다. 최근에는 립스콤 등이 아연 이온에 배위된 물 분자가 친핵체로 작용하며 이 때 Glu-270 이 일반 염기로 작용한다는 메커니즘 (18) 을 제안하였다 .38) CPA 에
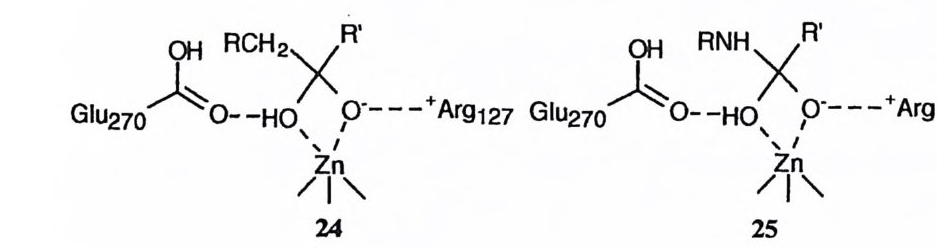 OH RCH2 R' OH RNH R'
OH RCH2 R' OH RNH R'
대하여 강력한 저해제로 작용하는 케톤들과 CPA 가 형성한 착화합물의 X- 선 결정 구조를 조사한 결과, 케톤이 수화되어 있고, 이 수화물의 두 산소가 Zn( ll ) 이온에 배위되어 있음 (24) 이 밝혀졌다. 케돈의 수화물이 CPA 의 활성자리에서 안정화되는 것은 특이한 사실이다. 립스콤 등은 이 케돈 수화물이 기질의 가수 분해에 관여하는 전이상태의 유사체라고 보고, 아미드 기질의 가수분해는 2 요 l 표시한 바와 같이 진행될 것이라고 제안하였다. 이러한 결정 구조로부터 메커니즘을 제안하는 데에는 몇 가지 중요한 한계점 이 있다. X - 선 결정학의 방법으로는 비생산적이며 정적인 효소-저해제 혹은 효소 - 가짜 기질의 착화합물의 구조를 밝힐 수 있을 뿐인 데 반하여 반응 메커 니즘에서는 생산적이며 동적인 전이상태의 구조와 관련되어 있음을- 염두에 두 어야 한다. 뿐만 아니라, X - 선 결정학이나 분광학적인 방법으로는 항상 가장 안정한 분자 형태에 대한 정보가 일차적으로 수립되는 데 비하여 수많은 화학 반응은 불안정한 분자 형태를 출발점으로 하여 진행되고 있다. 예를 들어 할로 겐화 시클로핵산의 안정한 형태에서 할로겐은 적도 위치를 점유하지만 이 화합 물의 가장 특칭적인 화학반응인 E2 제거반응은 할로겐이 축 위치를 점유한 불 안정한 형태에서 진행된다. X- 선 결정학으로부터 측정한 구조는 열역학적으로 가장 안정한 형태일 뿐이며, 실제 반응의 전이상태의 구조와 직접적인 관계는 없는것이다. 일반 염기 메커니즘을 주장하는 측에서 최근에 제시한 증거로 포스페이트 저 해제에 대한 자료가 있다. X-(Ala)n-Ala-Ala(n=l 혹은 2) 로 표시되는 일련 의 펩티드는 t a t /Kma pp가 102-l05s-1M-1 이고 Km 이 10-1-10-•M 정도인 기 질이이트다 .에 스그데런르데로 이 러바한꾼 기X질-(이A laC)P 2A-N 에H 의-C해H 가 (C수H분3)해 -되P (는02 아-) 미0-드C H결 (합CH을3 )포 c 스oo페- 는 K 가 10- 4- 10-1M 인 저해제이다. 이 저해제에 대하여 측정한 log Ki 는 상응하는 기질에 대하여 측정한 lo g K깁t a t에 대하여서는 기울기가 0 . 93 인 직선의 관계를 보이며, lo g Km 에 대하여 기울기가 0.98 인 직선의 관계를 보인 다. 39) 이 저해제들이 전이상태 유사체라면 lo g K 는 lo g K깁 &m 에 대하여 직선 관계를 보일 것이다. 한편 이러한 포스페이트 저해제와 CPA 가 형성한 착화합 물의 X 선 결정 구조를 측정한 결과, 24 및 E 와 유사하게 포스피닐기의 두
산소가 Zn(Il )이온에 배위되어 있음이 판명되었다. 40),4 이러한 일련의 자료로 부터 기질이 가수분해될 때의 전이상태는 포스페이트 저해제와 형성한 착화합 물과 유사하며, 따라서 전이상태에서 Zn(Il) 에 배위된 물 분자가 천핵체로 작 용한다고 해석하고 있는 것이다. 포스페이트 저해제로부터 얻은 결과를 해석하는 데에도 몇 가지 제약이 있 다. 첫째로, Io g K, 가 Io g Km 과 직선 관계에 있음에 비하여 K,가 1000 배 가량 변화할 때에 kca t은 거의 차이를 보이지 않는 점이다. 이는 포스페이트 저해제 의 결합 방식으로부터 전이상태보다는 기질의 결합 방식에 대한 정보를 일차적 으로 도출할 수 있음을 시사한다. 둘째로, 선형 자유 에너지 관계가 갖는 메커 니즘상의 의미이다. lo g K,는 저해제가 가지는 열역학적 자유 에너지를 대표하 고, lo g K깁 kca t은 기질이 가지는 속도론적 자유 에너지를 대표한다. 이 두 자 유 에너지 간에 직선 관계가 있다는 사실로부터 저해제와 효소가 이루는 구조 가 기질이 반응할 때 거치는 전이상태의 구조와 유사하다고 결론을 내리는 것 은 비합리적이다. 수많은 유기 반응에 대하여 여러 가지 종류의 선형 자유 에 너지 관계가 관찰되지만 선형 에너지 관계로부터 얻을 수 있는 전이상태 구조 에 대한 정보는 극히 제한되어 있다. 예를 들어, 벤조산 에스테르의 알칼리 가 수분해의 속도는 벤조산의 이온화 상수와 선형 자유 에너지 관계 (함게트식)를 보인다. 그러나 벤조산 유도체의 이온화 상수를 결정하는 요인인 벤조산 음이 온의 구조로부터 에스테르 가수분해의 전이상태의 구조를 예측할 수는 없는 것 0l 다. 친핵성 메커니즘을 주장하는 측에서 제시한 속도론적 자료도 대부분- 일반 염 기 메커니즘을 명백히 제거하는 데에는 미흡하다. 최근에 CPA 반응 도중에서 중간체가 축적 혹은 포획되고, 그 중간체가 아실 -CPA 중간체임을 시사하는 증거가 제시되기 시작하고 있다. 친핵성 메커니즘을 확고하게 증명하는 것은 Glu-270 이 친핵체로 작용하여 형성되는 산무수물 중간체를 분리하여 구조를 결정하는 것이다. 그러나 산무수 물이 화학적으로 매우 불안정한 화합물이며 그 파괴 과정에 관해서도 효소 반 응에서는 촉매 효과가 존재할 것이다. 따라서 산무수물 중간체가 존재하더라도 그 수명이 매우 짧울 것이 예측된다. 이러한 이유로 분광학적 방법에 의하여
중간체가 축적되는 것을 관찰하거나 포획재를 사용하여 중간체를 포획하는 연 구 방향에 주력하게 되었다. 샨무수물 중간체가 축적되는 반응국건을 찾으려면, 중간체의 파괴 속도가 형 성 속도보다 느려서 적절히 높은 농도로 중간체가 쌓여야 한다. 이것은 이러한 조건을 충족시키는 기질을 탐색함으로써만 해결할 수 있다 . 이의에도 축적된 중간체가 파괴되는 속도가 충분히 느려야지만 분광학적으로 중간체가 축적되어 있는 것을 확인할 수 있다. 더욱 중요한 것은 축적된 중간체가 단순한 효소-기 질의 착화합물이 아니고 산무수물 중간체임을 인정할 수 있는 증거가 수집되어 야한다는것이다. 0- p클로로신나모일 혹은 0- 신나모일 -L- /3페닐락트산의 가수분해에 대한 ~a t의 pH 의존도는 매우 특이하다. 23),42) 즉, pH 初}지는 pK a7} 6 . 5 인 작용 거가 이온화하여 염기성형일 때 활성을 나타내게끔 'ke a t이 변화하다가 pH 9 이 상에서는 급격히 증가한다. 이것을 9 와 10 으로써 다음과 같이 설명할 수 있다. 42) 解] %단계 , 즉 중간체 의 형성은 Glu-270 이 음이온으로 존재하여야 반응이 일어나므로 p Ka 가 약 6.5 인 작용기의 이온화가 그 pH 의존도에 반영될 것이다. 반면 %단계, 죽 중 간체의 파괴는 수산화 이온의 공격 경로가 우세한 pH 범위에서는 반응 속도가 수산화 이온의 농도에 비례할 것이고, 물 분자의 공격이 우세한 pH 범위에서 는 p H 에 무관한 경향을 나타낼 것이다. 식 15 낸에서 炭 < %이면 중간체의 형 성이 속도 결정 단계가 되어 'ke a t는 炭가 되며 'kea ~ pH 의존도는 %의 것과 부합할 것이다. 반면에 &>炭이면 &a t의 의존도는 %의 것이 되어 Glu-27~ p K갑 은 전혀 반영하지 않을 것이다. 0- p죽늄목·신나모일 혹은 0- 신나모일 L- /3페닐락트산의 ~a t의 pH 의존도는 %와 %의 크기가 비슷하다고 가정할 때 설명이 가능하다. 즉, 낮은 p H 에서는 'ke a t가 炭에 의해 좌우되어 (ki<~) kt의 p.H 의존도가 ~a t의 pH 의존도에 반영된다. 반면, 높은 p H 에서는 Glu-270 이 완전히 이온화됨으로써 'ke a t이 %에 의해 지배되고(ki>~) k.i의 pH 의존도가 ~a t의 pH 의존도에 직결되는 것이다. 이러한 분석에 의거하여 0- p픕늄로로신나모일 -L- /3-페닐락트산 (CICPL) 의 갸탄은해에 대한 CPA 의 촉매작용을· 저온에서 측정하게 되었다 .43) 죽 2 5° C 에
서는 &와 &가 서로 큰 ` 차이를 보이지 않지만, 각각의 활성화 에너지가 동일 하지는 않을 것이므로 저온에서 % > %의 조건이 충족될 가능성이 있다고 희망 한 것이다. 이에 따라 -40°C- ― 60°C 의 저온에서 CICPL 을 CPA 의 존재하에 반응시킬 경우 한 중간체가 축적되었다가 사라짐을 볼 수 있었다. 43) CICPL을 기질로 사용하여 저온에서 관찰한 중간체를 Glu-270 이 친핵체로 작용하여 형성된 산무수물이라고 주장하는 몇 가지 상황 증거가 제시되었 다. ”),45 ) 그러나 공명 라만 스펙트럼을 측정한 연구의 결과로부터 , 이 반응에서 산무수물 중간체가 축적되더라도 이 분광법에 의하여 확인될 수 있는 농도로는 쌓이지 않는다는 반론이 제기되었다 .46) 죽, CICPLl 료부터 관찰된 중간체는 산 무수물인 아실 -CPA 중간체가 아니고 단순한 비공유성 효소 - 기질 착화합물의 하나일 가능성이 커진 것이다. 라만 분광법에 의한 실험 결과를 비판하는 논평 이 발표되긴 하였어도, m CICPL을 사용한 저온 실험의 결과를 친핵성 메커니 즘의 증거로 채택하는 데에는 무리가 따른다. 댄실기를 포함하는 에스테르와 아미드 기질이 CPA 촉매작용하에 가수분해 되는 반응에 대하여 S 。 }> E 。의 조건하에서 정류 전 상태의 반응 속도를 측정한 바 있다. 48),4 9 ) 댄실기의 형광에 근거를 둔 형광 스펙트럼을 이용하여 반응 속도 를 측정하였으므로 매우 낮은 농도의 기질이 반응하는 것도 추적할 수 있었다. 이 실험 결과를 12ol] 따라 분석하여 k-1/kI, %, k-2 및 炭 값을 계산하였다. 이러한 속도 변수의 값을 결정하고 lEl 표시된 ES 및 ES1 의 두 중간체가 축 적된 것을 형광 분광학적으로 관찰하였지만, 중간체 ES 떡 구조에 대한 명확한 정보는 얻지 못하였다. 죽, ES 이 산무수물 중간체이어서 친핵성 메커니즘을 지지하는 증거를 제시할 수 있는지, 혹은 일반 염기 메커니즘이 작동하기 때문 에 ES 이 단순한 제器 1 비공유성 효소-기질 착화합물 중간체일 뿐인지에 관한 정보를 얻는 데에는 성공하지 못한 것이다. a- (아실 o }P]노)신나모일기를 아실기로 포함하는 에스데르 기질의 가수분해에 대한 CPA의 촉매작용에 대한 속도론적 연구의 결과로부터 친핵성 메커니즘을 지지하는 강력한 증거가 얻어졌다. 32),33),44) 이 연구에 사용된 기질의 구조를 표 2 에 요약하였다. 기질 a-d 에 대하여 측 정한 속도 변수의 값도 표 2 에 요약하였다.
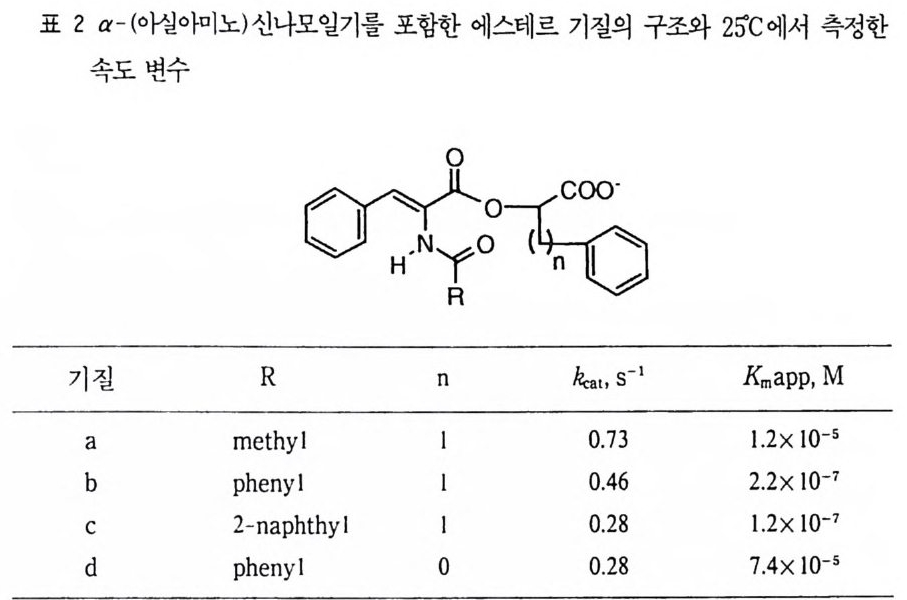 표 2 a - (아실아미노)신나모일기를 포함한 에스데르 기질의 구조와 2 5' C 에서 측정한
표 2 a - (아실아미노)신나모일기를 포함한 에스데르 기질의 구조와 2 5' C 에서 측정한
기질 b-d 에 대하여 &a t의 pH 의존도를 조사하였는데, pH 5.5-1 ()ol]서 k.:at 은 p H 에 무관한 경향을 나타내었다. 기질 b 혹은 c는 CPA 와 섞으면 안정한 중간체가 축적되는 것이 자의선 분광학적으로 관찰되었다. 32),51) 축적된 중간체 가 생성물로 변환되는 과정의 유사 일차 속도 상수는 갇은 조건하에서 정류 상 태 속도론적 측정에 의하여 구한 k.:at 값과 일치하였다. 기질 b 혹은 떠 가수분해 도중에 축적된 중간체는 다음과 같은 근거로 산 무수물 아실 -CPA 중간체라고 결론지어졌다. L- /3페닐락트산을 포함하는 기질의 KmaP Pi즌 대개 10-•M 근처이고, L- 만 델산을 포함하는 기질의 것은 대개 10-3M 이다. 이에 비하여 기질 a-d 의 Kmap ~ 이것들보다 훨씬 작은 값을 나타낸다. 이것을 1 책 반응식으로 설명 할 수 있다. 1 책 ES1 이 축적되는 경우에는 ESI>ES 이며 따라서 ki~ (I¾+ &)이다. 이 조건하에서 k.: a t과 KmaPPol] 관한 식인 1~ 1~ 곤 %과 27 과 같이 변환된다. 기질 a-d 에 대하여 관찰된 매우 작은 Kmap p 값은 27로 설명이 가 능하다. 죽 통상적인 에스데르에 대해서는 Kma pp가 Km 임에 비하여 a-d에 대 해서는 27 이 적용되는 것이다. 축적된 중간체가 생성물로 전환되는 과정의 속
도 상수 (1 책 &)가 kca t과 일치한다는 사실도 27 및 1 꺽끌 지지하여 준다. 幅 = ~ (26) Kmap p = Km (써 + %)/:K m = (k-1 냐 )/k1 (27) 福이 p H 에 무관하다는 사실은 축적된 중간체 ES1 이 Glu-27 個} 기질의 아 실 부분 사이에 형성된 산무수물이라는 것을 가리키고 있다. 만약에 tot 과정 에 Glu-270 이 친핵체 혹은 일반 염기로 참여한다면 Glu-27~ p Kn 가 t a t의 pH 의존도에 반영되어야 한다. 반면에 b-d 의 가수분해에서 산무수물 중간체의 파괴 (&)가 속도 결정 단계이고, 산무수물 중간체가 주로 물 분자의 공격에 의 해 파괴된다면 t a t은 p H에 무관하여야 하는 것이다. 아실기 이동 반응에서 다른 이탈기를 가진 기질에서 형성된 공통된 중간체가 동일한 속도로 분해되는 것 45) 을 중간체가 친핵성 메커니즘에 의하여 형성된 것 임을 입증하는 증거로 채택할 수 있다. 따라서 困斗 d 에 대하여 관찰된 거의 동 일한 tat 값도 이들 두 기질이 동일한 아실 CPA 중간체를 형성한디는 사실에 대한 강력한 증거가 된다. CPA 의 경우 이탈기 (L- /3페닐락트산 및 L- 만델산)의 결사술의 형수성의 차이에 따라 두드러진 반응성 차이를 보임에 비하여 b2-} d 의 t a t이 거의 동일하다는 것은 중요한 의미를 지닌다고 볼 수 있다. 효소 반응뿐만 아니라 모든 화학 반응에서 중간체의 존재를 확인하거나 분리 하여 구조를 결정하는 것은 반웅 메커니즘의 연구에 가장 중요한 과제가 된다. 그런데 어떤 중간체의 존재를 입증하더라도 그 중간체가 반응물과 생성물을 연 결하는 반웅 경로상에 위치한 진정한 반응 중간체인지 혹은 생성물의 형성과는 무관하게 존재하는 부차적인 평형 반응의 산물인지를 구별하지 않으면 메커니 즘의 연구에 결정적인 단서를 제공하지 못한다. 53) b-d 에 대하여 ES 이 축적되는 것을 관찰하고 그것이 산무수물 아실 -CPA 중 간체라는 것에 대한 강력한 증거가 제시되었다고 하지만, 이 중간체가 진정한 반응 중간체임을 입증하여야 친핵성 메커니즘이 정립된다. 죽 1kl 포함된 ESI 인지 28oll 표시된 부차적인 평형 반응의 산물로서의 ES1 인지를 구별하여야 하
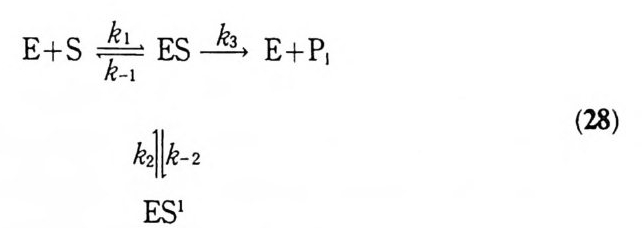 E+S ~k-1 ES A E+P1
E+S ~k-1 ES A E+P1
는것이다. 관찰된 중간체가 부차적인 평형 반응의 산물이 아니고 전정한 반응 중간체임 울 명백하게 밝힌 실험적인 사례는 모든 종류의 화학 반응을 통틀어서 매우 드 물다. CPA 의 경우에는 E 。 > S 。 조건하에서 d 의 가수분해를 속도론적으로 추 적하여 28 이 적용되지 않음을 밝힐 수 있었다. SO) 이 조건하에서 d 의 반응은 정 류 상태와 정류 전 상태가 구분되지 않고 섞여 있는 비정류 상태의 속도론적 행동을 보여주었다. 이 속도론적 자료를 지에 따라 분석한 결과, 측정된 자료 를 설명하려면 ES 가 ES1 쪽으로보다 E 쪽으로 훨씬 우세한 속도로 전환되어 야 함이 밝혀졌다. 즉 28o1l 따라 반응이 진행되면 관찰된 속도 자료로는 제粹] 중간체인 ES 까 축적될 수가 없는 것이다. 반면 1 約] 따라 실험자료를 분석한 결과, ES 이 상당량으로 축적될 수 있음을- 알게 되었다. 이상에서 설명한 a-d 에 대한 효소 반웅 속도론적인 일련의 실험 결과 산무수 물 중간체가 전정한 반응 중간체로 존재하며, Glu - 270 이 친핵체로 작동하는 메커니즘이 상당한 타당성을 지니고 있음을· 밝히게 되었다. 그러나 축적되는 중간체가 산무수물임을 명확하게 밝히는 증거가 제시되기 전에는 직접적인 증 거로 받아들이기 어렵다는 문제점이 있다. 이 연구의 결과로서 에스데르 기질 에 대하여 친핵성 메커니즘이 작동하는 것은 타당성을 어느 정도 인정받게 되 었지만, 펩티드 기질에 대하여서는 별도의 실험적 증거가 제시되기 전에는 메 커니즘을 단정짓기가 곤란하다. 54) 산무수물 중간체는 에스데르 혹은 아미드 기질보다 훨씬 불안정하므로 기질 에게는 아무런 영향을 미치지 못하는 친핵성 시약과 쉽게 반응할 수 있다. 이 것을 이용하여 산무수물 중간체를 포획하고자 하는 실험이 다수 수행되었다. 포획제를 외부에서 가해 주어서 산무수물 중간체를 포획하려는 연구로는 14c-
티로신 55) 및 14c- /3페닐락트산 56) 과 감은 동위원소로 표지된 화합물과 dl - a- 메 르캅토-/3대닐프로피온산 , 히드록실아민, 아지드이온, 메탄올, NaBH4 와 같 은 친핵성 시약을 5 7 ) 포획제로 사용한 경우가 보고되어 있다. 그러나 이러한 포 획 실험은 모두 성공하지 못하였다. 이것은 산무수물 중간체에서 활성자리가 기질의 아실 부분에 의하여 막혀 있어 포획제가 산무수물기에 접근하는 것이 봉쇄될 가능성과 중간체의 파괴도 효소에 의해 촉진되어 고속으로 진행될 가능 성으로써 설명할 수 있다.
 0 0
0 0
기질의 아실 부분에 포획용 작용기를 도입하여 산무수물 중간체의 분자내 포 획 반응을 시도한 예도 (29,30) 있다. 그러나 이러한 분자내 포획 반응도 성공 하지 못하였다. 32),57) 이는 효소 반응에 의한 중간체의 파괴 과정이 분자내 포획 반응보다 훨씬 빠르거나, 중간체내의 분자형태가 분자내 포획 작용기를 산무수 물기로부터 분리시키고 있기 때문으로 설명할 수 있다. 에스데르 혹은 아미드 기질을 _60°C-_75C 에서 CPA 와 섞고, 효소를 변 성시킨 뒤 NaCNBH3 및 히드록실아민 등으로 산무수물 중간체를 포획하는 데에 성공하였다는 결과가 보고된 바 있다. 58),59) 산무수물 중간체가 형성된다면 생성물로 변환되기 전에 이 중간체를 변성시켜 중간체의 파괴에 대한 효~ 반 응을 정지시킬 수 있다. 뿐만 아니라 변성에 의하여 활성자리 내부의 형태가 풀어져서 산무수물기가 포획제의 공격에 노출될 수도 있다. 이러한 이유로 이 포획 실험이 성공하였다고 볼 수 있다. 그러나 이 포획 실험 결과 도입된 방사 능의 수준이 그다지 크지 않아서 그 결론을· 신뢰하기 어렵다는 강한 이의가 제 기된 바있다 .54) 산무뀜 중간체의 존재를 지지하는 강력한 증거가 라만 분광의 기법에 의하 여 최근에 수집되었다. 60) L- /3-페닐락트산의 P- 디메틸아미노벤조산 에스데르
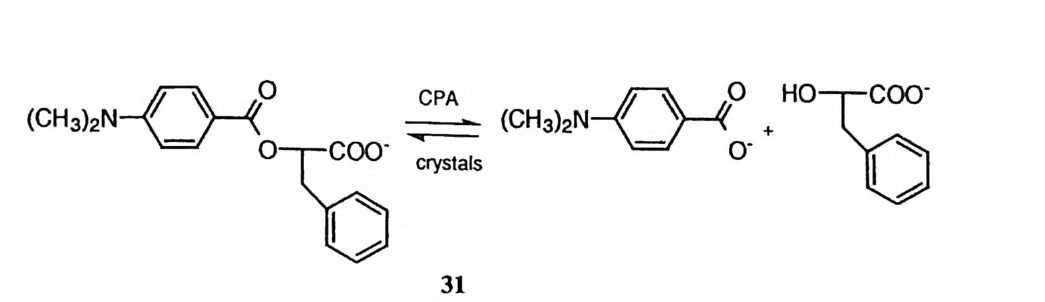 (CH3)2N::) ) 근Is (CH3)2N: HO) )
(CH3)2N::) ) 근Is (CH3)2N: HO) )
를 기질로 사용하여 CPA 의 결정과 반응시켰을 때 (31) 라만 스펙트럼상에 산 무수물의 형성이 관찰된 것이다. 한편 가수분해 반응의 생성물인 L- /3-페닐락 트산과 P- 디메틸아미노벤조산울 CPA 의 결정과 반응시켰을 때에도 산무수물이 형성되는 것이 라만 스펙트럼으로 관찰되었다. 이상의 결과로부터 평가할 때 적어도 에스테르 기질의 경우에는 산무수물 중 간체가 형성된다고 결론 내리는 것이 현재로서는 타당하다. 펩티드 기질의 61) 경우에는 펩티드로부터 산무수물이 형성되는 속도가 느려서 산무수물 중간체를 축적시키는 것이 에스테르 기질의 경우보다 훨씬 어려울 것이 예상되며, 따라 서 산무수물 중간체의 존재에 대한 증거를 수집하기가 쉽지 않을 것이다. 현재 로서는, 나중에 CPA 의 모형에 관한 결과를 논의할 때 언급할 것이지만, 에스 테르 기질이 무수물 메커니즘을 거쳐 가수분해된다면 펩티드 기질도 동일한 메 커니즘으로 가수분해될 것이라는 모형 연구의 결과가 펩티드 가수분해 메커니 즘에 대한 단서를 제공하고 있다. 제 4 절 Ty r-2 4~ 촉매 역할 Ty r-2 4~ 페놀기가 아미드 기질의 가수분해 과정에서 아민기가 이탈할 때 일반 산으로 작용할 가능성이 CPA 의 X- 선 결정학적 구조로부터 제기되었 다 .1),7) T yr -248 에 대한 연구는 화학적 수정에 의하여 본격적으로 개시되었다. 아세 틸 이미다졸 식초산무수물, I5 혹은 ICI-, 5- 디아조 -lH- 테트라졸, 데트라니 트로메탄, 디아조화된 아르사닐산 등을 가하여 T yr -248 을 화학적으로- 수정하
였다. 62)-74) T yr -248 이 화학적으로 수정될 때 에스데르에 대한 효소의 활성은 수배 증가하고 아미드에 대한 효소의 반응성은 거의 사라짐을 관찰하였다. 이 결과가 에스테르 및 아미드 기질에 대한 CPA 의 메커니즘이 다르다는 것을 가 리킨다고 해석하기도 하였다. 그러나 이러한 자료는 속도론적 측정이 미숙하기 때문에 얻어진 것이다. &a t과 Km 을 정확하게 측정하면 에스테르와 아미드에 대하여 큰 차이점이 없음이 밝혀졌다. 63) 효소의 작용기를 화학적 수정에 의하여 변환시키려는 경우에 항상 봉착하는 문제점은 수정 방법의 선택성의 결여이다. 즉 어느 한 개의 작용기만 수정되지 않고 여러 개의 아미노산 곁사슬이 동시에 수정되는 것이다. 이때에 얻어지는 속도론적 자료로부터는 메커니즘 결정에 유용한 정보를 도출해 내지 못하는 것 o] 다. T yr -24 짜 선택적으로 수정하는 것은 테트라니트로메탄에 의하여 Ty r-2 48 의 페놀을 니트로페놀로 변환시키는 방법과 디아조화된 아르사닐산울 사용하여 T yr -24 허 페놀을 아조 유도체로 바꾸는 방법이 알려져 있다. 이와 같은 방법 으로 얻은 니트로티로신 -24 8:ilt 아르사닐아조티로신 -24~ p Ka 가 각각 6 .4 및 7. 7 임이 알려져 있다. 이와 같이 수정된 CPA 에 의한 에스데르 기질 CICP~ 가수분해에 대하여 ~et 및 ka t IKma p펴 pH 의존도를 조사하였다. 42) 그 결과 p Ka 가 6.4 혹은 7.7 인 Ty r-2 4~ 수정체가 이온화하여도 반응성에는 아무런 변화가 없음이 밝혀졌다. 즉 에스데르 기질의 가수분해에 T yr -24~ 본 아무런 촉매작용을 하지 않는 것이다. 최근에는 유전자 재조합 기법을 사용하여 T yr-248 을 Ph~ 뉵i 바꾸고, 에스 데르 혹은 아미드 기질에 대한 반응성을 조사한 결과 두드러진 변화를 관찰하 지 못하였다. 75) T yr이 페놀을 결사슬로 가지고 있는 데에 비하여 Ph~ 결사 술인 페닐기에는 히드록시기가 결여되어 있다. 따라서 Ty r-2 4~ 페놀 히드록 시기는 에스테르와 아미드 기질에 대하여 중요한 촉매 역할을 담당하지 않는다 고할수있다. D- 티울예스데르의 가수분해에 대한 CPA 의 반응에서는 Ty r-2 4~ 친핵체 로 작용하는 것이 보고되어 있다. 5)
제 5 절 Zn(Il )이온의 촉매 역할 CPA 의 Zn(Il )이온을 다른 금속 이온으로 치환하면 에스데르 및 아미드 기 질에 대한 활성도가 변화한다. 76)- 8 3) 금속 이온에 따라 활성도가 유지 혹은 증 가되기도 하고 완전히 사라지기도 한다. 이와 갇이 수정된 CPA 에서의 금속 이온들의 배위 구조에 관한 여러 가지 정보가 수집되어 있기도 하다. 80),84)-89) 유기화학 혹은 무기화학적으로 효소가 수정되면 별개의 효소가 얻어전 것으로 간주할 수 있다. 수정된 효소에서 관찰되는 여러 가지 화학적 성질이 천연 효 소에 그대로 적용된다고 보기는 어렵다. 이러한 제한점을 감안하면서 수정된 효소로부터 얻은 정보를 천연 효소의 연구에 이용하여야 한다. CPA 의 Zn( Il )이온은 루이스산으로 작용하여 기질의 카르보닐기를 극성화 하거나 Zn (Il)에 배위된 물이 친핵체로 작용함으로써 촉매작용에 참여한다고 일반적으로 제안되고 있다. 이것은 아무런 실험적 근거를 가지고 있지 못하며, 단순히 X- 선 결정학적 연구의 결과로부터 제안된 메커니즘에 기인할 뿐이다. 현대 과학 수준으로는 CPA 의 촉매작용에서 Zn( Il )이온이 담당하는 정확한 역 할을 직접적으로 구명해 낼 기법이 존재하지 않는다고 할 수 있다. CPA 의 Zn ( Il ) 이온과 관련된 또 다른 촉매 작용기로 Zn (Il ) 이온에 배위된 물 분자를 들 수 있다. CPA 에 대한 핵자기 공명 연구와 81) 아르사닐아조 -T yr- 248-CPA 에 대한 가시광선 분광학적 연구의 30),31) 결과로 Zn( Il )이온에 배위된 물 분자의 pK a 값은 약 9 로 밝혀졌다. 그리고 전술한 바와 갇이 각종 기질에 대한 t a t /Kma p떠 pH 의존도에 이온화 상수가 반영되고 있다. 효소-기질 착화합물에서 Zn(Il )이온에 배위된 물 분자가 일반 산으로 작용 할 수도 있고, 이것이 이온화되어 수산화 이온으로 변하면 친핵체 혹은 일반 염기로 작용할 수도 있다. CPA의 Zn( Il )이온의 배위수는 정상치인 4 가 아니 고 5 이며, 이중 셋은 단백질의 극성 결사슬이 제공하는 염기성 작용기가 리간 드로서 점령한다. 89),91),92) 나머지 두 자리 중 하나를 기질이 차지하더라도 물 분 자 혹은 수산화 이온이 배위될 수 있는 자리 하나가 존재하는 것이다. 현재로 서는 Zn( Il ) 이온에 배위된 물 분자 혹은 수산화 이온이 CPA 의 작용에서 담당 하는 촉매 역할을 밝힐 수 있는 직접적인 증거를 제시하는 실험적 자료는 존재
하지 않는다. 제 6 절 기타 작용기의 역할 Ar g -14 핵 구가니디늄 OJ =o]온이 기질의 카르복실 음이온과 정전기적인 작용 울 함으로써 기질을 활성자리 속에 결합되게 할 것이라는 점은 X- 선 결정학 구조로부터 제안된 바 있다. Arg- 1 4~ 중요성은 단순히 X- 선 결정학적 구조 에 의하여만 지지받고 있을 뿐이며, 화학적 수정 93) 등의 화학적 연구에서는 별 다른 정보가 얻어지지 않았다. Glu-270, Ty r-2 48 및 Arg- 1 45 이의에도 Arg- 1 27, Ser-197, Ty r-1 98 등이 이차적인 중요 촉매 작용기로 존재한다는 것이 최근의 X- 선 결정학적 연 구의 결과로부터 제안되고 있다. 92) 제 7 절 모형 연구 금속 이온과 카르복실기가 분자내 촉매 작용기로 도입되어 괄목할 만한 협동 성을 실현한 예의 하나로 6- 카르복시아스피린의 가수분해에 대한 Fe( 매) 혹은 A l( lll) 이온의 촉매작용을 들 수 있다 •94) 이 촉매작용의 메커니즘은 제炳대서 논의한 바와 갇이 저해적인 역반응 경로를 금속 이온이 봉쇄하는 것을 포함하 고 있댜 따라서 이 반응에서 작용하는 금속 이온의 촉매 역할은 CPA 의 그것 과처이가난다.
 。
。
3 쩌 표시된 모형 반응에서 카르복실 음이온은 친핵체로, 금속 이온은 이탈 기의 이탈 능력을 제고하는 루이스산으로 작용한다고 제안되었다. 96) 그러나 이 반응의 속도 자료는 에스테르 결합에 카르복실기가 공격하는 것 이의에도 금속 이온에 배위된 물 분자가 친해체로서 공격하는 것과도 부합하는데, 이 두 메커 니즘이 명백하게 구별되어 해석되어 있지는 않다.
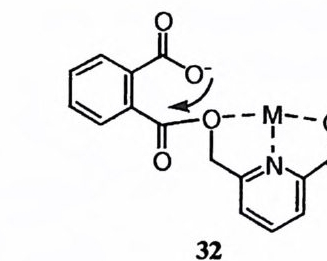
 H2NI 니`N H2 33 [M = Co(lll)l H2N: 니``N H2
H2NI 니`N H2 33 [M = Co(lll)l H2N: 니``N H2
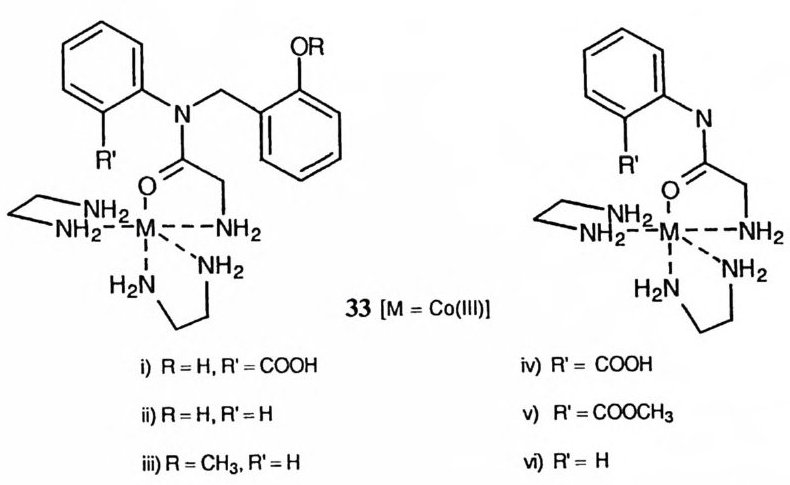
브레슬로 등은 아미드의 가수분해에 금속 이온과 카르복실기가 협동적으로 참여할 수 있는 모형으로 3~ 화합물을 연구하였다. 54) 이 모형 화합물에서 금 속 이온과 카르복실기가 협동하는 것은 속도론적으로 관찰되지 않았다. 이 모 형 반응에서 금속 이온과 의부 완충제에 의한 협동 작용은 관찰되었는데, 이 결과로부터 CPA 에 의한 아미드 기질의 가수분해 반응에서 Glu-270 이 일반 염 기로 작용할 것이라고 제안되었다. 그 이유는 Glu-270 이 일반 염기로 작용하 면 기질의 아민 부분이 이탈할 페에 질소를 양성자화하는 데에 필요한 양성자 공여체가 존재하지만 (34) , 천핵성 메커니즘인 경우에는 양성자 공여체의 역할 을 담당할 작용기가 없다는 것이다. 모형 반응에서 긍정적인 결과를 얻었으면, 그 촉매 요인이 효소계에서도 작 용할 것이라고 결론을 내릴 수 있다. 그러나 어느 촉매 요인이 모형 반응에서 관찰되지 않았으므로 효소계에서도 작용하지 않을 것이라고 단언할 수는 없다.
 주 n
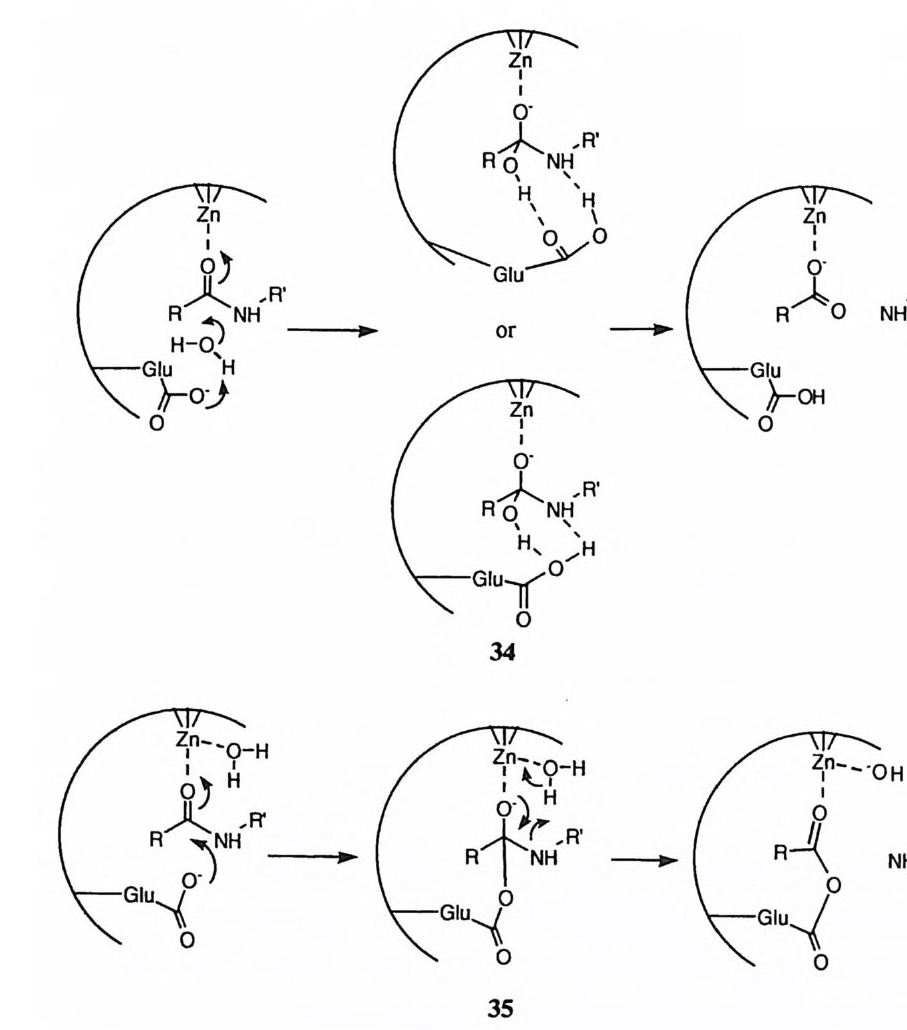
주 n
브레슬로의 해석은 모형 화합물게서 관찰되지 않았으므로 효소의 친핵성 메커 니즘을 부정한다는 논리이다. 그의 주장을 수용하면서도, CPA 가 아미드 가수 분해에서도 친핵성 메커니즘으로 작용할 가능성을 다음과 갇이 제안할 수 있 다. 전술한 바와 같이 CPA 와 기질이 형성한 착화합물에서 Zn( Il )이온의 배위 수는 57} 되어 물 분자가 배위될 수 있다. Glu-270 이 친핵체로서 아미드 기질 울 공격하면 정사면체 중간체가 형성된다. 이 중간체에서 아민이 이탈될 때에 Zn( Il )이온에 배위된 물 분자가 일반 산으로 작용하여 양성자 공여체의 역할을
 HN 〈。YX 214))) XXX === OONCC{CHHH333,, ) i, YY Y = == O OOHHH, , , ARA == = HC C HH2C2COOOOHH
HN 〈。YX 214))) XXX === OONCC{CHHH333,, ) i, YY Y = == O OOHHH, , , ARA == = HC C HH2C2COOOOHH
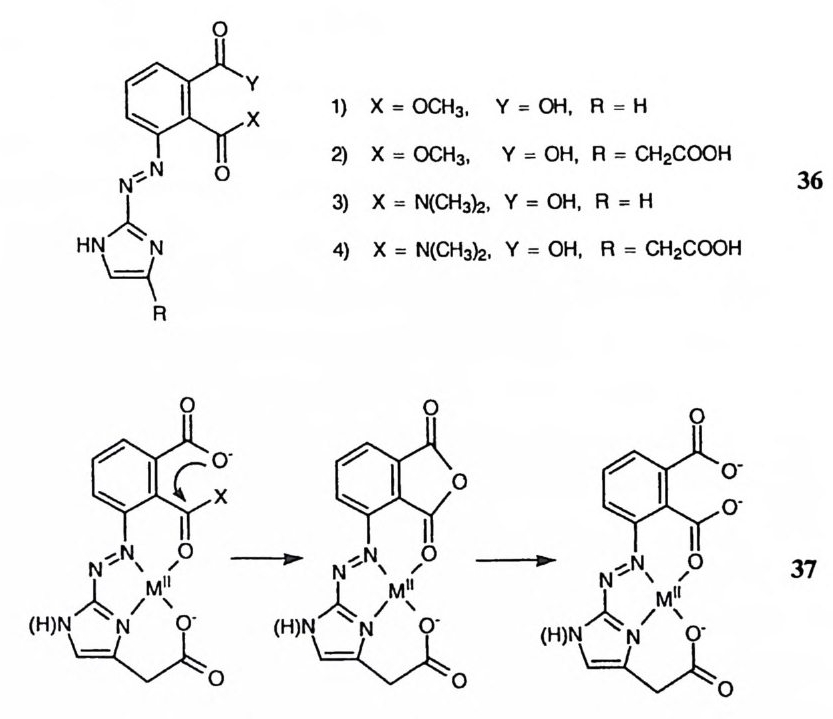
담당할 수 있다. 죽 3 요 l 요약된 바와 같은 메커니즘으로 . 반응이 진행된다면 브레슬로 등이 모형 연구에서 수집한 실험 결과와도 부합되는 것이다. 앞에서 소개한 각종 모형 연구에서 금속 이온과 카르복 4J 기가 CPA 에서 제 안된 촉매 역할을 담당하지는 않고 있다. %에 표시된 에스테르와 아미드의 가 수분해에 대한 금속 이온의 촉매작용에서 많은 수의 CPA 의 특칭이 성공적으 로 재현되었다. 96)-100) 특히 95% DMSO 를 용매로 사용하였을 때 Cu(Il ) 혹 은 N i(Il)이온이 효과적인 촉매로 작용하였는데, 이 반응의 메커니즘은 37과 갇이 카르복실 음이온이 친핵체로 작용하여 산무수물 중간체를 형성하는 것이 입증되었다. 이 모형에서 재현된 CPA 의 특칭을 나열하면 다음과 갇다. 1) 알킬 아미드 및 알킬 에스테르의 신속한 탈아실화. 2) 금속 이온, 카르복실기 및 매체의 협동. 3) 카르복실기가 음이온일 때에 활성 보유.
 )댜\ON. °:0
)댜\ON. °:0
4) 금속 이온과 카르복실기의 촉매 역할이 CPA 의 산무수물 메커니즘의 경 우와동일. 5) 에스데르와 비교하여 더 빠르거나 비슷한 속도로 아미드가 탈아실화. 특히 다섯번째에 언급한 바와 같은 아미드와 에스데르에 대한 반응성의 평준 화는 CPA의 중요한 특칭이다. 에스데르보다 아미드가 화학적으로 훨씬 안정 하기 때문에 키모트립신 등과 같은 통상적인 펩티다제는 아미드보다 에스테르 를 훨씬 신속하게 가수분해한다. 그런데 이 모형에서는 아미드와 에스데르의 반응성이 비슷한데, 그 이유는 꼬과 같은 메커니즘으로 아미드의 반응이 더욱 기속되기 때문으로 볼 수 있다. 이 모형 연구의 결과로부터, 에스데르 기질이 CPA 에 의하거 소뮤수물 메커니즘으로 가수분해되면 펩티드 기질도 동일한 메 커니즘으로 효과적으로 가수분해될 수 있다는 결론을 도출할 수 있다. 제 8 절 결론 CPA의 X- 선 결정학적인 구조가 발표된 이후 약 20 년간 수많은 논문이 CPA 의 메커니즘에 관하여 발표되었다. 그러나 현재로서는 Glu-270 이 에스테 르 기질의 가수분해에서 친핵체로 작용한다는 가설에 대한 강력한 증거가 속도 론적 및 라만 분광학적 자료에 의하여 제시되어 있을 뿐이다. 펩티드 기질의 가수분해에서 Glu-27 뼈 역할이 친핵체인지 혹은 일반 영기인지를 분간하는
증거는 아직 제시된 바 없다. Zn(Il) 이온의 정확한 촉매 역할을 밝히는 증거를 CPA 를 대상으로 칙접 실 험하여 수집하는 것은 현재의 과학 수준으로-는 곤란하다. 따라서 모형 화합물 의 연구에 의한 간접적인 접근 방법이 유용히다. 전술한 바와 같이 효소 반응 메커니즘을 결정하는 데에는 반응 속도론아 가 장 중요한 도구이다. 그러나 반응 속도론만으로.는 메커니즘을 결정할 수 없기 때 문 에 중간체의 축적 및 포획과 같은 화학적 방법, x- 선 결정학, 각종 분광 학 등의 기법을 종합적으로 사용하게 된다. X - 선 결정학은 효소의 활성자리 주위의 杯빗적 구조 를 밝혀주기 때문에 효소 반응 메커니즘의 연구에서는 필 수 불가결한 도구이다. 그러나 반응 메커니즘은 용액 속에서 일어니~ 동적인 과정에 관한 것임에 비하여 X- 선 결정 구조는 결정내에서 존재하는 정적인 구 조라는 점에서 큰 차이가 있다. 죽 X - 선 결정학으로부터 유용한 정보는 얻어 내지만, 이 정보가 실제 반응에서 작용하는 메커니즘에 반드시 적용이 가능한 것은 아니라는 것이다. 효소 반응 속도론에서 도출된 정보는 실제 반응의 메커 니즘에 반드시 적용된다는 점에서 X- 선 결정학과 두드러진 차이점을 보인다.
참고문헌 1) Lipsc omb, W. N. Acc. Chem. Res., 1 970, 3, 81 및 이 속새 인용된 문헌. 2) Kais e r, E. T. ; Kais e r, B . L. Acc. Chem. Res., 1 972, 5, 219. 3) 서정헌, 『 화학과 공업의 전보 ' ' 1977, 17, 84. 4) Kais e r, E. T. ; Chan, T.- W . ; Suh, J. I n Prote in- Meta l Inte r actio ns., fried -man, M. Ed., P lenum : N. Y., 1 974, p. 5 9. 5) Suh, J. ; Kais e r, E. T. Bio c hem. Bio p h y s . Res. Comm. , 1 975, 64, 863. 6) Camp be ll, P. ; Nashed, N. T. ]. Am. Chem. Soc., 1982, 104, 5221 . 7) Sug imo to , T. ; Kais e r, E. T. ]. Am. Chem. Soc., 1978, 100, 7750 ; 1979, 101, 3946.
8) Sp ra tt , E . ; Sug im oto , T. ; Kais e r, E. T. ]. Am. Chem . Soc., 1 983, 105, 3679. 9) Nashed, N. T. ; Kais e r, E. T. ]. A m. Chem. Soc., 1 981, 103, 3611 . 10) Nashed, N. T. ; Kais e r, E. T. ]. Am. Chem. Soc., 1986, 108, 2710. 11) Brown, J. R. ; Greeshie ld s, R. N. ; Yamasaki, M. ; Neurath , H. Bio c hemi stry, 1963, 2, 867. 12) Yamasaki, M. ; Brown, J. R. ; Cox, D. J. ; Greeshie ld s, R . N. ; Wade, R. D. ; Neurath , H. Bio c hemi stry, 1963, 2, 859. 13) Anson, M. L. ]. Gen. Phys i·oz ., 1 937, 20, 663. 14) Allan, B. J. ; Keller, P. J. ; Neurath , H. Bio c lze m is l1 y , 1964, 3, 40. 15) Cox, D. J. ; Bovard, F. C. ; Barge tz i, J . P. ; Walsh, K. A. ; Neurath , H. Bio - clzemi stry, 1964, 3, 44. 16) Bradshaw, R. A. ; Eric ss on, L. H. ; Walsh, K. A. ; Neurath , H. Proc. Natl. Acad. Sci . US .A., 1969, 63, 1389. 17) Lipsc omb, W. N. ; Hart su ck, J. A. ; Reeke, G. N. Jr., Qu io c ho, F. A. ; Beth -ge, P. H. ; Ludw ig, M. L. ; Ste i t z, T. A. ; Muir h ead, H. ; Cop po la, J. Brook- haven Sym p. in Bio l. , 1971, 21, 24. 18) Petr a, P. H. ; Neurath , H. Bio c hemi stry, 1969, 8, 2466, 5029. 19) Petr a, P. H. Bio c hemi stry, 1971, 10, 3163. 20) Hass, G. M. ; Neurath , H. Bio c hemi stry, 1971, 10, 3535, 3541 . 21) Kim, D. H. ; Kim, K. B. ]. Am. Chem. Soc., 1991, 113, 3200. 22) Yun, M. ; Park, C. ; Kim, S. ; Nam, D. ; Kim, S. C. ; Kim, D. H.]. Am. Chem. Soc., 1992, 114, 2281 . 23) Hall, P. L. ; Kais e r, B. L. ; Kais e r, E. T. ]. Am. Chem. Soc., 1969, 91, 485. 24) Glovsky, J. ; Hall, P. L. ; Kais e r, E. T. Bio c hem. Bio p h y s . Res. C omm., 1972, 29, 205. 25) Bunti ng , J. W. ; Mu rph y, J. ; My er s, C. D. ; Gross, G. G. Can. ]. Chem., 1974, 52, 2648. 26) Carson, F. W. ; Kais e r, E. T. J Am. Chem, Soc., 1964, 86, 2922. 27) Auld, D. S. ; Holm q떠 s t, B. Fed. P roc., 1972, 41, 435, Abst. No. 1230. 28) Auld, D. S. ; Vallee, B. L. Bio c hemi stry, 1970, 9, 4355 ; 10, 2892. 29) Chan, R. W. Ph. D. Thesis, Un ive rsity of Ch ica go , 1974.
30) Joh ansen, J. T. ; Vallee, B. L. Proc. Natl. A cad. Sci. US.A ., 1973, 70, 2006. 31) Qu io c ho, F. A . ; McMurray, C. H. ; Lip sc omb, W. N. Proc. Natl . A cad. Sci . U.S .A., 1972, 69, 2850. 32) Suh, J. ; Cho, W. ; Chung, S. J Am. Chem. Soc., 1985, 107, 4530. 33) Suh, J. ; Hong, S. B . ; Chung , S. J Bio l. Chem., 1 986, 261, 7112. 34) Breslow, R. ; Wern ick , D. L. J Am. Chem. Soc., 1976, 98, 259. 35) Breslow, R. ; Wernic k , D. L. Proc. Natl. A cad. Sci . U.S .A ., 1977, 74, 1303. 36) Suh, J. ; Cheong, M. ; Suh, M. P. J Am. Chem. Soc., 1982, 104, 1654. 37) Suh, J. ; Han, H. Bio o rg. C hem., 1984, 14, 177. 38) Chris ti a n son, D. W. ; Lipsc omb, W. N. Acc. Chem. Res., 1989, 22, 62. 39) Hanson, J. H. ; Kap la n, A. P. ; Bart let t , P. A. Bio c hemi stry, 1989, 28, 6249. 40) Kim , H. ; Lip sc omb, W. N. Bio c hemi stry, 1990, 29, 5546. 41) Kim, H. ; Lipsc omb, W. N. Bio c hemi stry, 1991, 30, 8171 . 42) Suh, J. ; Kais e r, E. T. ]. Am. Chem. Soc., 1976, 98, 7060. 43) Makin e n, M. W. ; Yamamura, K ; Kais e r, E. T. Proc. N atl. A cad. Sci. US.A ., 1976, 73 , 3882. 44) Makin e n, M. W. ; Kuo, L. C. ; Dy mo wski, J. J. ; Jaf f er , S. ]. Bio l. Chem., 1979, 254, 356. 45) Hoff m an, L. C. ; Fukuya m a, J. M. ; Makin e n, M. W. ]. Mal. Bio l. , 1983, 163, 63. 46) Hoff m an, S. J. ; Chu, S. S. -T. ; Lee, H. ; Kais e r, E. T . ; Carey, P. R. ]. Am. Chem. Soc., 1983, 105, 6971 . 47) Kuo, L. C. ; Makin e n, M. W. ]. Am. Cl 玩. Soc., 1985, 107, 5255. 48) Galdes, A. ; Auld, D. S. ; Vallee, B. L. Bio c hemi stry, 1983, 22, 1888. 49) Geog he ga n , K. F. ; Galdes, A. ; Mart ine lli , R. A. ; Holmq u is t, B. ; Auld, D. S. ; Vallee, B. L. Bio c hemi stry, 1983, 22, 2255. 50) Suh, J. ; Chung, S. ; Choi, G. B. Bio o rg. Chem., 1989, 17, 64. 51) Suh, J. ; Hwang, B. K. ; Jan g, J. ; Oh, E. ]. Bio c hem. Bio ph ys . Meth ., 1991, 22, 167. 52) Jen cks, W. P. Cata ly si s in Chemi stry, and Enzym olo gy , McGraw-Hi ll : New York, 1969, p. 5 0.
53) Jon es, R. A. Y. Phys i ca l and Meclzanis tic Orga nic Chemi stry, Cambrid g e Univ e rsity Press : Cambrid g e , 1984, 2nd ed., p . 9. 54) Schepa r t z, A. ; Breslow, R. ]. Am. Chem. Soc., 1987, 109, 1814. 55) Gin o dman, L. M. ; Malit sev , N. I. ; Orekhovic h , V. N. Bio k im iy a, 1966, 31, 1073 ; Cliem . Abst, 1966, 66, 43929q . 56) Hall, P. L. ; Kais e r, E. T. Bio c hem. Bio p h y s . Res. Comm., 1967, 29, 205. 57) Suh, J. ; Kais e r, E. T. ]. Korean. Clzem. Soc., 1978, 22, 164. 58) Sander, M. E. ; Wi tze l, H. Bio c hem. Bio p h y s . Res. Comm. , 1985, 132, 681 . 59) Sander, M. E. ; Wi tze l, H. In Zin c Enzym es, Bert ini , I. ; Luchin a t, C. ; Maret, W. ; Zepp e zauer, M. Eds., Bir k hauser : Bosto n , 1986, Chap . 13. 60) Bri tt, B. M. ; Petic o las, W. L. ]. Am. Chem. Soc., 1 992, 114, 5295. 61) Suh, J. ; Cho, M. Bio o rg. Chem., 1990, 18, 276. 62) Sim p so n, R. T. ; Rio r dan, J. F. ; Vallee, B. L. Bio c hem is try, 1963, 2, 616. 63) W 血 aker, J. R. ; Meng er , F. ; Bender, M. L. Bio c hemis try, 1966, 5, 386. 64) Davi es , R. C. ; Rio r dan, J. F. ; Auld, D. S. ; Vallee, B. L. Bio c hemis try, 1968, 7, 1090. 65) Rior dan, J. F. ; Vallee, B. L. Bio c hemi stry, 1963, 2, 1460. 66) Sim p so n, R. T. ; Vallee, B. L. Bio c hemi stry, 1966, 5, 1760. 67) Roholt , 0. A. ; Pressmann, D. Proc, Natl, Acad, Sci . U.S .A., 1967, 58, 280. 68) Sokolovsky, M. ; Vallee, B. L. Bio c hemis try, 1967, 6, 700. 69) Rior dan, J. F. ; Sokolovsky, M. ; Vallee, B. L. Bio c hemi stry, 1967, 6, 358. 70) Muszy ns ka, G. ; Rior dan, J. F. Fed. Proc., 32, 466, Abst. No. 1371 . 71) Kaga n , H. M. ; Vallee, B. L. Bio c hemis try, 1969, 8, 4223. 72) Kaga n , H. M. ; Vallee, B. L. Bio c hem. Bio p h y s . Res. Comm., 1969, 34, 654. 73) Joh ansen, J. ; Vallee, B. L. Proc. Natl. Acad. Sci . U .S.A ., 1971, 68, 2532. 74) Joh ansen, J. T. ; Living s t o n , D. M. ; Vallee, B. L. Bio c hemis try, 1972, 11, 2584. 75) Hil ve rt , D. ; Gardell, S. J. ; Rutt er , W. J. ; Kais e r, E. T. ]. Am. Chem. Soc., 1986, 108 , 5298. 76) Coleman, J. E. ; Vallee, B. L. ]. Bio l. Chem., 1961, 236, 2244. 77) Coleman, J. E. ; Pulid o , P. ; Vallee, B. L. Bio c hemis try, 1966, 5, 2019.
78) Davie s , R. C. ; Rio r dan, J. F. ; Auld, D. S. ; Vallee, B. L. Bio c hemi stry, 1968, 7, 1090. 79) Kang, E. P. ; Sto r m, C. B. ; Carson, F. W. Bio c hem. Bio p h y s . Res. Comm. , 1972, 49, 621 . 80) Dekoch, R. J. ; West, D. J. ; Cannon, J. C . ; Chaste e n, N. R. Bio c hemi stry, 1974, 13 , 4347. 81) Auld, D. S. ; Holmq u is t, B. Bio c hemi stry, 1974, 13, 4352. 82) Schneid e r, M. ; Suh, J. ; Kais e r, E. T.]. Chem. Soc, Chem. Comm., 1976, 98, 106. 83) Schaff er , A ; Auld, D. S. Bio c hemi stry, 1986, 25, 2476. 84) Latt , S. A. ; Vallee, B. L. Bio c hemi stry, 1971, 10, 4263. 85) Rosenberg, R. C. ; Root, C. A ; Wang , R. H. ; Gerdonio , M. ; Gray, H. B. Proc. Natl. Acad. Sci . U.S .A ., 1973, 70, 161 . 86) Rosenberg, R. C. ; Root, C. A ; Gray, H. B. ]. Am. Chem. Soc., 1975, 97, 21. 87) Kuo, L. C. ; Makin e n, M. W. ]. Am. Chem. Soc., 1985, 107, 5255. 88) Kuo, L. C. ; Lipsc omb, W. N. ; Makin e n, M. W. ]. Am. Chem. Soc., 1986, 108, 5003. 89) Hardman, K. D. ; Lipsc omb, W. N. ]. Am. Chem. Soc., 1984, 106, 463. 90) Ste p h e ns, R. S. ; Jen to f t, J. E. ; Bry an t, R. C. ]. Am. Chem. Soc., 1974, 96, 8041 . 91) Ch risti a n son, D. W. ; Lip sc omb, W. N. ]. Am. Chem. Soc., 1986, 108, 4998. 92) Chris ti a n son, D. W. ; David , P. R. ; Lip sc omb, W. N. Proc. Natl. Acad. Sci . U.S .A ., 1987, 84, 1512. 93) Rio r dan, J. F. Bio c hemis try, 1973, 12, 3915. 94) Suh, J. ; Chun, K. H. ]. Am. Chem. Soc., 1986, 108, 3057. 95) Fife , T. H. ; Przys t a s , T. J. ]. Am. Chem. Soc., 1982, 104, 2251 . 96) Suh, J. ; Chung, S. ; Lee, S. H. Bio o rg. Chem., 1987, 15, 383. 97) Suh, J. ; Hwang, B. K. ; Koh, Y. H. Bio o rg . Chem., 1990, 18, 207(1990). 98) Suh, J. Bio o rg. Chem., 1990, 18, 345. 99) Suh, J. ; Park, T. H. ; Hwa ng, B. K. ]. Am. Chem. Soc., 1992, 114, 5141 . 100) Suh, J. Acc. Chem. Res., 1992, 25, 273.
제 6 장 신의약설계 신의약 설계는 유기화학의 기법을 이용하여 의학적인 문제점을 해결하는 중 요한 예이다. 이 분야는 의약화학에서 광범위하게 다루어지고 있기 때문에 1) 여 기에서는 간단히 몇 가지 전형적인 사례를 소개하는 것으로 국한할 것이다. 제 1 절 효소저해 효소 저해제는 효소와 착화합물을 형성함으로써 효소가 기질과 반응하는 속 도를 저하시킨다. 어느 효소가 질병의 원인이 되는 물질을 발생시킨다면 이 효 소의 기능을 저해함으로써 질병을 치료할 수 있다. 효소 저해제가 의약품으로 개발된 사례로 고혈압 치료제인 캡토프릴을 들겠 다. 신장에서 분비된 레닌은 혈당 단백질인 안지오덴시노겐에 작용하여 1 (}7 R 의 아미노산으로 이루어전 안지오덴신 1 을 생성한다. 안지오덴신 1 은 안지오덴신 전환 효소 (ACE) 의 작용에 의하여 카르복실 말단에서 두 개의 아미노산울 방 출함으로써 안지오덴신 Il 를 생성한다. 안지오텐신 Il 는 혈압 상승의 원인이 되 기 때문에 AC~ 저해제를 두여하면 고혈압의 강하를 초래할 수 있다. AC 底 0 편 금속 효소이며 단백질 가수분해에 작용한다는 점에서 카르복시 펩티다제 A와 유사하다. AC& 기 구조가 알려지지 않았을 때에 카르복시펩티 다제 A의 강력한 저해제로 보고된 l- 벤질숙신산 (1) 을 모형으로 하여 loll 표시 한 과정을 거쳐 강력한 ACE 저해제인 캡토프릴의 구조를 도출하게 되었다. 1),2) 캡토프릴은 현재 전세계에서 가장 많이 팔리는 약 중의 하나인데, 캡토프릴울
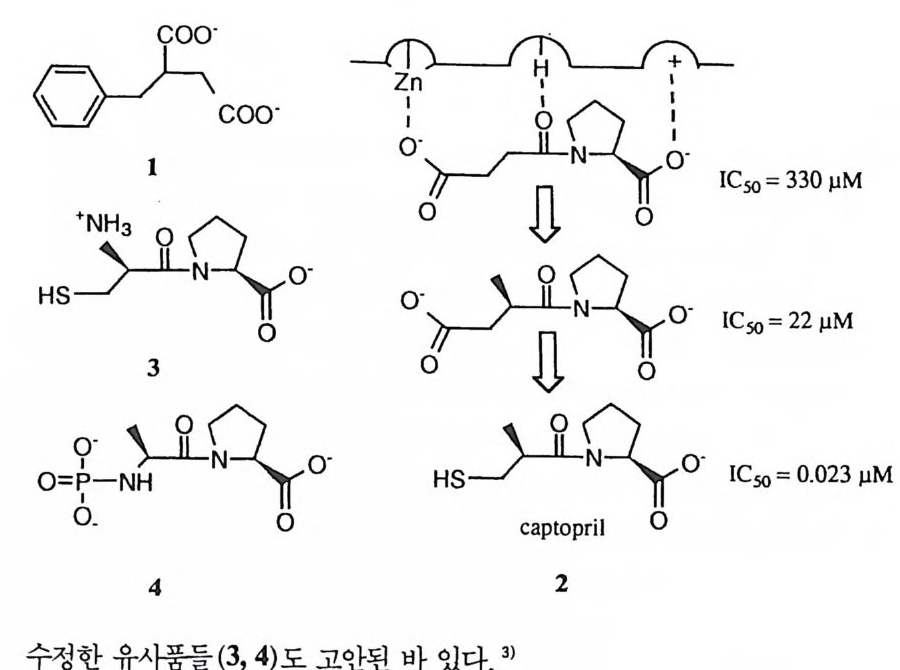 ~HSY c-1Co \os o Z&In ° 尸0~I\ ..,-,_ :。 I ICso= 330 µM
~HSY c-1Co \os o Z&In ° 尸0~I\ ..,-,_ :。 I ICso= 330 µM
제 2 절 수용체인식 약물은 체내 세포의 표면에 존재하는 일정한 구조와 형태를 가진 수용체와 결합하여 물리 화학적 작용을 동하여 세포내에서의 생화학적 변화를 유발하고 이는 결과적으로 거시적인 생리적인 반응으로 발전한다. 수용체와 결합함으로 써 생리적인 항전작용을 유발하는 약물을 효농약이라고 하며, 수용체와 결합은 하지만 생리 효과는 나타내지 못하며 효능약의 결합을 방해하는 물질을 길항약 이라고부른다. 신약을 개발하는 과정에서 수용체에 관한 정보를 수집하는 과정을 모르핀 수 용체를 예로 들어 살펴보기로 한다 .4)-6) 모르핀 (5) 은 강력한 진통제이지만 마 약성 물질로 많은 부작용이 있다. 모르핀보다 강력하면서 부작용이 없는 기적 의 진통제를 개발하려는 광범위한 노력이 기울여지고 있다. 아트로핀 (6) 은 콜 린 수용체에 작용하는 콜린 억제제인데 이것의 유사체로 합성한 데메롤 (7) 이
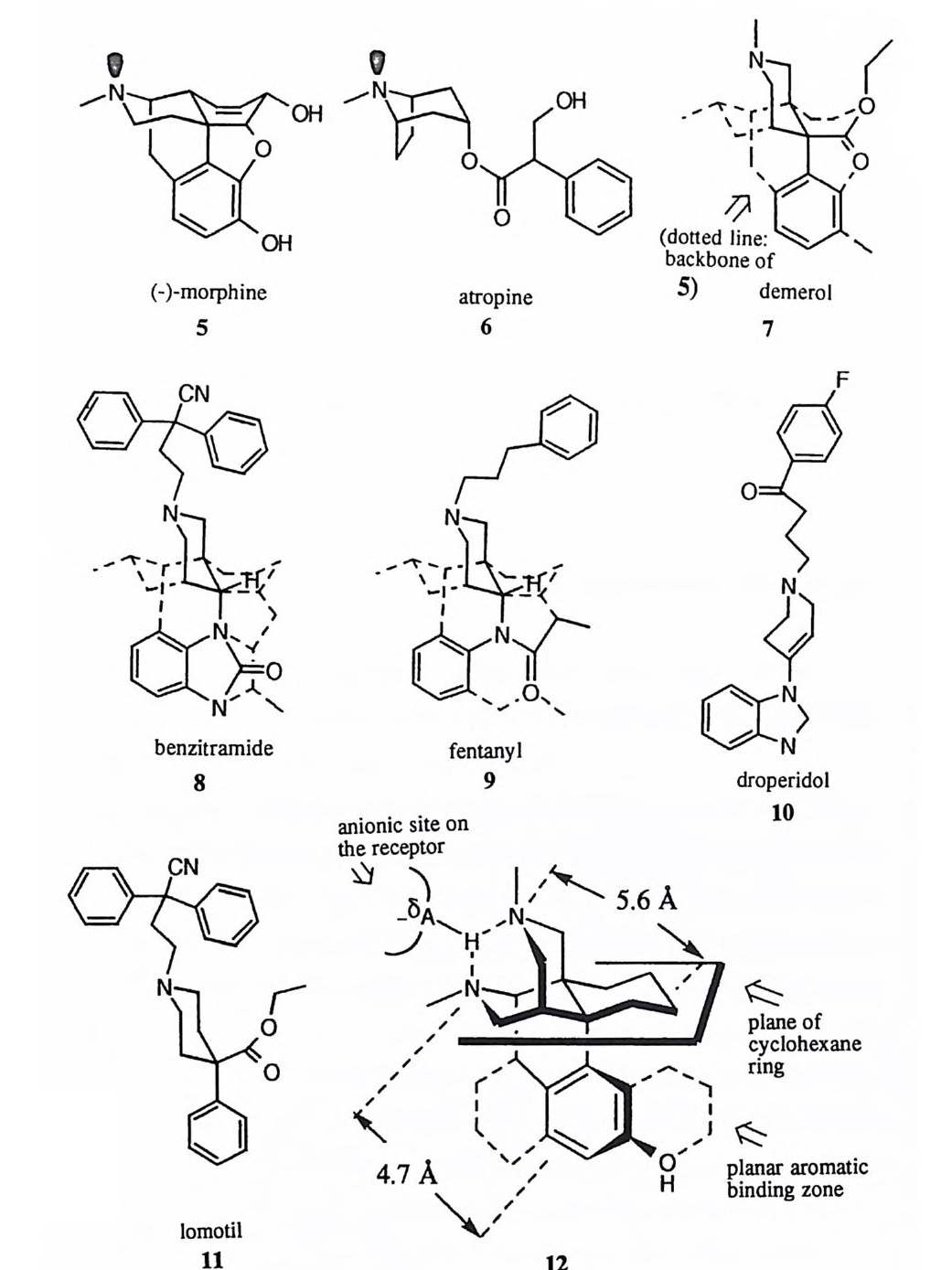 / 'N
/ 'N
진통 능력이 있음이 우연히 발견되었다. 7 에 점선으로 표시한 모르핀 구조와 데메롤이 잘 겹치는 것으로 보아 데메롤이 모르핀의 수용체를 인식할 수 있다 고되었 볼는데 수, 있이다와. 같아이트 로개핀발을된 선N도— C물H 질3 로영 역사의용 하협여수 성새을로 운개 선전하통기제 를위 하설여계 하합게성 된 벤지트라미드 (8) 는 모르핀보다 7 배가량 강력한 진통효과를 가지고 있다. 벤 지트라미드는 또 다른 진통제인 펜타닐 (9) 및 정신병 치료제인 드로페리돌 (10) 과 구조적인 유사성을 지니고 있다. 한편, 전통제인 벤지트라미드와 데메 롤의 구조를 섞어서 합성한 로모틸 (11) 은 전통 효과는 없고 진통제들의 부작용 만 극대화하여 설사약으로 효과가 있음은 재미난 시실이다. 이와 같은 정보와 그 밖의 여러 유도체가 보이는 약효로부터 모르핀 수용체의 구조를 n 와 같이 도식화할 수 있다. 수용체의 구조에 대한 정보가 구체적으로 수집됨에 따라 이 를 인식하는 신약을 합리적으로 개발하기가 용이해진다. 제 3 절 DNA 조절 DNA 의 箕변적인 형태를 파손하거나 DNA 의 복제를 저해하면 이 DNA 와 관련된 세포의 증식을 막을 수 있다. 이 세포가 암세포이면 이 방법으로 암울 화학적으로 치료할 수 있게 되고, 병원균의 DNA 의 복제가 저해된다면 이 균 에 의한 질병을 치료할 수 있게 되는 것이다. DNA 합성을 저해하는 의약은 뉴클레오시드계와 비뉴클레오시드계로 나눌 수 있다. 뉴클레오시드계의 약품으로 대표적인 것으로 시토신 아라비노스 (13)
 HO~< cyt os m e H,N
HO~< cyt os m e H,N
를 들 수 있다. 6) 이것은 생체내에서 포스포릴화된 후, DNA 중합을 저해하는 경우도 있고, 새롭게 합성된 DNA 에 끼여들어 가서 DNA의 기능을 파괴한 다. 또 다른 예를 들면, 5- 요도 -5' -아미노 - 2', 5' - 디데옥시유리딘 (14) 은 포유 류인 숙주의 DNA의 복제에는 영향을 미치지 않으면서 허피스 심플렉스 바이 러스의 DNA 복제를 저해한다.
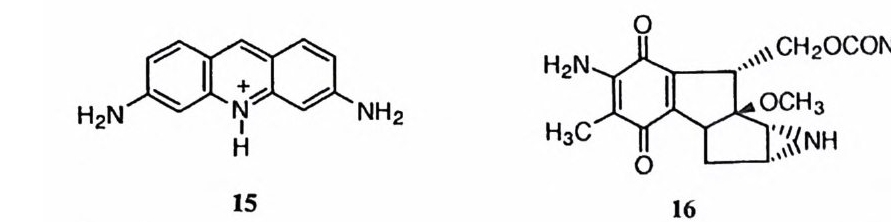 H2N:hN H2 ::广: :3HOCONH2
H2N:hN H2 ::广: :3HOCONH2
DNA 의 이중 나선구조에 결합함으로써 DNA의 합성을 저해하는 비뉴클레 오시드계 의약의 예로 프로플라빈 (15) 등과 같은 아크리딘 유도체와 미토마이 신 C(l6) 등과 같은 각종 항생제를 둘 수 있다. 프로풀라빈과 갇은 화합물의 평면 고리는 DNA 의 이중 나선구조에 있는 포개진 염기쌍 사이에 삽입됨으로 써 DNA와 결합하는데, DNA 에 이러한 화합물이 삽입되면 DNA 의 이중 나 선구조가 변형되어 DNA의 복제가 저해되는 것이다. 각종 항생제에 대하여서 도 비슷한 메커니즘이 작용하며, 미토마이신 G 근 항암효과를 나타낸다. 이 의 에도 4 배위 평면 구조를 가진 백금의 시스-디클로로디아민 착화합물등을 DNA 의 두 개의 구아니딘의 N 구원자에 배위됨으로써 이중 나선에 결합하여 항암효 과를보인다. DNA의 이중 나선구조를 인식하는 다양한 화합물이 미생물로부터 검색될 뿐 아니라 유기화학적으로 설계, 합성되 어 항암제, 항생제 등으로서의 효능을 시 험받고 있다. 특히 최근에는 DNA 의 가능한 한 긴 영기 순서와 특이적으로 결 합함으로써 특정 DNA를 선별적으로 인식하는 유기 화합물을 합성하려는 노력 이 상당한 성과를 거두고 있다. 또한, 단순히 DNA 염기 순서를 인식하여 결 합할 뿐 아니라 결합 부위에서 DNA 를 화학적으로 파괴하는 기능을 가진 분자 도 다양하게 설계되고 있는바, 이러한 연구가 진행되면 유기화학적인 기법으로 암과 같은 질병을 정복하게 될 것이다.
참고문헌 1) 김동한, r 의약화학 』 , 동일출판사. 1989. 2) Ondett i, M. A. ; Rubin , R. ; Cushman, D. W. Sci enc e, 1977, 196, 441 . 3) Polga r, L. Mecluznis ms of Prote a se Actio n , CRC Press : Boca Rato n , 1989, p. 199. 4) Henderson, G. ; McFedzean, I. Chem. Bri tain, 1985, 1094. 5) Hit e, G. J. In Pri nc ip le s of Medic ina l Clzernis tzy, Foy d, W. 0. Ed., L ea and Febig er , 1981, Chap ter 12. 6) Dug as , H . Bio o rga n ic C lzemi stry, Spr i n g e r -Verlag : New York, 1989, pp. 96- 102, 140- 15 3.
생물학의 기법을 제이 용2 부한 유기화학의 탐구 
제 7 장 유기 합성과효소 제 1절 입체 선택적인 유기 합성에 대한 요쇼의 이용 I) 이 효소 작용의 반응 및 기질 특이성 유기 반응의 거의 대부분의 유형에 대하여 효소가 촉매로 작용한다. 효소는 반응 속도를 괄목하게 제고할 뿐 아니라 개입하는 반응의 유형, 기질과 생성물 의 구조와 입체화학 등에 대하여 높은 선택성을 보인다. 유기 합성에서 응용도 가 높은 효소는 기질의 구조에 대한 특이성이 좁지 않아서 비교적 다양한 구조 룰 가진 기질을 수용하는 효소들이다. 일반적으로 포유류에서 추출되는 효소는 기 질 의 특 이성의 범위가 넓다. 미생물에서 추출되는 효소는 받아들이는 기질의 구조 의 범위가 좁은 대신 미생물의 종류가 매우 많다는 장점이 있다 .
 AR CHA O' 占 占XbR
AR CHA O' 占 占XbR
 5R HOHOOCh pIoCerr sOoOe xH rOi ad daH 'iss Qe h 5A 2 OH [R = CH(NH: JC OOH or
5R HOHOOCh pIoCerr sOoOe xH rOi ad daH 'iss Qe h 5A 2 OH [R = CH(NH: JC OOH or
효소를 여러 가지 사용하면 취급할 수 있는 기질의 구조 범위를 대폭 확대할 수 있다. 텨 예에서 보듯이 알콜을 카르보닐로 산화시키는 효소로 세 가지의 기알질콜에 데 대히하드여로 게효나소제 반를응 사을용 일하반면화 각하각여의 활 기용질할의 수 범있위다.를 ”중 첩시켜 매우 다양한 분자내에 화학적으로 유사한 반응 자리가 두 개 이상 존재하는 경우에 효소
를 사용하면 한 자리만 선택적으로 반응하게 할 수 있다. 산화에 관여하는 효 소를 예로 들어 효소의 효율성을 설명하여 보자. 2 2) 와 33 ) 에 표시한 선택적인 히 드록 시화 반응은 화학적인 산화 방법으로는 달성하기가 극히 어려운 반응이 다. 44) 와 같은 비닐 수소의 할로겐화도 효소를 이용하여 용이하게 진행시킬 수 있으며 , 5 5) 의 경우에는 세 개의 메독실기를 세 가지의 효소를 사용하여 선택적 으로 하나씩 가수분해하여 히드록시기로 변환하는데 이러한 합성 방법은 생리 환성 뭉질 의 합성에 매우 유용하게 사용될 수 있다. 이러한 합성 반응에 상응 하는 유기화학적인 방법아 있지만, 선택성이 효소 방법과 비교하여 크게 뒤지 고 격렬한 반응조건을 필요로 하며 수율이 낮다는 결점이 있다.
 12a-H y dr oxy st e r oid
12a-H y dr oxy st e r oid
효소를 이용한 입체 선택적인 산화 반응의 예로 66) 을 또 둘 수 있다. 양粹의 히드록시기 중에 한 개만 선택적으로 산화시키는 이 반응을- 비효소적인 방법으 로 달성하기는 쉽지 않다.
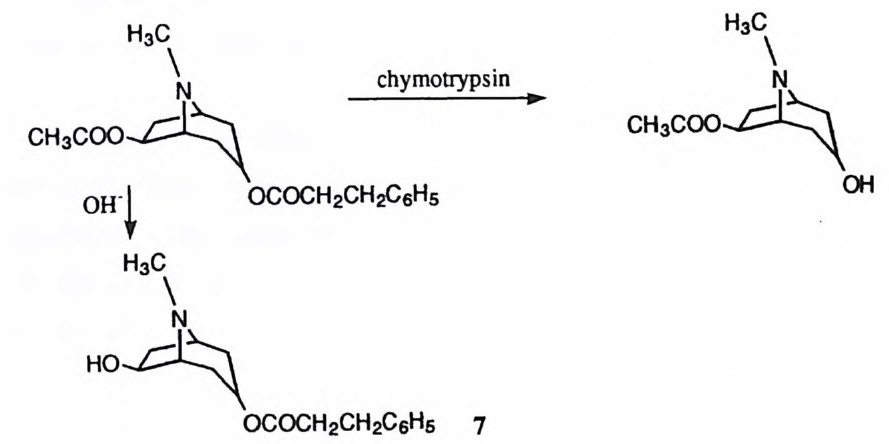 H3c\ H3c\
H3c\ H3c\
효소가 기질의 분자내예 존재하는 작용기들을 구별하여 방향 선택적으로 기 질의 구조를 변환시키는 것을 유기 합성에 자주 사용되는 탈보호 단계에 이용 할 수 있다. 예를 들어 77) 에 포함된 두 에스데르 중 각각을 선택적으로 제거하 는 방법이 존재하므로 원하는 히드록시기만 임의로 탈보호시킬 수 있는 것이다.
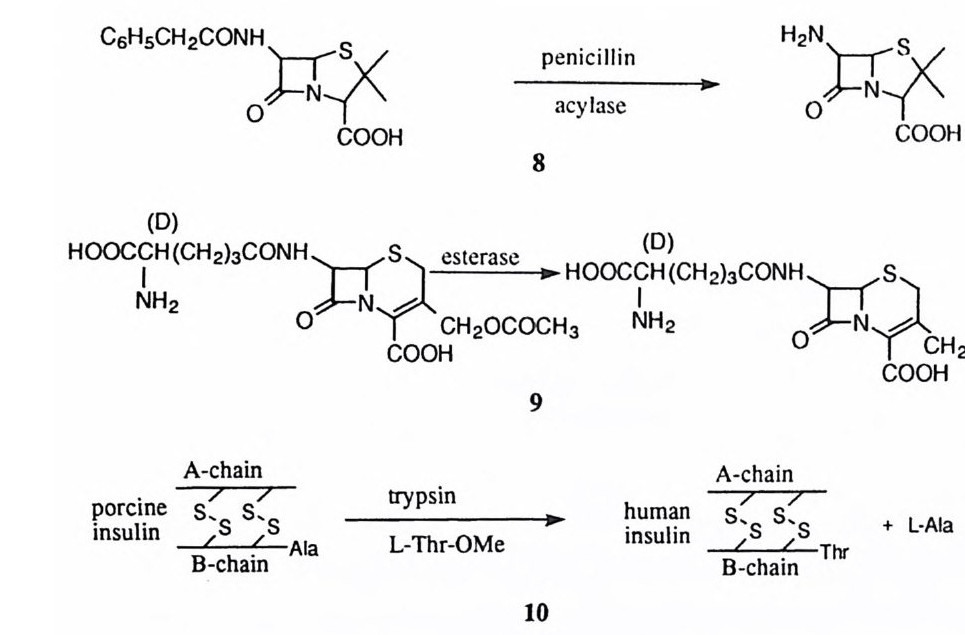 C6H5CH2CON。H 江〉 peacn yic f ai ls lei n H20N 二〉
C6H5CH2CON。H 江〉 peacn yic f ai ls lei n H20N 二〉
효소를 사용하여 아실기를 방향 선택적으로 가수분해하는 방법은 산업적으로 도 사용되는바, 88)과 99) 와 같이 페니실린 유도체 혹은 세파로스포린 유도체의 합성을그예로들수있다. 단백질 가수분해 효소를 이용하여 아실 유도체의 아실기를 물에 전달하면 가 수분해 반응이 일어나지만 아실기를 다른 친핵체에 전달하면 트란스아실화 반 응이 일어난다. 효소를 이용한 트란스아실화 반응은 효소의 특이성 때문에 기 질의 작용기를 보호할 필요가 없다는 이점이 있는데 이를 산업화에 응용-한 예 로 돼지의 인슐린의 알라닌 말단기를 트레오닌으로 바꾸어 인간 인슐린으로 전 환하는 반응 (10) J O) 을 들 수 있다. 효소 촉매 반응에서도 열역학적으로 안정한 생성물이 축적되게 되어 있지만
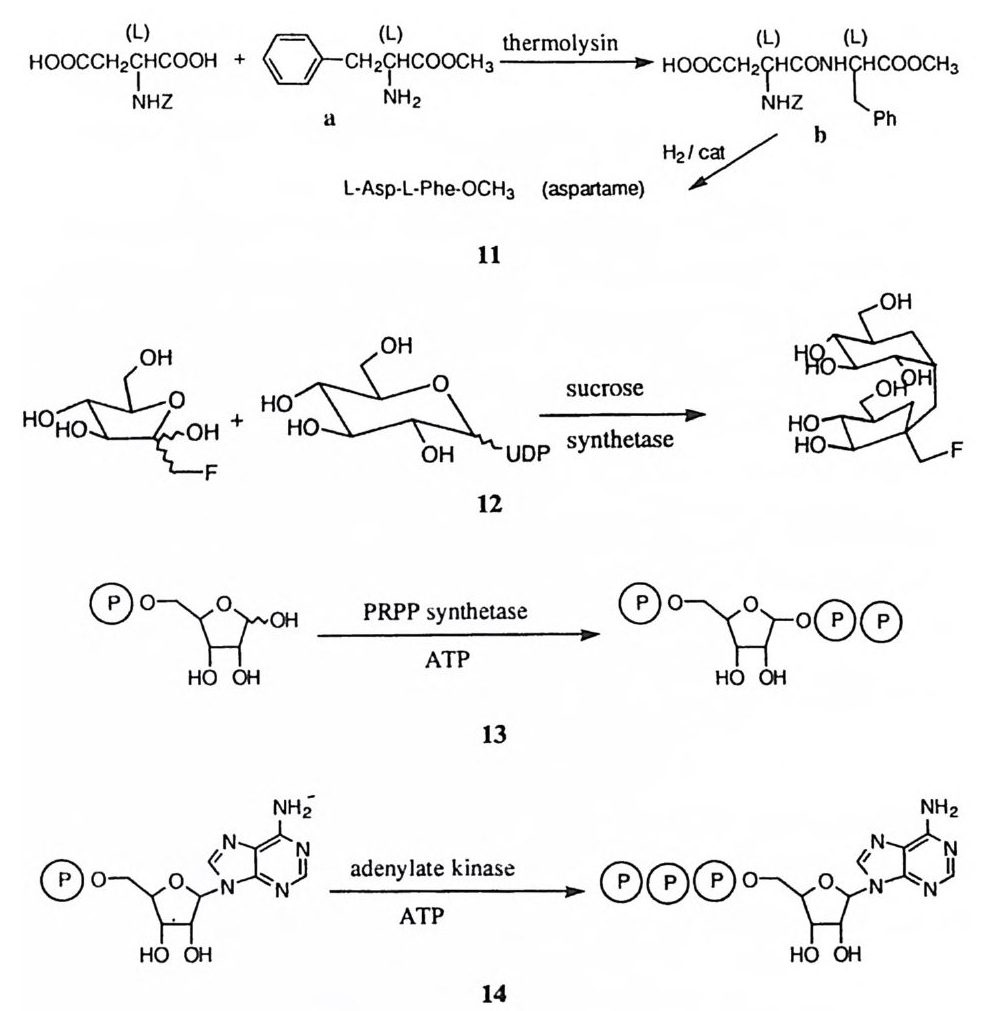 HOOCCH2\\cz00H + 0-CH:\\ :OOCH3 二노 HOOCCH2\\::H(\ \ PCh00CH3
HOOCCH2\\cz00H + 0-CH:\\ :OOCH3 二노 HOOCCH2\\::H(\ \ PCh00CH3
르 샤틀리에의 법칙을 이용하는 등의 방법으로 반응조건을 조절하면 열역학적 으로 불리한 쪽의 화합물을 분리하게끔 반웅의 방향을 전환시킬 수 있다. 이러 한 현상을 가장 자주 이용하는 분야가 펩티드 합성이다. 열역학적으로.는 펩티 드의 가수분해가 선호되지만 반응조건에 따라 아민과 카르복실산울 결합시켜 펩티드를 합성할 수도 있는데, 그 예를 11 에 표시한 감미료인 아스파르템의 합 성을 들 수 있다. 11) 여기에서 b 가 형성되면 b 가 a 와 불용성 염을 만들어 연속 적으로 제거되기 때문에 펩티드 결합의 형성이 일어나게 된다.
탄수화물을 이용한 유기 합성이 점점 중요시되고 있다. 탄수화물은 여러 가 지 히드록시기를 가지고 있는데 특정 히드록시기만 변환시키고자 할 때 효소가 매우 유용하게 사용될 수 있다. 예를 들어 llol] 서는 효소를 사용하여 비천연적 인 이량체 당화합물을 합성하였다. 12) 포스포릴화 및 탈포스포릴화 반응에 대하여서도 효소는 특이성을 가져서 유 기 합성에 유용하게 쓰이고 있다. 예를 들어, 1313) 과 1414) 에서는 여러 곳의 히 드록시기 및 포스포릴기가 포스포릴화되는 가능성이 있으나 효소를 사용하여 한 자리만 선택적으로 포스포릴화하고 있다. 뉴클레오티드 화학에서 효소를 촉매로 사용하여 염기 를 교환하는 방법이 유 기 합성에 유용하게 사용되는데, NAD 유사체 151s) 와 항바이러스제 1616) 의 합성이 그 좋은 예가 된다.
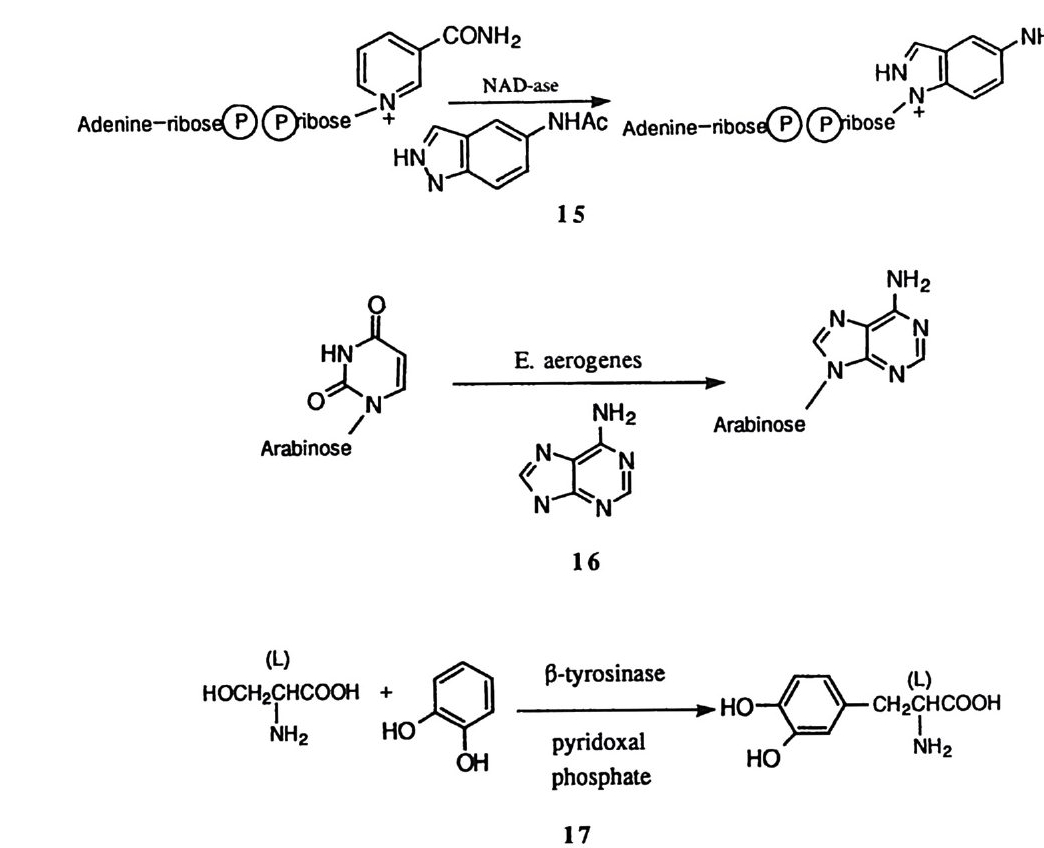 Adenne-nbos@@bose.,,,,D. 선C HNOTN 三:I).교.as e : Adenme-nbos 麟 boHNs e:I 그\ NHAc
Adenne-nbos@@bose.,,,,D. 선C HNOTN 三:I).교.as e : Adenme-nbos 麟 boHNs e:I 그\ NHAc
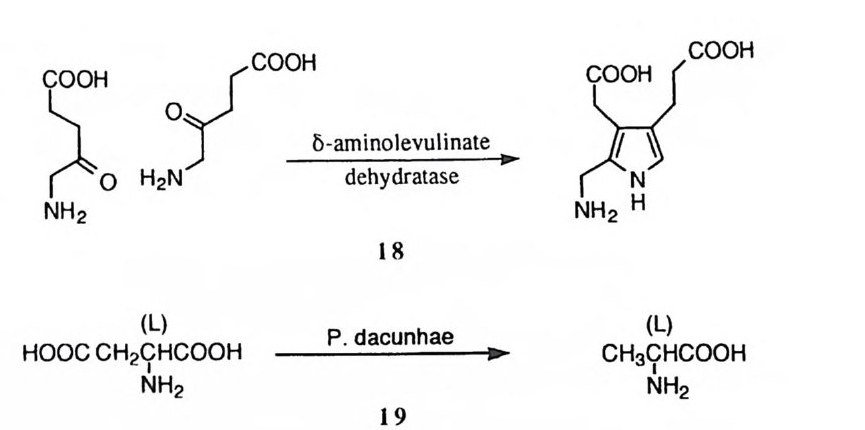 :Ho H2:< COO H6. ? elh?::t\\\ nate :>c。 。H
:Ho H2:< COO H6. ? elh?::t\\\ nate :>c。 。H
효소 를 사용하지 않고서는 달성하기가 매우 어려운 합성의 또 다른 예로 아 미노산의 결사슬을 교환하거나 (17 : 세린을 DOPA 로 전환함) 17) o- 레불린산을 축 합하여 포르포빌리노겐을 만드는 반응 (18)'18) 아스파르트산을 선택적으로 탈카 르복실화하여 알라닌으로 전환하는 반응 (19) 19) 등을 둘 수 있다. (2) 효소의 거울상 이성질 특이성의 이용 효소는 라세미 혼합물 기질의 거울상 이성질체를 구별하는 능력이 있다. 효 소가 거울상 이성질체에 대하여 특이성을 가지고 있으면 라세미 기질을 사용하 였을 때 반응은 정확하게 철반만 진행하게 될 것이다. 이때 생긴 미반응 물질 울 재활용하는 방법까지 함께 고안된 경우도 허다하다. 아실라제는 기질에 대한 구조적 요건이 넓어서 다양한 화합물을 기질로 받아 들이면서 동시에 촉매 반응에 대한 입체 특이성은 매우 좁아서 라세미 혼합물 을 구별하는 능력이 탁월하기 때문에 N- 아실 아미노산을 분할 (20) 하는 데에
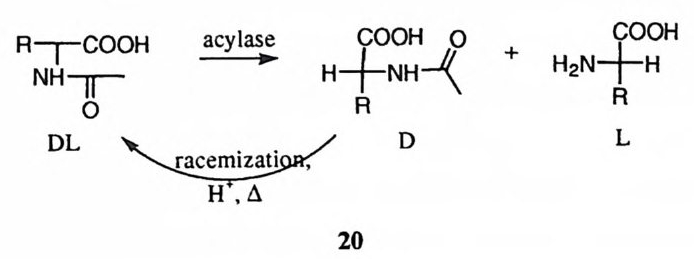 RN1~c-o;:o-H•• ~으꾹: H+CNOOHH -'o\ + H2N 十CO HO H
RN1~c-o;:o-H•• ~으꾹: H+CNOOHH -'o\ + H2N 十CO HO H
유용하게 사용된다. 20) L 이성질체만 아실라제에 의하여 가 수분 해되고 미반응 D- 이성질체는 화학적으로 라세미화 를 유도하여 기질로 재생 할 수 있다. 이 방법 은 산업적으로도 중요하게 사용된다.
 RCOOCH3 ~,.._ RCOOCH3 + RCOOH
RCOOCH3 ~,.._ RCOOCH3 + RCOOH
아미노산을 광학 분할하는 또 다른 방법으로 아미노산 에스데르를 입체 특이 적으로 가수분해하는 방법이 있다. 키모트립신과 갇은 효소는 넓은 범위 (21) 의 라세미 에스데르를 기질로 받아들이면서 입체 특이성을 나타낸다. 2” 에스데라 아제도 이와 같은 성질을 가지고 있어 합성에서 점점 광범위하게 활용되고 있 다 .22) 열역학적으로는 에스데르나 아미드의 가수분해가 자발적으로 일어나는 반응 이지만, 반응조건을 조절하여 역반왕긴 에스데르 혹은 아미드의 형성을 효소
 :了°CO뱌OH N 言 CCOH2OCH6 H5
:了°CO뱌OH N 言 CCOH2OCH6 H5
촉 매로써 쉽게 달성할 수 있다. 예를 들어 파파인을 촉매로 사용하여 222 1- 같 이 아미드 를 형성하면 생성된 아미드가 수용액에서 녹지 않기 때문에 생성 죽 시 침전되어 분리된다. 23) 키모트립신을 촉 매로 산과 알콜로부터 에스데르를 생성시킬 때 물과 물에 섞 이지 않는 유기 용매를 공동으로 사용하여 에스데르가 형성되면 유기 용매충으 로 추출되게끔 하여 평형의 위치를 에스테르 형성 쪽으로 바꾼 예도 있으며 ,24) 이와 유사하게 에스데라아제와 25) 리파아제를 26) 사용하여 에스데르 합성을 달성 하기도하였다. 키랄성 에폭시드를 합성하는 일은 유기화학에서 매우 중요하다 .27) 리파아제 를 사용하여 에폭시 에스테르를 거울상 이성질 선택적으로 가수분해함으로써
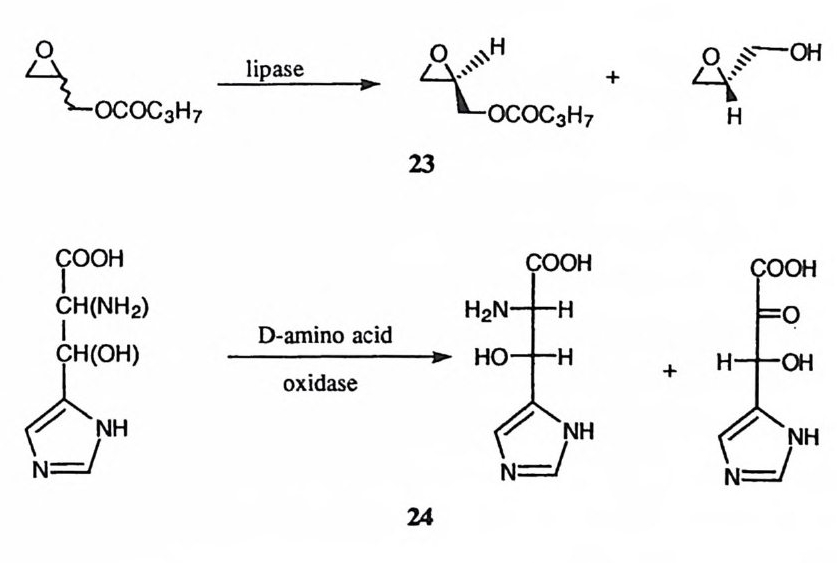 \ OCOC3H1 누 \LH-O C OC3H7 + 守O ~ -OH
\ OCOC3H1 누 \LH-O C OC3H7 + 守O ~ -OH
키랄성 에폭시드를 분리해 낸 작업 (23) 도 다수 보고되어 있다. 28) 목적물인 키랄성 화합물이 특히 중요하여 라세미 혼합물에서 목적물의 거울 상 이성질체를 구태여 분리하지 않아도 되는 경우에 입체 선택적으로 원하지 않는 이성질체를 파괴할 수도 있다. 꼬의 예에서는 한 이성질체 를 산화시켜 파 괴하였다 .29) (3) 효소의 프로키랄적 입체 특이성의 이용 앞 부분에서는 효소를 이용하여 라세미 혼합물 중에서 원하는 키랄성 화합물 을 분리하는 작업을 설명하였는데, 이때 원하지 않는 이성 질체를 재생하는 문 제가 뒤따르게 된다. 효소는 프로키랄적 입체 특이성을 가지고 있기 때문에 라 세미 혼합물 대신 원하는 키랄성 화합물만을 효소 를 사용하여 합성할 수 있다. 이 경우에는 원하지 않는 이성질체를 재생할 필요가 없다는 장점이 있다.
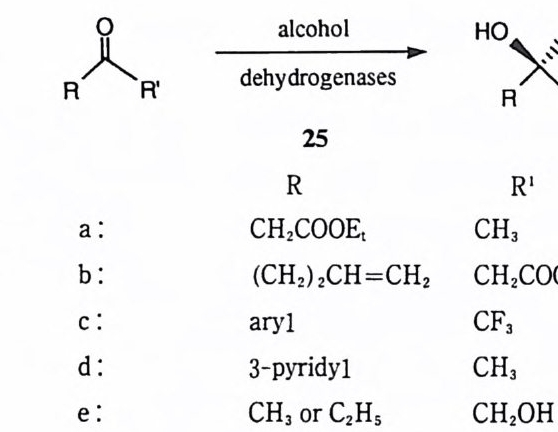 A 入 A' dehya ldcr oohgo eln ases HO_RxR '
A 入 A' dehya ldcr oohgo eln ases HO_RxR '
C=C, C=N 혹은 C=O 와 같은 평면형 이중결합의 양쪽 면이 비대칭일 때, 효소는 양쪽 면을 구별하는 능력이 있다. 예를 들어 케돈을 환원하면 케톤 의 카르보닐기가 키랄 중심으로 변환되는데 효소를 촉매로 사용하면 한 가지 거울상 이성질체만 생성 (25) 하여 각종 생리 활성 물질의 합성에 중간 물질로 사용할수있다 .1) 효소를 바꾸어 사용하면 한 가지 키랄 알콜의 두 거울상 이성질체 중 원하
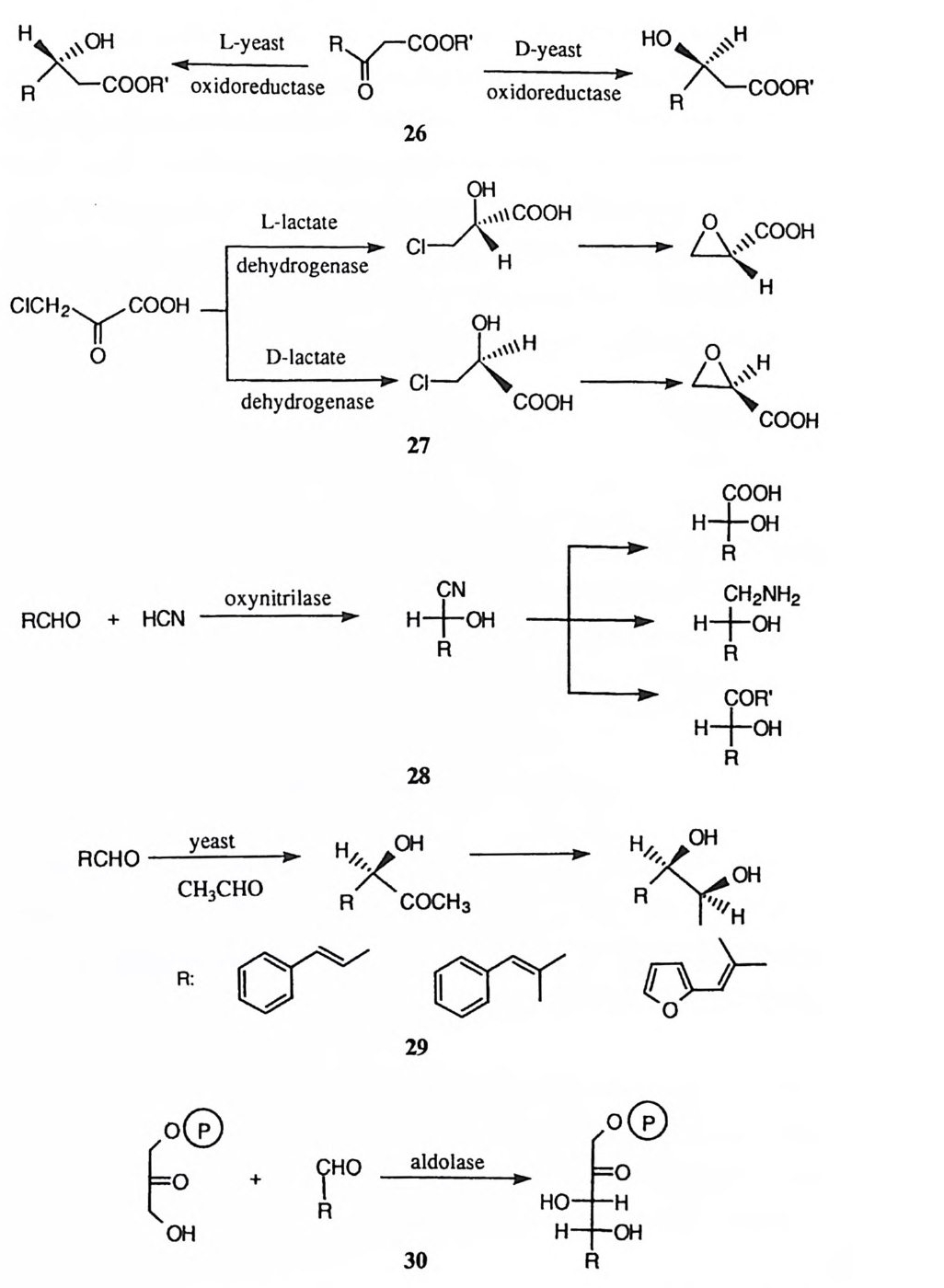 HR X_C OOR' 一oxid o reducta s e R< o COOR'교 교D-교ye 교a st - : •H·O -> <.-、` sC HO OR'
HR X_C OOR' 一oxid o reducta s e R< o COOR'교 교D-교ye 교a st - : •H·O -> <.-、` sC HO OR'
는 이성질체를 선택하여 생성물(예 : 26,27)30) 로 얻을 수 있다. 카르보닐기에 탄소 친핵체가 첨가되면 탄소-탄소 결합이 형성되어 유기 합성 에 유용하게 사용할 수 있다. 효소를 촉매로 사용하면 카르보닐기에 탄소 친핵 체가 첨가되어 한 가지 거울상 이성질체를 선택적으로 형성할 수 있다. 카르보 닐에 대한 시안산의 첨가 (28),31) 아실로인 축합 (29)'32) 알돌 축합 (30)33) 등을· 예로 둘 수 있다. 알돌 축합에 사용되는 알도라제의 경우에는 디히드록시 아세 돈 포스페이트를 기질 중의 하나로 사용하여야 하지만 공동 기질인 알데히드로 는 상당히 광범위한 구조를 가전 화합물을 받아들인다.
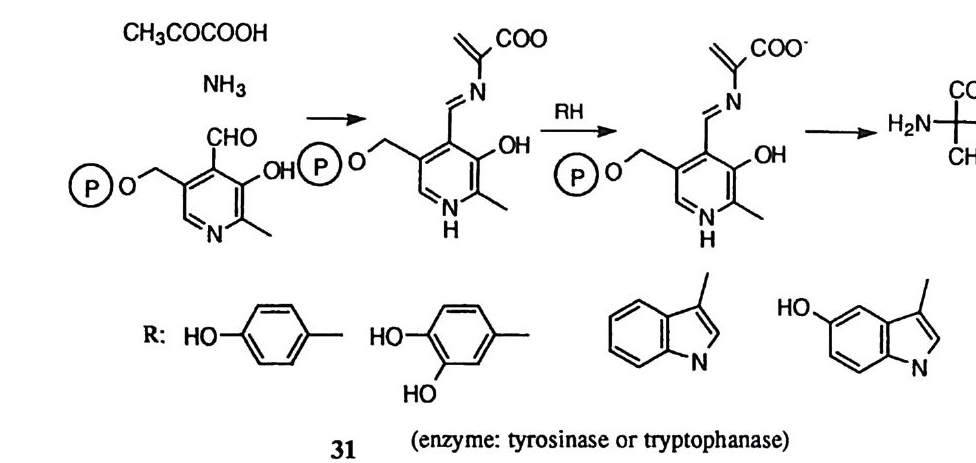 @oC 〈H3COCNOHO3 HO H 궁〈\c0O H0 言 〈\c0O H0 ·一 出 N 三
@oC 〈H3COCNOHO3 HO H 궁〈\c0O H0 言 〈\c0O H0 ·一 出 N 三
C= N 이중결합에 대한 첨가 반응도 효소의 영향 아래에서는 입체 선택적으 로 일어난다. 피루브산, 암모니아, 아렌 (RH) 을 조효소인 피리독살 포스페이 트와 함께 효소 촉매 반응에 따라 광학 활성인 아미노산으로 전환하는 반응 (31) 을 예로 들 수 있다. 35)
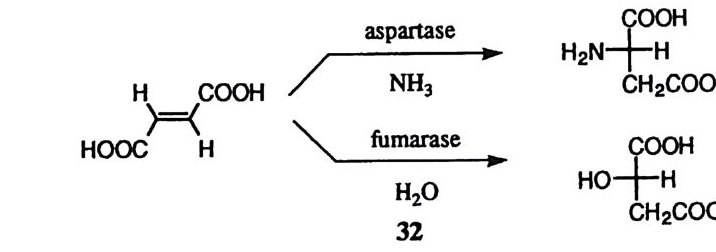 H>=
H>=
C= C 결합에 대하여 입체 선택성이 매우 높은 첨가 반응이 효소 촉매 존재 하에서 진행된다• 3~ 반응 예에 따라 아스파르트산 36) 과 말리산 37 ) 이 대량으로 재 조 되고있다.
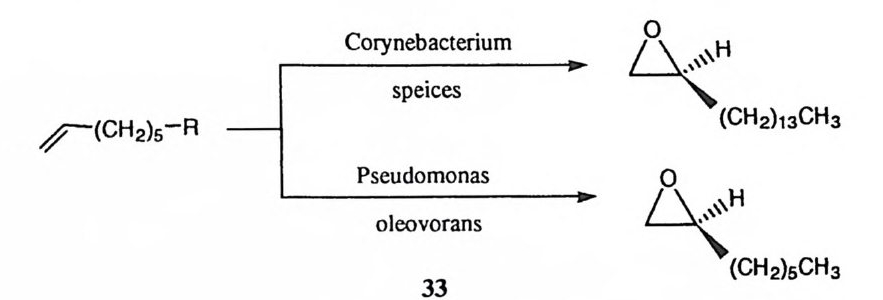 Cory n ebacte r i um 。
Cory n ebacte r i um 。
C= C 결합을 효소 를 사용하여 입체 선택적으로 에폭시화(예 : 33)38 벨· 수 있다. 샤프레스 에폭시화 방법 27 ) 이 알릴 알콜에 의하여 활성화된 알켄에 대하 여 효과적임에 비교하여 효소 반응은 비활성화 알켄도 어려움이 없이 입체 선 택적 으로 에폭시화하고 있다 .
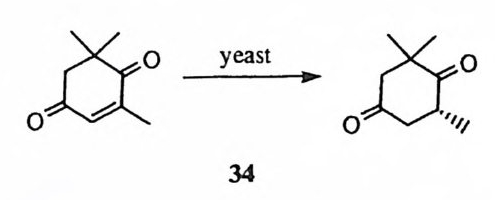 。ixo 二\D,,~
。ixo 二\D,,~
C=C 결합을 환원시키는 반응에도 효소가 촉매로 작용하여 입체 특이적으 로 두 개의 수소 원자를 첨가함으로써 광학 활성인 생성물을 형성 (예 : 34)39~ 다. 반응뭉게서는 두 위치가 동일하지만 각각이 수정되어 얻어진 두 가지의 생성 물은 서로 거울상 이성질체일 경우가 있다. 이 반응에서 효소를 촉매로 사용하 면 한 가지 생성물만 선택적으로 형성할 수 있다. 예를 들어 3 오 1 서 두 개의 동일한 히드록시기를 효소를 사용하여 산화함으로써 한 생성물만 선택적으로 얻어내고있다 .I)
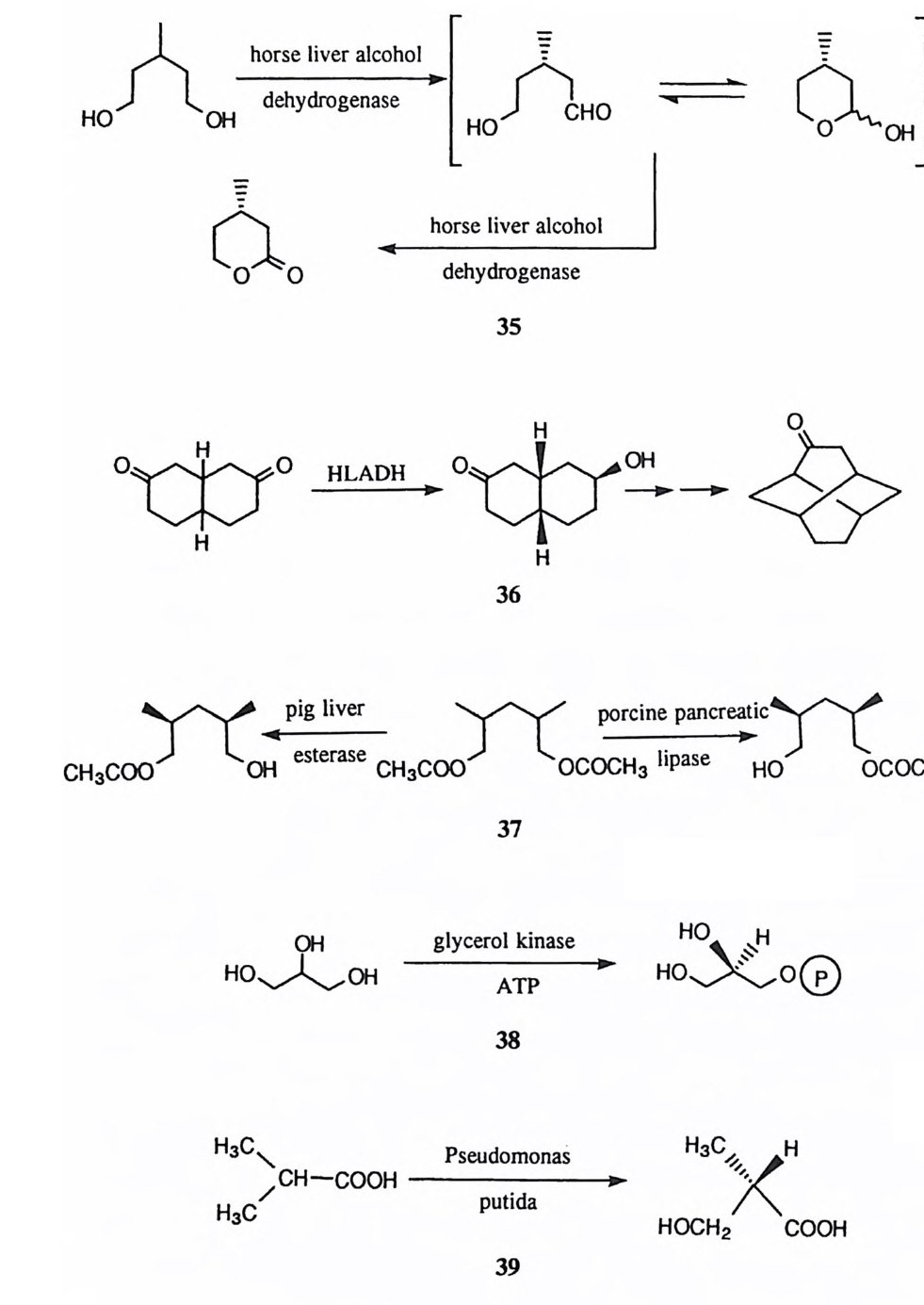 HO 占 OH hodr:y: : :::ehoI [ HO 占 HO ..... :OH]
HO 占 OH hodr:y: : :::ehoI [ HO 占 HO ..... :OH]
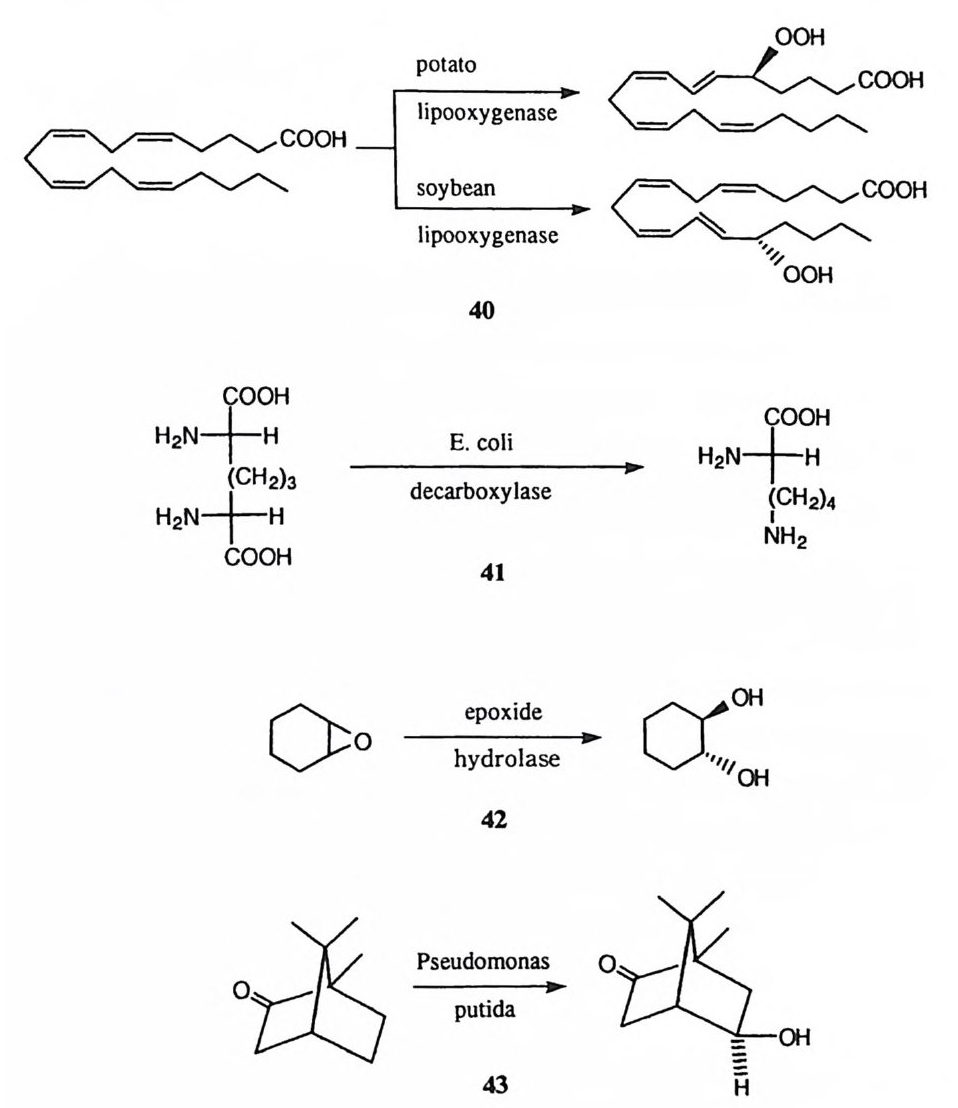 ’ pliop toa ot ox y ge nase 三C OOH
’ pliop toa ot ox y ge nase 三C OOH
디히드록시의 산화뿐 아니라 디케돈의 환원 (36) , 40) 디에스데르의 가수분해 (37) , 4 디히드록시의 포스포릴화 (38) ' 42) 디메틸의 히드록시화 (39) , 43) 디알케 닐의 퍼옥시화 (40),44) 디카르복실기의 탈카르복실화 (41),45) 에폭시드의 입체 선택적인 가수분해 (42)46) 에도 효소를 촉매로 사용하여 한 개의 반응 자리만 선 택적으로 반응시킬 수 있다.
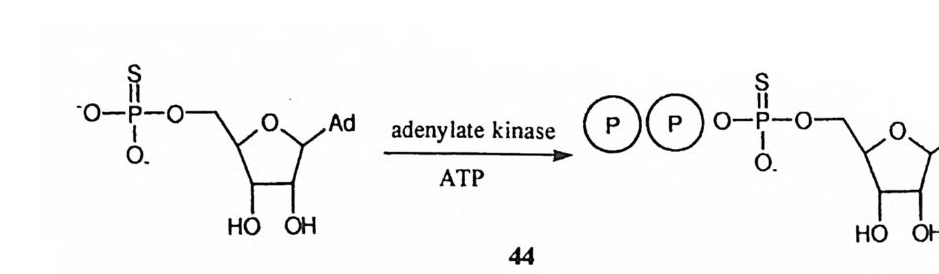 0\-O< Ad adeny la te kin a se OO 0\-0\〈 「 Ad
0\-O< Ad adeny la te kin a se OO 0\-0\〈 「 Ad
반응뭉게서는 두 위치가 동일하지만 각각이 수정되어 얻어진 두 가지의 생성 물은 서로 다이아스테레오머일 경우가 있다. 이 반응에서 효소를 촉 매로 사용 하면 한 가지 생성물만 선택적으로 형성할 수 있다. 예 를 들어 43ol] 서 는 환성 화되지 않은 메틸렌이 입체 선택적으로 히드록시화되며 , 47) ”에서는 인에 연결 된 산소 중 한 개만 선택적으로 디포스포릴화된다. 48) (4) 효소 특이성의 복합적인 이용 효소를 사용하여 비대칭 합성을 시도할 때에는 단일 단계 반응에서 효소의 특이적인 성질이 여러 가지가 복합적으로 조합될 수 있다. 예를 들어 4520) 에서 효소는 기질의 두 거울상 이성질체 중 한 가지만 수용하되 기질의 두 카르보닐 중 하나만 환원함으로써 두 가지 선택성을 복합적으로 시현한다.
 같 pdigeh lyiv der or ga el ncoahsoel 같H OH
같 pdigeh lyiv der or ga el ncoahsoel 같H OH
4649) 의 반응은 반응 자리에 대한 선택성과 거울상 이성질체에 대한 선택성이 조합되어 활용된 예이다.
 0 〉0 (C 바 )6COOCH3 uDniip n ou dc 4oles7au tsu s H。 ``` 。《니o` cCH 2 k~c 。 oc H3
0 〉0 (C 바 )6COOCH3 uDniip n ou dc 4oles7au tsu s H。 ``` 。《니o` cCH 2 k~c 。 oc H3
47 s o) 의 반응은 효소의 반응 자리에 대한 특이성과 이중결합의 면의 선택성이 조합되어 활용된 예이다. (5) 복수 효소의 이용 여러 개의 효소 를 순차적으로 사용하여 목적물을 합성하는 사례가 다수 보고 된 바 있다. 여기에서 관여 효소를 동시에 반응 용기에 집어넣고 합성할 수도 있고 고정화된 효소나 세포를 이용하여 연속 공정에 적용할 수도 있다. 예를 들어 4851) 의 반응에서는 프레드니솔론이 두 가지 효소의 연속적인 작용에 의하 여 합성되는데, 이 방법은 산업적으로도. 활용된다.
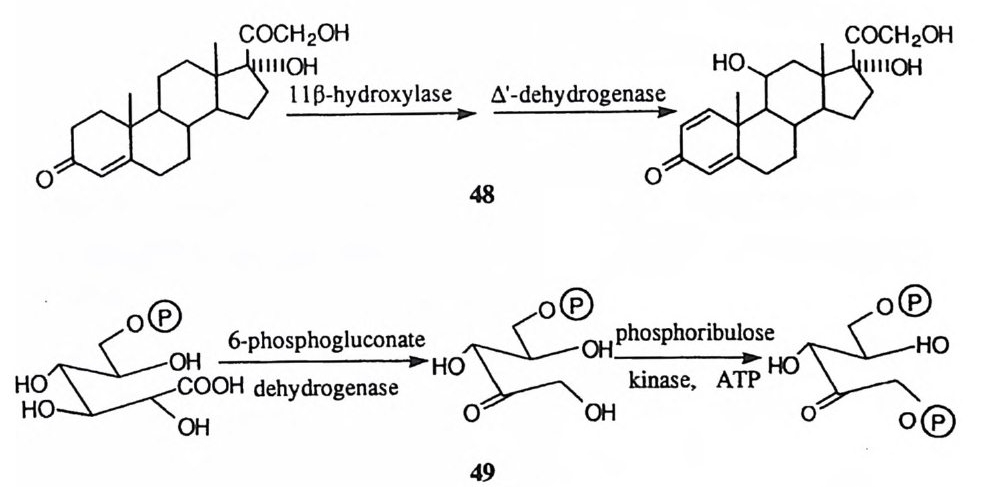 0:二 三三:십 OH
0:二 三三:십 OH
전통적인 유기 합성 방법으로는 당류에 포함된 작용기를 구별하여 선택성을 달성하기가 어렵지만 효소를 복합적으로 사용하면 용이하게 당류의 합성을 진 행할 수 있다. 예를 둘어 값이 비싼 리불로스 -1, 6- 디포스페이트를 복합적인
 HO:NH2 三 HO j〉° 二e 0?o®
HO:NH2 三 HO j〉° 二e 0?o®
효소 반응 (49) 52) 에 의하여 경제적으로 제조할 수 있다. 효소를 이용한 유기 합 성의 효과는 뉴클오레시드나 뉴클레오티드의 합성에서도 두드러지는데, 합성하 기가 어려운 항바이러스제인 D- 아라비노 뉴클레오시드 류의 합성 (50)53) 을 예 로들수있다. (6) 동위원소 라벨의 입체 특이적인 도입 효소의 입체 특이성을 이용하면 유기 화합물의 특정 위치에 동위원소를 도입 하는 것이 가능하다. 예를 들면, 알콜 데히드로게나아제를 사용하여 알데히드 롤 환원할 때 카르보닐기에 수소가 도입되는 입체 화학을 쉽게 예측할 수 있
 R YHo NYAADD2HH,. HRO X 、H 2H Rr3H 므NAD노H HOR\HH
R YHo NYAADD2HH,. HRO X 、H 2H Rr3H 므NAD노H HOR\HH
다. 54) 51 에서 보듯이 효모에서 얻은 효소 (YADH) 는 카르보닐기의 Re- 면에 琴 문]온을 첨가시킨다. 효소에 따라 입체 특이성이 다르기 때문에 5E] 서 와 갇이 효소를 바꾸거 사용함으로써 동위원소의 위치를 바꿀 수도 있다. 55)
 2H
2H
탈카르복실화 (53) ' 56) 일켄에 대한 첨가 (54) ' 57) 탈아실화 (55) 58) 반응 등을 이용하여서도 수소의 동위원소를 도입할 수 있다. 다효소계를 이용하여서 수소의 동위원소를 도입하는 예로 과당 유도체를 포 도당 유도체로 전환하는 반응 (56) ”딸 들 수 있다.
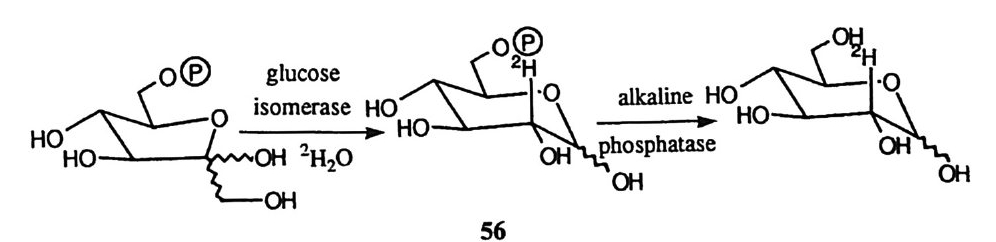 56
56
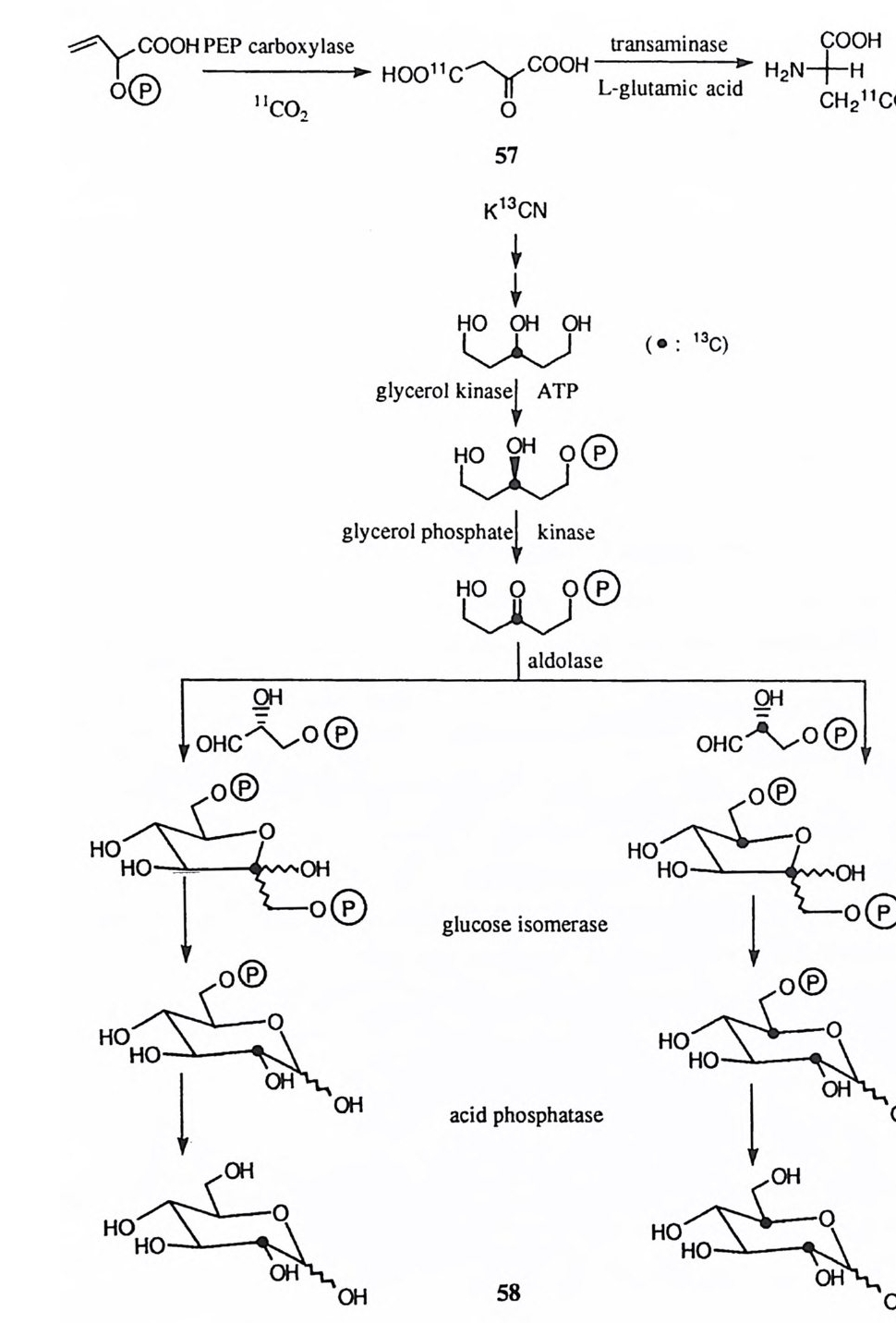 ~:OOH PEP carboxy la se tran sami na se COOH
~:OOH PEP carboxy la se tran sami na se COOH
효소를 사용하여 탄소 동위원소도 용이하게 도입할 수 있다. 반감기가 짧 은 11c 를 도입할 때에는 신속하게 합성 반응을 연결하여야 한다. 5760) 에 예로 든 효소 반응은 완결하는 데 15-2~ 냄뭐 소요되지 않는다. 효소를 사용하여 13C 를 도입한 반응의 예로 단당류의 특정 부분을 동위원소 로 라벨한 5838) 을 둘 수 있다.
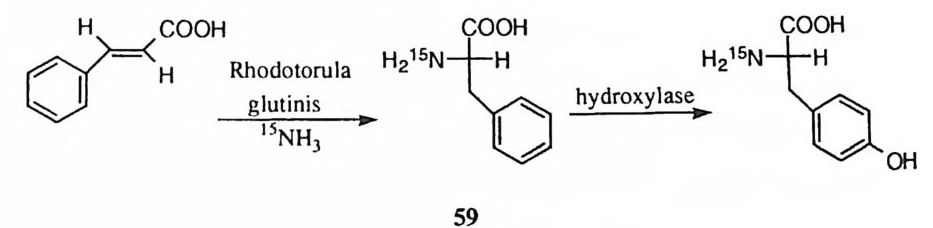 广 HCOOH二 간 N:-바 15N:OH
广 HCOOH二 간 N:-바 15N:OH
질소의 동위원소를 유기 화합물에 라벨하는 데에도 효소가 유용하게 서용된 다. 예를 들어, 신남산에 !SN- 암모니야를 첨가하여 ISN-L- 페닐알라닌을 만들 고 이를 히드록시화하여 “N-L- 티로신을 합성 (59)61 떨· 수 있는데, 이러한 반 응은 효소를 사용하지 않고서는 신속히 수행하는 것이 불가능하다. 이의에도 효소를 사용하여 산소의 동위원소, 인의 동위원소, 혹은 유황의 동 위원소 를 입체 특이적으로 도입한 사례가 다수 보고되어 있다. I) 제 2 절 효소와유기 용매 효소를 유기 합성에 사용할 때 가장 빈번하게 장애로 작용하여 유기화학자들 이 효소의 사용을 경원하게 하던 62) 것은 물을 전통적으로 용매로 사용하였다는 점이다. 유기 합성에서 관심을 끄는 화합물의 대부분은 물에 녹지 않으며, 때 로는 물이 원하지 않는 부반응을 일으키기도 하고 유기 합성용 시약을 파괴하 기도 할 뿐 아니라, 많은 반응게 대하여 열역학적인 평형이 물 속에서는 불리 하게 작용한다는 문제점이 있다. 최근 들어 유기 용매 속에서 효소가 활성을 상실하지 않는다는 점이 발견되
어 효소를 유기 용매에 현탁시킨 상태로 합성에 이용하는 연구가 활기를 띠고 있었다 .63)-66) 이때, 친수성인 유기 용매보다 협수성인 유기 용매를 사용하는 것이 바람직하며, 효소에 따라서는 건조된 유기 용매에 일부러 소량의 물을 첨 가해 주어야 하는 경우도 있고, 가루형 효소는 적정 pH.로 맞춘 효소 용액을 동결 건조함으로써 얻을 수 있고, 효소 가루의 입자 크기는 작을수록 좋으며, 반응 혼합물의 현탁액은 계속 저어주어 효소의 반응을 원활하게 하는 것이 필 요하다. 때로는 효소의 안정도가 수용액에서보다 유기 용매에서 더 높아지는 수도 있 고, 67) 용매를 바꿈에 따라 효소의 기질 특이성과 거울상 이성질체 선택성이 바 꾸어지기도 한다. 68)
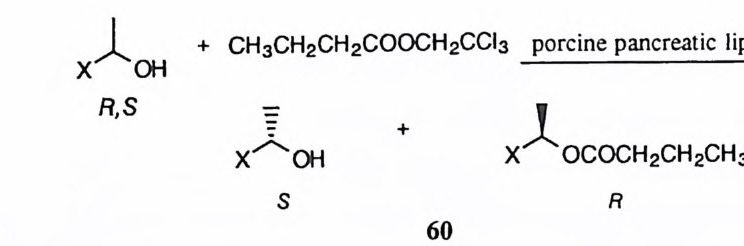 x J_ O__H . + CH3CH2CH2COOCH2CCl3 po rci ne pan creati c lipa se
x J_ O__H . + CH3CH2CH2COOCH2CCl3 po rci ne pan creati c lipa se
20- 팩 예에서 효소의 거울상 이성질 특이성을 이용하여 라세미 혼합물을 분할할 수 있음울 설명하였다. 이러한 작업에서도 위에 언급한 바와 같이 물을 용매로 사용하면 여러 가지 문제점이 뒤따르게 된다. 무수 에데르 혹은 헵탄 등의 유기 용매에서 리파아제를 사용하여 라세미 알콜을 트랜스에스데르화함으 로써 분할하는 방법 (60)69)은 이러한 문제점을 해소할 수 있는 예로 들 수 있 다. 트랜스에스데르화 반응에서 화학 평형을 생성물 쪽으로 완전히 치우치게 하
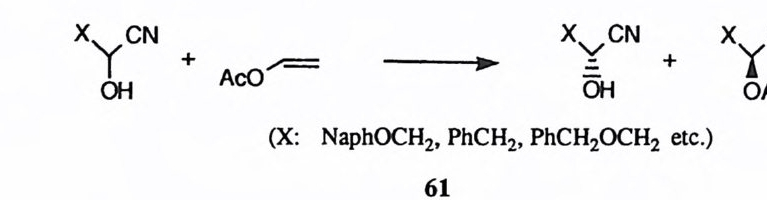 X CN X CN X CN
X CN X CN X CN
여 비가역적인 반응으로 전환시키는 방법으로 자주 사용되는 기법으로는 엔올 에스테르를 기질로 사용 (61)70) 하는 것이 있다. 엔을이 이탈기로 떨어져 나오면 알데히드 혹은 케톤으로 전환되어 버리기 때문에 엔을이 알콜로서 역반응을 일 으킬수없는것이다. 주진 효소에 대하여서도 용매를 바꿀 때 거울상 이성질체 선택성이 변화하 는 사례가 많이 보고되고 있기 때문에 효소의 선택뿐 아니라 용매의 선택에 따 라서도 라세미 혼합물의 광학 분할을 조절할 수 있다.
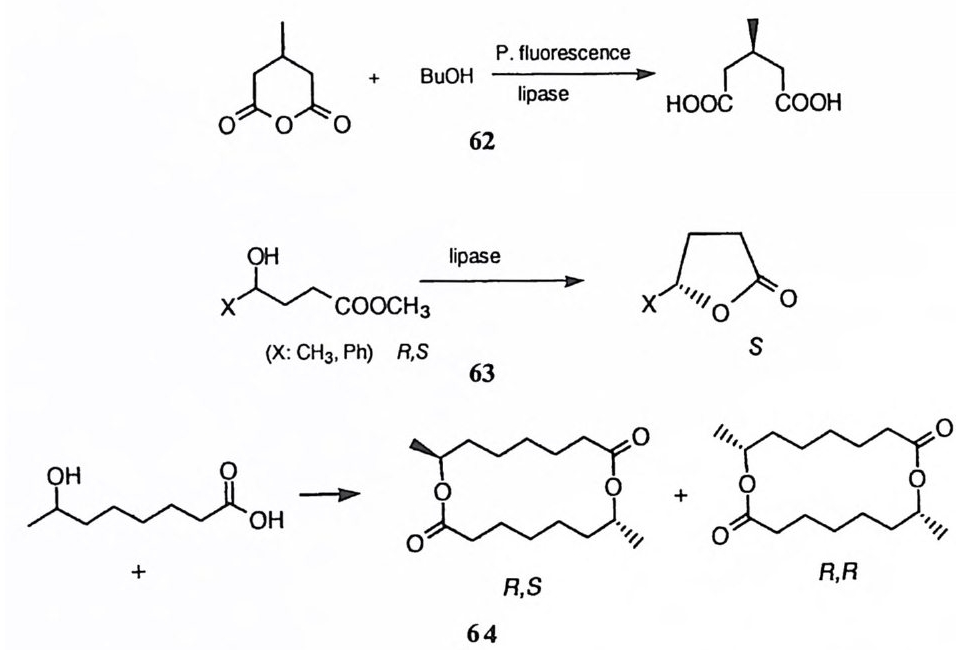 01 )0 + Bu0H62드 도 ooc占 OOH
01 )0 + Bu0H62드 도 ooc占 OOH
효소의 프로키랄적 입체 특이성을 이용하는 유기 합성 반응도 유기 용매 속 에서 진행시킨 사례 (62)71 끼}보고되고 있다. 리파아제를 이용하여 락돈화 반응 혹은 분자 간 에스데르화에 의한 거대 고 리 락돈의 형성을 유기 용매 ((63)72 본 에데르, (64)73)-i: -이 소옥탄)에서 수행한 예도보고된바있다. 유기 화합물이 여러 개의 작용기를 가지고 있을 때 효소는 한 작용기하고만 선택적으로 반응할 수 있음을 앞서 논의한 바 있다. 유기 용매 속에서도 이러
 OH ~ subti lisin
OH ~ subti lisin
한 효소의 특성을 유기 합성에 이용한 사례가 보고되어 있다. 6574) 와 같이 5a- 안드로스탄 -3 /3, 17 /3-디올은 트리플루오로에틸 부티레이트로 아실화할 수 있 는데, 무수 아세톤을 용매로 하여 섭티리신과 리파아제를 사용하면 각기 다른 히드록시기만 선택적으로 아실화할 수 있다. 6675) 에 예를 든 바와 갇이 탄수화 ½'ll 포 함된 히드록시기 중 하나만을 선택적으로 아실화하는 반응도 유기 용매 에서 수행할 수 있다. 탄수화물은 제한된 수의 천수성 유기 용매에만 녹는다는 제약이 있는데, 이 반응은 피리딘을 용매로 사용하였다. 제 3 절 효소의 가공 효소를 이용한 바이오 데크놀로지가 매우 발달하여 산업적인 응용이 발전함 에 따라 효소의 성질을 개선하는 일이 중요해지고 있다. 효소의 반응성과 특이 성뿐만 아니라 열에 대한 안정성, 격렬한 반응조건에 대한 저항성, 공정상의 간편성 등을 개선하려는 노력에서 효소를 여러 가지 방법으로. 가공하게 되었 다. 효소의 고정화는 효소의 안정성을 제고하며 공정상의 간편성을 확보하는 방 안으로 가장 자주 이용된다. 76) 각종 유기 혹은 무기 불용성 고분자에 포함된
작용기에 효소의 작용기를 화학적인 방법을 서용하여 연결함으로써 불용성 고 분자에 고정화된 효소를 얻을 수 있다. 이러한 고정화 효소는 열적 안정성이 높아지고 공정에 두입된 후 여과에 의하여 용이하게 회수할 수 있기 때문에 산 업적으로 많이 응용되고 있다. 화학적인 수정에 의하여 효소를 변형시킴으로써 효소의 성질을 개선할 수도 있다. 예를 들어, 아조벤젠기와 같이 빛에 민감하게 대응하여 구조 변화(아조벤 젠의 경우 트란스형과 시스형의 상호 변환)를 일으키는 작용기를 부착함으로써 효 소 활성을 빛에 의하여 제어하는 광감응성 효소를 얻기도 한다. 77) 유전자 조작에 의하여 효소의 특정 부위의 아미노산을 교체하는 단백질 공학 적인 기법에 의하여 효소를 가공함으로써 효소의 성질을 변화시키는 연구가 활 발히 진행되고 있다. 이 방법으로 효소의 열적 안정성 혹은 유기 용매에 대한 안정성을 제고한 사례도 다수 보고되고 있다. 78)-81 ) 강력한 용해력을 가지고 있 어 유기 합성에 매우 유용하게 사용되는 디메틸포름아미드 (DMF) 와 같은 친수 성 유기 용매는 효소로부터 물을 빼앗음으로써 효소의 삼처원 구조를 불안정하 게 만들어 효소의 활성을 파괴한다. 단지 섭티리신과 프로데아제 N 만이 DMF 에서 활성을 가짐이 알려져 있다. 82) 이들 효소도 안정성이 낮아서 섭티리신 BPN 은 DMF 에서 반감기가 2~ 예 불과하다. 그런데 단백질 공학에 의하여 얻은 섭티리신 변종 중에 물에서의 안정도가 수백 배 증가하고 DMF 에서의 안 정도가 50 배 중가한 사례가 보고되어 있다. 82)
 (PA A-ChT) PEI-ChT
(PA A-ChT) PEI-ChT
효소의 안정도는 삼차원 구조가 풀리는 것을 억제함으로써 개선할 수 있을 것이다. 이러한 목적으로 효소의 여러 부분을 한 개의 가용성 지지체에 연결하
여 열 안정성과 변성 시약에 대한 안정성 을 괄목하게 제 고할 수 있다. 83),8 4 ) 폴 리알릴아민 (PAA) 은 분자량이 약 10 만인 선형 고분자이고 폴리에 틸 렌아민 (PEI) 은 분자량이 硏 Rl 둥근 고분자이다. 키모트립신을 PAA 에 붙이거나 PEI 에 붙임 (67 : 큰 타원은 키모트립신, 검은 작은 타원은 활 성자리 를 표시함)으로 써 열 (50 . C) 에 대한 안정도와 변성제에 대한 안정도가 크게 증가하였다.
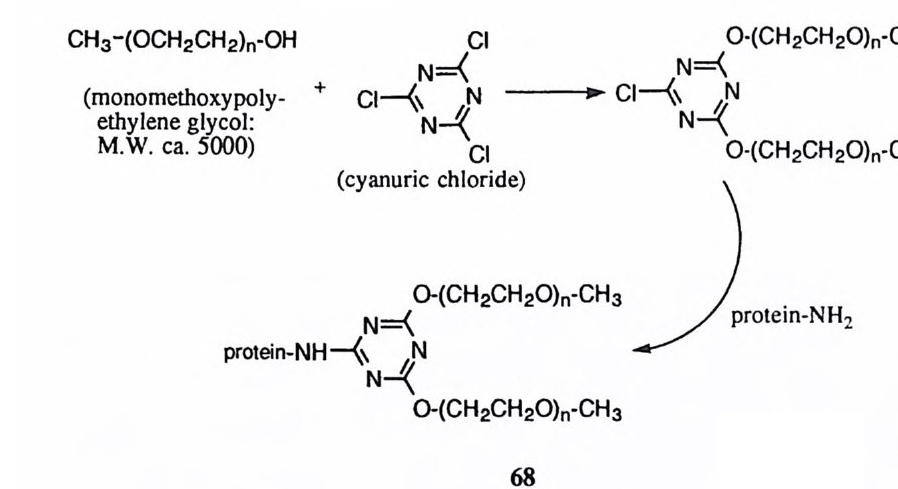 CH3-(0CH 후 )0-0H Cl O-(CH2CH20)0-CH3
CH3-(0CH 후 )0-0H Cl O-(CH2CH20)0-CH3
효소를 화학적으로 수정함으로써 유기 용매에 녹게 만든 사례도 보고된 바 있다. 트리아전 고리의 두 탄소에 분자량이 약 5000 정도 되는 폴리에틸렌글리 콜을 각기 부착한 다음 나머지 한 탄소에 단백질의 일급 아민을 연결 (68) 85 ), 8 6 ) 시켜 단백질을 유기 용매에 녹게끔 만든 경우가 알려져 있다. 이 방법으로 카 탈라아제, 퍼옥시아제, 콜레스테롤옥시다아제, 프로데아제, 리파아제 등이 수 정되어 유기 용매에서의 활성도가 검토되었다. 67 의 PAA-ChT 의 경우에도 PAA 의 아민기를 로로일화 (CuH 2 aCO- )하면 톨루엔에 녹게 된다 .87) 앞으로 유 기 용매에 녹는 효소를 이용함으로써 유기 용매 속에서 효소에 의한 균질상 촉 매작용이 상당한 관십을 끌 것으로 전망된다.
참고문현 1) Jon es, J . B. Te tr a h ed r on, 1986, 42 , 3351. 2) Klib a nov, A. M. ; Berman, Z. ; Alberti, B. N. J. Am. Chem. Soc., 1981, 103, 6263. 3) Ballard, D. G. ; Court is, A . ; Shir le y I. M. ; Tayl o r, S. C. J. C hem. Soc. Chem. Co mm., 1983, 954. 4) As hl ey , P. L. ; Griff in, B. W. Arch. Bio c hem. Bio p h y s ., 1981, 210, 167. 5) Booth ro y d, B. ; Nap ier , E. J. ; Somerf iel d, G. A. Bio c lzem. J, 1961, 80, 34. 6) Carrea, G. ; Bovara, R. ; Cremonesi, P. ; Lodi, R. Bio te c hnol. Bio e ng ng . , 1984, 26, 560. 7) Lin, Y. Y. ; Jon es, J. B . J. Org. Clze m., 1973, 38, 3575. 8) Lang er lof, E. ; Nath o rst- W estf ield t, L. ; Ekstr o m, B. ; Sjo b erg, B. Meth . EnZJ 'l n. , 1976, 44, 759. 9) Konecry , J. ; Sie b er, M . Bio te c hnol. B io e ng ng . , 1980, 22, 2013. 10) Markussen, J. ; Schaumberg, K. Pe ptide s, 1982, 387. 11) Oy am a, K. ; Nis h im ura, S. ; Nonaka, Y. ; Ki ha ra, Y. ; Hashim oto , T. ]. Org. Che m., 1981, 46, 5241 . 12) Fenselau, C. ; Palante , S. ; Batz inge r, R. P. ; Benson, W. R. ; Barron, R. P. ; Shein i n , E. B. ; Maie n th a l, M. Sci. e>1 ce, 1977, 198, 625. 13) Gross, A. ; Abril, 0. ; Lew is, J. M . ; Geresh, S. ; Whites id e s, G. M. ]. Am. Chem . Soc., 1983, 105, 7428. 14) Ladner, W. E. ; Whit es id e s, G . M. ]. Org. Chem., 1985, 50, 1076. 15) Tonooka, S. ; Sekik a wa, I. ; Azuma, I. Chem. Lett. , 1983, 805. 16) Mori sa wa, H. ; Uta ga wa, T. ; Mi jos h i, T. ; Yosh ina ga , F. ; Yamazaki , A. ; Mi tsu g i, K. Tet. L ett. , 1980, 21, 479. 17) Para, G. ; Fai, S. R. ; Baratt i, J . Bio t e c hnol. Lett. , 1980, 22, 2045. 18) Gurne, D. ; Shem in, D. Meth . Enzym ., 1976, 44, 844. 19) Yamamoto , K. ; Tosa, T. ; Ch iba ta , I. Bio te c hnol. B io engng., 1980, 22, 2045. 20) Jon es, J. B. ; Beck, J. F. Tech. Chem. (N. Y) , 1976, 10, 107.
21) Cohen, S. G. Trans. N Y. Acad. Sci., 1969, 3, 705. 22) Schneid e r, M. ; Eng el , N. ; Bonesmann, H. Ang ew . Chem . Int. E d. Eng l., 1984, 23, 64. 23) Aberneth y, J. L . ; Srulevit ch , D. ; Ordway, M. J. Jr. ]. Org. Chem., 1975, 40, 3445. 24) Mart ine k, K. ; Semenov, A. N. Bio c hem. Bio p lz y s. A cta ., 1981, 658, 90. 25) Cambou, B. ; Klib a nov, A. M. J Am. Che m. Soc., 1984, 106, 2687. 26) Inada, Y. ; Nis h im ura, H. ; Takahashi, K. ; Yoshim oto , T. ; Saha, A. R. ; Sait o, Y. Bio c lzem. Bio p h y s . Res. Commun ., 1984, 122, 845. 27) Kats u ki, T. ; Sharp le ss, K. B. J Am. Chem. Soc., 1980, 102, 5974. 28) Ladner, W. E. ; Whit es id e s, G . M. ]. A m. Chem. Soc., 1984, 106, 7250. 29) Hecht, S. M. ; Rup pre cht, K. M. ; Jac obs, P. M . ]. Am. Che m. Soc., 1984, 101, 3982. 30) Sih , C. J. ; Chen, C.-S. Ang ew . Chem . Int. Ed. E ng l. , 1 984, 23, 570. 31) Becker, W. ; Pfe il, E. ]. Am. Chem. Soc., 1966, 88, 4299. 32) Fug an ti , C. ; Grasselli , P. ; Servi, S. ; Spr e afi ca , F. ; Zir o tt i, C. ; Casati, P. ]. Org. Chem., 1984, 49, 4089. 33) Wong , C.-H . ; Whites id e s, G. M. ]. Org. Chem. , 1983, 48, 3199. 34) Wong, C.-H. Sci en ce, 1989, 244, 1145. 35) Fuku i, S. ; Ikeda, S. ; Fuji m ura, M. ; Yamada, H. ; Kumag ai , H. Eur. ]. Bio c hem., 1975, 51. 155. 36) Ch iba ta , I. ; Tosa, T. ; Sato , T. Meth . Enzym . , 1 976, 44, 739. 37) Yamamoto , K. ; Tosa, T. ; Yamash ita, K. ; Chib a ta , I. Eur. ]. Ap pl. Mi - crobio l. , 1976, 3, 169. 38) May, S. W. ; Ste l te n kamp , M. S. ; Schwart z, R. D. ; McCoy, C. J. ]. Am. Chem. Soc., 1976, 98, 7856. 39) Sim o n, H. ; Gunth e r, H. ; Bader, J. ; Tis c her, W. Ange w . Chem. Int. Ed. En gl., 1981, 20, 861 . 40) Dodds, D. R. ; Jon es, J. B. ]. Chem. Soc. Chem. Commun., 1982, 1080. 41) Wang, Y.-F. ; Chen, C.-S. ; Gir d aukas, G. ; Sih , C. J. ]. Am. Chem. Soc., 1984, 106, 3695.
42) Rio s -M ercadil lo , V. M. ; Whit es id e s, G. M. J Am. Chem. Soc., 1 979, 101, 5828. 43) Haseg aw a, J. ; Og ura , M. ; Kanema, H. ; Noda, N. ; Kawaharada, H. ; Wata n abe, K. ]. Ferment. Tee/ m ot., 1982, 60, 501 . 44) Corey, E. J. ; Lansbury , P. T. ]. Am. Cliem . Soc., 1983, 105, 4093. 45) Kanemi tsu, 0. Jap a n Pate n t, 1979, 75132179. 46) Annstr o ng , R. N. ; Levin , W. ; Jer in a , D. M. ]. Bio l. Chem., 1980, 255, 4698. 47) Yu, C.-A . ; Gunsalus, I. C. Bio c he m . Bio p h y s . Res. Commun., 1970, 40, 1431 . 48) Moran, J. R. ; Whit es id e s, G. M. ]. Org. Chem., 1984. 49, 704. 49) Ir.v in, A. J. ; Jon es. J.B . ]. Am. Che m. Soc., 1977, 99, 1625. 50) Sih , C. J. ; Heath e r, J. B. ; Peruzott y, G. P. ; Pric e, P. ; Sood, R. ; Hsu Lee, L.-F . ]. Am. Che m . Soc., 1973, 95, 1676. 51) Bih a ri, V. ; Goswami , P. P. ; Riz v i, S. H . M. ; Khan, A. W. ; Basa, S. K. ; Vora, V. C. ]. Ap pl. Mi cro bia l. Bio te c hnol., 1984, 26, 1403. 52) Sue, J. M. ; Knowles, J. R. ; Whit es id e s, G. M. Meth . Enzym ., 1982, 89, 108. 53) Krenit sk y, T. A. ; Koszalka, G. W. ; Tutt le, J. V. ; Rid e out, J. L. ; Eli on , G. B. Carbohyd . Res., 1981, 97, 139. 54) Prelog, V. Pz,re Ap pl. Clze ,n. , 1964, 9, 119. 55) Arig o ni, D. ; Luth y , J. ; Rete y , J. Natz tre, 1969, 221, 1213. 56) Belleau, B. ; Burba, J. J Am. Cliem . Soc., 1960, 82, 5751 . 57) Fie ld , S. J. ; Young , D. W. ]. Chem. Soc. Chem. Commun., 1979, 1190. 58) Batt er sby, A. R. ; Nic ol ett i, M. ; Sta u nto n , J. ; Vie g g e r, R. ]. Chem. Soc. Perkin Trans., 1, 1980, 43. 59) Sn ipes , C. E. ; Chang, C.-J. ; Gloss, H. G. ]. Am. Chem. Soc., 1979, 101, 701 . 60) Barr io, J. R. ; Eg be rt , J. E. ; Henze, E. ; Schelbert, H. R. ; Baum ga rt ner , F. J. ]. Med. Chem., 1982, 25, 93. 61) Sto c klein , W. ; Eis g r ub er, A. ; Schm idt, H.-L. Bio t e c hnol. Lett. , 1983, 5, 703.
62) Akiy am a, A. ; Bednarski, M . ; 斷 m , M.-J. ; Sim o n, E. S. ; Waldman, H. ; \\lni te s id e s, G. M. Che1nte c h., 1988, 18, 627. 63) Klib a nov, A. M. Chemt ec/ 1 ., 1988, 16, 354. 64) Halli ng , P. J. Bio te c hnol. A dv., 1987, 5, 47. 65) Dordic k , J. S. Enzym e Mi cro b. Tec h nol., 1989, 11, 194. 66) Klib a nov, A. M. Acc. Chem . Res., 1990, 23, 114. 67) Zaks, A. ; Klib a nov, A. M. Sci en ce, 1 984, 224, 1249. 68) 距t a gu ch i , H. ; Fit zpa tr ick , P. A . ; Huber, J. E . ; Klib a nov, A. M. ]. Am. Cl 奭. Soc., 1988, 110, 7236. 69) 距 rchner, G. ; Scollar, M. P. ; Klib a nov, A. M. ]. Am. Cli c11 1 . Soc. , 1985, 107, 7072. 70) Degu e il- C asta n g , M. ; DeJe s co, B. ; Brouil lar d, S. ; Mail lar d, B. Tet . L et t. , 1987, 28, 953. 71) Yamamoto , K. ; Nis h io k a, T. ; Oda, J. ; Yamamoto , Y. Tet . L et t. , 1988, 29, 1717. 72) Gutm a n, A. L. ; Zoubi, K . ; Bolta n ski, A . Tet . L e tt. , 1987, 28, 3861 . 73) Ng oo i, T . K. ; Sci lima ti , A . ; Guo, Z.-W . ; Sih , C. J. ]. Org . Chem . , 1 989, 54, 911 . 74) Riva , S. ; Klib a nov, A. M. ]. Am. Clzem. Soc., 1988, 110, 3291 . 75) Theri so d, M. ; Klib a nov, AM. ]. Am. Chem. Soc., 1986, 108, 5638. 76) Mosback, K. Ed. Metl w ds in Enzym olo gy , Academ ic Press ; New York, 1987, Vol. 135-137. 77) Wi ln er, I. ; Rubin , S. ; Rikl in , A. ]. Am. Chem. Soc., 1991, 113, 3321 . 78) Mats w nara, M. ; Yaswnara, S. ; Aiba , S. Natu re , 1986, 323, 356. 79) Imanaka, T. ; Sh ibr aya s h i, M. ; Takagi , M. Natu re , 1986, 324, 695. 80) Bry o n, P. N. ; Rollence, M. L. ; Panto l ia n o, M. W. ; Wood, J. ; Fin z el, B. C. ; Gil lilan d, G. L. ; Hamond, A. J. ; Panlos, T. L. Prote ins , 1986, 1, 326. 81) Wong, C.-H. ; Chen, S.- T . ; Hennen, W. J. ; Bib b s, J. A. ; Wang, Y.-F. ; Liu, ]. L.-C. ; Panto l ia n o, M. W. ; Whit low , M. ; Bry a n, P. ]. Am. Chem. Soc., 1990, 112, 945. 82) Wong, C.-H. Sci enc e,1 9 89, 244, 1145.
83) Suh, J. ; Hwang , B. K. Bio o rg. Chem., 1992, 20, 223. 84) Suh, J. ; Cho, Y. ; Kwang, G. Bio o rg. Chem., 1992, 20, 236. 85) Takahashi, K. ; Aji m a, A. ; Yoshim oto , T. ; Inada, Y. Bio c hem. Bio ph ys . Res. Commun., 1 984, 121, 261 . 86) Inada, Y. ; Mac;s u shi m a, A. ; Takahashi, K. ; Sait o, Y. Bio c ata ly si s , 1990, 3, 317. 87) Suh, J. ; Park, H. S. to be pu blis h ed.
제 8 장 항체효소 제 1 절 서론 효율적이고 고도의 선택성을 가진 촉매의 개발은 화학 전반에 걸쳐 중요한 위치를 차지하고 있다. 효소는 이러한 점을 충족하는 생체 촉 매이다. 그러나 효소는 생체 조건하에서 생명현상의 원인이 되는 화학반응에 대해서만 촉매로 작용하므로 제한된 종류의 반응에만 관여한다는 한계성을 가지고 있다. 이러한 점을 개선하여 생체 반응이 아닌 다른 어떠한 화학반응에 있어서도 효율적이며 고도의 선택성을 가진 생체 촉매를 개벌玲}려는 노력이 몇 가지 방향에서 진행 되고있다. 첫째로는 단백질 공학에 의한 변형 효소의 개발이다. I) 이 방법은 유전공학 기술을 사용하여 원하는 부분의 아미노산을 교체시켜서 새로운 변형 효소를 만 드는 작업이다. 둘째로는 혼성 효소이다. 2) 예를 들어, 슐츠에 의해 제작된 혼 성 효소는 효소에 올리고뉴클레오티드를 공유 결합으로 결합하여 RNA 나 DNA 에 결합 부위를 제공하여 특정한 위치를 절단하는 새로운 효소이다. 셋째 로는 항원을 설계하여 특정한 반응게 촉매로서 작용할 수 있는 항체 효소를 개 발하는 방법이다 .3)-27) 항체 효소는 화학의 각 영역에서 광범위한 잠재 응용도 를 보유하고 있는데, 1980 년대 중반부터 연구 결과가 발표되기 시작한 매우 새 로운 학문분야이다. 이 칭에서는 이중 항체 효소에 의한 촉매작용을· 소개하고 자한다•
제 2 절 항원과항체 항체는 척추동물의 면역계에 존재하는 B 림프구에 의해서 생산 분비되는 단 백질로 면역 글로불린 (lg) 이라 통칭된다. 항체는 각각 어떤 항원에 득이적으로 대응하는바, 수백만 종류의 다른 분자형을 갖는 분자의 집단이 생체에 존재하 는 것이다. 기본적으로 항체는 그림 1 처럼 동일한 업 H 의 L 사슬(아미노산 약 22 어로 구성됨)과 동일한 양 H 의 H 사슬(아미노산 약 4407» 로 구성됨), 도합 47H 의 폴리펩티드 사슬로 구성된 분자량 약 15 만의 단백질이며 기능적으로 독립된 몇 개의 부위로 구성되어 있다.
 가변부위 /.、 ^/ NH3+N H3+
가변부위 /.、 ^/ NH3+N H3+
항체의 L 및 H 사슬의 말단 쪽으로 각각 약 ll Q7 H 의 아미노산으로 구성된 가변 영역이라고 불리는 부위가 존재하고 이 부분의 아미노산 배열이 개개의 I g를 특징짓는다. 그리고 이 부분이 항원과의 특이적인 결합에 관계하고 있다. 따라서 하나의 항체 분자는 L 및 H 사슬 말단 양쪽에 의해서 형성되는 동일한 항원 결합 부위를 엄 H 가지고 있다. 한편 항체의 그 의 다른 영역은 항원과의 결합에는 직접 관여하고 있지 않다. 그리고 실제로 항원과의 결합에 직접 관계
하고 있는 부위는 가변 영역 중에서도 일부분으로, 3 - 4 개의 글루코오스 단위를 수용하는 정도의 크기를 갖는 아미노산 5-1 ()7 H 로 된 초가변 영역에 의해서 형 성되어 있다. 일반적으로 저분자 화합물을 항원으로 동물에 주사하면 면역 응답을 일으키 지 못한다. 일련의 면역 응답을 일으켜 특이적인 항체를 생산하는 림프구가 유 도, 분화되기 위해서는 상당히 큰 항원이 필요하다. 그래서 대상으로 하는 저 분자 화합물을 소의 혈청 알부민 등과 갇은 단백질에 공유 결합으로 연결하여 면역 응답을 유도한다. 이와 같이 단독으로는 면역원성을 갖지 못하나 적당한 고분자 화합물과 결합함으로써 면역원성을 갖게 되는 비교적 작은 크기의 화합 물을합텐이라한다. 항체는 통상 여러 항체 생산 림프구의 집단(이것을 〈 폴리클론 〉 이라고 한다)에 의해서 생산 분비되는 다종 다양한 분자로 구성된 집단으로 존재하고 있으나 개개의 항체 생산 임파구가 생산하는 l &i즌 단 한 종류로 단일한 분자이다. 따 라서 단 한 종류의 항체 생산 림프구(이것을 〈 모노클론 〉 이라고 한다)를 여러 항체 생산 림프구의 집단으로부터 선별하여 한 종류의 항원 결정기만 결합하는 모노 클론 항체를 얻을 수 있다. 19751d 에 증식 능력을 거의 갖지 않는 항체 생산 세포와 무한한 증식 능력을 갖고 있는 골수 세포를 융합시킴으로써 보다 안정 하고 무한한 증식 능력을 가진 항체 분비성의 하이브리도마 세포를 제작하는 방법이 개발되었다. 이후 현재에 이르기까지 모노클론 항체의 제작 기술이 확 립되어 원리적으로는 거의 모든 분자에 대하여 각각 특이적인 모노클론 항체를 만들수 있게 되었다 .28) 제 3 절 과거의 관련 연구보고 어떤 분자에 특이적으로 작용하는 항체가 그 분자의 화학반응에 미치는 영향 을 검사하기 위하여 1 을 항원으로 사용하여 토끼로부터 얻은 폴리클론 항체가 p-니트로 페닐 아세데이트 등 E} 유사한 구조를 가진 에스테르의 가수분해 속 도에 끼치는 효과를 조사한 결과가 1966 \1 발표되었다. 29) 그 결과 이 항체는
 5`o °<<
5`o °<<
면역되지 않은 토끼의 혈청에서 얻은 통상적인 l g와 비교할 때 에스데르의 가 수분해 를 오히려 저해한다는 결과를 얻었다. 이것은 이 항체의 항원 결합 부위 에는 반응에 관여할 수 있는 아미노산 잔기가 존재하지 않으며, 항원 결합 부 위가 일종의 보호 효과를 보여 에스데르가 가수분해를 받기 어렵게 된 것이라 고해석하였다. 그 후 스데로이드로부터 유도된 에스테르 E] 가수분해가 폴리클론 항체에 의해서 가속되는 것이 보고되었다. 즉 E] 가수분해는 이 항체에 의해서만 가 속되고 통상의 IgL -j-다 른 스테로이드로 면역되어 얻어전 l g에 의해서는 촉진되 지 않는 것이다. 또 이 항체는 다른 구조를 갖는 스테로이드의 에스테르 가수 분해에는 영향울 주지 않는 등의 특이성을 나타낸다는 것이 보고된 바 있다. 30) 그러나 이 촉매 반응은 화학양론적이어서 가해 준 항체의 당량에 상당하는 에 스데르가 가수분해된 이후에 반응은 정지해 버렸다. 결국 기질인 에스테르가 항체의 항원 결합 부위에 존재하는 친핵성 잔기에 접근하여 이것을 변형시키 고, 생성물에 의한 저해 작용 때문에 항체는 당량의 기질과 반응하며, 진짜 촉 매로서의 역할을 수행하지 못한 것이다. 이상의 연구는 어떤 분자의 화학반응 이 그 분자에 대한 항체의 항원 결합 부위에 의하여 가속될 수 있으며 그 작용 은 기질에 특이하게 적용될 수 있다는 사실을 시사하고 있다. 제 4 절 항체 효소의 개발 사례 (|) 전이상태와 상호보완적인 항체 194 쩌 폴링은 효소의 활성자리는 기저상태의 기질이 아니라 그 반응의 전이
상태와 상보적인 구조를 가지고 있으며 효소는 전이상태와 결합하여 전이상태 를 안정화시켜 활성화 에너지를 감소시켜서 반응이 가속된다는 기설을 발표하 였다. 31) 또 쟁크스는 어떤 반응의 전이상태와 상보적인 구조 를 가지는 분자를 인공적으로 만들면 그 분자는 반응의 전이상태와 특이적으로 결합하여 전이상 태를 안정화시켜서 촉매의 기능을 수행할 것이라고 생각하였다. 또한 전이상태 와 상보적인 결합 부위를 갖는 항체는 촉매 기능을 가질 것이라고 제안하였 다. 32) 그러나 화학적으로 매우 불안정하여 분리할 수 없는 전이상태 를 사용하 여 동물에 면역시키는 방법이 가장 큰 문제였다.
 02N- 0-0o \ 戶 + N三 /’ N -0-:\°6'。- 。H근 + N l/, 7 :J:
02N- 0-0o \ 戶 + N三 /’ N -0-:\°6'。- 。H근 + N l/, 7 :J:
이 점은 전이상태 유사체를 사용함으로써 해결되었다. 전이상태 유사체란 그 구조나 전하의 분포 상태가 실제의 전이상태와 매우 비슷하지만 자체로서 안정 하게 존재하는 화합물을 말한다. 에스테르 묘] 가수분해 반응의 전이상태 유체 인 포스페이트 에스테르를 그 예로 들 수 있다. 이와 갇이 전이상태 유사체를 합텐으로 사용하여 얻은 모노클론 항체가 에스테르의 가수분해를 촉진한다는 사실이 거의 감은 시기에 슐츠와 러너에 의해서 보고되었다. 34) 슐츠등은 p-니트로페닐 포스포린 콜린 4oll 특이적으로 결합하는 항체로 이
미 알려져 있던 MOPC 167 항체를 가지고 4ol] 대응하는 카보네이트 에스테르 책 가수분해 반응에 대한 이 항체의 영향을 조사하였다. 그 결과 MOPC 167 항체는 꾹끌 특이적으로 가수분해하고 촉매가 없을 때보다 770 배 (vIg= kIg[ Ig- S ] [OH], Vo1 1=kou[S][OH])) 가속함을 보였다. 이 항체를 펩신 프로테아제로 처 리하여 항원 결합 부위를 포함하는 Fab 조각(그림 l) 만을 분리하여 실험한 결 과 본래의 항체 MOPC 167 과 같은 활성을 보였다. 또한 데트라니트로 메탄으 로 처리한 결과 활성이 완전히 소실된 것으로 보아 항원 결합 부위에 존재하는 티로신 잔기가 반응에 관여하고 있음을 시사했다.
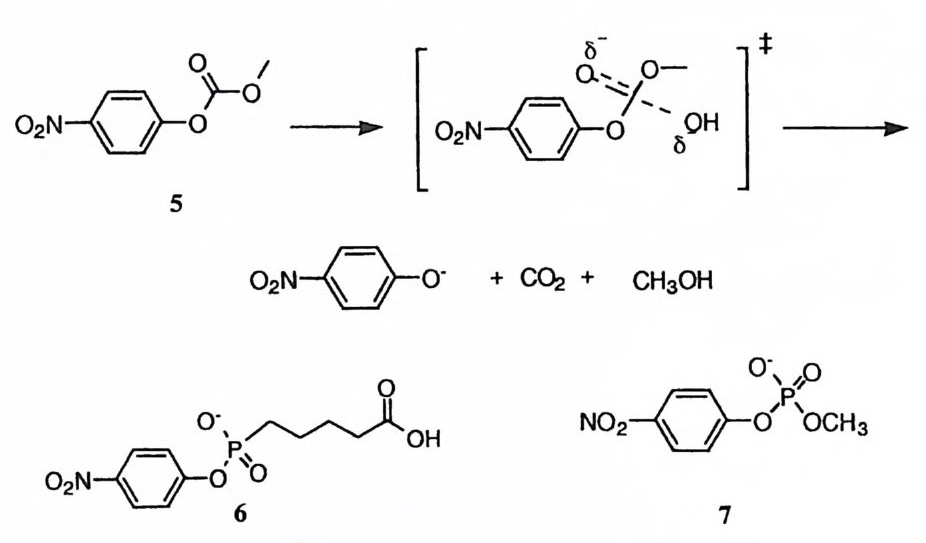 02N- 0-:\o1 一 [02N- d-:/?`~O H 一
02N- 0-:\o1 一 [02N- d-:/?`~O H 一
다음으로 그들은 카보네이트 에스데르 또 1 가수분해 반응에 전이상태 유사체 로서 같은 사면체 구조를 갖는 포스페이트 에스데르 qt 합성하여 그것을 합텐 으로 하여 얻어지는 모노클론 항체가 카보네이트 에스데르 허t 특이적으로 가 수분해하는 것을 보고하였다. 이 경우 항체 촉매 없이 반응했을 때와 비교하여 810 배 반응이 촉진되었다. 또한 실제의 전이상태와 매우 유사한 포스페이트 에 스테르 7 에 의해서 반응은 특이적으로 강력하게 저해되는 것이 밝혀졌다. 이상 은 항체가 기저상태의 기질 5가 아니라 전이상태에 대하여 상보적이며 가수분 해는 그 결합 부위의 작용에 의한 것임을 보여준다.
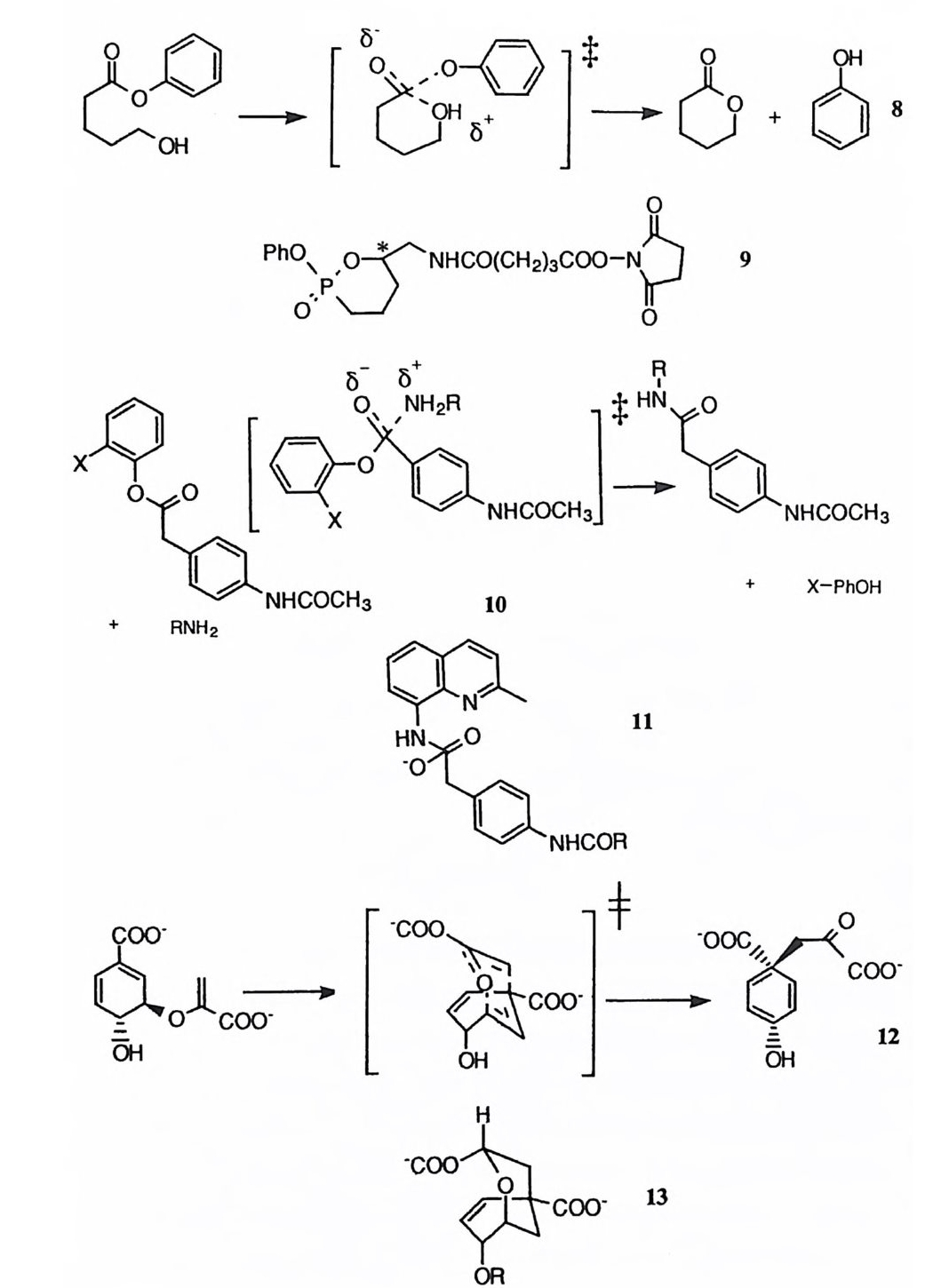 ::一[\;-P]〉 + 68
::一[\;-P]〉 + 68
지금까지 보고된 전이상태 유사체를 합텐으로 사용하여 얻은 항체를 사용한 촉매 반응의 예로 에스테르 (hea t/%야 I ; 10 드 105)4),6),8),27) 카보네이트 에스테르 (kco1/ k.i n 댜t ; 810)?) 및 아미드의 가수분해 (!«.1/k. inc ot ; 105), 15),23) 입체 특이성 락 본화 반응 (8) (硏 합텐임 : hea t/丸函〈 167)' S) 아미드 결합 형성 반응 (10) (11 이 합텐임 : kcnt /k.inc at : 102-105} , lOl,l4),lB) 클라이센 자리옮김 반응 (12) (lJo ] 합텐 임 : kcn t/k.i n 여t ; 10•) 9),11),19),20) 등을 들 수 있다. (2) 특정 촉매 작용기의 유도 효소와 같은 높은 촉매 효율을 보유하려면 단순히 전이상태와 상보적인 형태 를 제공할 뿐 아니라 각종 촉매 작용기를 반응 자리 주변에 위치하도록 유도하 여야 한다. 이에 따라 항체의 항원 결합 부위에 특정한 아미노산기를 도입하는 방법에 의하여 항체 촉매를 개발한 사례도 보고되어 있다.
 o F O o
o F O o
양이은을· 포함한 합텐 (14) 을 사용하여 유도한 항체 중에서 1~ fl-제거 반 응에 대하여 촉매로 작용하는 것이 분리되었다 (16).24) 이것은 합텐에 포함된 양이온에 인접한 위치에 상호 보완적으로 글루탐산 혹은 아스파르트산의 카르 복실 음이온이 항체 위에 유도되었으며, 이것이 제거 반응에 필요한 염기 역할 을 수행한 것으로 여겨진다. 자의선에 의하여 DNA 가 손상될 때 티민 두 개가 2+2 고리 첨가 반응을- 일
 hu .. 2 0人/r 17
hu .. 2 0人/r 17
으켜 이량체를 형성하게 된다. 이 경우에 생체내에서는 수선 효소가 존재하여 이량체를 원상태로 분해할 수 있다. 티민 이량체는 인돌, 퀴논, 혹은 풀라빈 등의 광감응제가 인접한 위치에 있으면 이들로부터 에너지를 전달받아서 단량 체로 변환될 수 있다. 티민 이량체가 가전 극성화된 1(계를 합텐으로 - 사용하여 항체를 유도하면 이에 대하여 상호 보완적으로 포갬 작용을· 할 수 있는 트립토 판이 항체의 가변 부위상에 위치한 아미노산으로. 유도될 수 있다. 이와 같은 방법으로 티민 이량체의 광분해 반응 (17) 을 촉매하는 항체가 얻어진바 ,20) 이 항체 효소의 활성자리에는 트립토판이 위치하여 광감응제 역할을 담당한다고 여겨진다. 합텐을 이용하여 특정 작용기를 항체 형성 과정에서 유도하는 방법 이의에도 항체를 화학적으로 수정하여 원하는 작용기를 도입할 수도 있다. 18ol] 예를 들
었듯이, 항체에 결합하는 리간드에 디술피드 결합을 포함시켜 항체에 연결한 다음 디술피드 결합의 티울을 손잡이로 사용하여 각종 작용기를 도입할 수 있 는 것이다. 22),25),33) (3) 유기 합성을 위한 항체 효소 유기 합성 반응에 유용한 항체 효소를 개발하게 되면 항체 효소의 용도를 대 폭 확대할 수 있다. 이러한 목적으로 상용될 수 있는 항체 효소의 예로 카르보 닐의 입체 선택적인 환원 (19, 20) 에 대한 촉매를 들 수 있다. 34),35) 케돈을 비대 칭적으로 환원하면 비대칭 유기 합성에 유용하게 사용할 수 있다. 비대칭 환원 은 한 광학 이성질체를 이것의 거울상보다 100 배만 더 빨리 형성되게 하여도 유기 합성상의 목적은 훌륭히 수행할 수 있다. 효소에 버금가는 촉매 능력을 달성하는 것이 목표라면 백만-억 배 이상의 가속 효과를 얻어야 하는 것에 비 하여 비대칭 합성에서는 단지 100 배의 가속만으로도 충분한 것이다. 항체 효소 를 촉매로 사용하면 이 정도의 가속 효과를 성취하는 것은 그다지 어려운 일이 아니므로, 비대칭 합성 분야에서의 항체 효소의 응용 전망은 밝은 편이라 할 수있다.
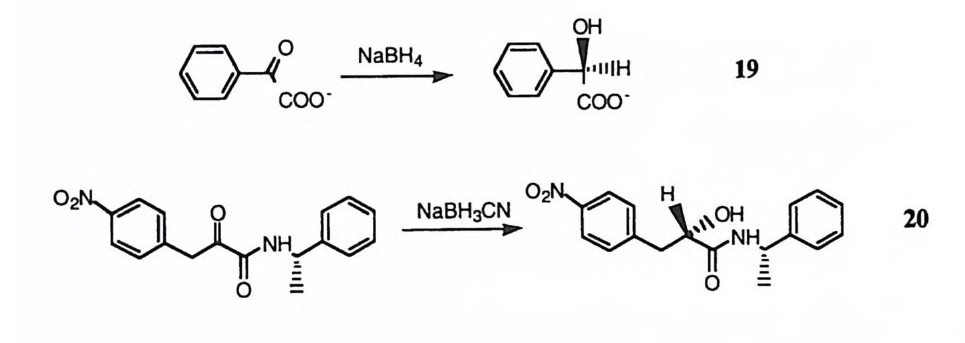 O~NOH°cpo o -드 노~ O呼旦~ (19 o
O~NOH°cpo o -드 노~ O呼旦~ (19 o
(4) 기타 이의에도 보조 효소(예 : 21) 의 결합 부위를 가지는 항체 촉매, 2S) 산화 환원 반웅 울 매개하는 항체 (22) 17) 등 다양한 종류의 항체 효소가 계속하여 고안되고 있다.
 HO~ ~。 OH
HO~ ~。 OH
참고문헌 1) CD Hilv ert , D. ; Gardell, S. J. ; Rutt er , W. J. ; Kais e r, E. T. ]. Am . Chem . Soc, 1 986, 108, 5298. @ Gerlt, J . A . Chem. Rev., 1 987, 87, 1079. @ Kais e r, E. T. Ang ew . Chem. Int . E d. En gl. , 1988, 28, 913. 2) (j) Zuckennann, R . N.; Corey, D. C. ; Schultz, P. G. ]. Am . Chem . Soc., 1 988, 110, 1614. @ Zuckennann, R. N. ; Schultz , P. G. ]. Am. Ch em. Soc., 1988, 110, 6592. 3) Pollack, S. J. ; Jac obs, J. W. ; Schultz, P. G. Sc ien ce, 1987, 234, 1570. 4) Tramonta n o, A. ; Jan da, K. D. ; Lerner, R. A. Sc ien c e ,, 1986, 234, 1566. 5) Tramonta no , A . ; Jan da, K. D ; Lerner, R.A. Proc. Natl . A cad. S ci . U.S .A., 1986, 83 , 6736. 6) Napp e r, A. D. ; Benkov ic, S. J. ; Tramonta n o, A. ; Lerner, R. A. Sc ien c e , 1987, 237, 1041.
7) Jac ob, J. ; Schultz , P. G . ]. Am. Chem . Soc., 1987, 109, 2174. 8) Tramonta n o, A. ; Ammann, A.A. ; Lerner, R. A. ]. Am. Chem. Soc., 1988, 110, 2282. 9) Jac kson, D. Y. ; Jac ob, J. W. ; Sug a sawara, R. ; Reic h , S. H. ; Bart let t, P . A ; Schultz , P. G. ]. Am. Chem. Soc., 1988, 110, 4841 . 10) Jan da, K. D. ; Lerner, R . A. ; Tramonta n o, A. ]. Am. Chem. Soc., 1988, 110, 4835. 11) Hilv ert, D . ; Nared, K. D. ]. Am. Chem. Soc., 1988, 110, 5593. 12) Hil v ert ! D . ; Carp e nte r , S . H. ; Nared, K. D. ; Maria - Teresa, M. Proc. Natl . Acad. S ci . U S .A ., 1988, 85, 4953. 13) Schultz , P. G . Sci en c e, 1988, 24 0 , 426 . 14) Benkovic , S. J. ; Napp e r, A. D. ; Lerner, R. A. Proc. Natl . Acad. S ci . U S .A., 1988, 85, 5355. 15) Jan da, K. D. ; Schloeder, D . ; Benkovic , S. J . ; Lerner, R. A. Sdence, 1988, 241, 1188. 16) Green, B. S. ; Zif fer, H. ; Tort en , M. ; Docto r , B.P. ; Balan, A.]. Chem. Soc. Chem . Commun., 1988, 106. 17) Schultz , P. G. ; Sug as awara, R. ; Leumann, C. ]. ; Shokat, K. M. An gew . Che m. Int. E d. Engl ., 1988, 27, 1172. 18) Tramonta n o, A. ; Lerner, R. A. ; Jan da, K. D. J Am. Chem. Soc., 1988, 110, 4835. 19) Schultz, P. G. ; Bart le tt , P. A. ; Reic h, S. H. ; Sug a sawara, R. ; Jac obs, J. W . ; Jac kson, D. Y. ]. Am. Chem. Soc., 1988, 110, 4841 . 20) Schultz, P. G. ; Sug a sawara, R. ; Cochran, A. G. ]. Am. Chem. Soc., 1988, 110, 7888. 21) Schultz, P. G. ; Jac obs, J. W. ; Massey, R. J. ; Sug a sawara, R. ; Bolin , R. J. ; Durf or , C. N. J Am. Chem. Soc., 1988, 110, 8713. 22) Schultz, P. G. ; Nakaya m a, G. R. ; Pollack, S. J. Sdence, 1988, 242, 1038. 23) Lerner, R. A. ; Iverson, B. L. Sci en ce, 1989, 243, 1184. 24) Schultz, P. G. ; Sug a sawara, R. ; Leumann, C. ]. ; Shokat, K. M. Natu re , 1989, 338, 269.
25) Schultz , P. G. ; Pollack, S. J. ]. Am. Clzem. Soc., 1989, 111, 1929. 26) Lerner, R. A. ; Audit or , M. T. M. ; Wein h ouse, M. I. ; Schwabacher, A . W. ]. Am. Chem. Soc., 1 989, 111, 2344. 27) Schultz, P. G . ; Hsiu rn , P. ; Pollack, S. J. ]. Am. Clzem. Soc., 1989, 111, 5961 . 28) CD Nim an, H. L. ; Elder, J. H. Proc. Natl. Acad. Sci . U.S .A ., 1980, 77, 4524. @ Kohler, G. Sci en ce, 1986, 233, 1281 . 29) Slobin , L. I. Bio c lzemi sh y, 1966, 5, 2836. 30) Kohen, F. ; Hollander, Z. ; Burd, J. F. ; Bog us laski, R. C. Fe1s. Lett. , 1979, 1000, 137. 31) Paulin g , L. Am. Sci ., 1948, 36, 51. 32) Jen cks, W. P. Cata lysis in Clzemi sh y and Enzym olo!J )',, McGraw-Hi ll : New York, 1969, p. 288. 33) Schultz , P. G. Acc. Clzem. Res., 1989, 22, 287. 34) Suh, J. ; Oh, E. ; Lee, C. S.; K im, S. H. ; Jeo n g, G. Bull. Korean Chem. Soc., 1991, 12, 352. 35) Nakaya m a, G. R. ; Schultz, P. G. ]. Am. Chem. Soc., 1992, 114, 780.
생체 제모 3방 부화 학 
제 9 장 분자인식 제 1 절 분자 인식의 연구 동향 분자 인식이라는 용어는 최근에 유행어처럼 널리 사용되고 있으며, 1980 년대 중반부터 미국, 유럽, 일본 등의 국가에서는 분자 인식에 관한 연구가 왕성하 게 전개되고 있다. 그러나 분자 인식의 의미는 엄밀하게 정의되어 있지 않은 상태여서 사람에 따라 조금씩 다른 의미를 내포하여 사용하고 있다. 분자 인식 울 좁게 정의하여 상호 보완적인 구조를 가진 두 분자가 여러 개의 약한 분자 간의 힘을 이용하여 서로를 선택적으로 인지하는 현상으로 간주하기도 한다. 반면에, 소형 분자끼리의 상호 작용뿐· 아니라 소형 분자와 거대 분자의 상호 작용(예 : 약과 이에 해당하는 생체 수용체) 혹은 거대 분자와 거대 분자의 상호 작용(예 : DNA와 단백질)까지 분자 인식의 범주에 포함시킬 수 있고, 표면 혹 은 계면에서 분자들이 질서 있게 배치되는 현상으로까지 분자 인식의 영역을 확대할 수 있다. I) 이에 따라 분자 인식은 비공유 결합에 의하여 연계된 두 개 이상의 화합물이 구성하는 다분자 집단에 관한 연구 분야로 간주하는 것이 적 절하다 .2) 생체계에서는 효소의 촉매작용, 물질 이동, 신경계의 신속한 전기적 신호의 전달, 유전 정보의 복제 등 대부분의 화학작용에서 분자 인식이 핵심적인 역할 을 담당하고 있다. 효소와 기질의 상호 작용은 분자 인식의 전형적인 예가 된 다. 효소는 기질의 구조를 인식할 뿐 아니라 기질을 생성물로 전환하는 과정에 대한 전이상태의 구조를 인식하여 선택적으로 안정화함으로써 효과적인 촉매작 용을 달성할 수 있게 된다. 이러한 분자 인식에 대한 정보는 효소 반응의 메커
니즘을 규명하는 것과 직결되어 있을 뿐 아니라, 각종 생리 현상에 핵심적인 역할을 하는 효소의 저해제를 설계함으로써 신약 혹은 신농약을 발견하는 작업 과도 연관된다. DNA 의 구조를 인식하여 선택적으로 절단하는 화합물은 암의 치료, 인간 게놈의 구조 결정 등에 이용된다. 거대 분자가 개입되는 계에서의 분자 인식에 대하여 기초적인 지식을 발굴하 고 분자 인식을 고도로 효율적으로 달성할 수 있는 계를 인공적으로 고안하려 는 노력이 최근에 유기 호스트 분자를 중심으로 활발하게 경주되고 있다. 이에 따라, 시클로덱스트린, 시클로판, 칼렉스아렌, 크러운에데르 등 큰 고리 화합 물의 구조를 다양하게 합성하거나 홈이 있는 분자를 고안하여 호스트 분자로 삼고, 이들과 상호 보완적인 구조를 거친 게스트 분자와의 작용을 연구하고 있 다. 분자 인식 분야의 연구에는 생물 유기화학적인 지식 이의에도 합성화학, 분 광학, 심지어는 계산화학의 기법이 사용된다. 효소의 저해제, 호스트 분자 등 의 구조를 설계하는 데에는 분자 간의 힘에 대한 지식과 생체내의 화학작용에 대한 지식이 필요하며 컵퓨터 그래픽이 매우 중요한 역할을 담당하게 된다. 또 한 이렇게 설계된 분자는 다단계 합성에 의하여 제작되기 때문에 유기합성이 큰 몫을 차지하게 된다. 분자 간에 일어나는 상호 작용을 측정하는 데에는 핵 자기 공명 , 형광 분광법 등 분광학적인 기법이 동원된다. 이 칭에서는 분자 인식의 기본원리를 규명하고 호스트 분자를 인공적으로 고 안하려는 노력에 대하여 중점적으로 논의하고자 한다. 제 2 절 시클로덱스트린 시클로덱스트린은 글루코오스 67fl (a- 시클로덱스트린) , 7개 (/3-시클로덱스트린) , 혹은 87fl (r- 시클로덱스트린)가 la-~ 합으로 연결되어 형성된 고리형 화합물로 믿이 없는 양동이와 같은 모양(그립 1) 을 가지고 있다. 3)-5) 넓은 쪽 테두리에는 이급 히드록시기가 위치해 있고, 좁은 쪽 테두리에는 일급 히드록시 기가 위치 해 있다. 가운대 구멍의 폭은 시클로덱스트린에 포함된 글루코오스의 수에 따
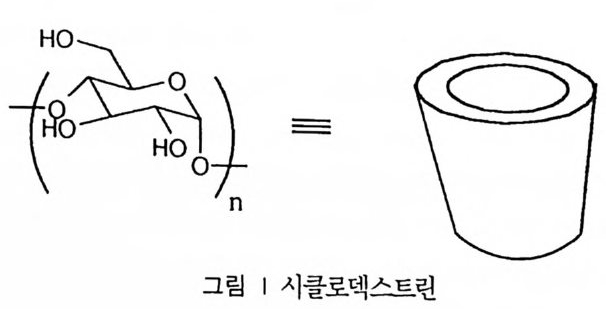 <\°0 \루
<\°0 \루
라 달라지며 벽은 형수성 부분으로 구성되어 있다. 시클로덱스트린의 구멍 속은 협수성 환경을 가지고 있기 때문에 물 속에서 형수성 물질은 시클로텍스트린의 구멍 속을 차지하여 착화합물을 형성하게 된 다. 구멍 속의 형수성 매질이 촉매작용으로 연결된 예로 페닐시아노아세트산의 탈카르복실화 반응을 들 수 있다. 6) 이 반응은 전이상태에서 음전하 밀도가 낮 아지기 때문에 형수성 매질 속에서 가속됨이 알려져 있다.
 °2N::t Bu °2N::t Bu : 2N:OHo 」 :u
°2N::t Bu °2N::t Bu : 2N:OHo 」 :u
시클로덱스트린 구멍 속에 분자가 삽입되면 이 분자내의 형태 자유도가 억제 되게 된다. 이에 따라 생산적인 형태의 농도가 증가하여 촉매작용으로 연결될 수도 있다. 예를 들어 시클로덱스트린 구멍 속에서 1~ 농도가 증가함으로써 1 률 형성하는 분자내 친핵성 반응속도가 5- 伊i 증가된다. 7) 27} 시클로덱스트
린과 결합함으로써 디엔과 울레핀 간의 상호 접촉 빈도가 커짐에 따라 딜스-알 더 반응의 89° 에서의 수율이 20% 에서 91% 로 증가한 사례도 보고된 바 있 다 .8) 시클로덱스트린이 가지고 있는 유일한 작용기인 일급 및 이급 히드록시기가 친핵체로 작용하여 에스테르와 같은 게스트 분자를 공격하는 반응에 대하여 많 은 연구가 되어 있다. 히드록시기가 친핵체로 작용하여 에스데르를 공격하면 비교적 안정한 에스테르(아실-시클로덱스트린)가 중간체로 형성되기 때문에 기 질인 에스데르의 가수분해에 대하여 시클로덱스트린이 촉매로 작용하지 않는다 는 점은 유의할 필요가 있다. 페닐 고리에 다양한 치환기를 가진 페닐 아세데 이트는 시클로덱스트린과 착화합물을 만든 뒤 시클로덱스트린의 이급 히드록시 기의 음이온의 공격을 받아 에스데르 결합이 단절된다. 이 반응에서 치환기가 메타 위치에 도입된 경우가 파라 위치에 도입된 것보다 훨씬 빠른 속도를 보여 준다. 이것은 뇨} 같이 메타 치환체가 도입된 경우 에스테르의 카르보닐기가 친핵체인 히드록시기에 인접한 위치에 놓이기 때문으로 해석된다. 4)
 三츨 °H 0 - 0-N02
三츨 °H 0 - 0-N02
시클로덱스트린은 물 속뿐 아니라 디메틸술폭시드 (DMSO) 를 상당량 포함하 는 물 속에서도 형수성 기질과 착화합물을 형성한다. /3-시클로덱스트린은 60%(v/v) DMSO 속에서 p-니트로페닐 페로신나메이트 (4) 와 착화합물을 형 성하며 이때 心] 탈아실화 속도는 7&1 J 배 가속된다 .9) 이에 비하여 m- t-부 틸페닐 아세데이트의 탈아실화 반응은 단지 260 배 가속된다. 이러한 차이는 에 스테르 기질의 구조 차이로 설명된다. 心] 에스테르기는 페로센 부분·과 칙각으 로 배열되어 있다. 따라서 47} 시클로덱스트린과 착화합물을 이룰 때 페로센에
서 직각으로 뻗은 부분이 팔과 같이 시클로덱스트린의 데 부분에 얹혀지게 되 고 에스테르 카르보닐이 정사면체 중간체로 전환될 때 형태 변화가 별로 일어 날 필요가 없다. 반면에 후자의 경우 페닐 부분이 구멍 안쪽으로- 내려앉게 되 면 정사면체 중간체로 전환하는 과정에서 불리한 형태 변화가 수반되는 것이 다.
 p-CD : X=H
p-CD : X=H
시클로덱스트린과 에스테르가 보다 생산적인 착화합물을 형성할 수 있게 함 으로써 반응성을 제고하려는 목적으로 시클로덱스트린에 바닥을 붙여서 구멍의 깊이를 조절하려는 연구가 시도되었다. 10) S 로 표시한 시클로덱스트린 유도체가 m - 니트로페닐 아세데이트 혹은 m- t부될페닐 아세테이트와 반응하여 에스테 르를 파괴하고 시클로덱스트린의 일급 히드록시기가 아세틸화되는 과정에 대한 속도론적 결과를 표 1 에 요약하였다.
 표| 바닥을붙인 시클로덱스트린유도체의 속도론적 행동
표| 바닥을붙인 시클로덱스트린유도체의 속도론적 행동
이 표에서 k1n t ra 는 시클로덱스트린과 에스데르가 형성한 착화합물의 속도상 수이고 Kd 는 착화합물의 해리싱수이다. 이 결과로부터 시클로덱스트린에 바닥 울 붙임으로써 구멍의 깊이가 얕아지면 기질과 착화합물을 형성하는 능력은 떨 어지지만, 이와 같이 형성된 착화합물에서 에스테르기가 시클로덱스트린의 일 급 히드록시기에 보다 인접한 위치를 점유하게 됨으로써 착화합물의 반응속도 는 제고된다. 따라서 /3 -CD-E t는 /3 -CD 와 비교하여 반응물인 에스데르 를 인 지하는 능력은 다소 떨어지지만 전이상태를 인지하는 능력은 약간 우세한 것이 다.
 AB 이성질체 AC 이성질체 AD 이성질체
AB 이성질체 AC 이성질체 AD 이성질체
시클로덱스트린의 히드록시기를 화학적으로 수정하여 시클로덱스트린에 여러 가지 구조적인 요인울 도입하였는데, 위에 언급한 적t 그 예로 들 수 있다. 히 드록시기 두 개만을 선택적으로 수정하려 할 때에는 수정되는 히드록시기의 상 대적인 위치에 따라 여러 가지 조합이 가능하다. 한 개의 분자가 두 개의 시클 로덱스트린 일급 히드록시기와 반응하여 시클로덱스트린에 모자를 씌울 때 그 모자의 위치는 6 처럼 세 가지가 가능하다. 디술포닐 클로리드 7 을 사용하면 AD 이성질체가, 읽끌 사용하면 AC 이성질체가 선택적으로 형성됨이 알려져 있다 .11)
 9 10
9 10
일급 히드록시기 혹은 이급 히드록시기를 수정하여 연결고리와 반응시키면 시클로덱스트린의 이량체를 합성할 수 있는데, 9 혹은 lfu !} 갇이 한 점 혹은 두 접 접촉에 의하여 형태 자유도를 제어할 수 있다. 이량체의 경우 게스트 분 자와 두 군데 구멍에서 접촉하여 착화합물을 형성하기 때문에 게스트를 매우 강하게 결합할 수 있으며, 연결고리의 길이를 변화시킬 때 결합되는 게스트의 구조가 변화하게 된다. 12), 13)
 11 12
11 12
시클로덱스트린의 히드록시기를 수정하여 여러 가지 기능성 작용기를 도입하 였다. 예를 들면 11 에서는 금속에 배위된 옥심기를 촉매 작용기로 도입하였 고, 14) 122 1-갇 이 아자크라운 에테르가 제 2 의 분자 인식 자리로 도입된 바도 있 다 .15) 시클로덱스트린을 이용하여 효소의 특성을 재현하는 효소 모형을 제작하려는 연구가 다수 보고된 바 있다. 예를 들어 13ol] 대하여서는 리보뉴클레아제의 모 형으로 카테콜 포스페이트 유도체의 가수분해에 대한 촉매작용이 검토되었으
 -H-O-PHOI- O/o -· H
-H-O-PHOI- O/o -· H
며 , 16) 1~ 아연 (Il ) 착화합물에 대하여서는 카르보닉 안히드라제의 모형으로 탄산 가스의 수화 반응에 대한 촉매작용이 측정되었다. 17) 키모트립신과 같은 세린 프로데아제의 활성자리에는 카르복실-이미다졸-히드록시 기의 조합이 포 함되어 있다. 이에 대한 모형으로 15 가 합성되어 에스데르 가수분해에 대한 활 성이 측정된 바 있다. 18)
 16二0s 回〉N 17 HO 占
16二0s 回〉N 17 HO 占
피리독살 포스페이트와 피리독스아민 포스페이트는 아미노산 대사에 개입하 는 중요한 조효소이다. 트란스아미나제의 반웅중에 전자의 알데히드기가 후자 의 아미노기로 전환되기 때문에 이 둘은 짝을 이루어 생체반응에 참여하게 된 다. 피리독스아민 부분을 시클로덱스트린에 연결시켜 트란스아미나제 촉매작용
울 모방한 연구가 16 및 17 을 사용하여 보고된 바 있다. 16ol] 서 피리독스아민 부분은 시클로덱스트린의 일급 히드록시기가 차지하는 면과 이급 히드록시기가 차지하는 면에 각각 위치하도록 두 가지가 합성되었다. 17 에 관련된 모형에서 는 피리독스아민이 시클로덱스트린의 일급 히드록시기가 차지하는 면에 이중으 로 연결되어 있다. 19), 2 0) 시클로텍스트린은 기질과 착화합물을 이루고 반응기와 기질 간의 반응속도가 증가하는 등 효소의 특칭을 다소간 재현하는 데는 성공하고 있다. 그러나 속도 의 증가폭은 대부분의 경우 괄목할 만한 수준까지 다다르지 못하고 있으며, 반 응성이 높은 기질에 대하여 주로 반응성이 연구되어 왔다는 21) 제약이 있다. 시 클로덱스트린에 도입되는 작용기의 반응성이 높지 않은 이유는 촉매계가 가지 는 형태상의 제약에 상당히 기인한다고 볼 수 있다. 시클로덱스트린에 기질이 끼여들어 형성한 착화합물에서 반응부와 촉매 작용기 사이에 유효농도가 높으 려면 작용기와 시클로덱스트린 사이에 충분히 긴 연결고리가 존재하여야 한다. 그러나 연결고리가 길어지면 합성이 어려워질 뿐 아니라, 연결고리가 가지는 형태 자유도가 매우 커지기 때문에 반응성이 높은 자리에 위치할 가능성이 작 아지는 것이다. 이러한 문제점을 해결하기 위하여 g-시클로덱스트린을 폴리에 틸렌이민 (PEI) 에 연결하여 PEl7t 보유한 아민기를 친핵체 혹은 친핵체를 연결 할 지지점으로 사용하려는 연구가 수행되었다. 22) 18로 표시하였듯이 시클로덱
 xg< g:
xg< g:
스트린의 구멍에 기질이 끼여들면 검은 원으로 나타낸 작용기가 기질의 반옹부 위에 인접한 곳에 위치하여 쉽게 반응을 일으킬 수 있을 것이다. PEI 에 연결 된 시클로덱스트린은 19 및 20 ½ 기질로 사용하였을 때 이 에스테르들의 털아 실화 반응속도를 매우 크게 가속하였다. 이는 21 £ 표시한 바와 감이 시클로덱 스트린에 결합된 기질의 에스테르 결합을 PEI 의 아민이 친핵체로 공격하였기 때문 0l 다.
 22
22
여러 개의 시클로덱스트린의 내부 원통 구조에 긴 사슬이 끼여들어 형성된 물질 (22) 에 대한 연구가 최근에 관심을 끌고 있다. 이러한 물질은 새로운 소재 로서 생체반응을 모방한 화학작용 이의에도 여러 가지 다양한 기능을 수행하게 될 것으로 기대된다. 23), 24) 제 3 절 시클로판 시클로판은 적어도 한 개의 방향족 고리의 양쪽이 적어도 한 개의 지빙족 고 리에 의하여 연결된 수용체 분자를 일컫는다. 2), 25) 시클로판의 고리 내부에 분 자가 포함되어 착화합물을 형성하는 최초의 예는 끄이 듀렌과 이룬 착화합물이 다. 그 이후에 시클로판의 구조를 다양하게 변화시키면서 여러 가지의 분자 간 힘을 이용하여 게스트 분자를 인식하여 착화합물을 효과적으로 형성시키려는 연구와 시클로판에 부착시킨 작용기를 이용하여 효소를 모방한 촉매를 제작하 려는 연구가 활발히 시도되었다. 시클로판이 호스트 분자의 역할을 효과적으로 수행하기 위하여 거대 고리 골
 CO2·
CO2·
격의 형태를 고정시켜 게스트 분자를 수용할 구멍이 무너지지 않도록 하여야 한다. 예를 들어 끄에서는 Ar ― CH2 ― Ar 의 각도가 고정되어 있어 벤젠 고리 를 수직 형태로 유지시켜 분자 중심부에 충분히 깊은 구멍을 보유하고 있다. 23 따 비교하여 23 J>i근 내부 구멍이 쉽게 붕괴되지 않는 형태상의 안정도를 가 지고 있어 게스트 분자와의 결합 능력이 훨씬 뛰어나다. 27) 시클로판의 효용성을 제고하려면 물에 대한 용해도를 증가시켜야 한다. 이를 위하여서 분자내에 친수성 부분을 도입하여야 한다. 한편, 물 속에서 게스트와 의 결합력을 높이려면 협수성 작용을 활용하여야 하는데, 이를 위하여서는 분 자내에 협수성 부분을 도입하여야 한다. 이러한 두 측면을 수용하기 위하여 분 자내의 친수성 부분과 형수성 부분을 멀리 떨어지도록 시클로판의 구조를 설계 하기도 하는데 그 예로 끄과 ”를 28) 들 수 있다 .
호스트 분자와의 결합 능력을 제고하기 위하여 전자 주게-받게 작용을 분자 간의 힘으로 활용할 수 있게끔 시클로판의 구조를 고안하기도 한다. 예를 들어 짜게서 페녹시벤젠 부분이 전자 주게로 작용하여 전자가 결핍된 나프탈렌 유도 체와 효과적으로 결합하는 것이 관찰되었다. 28) 벤젠 고리의 전자 밀도가 높은 23~ 전자 밀도가 높은 인돌 유도체보다 전자 밀도가 낮은 퀴늘린 혹은 이소 퀴놀린 유도체와 착화합물을 더 강하게 형성한다.
 二。 -~多- 0
二。 -~多- 0
생체계에서 분자 인식에 수소 결합이 매우 중요한 역할을 하는바, 단백질과 핵산의 나선형 삼차원 구조에서 그 전형적인 예를 볼 수 있다. 소형 분자에서 도 수소 결합을 사용하여 분자 인식을 효과적으로 성취하려는 노력을 자주 기 울이는데, 시클로판에서도 게스트의 구조를 인식하여 선택적이고 강한 착화합 물을 형성하는데 수소 결합을 효과적으로 활용한 사례가 자주 보고되고 있 다 .29) 그예로 E 을들수 있다 .30) 시클로판의 분자 인식 능력을 다앙화하기 위한 노력에서 의부 자극에 의하여 시클로판의 내부 구멍의 기하학적인 구조에 변화가 유도되어 게스트 결합능력 이 영향을 받는 경우가 연구되었다. 예를 들어 %은 가시광선과 자의선을 교차 하여 쬐어줄 때 가역적인 화학변화가 일어나서 구멍의 크기가 바뀌게 된다. 31)
 N02 `viU sVib l e. N02
N02 `viU sVib l e. N02
아조벤젠이 자의선을 쪼이면 불안정한 시스 형태로 바뀌었다가 가시광선을 쪼 이면 안정한 트란스형으로 복원된다는 사실을 이용하여 구조 변화를 유도하는 시클로판도 보고되었다 .32) 이의에도 플라빈을 모방한 시클로판을 합성하여 산 화환원 반응에 의하여 그 구조 변화를 가역적으로 유도하는 예도 알려져 있 다. 33)
 N~N
N~N
키랄성 게스트 분자의 구조를 인식하는 시클로판을 설계하려는 연구도 시작 되었다. 예를 들어 27 은 키랄성 시클로판이기 때문에 게스트의 키랄성을 인식 할 수 있어서 만델산의 두 광학 이성질체와 선별적으로 결합하는 능력을 가지 고 있다 .3 ◄) 시클로판의 내부 구멍을 결합부위로 사용하면서 제 E] 결합부위를 추가로 도 입하려는 연구도 진행되고 있는데, 예를 들어 꼬은 35) 형수성 탄화수소를 결합
 eEtr2 W :?:,... 文o― ――一 ( (CH/C2)~04 o o0노 一/o ^\ _Jo \
eEtr2 W :?:,... 文o― ――一 ( (CH/C2)~04 o o0노 一/o ^\ _Jo \
하는 시클로판 부분과 금속 이온을 인식하는 크라운 에데르를 포함하여 이중적 인 분자 인식을 수행할 수 있다. 시클로판이 게스트를 인식하는 능력을 개선하려는 목적으로 거대 고리 골격 의 형태 자유도를 억제하여 내부 구멍의 붕괴를 방지하고 내부 구멍의 형수성 을 제고하려는 노력에서 여러 개의 거대 고리를 축합한 구조를 고안하는 작업 도 추진되고 있다. 예를 들어 , 거대 이중 고리 구조를 가진 시클로판인 2 9-;:근 원판형의 내부 구멍을 가지고 있어서 디페닐메탄 부분의 메틸렌기 부분의 뾰족 한 각도에 수소 원자를 칙접 삽입할 수 있는 아렌 게스트를 선택성이 높게 인 식한다 .36) 시클로판에 긴 형수성 꼬리를 여러 개 붙여 미셀과 유사한 구조를 도입하여 강한 무극성 성격을 부여하려는 연구도 진행되고 있다. 예를 들어 3 0i곤 31 과 감기 표현할 수 있는데 이 화합물은 무극성인 게스트롤 만나게 되면 시클로판 의 구멍 부위와 여러 개의 팔을 사용하여 게스트를 껴안는 형태로 결합하여 착 화합물을 형성한다. 25)
 ~NR ~
~NR ~
시클로판을 이용하여 상호 보완적인 구조를 가진 게스트와 착화합물을 형성 하는 데 대하여서는 상당한 양의 연구 결과가 축적되었으나 물질 및 신호의 전 달 촉매작용 등 동적인 기능을 보유한 시클로판에 대한 연구는 상대적으로 미 미한 수준에 머물고 있다. 수용성 시클로판이 아렌을 비국성 작용에 의하여 결 합한 후 물 충을 가로질러 이동시키는 등 초보적인 전달 기능은 보고된 바 있 다. 2) 시클로판의 거대 고리 골격에 작용기를 부착시킨 뒤, 이 작용기가 간단한 유기반응에 촉매로 작용하는 예도 몇 가지가 보고 되었다. 2) 현재까지는 시클로 판에 관한 연구에서 효과적인 골격 구조를 설계하는 데 주력하였기 때문에 효 율적인 촉매를 고안하려는 연구는 시클로덱스트린과 비교하여 상당히 뒤지는 수준게 머물고있다. 제 4 절 칼릭스아렌 칼릭스아렌은 시클로판의 일종으로 복수 개의 아렌이 메틸렌기를 사이에 두 고 상호간에 메타 위치로 연결되어 형성한 거대 고리 화합물 (32) 을 일컫는 다. 37), 3 8) 칼릭스아렌 종류는 페놀 유도체와 포름알데히드의 축합에 의하여 본
 B3
B3
격적으로 합성되어 체계적안 연구가 가능하여졌다. 이에 따라 칼릭스아렌이라 는 명칭은 32 중에서도 A 가 히드록시기인 것에 대하여 사용하고 있다. 비교적 용이하게 합성되며 간단한 구조를 가진 칼릭스아렌의 예는 p - t유 L 틸 칼릭스 [4] 아렌 (33 : A=OH,B= t부릴)이다. 이 화합물은 33 처럼 깔때기 모양을 가질 수 있다. 그런데 벤젠 고리 중의 하나가 회전하면 묘와 같은 구조를 가질 수 있으며 회전하는 벤젠 고리 수가 엄 H 일 경우에는 여러 개의 형태가 가능해 진다. p-t냐낸칼릭스 [ 서아렌의 경우에는 결정 상태에서 깔때기 구조를 가지 고 있음이 X 선 회절 연구의 결과 알려졌다. 39) 칼릭스아렌이 깔때기 모양을 가지고 있을 때에는 내부에 구멍이 존재하여 호 스트 분자로서의 역할이 가능하다. 그러나 칼릭스아렌에 포함된 아렌 고리의 수가 많아지면 형태 자유도가 점점 증가하게 되고, 고리의 크기가 충분히 크면 접시 모양으로 벤젠 고리가 동일 평면상에 누우려는 경향까지 보일 수 있다. 그리고 용액상에서는 분자의 형태 자유도가 더욱 증가하여 깔때기 이의의 다양 한 형태가 가능하다. 칼릭스아렌을 깔때기 형태와 같이 내부 구멍을 가진 모양 으로 고정시키기 위하여 벤젠 고리 위에 여러 가지 작용기를 도입하는 작업이 수행된 바 있다. 깔때기의 두 쪽 테두리에 위치한 작용기의 크기를 확대하거나 이들을 연결 고리를 사용하여 접합함으로써 벤젠 고리가 회전하는 것을 방지하 려 시도한 것이다. 37) 칼릭스아렌이 게스트를 인식하는 능력을 제고하기 위하여서는 다양한 종류의 작용기를 도입하여야 한다. 칼릭스아렌의 각종 유도체를 합성한 계동도의 예를 35- %으로 표시하였다. 38) 깔때기 형태 (33) 를 가전 칼릭스아렌의 좁은 쪽 테두 리와 넓은 쪽 테두리에 수용성 작용기를 도입하여 물에 대한 용해도를 높이고
 43 (X = R, Y = CH2 P 0(0Hh _ 42. (X = R, Y = CH2 N Me3 ')
43 (X = R, Y = CH2 P 0(0Hh _ 42. (X = R, Y = CH2 N Me3 ')
협수성 꼬리 를 부착하여 무국성 상호 작용에 의하여 형수성 분자를 인식하도록 구조 를 설계한 예로 꼬과 %울 들 수 있다. 피렌을 게스트 분자로 사용하였을 때 38) 의 꼬의 결합상수는 R 이 부틸 (K : 약 106 M-I) 일 때가 메틸일 때보다 300 여 배 더 크고 핵실일 경우는 부틸인 경우와 비슷하였다. 이는 피렌을 감싸 안 듯이 결합하는 데에는 부틸 정도의 팔길이이면 충분하다는 것을 암시한다. 46 이 굽 AE 쿠 사용될 때, n 이 6 인 경우가 4 혹은 8 인 경우와 비교하여 피렌에 대한 친화력이 두드러지게 뛰어난데, 이는 피렌의 크기와 칼릭스아렌의 구멍의 크기가 n 이 6 인 경우에 가장 부합하기 때문이다. 37을 굽 AE 쿠 사용하고 47과 색을 게스트로 사용하였을 경우에 n 이 4 인 경 우에는 AS} 訪 L 다 작고 n 이 6 혹은 8 인 경우에는 AS} 僞i다 크다. 이것으 로부터 n 이 4 인 경우에는 정전기적인 상호 작용이, n 이 6 혹은 8 인 경우에는 협수성 힘이 호스트-게스트 착화합물을 형성하는 원동력이 된다고 해석한 바
 R
R
있다 .38) 카르복시메틸칼릭스 [n] 아렌 (49) 은 알칼리 금속 혹은 알칼리 토류 금속 이온 울 인식하여 착화합물 (50) 을 만든다 .38 ) 이것은 딱딱한 영기인 산소와 딱딱한 산인 알칼리 혹은 알칼리 토류 금속 이온 간의 상호 작용으로 볼 수 있다. 이 때 칼릭스아렌의 고리 크기에 따라 적합한 금속 이온의 크기가 정해진다. 호스트 분자가 금속 이온을 인식하는 능력은 란타니드 혹은 악타니드 계열의 희토류 금속 이온을 얼마나 찰 선별할 수 있는가로 시험할 수 있다. 이것은 희 토류끼리는 서로 구별하기가 어렵디는 측면과 바닷물에서 우라늄을 채집할 수 있는 경제적인 측면 때문이다. 우라닐 이온 (UO22 + )은 산소 리간드를 선호하며 리간드 원자 5 혹은 un 가 거의 동일한 평면 위에 놓일 때 착화합물을 잘 형성 한다는 성질이 알려져 있다. 크라운 에데르가 산소를 주게원자로 동원할 수 있 지만 이러한 기하학적인 구조는 제공하기 어렵다. 37(n=5, 6) 혹은 51(n= 5 , 6) 의 페놀 혹은 카르복시 산소 원자가 거의 동일한 평면상에 놓이는데 , 우라닐 이온에 대한 착물 형성 상수가 1016-20 으로 매우 큰 데 반하여 Cu( Il ) , Zn( Il ) , Ni (Il), M g (Il) 에 대하겨서는매우약한친화도를보여 이들과비교하여 우라 닐 이온에 대한 선택성이 매우 높다는 것이 관찰되었다. 38) 그러나 아직도 칼릭 스아렌 유도체를 우라닐 이온의 효과적인 추출제로 간주할 수는 없는데, 여타 의 희토류와 우리닐 이온을 구별하는 능력이 결여되었기 때문이다. 칼릭스아렌 유도체가 화학반응에 촉매작용을 일으킨다고 보고된 예는 40)-42) 아직매우드물다.
제 5 절 크라운 에테르 196 전에 최초의 크라운 에테르가 합성되고 금속 이온을 선택적으로 인식하 는 능력이 보고된 4 ~ ) 이후에 매우 다양한 구조를 가진 크라운 에테르 및 유사체 가 합성되었으며 ° .' 들에 의한 새로운 기능이 보고되었다. 5),44) 크라운 에데르는 적어도 한 개의 마크로 고리를 가지고 있으며 여러 개의 산소를 고리 원자로 포함하고 있는데, 산소 중 일부가 유황, 질소, 혹은 다른 헤데로 원자로 치환 되기도한다.
 ~°广00 \o~ 『0\_ \J° C 。bl _0o_ b _。j )
~°广00 \o~ 『0\_ \J° C 。bl _0o_ b _。j )
단순한 구조를 가전 12- 크라운 - 4 (52) , 15- 크라운 -5 (53) , 18- 크라운 -6 (54) 의 내부 구멍의 직경은 각각 1.2, 1.7, 2.6A 이다. 한편 알칼리 금속의 양이 온과 암모니움 이온의 직경은, 1. 4-3 . 3A 인바, 간단한 크라운 에데르는 크기 가 상호 보완적인 양이온을 선택적으로 결합한다. 유기 용매에 녹지 않는 염의 경우에 크라운 에테르를 가함으로써 양이온이 크라운 에테르와 착화합물을 형 성하여 용해도가 크게 증가하게 된다. 이러한 용해도의 증가가 반응속도를 증 가시키기도 한다. 음이온 친핵체의 카운터 양이온이 크라운 에테르에 의하여 배위되면 양이온의 유효 크기가 증가하게 되고 음이온 천핵체에 미치는 카운터 양이온의 정전기적 영향이 감소하게 되는데, 이 효과가 반응 메커니즘에 따라 반응속도를 증가시킬 수도 있고, 감소시킬 수도 있다. 크라운 에테르에 의한 반응속도의 영향에 따라 반응 메커니즘상의 문제점을 구명한 예도 있다. 45),46) 크라운 에데르는 5~ 예와 같이 정사면체 암모니움 양이온과도 안정한 착화 합물을 형성한다. 크라운 에테르를 키랄성인 구조를 가지게 고안하여 라세미 형의 아민을 분할하는 데 사용한 예가 다수 보고되어 있다. 47) 예를 들어 %은
 r\ 0o J\ \C
r\ 0o J\ \C
1, 1’- 비나프틸기의 2, 2'- 위치를 크라운 에데르에 연결한 구조를 가지고 있어 키랄성이다. %은 S 및 R- 메틸 페닐글리신 암모니움 양이온과 섞었을 때 S- 게스트 분자와 더 강한 착화합물을 형성함으로써 라세미 혼합물을 효과적으로 광학 분할할 수 있었다. S- 게스트는 57과 갇이 筑J에서 호스트와 접촉하여 착 화합물을 형성하게 되고, R- 게스트는 5~ 같이 4 점에서 호스트와 접촉하게 된다. 57 과 비교하여 5 8oJ]는 호스트와 게스트 사이에 입체 장애가 더 큰 반면 에 호스트의 나프틸과 게스트의 페닐 사이에 포갱에 의한 상호 작용이 존재한 다. 이 포갱 작용에 의한 안정도보다 입체 장애에 의한 불안정도가 더 우세하 여 57 이 58보 다 안정한 것이다. 크라운 에테르의 고리 위에 존재하는 산소 원자 중의 일부를· 질소로 바꾼 경 우를 아자 크라운 에데르라고 부른다. 한편, 크라운 에테르의 고리에 팔을 붙
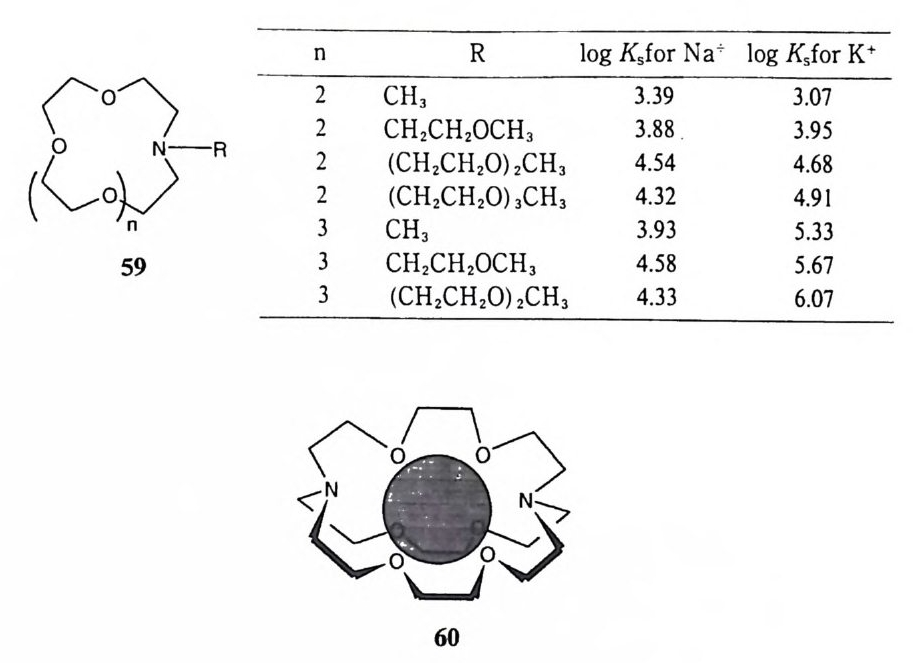 ((O\ 'o。~:N— R i2n222 33CCC((CC3HHHHH323 C2 2 CCH HH 2 022R 00C ))H 23C(3 HH 33 log K33443s.f.. ..35o839 984 32r Na log K34345......90936f o 157 83 r K+
((O\ 'o。~:N— R i2n222 33CCC((CC3HHHHH323 C2 2 CCH HH 2 022R 00C ))H 23C(3 HH 33 log K33443s.f.. ..35o839 984 32r Na log K34345......90936f o 157 83 r K+
이고 이 필에 배위자리를 도입한 일련의 화합물을 라리아트 크라운 에데르라 칭한다. 48) 59 의 예에서 보듯이 라리아트 크라운 에테르의 구조를 적절히 변경 함게 따라 금속 이온에 대한 결합능력이 크게 영향받는다. 크라운 에테르가 이차원적 구조를 가지고 있는 것을 발전시켜 삼차원적인 다 중 거대 고리구조를 가지게끔 설계한 화합물을 크립탄드라 부르고 크립탄드가 형성한 착화합물을 크립테이트라 한다. 49) 60 의 예와 같이 크립탄드의 내부에 OJ =o l 온, 음이온 등이 매우 강하게 결합된다. 크라운 에테르가 게스트의 구조를 인식하는 선택성을 제고하는 방안으로 한 분자가 두 개의 크라운 에데르를 가지고 있는 비스 크라운 에테르 종류도 다수 합성된 바 있다. 최근에는 두 크라운 에테르를 아조벤젠으로 연결하고 아조기 가 자의선에 의하여서는 트란스형이 열역학적으로 불안정한 시스형으로 바뀌고 가시광선에 의하여서는 시스가 트란스로 바뀌는 것 (61) 을 이용하고 시스형이 K 미온을 결합硏臣 능력이 월등히 양호하다는 점을 이용하여 광선에 의해 금 속 이온의 결합 능력을 조절할 수 있는 광감응성 크라운 에데르도 보고된 바
 61
61
있다 .38) 크러운 에테르 혹은 크립탄드는 게스트와 착화합물을 형성하였을 때에는 게 스트가 호스트의 내부에 끼여들어 구멍을 확보하게 되지만 호스트 자체로만 있 울 때에는 구멍이 붕괴된 형태를 가지고 있다. 착화합물을 형성하지 않은 호스 트만의 구조에서도 내부에 구멍이 존재하는 단단한 형태를 가전 크라운 에데르 도 합성되었는데 이러한 화합물을 스페란드라 부른다. SO) 스페란드는 구멍을 보 유하고 있고 그 속에 게스트가 끼여들기 때문에 게스트 결합 능력이 매우 뛰어 나다. 예를 들어 6~ 스페란드는 Li+이온의 결합상수가 1010_1011M-l 에 달한 다. 크라운 에테르 및 크립탄드 화합물이 게스트의 구조를 인식하여 안정한 착화 합물을 이루고 경우에 따라서는 게스트를 이동하는 기능까지 검토된 경우가 많 다. 이에 비하여 촉매로서 작용한 예는 훨씬 적다. 다른 호스트 분자의 경우와
 \<<[>>63 了 02〈 N -曲멸ONH 0 -0 군..H .C. H3
\<<[>>63 了 02〈 N -曲멸ONH 0 -0 군..H .C. H3
마찬가지로 전이상태를 인식하여 안정화시키는 연구는 안정한 화합물을 인식하 는 연구보다 뒤진 상태인 것이다. 크라운 에테르 및 크립탄드 종류가 반응속도 를 제고하는 데 사용된 예를 몇 가지 들어보자. 6 책 표시한 아자 크라운 에테르는 아크리딘 고리를 결사슬에 부착시켜 놓은 구조를 가지고 있는데 이온성 상호 작용과 방향족 고리 간의 포갬 상호 작용에 의하여 ATP 의 구조를 인식하여 착화합물을 형성한다. 5” 이 착화합물에서 이 급 아민이 친핵체로 작용하여 포스포릴기를 공격하여 치환반응을 일으켜 중간 체를 형성하며 이 중간체가 물의 공격을 받아 파괴됨으로써 AT P;가 ADP로 가수분해되는 반응의 촉매작용이 완결된다. 스페란드 64가 가지고 있는 세 개의 카르보닐 산소와 65( 알라닌의 P- 니트로페 닐 에스테르)의 암모니움 수소 간에 수소결합이 형성되며 ”는 6~ 강력한 착 화합물을 형성하며 이 착화합물에서 호스트의 벤질 알콜 부분의 알콕시기가 천
핵체로 게스트인 에스테르를 공격한다. 이에 따라 알콜의 아실화 반응성이 착 화합물을 형성할 수 없는 경우보다 101 대 정도 가속된다고 보고된 바 있다. 52) 이 반응은 에스데르의 가수분해가 아니고 호스트 분자의 아실화 반응이어서 호 스트 분자가 재생되지 않는다. 따라서 ”는 촉매로 작용하지는 않는다. 그러나 게스트와 강하게 결합하고 이에 따라 형성된 착화합물 속에서 게스트의 반응부 위와 호스트의 친핵체 사이에 매우 밀착된 입체 관계를 형성하였다는 점에서 효소_기질 착화합물의 특성을 재현한 모형으로 간주할 수 있다. 제 6 절 기타 이상에서는 구멍을 내부에 포함하고 있는 거대 고리 형태의 호스트 분자가 몇 가지 분자 간의 힘을 이용하여 상호 보완적인 구조를 가전 게스트 분자를 인식하는 예를 살펴보았다. 거대 고리의 구조를 가지지 않고서도 여러 개의 약 한 분자 간의 힘을 동원하여 게스트 분자와 비교적 안정한 착화합물을 형성하 는 호스트 분자들도 다수 알려져 있다. 2 책 표시한 시클로판은 수소결합을 다수 사용하여 선택적으로 게스트를 인 식하고 있다. 이것과 유사하지만 거대 고리가 아닌 호스트 분자가 수소결합을 디수 사용하여 게스트와 강한 착화합물을 형성하는 예로 “울 들 수 있다. 이
 C12H2s
C12H2s
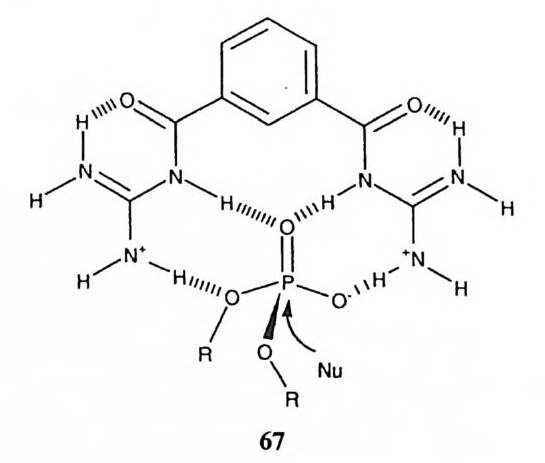 I'':0 〈三I o,IIII 『
I'':0 〈三I o,IIII 『
경우에 DMS 아문 용매로 사용하면 호스트와 게스트 간의 수소결합이 수용되지 않가 착화합물이 형성되지 않지만 1 : 1 CH2CI2 내円넷 l 을 용매로 사용하면 결 합상수가 1 X l06M-l 정도에 달한다. 53) 호스트 분자가 전이상태를 선택적으로 인식하면 효과적인 촉매작용을 달성할 수 있다. 반면에 전이상태보다 반응물을 선택적으로 인식하여 안정화시키면 호 스트 분자는 오히려 저해제로 작용하게 된다. 전이상태가 안정화되더라도 생성 물이 과다하게 안정화되면 호스트의 촉매작용은 생성물 저해작용에 의하여 효 과가 크게 감소한다. 전이상태를 선택적으로- 안정화하는 인공 촉매계로 제窮} 에서 논의한 항체 효소를 들 수 있다. 소형 유기 분자를 호스트 분자로 사용하 여 전이상태를 인식하여 촉매 효과를 실현한 예로 67을 들 수 있다. 여기에서 게스트의 인은 반응물의 상태에서는 정사면체 구조를 가지지만 친핵체의 공격 울 받으면 전이상태에서 5 배위 양삼각뿔의 성격을 강하게 지니게 된다. 호스트 분자는 정사면체 구조인 반응물보다 양삼각뿔 구조인 전이상태와 수소결합을 더 효율적으로 형성하면 활성화 에너지를 줄이고 속도를 증가시킬 수 있다. 이 반응게서 에틸 메르캅토 아세테이트의 유황이 친핵체로 작용할 경우 속도가 36 배 가속되는 것이 관찰되었다. 54) 磁 짜의 카르복실기가 모두 축 위치를 차지하는 특칭을 가지고 있다. 이 에 따라 이 세 작용기는 서로 모여드는 형상을 하고 있기 때문에 여러 개의 작
 CH3 CH3
CH3 CH3
용기가 한 지점에 집결하여 효과적인 다작용기 촉매작용을 일으키는 효소계를 쉽게 모방할 수 있다. 55) 68 을 이용하여 분자 인식에 사용한 예를 69ol1 들었다. 염료인 아크리딘 옐로우를 공간자로 사용하여 두 개의 鎬을 연결한 결과 양쪽 의 카르복실기가 서로 마주보게 된다. 이 두 카르복실기 간의 거리는 8-9A 으 로 6 혹은 꾸수형 고리형 디아민이 수소결합에 의하여 끼여드는데 69 에 표시한 피라진의 경우 결합상수가 l,400M-1 이다.
 H2N
H2N
懿 이용한 분자 인식 연구의 주목할 만한 결과로 호스트 분자에 의한 자기 복제를 둘 수 있다. 자기 복제는 DNA 에 저장된 유전 정보를 복제하는 것과 관련하여 생체의 주요 기능을 소형 분자로 재현하였다는 의의가 있다. 7 範t 71 울 결합시켜 7 2.i룹 얻는다. 7~ 근 7~ 71 을 동시에 결합하여 착화합물을 이룬 다. 이 착화합물 속에서 7~ 71 이 반응하여 (73) 7 꺽t 형성하게 되는데 , 727}
 H>2 N/fI >門°HC汀JNHNlr/_ H0\0 xI 2H I CHJ言
H>2 N/fI >門°HC汀JNHNlr/_ H0\0 xI 2H I CHJ言
자기 복제품을 제작하는 데 템플레이트로 사용된 것이다 생체의 유전 정보를 전달하는 과정을 모방한 이 연구의 후속 작업에서 생체의 돌연변이룰 모방한 수정 복제 기능까지 재현하게 되어 그 성과를 가중시키게 되었다. 크라운 에테르류를 사용하여 알칼리 금속 혹은 알칼리 토류 금속 이은을· 인 식하는 예는 앞서 논의한 바 있다. 전이 금속 등 기타 종류의 금속 이온을 안 식하는 것도 분자 인식의 중요한 부분이다. 금속 이온의 생체내에서 담당하는 다양한 역할에 비추어 선택적인 금속 이온의 인식은 많은 연구의 대상이 되고 있다. 그런데 금속 이온은 생물 무기화학에서 비중 있게 다루어지기 때문에 여 기에서는 전형적인 예를 하나만 둘기로 한다. 미생물에서 발견되는 Fe(DI) 이온 의 이동 물질로 엔데로박틴 (74) 이 있다. 엔테로박틴은 철이 결핍된 상태에서 분비되는 물질인데 UH 의 카데콜 히드록시기를 리간드로 사용하여 Fe( 매)이온
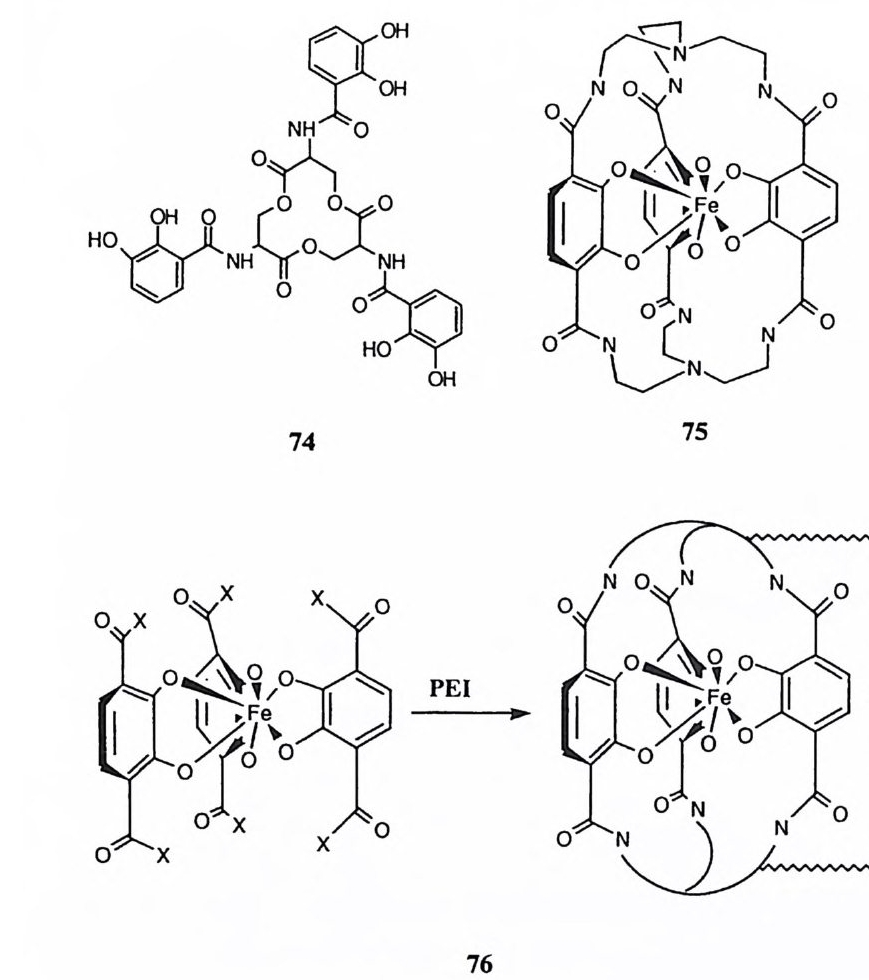 HO 군 NoHyo N ~`\~HoO O: OH~HoH ~ \
HO 군 NoHyo N ~`\~HoO O: OH~HoH ~ \
과 결합하는데 그 결합상수가 10s2M-1 이나 된다. 엔테로박틴이 지니고 있는 트리스 카테콜에이트 구조를 보존하면서 강력한 Fe(lll) 이온의 리간드를 제작하 려는 연구가 시도되었다. 세 개의 카데콜 분자를 동시에 엮어주는 연결자로 여 러 가지 트리아민을 사용하였는바 그 예가 75o] 다. 56) 그런데 인공적인 연결자 를 사용하여서는 리간드로서의 효과가 천연의 엔데로박틴을 능가하는 것이 매 우 어렵다. 이것은 Fe(lll )이온의 결합에 적합한 기하학적인 구조를 제작하려면 연결자의 크기를 바꿈으로써 결합자리의 크기를 매우 미세하게 조정할 수 있어 야 하는데 연결자의 탄소수를 변화함으로써 이러한 효과를 얻기가 용이하지 않
기 때문으로 볼 수 있다. 그런데 Fe(III )이온의 트리스 카테콜 착화합물을 먼저 형성한 뒤에 세 카데콜을 폴리에틸렌이민(제 11 장 참조)이 가지고 있는 아민들로 연결한 결과 (76) 새로운 인공 엔테로박틴이 얻어졌다. 57) 여기에서 풀리에틸렌 이민이 곁사슬이 많은 둥근 구조를 가지고 있기 때문에 적합한 기하학적 구조 를 만들어줄 수 있는 아민들이 연결자로 사용된 것이다. 아상에서 비교적 작은 크기의 호스트 분자가 게스트 분자의 구조를 인식하여 착화합물을 형성하거나 유기 반응의 속도를 증가시키는 예를 살펴보았다. 호스 트와 게스트 간에는 여러 종류의 약한 분자 간의 힘이 활용되어 다분자 결합력 으로 작용하는데, 이러한 분자 간의 힘이 효과적으로 작용하기 위하여서는 호 스트와 게스트의 구조가 상호 보완적이어야 한다. 분자 인식에 이용되는 분자 간의 힘은 이온성 상호 작용, 수소결합, 쌍극자 간의 상호 작용, 전하 이전 상 호 작용 등 국성인 힘과 형수성 상호 작용과 포갬 상호 작용과 갇은 무국성인 힘으로 나눌 수 있다. 이러한 분자 간의 힘에 대하여서는 물리화학, 유기화학, 생화학 등의 분야에서 집필된 기초적인 교과서에서 정보를 쉽게 얻을 수 있다. 단지 포갬 상호 작용에 대하여서는 정보를 얻기가 쉽지 않다. SS tj-6 3oJ]서 예 롤 든 바와 같이 방향족고리가포개질 때 두고리 간에 작용하는 약한 힘을포 갬 상호 작용이라고 한다. 이때 두 방향족 고리는 서로 면끼리 맞대면서 포개 질 수 있고 두 면 간에 각도를 이루면서 포갤 수도 있다. 일부 단백질의 삼차 원 구조에서는 두 방향족 고리가 직각으로 포개지는 예도 발견된 바 있다. 58)
참고문헌 1) Roberts , S. M. ed., Moleculalr Recog n it ion , Chemi ca l and Bio c Jz emi ca l Problems, Roy al Soc iet y of Chem istr y : Cambri dge, 1989. 2) Dietr ich, F. Cyc l op ha nes, Roy al Soc iet y of Che mist ry : Cambri dge, 1991. 3) Bender, M. L. ; Kom iyam a, M. Cyc l odex trin Chemzstry , Sp ringe r Verlag :
New York, 1978. 4) Bender, M . L. In Enzym e Meclzami sms, Page , M.I. ; Wi lliam s, A. Ed., Roy al Soc iety of Chem ist ry : Cambri dg e , 1987 ; Chap ter 4. 5) Dug as , H. Bio o rga n ic Cliem is try, A Clzemi ca l Ap pro ach to Enzym e Acti on , Sp ring e r-Verlag : New York, 1989, 2nd ed., Chap ter s 4 and 5. 6) Str au b, T, S. ; Bender, M. L. J Am. Clzem. Soc., 1971, 84, 8875. 7) Gri ffiths, D. W. ; Bender, M. L. J Am. Clzem. Soc., 1973, 95, 1679. 8) Ste r nbach, D. D. ; Rossana, N. K. ]. Am. Clzem. Soc., 1 982, 104, 5853. 9) Breslow, R. ; Czarn iec ki, M. F. ] . Am. Clzem. Soc., 1978, 100, 7771 . 10) Czarnie c ki, M. F. ; Emert , J . H amagu c hi, H. ; Breslow, R., ]. Am. Chem. Soc. 1975, 97, 670. 11) Tabushi, I. ; Kuroda, Y. ; Mi zu ta n i, T. ]. Am. Che m. Soc., 1981, 103, 711 . 12) Breslow, R. ; Greenspo o n, N. ; Guo, T. ; Zarzyc k i, R . ]. Am. Clze m. Soc., 1989, 111, 8296. 13) Breslow, R. ; Chung, S. J Am. Chem. Soc., 1990, 112, 9659. 14) Breslow, R. ; Ovennan, ]. Am. Chem. Soc., 1970, 92, 1075. 15) Wi lne r, I. ; Goren, Z. Chenz. Commun., 1983, 1469. 16) Breslow, R. ; Dohert y, J. B . ; Gull ilo t, G. ; Lipse y, C. ]. Am. Chem. Soc., 1978, 100, 3277. 17) Tabush i, I. ; Kuroda, Y. ; Moch izu ki, A. J Am. Chem. Soc., 1980, 102, 1152. 18) D'S o uza, V. T. ; Hanabusa, K. ; O'Leary , T. ; Gadwood, R. C. ; Bender, M . L. Bio c hem. Bio ph ys . Res. Commun., 1985, 129, 727. 19) Breslow, R. ; Czarn ik, A.W . ; Lauer, M. ; Lepp k es, R. ; Wi nk ler, J. ; Zim - mennan, S. ]. Am. Chem. Soc., 1 986, 108, 1969. 20) Breslow, R. ; Canary , J. W. ; Varney, M. ; Waddel, S.T.;Yang, D. ]. Am. Chem. Soc., 1990, 112, 5212. 21) Meng er , F. M. ; Lad ika , M. ]. Am. Chem. Soc., 1987, 109, 3145. 22) Suh, J. ; Lee, S. H. ; Zoh, K. D. ]. Am. Chem. Soc., 1992, 114, 7916. 23) Wenz, G. ; Keller, B, Ange w . Chem. Int. Ed. Eng l., 1992, 31, 197. 24) Harada, A. ; Li, J. ; Kamachi , M. Natu re , 1992, 356, 325. 25) Murakam i, Y. ; Kiku chi , J-1 .;0hno, T. Adv. Supr a molecular Chem., 1990, 1, 109.
26) Odashim a, K. ; !tai l, I. ; Iitak a, Y. ; Kog a, K. ]. Am. Chem. Soc., 1980, 102, 2504. 27) Shepo d d, T. J. ; Pett i, M. A. ; Doug he rt y, D. A. ]. Am. Chem. Soc., 1986, 108, 6085. 28) Fergu s on, S. B . ; Die d eric h , F. Ang ew , Chem. , 1 986, 98, 1127 ; Andge w , Chem. Int. E d. E ng l., 1986, 25, 1127. 29) Hami lto n , A. D. Adv. Supr a molecular Chem., 1990, 1, 1. 30) Chang, S. K . ; Hami lto n , A. D. ]. A m. Chem. Soc., 1988, 110, 1318. 31) Wi nk ler, J . D. ; Deshay es , K. ]. Am. Chem. Soc., 1987, 109, 2190. 32) Losensky , H . W. ; Sp el th a nn, H. ; Ehlen, A. ; Vog tle, F. ; Bargo n , J. Ange w . Chem. Int. Ed. E ng l. , 1988, 27, 1189. 33) Seward, E. ; Die d rich , F. Tei. Lett. , 1987, 28, 5111 . 34) Takahashi, I. ; Odashim a , K. : Kog a, K. Tet. Lett. , 1984, 25, 973. 35) Diet r ich , F. ; Dic k , K. Ang ew . Chem. Int. Ed. Engl . , 1 988, 27, 1705. 36) Vog tle, F. ; Muller, W . M. ; Werner, U . ; Losensky, H. W. Ange w . Chem. Int. Ed. Engl ., 1987, 26, 901 . 37) Guts c he, C. D. Caliza renes, R oy al Socie ty of Chem istr y ; Cambri dg e , 1989. 38) Shi nk ai, S ., In Bio o rga n ic Chemi stry Fronti ers , Dug as , H. Ed., S p ring e r- Verlag : Berlin, 1990, Vol. 1, pp. 161-195. 39) Coruzzi, M. ; Andreett i, G, D. ; Bocchi , V. ;Pochin i, A. ; Ung ar o, R. J Chem. Soc. Perkin Trans. 2 , 1982, 1133. 40) Shin k ai, S. ; Mori, S. ; Koreis h i , H. ; Tsubaki, T . ; Manabe, 0 . J Am. Chem. Soc., 1986, 109, 2409. 41) Shin k ai, S. ; Shir a hama, Y. ; Tsubaki, T. ; Manabe, 0. J Chem. Soc. Perkin Trans. 1, 1989, 1859. 42) Shin k ai, S. ; Shi ra hama, Y. ; Tsubaki, T. ; Manabe, 0. ]. Am. Chem. Soc., 1989, 111, 547 7. 43) Pederson, C. J . ]. Am. Chem. Soc. 1967, 89, 2495, 7017. 44) Gokel, G. Crown Eth e rs and Cry ptan ds, Roy al Chem ica l Soci et y : Cam- brid g e , 1991. 45) Sun, J. ; Moon, B. S. ]. Org. Chem., 1989, 54, 2009.
46) Suh, J. ; Heo, J. S. ]. Org. Chem., 1990, 55, 5531 . 47) Cram, D. J. In Ap p!ica tz· o ns of Bio c hemi ca l Sys t em s in Orgn ic Che m is try, Jon es, J. B. ; Sih , C. S. ; Perlman, D. Ed., Wi le y ; New York, 1976, Part Il , Chap. 5. 48) Gokel, G. W. Chem. Soc. Rev., 1992, 21, 39. 49) Lehn, J.-M . Sci en ce, 1985, 227, 849. 50) Cram, D. J. Sci en ce, 1983, 219, 117 7. 51) Hossein i , M. W. ; Blacker, A. W ; Lehn, J. M. ]. Am. Clze m. Soc., 1 990, 112, 3896. 52) Cram, D. J. ; Katz , H . E. ]. Am. Che in. Soc., 1983, 105, 135. 53) Kelly, T. R. ;Magu ire , M. P. J Am. Chem . Soc., 1987, 109, 6549. 54) Jub ia n , V. ; Dixo n. R. P. ; Hami lto n , A. D. ]. Am. Clze m. Soc., 1992, 114, 1120. 55) Rebek, J. J r. Sden ce , 1987, 234, 1478. 56) Garret, T. M. ; McMurr y, T. J. ; Hossein i , M . W . ; Rey es , Z. E . ; Hahn, F. E. ; Raym o nd, K. N. ]. Am. Chem. Soc., 1991, 113, 2965. 57) Suh, J. ; Lee, S. H. ; Paik , H. C. to be pub li sh ed. 58) Ha milto n , A D. Adv. Sup ra mol. C hem., 1990, 1, 1.
제 10 장 막모방화학 제 1 절 생체막 세포는 핵, 미토콘드리아, 리보솜, 골지체 등 여러 가지의 세포기관을 포함 하고 있다. 세포와 이러한 세포기관은 특칭적인 형태를 가지고 있으며 이는 각 각의 기능과 밀접한 관계를 가지고 있다. I) 세포와 세포기관이 일정한 형태와 개체성을 가질 수 있는 것은 막에 의하여 환경과 분리되기 때문아다. I) 생체 막은 주로 단백질과 지방질로 이루어전 조직 호변 조립체이다. 2) 생체 막은 막 지방질이 자체 배열함으로써 얻어지며 두께가 60-lOOA 인 얇은 판과 같은 구조를 하고 있다. 막 지방질 사이에 특이한 단백 질이 존재하여 막들의 독특한 기능을 매개하고 있다. 막을 형성하는 막 지방질은 포스포글리세리드, 스핑고미엘린, 당 지방질, 콜
 [IIIII IIIIIIIIII
[IIIII IIIIIIIIII
 ~一o。 0 \_ 0g. - 0 三戶
~一o。 0 \_ 0g. - 0 三戶
레스테롤 등인데, 이들은 형수성 부분과 친수성 부분을 동시에 보유하고 있는 양쪽성 화합물이다. 이들은 자체 배열에 의한 집적의 결과로 이분자 층 (1: 둥 근 부분은 천수성 머리, 긴부분은 협수성 꼬리)을 형성한다. 1 에서는 편의상 이분자 충을 1 차원적으로 표시하였지만, 이것을 확장하여 2 차원적으로 배열하 면 막의 면이 형성된다. 그리고 이것을 杯 R} 적으로 확장하여 배열하면 주머니 와 같은 구조가 생기는데 이러한 주머니를 리포솜 혹은 베시클이라고 부른다. 포스포글리세리드는 글리세롤 뼈대, 두 개의 지방산 사슬, 인산화된 알콜로 구성되는데, 포스파티딜콜린 (2) 을 예로 들 수 있다. 스핑고미엘린 (3) 은 스핑 고신 (4) 을 뼈대로 하여 형성되는데, 포스포글리세리드와 더불어 인산 지방질에 속한다. 당 지방질은 스핑고신에서 유래되며 당을 친수성기로 함유하고 있는 데, 세레브로시드 (5) 를 예로 둘 수 있다. 콜레스테롤 (6) 은 C 궁의 히드록시기 가천수성부분이다.
제 2 절 미셀 제 1 절에서 디룬 바 있듯이 분자내에 형수성 부분-과 친수성 부분을 둘다 포함 하고 있는 양쪽성 화합물인 계면활성제를 일정한 농도 이상으로 녹이면 분자끼 리 회합하여 대형 집합체를 형성하는데 분자의 구조에 따라 미셀, 베시클 혹은 더 큰 집단을 형성하게 된다. 협수성 사슬과 천수성 머리가 한 개씩 포함된 경 우에는 흔 히 미셀을 형성한다. 물 속에서 형성되는 미셀의 경우에는 계면활성 제 분자가 임계 미셀 농도 이상으로 물에 용해될 때 20-10 ()7 H 의 분자가 집적되 어 반경 12 - 30A 의 미셀이 얻어진다. 3)-5) 이 집적체에서 형수성 부분은 내부로 향하고 친수성 부분은 용매 쪽으로 향하여 7 과 같은 구조를 가지게 된다. 유기 용매에서는 친수성 부분이 내부로, 형수성 부분이 용매 쪽으로 향하여 역전 미 셀을 형성하기도 한다. 미셀의 공 모양 구조에 대하여서는 7보 다 더 개선된 모 델들이 제시된 바 있다. 6) 미셀의 형태는 이러한 구형 이의에도 계면활성제의 구조와 농도에 따라 막대 모양 등 더 복잡한 형태를 가지기도 한다. 임계 미셀 농도는 계면활성제의 구 조 용매, 전해질의 종류와 농도 등에 크게 의존한다. 물 속에서 형성된 미셀의 내부는 액체 탄화수소와 같은 성질을 띠게 되고, 미셀 표면의 친수성 부위에 인접한 부분은 스턴 충이라고 불리는 특이한 성질
 7
7
울 가진 매질을 제공하게 된다. 미셀의 친수성 부분이 이온성이면 스턴 충에서 는 이것의 반대 이온의 전체 양 중 50 - 70% 가 수용된다. 스턴 충은 물과 탄화 수소의 중간 성질을 가지게 되는데, 스턴 충 내부의 용매로서의 성격은 미셀 표면에서의 거리에 따라 달라지게 된다. 소형 분자가 미셀과 혼합되면 미셀의 내부 혹은 표면에 흡착되어 착화합물을 형성한다. 이때에 이러한 소형 분자와 미셀 간의 형수성 작용과 극성 작용이 착화합물의 형성에 개입된다. 미셀에 의한 촉매작용은- 기질과 착화합물을 형성한 뒤 진행된디는 점에서 효 소 반응과 공통점을 지니고 있다. 그러나 효소-기질 착화합물에서는 기질의 자 유도가 크게 구속되어 촉매 작용기와 기질의 반응부위 사이에 밀접한 인접 효 과를 성취할 수 있음에 바하여 미셀-기질 착화합물에서는 기질이 비교적 자유 로이 움칙일 수 있다는 접에서 차이가 있다. 미셀-기질 착화합물 속에서 반응부위 간의 유효 농도의 증대와 바닥 상태와 전이 상태의 상대적인 안정성에 대한 용매 효과가 주요한 요인으로 작용하여 촉매작용을 일으키게 된다. 형수성 혹은 극성 효과에 의하여 미셀의 내부 혹은 표면에 반응물이 선택적 으로 흡수되어 반응부위 상호 간의 유효 농도가 증가할 수 있다. 양이온성 미 셀 표면에 수산화 이온이 선택적으로 흡수됨으로써 미셀 표면의 유효 p H 가 증 가하는 것도 이것의 예로 들 수 있다. 촉매 작용기를 계면활성제에 공유 결합 에 의하여 연결한 경우에는 미셀 표면에 선택적으로 흡착된 기질과 이 촉매 작 용기 간의 유효 농도가 증가되어 반응속도를 제고할 수 있다. 이미다졸이 친핵성 촉매로 작용하는 p-니트로페닐 아세데이트 (PNPA) 의 에 스테르 가수분해 반응의 속도에 대한 미셀의 영향을 예로 들어 보자. 양이온성 미셀의 한 부분으로서 이미다졸이 존재하는 C,sH31N+ (CH3) 2CH2Im (Im : 이미 다졸릴)의 반응성은 이미디졸보다 약 3(}l lI 높은 값을 보인다. 7),8) 이것은 PNPA 가 형수성 작용에 의하여 미셀과 착화합물을 형성하면 미셀의 친수성 표 면에 노출된 이미다졸기와 PNPA 간의 유효 농도가 증가한다는 것 (8) 만으로 도설명이 가능하다. 미셀과 기질 간의 형수성 작용을 증가시키면 촉매 작용기와 기질 간의 유효
 8
8
농도를 더욱 증가시킬 수 있다. 예를 들어 미셀인 N- 미리스토일 -L- 히스티딘 에 포함된 이미다졸기와 P- 니트로페닐 핵사노에이트와의 반응성은 미셀이 아닌 N- 아세틸 -L- 히스티딘의 이미다졸기와 p-트로페닐 핵사노에이트와의 반응성보 다 3,300 배가 더 크다 .9) 중성인 분자가 이온화하여 음이온으로 전환된 후에 촉매 작용기로 행동하는 경우가 있다. 옥십이 옥심 음이온으로 . 이온화되어야 친핵체로 작용하는 것 (9) 10) 을 그 예로 들 수 있다. 이러한 촉매 작용기가 양이온성 미셀의 표면에 위치하는 경우 미세 환경의 정전기적 효과에 의하여 촉매 작용기의 이온화가 촉진되고 이에 따라 촉매 작용기의 반응성이 제고될 수 있다. 미셀의 특성과 친핵성 치환기의 효용을 조합하여 효과적인 촉매를 개발한 사 례로 인산 트리에스테르의 가수분해에 대한 계면활성제 촉매를 둘 수 있다. 인 산 트리에스테르는 인산 에스테르 유도체인 신경 가스와 유사한 반응성을 가지 고 있기 때문에 인산 트리에스테르 가수분해에 대한 촉매는 신경 가스 파괴를
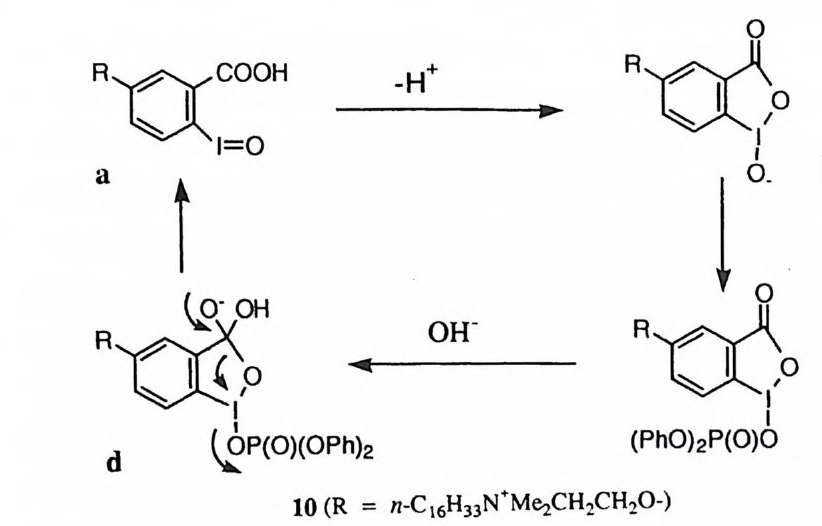 RUCOOH -H+ R~O
RUCOOH -H+ R~O
위한 촉매를 개벌능}기 위하여 각국에서 연구되고 있다. 인산 트리에스데르의 가수분해에 대하여 0 갑 요도소벤조산을 촉매 작용기로 장 착한 양이온성 미셀이 뛰어난 촉매 효과를 보이고 있다. 11) 이 촉 매작용에는 10 에 표시한 바와 같은 메커니즘이 작동하는데, 요도소기에 카르복실 음이온이 첨가되어 음이온성 친핵체 b 가 생기면 이 친핵체가 기질인 인산 트리에스테르 룰 공격하여 증간체 C 를 형성하게 되며, 이 중간체는 수산화 이온의 공격을 받 아 파괴되면서 촉매를 재생하게 된다. 이 반응의 촉매 효과가 높으려면 중간체 의 생성 단계의 속도와 파괴 단계의 속도가 모두 높은 수준으로 가속되어야 한 댜 미셀의 스턴 충 속으로 협수성 기질이 몰려 들게 됨으로 친핵체와 기질 간 의 유효 농도가 증가하여 중간체 생성 단계가 가속된다. 또한, 미셀의 양이온 성 매체내에서 수산화 이온의 유효 농도가 증가하게 됨으로 중간체의 파괴 단 계가 가속된다. 이에 따라, 중간체 생성 및 파괴 단계가 모두 증속되어 전체적 으로 효율적인 촉매 효과를 시현하게 되는 것이다. 이와 유사하게 미셀에 장착된 카르보닐기의 수화물이 친핵체로 작용하여 카 르복실 에스테르와 인산 트리에스테르를 공격하는 단계와 이로 인하여 형성된 중간체의 파괴 단계가 효율적으로 진행됨으로써 효과적인 촉매작용이 실현된 예 (11) 도 보고되어 있다. 12) 미셀 촉매계는 효소의 특성 중 몇 가지를 재현할 수 있고 이에 따라 상당한
 yH3 。 fH3 0H
yH3 。 fH3 0H
수준의 촉매 효과를 보인 예가 다수 있다. 그러나 미셀을 실용성이 있는 촉매 로 사용하기에는 제약이 있다. 크기가 제한되어 있기 때문에 여러 가지 촉매 요인을 동시에 도입하는 것이 용이하지 않다. 특히 매체에서 유기 용매의 함량 이 증가하면 미셀의 구조가 파괴되기 때문에 높은 농도의 기질을 녹일 수 없는 예가 허다하여 실용적인 사용이 어렵다. 이러한 단점은 고분자 골격 위에 미셀 과 같은 미세 환경을 조성하거나, 대규모로 분자가 집적되어 형성되는 베시클 울 사용함으로써 개선할 수 있다. 제 3 절 인공 막 생체내에서 발견되는 양쪽성 분자 이의에도 인공적으로 합성한 앙쪽성 분자 를 사용하여 막을 형성하고 베시클을 제작한 뒤, 이를 이용하여 여러 가지 기 능을 수행하는 연구가 활발히 진행되고 있다. 디옥타데실 디메틸 암모니움 클 로리드 (12) 는 양이온성 극성 머리에 형수성 꼬리가 두 개가 연결된 구조를 가 지고 있고, 디핵사데실 포스페이트 (13) 는음이온성 극성 머리에 형수성 꼬리가 두 개 연결된 구조를 가지고 있다. 이들은 꼬리가 한 개인 유사 구조체가 미셀 울 형성함에 반하여 수용액 상에서 베시클을 형성한다. 13) 양쪽성 화합물로부터
 CH3(CCHH:3 v(1C sH— : v-N1' IsH+ —3 CH3 er CH3(CCHH3 기(C 1H5. v01- sRPOI ― -o·
CH3(CCHH:3 v(1C sH— : v-N1' IsH+ —3 CH3 er CH3(CCHH3 기(C 1H5. v01- sRPOI ― -o·
베시클을 형성시킬 때에는 상전이 온도 아상에서 초음파로 처리하게 된다. 상 전이 온도 이하에서는 겔과 같은 구조를 가지고 있다가 상전이 온도 이상에서 탄화수소 사슬이 서로 융화하면서 유동적인 구조를 가지게 된다. 12 혹은 133 1} 같은 디알킬 양쪽성 화합물 이의에도 긴 사슬을 한 개만 가진 양쪽성 화합물로도 여러 가지 분자 집적체가 형성되는 것이 알려져 있다. 14) 이 경우 자체 배열에 의하여 이분자 충을 이루고 이를 통하여 거대한 집적체를 형 성하려면 14ol] 표시된 것과 같은 구조적인 요건을 충족시켜야 한다. 견고 부위 의 예로는 디페닐아조메틴 (15) , 비페닐 (16) , 아조벤젠 (17) 등이 사용될 수 있 다. 한편 견고 부위 양쪽에 알킬 사슬을 연결한 뒤, 그 두 끝에 천수성 머리를 붙일 경우에는 일분자 충의 막이 형성될 수도 있다. l4) 인공 양쪽성 분자에 의하여 형성된 분자 집적체는 여러 가지 형태를 가질 수 있다. 둥근 주머니, 길쭉한 주머니, 양파와 같은 여러 겹의 껍질 구조, 원판 형, 튜브형, 막대형, 리본형, 꼬인리본형등다양한구조가보고된바있다. 생체 모방 막을 기능적으로 응용한 사례의 일부를 소개하면 다음과 같다.
비균질성 촉매의 촉매 효율은 촉매 입자의 크기를 줄이고 표면적을 넓힐수록 증가할 수 있다. 이에 따라 수소화 반응의 촉매로 쓰이는 금속을 미세한 크기 로 생체 모방 막 사이에 분산시킴으로써 촉매 효과를 제고할 수 있다. 15) 반도체 금속염 (예 : CdS) 을 생체 모방 막에 미세한 크기로 분산시킨 뒤 빛을 쪼여 여기시킨 뒤 아를 이용하여 물을 환원하여 수소를 발생시키는 것과 같은 산화환원 반응을 유도하는 연구도 수행되고 있다. 15) 막에 분산된 반도체는 표 면적이 넓어 빛을 받아들이는 효율이 높고, 크기가 작아서 분산된 빛으로 인한 간섭 효과를 최소화함으로써 전자 이동예 관한 연구에 상당한 장점을 제공하기 도한다. 자성 입자를 생체 모방 막에 분산시킴으로써 자성 물질이 삼차원적으로 적절 히 배열됨으로써 각종 홍미 있는 현상을 일으킬 수도 있다. 15) 생체 막이 양쪽을 격리시키고 환경을 차별화할 수 있고 이에 따라 물질과 에 너지를 이동시킬 수 있는 성질을 생체 모방 막을 이용하여 연구하는 활동도 왕 성히 전개되고 있다. 베시클의 이분자 충을 통과하는 에너지와 전자 이동은 광 에너지를 화학 에너지로 변환하는 데 응용될 수 있다. 16) 생체 모방 막의 양쪽 에 전자 주게와 전자 받게를 격리시키고 광화학적으로 전자 주게를 여기시킨 뒤 이들 간에 전자가 이동되는 연구도 수행되었다. 13) 최근에는 물의 광화학적 인 분해로 배시클의 양쪽에서 각각 산소와 수소를 별도로 발생시키는 응용 연 구도 추진중에 있다. 16) 이러한 연구가 결실을 보면 에너지 문제의 해결에 상당 한 도움을 줄 수 있을 것이다. 한편 베시클내에 의약품을 포획시켜 의약 전달에 사용하는 연구도 활발히 진 행되고 있다. 18) 이 방법을 사용하면 무여량을 감소시킴으로써 각종 부작용을` 감소시키는 이접이 있다. 베시클의 여러 가지 구조적 요인에 따라 베시클에 포 획된 의약은 특정 조직의 세포에 이동된 뒤 의약을 방출하게 된다. 베시클이 갖는 가장 큰 결함은 낮은 안정성이다. 이를 보완하는 방법으로 최 근에 제시되고 있는 것이 베시클의 중합이다. 베시클을 형성하는 계면활성제의 형수성 사슬 부위 혹은 국성 머리 부위에 에틸렌 아중결합을 도입하고 이러한 계면활성제를 자체 배열하여 베시클을 형성한 뒤 자의선을 쪼여 주거나 중합 개시제를 가함으로써 에틸렌 이중결합 사이에 중합을 유도할 수 있다. 17)-19) 베
시클을 형성하는 계면활성제 중 일부분을 공유결합으로 연계시킴으로써 베시 클을 안정화하여 30% 에탄올 속에서도 파괴되지 않음이 관찰되었다. 중합된 베시클은 고분자의 안정성 및 견고성을 베시클의 유동성 및 두과성과 조합시킴 으로써 실용적인 면을 제고할 수 있는 것이다. 베시클내에 존재하는 에틸렌 이중결합 간에 중합이 일어나는 정도는 반응조 건에 따라 차이가 있지만 부분적인 중합에 의하여서 베시클의 안정도를 증가시 키면서 베시클 안의 침투성을 유지하게끔 조절할 수도 있다. 에틸렌 이중결합 이 계면활성제의 협수성 사슬의 끝 부분에 위치하고 있으면 이분자 충 베시클 내에서 중합이 일어날 경우에 같은 분자 충내에 있는 계면활성제끼리 연결될 수도 있고 다른 분자 충에 있는 계면활성제 간에도 교차 중합이 일어날 수 있 다.
 O_/
O_/
중합 반응은 비금속 원자 간에 공유결합이 형성됨으로써 진행될 수도 있지 만, 금속 이온을 사이에 두고 리간드가 금속에 배위됨으로써 배위 중합체를 형 성할 수도 있다. 예를 들어 2, 2' -디히드록시아조벤젠은 18 혹은 19ol] 표시한 바와 같이 야조 질소와 페놀 산소를 주게원자로 이용하여 금속 이온과 배위 중 합체를 형성한다. 20) 2, 2' -디히드록시아조벤젠 부분을 친핵성 머리 부분에 포함한 계면활성제 (20) 를 Fe( Il ) 혹은 Fe (lll)이온에 배위시킴으로써 배위 중합 베시클 (21) 울 합 성한 사례가 있다. 이와 같이 합성된 배위 중합 베시클은 매우 뛰어난 안정성 울 가진바, 에탄올 속에서 3~ 손간 5 0 C 에서 가열하여도 분자 집적체의 구조를 유지하고 있다. 21) 구조 21 에는 계면활성제 분자 y n 가 철이온에 의하여 배위
 HO:HO O
HO:HO O
중합되는 과정을 도식적으로 표시하고 있다. 이와 같이 얻은 배위 중합 베시클은 안정도가 뛰어날 뿐 아니라 중합 과정에 개입한 금속 이온이 가지는 전자적, 자기적, 촉매적 성격을 활용할 때 여러 가 지 기능을 수행할 수 있을 것으로 예측된다. 한 예로, Fe(][ )이온과 Fe( 風)이 온을 섞어서 2 範} 배위 중합체를 형성하면 반도체 정도의 전기 전도도를 가지
게 된다. 이러한 전기 전도성은 22oll 도시한 바와 같이 철이온 간에 전자가 이 동하기 때문으로 설명된다. 20) 이와 같은 전도성 물질은 새로운 유형의 전도성 고분자로서 다양한 실용적인 용도를 가질 것이 예상된다. 1970 년대 종반 아후 에 전도성 고분자에 대한 연구가 폭발적으로 진행되고 있지만, 전도성 고분자 를 실용적인 용도에 사용하는 것은 현재로는 많은 난관이 있다. 지금까지 개발 된 전도성 고분자는 도핑 괴정을 통하여 카르보 양이온 등의 불안정한 구조를 도입하여야 하기 때문에 환경적인 안정성이 결여되고 많은 경우에 가공성에 문 제가 있었다. 21 과 갇은 배위 중합 베시클은· 도핑이 필요없는 본원적인 전도성 울 보이고 있으며 에탄문세 녹기 때문에 가공성이 문제시되지 않는다.
참고문헌 1) Lehninge r, A. L. B io c hemis try, Wort h Publi sh ers : New York, 1973. 2) Str yer , L. Bio c hemis try, Freeman : San Franci sc o, 1975, Chap ter 10. 3) Cordes, E. H . ; Git ler , C. In Progr e ss in Bio o rga n ic Chemi stry, Kais e r, E. T . ; Kezdy, F. J. Ed., Wi le y : N. Y., 1973, Vol. 2, pp. 1-54. 4) Fendler, J. H. ; Fendler, E. J . C ata ly si s in Mi ce llar and Macromolecular Sys - tern s, Academ ic P ress : N. Y. , 1975. 5) Ku nitak e, T. ; Shi nk ai, S. Adv. Phys . Org. Chem., 1980, 17, 435. 6) Meng er , F. M. Acc. Chem. Res., 1979, 12, 111 . 7) Taga ki, W. ; Chig ira , M. ; Amada, T. ; Yano, Y. ]. C hem. Soc. Commun., 1972, 219. 8) Taga ki, W. ; Fukushim a, D. ; Eik i , T. ; Yano, Y. ]. Org. Chem., 1979, 44, 555. 9) Git ler , C. ; Ochoa-Solano, A. ]. A m. Chem. Soc., 1968, 99, 5004. 10) Yats ilnirs ki , A. K ; Mart ine k, K ; Berezin , I. V. Tetr ah edron, 1971, 27, 2855. 11) Moss, R.A.;Kim, K.Y .;Swarup, S. JA m. Chem. Soc., 1986, 108, 788.
12) Meng er , F. M . ; Whit es ell, L. G. ]. Am. Chem. Soc., 1985, 107, 707. 13) Fendler, J. H. Acc. Chem. Res., 1980, 13, 7. 14) Kunit ak e, T. ; Okahata , Y. ; Shim omura, M. ; Yasunami, S.-I. ; Takarabe, K. ]. Am. Chem. Soc., 1981, 103, 5401 . 15) Fendler, J. H. Chem. Rev., 1987, 87, 877. 16) Robin s on, J. N. ;Cole-H am ilto n , D. J. Che1n. Soc. Rev., 1991, 20, 49. 17) Reg en , S. L. ; Czech, B. ; Sin g h , A. ]. Am. Chem. Soc., 1980, 102, 6638. 18) Kunit ak e, T. ; Nakashim a, N. ; Takarabe, K. ; Nag ai , M. ; Tsug e, A . ; Uanagi , H. ]. A m. Chem. Soc., 1981, 103, 5945. 19) Fendler, J. H. Acc. Chem. Res., 1984, 17, 3. 20) Suh, J. ; Oh, E. Syn th. Met., 1990, 39, 177. 21) Suh, J. ; Bae, K. T. ; Kwon, 0.B . ; Lee, K . J. to be pub lish ed. 22) Sun, J. ; Oh, E. ; Kim , H. C. Syn th. Met., 1992, 48, 325.
제 11 장 합성 고분자와인공효소 제 1 절 미세 환경 합성 고분자가 형수성기와 전하를 동시에 보유하고 있으면 미셀과 폴리 전하 질의 구조적 요인을 동시에 보유하게 된다. 이에 따라 미셀계에 대하여 단점으 로 간주되는 구조의 불안정성이 해소된다. 뿐만 아니라, 고분자에 별도로 여러 가지 촉매 요인을 도입함으로써 미셀이 가지는 촉매로서의 한계점을 극복할 수 도있다.
 \^〈三
\^〈三
이러한 목적을 위하여 사용된 고분자 골격의 예로 폴리 (비닐피리딘)의 유도체 (예 :1)' 폴리(비닐이미다졸)의 유도체(예 :2) 및 이들의 공중합체를 들 수 있 다. 1),2) 이러한 비닐 유도체의 고분자는 직선형의 골격 구조를 가지고 있다. 이 골격 위에 협수성 시술과 전하를 도입하면 국부적으로 미셀과 같은 구조를 가 지게 되어 구형의 형태를 가질 수 있다. 그러나 분자 전체로는 이러한 미셀 덩 어리가 선형으로 배열된 구조를 가지게 된다. 이러한 고분자들을 가지고 작업 할 때에는 용해도가 낮은 점이 현실적으로 봉착하는 애로사항이다.
 :SN H2 •••• , , H .iNH H ~
:SN H2 •••• , , H .iNH H ~
폴리 에틸렌이민 ”(PEI , 3)3) 은 에틸렌이민의 중합체이어서 에틸아민기가 반복 되는 구조를 가지고 있다. 인공 효소의 골격으로 가장 많이 사용된 것은 분자 량기 약 硏R l 것인데, 여기에는 단량체가 약 1,4 0 () 7I 포함되어 있다. 1,40()7 l 의 아민 질소 중 약 25% 는 일급 아민이고, 50% 는 이급 아민, 25% 는 삼급 아민이다. 삼급 아민은 이 고분자의 골격에서 결사슬이 생기는 부분이 된다. 따라서 이 고분자는 결사슬이 매우 많아서 전체적으로 둥근 형태를 가진다. 아 민 질소가 차지하는 중량비가 커서 물에 대한 용해도가 매우 크며, 여러 가지 형수성기나 유기 작용기를 도입하는 경우에도 물에서의 용해도로 인한 큰 문제 점이 없다는 장점이 있다. PEI 는 아민 질소를 알킬화, 아실화, 혹은 이민화 반응게 의하여 수정함으로써 협수성 사슬뿐 아니라 각종 작용기를 용이하게 도 입할 수 있다는 이점이 있다. 비닐 유도체의 중합체의 경우에는 모노머의 구조 를 수정한 뒤 중합 혹은 공중합에 의하여 새로운 고분자를 흔히 제작하게 되는 바, 분자 크기를 일정하게 유지할 수 없는 문제점이 있으며 고분자의 골격에 일관성을 기할 수 없다는 단점이 있다• 고분자를 곱격으로 제작한 폴리 비누의 미세 환경은 미셀의 경우에 사용한 기법에 따라 메틸 오렌지, 디클로로페놀-인도페놀 등의 흡수 스펙트럼이나 아 닐리노나프탈렌 술포네이트 이온의 형광 스펙트럼으로 조사하기도 한다. 2) 한 편, 미세 환경에 민감한 화학반응을 이용하여 환경에 대한 정보를 수집하기도 한다. 예를 들어 心] 반옹은 미셀의 존재하에서는 가속되지만, 폴리 전해질의
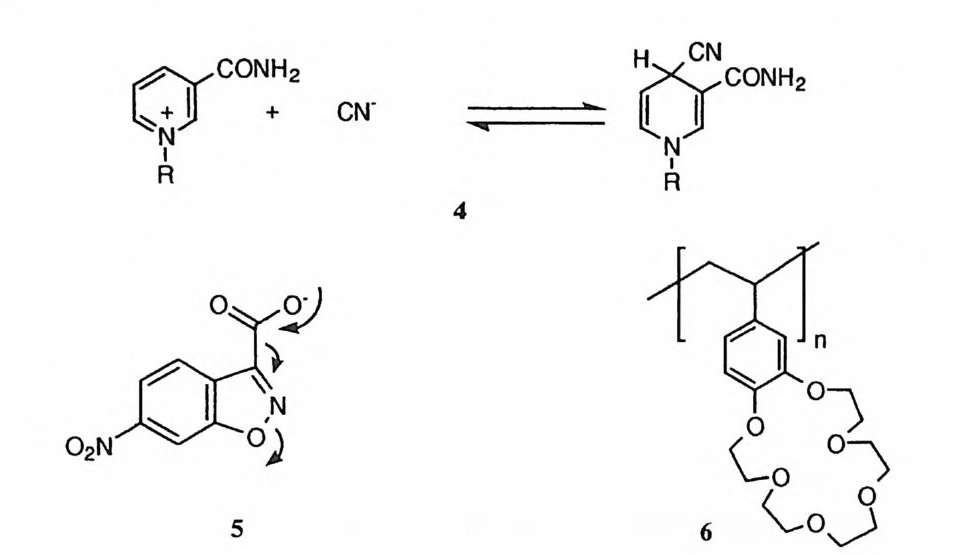 IN訂c 。N H 2 + c N 4 ` ~CR ONH2
IN訂c 。N H 2 + c N 4 ` ~CR ONH2
존재하에서는 감속되는 것이 알려져 있다. 이에 착안하여 E| 유도체의 구조에 따라 미셀의 성격 혹은 폴리 전해질의 성격을 가지는가를 판별할 수도 있었 다. 2) 이상에서 거론된 조사용 분자는 조사 대상 고분자와 착화합물을 만든 뒤 그 자신이 위치한 지점에서의 매질의 성격에 대한 정보를 제공하게 된다. 이러 한 제약점을 극복하기 위하여 조사용 분자를 포함하는 모노머를 제작한 뒤 이 들을 조사 대상인 고분자 속에 공중합에 의하여 포함시킨 다음 고분자 골격 위 에서 매질의 성격에 대한 정보를 수집하도록 하는 경우도 있다. PEI 의 경우에도 아민기를 긴 사슬로 아실화 혹은 알킬화할 경우 이 사슬들 이 집적되어 미셀과 같은 구조를 형성하게 된다. 이에 따라 소형 유기 분자에 대한 친화력이 매우 증대되어 용이하게 착화합물을 형성한다. 4> PEI 유도체의 형수성 미세 환경은 불소 핵자기 공명으로 측정한 바 있다. 5) 또한 5로 표시한 털카르복실화 반응은 음전하의 밀도가 전이상태에서 분산되는 유형의 반응으로. 그 속도가 매체의 성격에 민감하게 의존하는바, 여러 PEI 의 알킬화 유도체 6) 및 기타 폴리머 유도체에 대하여 그 속도를 측정한 바 있다. 3) 이 탈카르복실화 반응속도에 따르면 도데실기 등으로 알킬화된 PEI 유도체는 /3-시클로덱스트 린, 0詞 온성 미셀, 알킬화된 폴리 (바닐이미다졸) 유도체 , 폴리 (크라운 에데르) 유도체 6 등과 비교하여 형수성이 더 큰 미세 환경을 가지고 있다.
 『H NN 느 N
『H NN 느 N
PEI 의 아민 질소 중 10% 를 3-(2 - 이미다졸릴아조)벤조일(이마벤)기로 아실화 하여 얻은 이마벤 -PEI 에서는 이마벤기끼리 포갬 상호 작용에 의하여 서로 회 합하여 콤팩트한 형태 (7) 를 가지고 있다. 7) 이에 따라 폴리머의 의부로 노출된 아민의 양이 PEI 나 PEI 의 아민 중 10% 가 도데실화된 도데실 -PEI 와 비교하 여 훨씬 적다. 그리고 9- 안트라센카르복실산 음이온이 도데실 -PEI 에는 거의 결합하지 못함에 비하여 이마벤 - PEI 와는 매우 효율적으로 결합하는데 , 이는 이마벤 부위와 6- 안트라센카르복실 음이온 간의 포갱 상호 작용 (8) 에 기인한 다. 제 2 절 결합부위 제 9 칭에서 논의한 각종 호스트 분자를 사용하여 인공 효소를 제작하는 것은 호스트 분자의 크기 때문에 파생되는 제약점 때문에 용이하지 않다. 즉, 효~ 와 같이 다양한 촉매 요인을 인공 촉매에 도입하기 위하여서는 상당히 큰 분자 골격을 필요로 하는 것이다. 이러한 관점에서 인공 고분자를 골격으로 사용하 는 것이 인공 효소를 더 용이하게 제작하는 방법이 될 수 있다. 비닐 유도체의 중합체의 경우에는 모노머에 소기의 촉매 요인을 도입한 뒤 모노머를 중합함으로써 폴리머에 각종 촉매 작용기를 도입한다. PEI 의 유도체
의 경우에는 PEI 골격 위에 있는 아민 질소를 화학적으로 수정하여 각종 촉매 요인을 도입하게 된다. 이러한 전략에 따라 폴리머를 가공하게 되면 구조적 요 인을 계획성 있게 도입하기가 불가능하다. 뿐만 아니라 이러한 고분자를 촉매 로 사용하면 미셀 촉매계에서와 같이 고분자의 표면에 기질이 흡착되어 반응을 일으키게 되므로 효소와 같이 정교한 구조적 환경 속에 기질을 결합시킬 수가 없다. 고분자에 특정한 구조를 가진 기질만 수용하는 결합 부위를 창출한 최초의 성공적인 사례로 /3-시클로덱스트린(제筑l 참조)을 PEI 에 연결하여 얻은 CD-
 尸
尸
PEI 를 들 수 있다. 8) /J-시클로덱스트린의 일급 히드록시기를 토실화하여 얻은 토실-{J -C D-를 PEI 의 아민으로 친핵성 치환반응을 일으켜 CD-PEI 를 제작한 다. CD-PEI 는 PEI. £. 하여금 고분자로서는 특이하게 구체적인 결합 부위를 보유하게 하고, g-시클로덱스트린으로 하여금 자신의 구멍 위에 각종 반응기 를 도입할 수 있도록 하는바, 이를 歸 l 도식적으로 설명하였다. 에스데르 10 혹은 11 을 CD-PEI 와 반응시킨 결과, 이들의 p-t유 L 틸페닐기가 시클로덱스트 린의 구멍에 결합한 뒤, PEI 골격 위에 위치한 아민이 친핵체로 공격함 (12) 으 로써 매우 효과적으로 탈아실화 반응이 일어남을 관찰하였다.
 13
13
후술(제 4 절)하게 될 PEI 에 구축된 폴리아자마크로 고리 금속 착화합물에서는 금속 이온이 정전기적 작용에 의하여 음이온에 대한 결합 부위 역할을 담당하 고있다. 효소와 갇은 특성을 가지면서 효소와 같은 효율을 가전 촉매를 합성 고분자 를 골격으로 사용하여 제작하려면 특정 구조를 인식하는 결합 부위 이외에도 이 부위와 협동적으로 작용할 수 있는 촉매 작용기를 인접한 위치에 도입할 수 있어야 한다. CD-PEI 에서 출발하여 원하는 위치에 촉매 작용기 를 도입하 는 작업이 시도되었다. 즉, 시클로덱스트린의 구멍에 친화력이 있는 p - t나 L 틸페 닐기와 소기의 촉매 작용기를 포함하고 있는 분자(예 : 13,9) 14 10) ) 를 CD-PEI 와 반응시켜 1 쩌] 표시한 바와 같이 PEI 골격에 위치한 아민기에 아실기 를 이동 시킴으로써 원하는 작용기를 결합 부위 주위에 도입할 수 있는 것이다. 합성 고분자 위에 구체적인 결합 부위를 도입하고 그 인접 위치에 각종 촉 매 작용기 를 도입하는 기법이 개발되면 합성 고분자를 골격으로 사용하여 인공효소를 제 작하는 작업이 전일보하게 될 것이다. 제 3 절 유기 촉매 작용기 앞 철에서 설명한 바와 같은 결합 부위를 보유한 고분자 촉매는 극히 최근에 제작되기 시작하였다. 종전에는 폴리 비누로서 고분자를 가공하여 여러 가지 유기 반응에 대하여 촉매로서의 효율을 조사하였다. 폴리 비누로서 고분자가 촉매로 작용할 경우에는 미셀 촉매작용에서 관찰된 촉매 요인이 주로 작동한 다. 예컨대, 형수성 미세 환경을 이용하여 반응속도를 제고한 사례가 자주 보고 된 바 있다. 앞서 논의한 바 있는 모] 탈카르복실화, 친핵성 방향족 치환반 응 11) 등은 형수성 매질 속에서 상당한 속도 증가를 보이는 반응인바, 합성 고 분자의 미세 환경 속에서 촉매 효과를 관찰한 바 있다. 미셀 촉매계에서와 마찬가지로 폴리 비누의 미세 환경은 각종 약산의 이온화 에 정전기적인 효과를 미치게 된다. 양이온성 매질은 음이온이 형성되는 것을
돕게 된다. 경우에 따라서는 유효 p H 가 증가하여 유효 수산화 이온 농도가 증 가함으로써 수산화 이온에 의한 반응속·도가 제고되어 촉매작용을 실현하기도 하는데, 트리플루오로아세트 아닐리드 류의 가수분해가 PEI 유도체에 의하여 촉매작용을 받는 것을 그 예로 들 수 있다. 12) 폴리 비누의 미세 환경이 가지고 있는 협수성과 정전기적 특성 때문에 각종 분자가 선택적으로 흡착되면, 이들 간에 유효 농도가 증가하게 되고 이로 인하 여 이 들 간의 반응속도가 증가하게 된다. 폴리 비누에 공유 결합에 의하여 각 종 촉매 작용기가 도입되기도 하고 단순한 흡착 과정을 통하여 유입되기도 한 다. 폴리 비누계에 사용된 각종 유기 촉매 작용기를 살펴보기로 하자. 효소의 촉매 작용기로 사용되는 국성 유기 작용기는 이미디졸, 카르복실, 페 눌 히드록실, 술피히드릴, 아미노 기 등이다. 이와 관련된 작용기들이 고분자 에 의한 촉매작용에서 활용된 바 있다. 이미다졸이 탈아실화 반응에서 친핵성 공격을 통하여 아실-이미다졸 중간체 를 형성하는 경우 이 중간체의 파괴가 원활히 일어나기 때문에 탈아실화 반응 에 대한 진정한 촉매로 작용할 수 있다. 이 점에서 피리딘은 이미다졸의 유사 체로 볼 수 있다. 양이온성 미셀의 존재하에서 P- 니트로페닐 아세데이트에 대 햐겨 이미다졸 자체의 반응성은 두드러지게 제고되지 않고 이미다졸 음이온에 의한 반응성은 매질 효과에 의하여 상당히 증가한다. 13) 폴리 비닐이미다졸이나 폴리 비닐피리딘의 경우에 14),15) 이미다졸이나 피리딘의 대부분이 중성인 염기형 으로 존재하면 에스데르에 대한 반응성이 상응하는 모노머 친핵체와 비교하여 큰 차이를 보이지 않는다. 단지 p H 가 낮아져서 고분자 속의 일부 염기가 양성 자화되었을 때에는 음이온성 에스데르에 대한 친화도가 커져서 고분자 표면에 서 피리딘 혹은 이미다졸과 음이온성 에스데르 간의 유효 농도가 증가하여 속 도가빨라진다. PEI 에 이미다졸기와 형수성 알킬기가 도입되면 PEI7} 협수성 에스테르와 착 화합물을 형성하는 능력이 커지고 이에 따라 에스테르 가수분해에 대한 상당한 촉매 효과를 보인다. 16) 이미다졸메틸기가 PEI 에 도입되면 이미다졸은 협수성 영역에서 위치하게 되며, 고분자에 흡착된 형수성 기질과 높은 유효 농도와 이 에 따른 반응성의 증가를 시현하게 된다.
피리딘 유도체 중에서는 4- 알킬아미노피리딘이 상당히 높은 염기성과 강력한 친핵성을 보인다. 로릴 (도데실)화된 PEI( 로릴 -PEI) 에 4 - 알킬아미노피리딘이 공유 결합으로 연결되면 협수성 에스테르에 대한 피리딘의 공격력이 크게 증가 한다 .17) 4 - 니트로페닐 카프로에이트의 가수분해에 대하여, p H7.3 에서 4- 디메 틸아미노피리딘보다 로릴 -PEI 에 연결된 4- 알킬아키노피리딘은 1,000 배 정도 높은 반응성을 보인다. 그런데 이러한 반응성의 증가는 주로 PEl7} 가지고 있 는 양이온성 환경 때문에 4 - 알킬아미노피리딘의 질소 원자가 양성자화되는 것 이 억제되고 p Ka 값이 9 . 7 에서 7.3 내의로 저하한 것에 기인한다. 즉, pH 7.5 에서 로릴 - PEI 에 연결된 4- 알킬아미노피리딘은 약 절반이 염기성 형태로 존재 하는데 4- 디메틸아미노피리딘은 1% 미만만이 염기성 형태로 존재하는 것이 다. 그렇지만 4- 알킬아미노피리딘이 전부 염기성 형태로 존재할 때의 반응성도 로릴 -PEI 골격 위에서 5-15 배 정도 증가하는데 이는 협수성 작용에 의하여 기 질이 PEI 와 착화합물을 형성할 수 있기 때문인 것으로 풀아할 수 있다.
 0\?H 군 /C12 H2s
0\?H 군 /C12 H2s
로릴 -PEI 에 카르복시기를 추가로 도입하여 제작한 로릴 - 카르복시 -PEI 는 (15) 표시된 이민 양이온이 물 분자의 공격에 의하여 가수분해되는 속도를 102 배 가속한다. 18) 이 정도의 가속 효과를 일반 염기 촉매작용에 의하여 성취하려 면 아세트산의 농도가 70M 에 달하겨야 한다. 이 수치는 일반 염기의 유효 농 도로서는 상당히 높은 편에 속한다. 로릴구}르복시 -PEI 위에서는 형수성 미세 환경 속에서 반응이 일어나는데, 카르복시 음이온에 의한 다음의 반응은 전하 가 전이상태에서 파괴되고 있는 유형의 반응이기 때문에 형수성 매체에 의하여
본원적인 반응성이 제고된 것으로- 볼 수 있다. 산소 친해체로 고분자 골격 위에 장착된 작용기로 히드록삼산기와 19) 욱심기 를 20) , 21) 들 수 있다. 히드록삼산과 옥심은 이온화하여 음이온으로 전환되어야 친핵성 공격을 효과적으로 수행할 수 있다. 따라서 양이온성 고분자 환경에서 는 그 이온화가 촉진되어 반응속도가 증가할 수 있다. 이러한 매체 효과 이의 에도 기질과 착화합물을 형성함으로써 친핵체의 유효 농도가 제고되는 이유로 인하여 고분자 골격에 이러한 작용기가 도입될 때 반응성이 증가한다. 이에 따 라 니트로페닐 에스데르를 기질로 사용하였을 때 기질의 탈아실화는 가속된다. 그러나 이 결과로 형성된 아실-히드록삼산 혹은 아실크수심의 가수분해가 충분 히 빠르지 못하기 때문에 촉매 작용기로서 만족스러운 결과는 얻지 못한 형편 O] 다. 고분자에 도입된 술피히드릴기의 22),23) 경우에도 히드록삼산 혹은 옥심과 같이 음이온형일 때 활성화되지만 에스테르를 공격하여 안정한 아실-천핵체 중간체 롤 형성한다는 한계가 있다.
 R-NH2 + o=(C 02H 노 R- +N곡I kHc 0 2HcO_ 2
R-NH2 + o=(C 02H 노 R- +N곡I kHc 0 2HcO_ 2
고분지에 아민기가 도입되면 친핵성 작용기로 활용할 수 있다. PEI 의 경우 에는 고분자 골격에 수많은 아민기가 존재하며, 에스테르의 탈아실화 반응에 이들이 활용되어 인공 효소로서의 각종 측면을 시험하는 데 사용되기도 하였 다. 8),24) 아민이 에스테르를 공격하면 에스데르보다 더 안정한 아미드가 형성되 기 때문에 에스테르의 가수분해 반응에 대하여 아민기가 촉매작용을 담당하기 는 불가능하다. PEI의 아민기가 친핵체로 작용하면서 촉매작용을 유발하는 예 로 옥상가세트산의 탈카르복실화 반응을- 들 수 있다. 25) 16o jj 표시한 바와 같이
PEI 의 일급 아민기가 기질과 반응하여 이민 중간체를 형성하면 이민 양이온의 상태에서 탈카르복실화가 신속히 일어나며 이 결과로 형성된 제 2 의 아민이 가 수분해됨으로써 촉매작용이 완결된다. 효소 작용에서는 아마노산의 곁사슬에 위치한 유기 작용기만으로 원활한 촉 매 효과를 달성하기 어려울 경우에 조효소라고 불리는 일련의 유기분자를 활용 하고 있다. 고분자 촉매계에 사용된 조효소의 예로 풀라빈을 들 수 있다. 26),27) 17 의 A 혹은 B 를 통하여 풀라빈 유도체를 PEI 혹은 양이온성 폴리스티렌 유 도체 등에 연결한 후, NADH, 카르보 음이온, 티올 등의 산화 반응에 대한 촉매 효과를 조사한 바 있다.
 A>
A>
고분자 위에 도입된 두 가지 종류의 유기 작용기가 협동하여 촉매 효과를 시 현하는 예는 혼하지 않다. 히드록삼산과 이미다졸을 포함하는 공중합체를 제작 한 결과 에스데르의 탈아실화 속도가 10 배 가속되었는데, 히드록삼산이 에스 테르를 공격하여 형성한 중간체의 파괴 과정에서 이미디졸이 촉매로 관여하는 것 (18) 으로 이 현상이 설명되었다. 29) 합성 고분자를 이용하여 생체 모방 촉매를 제작하는 작업이 1970 년대에는 왕 성하게 전개되다가 1980 년대에는 상당히 침체해졌다. 그 주된 이유는 폴리 바 누의 효과보다 더 정교한 촉매 요인을 도입하기가 어려웠기 때문으로 보인다. 이러한 한계를 극복하려면 고분자 골격 위에 분자 인식에 의하여 기질과 착화 합물을 형성할 수 있는 결합 부위를 창출하고, 이 결합 부위에 인접한 위치에 촉매 작용기를 도입하며, 촉매 작용기 간의 효과적인 협동을 유도하는 작업이 필요하다. 1980 년대까지는 이러한 돌파구를 열 수 있는 계기가 마련되지 못하 였다. 그러나 1990 년대에 들어와서 전술한 바와 같이 PEI 골격 위에 마크로고
리 금속 착화합물을 형성하거나 시클로덱스트린을 도입함으로써 결합 부위를 창출하는 데 성공하였기 때문에 합성 고분자를 이용한 인공 효소의 제작이 활 기를 띠게 될 것으로 기대된다. 소형 호스트를 이용하여 분자 인식에 의하여 착화합물을 형성하는 연구가 1980 년대에 시작되어 큰 발전을 보이고 있는데, 이러한 분자 인식의 연구는 다음 단계에서 인공 효소의 고안으로 발전될 전망 이다. 인공 효소를 설계하는 데에는 결합 부위와 촉매 작용기의 조합이 요구되 는데, 이러한 목적으로는 합성 고분지를 골격으로 사용하는 것이 여러 가지 이 점이 있다. 이러한 견지에서 합성 고분자를 이용한 인공 효소의 제작은 더욱 활기를 얻게 될 것으로 예상된다. 제 4 절 인공금속효소 금속 효소는 효소의 활성도를 위하여 금속 이온이 필수적인 역할을 담당하는 효소이다. 30) 금속 효소에서 금속 이온은 루이스산 촉매로 작용하거나 산화환원 촉매로 작용한다. 금속 이온이 유기 반응에 루이스산 촉매로 작용할 때 다양한 촉매작용을 수행함을 제 4 장에서 소개한 바 있다. 인공 금속 효소는 금속 이온이 기질의 구조를 인식하거나 기질의 구조를 변 환시키는 데 핵심적인 역할을 담당하는 인공 효소라고 정의할 수 있다. 효과적 인 인공 금속 효소를 개발하기 위하여서는 일차적으로 금속 이온이 매우 강력 하게 결합되어 있는 분자 골격을 창출하여야 한다. PEI 는 에틸렌디아민이 반복되는 구조를 가지고 있으므로 금속 이온에 대한 높은 친화력을 가지고 있을 것으로 기대된다. 그러나 매우 높은 p H 를 제의하 면 상당한 부분의 아민이 양성자화되어 있기 때문에 금속 이온의 결합이 정전 기적으로 저해될 수 있다. Ni( Il )이온에 대하여 PEI 는 104 정도의 결합싱수를 가지고 있는데 ,24) 이 정도의 결합상수로는 인공 금속 효소를 제작할 수 있을 만큼금속 이온이 충분히 강하게 결합된다고보기 어렵다. 무기화학계에서는 폴리아자마크로고리 착화합물에 관한 연구가 매우 발전되 어 있다. 31)-33) 폴리아자마크로고리 착화합물 중의 일부분은 D 에서 예로 든 바
 二~<二 +C H\NNH~2:戶 :H:H H 2NN 1)9 cH~~:,H: (군cN曰2二 〔N) )
二~<二 +C H\NNH~2:戶 :H:H H 2NN 1)9 cH~~:,H: (군cN曰2二 〔N) )
와 같이 금속이온 존재하에 디카르보닐 화합물을 폴리아민과 축합시킴으로써 합성된다. 32) PE 뇨. 이와 같이 금속 이온 존재하에 디카르보닐 화합물과 축합 되어 마크로고리 착화합물을 형성한다. 24) 그 예를 20-23으 로 들 수 있다. 20- 쩍 PEI 위에 제작한 마크로고리 착화합물의 정확한 구조를 표시하지는 않 고, 단지 마크로고리의 제작 과정을 밝히고 있다. 예를 들어, 꼬은 Ni (Il )이 온 존재하에서 PEI 를 부탄디온과 축합하겨 제작한 것이다 . 이와 같이 제작한 고분자형 마크로고리 착화합물은 두석을 여러 번 반복하여도 금속 이온의 함량 이 감소하지 않을 정도로 강하게 금속 이온을 결합하고 있다. 이러한 PEI 유
도체는 금속 중심이 강하게 결합되어 있고, 금속 중심이 +2 혹은- + 隣 1 전하 를 가지므로 p H 에 무관하게 거대한 양전하를 가지는 고분자이다. 따라서 음이 온을 인식하는 능력을 보유하고 있을 것으로 예상되는바, 벤조산 유도체의 음 이온은 104 정도의 결합상수를 가지고 있다. 한편, 마크로고리 중심을 보유한 PEI 유도체는 음이온성 카르복실 페닐 에스데르의 탈아실화를 두드러지게 가 속화하는데, 속도론적인 연구 결과에 따르면 금속 중심이 기질의 카르복시기를 인식하여 고정하면 고분자 골격에 위치한 아민이 천핵체로 에스테르를 공격 (24) 한다. 이 반응에서 금속 이온은 촉매 작용기로서 행동하지는 않고 기질과 착화합물 을 형성하는 데 루이스산으로 작용하였다. 인공 금속 효소로서 좀더 진보된 형 태는 금속 이온이 촉매 작용기로 참여하는 경우가 될 것이다. 금속 이온이 음 이온성 기질의 결합에도 참여하면서 동시에 촉매 작용기로도 개입하는 예로 인 산 모노에스테르 및 인산 디에스데르의 가수분해 반응을 둘 수 있다.
 M:、 ,O O-PH,:유--.). /.. ._O 。A Arr M、~,'O O' -HP유 I· J /o'O· Ar M:、 O o〉 ( P〈 \ \\>
M:、 ,O O-PH,:유--.). /.. ._O 。A Arr M、~,'O O' -HP유 I· J /o'O· Ar M:、 O o〉 ( P〈 \ \\>
인산 디에스데르는 가수분해에 대하여 매우 높은 안정성을 가지고 있어서, 2 5 C 와 pH 7 에서 디메틸 포스페이트의 반감기는 200 억 년, DNA 의 반감기는 8 억 년으로 추산되며, 활성화된 비스 (4- 니트로페닐) 포스페이트 (BNPP) 의 반감 기도 2000 년에 달한다. 인산 모노에스테르는 디에스테르보다 가수분해에 대한 저항이 약하지만 여전히 높은 수준이어서 4- 니트로페닐 포스페이트 (NPP) 의 경우 같은 조건에서 반감기가 4 년 정도이다. 인산 디에스데르와 인산 모노에스 테르의 가수분해 반응에 대하여 몇 가지 금속 착화합물이 촉매작용을 나타내는 것이 보고된 바 있다. 32)-35) 25, %에 표시한 바와 같이 금속 이온의 배위자리에 인산 에스데르의 음이온성 산소가 배위하면 이 배위자리와 시스 위치에 결합된
수산화 이온이 분자내 친핵체로 공격하여 F才 형 전이상태를 형성하게 된다. 27 에 표시한 바와 갇이 인산 모노에스테르의 경우에는 금속에 배위하여 止才형 고 리를 먼저 형성한 뒤 의부의 수산화 이온에 의하여 공격받기도 한다. PEI 를 Co(Il )이온 존재하에 글리옥살과 축합하여 합성한 꼬은 NPP 와 BNPP 의 가수분해에 대하여 효과적인 촉매작용을 보인다. 36) 특히 NPP 의 가 수분해에 대하여서는 포화 속도론적 행동을 시현하는바, NPP 와 착화합물을 먼저 형성한 뒤 효율적인 촉매 단계를 거치며, 가수분해 반응을 완결하는 진정 한 촉매로 작용한다. NPP의 가수분해에 대하여 28 이 보이는 촉매 효과는 효 소의 특성을 다수 포함하고 있으므로 乃울 인공 금속 효소로 간주할 수 있는 것 0] 다. CD-PEI를 8) 이용함으로써 더욱 효과적인 인공 금속 효소를 제작할 수 있다. 죽, CD 를 결합 부위로 이용하고 촉매 부위로 CD 의 고리 위에 금속 중심이 도 입되게끔 설계함으로써 결합 부위와 촉매 부위가 분리되어 협동하는 생체 모방 촉매를 설계할 수 있을 것이다. 제 5 절 분자 형태 조정 효소는 분자량이 수만 혹은 수십만이 되는 거대 분자이지만, 활성자리는 분 자량이 수백에 불과한 분자를 수용하는 것이 보통이다. 비교적 작은 크기의 기 질 분자가 일으키는 화학반응을 촉진하기 위하여 그보다 수십 배 이상이 더 큰 촉매를 사용하는 것은 비경제적인 것처럼 보이기도 한다. 그러나 이것은 효소 의 활성자리 속에 위치한 각종 촉매 작용기와 구조적인 요소들을 가창 적절한 위치에 놓이게끔 하기 위한 것이다 . 거대한 고분자인 단백질의 곁사슬에 이러 한 요인들을 장착하면 단백질의 삼차원 구조가 결정됨에 따라 활성자리에 이러 한 요인들이 결집하게 되는 것이다. 죽, 이러한 촉매로서의 구조적 요소가 적 절한 위치에 놓이게끔 분자 형태를 조정하는 데 효소의 분자 골격을 사용하는 것이다. 합성 고분자를 사용하여 인공 효소를 제작할 때에도 합성 고분자의 골격을
 o?:0 0\二 rn 。 갔°。
o?:0 0\二 rn 。 갔°。
이용하여 분자 형태를 조정함으로써 다양한 촉매 요인을 적절한 위치에 배치할 수 있는가가 관건이 된다. 합성 고분자를 이용하여 분자 형태를 조정하여 분자 인식이나 촉매 작용에 사용한 예가 몇 가지 보고된 바 있다. 모노머에 기질을 결합시킨 뒤 중합하여 고분자를 제작하고 결합된 기질을 방 출하면 고분자에 틈이 형성되는데 이 툼이 기질의 구조를 기억하여 기질을 선 택적으로 결합할 수 있다. 이러한 접근 방식으로 분자 인식 능력이 있는 고분 자를 합성한 사례를 소개하면 29,37) J038 ) 등을 둘 수 있다. 제 筑} 제 硏길에서 소개한 구조 7~ 즌 금속 이온을 인식하는 데 적합한 리간드 의 형태를 고분자를 이용하여 조정하는 사례이다. 여기에서는 금속 이온과 리 간드 간에 착화합물을 먼저 형성한 뒤 각 리간드에 도입된 작용기끼리 를 PEI 로 연결함으로써 먼저 형성된 착화합물이 가지고 있는 기하학적인 구조를 보존 하려는 것이 시도된 것이다. 제 7 장 제筑길에서 소개한 구조 67은 폴리(알릴아민) 혹은 PEI 유도체를 연결 자로 사용하여 키모트립신의 삼차원적 구조를 안정화시킴으로써 열, 변성제 등 에 의한 활성도의 상실을 억제한 반합성 효소이다.
참고문헌 1) Overberge r , C. G . ; Salamone, J. C. Acc. Chem. Res., 1969, 2, 217. 2) Ku nitak e, T. ; Shi nk ai, S. Adv. Phys . Org. Chem., 1980, 17, 435. 3) Klotz . I. M. In Enzym e Mechanis ms, Page , M. I., Wi lliam s, A., Ed. , Roy al Che mica l Soci et y : London, 1987, Chap ter 2. 4) Klotz , I. M. ; Roy er , G. P . ; Slon ies ky, A R. Bio c hemis try, 1969, 8, 4752. 5) Joh nson, T.W. ; K.lot z , I. M. Macrorrwlecules, 1974, 7, 618. 6) Suh, J. ; Scarp a , I. S. ; Klotz , I. M . J Am. Chem. Soc., 1976, 98, 7060. 7) Suh, J. ; Kim, M. J. B io o rg. Chem. , 1 992, in pre ss.
8) Suh, J. ; Lee, S. H. ; Zoh, K. D. ]. Am. Chem. Soc., 1992, 114, 7916. 9) Suh, J. ; Zoh, K. D. ; Lee, S. H. to be pu blish ed. 10) Suh, J. ; Lee, S. H. to be pub lish ed. 11) Suh, ]. ; Klotz , I. M. Bio o rg. Chem., 1977, 6, 165. 12) Suh, J. ; Klotz , I. M. ]. Am. Chem. Soc., 1984, 106, 2373. 13) Mart ine k, K. ; Osip o v, A. P. ; Yati sim i rsk i, A. K. ; Dadall, V. A. ; Berezin , I. V. Tet. Lett ., 1 975, 1279. 14) Lets i n g e r , R. L. ; Savereid e , T. ]. ]. Am. Chem. Soc., 1962, 84, 3122. 15) Kir s h, Y. E. ; Pluzhnov, S . K. ; Shom ina , T. S. ; Kabanov, V. A. ; Kargi n, V. A. Vys o komol. Soedin . Ser. A., 1970, 12, 186 ; Chem. Abst., 1970, 72, 86675. 16) Klotz , I. M. Adv. Chem. Phys ., 1978, 39, 109. 17) Delaney, E. J. ; Wood, L. E. ; Klotz , I. M. ]. Am. Chem. Soc., 1982, 104, 799. 18) Suh, ]. ; Klotz , I. M. ]. Pol. Sci . P ol. C hem. Ed., 1978, 16, 1943. 19) Okahata , Y. ; Kunit ak e, T. ]. Pol. Sci . P ol. Chem. Ed., 1 977, 15, 2571 . 20) Kirsh , Y. E. ; Lebedeva, T.A . ; Kabanov, V. A. ]. Pol. Sci . Pol. Lett. E d., 1 975, 14, 207. 21) Klotz , I. M. ; Drake, E. N. ; Sis id o , M. Bio o rg. Chem., 1981, 10, 63. 22) Bir k , Y. ; Klotz , I. M. Bio o rg, Chem., 1971, 1, 175. 23) Murachi, T. ; Okamura, K. ]. Pol. Sci . P ol. Lett. Ed., 1976, 14, 361 . 24) Suh, ]. ; Cho, Y. ; Lee, K. J. ]. Am. Chem. Soc., 1991, 113, 4198. 25) Sp et n a ge l , W. J. ; Klotz , I. M. ]. Am. Chem. Soc., 1976, 98, 8199. 26) Sp et n a ge l , W. J. ; Klotz , I. M. Bio po lym ers , 1978, 17, 1657. 27) Shin k ai, S. ; Yamada, S. ; Ku nitak e, T. Macromolecules, 1978, 11, 65. 28) Shin k ai, S. ; Yamada, S. ; Ando, R. ; Ku nitak e, T. Bio o rg. Chem. , 1 980, 9, 238. 29) Kunit ak e, T. ; Okahata , Y. ]. Am. Chem. Soc., 1976, 98, 7793 ; Adv. Poly m. Sci ., 1976, 20, 217. 30) Suh, J. Acc. Che m. Res., 1992, 25, 273. 31) Mashi ko , T. ; Dolph in, D. In Com pr ehen si,ve Coordin a tio n Clzemi stry, Wi lkin - son, G., Ed., Perga m on : Oxfo rd , 1987, Vol. 2, Chap ter 21. 1. 32) Curt is, N. F. In Comp re hen si,ve Coordin a tio n Chemi stry, Wi lkin so n, G., Ed., Perga m on : Oxfo rd , 1987, Vol. 2, Chap ter 21. 2.
33) Mert es , K. B. ; Lehn, J.-M . In Comp rehe? 函 ve Coordin a ti on Cheniis try , Wi lki n - son, G., Ed., Perga m on : Oxfo r d, 1987, Vol. 2, Chap ter 21. 3. 34) Ch in, J. Acc. Chem. Res., 1991, 24, 145. 35) Chin , J. ; Banaszezyk , M. ; Jub ia n , V. ; Zou, X. J Am. Chem. Soc., 1989, 111, 186. 36) Jon es, D. R. ; Lin d oy, L. F. ; Sarge s on, A. M. J Am. Chem. Soc., 1983, 105 , 7327. 37) Koik e , T. ; Kim ura, E.]. Am. Chem. Soc., 1 991, 113, 8935. 38) Suh, J. ; Kim, N. to be pub li sh ed. 39) Shea, K. J. ; Thomp so n, E. A . ; Pandey, S . D. ; Beauchamp , P. S. J Am. Cliem . Soc., 1980, 102, 3149. 40) Fuji i, Y . ; Mats u ta m i, K. ; Kik u chi, K. Chem. Commun., 1 985, 415.
-l 가변 영역 139 가수분해, 말레암산유도체- 24 아미드의 一 23, 47, 55, 145 아세탈 ― 23 에스데르- 47 인산 디에스데르— 56 카보네이트 에스테르― 143 가시광선 164 거울상 이성질 113 게스트 30 고정화 131 광감응성 131, 175 광분해 147 광 에너지 193 글리코시드 111 금속이온 이핵체- 57 금속효소 46 길항약 99 L 뉴클레오시드 124 뉴클레오티드 33, 112, 124, 138
E: 다중 작용기 촉매효과 27 단백질 공학 131 당 지방질 185 데옥시리보오스 33 돌연변이 179 동위원소 라벨 124 딜스-알더 반응 155 근 라리아트 크라운 에테르 173 라만스펙트럼 85 라세미화 113 B- 락탐 49 락돈화 54, 145 루이스 산 21, 28, 46 리보뉴클리아제 39, 159 리보오스 33 리파아제 115, 129, 132 리포솜 186 口 막 185 면역 글로볼린 139 물, 금속에 배위된― 53 물리 유기화학 19, 66
모노클론 140 모노클론 항체 140 미세 환경 189, 198 미셀 23, 187 미카엘리스-멘텐 식 71 닙
반도체 193 반응속도론 93 방향 선택 110 배위 중합 베시클 194 베시클 186 변성 131 변형 52 변형 효소 138 분자 인식 153, 212 분자 집적체 192 분할 113 불소 핵자기 공명 200 비균질성 촉매 193 비대칭 환원 147 비스크라운 에테르 173 비정류상태 82 人 산성도 50 산화 109, 207산화환원 46 생물유기화학 ―의 연륜 15 ―의 영역 13 _의 정의 13 생체 막 185 생체 모방막 193 생체 모방촉매 211 생체 분자 33 섭티리신 131 세린 프로데아제 160 수산화 이온, 금속에 배위된_ 49, 51 수선 효소 147 수소 결합 164 수용체 99, 151 수화 니트릴— 47 알데히드 혹은 케톤— 47 이중결합― 56 탄산가스의― 160 스던 충 188 스페란드 174 스핑고미엘린 185 시클로덱스트린 154 시클로판 162 신경 가스 189 신경계 153 신의약 98
。 아미노산의 견사슬 20 아미드 55, 112, 145 아실로인 축합 118 아실리움 양이온 43 아실 유도체 41 一 치환반응 메커니즘 42 아자 크라운 에데르 159, 172 아조벤젠 165 안지오덴신 전환 효소 99 알도라제 118 。강돌 축합 51, 118 알콜 데히드로게나제 108 암 101 양성자공여체 89 양성자교환 47 에너지 이동 193 에스데라아제 115 에폭시화 119 에피머화 51 엔을 에스테르 129 엔을화 51 엔테로박틴 180 역반응, 저해적인- 52 열안정성 132 옥십 49 왜력 52 용매 효과 27
우라닐 이온 170 유기 금속화합물 46 유기 용매 128 유기 합성 104 유사 회전 36 유전자 재조합 86, 138 유전 정보 151 유효 농도 188 유효물농도 24 유효 pH 188 의약 전달 193 이분자층 186 이성질체 효소 68 이탈기 49 인공 금속 효소 61, 208 인공 막 191 인공효소 198 인접 효과 24 인산 ―디에스테르 34, 56, 211 一모노에스데르 34, 211 ―에스테르가수분해 메커니즘 35 _트리에스테르 34, 190 일반 산 21, 54, 84 일반 영기 22, 56, 74 일분자층 192 임계 미셀 농도 187 입체 선택 53, 107 입체 압축효과 26
x: 자기 복제 178 자성 입자 193 자의선 164 저해 작용 70 저해제 99 전도성 고분자 196 전이 상태 유사체 142 전자 이동 193 정류 상태 81, 83 정류전 상태 83 정사면체 중간체 42 정전기적 작용 23 &제거 반응 66, 145 구휴수 17 중합된 베시클 194 진통제 100 •K 착물 형성 28 첨가 117, 118, 125 초가변 영역 140 천전자체 21,47 친핵성 방향족 치환반응 204 친핵체 22, 54, 56, 75
구 카르보 음이온 중간체 51 카르보닉 안히드라제 56, 160 카르보닐 기 117 카르복시펩티다제 A 56, 66, 98 —모형 56, 87 ―에폭시드 저해제 70 ―중간체 76 ―중간체 포획 78 —포스페이트 저해제 76 —一——— ASTGAer lyru r g gr-- - --2 1111 7 994 207857 8688889888 — Ty r- 248 85 — Zn( Il )이온 86 카탈라아제 132 카데콜 포스페이트 159 칼릭스아렌 167 콜렉스테롤 185 콜레스테롤 옥시다제 132 크립탄드 173 크립테이트 173 클라이센 자리 옮김 반응 145 키랄성 에폭시드 115 키모트립신 115, 159, 212
E 탈보호 110 탈아실화 91, 125 탈카르복실화 47, 113, 125, 204, 207 템플레이트 효과 50, 52 트립토판 146 특이성 107 특정 산 21 특정 영기 22 티민 146 고 파파인 115 퍼옥시다제 131 펩티드 합성 111 평형 반응, 부차적― 82 포갱 작용 146, 171, 180, 200 포스파타제 40 포스포글리세리드 185 포화속도론 212 폴리 비누 199, 204 폴리아자 마크로고리 209 폴리알릴아민 212 폴리에틸렌이민 161, 181, 198 폴리 전해질 198 푸린 33
프로카르복시펩티다제 A 68 프로키랄적 입체 특이성 116 프로테아제 132 피리독살 포스페이트 118, 160 피리독스아민 포스페이트 160 _o 할로겐화 33 함메트 식 107 합텐 78 항바이러스제 140 항생제 112, 124 항암제 104 항원 139 항체 139 항체 효소 139 핵산 33 헤미아세탈 26 형수성 작용 23 형태, 생산적인- 52 형태 변화 53 활성 자리 19 굽人一§. 29 혼성 효소 139 화학에너지 193 화학적 수정 69, 85 환원 47 효능약 99
효소·촉매 반응의 특칭 19 희토류 170 히드록시화 109 기타 c-AMP 33 ATP 33, 175
DNA 33, 37, 101, 138 c-G MP 33 GTP 33 NAD+ 33 NADP+ 33 RNA 33, 37, 138 t-R NA 40 X 선 결정학 93
서정헌 1948 년 출생 1971 년 서울대학교 문리과대학 화학과 졸업 (이학사) 1975 년 미국 시카고대학 화학과 졸업 (Ph. D) 미국 노스웨스턴대학 연구원 1987 년 제 1 회 한국과학상 화학부문· 수상 l993 년 제 4 회 한국과학상 대상 수상 현재 서울대학교 자연과학대학 화학과 교수 IO 여 편의 논문이 있음 생물유기화학 대우학술총서 • 자연과학 92 11 판판 볘몌 펴찍냄음 -—— 11999944 년년 33 월월 2100 일일 지은이-서정헌 펴낸이-朴孟 浩 펴낸곳- (주)民 音 社 출판등록 1991 . 12. 20 제 16-490 호 135-120 서울 강남구 신사동 506 강남출판문화센터 5 층 대표전화 515-2000 팩시밀리 515-2007 Pri nt e d in Seoul, K orea © 서정헌, 1994 순수과학 , 유기화학 KDC/437.9 값 8,500 원 ISBN 89-374-3592-6 94430 ISBN 89-374-3000-2 (세트)
대우학술총서(자연과학) 1 소립자와 게이지 상호작용 김진의 47 생체에너지 주충노 2 동역학특론 이병호 48 리이만 기하학 박을룡 3 질소고정 송승달 49 군표현론 박승안 4 상전이와 임계현상 김두칠 50 비선형 편미분 방정식론 하기식 5 촉매작용 진종식 51 생체막 김형만 6 외스바우어 분광학 옥항남 52 수리분류학 고철환 7 극미량원소의 영양 승정자 53 찰스 다윈 정용재 8 수소화봉소와 유기봉소 54 금속부식 박용수 화합물 윤능민 55 양자광학 이상수 9 항생물질의 전합성 강석구 56 효소반응속도론 서정헌 10 국소적 형태의 Ati ya h-Si nge r 57 화성암 성인론 아만성 지표이론 지동표 58 확률론 구자홍 11 Muco p ol y sacchar i des 의 59 분자 분광학 소현수 생화학 및 생물리학 박준우 60 벡터속 이론 양재현 1123 프천로체스물타리글학란 홍딘승 수합 성 김성각 6612 곤에충너신지경띠 생이리론학 모 혜부정경 생 11116574 고지결천방정분연자물화영의유화양리 학 화김 연숙학김구희병반법 호응 우조원의식환 666543 SB수신oC학경lHo 과부m기o학호초n 와 부론박 호찬 R김 e웅e상아김d만문-승영 업 18 과학혁명 김영식 66 양자 전기역학 김영덕 19 한국지질론 장기홍 67 군환론 박재걸 20 정보이론 한영열 68 대수기하학 조영현 21 원자핵반응론 정운혁 69 양자 장이론 이재형
