
현명호 서울대학교 문리과대학 화학과 ( 이학사 ) 한국과학원 화학과 ( 이 학석사 ) 미국 Uni ve rsit y of 111i no i s at Urban a- C ham pa ig n 대학원 화학과 (Ph . D.) 미국 Un iv e rsi ty of 111i no i s at Ur bana- C ham pa ig n 박사후 연구원 미국 Harvard Un iv e rsit y 박사후 연구원 미국 Un iv e rsi ty of 111i no i s at Urb ana- C h am pa ig n 연구원 현재 부산대학교 자연과학대학 화학과 부교수 ctiv e Phases 외 다수
 LC 에 의한 광학 이성질체의 분리
LC 에 의한 광학 이성질체의 분리

 LC 에 의한 광학 이성질체의 분리
LC 에 의한 광학 이성질체의 분리

책 머리에 서로 거울상의 관계에 있는 한 쌍의 광학 이성질체는 동일한 물리 적 • 화학적 성질을 가지고 있어서 보통의 혼합물을 구별하는 방법으로 는 구별이 불가능하며 오로지 키랄환경 (chir a l env i ronmen t)하에서만 구별이 가능하다. 키랄환경하에서는 두 개의 광학 이성질체가 키랄환 경과 작용하여 물리적 • 화학적 성질이 서로 다른 두 개의 부분 입체 이성질체 (d i as t ereomer) 의 관계에 있게 되기 때문이다. 생체는 본질적으로 비대칭이기 때문에 키랄환경으로- 작용하며 따라 서 한 쌍의 광학 이성질체는 생체내에서 서로 디른· 물리적 • 화학적 성 질을 나타내고 결국 다론 생리활성을 나타낸다. 예를 들면 라세미형태 로 유통되는 의약품의 두 광학 이성질체 중 종종 하나의 광학 이성질 체만이 우리가 원하는 약리작용 혹은 생리활성을 나타내며 다른 절대 배열을 가전 광학 이성질체는 아무런 생리활성을 나타내지 않거나 혹 은 생체에 심각한 부작용을 나타내는 경우가 많이 알려져 있다. 이와 같이 서로 거울상의 관계에 있는 한 쌍의 광학 이성질체가 생체내에서 서로 다른 생리활성을 나타내기 때문에 서로 거울상인 두 개의 광학 이성질체를 분리하거나 혹은 구별하는 노력은, 1848 년 파스퇴르가 타 르타르산의 나트륨 • 암모늄염의 광학분할울 보고한 이래로 다양하게 이루어지고 있다. 서로 거울상인 두 개의 광학 이성질체를 분리하거나 구별하는 방법 은 어느 경우에나 적절한 키랄환경을 제공함으로써 두 개의 광학 이성 질체가 키랄환경과 작용하여 물리적 • 화학적 성질이 서로 다른 부분
입체 이성질 관계가 되게 하는 것으로서 최근에 이 르 러 화학, 약학` 의학, 생물학 등 입체화학에 관련된 분야의 연구자 들 및 광학적으로 순수한 키랄 의약품을 제조하고자 하는 제약학 등의 분야에서 큰 관심 의 대상이 되고 있는 방법은 액체 크로마토그래피 (LC) 용 혹은 고압 액체 크로마토그래피 (HPLC) 용 키랄고정상을 이용하여 라세미화합물 울 광학분할하는 방법이다. 이 방법은 액체 크로마토그레피용 칼럼에 채워진 고정상을 비대칭으로 만들어 서로 거울상인 한 쌍의 광학 이성 질체가 칼럼을 통과함에 따라 키랄환경으로- 작용하는 비대칭 고정상과 서로 다른 상호작용을 하게 함으로써 두 광학 이성질체 를 분리하는 방 법이다. 크로마토그램의 두 피이크를 분리수거하여 두 개의 순수한 광 학 이성질체를 얻을 수 있을 뿐만 아니라 두 피이크의 면적 혹은 크기 의 비로부터 광학활성 물질의 광학순도를 측정할 수 있고 두 광학 이 성질체와 키랄고정상 사이의 상호작용 메커니즘(키랄성인지 메커니즘) 울 정확히 이해할 경우 분리순서로부터 광학 이성질체의 절대배열을 동시에 결정할 수 있기 때문에 액체 크로마토그래피에 의한 키랄고정 상법은 입체화학에 관련된 문제를 해결하는 데 이용될 수 있는 거의 이상적인 방법으로 생각되고 있다. 액체 크로마토그래피용 키랄고정상에 의한 라세미화합물의 광학분 할방법은 1950 년대부터 자연상태의 광학활성 물질을 키랄고정상으로 사용하여 시도되기 시작하였으나 크게 성공적이지 못하였다. 그 후 1970 년대 후반기부터 본격적으로 이 분야에 대한 획기적인 연구 결과 가 발표되기 시작하였고 특히 1980 년대에 들어서는 일리노이대학의 Pir k le 교수가 아미노산의 亢-산성 유도체를 기저로 하는 고압 액체 크로마토그래피용 키랄고정상을 개발하고 또 성공적으로 상품화함에 따라 고압 액체 크로마토그래피용,키랄고정상에 의한 광학분할의 연구 결과가 폭발적으로 발표되기 시작하였다. 이와 같이 비교적 최근인 지 난 10 여 년 동안에 액체 크로마토그래피를 이용한 광학 이성질체 분리 의 연구가 많이 진행되었기 때문에 이 분야는 국내에서는 비교적 새로
운 분야이며 이 분야에 대한 국내 저서는 현재 전무한 상태이다. 세계 적으로 볼 때도 이 분야에 대한 전문도서는 1987 년 이후에 몇 권이 출 판되었을뿐이다. 이 책자에서 저지는 액체 크로마토그래피를 이용한 광학 이성질체 분리 방법 중 특히 최근에 많은 관심의 대상이 되고 있는 키랄고정상 법을 집중적으로 디룸으로써 키랄고정성을 이용한 광학분할 방법을 국 내에 소개하여 화학, 의학, 약학, 생물학 등의 분야에서 입체화학에 관 련된 연구 를 수행하고 있는 대학원생 및 연구기관이나 제약회사에서 연구에 종사하고 있는 전문연구가들에게 도움이 되도록 하였다. 특히 최근의 연구결과까지를 이 책에 포함시키기 위하여 1990 년 초반기까 지의 연구문헌들을 조사하여 인용하였다. 이 책의 제 1 장 〈 서론 〉 에서는 탄소화합물의 광학 이성질현상 및 광학 이성질체 분리의 중요성에 대하여 기술하였다. 제 2 장 〈 광학 이성질체 분리의 일반적 고찰〉에서는 서로 거울상인 한 쌍의 광학 이성질체의 분리가 이루어지기 위한 일반적인 조건들을 논 의하였고 가능한 여러 방법들에 대하여 구체적인 예를 들어 기술하였 다. 죽 라세미화합물의 결정화에 의한 광학분할, 속도론적 광학분할 (kin e ti c resoluti on ), 부분 입체 이성질체의 결정화에 의한 광학분할, 핵자기공명 스펙트럼에 의한 광학분할(광학순도 측정) 및 액체 크로마 토그래피에 의한 광학분할에 대하여 기술하였다. 액체 크로마토그래피용 키랄고정상은 크게 두 가지로 분류할 수 있 다. 키랄고정싱을 구성하는 여러 키랄단위들이 모여서 일종의 키랄공 동 (ch i ral cav ity)을 형성함으로써 이 키랄공동과 라세미 화합물의 두 광학 이성질체가 입체선택적인 상호작용을 하여 키랄성의 인지가 가능 한 복합방식의 키랄고정상과 키랄고정상을 구성하는 개개의 키랄단위 가 광학분할하고자 하는 라세미화합물의 두 광학 이성질체와 입체선택 적인 상호작용을 함으로써 키랄성의 인지가 이루어지는 독립방식의 키 랄고정상이다. 제 3 장과 제 4 장에서는 복합방식의 키랄고정상에 대하여
기술하였으며 제 5 정부터 제 8 장까지는 독립방식의 키랄고정상에 대하 여 기술하였다. 제 9 장 〈 상품화된 카랄고정상 〉 에서는 현재 싱품-화되어 있어서 구입 이 가능한 여러 키랄고정상들에 대한 그들의 특성, 한계, 분리가능한 라세미체들의 예, 사용하여야 할 유동싱 - 등을 자세히 제시하고 이들의 제조회사 혹은 공급회사에 대한 정보를 제공함으로써 액체 크로마토그 래피를 이용하여 키랄고정상에서 라세미체를 광학분할하려는 모든 연 구자에게 도움이 되도록 하였다. 특히 제 9 장만을 참고하더라도 상품 화된 키랄컬럼의 선택 및 사용에 도움이 되도록 노력하였다. 학술용어는 가능한 한 우리말로 옮기도록 노력하였으며 이 과정에서 대한화학회의 『 화학술어집 』 과 이우주 엮음 『 영한 의학사전 』 (아카데마 서적) 및 이우영 저 『 영핫l-한영 최신 화학용어사전 』 (담구당)을 참고 하였다. 이 책자의 원고가 1990 년 12 월에 완료되었으나 저자의 사정에 의하 여 출간이 늦어전 관계로 1990 년 중반기 이후의 이 분야 연구결괴들· 이 본 책자에서 누락되었으며 이 점에 대하여 독자들에게 사과를 드린 다. 이 책자에는 아직도 미비하고 부족한 점이 많음을 통감하며 앞으 로 기회가 있을 때 보충하고 보완하여 더욱 충실한 내용이 되도록 노 력하고자한다. 끝으로 원고를 타자하느라 수고하신 전일련 씨와 교정에 도움을 준 류재정, 양덕호, 김현주, 임납언 군에게 감사하며 이 책의 출간을 지 원해 준 대우재단 및 이 책의 출간을 맡아 준 민음사 편집부 여러분에 게 감사드린다. 1992 년 5 월 현명호
LC 에 의한 광학 이성질체의 분리
키랄고정상법을 중심으로 차례 책 머리에 • 5 제 1 장 서론 1 탄소화합물의 광학 이성질현상 -- 15 1.1 이성질체의 분류 • 15 1.2 분자의 키랄성 및 키랄분자의 광학적 성질 • 18 1.3 분자 키랄성의 조건 및 분자의 대칭성 • 21 1.4 키랄 중심을 가지는 · 분자의 광학 이성질현상 • 21 1.5 키랄 중심을 갖지 않은 화합물들의 광학 이성질현상 • 27 1.6 광학 이성질현상의 역사적 배경 • 33 2 광학 이성질체 분리의 중요성 -- 36 참고문헌 (제1장) • 42 제 2 장 광학 이성질체 분리의 일반적 고찰 1 서론 -- 45 2 라세미 혼합물의 결정화에 의한 광학분할 -- 473 속도론적 광학분할 -- 50
4 부분 입체 이성질체의 결정화에 의한 광학분할 -- 53 5 NMR 에 의한 광학분할(광학순도측정) -- 60 6 액체 크로마토그래피에 의한 광학분할 -- 65 6.1 서론 • 65 6.2 간접분리 방법 • 68 6.3 직접분리 방법 • 90 참고문헌 (제2장) • 112 제 3 장 광학활성 천연물을 이용한 키랄고정상 1 서론 -- 121 2 셀룰로오스를 기저로 한 키랄고정상 -- 122 2.1 셀룰로오스의 특성 및 역사적 배경 • 122 2.2 미세결정형 셀룰로오스 트리아세데이트 키랄고정상 • 124 2.3 실리카 겔에 흡착된 셀룰로오스 유도체 키랄고정상 • 132 3 시클로덱스트린 키랄고정상 -- 150 3.1 시클로덱스트린의 구조 및 특성 • 150 3.2 시클로덱스트린 키랄고정상의 제법 및 특성 • 153 3.3 시클로덱스트린 키랄고정상을 이용한 광학분할의 예, 특성 및 키랄성인지 메커니즘 • 157 3.4 광학분할에 영향을 미치는 요인들 • 170 4 단백질을 기저로 한 키랄고정상 -- 179 4.1 서론 • 179 4.2 AGP 키랄고정상 • 1804.3 BSA 키랄고정상 • 203
4.4 기타 단백질을 기저로 한 키랄고정상 • 217 참고문헌 (제3장) • 223 제 4 장 광학활성 합성 고분자를 이용한 키랄고정상 1 서론 -- 233 2 광학활성인 나선성 고분자를 기저로 한 키랄고정상 • 234 2.1 광학활성인 나선성 고분자의 합성 및 광학활성인 나선성 고분자를 기저로 한 키랄고정상의 제조 • 234 2.2 광학활성인 나선성 고분자 키랄고정성에서의 광학분할의 예 • 237 2.3 광학활성인 나선성 고분자 키랄고정성에서의 광학분할의 특성 및 조건 • 244 3 광학활성 폴리아미드를 기저로 한 키랄고정상 • 250 3.1 키랄고정상의 제조 • 250 3.2 폴리아미드 키랄고정상에서의 광학분할 및 특성 • 255 4 키랄공동을 가전 고분자 키랄고정상(주문자방식의 키랄고정상) -- 266 참고문헌 (제4장) • 280 제 5 장 리간드 교환 키랄고정상 1 서론 -- 283 2 리간드 교환 키랄고정상의 제조, 광학분할의 특성,광학분할메커니즘 -- 286
2.1 키랄 리간드화합물을 고분자에 고정시킨 키랄고정상 • 286 2.2 키랄 리간드화합물을 실리카 겔에 고정시킨 키랄고정상 • 298 2.3 키랄 리간드화합물을 실리카 겔에 흡착시킨 키랄고정상 • 311 3 리간드교환 키랄고정상에서의 광학분할에 대한 이동상의 조성, pH 및 온도의 영향 -- 314 참고문헌 (제5장) • 317 제 6 장 크라운 에테르 키랄고정상 1 서론 -- 321 2 크라운 에테르 키랄고정상의 제조 및 광학분할의 특성 • 324 참고문헌 (제6장) • 335 제 7 장 전하이동 착물형성 키랄고정상 1 서론 -- 337 2 카르비놀 (carbinol) 키랄고정상 -- 343 3 Ir-산성 키랄고정상 -- 347 3.1 키랄성인지의 상호성 개념 • 347 3.2 페닐글리산-DNB 및 루이신-DNB 키랄고정상의 제조 및 광학분할의 특성 • 3483.3 키랄성인지의 메커니즘 • 361
3.4 (R)-페닐글리신-DNB 키랄고정상에 의한 제조목적의 광학분할• 372 3.5 다른 종류의 r-산성 키랄고정상 • 376 3.6 광학분할에 대한 이동상의 영향 • 385 4 t-염기성인 a-아릴알킬아민 키랄고정상 -- 391 4.1 a-아릴알킬아민 키랄고정상의 디자인 • 391 4.2 a-아릴알킬아민을 기저로 한 제3세대 상호성 키랄고정상의 제조, 개발 및 광학분할의 특성 • 393 4.3 a- 아릴알킬아민을 기저로 한 r-염기성 키랄고정상의 다른 예 • 417 5 a-아릴알킬아민을 기저로 하지 않은 r-염기성 키랄고정상 -- 421 5.1 제3세대 T -염기성 키랄고정상의 제조 및 광학분할의 특성 • 421 5.2 a-아릴알킬아민을 기저로 하지 않은 T-염기성 키랄고정상 의 다른 예 • 428 참고문헌 (제7장) • 435 제 8 장 수소결합 키랄고정상 1 서론 -- 443 2 아미노산 아미드 키랄고정상 -- 444 3 기타의 수소결합 키랄고정상 -- 455 참고문헌 (제8장) • 461제 9 장 상품화된 키랄고정상
1 서론 -- 463 2 복합방식의 키랄고정상 -- 465 2.1 셀룰로오스를 기저로 한 키랄고정상 • 465 2.2 시클로덱스트린 키랄고정상 • 468 2.3 단백질 키랄고정상 • 469 2.4 고분자 키랄고정상 • 472 3 독립방식의 키랄고정상 -- 474 3.1 리간드교환 키랄고정상• 474 3.2 크라운 에데르 키랄고정상 • 476 3.3 전하이동 착물형성 키랄고정상 • 477 3.4 수소결합 키랄고정상· 484 4 상품화된 키랄고정상 제조회사의 요약 -- 485 5 키랄 HPLC컬럼 제조회사 혹은 공급회사의 주소 -- 489 참고문현 (제9장) • 491 찾아보기 • 493제 1 장 서론 1 탄소화합물의 광학 이성질현상 (1-4) 1.1 이성질체의 분류 같은 종류, 같은 수의 원소로 구성되어 있으나 물리적 • 화학적 성질 이 서로 다론 화합물들을 이성질체(i somer) 라고 하며, 이성질체는 그 립 1 - 1 에서 보는 바와 같이 여러 종류로 분류할 수 있다. 분자식이 같 은 두 화합물이 서로 이성질체의 관계에 있는가 없는가 하는 것은 두 화합물을 서로 포개어 놓았을 때 두 화합물이 서로 완전히 겹치는가 겹치지 않는가에 의하여 결정된다. 두 화합물이 완전히 겹치면 두 화 합물은 동일 화합물이며 두 화합물이 겹치지 않으면 두 화합물은 서로 이성질체이다. 두 화합물을 구성하는 원소와 원소들간의 결합방식 혹 은 결합순서가 다르기 때문에 서로 겹치지 않는 이성질체를 특히 구조 이성질체 (str u ctu r al i somer 혹은 consti tut i on al i somer : 문헌에 따라서 는 str u ctu r al i somer와 consti tut i on al i somer를 구별하는 경우가 있음 (1) )라고 한다. 서로 구조 이성질체의 관계에 있는 두 화합물은 에탄
두 화합물(l같은 서 로분 자겹식 침) (suIp e rim p o sable) 여l ’ 아니오 동일 화합물 이성질체 (iso mer) 동일한 결합방식 (same consti tut i on ) 예아니오 (입st체e r eo이is성 o m질e체r) (co구ns조ti tu 이tio성n 질al 체is o mer) 서로 거울상의 관계 예 아니오 광(en학an이ti성 om질 e체r) 부(분dia s입 te체 r e o이m성er질) 체 그림 1기 이성질체의 분류 울 (CH3CH20H) 과 이것의 구조 이성질체인 디메틸에테르 (CH30CH3) 에서 보는 바와 같이 통상적으로 완전히 다른 화합물로 취급되고 있 다. 반면 두 화합물을 구성하는 원소와 원소들간의 결합방식 혹은 결 합순서는 동일하나, 원소들의 공간배열의 차이 때문에 서로 완전히 겹 치지 않는 두 이성질체를 서로 입체 이성질체 (s t ereo i somer) 라고 하며 입체화학 (st ereochem i s try)의 연구대상이 된다. 서로 입체 이성질체의 관계에 있는 두 화합물은 서로 거울상의 관계 에 있을 때 광학 이성질체 (혹은 거울상 이성질체 혹은 에난시오머 : op tica l iso mer, mi rr or im ag e iso mer, enan ti omer) 라고 하며 , 서로 거
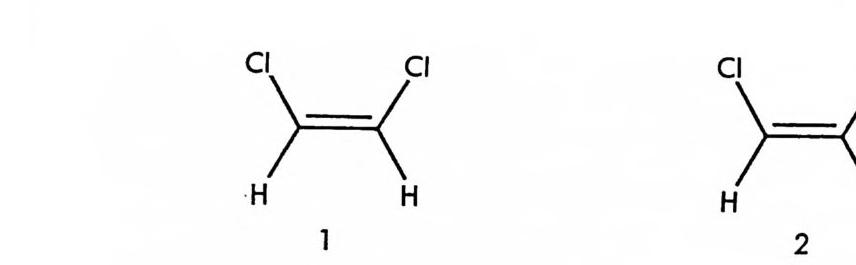
울상의 관계가 아닐 때 부분 입체 이성질체 (d i as t ereomer) 라고 한다. 따라서 광학 이성질체의 관계에 있지 않은 입체 이성질체들은 모두 부 분 입체 이성질체라고 할 수 있다. 예를 들면 일반적으로 기하 이성질 체 (ge ometr i c a l i somer: 구성하는 원소들의 기하학적 위치가 다르기 때문 에 붙여진 이름으로서 통상적으로 많이 사용되나 특별한 의미는 없음)로 알 려진 시스 - 1, 2_ 디클로로에틸렌 1 과 트란스 -1, 2- 디클로로에틸렌 2 은 서 로 거울상관계에 있지 않은 입체 이성질체이므로 서로 부분 입체 이성 질체라고할수있다. 입체 이성질체들을 서로 겹칠 수 없는 거울상의 관계에 있나 없나에 따라서 분류(대칭성에 따른 분류 : sym metr y class ifi ca ti on) 하는 방법 의에 두 입체 이성질체간의 전환이 얼마나 용이한가에 따라서 입체 이 성질체들을 배열 이성질체 (confi gur ati on al i somer) 와 형태 이성질체 (confo r mati on al i somer) 로 분류(전환용이도에 따른 분류 : energy clas- s ifi ca ti on) 할 수 있다• 두 입체 이성질체간에 전환이 불가능하거나 (결합을 끊어야 하는 경우) 전환에 극히 높은 에너지가 필요한 경우(단 일결합을 축으로 하는 회전이 큰 입체장애 때문에 억제된 경우나, 이중결합 을 축으로 회전하여야 할 경우)에 두 입체 이성질체를 서로 배열 이성질 체라고 하며, 단일결합을 축으로 하는 회전에 의하여 쉽게 전환이 가 능하거나 혹은 다른 방법에 의하여 전환이 쉽게 이루어지는 두 입체 이성질체를 형태 이성질체라고 한다. 따라서 배열 이성질체들은 안정 한 화합물로서 분리가 가능하나 형태 이성질체들은 실온에서 서로간의 전환이 너무 빠르므로 분리가 불가능하다. 보통 실온에서 전환에 필요 한 활성화 에너지가 20kcal/ mo l 이상인 경우 배열 이성질체라고 할 수 있으며 두 이성질체의 분리가 가능하다(1). 서로 거울상의 관계에 있는 두 개의 광학 이성질체는 상호전환의 용 이도에 따라 배열 광학 이성질체 (confi gur ati on al enan ti omers) 와 형 태 광학 이성 질체 (con for mati on al enan ti omers) 로 나눌 수 있으며 부 분 입체 이성질체의 경우에도 배열 부분 입체 이성질체 (con figur-
 Cl>=
Cl>=
ati on al d i as t ereomers) 와 형태 부분 입체 이성질체 (confo r mati on al d i as t ereomers) 로 나눌 수 있다. 그러나 보통 광학 이성질체 혹은 부 분 입체 이성질체로 표현할 때는 배열 광학 이성질체나 배열 부분 입 체 이성질체를 의미한다. 1.2 분자의 키랄성 및 키랄분자의 광학적 성질 우리의 왼손과 오른손이 서로 겹칠 수 없는 거울상과 같음에 비유하 여 어떤 분자가 서로 겹칠 수 없는 거울상을 갖고 있을 때, 이 분자는 그리이스어로 손을 의미하는 〈키 랄 (ch i ra l) ) 이라는 말을 이용하여 키 랄성 (ch i ra lity)이 있다고 한다. 따라서 서로 겹쳐지지 않는 거울상의 관계에 있는 한 쌍의 광학 이성질체는 키랄성이다. 키랄성을 가지는 키랄분자의 특칭은 평면편광(p lane po lariz e d lig h t) 1) 의 편광면을 어느 한쪽 방향으로 회전시키는 것으로서, 키랄분자가 평면편광의 편광면 을 회전시 킬 때 이 분자를 광학활성 (op tica lly acti ve ) 이 라고 한다 (그 러나 모든 키랄분자가 전부 광학활성은 아님). 키랄분자 중 평면편광의 편광면을 오른쪽으로 회전시키는 분자를 우선성 (dextr o rota t o r y) 이라 하고 d- (十)로 표시하며, 평면편광의 편광면을 왼쪽으로 회전시키는 분자를 좌선성 (l evoro t a t o ry)이라 하고 l - (- )로 표시한다. 서로 겹쳐
1) 방해석으로 된 Nic o l 프리즘이나 폴라로이드 렌즈를 동과한 빛의 전자파는 한 면에서만 진동하는데 이 때 한 면에서만 진동하는 전자파로 이루어진 빛을 평면 편광이라고한다.
지지 않는 거울상의 관계에 있는 한 쌍의 광학 이성질체 중 하나의 키 랄 분자가 평면편광의 편광면을 오른쪽으로 회전시킨다면 다른 하나의 키랄분자는 평면편광의 편광면을 똑같은 양만큼 왼쪽으로 회전시킨 다. 이와 같은 광학적 성질 때문에 겹칠 수 없는 거울상 관계에 있는 한 쌍의 입체 이성질체를 특히 광학 이성질체 (op tica l i somer) 라 하게 되었다. 한 쌍의 두 광학 이성질체가 같은 양 섞여 있는 혼합물은 평면편광 의 편광면을 회전시키지 않는다. 각각의 광학°이성질체가 평면편광의 편광면을 똑같은 양만큼 오른쪽, 왼쪽으로 회전시키기 때문에 서로 상 쇄되어 광회전 (op tica l ro t a ti on) 이 없는 것으로 관찰되는 것이다. 이 와 같이 두 광학 이성질체가 갇은 양만큼 섞여 있어 광회전을 관찰할 수 없는 혼합물을 특별히 라세미체 2) 라 하며 (士)- 혹은 dl- 로 표시하 고 라세미체를 순수한 두 개의 광학 이성질체로 분리하는 것을 광학분 할 (o pti cal resolu ti on) 이라고 한다. 반대로 광학적으로 순수한 하나의 광학 이성질체가 적당한 과정을 거쳐서 광학 비활성인 라세미체로 되 는 과정을 라세미화 (racem i za t i on) 라 한다. 광학활성 물질이 평면편광의 편광면을 회전시키는 회전량을 광회전 도 (o pti cal ro t a ti on) 라고 하며 a 로 표시하고 편광계를 서용하여 측정 이 가능하다. B i o t의 법칙에 의하면 일정온도 (T) 와 일정파장 (A) 에서 광회전도 a 는 광학활성 물질의 농도 (c) , 평면편광이 통과하는 빛의
2) 라세미체는 영어로 racemi c fo rm 혹은 racemi c mod ifi ca ti on 이라고 한다. 라세미체가 기체상이나 액체상(용액포함) 일 때는 일반적으로 순수한 광학 이 성질체와 같은 물리적 성질을 나타내는 하나의 형태로 존재하나 고체상으로 존 재할 때는 세 가지의 디른 형태 죽 라세미 혼합물 (racem ic m i x ture 혹은 cong lo merate ) , 라세미 화합물 (recemate 혹은 racemi c comp o und) , 라세 미 고용체 (racemi c solid solu ti on) 로 존재하며 각각의 차이점은 참고문헌 1-5 에 잘 나타나 있다. 보통 라세미체라고 할 때에는 두 광학 이성질체가 같은 양 섞여 있어 광회전이 없는 경우를 총칭하며 라세미 혼합물 혹은 라세미 화합물이 라세미체의 동의어로 사용되기도 한다.
통과 길이 (cell pa th len gt h : I) 에 비례하며 사용하는 용매에 따라 달라 지는 값이다. 광회전도는 측정하는 조전에 따라서 달라지므로 단위농 도와 단위통과길이에 해당하는 광회전도를 고유 광회전도 (s p e cifi c op tica l rota t i on ) 라고 정의 하여 사용하고 있다. 고유 광회전도 : [a n= 丁틀 (용매 . C) 이때 농도의 단위는 g/ml 이며 빛의 통과 길이의 단위는 dm 이다. 흔히 광회전도 측정에 있어서 사용되는 광원은 나트륨 D 선 (파장 : 589 run) 이고 광회전도는 실온에서 측정되므로 고유 광회전도는 [따 D 로 표시하기도한다. 순수한 광학 이성질체의 고유 광회전도는 이 키랄분자가 나타낼 수 있는 최대값을 나타내며 흔히 문헌에 보고되어 있는 고유 광회전도, [리 T 값은 순수한 광학 이성질체에 대한 것이다. 그러나 두 광학 이 성질체가 다른 비율로 섞여 있을 때의 고유 광회전도의 값은 〔 a]T 보 다 작은 값을 나타낸다. 따라서 두 광학 이성질체가 섞여 있는 혼합물 의 광학순도 (o pti cal p ur ity)는 다음과 같이 정의된다. % 광학순도 = 占합 :X lOO 여기서 [a 〕는 두 광학 이성질체가 섞여 있는 혼합물의 고유 광회전 도이며 [a 〕。는 순수한 광학 이성질체의 고유 광회전도이다. 1.3 분자 키랄성의 조건 및 분자의 대칭성 분자가 키랄성이기 위한 필요하고도 충분한 조건은 이 분자가 겹칠
수 없는 거울상을 갖는다는 것 이다. 분자의 대 칭 성 (molecular s y mme t r y)을 고려할 때 분자가 겹칠 수 없는 거울상을 가지는 경우는 두 가지이다. 우선 어떤 분자가 대칭성을 전혀 가지고 있지 않을 때 이 분자의 거울상은 이 분자와 겹치지 않으며 이 분자는 키랄성이다. 분자의 대칭성을 나타내는 대칭요소는 대칭면(
3) 대칭면 (tr)은 SI 반전축과 동일하며 대칭중심 (i)은 S2 반전축과 동일함을 인식 한다면 분자의 비키랄성은 반전축 (Sn) 의 존재에 기인하는 것으로 일반화할 수 있다.
1. 4 키랄중심울 가지는 분자의 광학 이성질 현상 분자가 키랄성이기 위한 조건들 중에서 대칭요소를 전혀 갖고 있지
않아 C1 점군에 속하는 비대칭 분자이기 때문에 키랄성인 분자의 예는 키랄중심을 가지는 분자에서 찾아 볼 수 있다. 키랄중심 중 가장 흔한 것은 하나의 탄소 (s 업 혼성 탄소) 중심에 모두 다른 네 개의 치환기가 붙어 있는 경우로서 이 탄소를 키랄 탄소 (ch i ral carbon) 혹은 비대칭 탄소 (asym m etr i c carbon) 라고 한다. 키랄 탄소에 붙어 있는 네 치환기의 공간배열을 평면에 표시할 때는 일반적으로 두 가지 구조식 즉 쐐기 구조식 (flying -w edg e fo rmula) 과 F i sher 투영식 (Fis h er pro je c ti on fo mula) 을 이용한다. 키랄 탄소를 하 나 가지고 있는 락트산(l ac tic a ci d) 의 입체구조를 쐐기 구조식과 F i sher 투영식을 사용하여 그림 1-2 에 나타내 었다.x eoo_ H 。 0 0H H H:x::30H 凡 H 晶CI O OOHH H 0 _H_H? R 그림 |-2 락트산의 쐐기 구조식과 F i sher 두영식
쐐기 구조식은 일종의 원근법 그립 (pe rspe cti ve draw i n g)으로서 결 합을 나타내는 보통의 실선은 지면상에 놓여 있는 결합을 표시하며 실 선 쐐기 ( ..... )와 점선 쐐기 ( (llu••· ) 혹은 점선은 각각 지면 앞으로 튀어 나온 결합과 지면 뒤로 들어간 결합을 의미한다. F i sher 투영식에서 수평선과 수직선의 교점은 키랄 탄소 원자를 나 타내며 수평 결합들은 지면 앞으로 튀어나온 결합을, 수직 결합들은
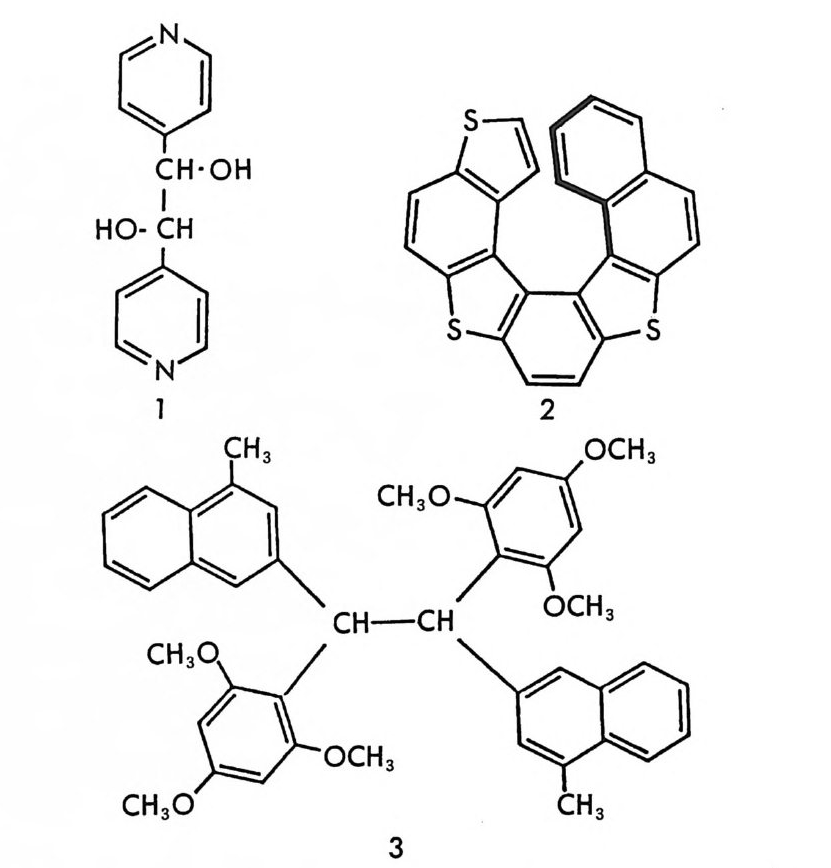
지면뒤로 들어간 결합을 의미한다. 그림 1-2 에서 보듯이 하나의 키랄 탄소를 가진 화합물의 거울상은 결코 서로 겹칠 수 없으며 따라서 두 개의 거울상 분자는 각각 키랄성이고 광학활성이다. 키랄탄소 주위의 네 개의 다른 치환체들의 공간 배열 순서를 절대배 열 (absolute confi gu rati on ) 이라고 하며 서로 거울상인 두 광학 이성질 체는 서로 다른 절대배열을 가진다. 키랄 탄소 주위의 네 치환체들의 절대배열을 나타내기 위하여 Cahn-In g old-Prelo g에 의하여 제안된 (R, S) 명명법이 사용되고 있다 (6.7). (R, S) -명명법에 의하면, 우선 키랄중심에 결합된 네 개의 치환기를 Cahn-In g old-Prelo g의 순위결 정 규칙 (seq u ence rule) 에 따라 우선순위 (p r i or ity)를 정하며 키랄중심 에 직접 붙어 있는 원소의 원자번호가 클수록 높은 순위를 가진다(자 세한 우선순위 결정규칙은 제 1 장 뒤의 참고문헌에 수록된 단행본들과 참고 문헌 3 을 참조할 것). 일반적인 치환기들의 우선순위는 그림 1-3 과 같 다. l ---Br --Cl----S03H----SCH3 ---SH ----F -- OCH3 ----O H _---N02 ----N (CH3) 2 ---NH 2 _一 CF3 _---C OOCH3----COOH ----C ONH2 ----cocH3-- CHO ---CH OH-CH3 ----CH20H ----C N --- C6H5 ----C (CH3) 3 ----C H (CH3) 2 ----c 2H5 -- CH3 --Li — --- D —--H ----L one Pai r Electr on 그림 |-3 키랄중심에 결합된 치환기의 (R,S) _명명 우선순위
네 치환기에 대한 우선순위를 정한 후(편의상 우선순위가 높은 것을 1 로 하자), 키랄 중심에서 우선순위가 가장 낮은 우선순위 4 번 치환기 를 쳐디볼 때, 다른 세 개의 치환기 1-2-3- 의 배열이 시계방향이면
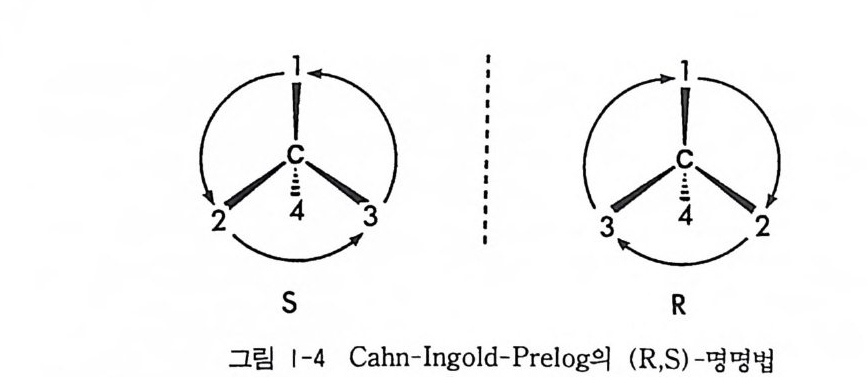 IIIIIIIIIIIII />1_\'c 2
IIIIIIIIIIIII />1_\'c 2
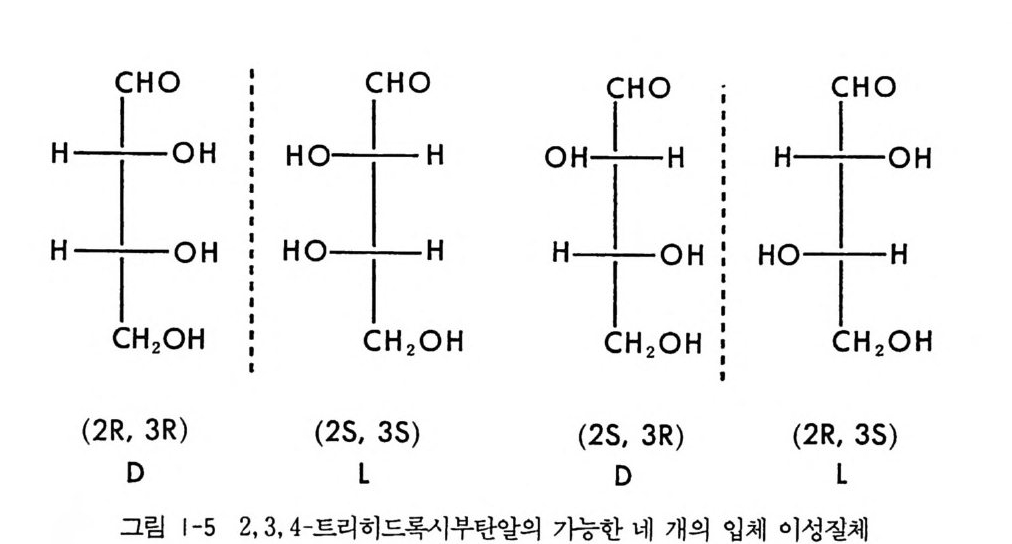
R, 시계 반대 방향이면 도료 절대배열을 표기한다(그립 1-4 ). 따라서 (R, s) -명명법을 이용하여 그림 1-~ l-(-) -락트산의 절대배열은 R 로 표시할 수 있고, d-(+) -락트산의 절대배열은 도료 표시할 수 있 다. Cahn-In g old-Prelo g의 (R, S) -명명법이 키랄분자의 절대배열을 나타내는 데 흔히 사용되나 특히 탄수화물이나 아미노산의 절대배열을 나타내는 데는 (D, L) -명명법이 사용되기도 한다. 탄수화물의 Fis h er 투영식을 그릴 때는 가장 긴 탄소사슬을 수직으로 두고, 사슬의 두 끝 중 산화상태가 더 높은 탄소를 위에 오도록 한다. 이렇게 F i sher 두영 식을 그렸을 때, 키랄 탄소에 붙어 있는 치환기(보통 -OH 혹은 -NH2), 혹은 탄수화물에 있어서 각 키랄탄소에 수직사슬의 위에서부 터 밀으로 번호를 붙일 때 , 가장 높은 번호의 카랄탄소에 붙어 있는 치환기가오른쪽에 있으면 D, 왼쪽에 있으면 L 로표기한다. 그림 1- 쩍 락트산의 F i sher 투영식에서 (-)-락트산의 절대배열은 D 로, (+)-락트산의 절대배열은 止료 표시할 수 있다. 이상에서 기술한 바와 같아 키랄 분자의 절대배열을 R, S 혹은 D, 더 기호를 사용하여 표기할 수 있으나 이것은 다만 규약에 불과하 며, 분자의 광학적 성질 죽 우선성이나 좌선성과는 전혀 관계가 없다. 한 분자내에 n7l 의 키랄중심이 존재할 경우에는 일반적으로 2n 개의 입체 이성질체가 가능하다. 한 분자내에 두 개의 키랄중심 (n=2) 을
 HH CCIH H2 O0OO HHH HHOO CCIIIHH 2O0HH H OHHJ CCII_HH 2OO0H HH HHO CC|IIHH O2OH0 HH
HH CCIH H2 O0OO HHH HHOO CCIIIHH 2O0HH H OHHJ CCII_HH 2OO0H HH HHO CC|IIHH O2OH0 HH
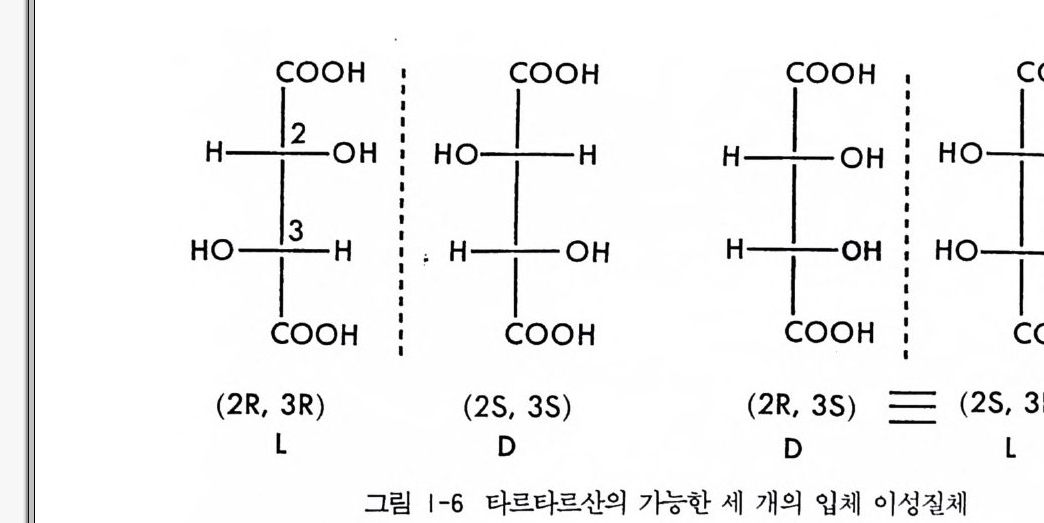
가전 경우의 예로서 2, 3, 4- 트리히드록시부탄알 (2, 3, 4- tr i h y d rox- y bu t anal) 은 네 개의 입체 이성질체가 가능하다(그림 1-5 ). 가능한 네 개의 입체 이성질체들중 그림 1 - 5 의 (2R , 3R) -이성질체와 (2S, 3S) -이성질체 , (2S, 3R) -이성질체와 (2R, 3S) -이성질체는 서로 겹치지 않는 거울상의 관계에 있으므로 (2R, 3R) -이성질체와 (2S, 3S) -이성 질체, (2S, 3R) -이성질체와 (2R,3S) -이성질체는 서로 광학 이성질체 의 관계에 있으며, 나머지 다른 입체 이성질체들은 서로 거울상의 관 계에 있지 않으므로 모두 부분- 입체 이성질체들이라고 할 수 있다. 일 반적으로 한 분자내에 n 개의 키랄중심이 존재할 때 2n_1 개의 광학 이 성질체 쌍이 존재하며 각 입체 이성질체는 2n_27H 의 부분 입체 이성 질체를가진다. 그림 l- 碑] 타르타르산(t a rt ar ic ac i d) 도 역시 업 R 의 키랄중심을 가 전 화합물이다. (2R , 3R) -이성질체와 (2S, 3S) -이성질체는 서로 겹칠 수 없는 거울상의 관계에 있으므로 (2R, 3R) -이성질체와 (2S, 3S)- 이 성질체는 서로 한 쌍의 광학 이성질체이다. 그러나 (2R, 3S) - 이성질 체와 (2S, 3R) -이성질체는 서로 거울상의 관계에 있으나 겹칠 수 있으 므로 (2R, 3S) -이성질체와 (2S, 3R) - 이성질체는 서로 이성질체가 아
 HHO CI1132O OOHH H ‘HHO CIIIO OOHHH HH CIIIO OOOHHH HHOO COOHHH
HHO CI1132O OOHH H ‘HHO CIIIO OOHHH HH CIIIO OOOHHH HHOO COOHHH
니라 동일화합물이다. 이들이 서로 겹칠 수 있는 것은 분자내에 대칭 면 (6) 이 존재하기 때문이며 따라서 (2R, 3S) - 이성질체 혹은 (2S, 3R) -이성질체는 비키랄성이며 광학불활성이다. 이처럼 키랄중심을 가지 고 있으나 분자내에 대칭면이 존재하기 때문에 광학 불활성인 이성질 체를 메조 (meso) 형이라고 한다. 메조 이성질체와 (2R, 3R) -이성질체 혹은 (2S, ~S) -이성질체는 서로 거울상의 관계에 있지 않으므로 서로 부분 입체 이성질체들이다. 타르타르산처럼 구조적으로 대칭인 분자 에 있어서 n 개의 키랄중심이 존재할 때, n 이 찍~ 2( n - I)+ 2(n-2)/ 2 개의 입체 이성질체가 가능하며 n 이 홀수이면 2 ( n - 1) 개의 입체 이성질 체가 가능하다. 타르타르산은 두 개의 키랄중심을 가지므로, 그림 1_ 6 에서 보는 바와 같이 세 개의 입체 이성질체가 가능하다. 특히 두 개의 키랄중심을 가전 화합물에 있어서 두 키랄중심 사이의 상대배열 (rela ti ve co nfigur a ti on) 을 나타내는 말로서, 가리워진 형태 (eclip s ed co 쿄 orma ti on) 의 화합물을 그렸을 때 , 같거나 혹은 비슷한 치환체끼리 서로 가리워진 구조를 가지면 er yth ro 라 하고, 같거나 비 슷한 치환체들 중 서로 가리워진 위치에 있지 않은 구조를 가지면 thr eo 라고 명명하기도 한다.
 r +/ CHJ
r +/ CHJ
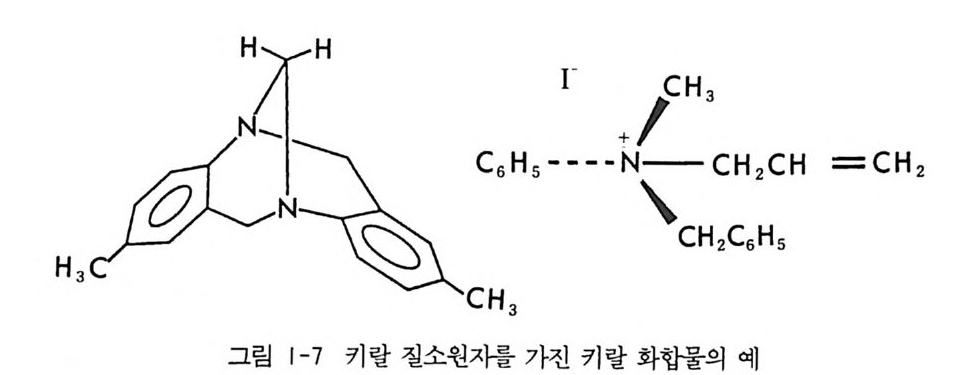
키랄중심으로는 키랄 탄소원자 의에, 키랄 질소원자, 키랄 황원자, 키랄 규소원자, 키랄 인원자, 키랄 비소원자 등이 있다. 예로서 키랄 질소가 존재하여 광학활성인 물질에는 그림 1-7 의 Tro g er 염기와 키 랄 사차암모늄염 화합물이 있다. 1.5 키랄 중심을 갖지 않은 화합물들의 광학 이성질현상 중심원자 X 에 네 개의 다른 치환기 a, b, c, &卜 붙어 있는 키랄 정 사면체의 키랄중심 X 를 그림 l_ 料} 같이 어느 한쪽 방향으로 늘렸을
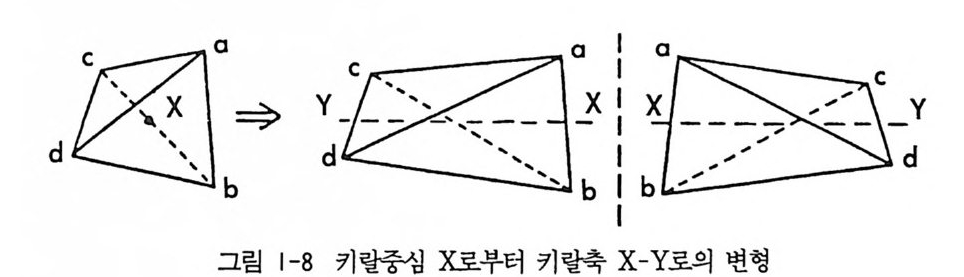 Yd x_b
Yd x_b
때, 치환기 a,b,c,d 는 키랄중심 X 를 늘려서 생기는 축 X-Y 주위 에 배열되는 새로운 형태의 분자를 생각할 수 있다. 이 새로운 분자는 어떤 대칭요소도 포함하지 않으며 거울상과 겹칠 수 없으므로, 비록
키랄중심을 갖고 있지는 않으나 바대칭 분자로서 키랄성이며 C1 접군 에 속하는 분자이다. 이 때의 축 X ― Y 가 키랄축이다. 이와 같이 키 랄축 X ― Y주 위에 네 개의 치환체를 적절히 배열함으로서 생기는 키 랄 분자의 경우, X― Y 축의 X 끝에 있는 두 치환체가 서로 다르고 Y 끝에 있는 두 치환체가 서로 다르기만 하면 이 분자~ 키랄 분자이다. 실제로 그림 1-8 에서 c7} aJ료 바뀌고 &} b 로 바뀌는 경우에도 두 거 울상은 겹치지 않으므로 이 분자는 키랄성이다. 그러나 이 경우에 분 자는 C2 대칭요소를 가지므로 반대칭 분자이다. 키랄축에 의한 키랄 분자들은 다양하다. 알렌 (allene) 화합물 앉은 키랄축 주위에 네 개의 다론 치환체를 가전 화합물의 예로서 비대칭이 며 서로 겹치지 않는 거울상인 광학 이성질체 쌍으로 존재한다. 그러 나 한 쌍의 광학 이성질체로 존재하는 알렌 화합물인 글루티닉산 (glu ti ni c acid ) 산는 C2 대칭요소를 가지고 있으므로 반대칭인 키랄분 자로서 C2 점군에 속한다. 알렌 화합물 의에 키랄축에 의한 키랄 화합물들은 스피란 (s pi rane) 화합물, 아다만탄 (adamant an ) 화합물, 비 페 닐 (bip h eny l) 화합물 등
 Cl IIIIII,'3 Cl
Cl IIIIII,'3 Cl
다양하다. 특히 비페닐 계통의 화합물에서는 두 개의 페닐고리가 단일 결합에 연결되어 있으나 페닐고리에 붙어 있는 치환기 때문에 단일결 합을 중심으로 하는 자유회전이 제한을 받게 되어 이성질 현상이 가능 하게 된다. 이와 갇이 단일결합 주위의 자유회전이 제한을 받아 생기 는 이성질체를 회전장애 이성질체 (a t ro pi somer) 라고 한다. 화합물 § 는 회전장애 이성질체로 존재하는 화합물의 예로서 페닐고리에 붙어 있는 디른 치환기들 때문에 단일결합 주위의 회전이 제한을 받으며 거 울상과 겹치지 않는 키랄 분자이다. 단일결합 주위의 자유회전이 제한 을 받아서 생기는 광학활성 분자 중 고시폴(g oss yp ol) 이라는 화합물 § 은 면화씨 기름에서 얻을 수 있는 천연화합물로서 중국에서는 남성용 피임제로 사용되어온 화합물이다.
 N020 2N N0022 N
N020 2N N0022 N
키랄축에 의한 키랄 분자들의 절대배열은 (R, S) -명명법에 따라 표 기할 수 있다. 키랄축에 의한 키랄 분자를 축의 어느 한쪽 방향 (X 쪽 혹은 Y 쪽)에서 쳐다 볼때, 가까운 두 치환기의 우선순위를 먼 위치에
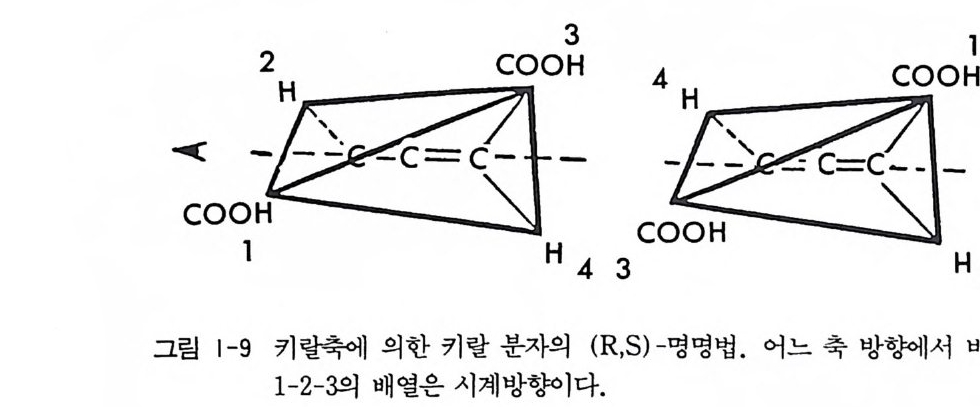 COOH3 COOH1
COOH3 COOH1
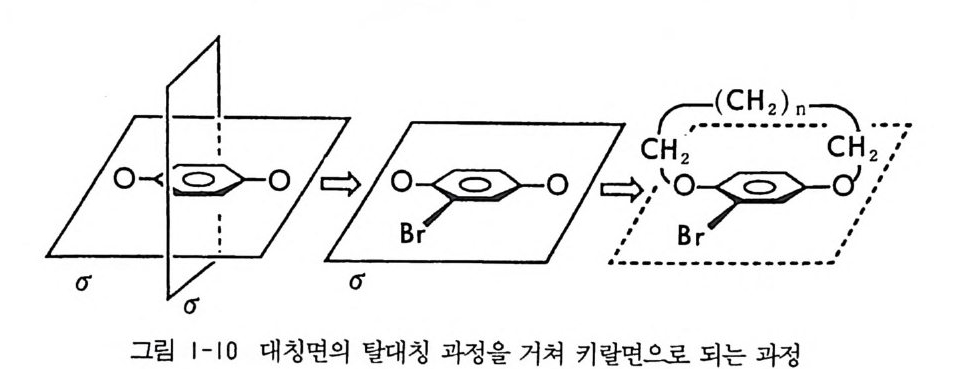
있는 두 치환기의 우선순위보다 높게 정하고, 우선 순위가 가장 낮은 4 번 치환체를 시야의 뒤에 둔다. 이 때 우선순위가 1, 2, 3 인 세 개 치 환체의 배열순서가 시계방향이면 R, 시계 반대 방향이면 오료 표시한 다. 그림 1-9 에서 보는 바와 같이 키랄축에 의한 키랄 분자의 (R)- 혹은 (S)-표 기는 키랄축의 X 쪽에서 바라보나 Y 쪽에서 바라보나 그 결과는같다• 화합물 1은 키랄중심을 갖고 있지 않으며 키랄축도 갖고 있지 않으 나 겹칠 수 없는 거울싱을 가지고 있는 키랄 분자이다. 그림 1-10 에 서 보는 바와 같이 두 산소원자를 파라(p ara) 위치에 가진 벤젠고리 는 두 개의 (대CH广l칭C,)면 H, 김을 값) 6가 1. 지B r나 벤젠고리B問 에i C브C HH롬, j1원 자 를 붙임으로써 벤젠 - 2 R s 7 으 7~
 三三三二二,:,-':',-
三三三二二,:,-':',-
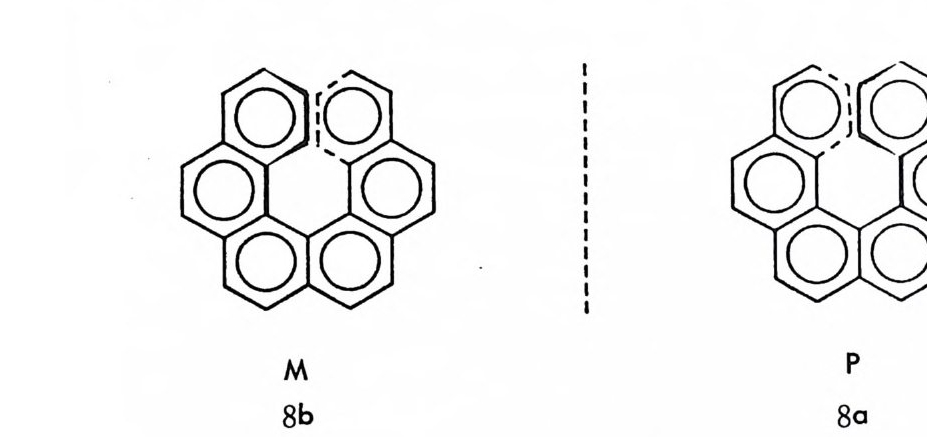
고리와 브롬원자 및 두 산소원자를 포함하는 하나의 대칭면만을 가지 게 된다. 이제 이 대칭면의 위 혹은 아래로 여러 개의 메틸렌 가교를 연결함으로서 서로 거울상의 관계에 있는, 겹칠 수 없는 한 쌍의 광학 이성질체를 얻게 된다. 이때 벤젠고리, 두 산소원자 및 브롬원자를- 포 함하는 면을 키 랄면 (pla ne of chir a li ty) 이 라 한다. 키랄면에 기인한 키랄분자의 (R, S) -명명법에는 우선 지침원자 (pilo t a t om) 의 선택이 필요하다. 지침원자는 키랄면에 가장 인접한 원자들 중에서 선택하며, 화합물 7_ a 의 경우 아래쪽의 메틸렌탄소가 브롬원자에 가까워 우선순위가 높으므로 지침원자가 된다. 지침원자 가 결정되면, 지침원자에 붙어 있는 키랄면의 원자가 1 번 원자가 되 며 , 1 번 원자 다음의 원자가 2 번 원자, 2 번 원자 다음의 원자가 3 번 원자가 된다. 이때 2 번 원자에 붙어 있는 원자가 하나 이상이므로 Cahn-In g old-Prelo g의 우선순위결정 규칙에 따라 우선순위가 높은 원자가 3 번 원자가 된다. 이렇게 하여 1 번, 2 번, 3 번 원자의 배열이 시계 방향이면 R, 시계 반대 방향이면 도료 명명한다. 따라서 화합물 7_ a 의 절대배열은 R 로 표기되며, 화합물 7_~긱 절대배열은 또로 표기한 다. 특히 키랄면에 의한 키랄분자의 절대배열임을 분명히 하기 위하여 pR 혹은 pS (武근 pla ne of ch i ra lity의 약자)로 명명하기도 한다. 헥사헬리센 (hexaheli ce ne) 對본 서로 겹치지 않는 거울상 관계의 두 이성질체로 존재하므로 키랄분자이며 §_a 와 참注곤 서로 광학 이성질체
이다. 핵사헬리센 對존 키랄축을 가진 반대칭 분자이므로 키랄축에 의 한 키랄 분자라고 생각할 수도 있으나 분자의 나선성 (hel icity)에 기인 한 키랄 분자라고 생각하는 것이 편리하다. 나선 (he li x) 은 서로 겹칠 수 없는, 거울상 관계인 오른쪽 회전 나선 (rig h t handed he li x) 과 왼쪽 회 전나선 (lef t handed heli x) 이 가능하므로 나선성 인 분자는 키 랄성 이 다. 화합물 쁘와 같이 오른쪽 회전 나선성 키랄분자는 P 로 표시하며 , 화합물 §臣斗 같이 왼쪽 회전 나선성 키랄분자는 M 으로 표시한다. 헥 사헬리센의 경우 우선성 (dextr o rota t a r y) 광학 이성질체는 (P) -나선 성을 가지며 고유 광회전도는 아주 큰 것으로 알려져 있다 ( [ a 〕 D = 3700°) .
 M P
M P
핵사헬리센이 나선성으로 존재하는 이유는 벤젠고리 여섯 개가 밀집 하여 있을 경우, 마지막에 놓이게 되는 두 벤젠고리는 입체장애 때문 에 같은 평면상에 놓일 수 없고 꼬인 상태로 놓이게 되기 때문이다. 핵사헬리센 이의에도 분자의 밀집구조로 인한 입체장애 때문에 꼬인 상태로 놓이게 되는 화합물들은 모두 나선성에 의한 키랄분자로 생각 할수있다.
1.6 광학 이성질 현상의 역사적 배경 (8-11) 분자의 광학 이성질 현상에 대한 현대적인 개념의 기초는 대부분 19 세기 몇 명의 위대한 과학자들에 의하여 이루어졌다. 19 세기 초 편광계를 만든 B i o t는 수정결정의 결정축을 따라 잘라낸 수정판에 평면편광을 쪼였을 때 수정판을 만든 수정결정의 형 (수정결 정은 서로 거울상인 왼쪽 형과 오른쪽 형이 존재함)에 따리- 평면편광의 편광면이 오른쪽 혹은 왼쪽으로 회전하는 것을 관찰하였을 뿐만 아니 라 테르펜유, 레몬유 등 많은 유기화합물들도 평면편광의 편광면을 회 전시킴울 처음으로 관찰하였다. 이와 같이 평면편광의 편광면을 회전 시키는 성질은 액체뿐만 아니라 용액 상태 혹은 고체 상태에서도 관칠 되기 때문에, B i o t는 이와 같은 성질이 분자 자체의 구조에 기인하는· 것으로생각하였다. 한편 l 앙 1] 기 후반, 포도주를 만드는 과정에서 생긴 포도주 앙금에 서 얻어전 타르타르산(t ar t ar ic a ci d) 과 1820 년에 타르타르산의 정제 과정에서 분리된 라세미산 (racem i c a ci d) 은 1831 년에 이르러 Ber· zel i us 에 의하여 같은 분자식 (C4H606) 울 가진 화합물임이 밝혀침으로 써, 같은 분자식을 가지나 다른 화합물임을 나타내는 이성질 현상 (i somer i sm) 이라는 말이 처음 도입되었다. 1844 년, M it schelr i ch 는 타르타르산과 라세미산의 나트륨-암모늄 염의 결정구조를 연구한 결 과 이들 염의 결정은 같은 형태이며, 결정각, 비중, 굴절률 등 모든 물리적 성질이 같음을 발표하였고 B i o t는 타르타르산의 염은 평면편 광의 편광계의 편광면을 오른쪽으로 회전시키는 우선성을 가지는 반면 라세마산의 염은 광학불활성이라는 사실을 발표하였다. 1846 년, Pas t eur 는 1 9'1엽추의 타르타르산염 결정들이 모두 같은 방향성을 가진 반면상 (hem i hedral) 임을 관찰하였고 타르타르산염 결정의 반면상 구 조는 타르타르산염의 광학활성과 관계가 있어야 한다고 생각하였으며 따라서 광학불활성인 라세미산염은 반면상 구조를 가지지 않을 것으로
기대하였다. 그러나 Pas t eur 는 라세미산염들의 결정구조를 관찰한 결 과 기대와는 달리 반면상 구조를 가짐을 확인하였으며 이것은 Mi tsc helric h 7} 발표한 내용과 일치하는 것이다. 그러나 Pas t eur 는 라세미산염의 결정은 두 가지 형태의 반면상 결정의 혼합물이라는 중 요한 사실을 관찰하였다. 1848 년, Pas t eur 는 광학불활성인 라세미산 의 나트륨-암모늄염을 27C 보다 낮은 온도에서 결정화를 반복함으로 써 서로 겹치지 않는 거울상의 관계에 있는 두 가지 형태의 반면상 결 정 (그림 1-11) 을 만들어 현미경 하에서 직접 두 가지 형태의 결정을 분 리해낼 수 있었다. 두 가지 형태의 결정 중 하나는 타르타르산 나트륨 -암모늄염의 결정구조와 똑같으며 이 결정을 물에 녹여 광회전을 관찰 하였을 때 평면편광의 편광면을 오른쪽으로 회전시킴이 확인되었다.
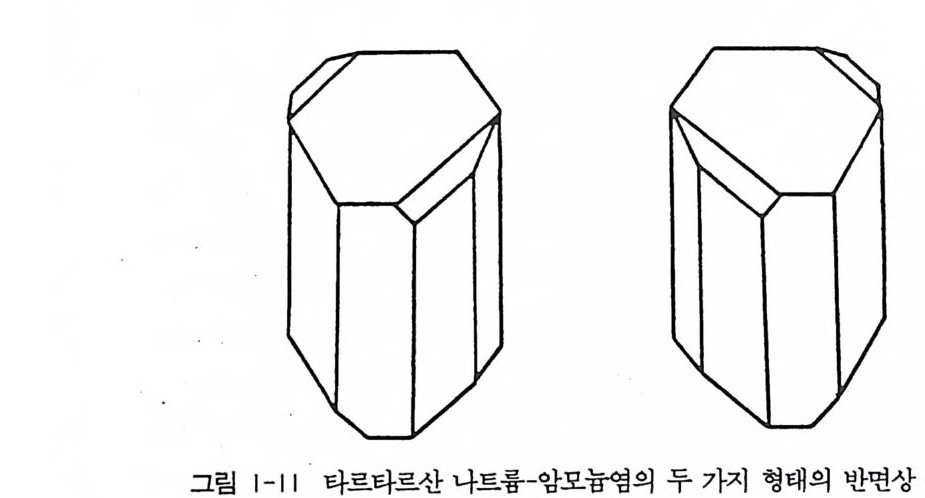 그림 1-l| 타르타르산나트륨-암모늄염의 두가지 형태의 반면상구조
그림 1-l| 타르타르산나트륨-암모늄염의 두가지 형태의 반면상구조
한편 다른 형태의 결정을 물에 녹여 광회전을 관찰하였을 때 평면편광 의 편광면은 왼쪽으로 회전됨이 확인되었으며, 두 형태의 결정을 같은 무게만큼 녹인 수용액은 광학불활성이라는 사실이 확인되었다. Pas t eur 는 두 가지 형태의 결정이 같은 양으로 섞여 있는 광학불활성 인 혼합물을 라세미체 (recema t e) 라고 표현하였으며 이와 같은 표현은
0��X���. ��췘� Pas t eur �� |�8��������X� ���l�p�|� �0�\� �� �� 0��@Ɣ� 첬� �t��� l�p�|� ���D� U�x�X��<�p� tǃ�@ � Mitschelrich7} �\�\ � ������ |�X�XՔ� ��t��. ��췘� Pas t eur �� |�8�������X� ���@� P� ��� ���X� �t
��� ���X� <�i�<�t�|��� � ��\� ����D� �0�X���. 1848 D�, Pas t eur �� �YՈ�\�1�x� |�8����� X� ����h�-TŨ�����D� 27C �� ��@� (�ij��� ���T�|� ���h�<�\� h� �\� ��X��� JŔ� p�����X� �Ĭ�� �ǔ� P� ��� ���X� �t ��� �� � (���� 1-11) D� ̹䴴� ����� X���� ��� P� ��� ���X� ���D� �� ��tռ� � �����. P� ��� ��X�� �� � � X��� ��t���t��� ����h� -TŨ�����X� ���l�p�@� ��<�p� t� ���D� <��� y��� ����D� �0� X��D� L� ��t����X� ���t�D� $�x���<�\� ����´�t� U�x�����. ���� 1-l| ��t���t�������h�-TŨ�����X� P���� ���X� �t���l�p � \ո� �x� ���X� ���D� <��� y��� ����D� �0X���D� L� ��t���� X� ���t�@� |ƽ�<�\� ���(�t� U�x����<�p�, P� ���X� ���D� �@� 4���̹|� y�x� ©�a�@� �YՈ�\�1�t�|��� ����t� U�x�����. Pas t eur �� P� ��� ���X� ���t� �@� ��<�\� ��� �ǔ� �YՈ�\�1� x� <�i�<�D� |�8����� (recem a t e) |�� \��X��<�p� t�@� �@� \�@�� 34 하였으며 이와4) 이 당시에 독일의 화학자 Kekul 려 의하여 유기화합물을 구성하는 탄소원자는 4 개의 원자나 혹은 夕脂의 치환체에 직접결합된 47}로 존재함이 알려져 있었다. 1869 년 Pa t em 야근 이미 탄소화합물의 정사면체 구조를 제안하였으나 유기화합 물의 광학활성을 합리적으로 설명하는 데까지는 이르지 못하였다.
구실에서 연구하던 Le Bel£ Pas t eur 의 연구에 기초하여 독자적으로 탄소원자 주위의 네 개 치환체의 배열에 대한 개념을 제안하였다. 이 러한 비대칭 탄소원자의 개념은 그후 계속 발전하여 1888 년 Me y er 에 의하여 입체 이성질체라는 용어가 사용되기 시작하였으며, 1894 년 F i sher 는 당(특히 단당류)과 시안화수소를 반응시켜 다른 성질을 가전 두 개의 시아노히드린 에피머 (ep im er) 5) 를 합성함으로써 비대칭 반응 의 효시를이루었다.
5) 여러 개의 키릴중심을 가전 화합물의 입체 이성질체들 중 단 하나의 키랄중심의 절대배열만 다른 두 개의 입체 이성질체울 서로 에피머라고 한다.
2 광학 이성질체 분리의 중요성 서로 거울상의 관계에 있는 두 광학 이성질체의 물리적 • 화학적 성 질은 동일하나 키랄환경 (chir a l env i ronmen t)에서는 다르다• 키랄환경 과의 작용에 의하여 두 광학 이성질체는 더 이상 광학 이성질체의 관 계가 아닌 부분 입체 이성질체의 관계에 있게 되기 때문이다. 키랄환 경으로는 평면편광, 키랄시약 등이 있으며 생체 역시 본질적으로 키랄 성이므로(1 3, 12) 키랄환경을 제공한다고 할 수 있다. 따라서 한 쌍의 광학 이성질체는 생체내에서 다른 물리적 • 화학적 성질을 나타내며 따 라서 다른 생리활성을 나타낸다. d- 아스파라전 혹은 d- 클루코오스는 단맛을 나타내나 l- 아스파라진 혹은 l- 글루코오스는 단맛을 나타내지 않는다는 것은 한 쌍의 광학 이성질체가 생체내에서 다른 생리활성을 나타내는 좋은 예라고 할 수 있다 (3).
특히 한 쌍의 광학 이성질체로 존재하는 의약품에 있어서 두 광학 이성질체가 나타내는 인체내의 다른 생리활성은 아주 중요하다. 종종 한 쌍의 광학 이성질체 중 단지 하나의 광학 이성질체만이 약리활성을 나타내며 다론 광학 이성질체는 아무런 약리활성을 나타내지 않거나 혹은 독성을 갖고 있는 경우가 많이 보고되어 있다.
 니9 广 ° :\鬪二 COOH
니9 广 ° :\鬪二 COOH
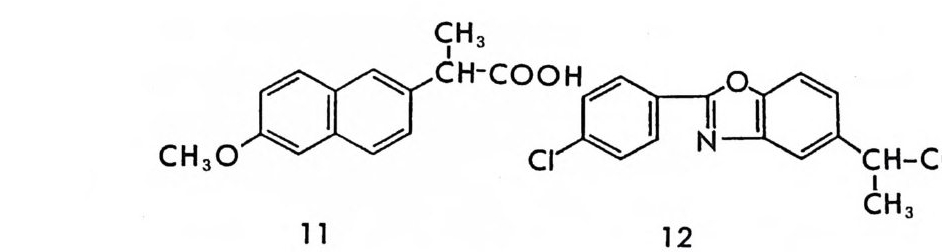
의약품에 있어서 두 광학 이성질체의 생리활성이나 독성효과를 고려 하지 않고 라세미 의약품울 상품화함으로써 비극을 초래한 예는 탈리 도미드(t hal i dom i de) ~에서 찾아 볼 수 있다. 1950 년대 말, 진정제 혹은 수면제로 유럽에서 상품화된 탈리도미드는 독성 혹은 부작용이 없는 것으로 주장되었으나 상품화된 후 곧 말초신경영을 일으키는 신 경독성 및 태아기형 (t era t o g ene city)울 유발시키는 부작용을 가진 것 으로 밝혀졌다. 탈리도미드의 심각한 부작용이 알려진 후 많은 연구에 의하여, 태아기형을 일으키는 탈리도미드의 부작용은 오로지 탈리도 미드의 s- 이성질체에 기인함이 밝혀졌다(1 4). 만일 탈리도미드가 상 품화될 당시에 광학 이성질체의 분리가 손쉽게 이루어져서, 두 광학 이성질체에 대한 임상효능 및 부작용에 대한 실험을 실시할 수 있었다 면 아마도 많은 기형아를 출생시키는 비극을 막을 수 있었을 것이다. 라세미 의약품의 두 광학 이성질체는 생체내에서 다른 약물학적 효 과(임상효과 및 독성 등)를 나타낼 뿐만 아니라, 생체내 약물반응속도 (p harmacok i ne tic s) 에 있어서도 다른 성질을 나타낸다• 비스데로이드 계 소영제로 광범위하게 사용되는 2- 아릴프로피온산 계통의 화합물들 은 한국내에서도 여러 종류가 시판되고 있다. 이들 2- 아릴프로피온산
 CH30 二11 언CH 3 -co:b121 U~H-CCH3O OH
CH30 二11 언CH 3 -co:b121 U~H-CCH3O OH
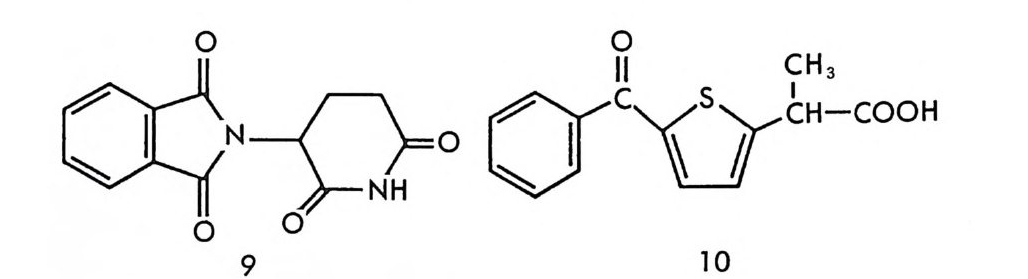
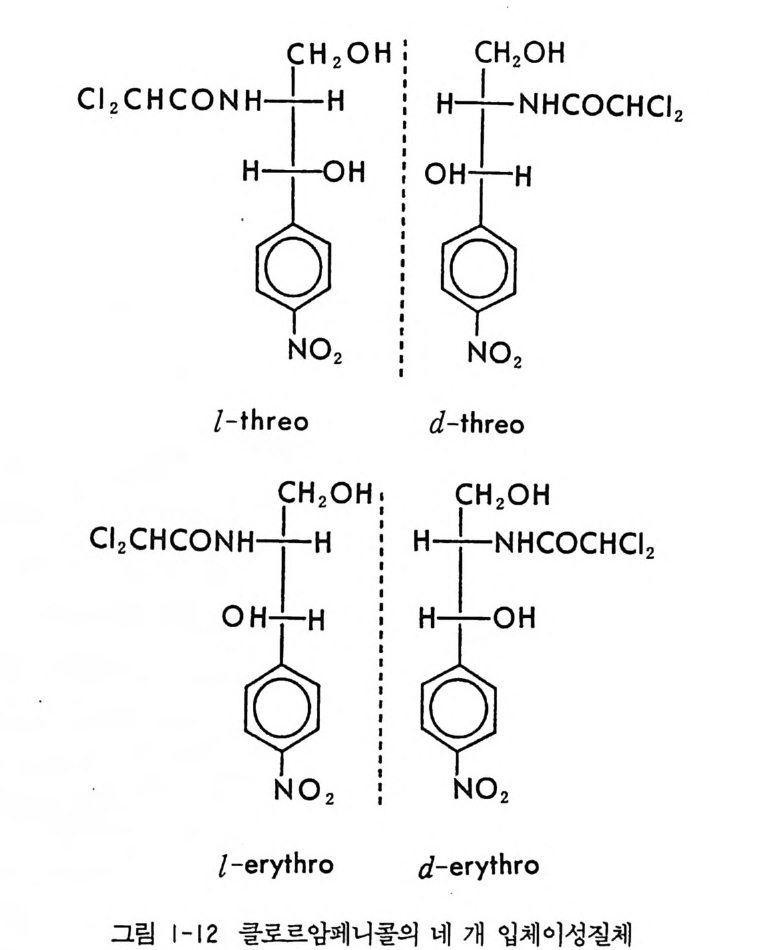
계통의 소영제들은 생체내 대사과정에서 거의 모두 R_ 이성질체가 S -이성질체로 바뀌는 것으로 알려져 있다(최근 티아프로페닉산(ti a pro te r ic aci d) 핀 에서는 이와 갇은 생체내 이성질체 변환이 없는 것 으로 알려져 있음) (15). 상품화되어 팔리고 있는 대부분의 2- 아릴프로 피온산 계통의 소영제들은 라세미체이며 유일하게 나프록센 (na p rox en) 브이 S- 이성질체의 형태로 시판되고 있다. 2_ 아릴프로피온산 계 통의 화합물인 베녹사프로펜 (benoxa p ro f en) 브의 경우에 있어서도 생체내에서 R- 이성질체가 S_ 이성질체로 변환되는 사실이 알려져 있 으나 (16), 생체내에서의 이성질체 전환이 나이 많은 환자에게 있어서 이 화합물의 생체내 대사 및 배설속도를 느리게 하여 간장장해 (he p a t o t ox icity)의 원인이 되며 이로 인하여 미국에서 26 명이 사망하 였고 제약회사 Eli L illy에서 상품화된 Ora fl ex( 베녹사프로펜의 성품 명)의 시판이 금지된 일이 있다 (17). 이들 의에 의약품의 두 광학 이성질체가 생체내에서 다른 생리활성 울 나타내는 예는 무수히 많다 (18). 혈액응고 방지제로 쓰이는 와파린 (Wa rf ar i n) 은 라세미체로 투여되고 있으나 (S)-(— ) -이성질체가 R -(+)-이성질체보다 5 배 정도 효력이 더 좋은 것으로 알려져 있으며 (19 ) ' /3-차단제 (/3- adrenergi c b lockin g ag e nt) 인 프로프라놀놀 (pro - p ranolol) 의 경우도 라세미체 상태로 투여되고 있으나 (S)-( ―)-이성 질체가 (R)-(+) -이성질체보다 100 배 정도 효력이 더 좋은 것으로 알 려져 있다 (20). 항생제인 클로로암페니콜 (chloram p he ni col) 은 두 개 의 키랄중심을 가진 화합물로서 네 개의 입체 이성질체가 가능하나(그
림 1-12) 네 개의 입체 이성질체 중 오로지 l-(-)-th r eo 만이 강력한 항생제의 활성을 가지며 d- (+ )-er yt h 어근 아주 약한 항생제 활성을 가지고 있고 d-(+) - t hreo 나 l-( ― )-er yt h 야근 항생제 활성이 전혀 없다 (3).
 OH : CH20H
OH : CH20H
위의 여러 예에서 보는 바와 갇이 많은 라세미 의약품들이 임상효능 이나 독성 동에 있어서 문제점을 내포하고 있음에도 불구하고, 그립 1 -13 에서 보는 바와 같이 현재 전세계적으로 시판되고 있는 528 개의
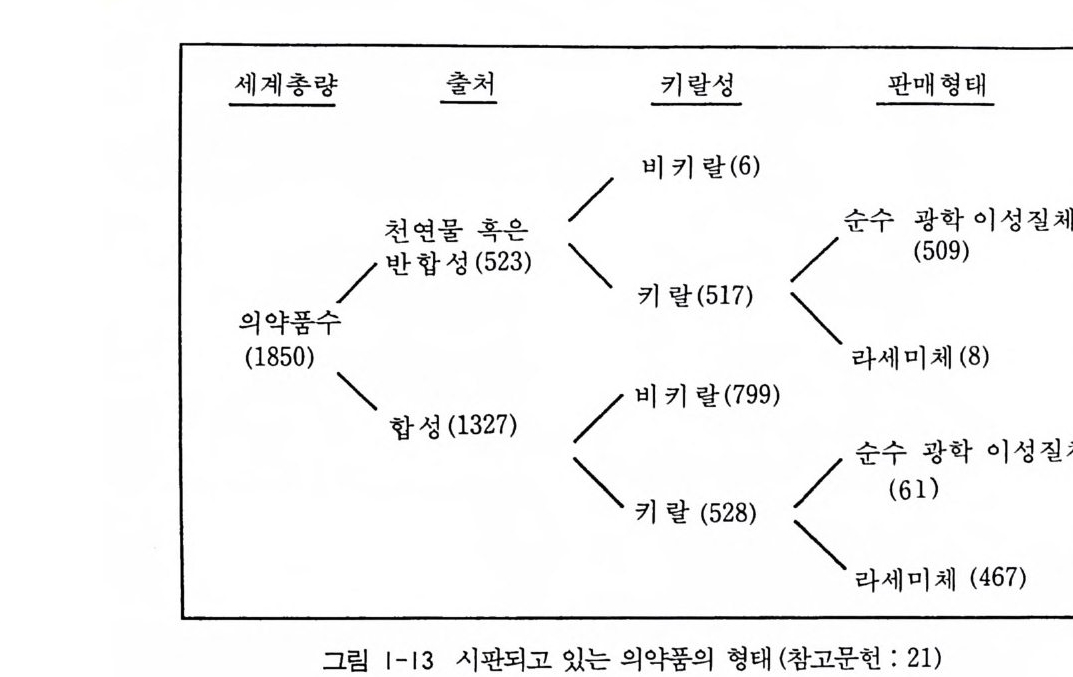 세계총량 출처 키랄성 판매형태
세계총량 출처 키랄성 판매형태
합성 키랄 의약품들 중 단지 61 개만이 순수한 하나의 광학 이성질체 형태로 공급되고 있다 (21). 특히 천연물 혹은 반합성 의약품이 대부분 순수한 광학 이성질체의 형태로 시판되고 있음에 비하여 순수하게 합 성에 의하여 얻어진 의약품의 경우에는 전체 키랄 합성 의약품 중 라 세미체의 형태로 시판되고 있는 의약품이 88% 나 차지하고 있는 것은 의약품을 순수한 광학 이성질체의 형태로 합성하는 것이 어려울 뿐 아 니라 광학활성 의약품들의 광학순도를 정확하게 측정하기가 어려웠기 때문인 것으로 생각된다. 그러나 라세미체 형태로 공급되는 의약품들 의 가능한 문제점들 때문에 조만간 미국의 식품 및 의약 관리국 (FDA) 에서는 새로운 의약품을 허가하는 과정에서 이 의약품이 라세 미체 형태로 사용해도 좋은 충분한 근거가 없는 한 라세미체 형태로는 허가하지 않을 새로운 규정을 발표할 예정이며 이러한 추세는 전세계 여러 나라의 의약품 관리당국에까지 파급될 전망이다. 따라서 앞으 로, 요구되는 효과는 최대이면서 부작용은 최소인 의약품(이 경우에는
특히 비대칭 혹은 반대칭 분자로서의 의약품이며, 의약품 이의에도 농약이 나 식품 첨가물을 개발하는 과정에도 똑같이 적용된다)을 개발하고 상품 화하는 과정에서 필수적으로 요구되는 과정은 라세미 화합물과 광학적 으로 순수한 두 광학 이성질체의 효능 및 독성 등을 비교검토하는· 것 이다. 이러한 비교검토 실험을 실시하기 위해서는 라세미 형태의 화합 물뿐만 아니라 광학적으로 순수한 두 광학 이성질체를 얻을 수 있어야 한다. 광학 이성질체 분리의 필요성과 중요성은 라세미체의 광학분할에 의 하여 광학적으로 순수한 광학 이성질체를 얻는 데 있을 뿐만 아니라 다른 방법 즉 비대칭 합성 (asy m metr i c s ynt hes i s) 에 의하거나 혹은 동 • 식물 등의 생체 천연물에서 추출하여 광학활성 물질을 얻는 경우 에도 이들 물질의 광학순도를 정확히 측정하기 위하여 광학 이성질체 분리의 방법이 사용된다는 점에 있으며, 이들 광학 이성질체둘의 생체 내 변화과정을 추적하는 데 광학 이성질체 분리의 방법이 효율적으로 사용된다는점에 있다. 생체내에서 생리활성을 나타내는 물질들이 많은 경우에 있어서 광학 활성 물질임이 밝혀지고 동 • 식물로부터 추출하여 얻을 수 있는 많은 생리활성 천연물들이 광학활성 물질임이 밝혀짐에 따라 유기화학에 있 어서도 최근에는 광학활성 물질을 직접 얻기 위하여 비대칭 합성에 대 한 연구가 많이 전행되고 있다 (22). 비대칭 합성의 대상이 되는 물질 은 생리활성을 가진 광학활성 물질 뿐만 아니라 이들 물질을 합성하는 과정에서 필요한 키랄 중간체들이다. 비대칭 합성에 의하여 얻어지는 광학활성 물질들의 광학순도를 결정하고 또 이들의 절대배열을 결정하 는 데 있어서도 광학 이성질체분리의 방법은 적절히 요구되며 응용되 고있다. 결론적으로 광학 이성질체 분리의 필요성은 약제학, 약리학 뿐만 아 니라 의학, 식품과학, 농학, 유기화학, 분석화학 등 입체화학에 관련 된 모든 분야에 있어서 아주 크다고 할 수 있다. 광학 이성질체 분리
의 중요성 혹은 필요성이 이와 같이 크게 요구됨에도 불구히고. 광학 이성질체 분리가 문제점으로 남아 있는 것은, 두 광학 이성질체의 동 일한 물리적, 화학적 성질로 인하여 일반적인 혼합물 분리의 방법이 적용될 수 없기 때문이다.
참고문헌(제 1 장) 1. B. Testa , Pr inc ip le s of Orga nic Ste r eoschemi stry, Marcel Dekker, New York, 1979. 2. H. Kag a n, Orga nic Ste r eochemi stry, Joh n Wi le y and Sons, New York, 1979. 3. G. Natt a and M. Farin a , Ste r eochemi st r y, Harpe r and Row, New York, 1972. 4. K. Mi sl ow, Intr o ducti on to Ste r eochemi stry, W. A. Benja m i n, Inc., New York, 1966. 5. J. Jac qu es, A. Collet and S. H. Wi len , Enanti om ers, Racemate s and Resoluti on s, Wi le y Inte r sci en ce, New York, 1981. 6. R. S. Cahn, C. Ing o ld and V. Prelog , Ang e w. Chem. Int. Ed. Eng l. ~. 385 (1966). 7. R. S. Cahn, J. Chem. Ed., 산, 116 (1964). 8. Y. Izumi and A. Tai, Ste r eodif fere nti ating Reacti on s the natu r e of asymm etr ic reacti on s, Academi c Press, New York, 1977, chap 1. 9. G. Natt a and M. Farin a , Ste r eochemi stry, Harpe r & Row, New York, 1972, chap 1. 10. 이길상, 『화학사상사』, 연세대학교 출판부, 1981 . 11. G. B. Kau ffm an and R. D. My er s, J. Chem. Ed., 쁘, 777 (1975). 12. J. H. Brewste r , J. Chem. Ed., 템 667 (1986). 13. 0. J. Gonzalez, J. Chem. Ed., ~ . 503 (1985). 14. W. H. De Camp , in Chir a l Liq u id Chromato g r aph y , W. J. Lou gh , Ed., Blackie , Glasgo w and London, 1989, chap 2. 15. N. N. Sin g h , F. Jam ali, F. M ..P asutt o, A. S. Russell, R. T. Coutt s
and K. S. Drader, J . Pharm. Sci. , ~ . 439 (19 86). 16. A. J. Hutt and J. Caldwell, J. P harm. Pharmacol., 완, 693 (1983). 17. E. Marshall, Sc ien ce, ~ . 1071 (1985). 18. D. E. Dray e r, C lin . Pharmacol. Ther. , 썬 , 125 (1986). 19. C. Banfi el d and M. Rowland, J. Pharm. Sc i., T!:_, 921 (1983). 20. A. M. Barrett and V. A. Cullum. Br. J. Pharm., ~. 43 (19 68). 21. Chem i ca l and Eng en ee r i ng News, Americ a n Chemi ca l Socie t y , March 19, 1990. pp 38-44. 22. J. D. Morris o n, Ed., As ym metr ic Syn th e sis , vol. 2-5 , Academi c Press, New York, 1983- 19 85.
제 2 장 광학 이성질체 분리의 일반적 고찰 1 서론 넓은 의미에 있어서 광학 이성질체의 분리는 라세미체를 순수한 두 광학 이성질체로 분리 (광학분할)하는 ' 방법뿐만 아니라 두 광학 이성질 체를 구분하는 모든 방법을 포함한다고 할 수 있다. 제 1 장에서 기술한 바와 같이 한 쌍의 광학 이성질체는 물리적, 화학적 성질이 동일하다. 따라서 혼합물의 각 성분 화합물들을 분리하거나 구별하는 보통의 방 법으로는 광학활성 물질의 광학순도나 두 광학 이성질체의 조성 (enanti om eric com p os iti on) 을 밝혀낼 수 없으며 두 광학 이성질체를 분리할 수 없다. 죽 보통의 방법으로는 두 광학 이성질체의 구분이 불 가능하다. 한 쌍의 두 광학 이성질체를 구분할 수 있는 경우는 오로지 키랄환경 (chir a l en vi ronmen t)하에서, 두 광학 이성질체가 키랄환경과 작용하여 물리적 성질과 화학적 성질이 서로 다른 부분 입체 이성질체 의 관계에 있을 때이다. M i slow는 그의 저서에서 한 쌍의 광학 이성 질체가 키랄환경하에서 키랄환경과 작용하여 서로 부분 입체 이성질체 의 관계에 있게 되는 것이 두 광학 이성질체를 구분히~근 필요조건이
될 뿐만 아니라 충분한 조건이 된다고 하였다(1). 그러나 라세미체가 고체상일 때 라세미 혼합물 (con g lomera t e) 로 존재한다면 키랄환경이 아니더라도 보통의 결정화 방법에 의하여 라세미체의 광학분할이 가능 할 수 있다. 라세미 혼합물은 서로 거울상의 관계에 있는 두 개의 광 학 이성질체 중 하나의 광학 이성질체만을 포함하는 두 종류 결정의 기계적인 혼합물로서 직접분리가 가능할 만큼 두 종류의 결정을 충분 히 크게 만들 수 있다면 Pas t eur 가 타르타르산의 나트륨 - 암모늄염을 분리한 방법과 마찬가지로 직접 분리해낼 수 있다. 그러나 이 경우에 도, 광학 이성질체의 두 종류 결정을 구분해 낼 수 있는 사람의 시각 이 키랄환경이라고 생각한다면 키랄환경인 시각과 두 광학 이성질체 결정이 작용하여 부분 입체 이성질체의 관계에 있게 되기 때문에 두 종류 결정의 구분이 가능하다고 생각할 수 있다 (2). 한 쌍의 두 광학 이성질체가 키랄환경 하에서 키랄환경과 작용하여 부분 입체 이성질체의 관계에 있게 될 때 이것을 구별하는 방법은 다 양하다. 물리적 성질의 차이 중에서 용해도의 차이를 이용할 경우 재 결정 방법에 의하여 구분이 가능하며, 흡착성질의 차이를 이용할 경우 여러 종류의 크로마토그래피 기법에 의하여 구분이 가능하다. 두 광학 이성질체가 키랄 분자와 반응하거나 키랄 촉매 존재하에서 반응할 때 이들이 구분되는 것은 화학적 성질의 차이 때문이다. 한 쌍의 광학 이 성질체를 구분하는 이들 모든 방법은 크게 크로마토그래피를 이용한 분리방법과 크로마토그래피를 이용하지 않은 분리방법으로 나눌 수 있 다. 제 2 장에서는 라세미 혼합물의 결정화에 의한 광학분할, 속도론적 광학분할법 등 크로마토그래피를 이용하지 않은 광학분할 방법과 크로 마토그래피를 이용한 광학분할 방법들에 대하여 간단히 소개하고자 한 다. 라세미체를 순수한 두 광학 이성질체로 분리하지 않고서 광학활성 물질의 광학순도를 측정하는 방법으로서 열량계 방법 (calorim etr i c meth o d) (3, 4) 과 동위원소 희석법 {iso to p e dil ut i on meth o d) (3, 5)
이 있으나 이 책의 내용과는 다소 거리가 있으므로 기술되지 않을 것 이다. 2 라세미 혼합물의 결정화에 의한 광학분할 라세미체가 고체 상태에서 라세미 혼합물로 존재할 때 결정화에 의 해 광학분할이 가능하며 세 가지의 방법이 알려져 있다. 모든 라세미 체들이 고체 상태에서 라세미 혼합물로 존재할 수 있는 것은 아니기 때문에 결정화에 의한 광학분할은 특정한 라세미체의 광학분할에만 가 능하며 라세미 혼합물로 존재하는 라세미체들은 많이 알려져 있다 (4). 라세미 혼합물의 결정화에 의한 광학분할 방법 중 역사가 가장 오랜 방법은 1848 년 Pas t eur 가 타르타르산의 나트륨-암모늄염 광학분할에 사용한 방법으로서 자발광학분할 (sp o nta n eous resoluti on ) 이라고 한 다. 순수한 광학 이성질체만을 포함하는 두 결정이 충분히 자라고 (1m g 이상), 좌선성 결정과 우선성 결정이 뚜렷이 구별되면 Paste u r 가 했던 것처럼 칙접 손으로 두 결정을 골라낼 수 있다. 좌선성 결정 과 우선성 결정이 뚜렷이 구별되지 않으면 결정 하나 하나롤 용매에 녹여 편광계를 사용하여 광회전도를 측정하는 방법으로 구별이 가능하 다. 그러나 이 방법은 많은 노동력이 필요한 방법으로서 다만 씨결정 (seedcr y s t al) 을 얻는 방법으로서 유용하게 쓰인다. 좌선성 결정과 우 선성 결정을 키우는 과정에서, 라세미 혼합물의 포화용액이 들어 있는 용기내의 다른 두 부위에 우선성 씨결정과 좌선성 씨결정을 넣어 결정 화할 경우에는 좌선성 결정과 우선성 결정이 용기내의 두 위치에서 자 라게 되므로 두 결정을 충분히 키운 후 쉽게 분리해낼 수 있다. 실제 로 Zau gg는 이 방법을 이용하여 50 g의 라세미 메타돈 (me th adone) 이 녹아 있는 용액으로부터 13.0 g의 (+)-메타돈 결정과 13.l g의 (-)-메 타돈 결정을 얻을 수 있었다 (7). 여기서 논의된 자발광학분할의 특칭
은 라세미 혼합물의 포화용액에서부터 좌선성 결정과 우선성 결정이 동시에 얻어지며 결정을 얻고 남은 모액 (moth e r l iq uor) 은 여전히 라 세미 혼합물을 포함한다는 것이다. Pas t eur 의 방법과 Zau gg의 방법 의에, 자발광학분할을 산업적인 규모로 이용하기 위하여 적절히 배열 된 좌선성 씨결정과 우선성 씨결정 주위로 라세미 혼합물의 과포화 용 액을 순환시킴으로써 좌선성 결정과 우선선 결정을 크게 자라게 해서 광학분할하는 방법 등 여러 가지 방법이 있으며 Jac q u es9 -j 저서에 자 세히 서술되어 있다 (4). 라세미 혼합물의 과포화 용액을 결정화하여 두 종류의 결정을 동시 에 얻는 자발광학분할과는 달리 , 유발광학분할 (ind uced resolu ti on) 은 라세미 혼합물의 과포화 용액에 우선성 씨결정이나 좌선성 씨결정 하 나만을 넣어 적절한 조건하에서 결정화함으로써 두 광학 이성질체의 결정화속도 차이에 의해서 선택적으로 하나의 광학 이성질체 결정만을 얻는 방법이다. 예롤 들면 알라닌 벤젠술폰산염의 과포화용액에 좌선 성 씨결정이나 혹은 우선성 씨결정을 넣어 결정화함으로서 D- 광학 이 성질체 혹은 L- 광학 이성질체를 선택적으로 얻을 수 있다 (8). 라세미 혼합물의 과포화 용액에 만일 좌선성 씨결정을 첨가하여 적 절한 조건 하에서 결정화하면 좌선성 결정만을 얻게 되며, 모액에는 우선성 광학 이성질체가 과량 존재하게 된다. 이 모액에 우선성 씨결 정을 넣어 결정화하면 이번에는 우선성 결정을 얻게 된다. 이러한 과 정에서 순수한 광학 이성질체 결정으로 분리해낸 양만큼의 라세미 혼 합물을 계속 공급하면서 좌선성 씨결정과 우선성 씨결정을 반복적으로 첨가하여 결정화하면 두 광학 이성질체 결정을 연속적으로 얻을 수 있 다. 이렇게 반복적인 과정에 의해서 광학분할하는 것을 동반에 의한 광학분할 (resoluti on by entr a in m ent) 이라 한다. 동반에 의한 광학분 할의 좋은 예는 히드로벤조인의 광학분할에서 볼 수 있다. (+)-히드 로벤조인 ll g을 0.37 g의 (-)-히드로벤조인과 함께 85 g의 95% 에탄 올에 녹여 온도를 1 5° C 로 유지하고 10m g의 (-)-히드로벤조인 씨결
정을 가하여 결정화하면 0.87 g의 (-)-히드로벤조인 결정을 분리해낼 수 있다. 모액에 분리해낸 만큼의 라세미 혼합물을 가하고 여기에 10m g의 (+) - 히드로벤조인 씨결정을 가하여 l S' C 에서 결정화하면 0. 9g 정도의 ( + )-히드로벤조인 결정을 분리해낼 수 있고 이와 같은 과 정을 15 번 반복하였을 때 , 6.5 g의 (-) _ 결정과 5.7 g의 (+ ) -결정을 얻 울수 있었다 (9).
 H 『》〈IN〉〔〔 H C30CH 2
H 『》〈IN〉〔〔 H C30CH 2
라세미 혼합물의 결정화가 키랄용매에서 이루어지거나 키랄용질이 녹아 있는 용액에서 이루어지면 키랄용매나 키랄용질이 키랄환경으로 작용하므로 두 광학 이성질체의 결정화 속도가 달라서 광학분할이 가
능할 수 있음을 짐작하기는 어렵지 않다. 특히 키랄용매 혹은 키랄용 질과 두 광학 이성질체의 강한 상호작용으로 부분- 입체 이성질 관계를 유지할 때 결정화에 의한 광학분할은 더욱 용이하다. 예를 들면 디올 화합물 !이 (d) -타르타르산 이소프로필에스테르에서 단순한 결정화에 의해 광학활성인 형태로 얻어지며 (10), 헵타헤데로헬리선 (he pt a he t erohe li cene) 인 화합물 2 가 (― )-a- 피넨 (pi nene) 에서 결정화에 의 해 광학분할된다 (11). 라세미 혼합물인 형태로 존재하는 화합물 妖는 (+) - a- 피넨내에서 (+)_a- 피넨과 함께 1 : 1 착물인 내포화합물 (inc lusio n com p ound) 을 형성하며, 분리후 키랄용매인 ( + )-a- 피넨 울 제거하였을 때 부분적으로 광학분할된 화합물 청이 얻어짐이 알려 져 있다(1 2). 라세미 혼합물의 결정화에 의한 광학분할에 대한 이론적인 배경은 참고문헌 (4) 와 (26) 에 잘 기술되어 있다. 3 속도론적 광학분할 (k in e tic resoluti on ) 라세미체가 키랄시약과 반응하여 다른 화합물로 변형될 때 라세미체 롤 구성하는 두 광학 이성질체와 키랄시약과의 반응속도에 차이가 있 으면 두 광학 이성질체의 구별이 가능하다. 라세미체가 키랄시약과 반 응하여 다른 화합물로 변형될 때, 전이상태에서 두 광학 이성질체와 키랄시약의 착물형성으로 인하여 이들은 서로 부분 입체 이성질관계에 있게 되며 따라서 반웅 과정에서 두 광학 이성질체의 활성화 에너지가 달라지고 반응속도에 차이가 있게 된다. 라세미체 (士 )-E 가 키탈시약 R* 와 반응할 때 (식 2-1) (+)-£!가 R* 의 반응에 대한 반응 속도상수 k1 이 (― )-E 라 R* 의 반응에 대한 반응 속도싱수 k2 보다 크다면 소량의 R* 롤 사용하여 반응할 경우, 반응하 지 않고 남아 있는 출발물에는 (― )-E 가 과량 존재할 것이며 당량의
R* 를 사용하여 반응할 경우 반응이 완전히 종결되기 전의 적당한 순 간에 반응을 차단하면, 반응속도상수 k1 과 k2 의 비에 따라 출발물에 는 (-)-E 가 과량 존재하게 되어 광학분할이 이루어진다. 이와 같이 거울상인 두 광학 이성질체와 키랄시약 사이의 반응속도차이를 이용하 여 라세 미 체 를 광학분할하는 것 을 속도론적 광학분할 (kin e ti c resoluti on ) 이 라고 한다.
 ~E + R *\ / kk21 ►► -+EE' ' (2-1)
~E + R *\ / kk21 ►► -+EE' ' (2-1)
엄밀한 의미에서 속도론적 광학분할은 라세미체를 구성하는 두 광학 이성질체를 따로 분리하는 방법이 아니고 하나의 광학 이성질체를 다 른 화합물로 변형시킴으로써 변형되지 않고 남아 있는 광학 이성질체 를 분리하는 방법이므로 이 방법을 비대칭 파괴 (asym m etr i c destr u c- ti on) 라고 하기도 한다(1). 속도론적 광학분할에 의하여 광학활성 물질을 얻은 예는 많이 알려 져 있다(1 3). 최근 국내에서도 키랄환원 시약인 칼륨 글루코리드 (K
 나。
나。
glu corid e ) 1 를 0. 5 당량 사용하여 라세미 케톤을 환원한 후 반응하지 않은 케돈을 회수하였을 때 회수된 케돈아 광학활성을 가짐을 확인하 였다. 특히 2- 누 근 님 근나논 (2-norbornanone) 을 환원할 경우, ee1) 가 66% 인 (1S, 4R)-2 적료나냐크나논을 분리할 수 있음이 보고되었다 (14) 효소 중에는 라세미체의 두 광학 이성질체 중 특별히 하나의 광학 이성질체만을 정량적으로 다론 화합물로 변형시키는 것들이 있다. 이 때 반응하지 않은 화합물은 광학적으로 순수한 형태로 남아 있게 되므 로 순수한 광학 이성질체를 얻을 수 있다. 역사적으로는 1858 년 처음 으로 Pas t eur 가 라세미산의 나트륨-암모늄염을 페니실리움 글라우컴 이리는 곰팡이와 함께 발효시켰을 때 (d) -이성질체만이 소모되고 순 수한 (l)-이성질체를 얻을 수 있었던 사실이 효소를 이용한 속도론적 광학분할의 효시를 이룬다. D 쵸~ L- 아미노산 산화효소는 라세미 아미노산 중 D- 아미노산이나 L- 아미노산만을 a- 케토산으로 변형시 키므로(식 2-2) 효소반응 후 반응하지 않은 광학적으로 순수한 D- 아 미노산이나 L- 아미노산을 분리해 낼 수 있으며, 반응중에 소모되는 산소의 양을 정확하게 측정함으로써 출발 아미노산의 광학순도를 정확 히 알 수 있다(1 5).
1) enanti or uneric excess 의 약어로서 광학순도(제 1 장)와 마찬가지로, 광학활성 물질의 조성을 나타내며 다음과 같이 정의된다• %ee= 盟:갭 XlOO 광학활성 물질의 광회전도 a 와 광학활성 물질의 조성이 정확히 비례하고, 광 회전도 측정시 실험 오차가 없으면 %ee9 .} % 광학순도는 정확히 일치한다•
? N|H 2 D- 산아화미효노소산 R+ 一( LC) 一-R C— OC깁OH HH 2 —+ NCHO 3O H (D)-+ (L)-R-CH-COOH (2-2)
L- 아미노산 R-CH2 -N H2+C02 (D)-+ L )-R-CH-COOH 1 카르복시 이탈효소 + (D)- R -CH-COOH NH2 깁 H2 (2-3) L - 아미노산카르복시 이탈효소는 오로지 L- 아미노산의 카르복시기 룰 제거하여 아민 및 이산화탄소를 생성하므로(식 2-3) 이 경우에도 반응하지 않은 D- 아미노산을 분리할 수 있을 뿐만 아니라 생성되는 이산화탄소의 양을 정확하게 측정하여 출발 아미노산의 광학순도를 알 수 있다 (15). 4 부분 입체 이성질체의 결정화에 의한 광학분할 라세미 혼합물과 키 랄시 약 혹은 광학분할 시 약 (resolvin g a g en t)의 상호작용에 의하여 형성되는 한 쌍의 부분 입체 이성질체는 물리적 성 질과 화학적 성질이 다르므로- 여러 가지 방법으로 분리가 가능하며 분 리 후 적절한 방법으로 키랄시약을 제거함으로서 광학적으로 순수한 광학 이성질체를 얻을 수 있다. 한 쌍의 부분 입체 이성질체의 서로 다른 물리적 성질 중 용해도 차이를 이용할 경우 결정화에 의하여 분 리가가능하다. 결정화에 의하여 분리할 수 있는 부분 입체 이성질체의 형태는 다양 하다. 이들은 크게 부분 입체 이성질 염, 키랄 리간드를 포함한 전이 금속 착물, 전하이동 착물, 결정성 내포 화합물 등과 감이 분리 후 광 학분할 시약의 제거가 용이한 순간 부분 입체 이성질체 (tra nsie n t dia s te r eomer) 및 라세미화합물과 광학분할 시약 사이의 공유결합에 의하여 형성된 공유결합 부분 입체 이성질체 (covalent bond dia s te r - eomer) 로 나눌 수 있다.
 5
5
부분 입체 이성질체의 결정화에 의한 광학분할 방법 중 가장 폭넓게 사용되는 방법은 라세미 물질과 광학활성인 광학분할 시약 사이예 생 긴 부분 입체 이성질 염의 결정화에 의한 광학분할이다. 일찍이 Pas t eur 는 dl- 타르타르산과 천연산 알칼로이드 계통의 광학활성 염 기인 l- 신코니신 (l-ci nc honic ine ) 發} 반응시켰을 때 생기는, 서로 부 분 입체 이성질 관계에 있는 dl- 염 및 ll- 염 중에서 먼저 결정화 되는 ll- 염을 분리한 후 산 혹은 영기로 처리함으로써 l- 타르타르산을 얻을 수 있었다. 그 후 많은 라세미 카르복실산들이 광학활성 염기에 의하 여 광학분할 되었으며 (16 ) 많은 라세미 염기들이 광학활성 산에 의하 여 광학분할되었다 (17). 부분 입체 이성질 염의 결정화에 의한 광학분 할 방법이 널리 이용되는 이유는 부분 입체 이성질 염이 라세미산과 광학활성 염기 혹은 라세미 염기와 광학활성 산의 반응에 의하여 쉽고 빠르게 형성될 뿐만 아니라, 상황에 따라 무기 산 혹은 무기 염기로 처리하거나 알루미나가 채워진 컬럼을 통과시키거나 혹은 이온교환수 지를 통과시킴으로써 쉽게 본래의 산이나 영기로 분해되기 때문이다 (18 ) . 부분 입체 이성질 영의 결정화에 의한 광학분할에 사용할 수 있는
광학분할 시약은 안정한 화합물이어야 하며 경제적으로 쉽게 구입할 수 있어야 하고 특히 중요한 것은 광학순도가 아주· 높아야 한디는 것 이다. 광학분할 시약의 광학순도는 광학분할하여 얻은 광학활성 물질 의 광학순도의 한계가 되기 때문이다. 예로서 라세미 혼합물인 R S-를 광학순도가 100% 가 아닌 광학분할 시약 rS' (소량의 R' 를 포함한 시약) 와 반응시켜 생기는 부분 입체 이성질체, RS' 와 SS' 를 분리할 때 RS’ 에는 이것의 거울상 이성질체인 Sr 이 섞이게 되며 SS’ 에는 이것의 거울상 이성질체인 Rr 이 섞이게 된다(식 2-4). RS+rS'---R-S- S=r -- '- - +• ..S-R :.S- :r . ' . . (2-4) 따라서 최종적으로 염의 분해에 의하여 R 을 분리해낼 경우 R 에는 r 에 해당하는 양의 S 가 섞이게 되고 마찬가지로 S 에는 r 에 해당하는 양의 R 이 섞이게 된다. 표 2-1 은 부분 입체 이성질 염을 형성할 때 흔히 사용되는 광학활성 산과 광학활성 염기들울 요약한 것이다. 광학분할 시약은 이 d- 형 및 l- 형으로 구할 수 있을 경우에 특히 유용하다. 예를 들어 l_ 브루신 (/ - bru ci ne) 을 사용하여 라세미 카르복실산울 광학분할할 경우려] 브루 신은 l ~ 형으로만 존재하기 때문에 l- 브루신과 반응하여 결정으로 분 리되는 한 종류의 광학 이성질체만을 얻을 수 있는 반면 합성 키랄아 민인 a- 페닐에틸아민은 d- 형과 l- 형 모두 쉽게 구할 수 있기 때문에 원하는 형의 광학 이성질체를 모두 얻을 수 있다. 일반적으로 라세미 형태의 산 (dlA) 과 광학활성 염기 (dB) 가 반응하여 형성된 두 개의 부 분 입체 이성질 염 dAd& 가 lAdB 중에서 dAdB 가 선택적으로 결 정화하여 분리되며, 염의 분해시 순수한 dA 를 얻을 수 있다면 라세 미 형태의 산 (dlA) 과 광학활성 염기 (l B) 가 반응하여 형성된 두 개의 부분 입체 이성질체 염 dAl& 나 lAl W..근 lAdB 및 dAd& 가 거울상
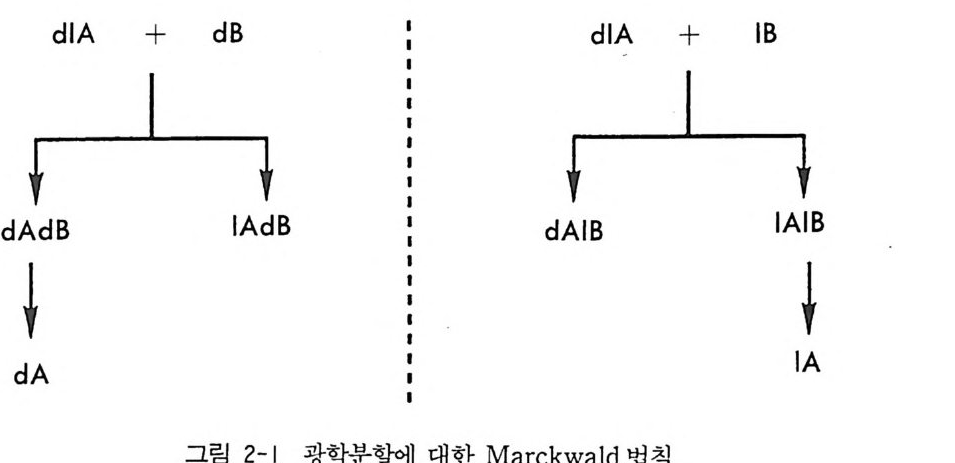
표 2-1 부분 입체 이성질 염의 형성에 사용되는 광학 활성 화합물의 예 명칭 분자식 분자량 [ 리。 (deg ) Bruc ine c23H2 6N20 4 394.4 -127(CHCb) Str ych in i n e C21 H 2 2N20. 354.4 一 139(CHCl3) Qu in i n e C20 H 2.N202 324.4 -117(CHCJ 3) Cin c honid i n e C1 9H 22N2 0 294.4 -109(Et 0 H) Cin c honin e C19H22 N20 294.4 +2 29(Et O H) Morpi ne C11H19N03 285.3 -132(Me0H) Dehy d roabie t y la mi ne C2 0H 3 ,N 285.5 +4 6(MeOH) Ep h edrin e (-), (+) C10H1s NO 165. 2 土 6 .3(E t OH) Amp h eta m i ne (-), (+) C9 H 13 N 135. 2 土 38(C6H6) a-Pheny le th yla mi ne (— ) , (+ ) Cs H 11N 121.2 土 40(Nea t) cr-(1- N aph th yl) eth yla mi ne (— ) , (+ ) C12 H 13N 171. 2 土 82(Nea t) a-(2-Naph th yl)et h yla mi ne (키 , (+ ) C12H13N 171 .2 土 19(Ne~t) Tart ar ic ac id (—), (+) C4H606 150.1 土 12(H20) 0(+, )0 '-Di be nzoy l ta rta ric acid (-) , c18H1408 358.3 土 118(E t 0H) Mandelic acid (-) , (+ ) c8H8°3 152.2 土 157(H20) Mali c a c id (키, (+) C4H60s 134. 1 土 2.3(H20) 2-Phenoxy pr op ion ic ac id (+ ) C9HIOo3 166.2 +40(Et O H) N-Acety ll euc ine (-) , (+ ) CsH1sN03 173.2 士 24(MeOH) 의 관계에 있으므로 이번에는 lAlB 가 선택적으로 결정화되어 분리되 며, 이 염을 분해하였을 때 lA 를 얻을 수 있다. 이것을 광학분할에 대 한 Marckwald 법 칙 (Marckwald pr in c ip le ) 이라고 한다 (그림 2 一 1) .
 dlA + dB dlA + 18
dlA + dB dlA + 18
예를 들면 l - a- 페닐에틸아민 혹은 d-a- 페닐에틸아민을 이용하여 라세미 이부프로펜(i bu p ro f en) 을 광학분할할 경우에 정확히 같은 방법으로 (R) -이부프로펜과 (S) -이부프로펜을 얻을 수 있다 (19). 라세미 알코올, 라세미 케톤, 라세미 알데히드 등은 산이나 염기 형 태의 광학분할 시약과 반응시켜 직접 부분 입체 이성질 염을 만들 수 없기 때문에, 부분- 입체 이성질 염의 결정화에 의한 광학분할이 불가 능하다. 그러나 이 경우에도 이들 화합물들을, 염을 쉽게 형성할 수 있는 유도체로 변형시킴으로써 부분- 입체 이성질 염의 결정화에 의한 광학분할이 가능하며 실제로 이 방법에 의하여 많은 종류의 라세미 알 코올과 라세미 카르보닐 화합물들이 광학분할되었다(1 3, 18, 20) . 그러 나 부분 입체 이성질 영의 분리 후 염의 분해가 필요할 뿐만 아니라 최종적으로 유도체 부분을 떼어내야 하므로 번거로운 과정이 많다는 결점이 있다. 쉽게 분해되는 결정성 부분- 입체 이성질체를 형성함으로써 광학분할 이 가능한 다른 예는 라세미 물질과 광학분할 시약 사이의 전하이동 착물 (charge tra nsfe r com p le 지울 형성할 때이다. 예를 들면 a- 아미 노프로필리덴아미노옥시프로피온산과 2, 4, 5, 7 -테 트라니트로풀
루레논 사이의 옥심 교환반응에 의하여 생긴 (+ )- 혹은 (- )-a-(2, 4, 5, 7- 테트라니트로플루오레닐리덴아미노옥시 )프로피온산 (2, 4, 5, 7- tet r a nit ro bfl uo reny li d e neami n ooxy ) pr op ion ic acid : TAPA) _§ 은 (21) 전하부족 화합물로서 헬리센 계통의 방향족 화합물 혹은 방향 족 아민 등과 전하이동 착물을 형성하며 이때 두 전하이동 착물이 선 택적으로 결정화될 때 광학분할이 가능하다 (22 , 23).
 02N ?/' 0-C언H-C ONO0H2
02N ?/' 0-C언H-C ONO0H2
광학활성 물질이 리간드로 사용된 전이금속 착물이 라세미 물질과 반응하여 부분 입체 이성질 착물을 형성할 때는 부분 입체 이성질 착 물의 용해도 차이를 이용하여 선택적으로 결정화함으로써 광학분할이 가능하다(1 3). 예를 들면 광학활성인 a- 페닐에틸아민이과 에틸렌 리 간드로 사용된 백금 착물과 라세미체인 트란스 - 시클로욱텐이 반응하 여 에틸렌 대신 트란스-시클로옥텐이 치환될 경우 두 개의 부분- 입체 이성질 착물이 형성되며 (그림 2-2) 선택적 결정화에 의하여 분리가 가 능하고 트리페닐포스핀으로 처리함으로 광학활성인 트란스-시클로옥 텐을 얻을 수 있다 (24). 시클로덱스트린 (c y clodex t r i n: 제:w, 3 . 1 절 참조)이나 광학활성인 트리 -o- 티모티드 (tri- o -th ymo -t i de : TOT) Z 은 키 랄공동 (chir a l cav- ity)을 가지고 있으므로 키랄공동에 맞는 크기의 라세미 물질이 이 구
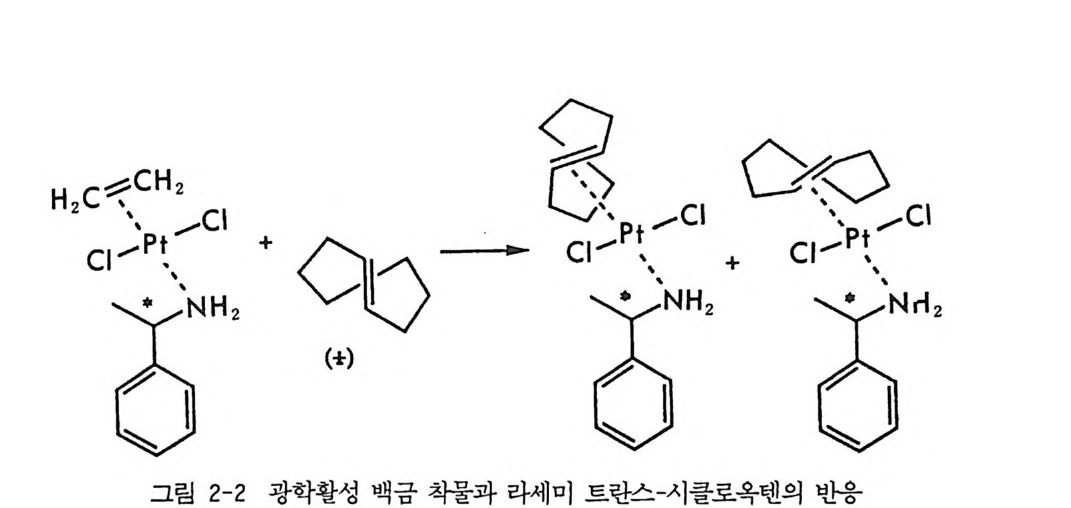 H2CCl~.,.C`,`P H!O~2/ ` C IH 2 N+ 5° 一 言b `P\to 2 H:+ 오: Nc 2 ld
H2CCl~.,.C`,`P H!O~2/ ` C IH 2 N+ 5° 一 言b `P\to 2 H:+ 오: Nc 2 ld
조에 끼어들어갈 경우 결정성 내포 화합물이 형성되며 결정화에 의하 여 분리가 가능하다. 시클로덱스트린의 키랄공동을 이용한 광학분할 은 특히 액체크로마토그래피를 이용한 광학분할에 광범위하게 이용되 고 있으며 제 3 장 3 절에서 자세한 논의가 있을 . 것이다. 특히 키랄공동 이 특수한 키랄환경을 제공하기 때문에 라세미 화합물이 이 공동에 내 포될 때에는 두 광학 이성질체가 모두 내포되지 않고 자물쇠 - 열쇠 관 계처럼 특정한 절대배열을 가전 광학 이성질체만 내포가 가능하여 결 정성 내포 화합물을 형성하기 때문에 광학분할이 가능하다. 라세미체 인 트란스 -2,3- 에폭시부탄은 광학활성인 트리 -0- 티모티드의 키랄공
 。
。
동과 자물쇠―열쇠 관계에 의하여 광학분할이 가능한 것으로 알려져 있다 (25). 라세미 물질과 광학분할 시약이 반응하여 두 개의 공유결합성 부분 입체 이성질체가 쉽게 만들어지며 이렇게 만들어진 두 개의 부분 입체 이성질체의 용해도 차이가 충분히 크고 적어도 하나의 부분 입체 이성 질체가 결정성일 때, 결정화에 의하여 두 개의 공유결합성 부분- 입체 이성질체의 분리가 가능하다. 그러나 공유결합성 부분 입체 이성질체 의 결정화에 의한 광학분할의 문제점은 부분 입체 이성질체의 분리 후 순수학 광학 이성질체를 얻기 위하여 부분 입체 이성질체로부터 광학 분할 시약을 떼어낼 때 공유결합을 끊어야 하므로 어느 정도 강한 반 응조건이 필요하고 따라서 광학 이성질체가 라세미화할 위험성이 있다 는 것이다. 이러한 문제점에도 불구하고 많은 라세미 물질들이 공유결 합성 부분 입체 이성질체로 변형된 후 결정화에 의하여 광학분할되었 다(1 8). 5 NMR 에 의한 광학분할(광헉당도 측정) 라세미체를 순수한 두 광학 이성질체로 직접 분리하는 것은 아니지 만, 두 광학 이성질체의 비율 혹은 조성에 대한 정보롤 쉽게 얻을 수 있는 기법이 NMR 에 의한 광학분할이다. 서로 거울상인 한 쌍의 광 학 이성질체는 NMR 상에서 똑같은 화학적 이동 (chem i cal sh ift)을 보 이나 적절한 방법으로 키랄환경을 제공할 경우 한 쌍의 광학 이성질체 는 부분 입체 이성질 관계에 있게 되며 따라서 다른 화학적 이동을 보 이게 된다. NMR 측정시 서로 거울상인 한 쌍의 광학 이성질체가 다 른 화학적 이동을 나타내도록 하기 위하여 키랄환경을 제공함으로서 부분 입체 이성질 관계를 갖도록 하는 방법으로는 키랄유도체 시약 (chir a l deriv a ti zin g a g en t :CDA) 과 라세미 물질을 반응시켜 공유결합
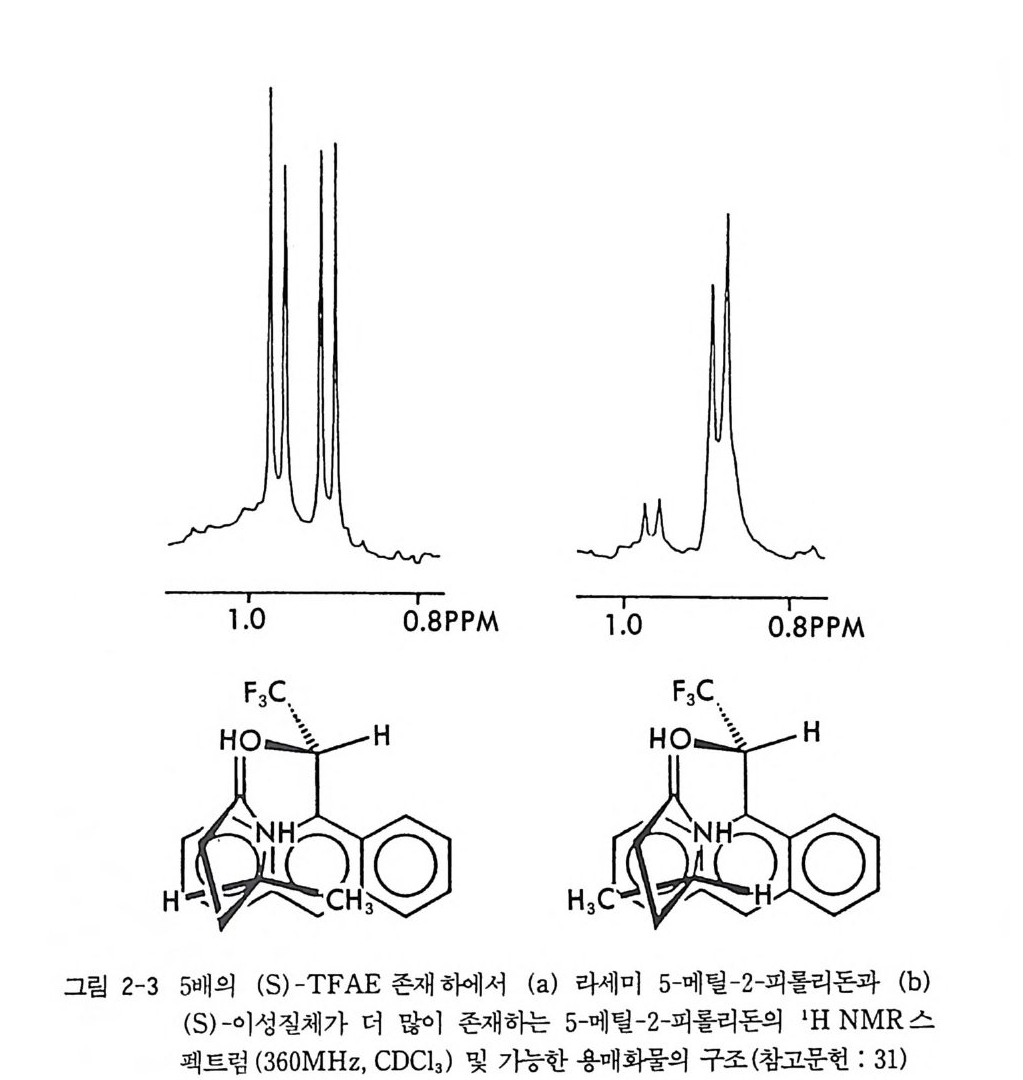
성 부분 입체 이성질체를 만드는 것이다. 한 쌍의 공유결합성 부분- 입 체 이성질체는 서로 물리적 성질이 다르므로 NMR 의 화학적 이동도 다를 것이며 따라서 NMR 에 의한 구분이 가능하다 (27). 한 쌍의 공 유결합성 부분 입체 이성질체의 NMR 연구는 액체 크로마토그래피에 의한 공유결합성 부분 입체 이성질체 분리와 연관되어 연구가 많이 되 어 있으므로 액체 크로마토그래피에 의한 부분- 입체 이성질체의 분리 와 함께 제 2 장 6.2 절에서 다루게 될 것이다. 키랄환경을 적절히 제공함으로써 NMR 을 이용하여 광학활성 물질 의 광학순도를 측정할 수 있는 다른 방법은 키랄 용매화 시약 (ch i ral solvati ng age n t :C SA) 방법과 키랄 란타나이드 이동 시약 (ch i ral lanth a nid e shif t reag en t :C L SR) 방법이 있다. CSA 방법과 CLSR 방 법은 주로 1970 년대에 많이 연구되어 발표되었으나 현재에도 비대칭 합성시 얻게 되는 광학활성 물질의 광학순도를 결정할 때 유용하게 사 용되는 중요한 NMR 방법이라고 할 수 있다. CSA 방법의 중요한 부분은 1970 년대 P i rkl 러] 의하여 주로 연구되 었다 (28, 29). 적절한 작용기 (보통 히드록시기, 카르복시기, 아민기 등) 롤 가전 광학적으로 순수한 키랄용매와 라세미 물질을 혼합하였을 때 라세미 물질은 키랄용매에 의해 용매화되어 NMR 상에서 다른 화학 적 이동을 나타내는 부분 입체 이성질 용매화물(di as t ereomer i c solva t e) 이 되므로 NMR 의 화학적 이동으로부터 구분이 가능하다. P i rkl 쑥 여러 종류의 CSA 를 개발하였으나 (28) 특히 현재 상업적으 로 구할 수 있는 2,2,2_ 트리플르오르 -1-(9- 안트릴)에탄올 (2,2,2 -trif luo ro-1-(9 -anth r yl ) eth anol : TFAE) §이 광학활성 물질의 광 학순도 결정에 많이 사용되고 있을 뿐만 아니라 광학 이성질체의 절대 배열 결정에도 사용되고 있다 (30). 예를 들면 라세미 5- 메틸 -2.- 피롤 리돈 (5-me t h y l-2- pyrr o li done) 은 (S) -TFA& 라 그림 2 궁과 같은 상 호작용을 한다. (S,S) 섬卜매화물의 경우에는 5- 메틸 -2- 피롤리돈의 5 -메틸기가 TFA 탸 안트릴기 위에 위치하기 때문에 NMR 상에서
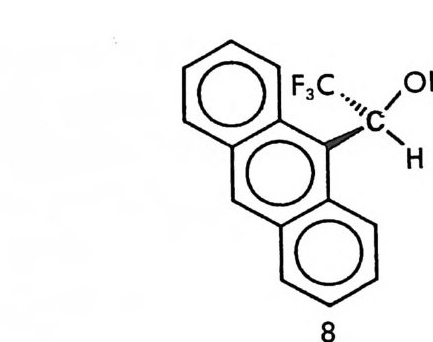 C. ·:.c/, OH
C. ·:.c/, OH
5- 메틸기에 해당하는 이중선이 안트릴기의 비등방성 효과 (an i so t ro pi c e ff ec t)에 의하여 높은 장 쪽으로 이동한다. 반면 (R,S)-%- 매화물의 5 -메틸기는 안트릴기의 의부에 위치하므로 5 - 메틸기에 해당하는 이중 선이 NMR 상에서 낮은 장 쪽으로 이동하여 그림 2-3 의 NMR 스펙트 럼에서 보는 바와 같이 5- 메틸 -2- 피롤리돈의 (R) -이성질체와 (S)- 이 성질체가 구분된다 (31). 그림 2-3 에 제시되어 있는 부분 입체 이성질 용매화물의 구조가 실제구조와 동일하다면 5- 메틸기의 상대적인 화학 적 이동으로부터 자동적으로 두 광학 이성질체의 절대배열이 결정된 다. 또한 5- 메틸기에 해당하는 피이크의 상대적인 세기를 측정한다면 두 광학 이성질체의 존재비율 죽 광학순도를 측정할 수 있다. 최근에 보고된 연구에 의하면 MTPA(a-meth oxy - a-(tri f luo r- ometh yl)ph eny la ceti c acid ) ~(32), BNPPA ((R) 혹은 (S)-1, l' -bin a p h th yl- 2, 2'-diy lp h osph oric ac id) .!Q( 33) , 만델산 브 (34) 등 이 광학활성 아민의 광학순도를 측정하는 NMR 의 CSA 로 사용되고 있广다. : :Ho3 °o`/P i,O o H ifcooH H._ ->OH 10 l l
 1.0 0.8PPM 一1.0 0.8PPM
1.0 0.8PPM 一1.0 0.8PPM
CLSR 방법은 1970 년대 Whit es id e s 등에 의해서 많이 연구되었다 (35, 36). 흔히 시용되는 CLSR 로는 착화합물 g, 11 등과 같이 보통 키랄 디케톤 화합물과 란탄계열금속 중 득히 유로품 (Eu) 사이의 육배 위 착화합물들이다. 이들 착화합물들은 루이스 산으로써 알코올, 에 스테르, 아민, 케톤, 에테르, 에폭시드, 아미드 등과 같은 루이스 염 기인 유기화합물들과 착물을 형성할 수 있다. · 루이스 영기가 라세미
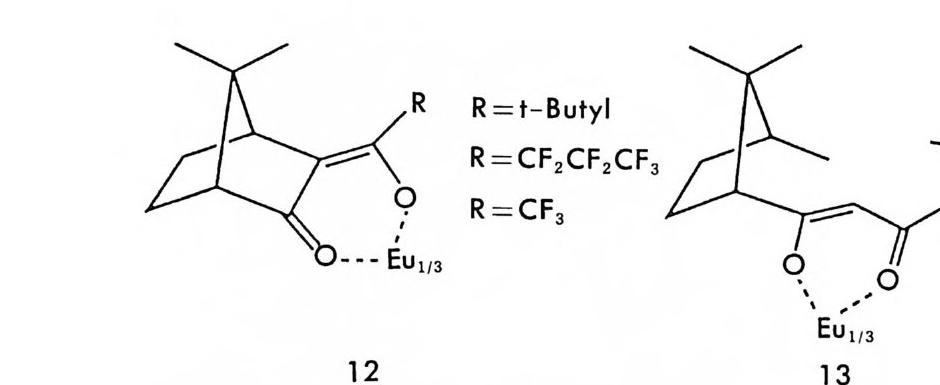 R R= t-B uty l
R R= t-B uty l
화합물일 경우 CLSR 는 라세미 화합물과 반응하여 두 개의 부분 입체 이성질 착화합물을 형성한다. 이 때 두 개의 부분- 입체 이성질 착화합 물의 안정도에 차이가 있으면 보다 안정한 착화합물의 경우 광학 이성 질체가 금속에 보다 강하게 구속되어 있을 것이므로 NMR 의 화학적 이동(낮은 칭쪽으로)이 크게 영향을 받게 된다. 만일 두 개의 부분 입 체 이성질 착화합물이 다른 기하 구조를 갖는다면 이 경우에도 당연히 두 착화합물의 NMR은 다른 화학적 이동을 보일 것 이며 따라서 두 개의 광학 이성질체의 구분이 가능하다 (37 ) . CLSR 방법이 CSA 방법에 비해 광학순도의 결정에 있어서 사용범 위가 일반적으로 더 넓은 것은 CLSR 이 모든 종류의 루이스 염기인 라세미 물질과 착화합물을 형성할 수 있을 뿐만 아니라 CSA 방법에 서 보다 더 큰 화학적 이동의 차이를 보이기 때문이다. 그러나 CLSR 과 라세미 물질 사이에 형성되는 착화합물의 구조가 확실히 밝혀지지 않았고 복잡하기 때문에 화학적 이동으로부터 광학 이성질체의 절대배 열을 결정하는 것이 간단하지 않을 뿐만 아니라 화학적 이동의 차이 룰 크게 하기 위하여 CLSR를 많이 사용할 경우에 NMR 의 선폭증가 (line broadenin g ) 현상이 관찰되며 , CLSR 들은 흡습성 이므로 보관이 나 취급에 주의하여야 하는 문제점이 있다. 결정화에 의한 광학분할에서는 광학분할 시약의 광학~ 분할되 는 광학 이성질체의 광학순도의 한계가 되나 CLSR 방법 및 CSA 방
법과 같은 NMR 방법에서는 CLSR 이나 CSA 의 광학순도가 조금 떨 어지더라도 시료의 광학순도를 결정하는 데 아무런 문제가 없다. CLSR 이나 CSA 가 광학적으로 순수하지 않더라도 이것은 다만 두 광 학 이성질체의 화학적 이동의 차이 (AA8) 만 감소시키기 때문이다 (36). 6 액체 크로마토그래피에 의한 광학분할 6.1 서론 다른 광학분할 방법과 마찬가지로 크로마토그래피를 이용하여 서로 거울상의 관계에 있는 한 쌍의 광학 이성질체를 분리하기 위하여서는 적절한 방법으로 키랄환경을 제공하여야 한다. 크로마토그래피를 이 용한 광학분할의 한 가지 방법은 서로 거울상인 한 쌍의 광학 이성질 체를 키랄유도체 시약 (ch i ral deriv a ti zi n g a g en t :CDA) 과 반응시켜 물 리적, 화학적 성질이 서로 다른 한 쌍의 공유결합성 부분 입체 이성질 체를 만듦으로써 고정상에 의한 두 부분 입체 이성질체의 흡착의 차이 를 이용하여 분리하는 방법이다. 이 방법은 한 쌍의 광학 이성질체를 칙접 분리하지 않고 부분 입체 이성질체를 만들어 분리하므로 간접분 리 방법 (ind ir e ct resolu ti on) 이라 한다. 반면에 한 쌍의 광학 이성질 체를 공유결합성 부분- 입체 이성질체로 만들지 않고 한 쌍의 광학 이 성질체와 키랄선택자 (ch i ral selecto r ) 사이의 상호작용에 의하여 일시 적인 부분 입체 이성질체 (tra nsie n t d i as t ereomer) 를 만듦으로써 광학 분할이 이루어지는 방법을 직접분리 방법 (dire ct resoluti on ) 이라 한 다. 직접분리 방법에는 여러 가지가 있으나 광학분할 시약 혹은 키랄 선택자를 크로마토그래피에 쓰일 수 있는 고정상에 적당한 방법으로 결합시킴으로써 고정상 자체가 비대칭성을 갖게 되어 두 개의 광학 이
성질체를 구별할 수 있는 키랄고정상 (ch i ral sta t i on ary p hase : CSP) 을 이용한 광학분할 방법이 1980 년대에 둘어선 이후 아마도 가장 이상적 인 광학분할 방법으로 인식되고 있다. 이 책의 제 3 장부터는 여러 종 류의 CSP 에 대한 자세한 기술이 있게 될 것이다. 간접분리 방법이나 직접분리 방법이나 모두 모든 종류의 크로마토그래피 방법 (기체 크로 마토그래피, 얇은 막 크로마토그래피 등)에 응용할 수 있으나 기술적이 고 기계적인 내용만 다를 뿐 광학분할의 내용은 동일하므로 여기서는 광범위하게 사용되는 액체 크로마토그래피 (Liq u id Chromato g ra p h y : LC) 를 이용한 광학분할에 대하여 주로 기술하게 될 것이다. 제 2 장 6.1 절 서론에서는 앞으로 계속 논의하게 될 몇 가지의 크로 마토그래피 용어에 대하여 간단히 정의하고자 한다. 두 종류의 물질(한 쌍의 광학 이성질체 혹은 한 쌍의 부분 입체 이성질 체 포함)이 액체 크로마토그래피 (혹은 기체 크로마토그래피)상에서 그림 2-4 와 같이 분리될 때, 두 물질의 분리 정도를 정량적으로 표현하기 위하여 보통 용량인자 (ca p ac ity fac to r), 분리인자 (se p ara ti on fac - tor ), 분해인자 (resolu ti on fa c t or) 를 정의하여 사용되고 있다. 컬럼의 용량인자 k' 는 고정상에 있는 물질의 총몰수 (n s ) 와 이동상에 있는 용질의 총몰수 (n 니의 비율, 즉 n s/ nm 과 같은 값으로 정의된다 (38). 그림 2- 伴 물질 A 에 대한 용량인자 kA’ 는 크로마토그램으로 부터 쉽게 측정이 가능한 양으로부터 식 (2 - 5) 와 같이 계산된다. 여기 서 t o 는 지체되지 않는 용질의 머무름 시간 (re t en ti on tim e) 혹은 이동 상이 컬럼울 빠져나오는 데 걸리는 시간이며, t A 는 물질 A 의 머무름 시간으로서 시료주입시간부터 물질 A 가 칼럼에서 빠져나올 때 (보통 피이크의 꼭지점)까지 걸리는 시간이다. 물질 B 에 대한 용량인자 kB, 는 용량인자 kA’ 와 마찬가지로 식 (2-5) 와 같이 계산된다. kA'=t~。 。 ’ kB' = t Bt-。 t。 (2-5)
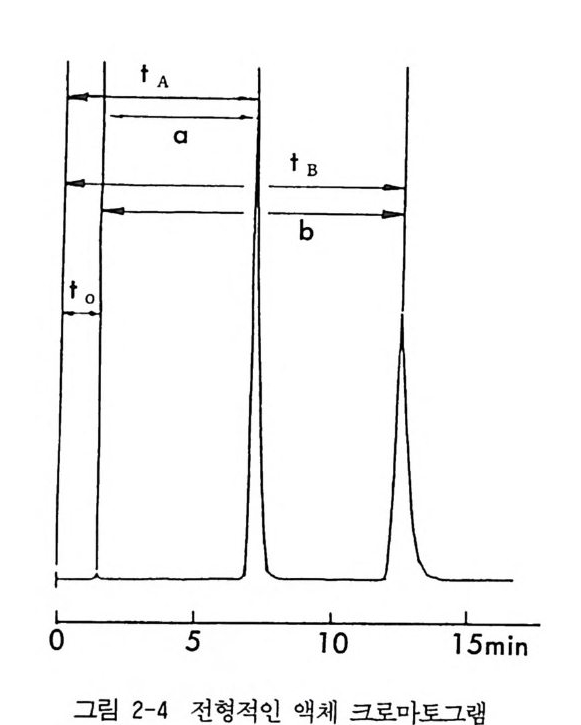 t A
t A
특히 물질 A 혹은 물질 B 의 머무롬 시간에서부터 t o 를 뺀 값 즉 tA — to 나 tB ―t o 를 물질 A 혹은 물질 탸 1 보정된 머무롬 시간 (ad j us t ed rete n ti on ti me) 이라고 하기도 한다. 일반적으로 컬럼의 길이가 길어 지거나, 고정상과 용질분자 사이의 상호작용이 효과적으로 강하게 이 루어지면 용량인자는 증가한다. 고정싱에서 두 물질의 분리정도를 나타내기 위하여 분리인자 a 가 정의되어 사용되고 있다. 분리인자 a 는 크로마토그래피 시스템의 기 계적 성질이나 컬럼의 충전상태 등과 상관없이 오로지 고정상과 두 용 질분자 사이의 상호작용 에너지 차이에 의하여 결정된다. 키랄고정상 과 두 광학 이성질체의 상호작용 에너지 차이와 a 값과의 관계는 6. 3.3 에서 기술될 것이다. a 값은 물질 A 와 E 긱 용량인자의 비와 같으 며 크로마토그램으로부터는 물질 A 와 물질 E 긱 보정된 머무름 시간 울 이용하여 식 (2-6) 과 같이 쉽게 계산할 수 있다.
a= 노kA: ' = t도^t―t°。 =브a (2-6) 두 물질 A 와 B 의 머무름 시간에 충분한 차이가 있다고 하더라고 컬럼내 물질전달의 문제점 등에 의하여 각 피이크가 길게 끌리거나 넓 어진다면 두 피이크롤 확실하게 구별하지 못하는 경우가 있을 수 있 다. 두 피이크를 두 개의 뚜렷한 다른 피이크로 분해 (resolu ti on) 할 수 있는 정도를 타나내기 위하여 분해인자 R s 가 식 (2 구)과 같 이 정의 되어 사용되고있다. Rs = ½wt,\B+— ½t A w B -= 2w (AtB +— wtBd (2- 7 ) 여기서 W 려 W 솥 물질 A 와 물질 B 에 해당하는 피이크 A 와 피이 크 표키 띠 폭 (band wi d t h) 으로서 tA 및 tB 와 같은 단위로 측정된 값 아다. Rs 가 보통 1. 00 보다 크면 두 피이크를 구분하여 확인할 수 있 기에 충분한 것으로 생각되고 있다. 지금까지 기술한 용량인자, 분리인자, 분해인자 등 크로마토그래피 에 흔히 사용되는 용어들에 대한 더 자세한 정의가 필요하면 참고문헌 (38) 이 크게 도움이 될 것이다• 6.2 간접분리 방법 6.2.1 특성 및 범위 서로 거울상인 한 쌍의 광학 이성질체를 CDA 와 반응시켜 두 개의 공유결합성 부분 입체 이성질체를 만들고 이들의 흡착성 차이를 이용 하여 크로마토그래피로 분리하는 간접분리 방법은 직접분리 방법과는 달리 우리 주위에서 쉽게 구할 수 있는 보통의 컬럼을 사용한다는 이
점을 가지고 있다. 그러나 직접분리 방법과 바교하여 간접분리 방법은 여러 문제점들을 내포하고 있다. 한 쌍의 거울상 이성질체를 CDA 와 반응시켜 공유결합성 부분- 입체 이성질체를 만든 후 두 개의 공유결합성 부분 입체 아성질체를 분리하 더라도 순수한 광학 이성질체를 얻기 위하여 CDA 를 제거하여야 할 경우에 사용한 화학적 방법의 반응조건이 격렬하면 라세미화의 위험성 이 있다. 광학활성 물질의 광학순도를 결정하는 데 있어서도 LC 에 의 한 간접분리 방법에는 여러 문제점이 있다. 라세미 물질(혹은 어느 한 쪽 이성질체가 과량으로 존재하는 광학활성 물질)과 CDA 를 반웅시키는 과정에서 반응조건에 따라 라세미화의 문제점이 있을 뿐만 아니라, 두 광학 이성질체와 CDA 의 반응이 정확히 같은 속도로 일어나지 않는다 면 생성되는 두 개의 부분 입체 이성질체 비율은 본래의 두 광학 이성 질체의 비율과 다를 수 있으며 따라서 생성된 두 개의 부분 입체 이성 질체의 비율(크로마토그래피에서 두 부분- 입체 이성질체에 해당하는 피이 크의 넓이 비율)로부터 본래 두 광학 이성질체의 비율 혹은 조성 (광학 순도)을 결정할 수 없다. 또한 LC 를 이용하여 두 개의 부분」 입체 이 · 성질체를 분리할 때 두 부분 입체 이성질체의 물리적 성질의 차이 때 문에 두 부분 입체 이성질체에 대한 검출기의 감응도 (de t ec t or res p onse) 가 일치하지 않을 수 있으며 이 경우에도 두 부분 입체 이성 질체에 해당하는 크로마토그램 피이크의 넓이로부터 본래의 두 광학 이성질체의 바율을 결정할 수 없게 된다. 또 다른 문제점으로는 사용 하는 CDA 의 광학순도가 100% 가 아닌 경우에 LC 를 이용하여 라세 미 물질의 CDA 유도체를 완전히 분리하더라도 최종적으로 얻은 광학 이성질체의 광학순도는 CDA 의 광학순도를 능가할 수 없으며, CDA 의 광학순도가 광학활성 물질의 광학순도를 결정하는 데 있어서 곧바 로 제한요소가 된다는 것이다. 예를 들면 R/ 않 1 비가 99 : 1 인 광학 활성 물질을 광학순도가 98% 인 CDA(99% 의 R', 1% 의 S' )와 반응시 켜 부분 입체 이성질체를 얻었을 때 LC 상에서 두 피이크의 비 (검출기
표 2-2 LC 를 이용한 광학 이성질체의 간접분리 방법에 사용되는 키랄 유도체 시약 (CDA) 의 예
CDA 의 구조 명칭 특징 아o민H, CCo-D-
티오이소시아네이트 CDA 주로라세미 알코올및 라 쌩三OA< IGITC 세미 아민의 광학분할에 이용됨 OA, 군 CNCS IAITC 0^' 카르복실산 CDA 주로라세마 알코올및 라 ©fF, 세이미용 됨 아민의 광학분할에 CH,0-C- CO OH a- 메독시 -a-( 트리플르오 르메틸 )-a 페닐아세트산 c H`0 千IHC Dc 。 。 H 산a- 메톡시 -a 대닐아세트 〈
산p여 화 물:广AY广》 H 2 OIIcc r(mid ee n) th o xy a cety l chlo-주세미로 라아세민미의 알광코학올분및할 에라 영화 멘록시아세틸 | 이용됨 H :問 o,c, (영c화am캠p 퍼h 술or포su닐l ph ony l chlorid e ) ::[ 클로로포름산멘틸 OIIc (ment h y l chlorofo r - c mate ) H감응도는 동일하다고 가정 할 경우)는 99 : 1 이 되는 것 이 아니라 98 : 2 가 된다. 왜냐하면 유도체화 과정을 통하여 RR' 부분- 입체 이성질체와 SR' 부분 입체 이성질체만 얻어지는 것이 아니라 이 들 과 함께 CDA 의 광학불순물인 S ' 이성질체와 광학활성 물질이 반응하여 생긴 RS ' 부 분 입체 이성질체 및 SS' 부분 입체 이성 질 체도 함께 얻어지며 RR ' 와 SS' 및 SR' 와 RS' 는 서로 거울상의 관계에 있는 광학 이성 질 체이므로 보통의 LC 컬럼상에서 구분되지 않고 같 은 피이크로 나타나기 때 문 이 다. 간접분리 방법의 이러한 여러 문제점에도 불구하고, 원리적으 로는 라세미체의 CDA 유도체인 한 쌍의 부분 입체 이성 질 체 는 크 로 마 토 그 래피 방법에 의하여 보통의 컬럼상에서 모두 분리가능한 것 으로 이해 되고 있기 때문에, 크로마토그래피를 이용한 라세미 화합물의 간접광 학분할 방법은 현재에도 광범위하게 쓰이고 있으며 지난 10 여 년간 이 분야에 대한 많은 논문이 발표되었을 뿐만 아니라 Pir k le(3 9 ), Soute r (40) , Li n dner (41) , Gal (42, 43) , Ahno ff와 Ei na rsson (44) 등에 의 하여 총설 논문들이 쓰여졌다. CDA 와 반응시켜 공유결합성 부분 입체 이성질체 를 만듦 으로써 LC 상에서 분리되는 라세미 물질들은 CDA 와 온화한 조건에서 쉽게 반응 할 수 있는 작용기를 가지고 있어야 할 뿐만 아니라 CDA 또한 이 들 과 쉽게 반응할 수 있는 작용기를 가진 광학적으로 순수한 키랄 화합 물이어야 한다. 따라서 LC 간접분리 방법에 의하여 광학분할아 가능 한 라세미 물질들은 보통 히드록시기, 아민기, 카르복시기, 카르보닐 기 등과 같은 작용기를 가전 물질들이며, CDA 들은 이들과 쉽게 반응 할 수 있는 작용기를 가전 광학적으로 순수한 광학활성 물질로서 보통 키랄 아민, 키랄 카르복실산, 키랄 이소시아네이트(i soc y ana t e), 키랄 이소티오시아네이트(i so t h. i oc y ana t e), 키랄 아실화 시약 등이 CDA 로 시용된다. 지금까지 CDA 로 사용된 바 있는 광학활성 물질들 이 많이 알려져 있으나 이중 흔히 쓰이는 CDA 둘을 표 2-2 에 요약하
여 놓았다. CDA 로 쓰이는 광학활성 물질들의 더 많은 예는 Soute r (40), Lin d ner(41), Ahnho ff와 E i narsson(44) 의 문헌을 참고하면 될 것이다. 표 2-2 의 CDA 들 중 키랄 아민들은 주로 라세미 카르복실산들의 광 학분할에 사용된다. 키랄 아민인 CDA 와 라세미 카르복실산의 반응에 의하여 부분 입체 이성질 아미드가 형성되는데, 이 때 아미노기와 카 르복시기 사이의 아미드 형성반응을 쉽게 하기 위하여 흔히 유기합성 에서 사용되는 많은 방법들이 시도된다. 카르복실산을 SOCl2 와 반응· 시켜 산염화물을 만든 후 아민과 반응시켜 아미드를 만들 수 있으나 산과 SOCl2 의 반응시 카르복실산이 광학활성일 경우 라세미화가 일어날 가능성이 있다. 따라서 온화한 조전에서 간단하게 반응 시키기 위하여 짝지음 시약 (coup ling a g en t)인 CDI(1,1'-carbony l• dii m i da zole) (45, 46) , DCC (N,N'-dic y c l ohexy lc arbodii m i de ) (47) , 혹 은 HDBT(l-h y d roxy b enzotr i a z ole) (48) 를 사용하여 라세미 카르복실 산과 키랄 아민 CDA 사이에 아미드 부분 입체 이성질체들을 만들어 광학분할한 예들이 보고되어 있으며 저자의 실험실에서는 EEDQ ( 2 -eth o xy - l-eth o xy c arbony l -1, 2-d i hy dro qu i nol i ne) 를 짝지음 시 약으로 사용하여 아미드 부분- 입체 이성질체를 쉽게 만들고 있 다. 키 랄 아민 CDA 들 중 특히 DANE (1-( dim eth yla mi n o-1- nap h th yl)e th y l ami ne ) (49) 과 1- (안트릴)에틸아민 (50) 은 형광성이 있 으므로 부분 입체 이성질 아미드를 만든 후 LC 분리시에 검출이 훨씬 용이하다. 여러 키랄 아민들 중 상업적으로 쉽게 구할 수 있으며 가격 이 바교적 저렴하기 때문에 가장 많이 사용되는 것은 a- 페닐에틸아민 이다. 이들 키랄 아민 CDA들 은 카르복실산의 광학분할뿐만 아니라, 라세 미 카르보닐화합물과 쉽게 이민결합을 형성하여 쉽게 부분 입체 이성 질체를 만들 것이므로(이민결합의 시스, 트란스 이성질 현상때문에 조금 복잡하리라고 예상됨) 라세미 케톤이나 라세미 알데히드의 광학분할에
도 사용될 수 있을 것이다. 실제로 최근 고시폴(g oss yp ol ) 의 두 광학 이성질체와 (+)-페닐알라닌에틸에스데르가 반응하여 생긴 부분- 입체 이성질 이민 브가 LC 상에서 제조의 목적 (pr ep a rati ve resolu ti on) 으로 분리된 예가 보고되어 있다 (51).
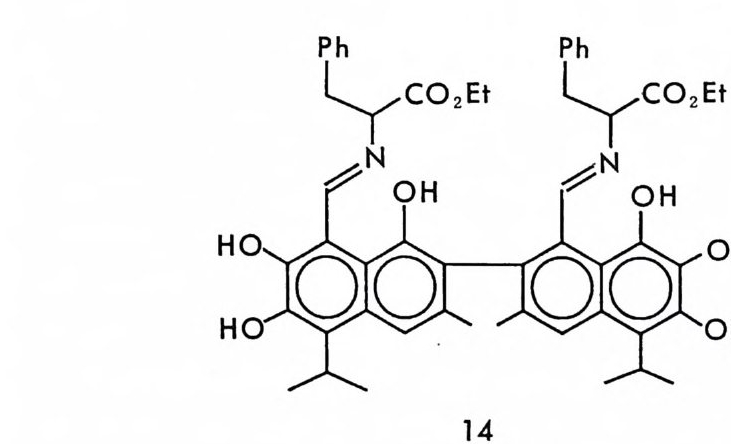 H0` 广Ph 2E t Plhy ~-co ,E- O, H
H0` 广Ph 2E t Plhy ~-co ,E- O, H
라세미 알코올, 라세미 아민의 광학분할에 가장 광범위하게 사용되 는 CDA들 은 키랄이소시아네이트 화합물들로서 대표적인 것들은 표 2 -2 에 그 구조가 나와 있다. 페닐에틸이소시아네이트 (PEIC) 와 1- 나프 틸에틸이소시아네이트 (NEIC) 는 상업적으로 구할 수 있을 뿐만 아니 라 페닐에틸아민이나 1- 나프틸에틸아민으로부터 쉽게 합성되며 (52) 데 히드로아비틸아민 (deh y droab i ety lam i ne) 또한 상업적으로 구할 수 있 기 때문에 표 2- 除] 세 가지 키랄 이소시아네이트 화합물들이 CDA 로 많이 사용되고 있다. 이들 키랄이소시아네이트 CDA들 은 라세미 알코 울과 반응하여 카르밤산 에스데르 부분 입체 이성질체가 되며 (53-56) 라세미 아민과 반응하여 우레아 부분 입체 이성질체가 되어 (57) LC 상 에서 분리가 가능하다. 키랄 아소시아네이트와 라세미 아민의 반응은 빠른 시간내에 종결되는 반면 키랄 이소시아네이트와 라세미 알코올의 반응은 천천히 진행되며 조금 높은 온도가 필요할 뿐만 아니라 촉매량 의 N, N- 디메틸에탄올아민 등을 필요로 한다 (58). 아민과 알코올의
이와 같은 반응성의 차이 때문에 /3 一 차단제와 같이 아미노기와 히드록 시기를 둘 다 가진 아미노알코올 라세미체의 경우 아미노기만을 선택 적으로 반응시켜 부분 입체 이성질체를 만들 수 있다 (59). 특히 카르 밤산 에스테르 부분 입체 이성질체는 자동화된 MPLC(mediu m pre ssure liq u id chromato g rap h y ) 시스템을 이용하여 다량으로 분리할 수 있을 뿐만 아니라 (52 , 60) LC 분리 후 트리에틸아민 존재하에서 삼 영화실란과 반응시킴으로써 온화한 조건하에서 광학활성 알코올을 얻 울 수 있고 CDA 인 키랄 이소시아네이트가 그대로 회수되기 때문에 라세미 알코올의 광학분할에 아~노 유용동}게 쓰인다 (61) . 라세미 락탐 또한 페닐에틸이소시아네이트 혹은 1_ 나프틸에틸이소 시아네이트를 CDA 로 이용하여 광학분할이 가능하다. 라세미 락팀을 페닐에틸이소시아네이트와 반응시켜 얻은 우레이드 (ure i de) 유도체는 그립 2-5 와 같이 실리카 겔 컬럼상에서 분리가 잘 될 뿐만 아니라
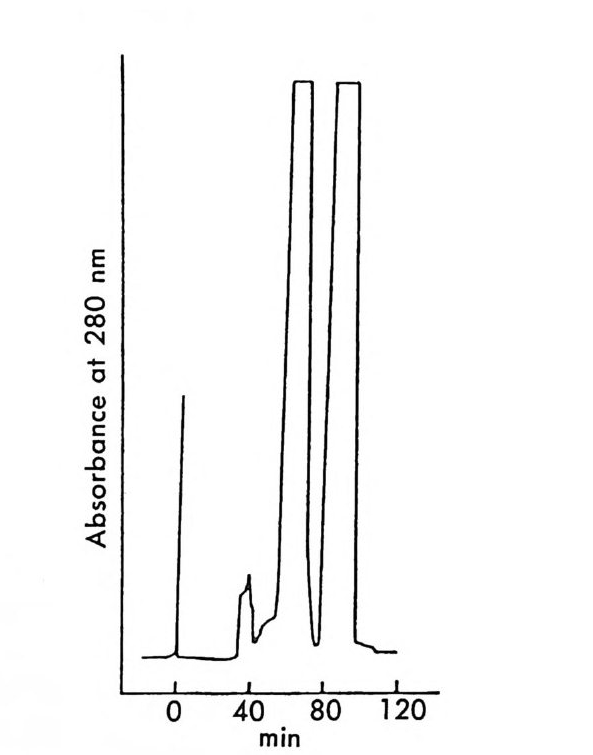
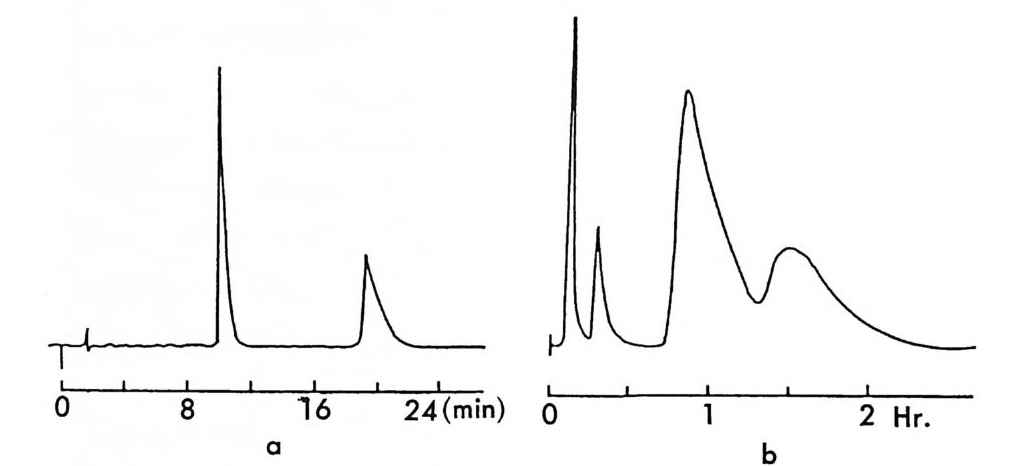 。 8 a 16 24(mi n) O b 2 Hr.
。 8 a 16 24(mi n) O b 2 Hr.
그림 2-5 실리카 겔 컬럼상에서 라세미 5- 페닐피를리돈의 (S) -페닐에틸우레이드 유도체에 해당하는 두 부분· 입체 이성질체의 분리. (a) 분석용 분리, 이동상 : 이소프로판을-핵산 (1/ 99) , 유속 : 2mL/mi n. (b) 제조용 분리 (MPLC 사용) , 컬 럼 크기 : 2.5X 30cm, 시료크기 : 1.8g , 이 동상 : CH2 Cl2, 유속 : 30mL/mi n. (참고문헌 : 31)
MPLC 시스템을 이용할 경우 2.5 X 30cm 의 컬럼상에서 1. 8 g까지 분 리가 가능하다 (31). 분리된 순수한 부분· 입체 이성질 우레이드 유도체 는 나트륨메독시드 (sod i um me t hox i de) 로 처리하면 광학적으로 순수 한 락탐이 얻어진다 (31). 한편 데히드로아비틸 이소시아네이트 (deh y droab i ety l i soc ya t e) 는 주로 히드록시아이코사데트라엔산 (h y drox y e i cosa t e t raeno i c acid : HETE) 의 광학분할에 사용된 바 있다 (62). 키랄 이소티오시아네이트인 CDA 둘로서 광범위하게 사용되는 것은 GITC (2, 3, 4, 6-te t r a -o-acety l- /3 -D -g l ucop yr anosyl iso th i o c y a nate ) 와 AITC(2, 3, 4-tr i - o -acety l- a-D-arabin o p yra nosy l iso th i o c y a nate ) 로서, Ni m ura 등에 의하여 티오시안산 은 (s il ver t h i oc y ana t e) 과 아 세 뚜 l:I 쿠 F 슈가 (ace t obromosu g ar) 를 반응시켜서 얻은 것이다 (64, 65) . 본래 GITO --l-A ITC 는 아미노산의 광학분할에 사용되 었으나 라 세미 일차아민이나 라세미 이차아민과 반응하여 부분 입체 이성질 티 오우레아를 형성함으로써 라세미 아민이나 라세미 아미노알코올의 광 학분할에 사용되고 있다 (57, 65). 그림 2-6 은 일차아민인 라세미 파라 클로로암페타민이 GIT~ 부분 입체 이성질 티오우레아 유도체로서 LC 에 의하여 분리된 크로마토그램의 예를 제시한 것이다. CDA 로 사용되는 키랄 카르복실산들은 라세미 아민이나 라세미 알 코올과 반응하여 부분 입체 이성질 아미드나 에스데르를 만듦으로써 광학분할에 이용된다. 그러나 카드복시기의 반응성 때문에, 짝지음 시약인 DCC 를 매개로 하여 반응이 일어나거나 (66, 67) 혹은 카르복실 산의 산염화물 형태로 라세미 아민이나 라세미 알코올과 반응하여 부 분 입체 이성질체를 만든다 (57, 68). CDA 로 사용되는 키랄카르복실산 이 a- 위치의 키랄중심에 수소를 갖고 있을 경우는 염화티오닐과 반응 하여 산염화물을 만드는 과정에서 혹은 유도체를 만드는 과정에서 라 세미화가 일어 날 수 있으나 a- 메목시 -a- (트리플르오로메틸 )-a- 페닐 아세트산(혹은 산염화물 : MTPA) 은 a 려난遷一 가지고 있지 않으므로
 R s
R s
라세미화의 위험성을 염려할 필요가 없다 (57). 염화 멘톡시아세틸 (menth o xy ac ety l chlor i de) 은 라세미 디올 화합 물과 반응하여 비스-(키_멘톡시아세트산에스데르를 형성함으로써 LC 상에서 광학분할이 가능하다(그림 2-7) (69). 특히 NMR 상에서 멘 톡시아세트산에스테르의 아세틸기에 있는 두 수소(그림 2 구에 a 와 困료 표시된 수소)의 화학적 이동이 다르기 때문에 화학적 이동의 차이를 이 용하여 디올의 광학순도를 측정할 수 있다. 키랄 알코올울 포스젠(p hos g ene) 과 반응시킴으로써 라세미 아민이 나 라세미 알코올과 쉽게 반응하여 부분 입체 이성질 카르밤산 에스테 르나 탄산 에스데르를 형성하는 클로로포름산 에스데르 (chloro fo r mate ) 계통의 CDA 들을 얻을 수 있다. 특히 멘톨 (men t hol) 은 광학활 성인 형태로 쉽게 구할 수 있기 때문에 이것의 클로로포름산 에스테르 인 (+)들t로로포름산 멘틸 ((-)-menth yl chloro fo rma t e) 은 라세미
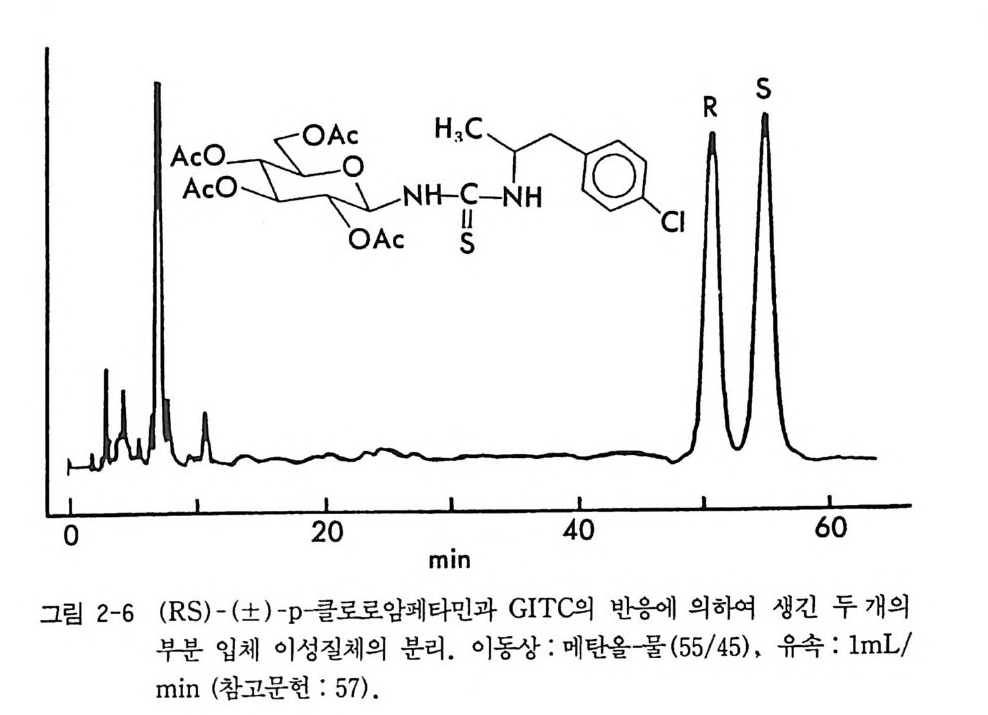
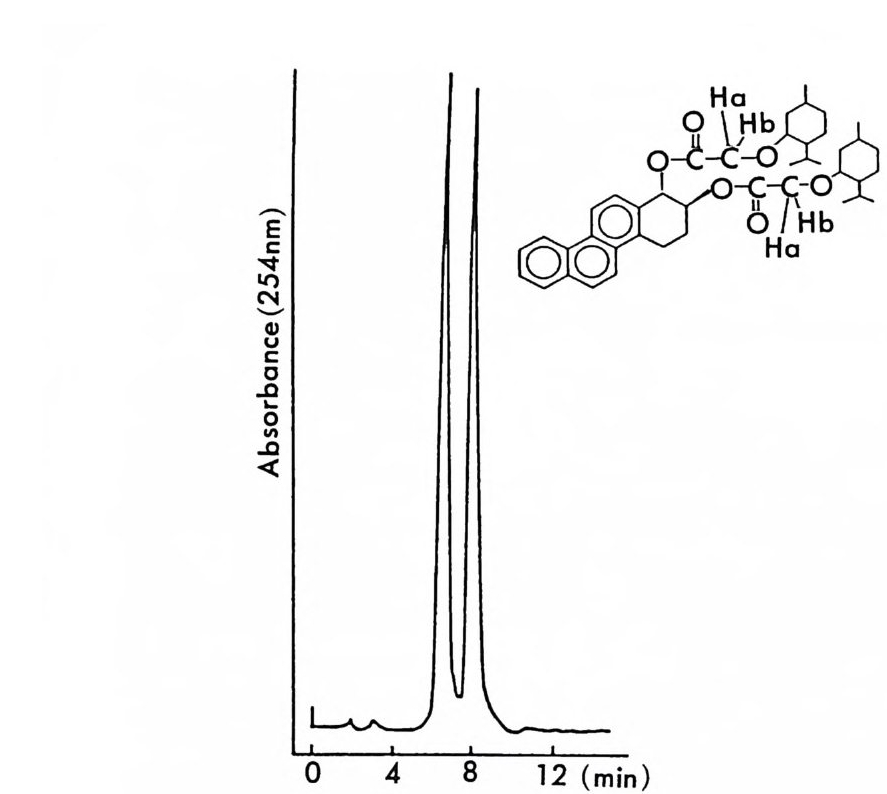 U
U
그림 2-7 1, 2, 3, 4- 데트라히드로크리센-트란스-1, 2- 디올 (-) -멘톡시 아세트산에스테르의 두 부분· 입체 이성질체 분리. 이동상 : 에테르-시클로핵산 (8/92) , 유속 : 2mL/mi n (참고문헌 : 69) 알코올이나 (70) 라세미 아민의 (71) 광학분할에 흔히 사용되는 CDA 이다. 위에 기술한 CDA 들 의에도 Pir k le<> !] 의하여 연구된 염화 옥사졸 리돈카르바모일 (oxazoli do ne carbamoy l chlorid e ) 이 라세 미 아민과 반응하여 부분- 입체 이성질 우레이드유도체를 만듦으로써 이 유도체 가 실리카 겔 컬럼상에서 분리가 아주 잘 됨이 확인되었으며 (72) ' San g er 시약의 일종인 키랄 1 국문르오르 -2, 4- 디니트로페닐 -5-L- 알라 닌 아미 드 (l-fluo ro-2, 4-di nitro p h eny l- 5-L-alan ine ami de : Marley 's rea g en t)가 아미노산의 광학분할에 좋은 CDA 로 사용될 수 있음이 확 인되었다 (73).
위에 기술한 CDA 들 이외에도 더 많은 CDA 들이 라세미 화합물의 광학분할에 이용된 바 있으며 (40- 44 ) 앞으로도 더 다양한 CDA 둘이 계속 개발될 것으로 생각된다. 그러면 위에 기술한 다양한 CDA 들 중 주어진 라세미 물질을 LC 상 에서 광학분할하기 위하여 어느 것을 선택하여야 할 것인가? 우선 CDA 와 주어진 라세미 물질 사이의 반응이 빠르게 진행되고 정량적으 로 이루어져야 한다는 조건을 고려하더라도 선택 가능한 CDA 들들은 다양하다. 예를 들면 라세미 알코올은 이소시아네이트 형태의 CDA 과 반응하여 카르밤산 에스테르유도체로 광학분할될 수 있으며 이소시아 네이트 형태의 CDA 들과 반응하여 티오카르밤산 에스테르 유도체로 광학분할될 수 있을 뿐만 아니라, 키랄 카르복실산 혹은 키랄 카르복 실산 영화물 형태의 CDA 들과 반응하여 에스데르 유도체로, 키랄 클 로로포름산 에스데르 형태의 CDA 들과 반응하여 탄산 에스테르 유도 체로 광학분할이 가능하다. 이들 여러 종류의 CDA 등 중 최선의 CDA를 선택하는 요건은, 어느 형태의 부분 입체 이성질체가 가장 좋 은 분리능을 보이는가, 부분 입체 이성질체를 만들기 위한 CDA 는 쉽 게 구할 수 있는가 특히 상업적으로 저렴한 가격에 구할 수 있는가 하 는것 등이 될 것이다. 물론 어느 형태의 부분 입체 이성질체가 분리능이 가장 좋은가 하는 것은 일반적으로는 이론상으로 결정할 수 없으며 다량의 라세미 물질 울 광학분할할 경우에는 여러 형태의 부분 입체 이성질체들을 소량 만 들어 분리능을 확인해 보는 수밖에 없다. 라세미 락탐의 경우 NEIC 와 반응하여 얻어지는 부분 입체 이성질 우레이드 유도체가 PEIO 라 반응하여 얻어지는 유도체보다 LC 상에서 더 큰 a- 값을 보이기 때문 에 락탐의 광학분할을 위하여서 우선 선택할 수 있는 CDA 는 NEIC 이다. 그러나 다량의 락탐울 광학분할하고자 할 때에는 경제적인 문제 를 고려하여야 할 필요가 있으며 PEIO 긱 가격이 NEIO 긱 가격에 비 교하여 훨씬 저렴하므로 분리능은 조금 떨어지더라도 PEIC를 CDA
로 선택하여 락탐의 광학분할을 실시하는 것이 바람직하다고 할 수 있 다 (31). 마지막으로 CDA 를 선택할 때 고려하여야 할 점은 부분 입체 이성질체 분리후 온화한 조건에서 CDA 를 제거할 수 있어서 광학적으 로 순수한 광학 이성질체를 얻을 수 있는가 하는 점이다. 물론 이것은 분석이 목적인 경우에는 필요없는 과정이나 순수한 광학 이성질체를 얻고자 할 때는 필수적인 과정이며 이때 라세미화가 없고 다른 화합물 로 변화됨이 없이 CDA 를 회수하여 다시 사용할 수 있다면 이 CDA 는 다량의 라세미 화합물을 광학분할하고자 할 때 가장 선택가능성이 높은 CDA 가 될 것이다. 예를 들면 라세미 알코올의 광학분할에 사용 가능한 CDA 중에서 NEIC 는 라세미 알코올과 쉽게 반응할 뿐만 아 니라 LC 상에서 분리가 찰 되며 분리 후 삼염화실란과의 반응에 의하 여 광학활성 알코올울 얻을 수 있고 (54, 55, 56) 경우에 따라서는 NEIC 가 회수되기 때문에 가장 바람직한 CDA 라고 할 수 있다 (61). 6.2.2 간접분리 방법의 분리 메커니즘 라세미 화합물과 CDA 의 반응에 의하여 얻어전 두 개의 부분 입체 이성질체가 LC 상에서 분리되는 분리 메커니즘을 이해하는 것은 매우 중요하다. 분리 메커니즘을 이해함으로써 두 부분 입체 이성질체의 용 리순서 (elu ti onorder) 를 예측할 수 있을 뿐만 아니라, LC 에 의하여 두 부분 입체 이성질체의 용리순서를 측정하였을 때 용리순서로부터 최종적으로 얻게 될 광학 이성질체의 절대배열이나 혹은 분리된 부분 입체 이성질체의 시료부분에 대한 절대배열을 결정할 수 있기 때문이 다. 그러나 일반적으로 두 개의 부분 입체 이성질체가 LC 상에서 분리 되는 분리 메커니즘을 이해하는 것은 쉬운 일이 아니다. 흡착 크로마토그래피에 의하여 두 부분 입체 이성질체의 분리가 이 루어질 때 두 가지의 메커니즘이 가능하다. 첫째는 두 부분 입체 이성 질체가 이동상에 의하여 용매화될 때 용매화의 정도에 차이가 있으면 분리가 가능한 것으로 생각할 수 있다. 그러나 이 경우에는 용매화의
차이뿐만 아니라 용매화된 두 부분 입체 이성질체의 흡착성의 차이 등 여러 요인이 용리순서에 관여할 것이므로 일반적인 메커니즘을 제시하 기가 곤란한다. 둘째는 이동상과는 무관하게 두 부분 입체 이성질체와 고정상 사이의 상호작용에 에너지 차이가 있을 때 분리가 가능하다고 생각할 수 있다. 그러나 부분- 입체 이성질체는 다양한 이형태체 (con fo rmer) 로 존재할 수 있으며 각 이형태체간의 에너지 차이가 크 지 않을 경우 실제로 어떤 이형태체가 고정상과 상호작용하는지 알 수 없기 때문에 분리 메커니즘을 아는 것은 쉽지 않다. 그러나 부분 입체 이성질체가 어느 정도 고정된 이형태체로 존재할 때는 두 부분 입체 이성질체의 고정된 이형태체와 고정상간의 상호작용을 고려함으로써 분리 메커니즘을 제안할 수 있다. 여기서는 두 부분 입체 이성질체의 용매화는 전혀 고려하지 않고 두 부분 입체 이성질체와 고정상이 상호 작용할 때 상호작용 에너지 차이에 의해 분리가 이루어지는 분리 메커 니즘에 대하여 논의하고자 한다• 아미드 카르밤산 에스테르 혹은 우레이드 (ure i de) 형태의 부분- 입체 이성질 유도체들은 분자내 수소결합을 형성함으로써 어느 정도 고정된 이형태체로 존재하고 따라서 그림 2-8 에서 보는 바와 같이 어느 정도
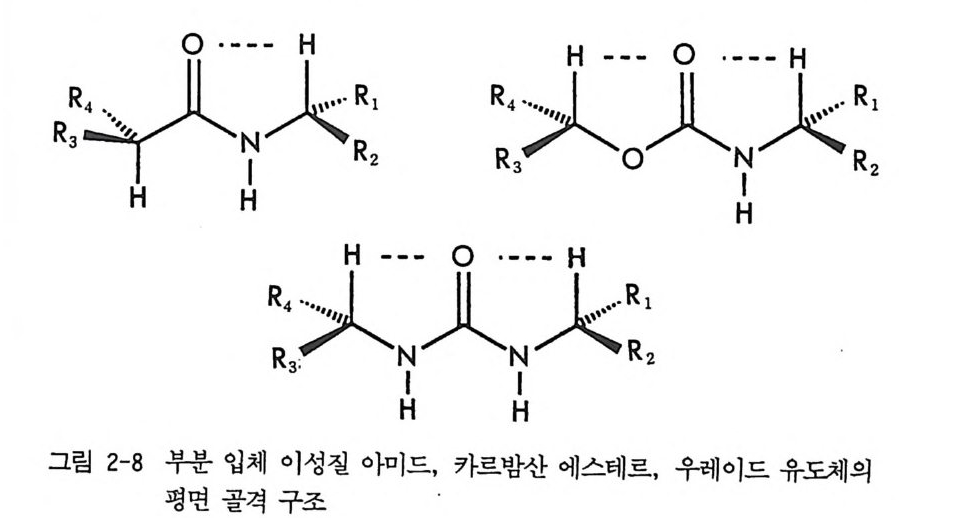 R: 」H? 0 ~·-H-: -:R·3) .H : (. NI, )RlR4.N3츠 I , XH - :-0入R:- 2 0 ·-N冬l- -H RRI2
R: 」H? 0 ~·-H-: -:R·3) .H : (. NI, )RlR4.N3츠 I , XH - :-0入R:- 2 0 ·-N冬l- -H RRI2
고정된 평면골격 (semi ri g id backbone) 을 가진다고 생각할 수 있다 (58, 74, 75, 76, 77). 이 때 네 개의 치환기 R1, R2, RJ , R4 는 평면골격의 평 면에서부터 앞으로 나와 있거나 혹은 뒤로 둘어가 있다. 어느 정도 고 정된 평면골격 구조를 가진 아미드 부분 입체 이성질체가 알루미나나 실리카 겔과 같은 극성 고정성에서 분리될 때 분리되는 메커니즘은 Helmchen 에 의하여 제안되었으며 (75 - 77) 그 후 라세미 알코올의 부 분 입체 이성질 카르밤산 에스데르 유도체와 (53 - 56) 라세미 옥사졸리 돈 (oxazo li done) (72) 혹은 라세미 락탐 (31) 의 부분」 입체 이성질 우레 이드 유도체의 분리 및 그 메커니즘에 대하여 Pi rk le 둥이 자세히 연 구하였다. 실리카 겔에 의한 부분 입체 이성질체의 흡착이 평면골격의 평면상 에 있는 부분 입체 이성질체의 국성 부분七(카르보닐 산소 및 질소)과 실 리카 겔의 실라놀기 사이의 수소결합에 의하여 이루어지고, 이 과정에 서 부분 입체 이성질체의 형태가 변화하지 않는다는 Helmchen 의 가 설을 따르면 (75-77), 두 부분 입체 이성질체의 분리는 치환기 R1, R2, R3, R4 의 크기 (입체장애) 및 방향에 의하여 결정된다. 그림 2 - 9 에 평 면골격 구조를 가지는 한 쌍의 부분 입체 이성질체 모형도와 흡 착의 상대적 세기를 표시하였다. 그림 2-9a 에서 보는 것처럼 두 개의 큰 기 (L, L' )와 작은 기 (S, S' )가 평면골격에서부터 같은 방향으로 나와 있으면 실리카 겔 표면에서의 흡착은 두 개의 작은 기가 나와 있는 방 향으로 일어나서 강한 흡착이 될 것이고 반면에 그립 2-9b 에서처럼 큰 기와 작은 기가 같은 방향으로 나와 있으며 실리카 겔 표면에의 홉 착이 상대적으로 약하게 된다. 결국 부분 입체 이성질체 a 는 실리카 겔 표면에 오래 머무르고 困즌 컬럼에서 빨리 나오게 되어 두 부분 입 체 이성질체의 분리가 이루어 진다. 이 모델이 맞다면 두 부분 입체 이성질체의 평면구조로부터 두 부분 입체 이성질체가 컬럼으로부터 분 리되어 나오는 용리순서를 예측할 수 있으며, 역으로 용리순서로부터 부분 입체 이성질체의 구조를 예측할 수 있고, CDA 의 절대배열은 이
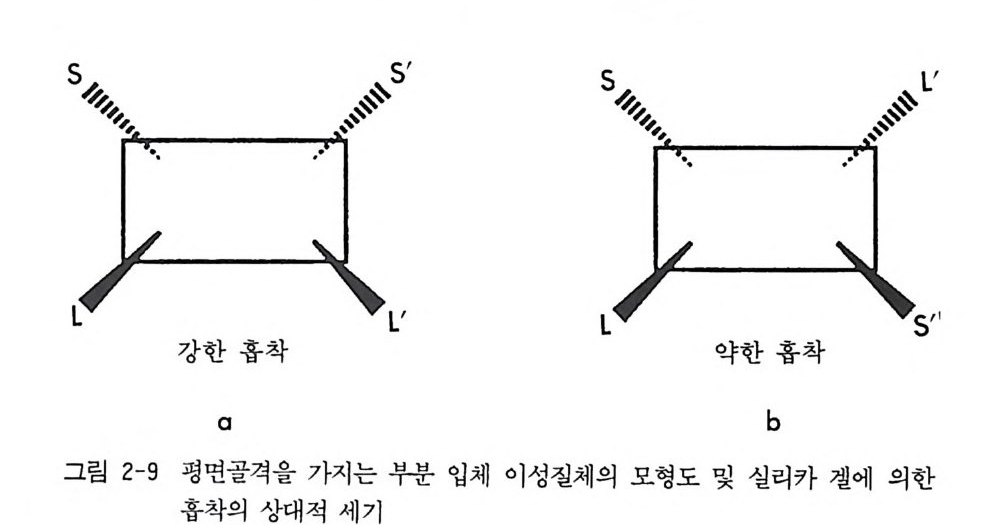 l' S''
l' S''
미 알려져 있으므로 절대배열을 모르는 시료의 절대배열을 결정할 수 있다 (58, 72). 이 분리 메커니즘으로부터 예측할 수 있는 또 하나의 중요한 사실은 그림 2 선에 있어서 L 기와 S 기 (혹은 L' 기와 S' 기)의 크기 (입체장애) 차 이가 크면 클수록 분리가 더 잘 되리라는 것을 예측할 수 있디는- 것이 다. 라세미 락탐과 (S)-1- 페닐에틸이소시아네이트 (PEIC) 의 반응에 의하여 생긴 부분 입체 이성질 우레이드 유도체 旦 a 와 1§.b 가 실리카
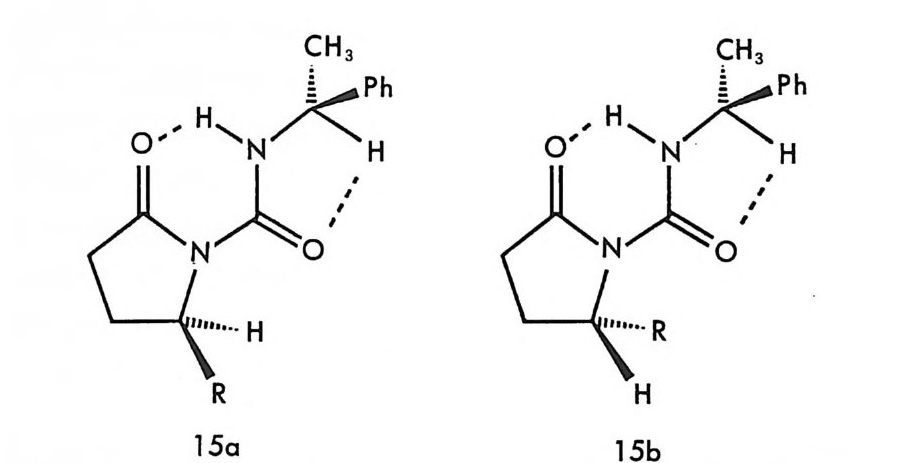 CHa CH3
CHa CH3
겔 컬럼에서 분리될 경우 R 기의 입체장애가 크면 클수록 분리가 더 잘 되리라고 예측할 수 있으며 실제로 0.5% 의 2 - 프로판올이 섞여있 는 핵산용액을 이동상으로 사용하여 실리카 겔상에서 이들을 분리할 경우 표b 가 표갇虹다 컬럼에서 먼저 용리되며 R 이 메틸기일때 a 값은 1. 17 이나 R 이 페닐기일 때는 a 값은 2.24 임이 보고된 바 있다 (31). 그러나 R 기의 입체장애만을 고려하여 용리순서를 예 측 하였을 때 실 지의 용리순서와 상이한 경우가 있다. 한 쌍의 카르밤산 에스데르 부 래분 ph序 머입 무체a 르이게성- O되질;丈체고1 6 a-1. N!§§._b a 와가_내人`’ , H 먼표ca 저比b 의 N용 a 경 p리h 우되 어페罰효 닐나a기울H의广J`A 것 0I 입 으 체丈로1장6 b예_H애 상가 된나〈 다C.F ? 3 의그 N 러입 a p 체장애보다 크므로, 그림 2-9 의 모델에 의하여 .!§_a 가 컬럼상에 더 오 나 실제로는 登b 가 컬럼상에 더 오래 머무르고 l§.a7 } 먼저 용리되어 나온다 (58). 이와 같이 용리순서가 예측하는 바와 디론· 이유는 CF3 가 특별히 소수성이 크기 때문이다 (78). CF3 의 소수성이 크므로 부분 입 체 이성질체 표이 실리카 겔 표면이나 알루미나 표면과 상호작용할 경 우에 CF3 가 나와 있는 면과 상호작용하기 보다는 반대면과 상호작용 하는 것이 훨씬 유리하다. 이렇게 되면 旦b 에서는, 페닐기와 메틸기 가 같은 방향으로 나와 있는 면과 실리카 겔 표면이 상호작용할 것이 므로 페닐기와 1- 나프틸기가 같은 방향으로 나와 있는 拉킥i다 더 오 래 컬럼에 머무르게 된다. 치환기의 입체장애 효과 및 소수성을 모두 고려할 때 실리카 겔이나 알루미나와 같은 고정상과 치환기 사이의 반 발효과의 순서는 여러 종류의 치환기에 대하여 다음과 같다 (39, 58).
H < 메틸 < 페닐 = 에틸 < 프로필 < t유너i < 트리플루오르메틸 (CF3) < a -나프틸 < 9- 안트릴 < 펜타플루오르에틸 (CF3C F 2) < 헵타플루 ? 근 立 쿠 필 (CF3CF2C F 2 ) 카르밤산 에스데르 유도체 L統 든 CF3 가 CH3 로 바뀐 카르밤산 에스데 르 유도체와 비교하여 용리순서가 바뀔 뿐만 아니라 a 값도 훨씬 크다 (a 값은 1. 58 대 1. 30 임 : 중성 알루미나, 벤젠 이동상) (58). 카르비닐 수소 (carbin y lh y d rog e n : 브러 Ha) 의 산성도가 CF3 때문에 크게 증가 하며 따라서 분자내 수소결합의 세기가 증가하고 형태 (confo rm at ion ) 가 더욱 고정되기 때문이다. 만일 이동상의 극성을 증가시킨다면 분자 내 수소결합을 약화시킬 것이므로 형태의 고정도는 감소하고 a 값이 감소하리라고 예측할 수 있다 (58). 그립 2-9 의 분리 메커니즘 모델울 이용할 경우에 두 부분- 입체 이성 질체의 NM~ 스펙트럼을 분석하여 키랄중심의 절대배열을 예측할 수 있을 뿐만 아니라 용리순서로부터 예측한 절대배열과 비교함으로써 절 대배열의 결정에 있어서 서로 보완적인 역할을 하게 된다. 예를 들면 락탐의 우레이드 유도체 .!_§_a 와 .!_§_b 의 경우 페닐기가 락탐부분의 키랄 중심에 붙어 있는 R 기나 H 의 화학적 이동에 디른- 영향을 줄 수 있 다 . .!_§_a 에서 R 기는 페닐기와 같은 방향에 있으므로 R 기에 미치는 페 닐기의 가려막기 효과 (sh i eld i n g e ff ec t)는 브b9.l R 기에 비해서 더 크 다. R7] 가 CH3 일때 .!_§_a 에 있어서 CH3 의 화학적 이동은 표 b 에 있어 서 CH 려 화학적 이동보다 0.05 pp m 만큼 작은 것이 관찰되었다. 따 라서 한 쌍의 부분 입체 이성질체를 분리한 후 각각의 NMR 스펙트 럼을 얻었을 때 R 기의 화학적 이동의 상대적인 크기로부터 락탐 부분 의 절대배열을 예측할 수 있다. 이렇게 하여 예측된 철대배열은 LC 상 의 용리순서로부터 예측한 철대배열과 동일함이 보고되었고, 또 .!_§_a 와 旦낡룹 LC 에 의하여 분리한 후 적절한 화학적 방법을 통해 얻은 순 수한 락탐의 광회전도 [a]값 은 문헌과 서로 일치함이 보고되었다 (31).
결론적으로 간접분리 방법에 의한 라세미 물질의 LC 광학분할을 완 전하게 설명할 수 있는 메커니즘은 아직 없으나 그림 2 - 9 의 입체장애 모델은 부분 입체 이성질체의 구조 혹은 입체화학 및 용리순서를 연관 시켜 설명할 수 있는 유용-한 모델이라고 할 수 있다. 6.2.3 응용 간접분리 방법에 의한 LC 광학분할을 응용하여 광학활성 물질의 광 학순도를 결정한 예는 무수히 많다. 특히 라세미 의약품들의 광학분할 이나 광학활성 물질의 생체내 대사과정에 따르는 광학활성의 변화를 추적하는 데 이 방법이 광범위하게 사용되었고 많이 보고되어 있다 (40 -44). 여기서는 간접분리에 의한 LC 광학분할방법이 광학활성인 화합 물들을 합성할 때 사용된 예를 제시함으로써 그 응용의 다양성을 보이 고자한다.
 OH
OH
 Eu
Eu
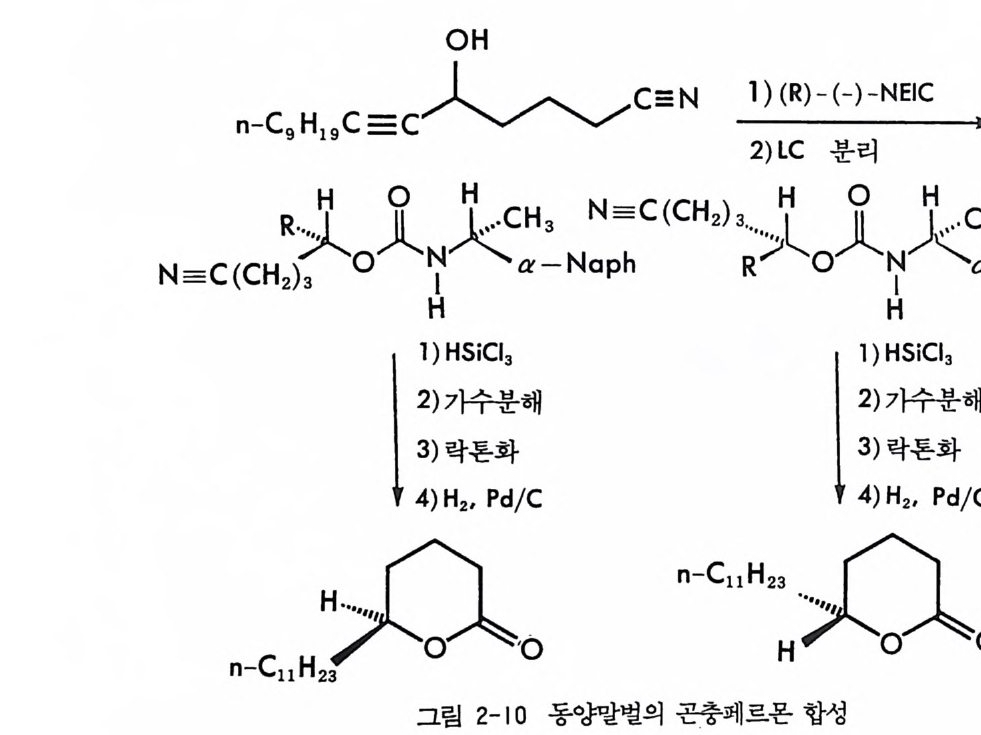
그림 2-11 자동화된 MPLC 시스템을 이용한, 그림 2-1 뼈 두 부분 입체 이성질체 분리. 컬럼크기 : 2in x 4.5f t (5.08cmx 137cm), 시 료크기 : 1.0- 2. Og (참고문헌 : 79) Pir k le 등은 라세 미 시 아노알코올 (cy an oalcohol) 을 (R) -NEIO 나 반응시켜 얻은 한 쌍의 부분 입체 이성질 카르밤산 에스테르를 LC로 분리한 후 트리클로로실란으로 처리하여 광학적으로 순수한 시아노알 코올울 얻음으로써 생리활성을 가진 키랄 락톤 화합물들을 합성할 수 있었다. 예를 들면 시아노알코올을 (R)-(-)-NEIO 가 반응시켜 한 쌍의 부분 입체 이성질 카르밤산 에스데르를 얻었을 때 (그립 2-10) 이 둘은 자동화된 MPCL 시스템을 (52) 사용하여 그림 2-11 과 같이 분리 되며 트리클로로실란으로 처리하여 얻은 광학적으로 순수한 시아노알 코올을 가수분해하고 락론화한 후 락돈의 옆가지에 있는 삼중결합을
환원하면 동양말벌의 곤충페르몬(한 쌍의 광학 이성질체)을 얻을 수 있 다 (79). 비슷한 방법으로 고리형의 라세미 시아노알코올을- 광학분할하여 Pir k le 등은 광학활성 인 다중고리 락탐 끄을 합성하였고 (55) , 라세미 히드록시아세틸렌카르복실산 에스데르를 광학분할함으로써 천연화합 물의 일종인 광학활성 락돈 登를 합성하였다 ( 56).
 H_µ 。 ::nc广 3C5HII
H_µ 。 ::nc广 3C5HII
이 의에도 P i rkle 등은 액체 크로마토그래피를 이용한 광학 이성질 체의 간접분리 방법에 의하여 매미나방의 성페르몬으로 알려져 있는 광학활성 에폭시 화합물을 합성할 수 있었으며 (54, 80), 콩바구미 수컷 의 성유인페르몬인 광학활성 알렌 화합물을 합성할 수 있었다 (53, 81). (그림 2-12 및 2-13) .
 SPh 1 ) (R) -( -)- N EIC
SPh 1 ) (R) -( -)- N EIC
 OH l ) R-( -)- N EIC
OH l ) R-( -)- N EIC
위에 제시한 예 의에도 LO 긱 간접분리 방법에 의한 광학분할을 이 용하여 광학활성 물질들을 합성하는 예는 무수히 많으며 앞으로도 이 에 대한 연구는 계속될 것이다. 부분 입체 이성질체의 LC 용리순서와 분리된 부분 입체 이성질체의 NMR 스펙트럼으로부터 광학분할하고 자 하는 광학활성 물질의 철대배열을 결정할 수 있는 경우에는 최종 생성물의 절대배열을 예측할 수 있으므로, 라세미 물질의 LC 간집광 학분할에 의한 광학활성 물질의 합성법은 특히 유용하다고 할 수 있 다.
6.3 직접분리 방법 6.3.1 특성 라세미체를 CDA 와 반응시켜 한 쌍의 공유결합성 부분 입체 이성질 체를 만들어 LC 상에서 분리하는 간접분리 방법과는 달리 직접분리 방 법에서는 라세미체와 키랄 선택자 (ch i ral selecto r ) 사이의 상호작용에 의하여 일시적인 부분 입체 이성질체 (tra nsie n t d i as t ereomer) 를 형성 함으로써 라세미체를 직접 광학분할한다. 직접분리 방법에서는 공유 결합성 부분 입체 이성질체를 만들 필요가 없이 일시적인 부분- 입체 이성질체를 통하여 순수한 광학 이성질체를 직접분리하므로, 간접분 리 방법에서 공유결합성 부분 입체 이성질체를 만들기 때문에 생기는 여러 문제점들이 직접분리 방법에는 없다. 직접분리 방법에서도 고정 상에서 분리의 용이성을 증진시키고 검출기의 감응도를 증진시키기 위 하여 라세미체를 비키랄성 유도체로 만든 후 분리하기도 하나, 이 경 우에는 유도체 시약이 비키랄성이므로 라세미체의 두 광학 이성질체와 정확히 같은 반응속도(같은 바율)로 반응하며 반응하여 생성된 바키랄 성 유도체에 대한 검출기의 감응도가 일치하기 때문에 크로마토그래피 상의 두 피이크의 크기로부터 두 광학 이성질체의 존재비율을 정확히 측정할수있다. 간접분리 방법과 비교하여 직접분리 방법의 또 한 가지 큰 장점은 키랄 선택자의 광학순도가 100% 에 못미치더라도 광학 이성질체를 광 학적으로 순수한 형태로 분리할 수 있디는 사실이다. 키랄 선택자의 광학순도는 다만 . a 값에만 영향을 미친다. 이 영향의 정도는 식 (2-8) 으 로 주어짐이 알려져 있다 (82, 83). %s = aa P(1+0 (01-0P0)— +P P) (2-8)
여기서 a 는 키랄 선택자의 광학순도가 100% 일 때의 분리인자이며 O! ob s 는 광학순도가 100% 가 아닌 키랄 선택자에서 과량 존재하는 광학 이성체의 바율이 P(% )일 때의 분리인자이다. P 는 광학순도와는 다 른 값이며 P=5 0i픈 두 광학이성질체가 같은 비율로 섞여 있음을 의미 하고 이때 광학순도는 0 이다. 따라서 이론적으로는 P > 50 이기만 하면 O! ob s> 1 이므로 광학분할이 가능하며 키랄 선택자의 광학순도를 정확히 알아야 할 필요는 없다. 물론 더 좋은 광학분할(더 큰 a 값)을 얻기 위 하여서는 순도가 높은 키랄 선택자를 사용하는 것이 좋을 것이다. 직접분리 방법은 키랄 선택자의 형태에 따라 CMP(Chir a l Mobil e Phase : 키 랄 이동상) 방법 , CMPA (Chir a l Mobil e Phase Addit ive : 키 랄이 동상 첨가제)방법 및 CSP(Chir a l Sta t i on ary Phase: 키랄고정상)방법으 로 나눌 수 있다. CMP 방법은 말 그대로 이동상으로서 키랄 용매를 사용하는 방법이다. 그러나 이 방법은 다량의 키랄 용매를 사용하여야 하는 어려움 때문에 보편화될 수 없는 방법이다. 이러한 어려움을 극 복하기 위하여 라세미체와 서로 작용하여 일종의 일시적인 부분 입체 이성질 착물 혹은 복합체 (tra nsie n t dia s te r eomeric com p lex) 를 형성 할 수 있는 키랄 물질을 이동상에 가함으로서 라세미체를 광학분할하 는 방법을 키랄이동상 첨가제법 (CMPA 법)이라고 한다. CMP 방법이 나 CMPA 방법이나 키랄 선택자가 이동상에 있다는 점에서 두 방법 은 비슷하며 때로는 같은 방법으로 취급되기도 한다 (84). 반면 CSP 방법에서는 키랄 선택자를 고정상에 공유결합(화학적 방법)시키거나 혹은 물리적 방법으로 고정상에 흡착시켜 고정되게 함으로서 키랄 선 택자는 고정상의 일부가 되며 고정상 자체가 비대칭성을 가지게 된다. CMPA 방법의 가장 큰 장점은 비키랄성인 보통컬럼을 사용하여 라 세미체의 광학분할이 가능할 뿐만 아니라 용매 선택이나 pH 조정 등 여러 방법으로 여러 가지 다양한 분석방법의 개발이 가능하다는 것이 다. 따라서 CMPA 방법에 의한 광학분할의 예는 무수히 많이 보고되 어 있다 (84, 85). 그러나 Davankov 와 Kur g anov가 지적한 것처럼
(86) CMPA 방법에서는 광학분할의 메커니즘이 복잡하여 용리순서를 설명하기가 어려우며 따라서 용리순서로부터 절대배열을 결정하기가 어렵다. 한 쌍의 광학 이성질체 A 와 A* 가 CMPA 와 상호작용하여 두 개의 부분 입체 이성질 착물을 만든다면 다음과 같이 두 개의 평형식을 쓸 수있다. A + CMPA [A-CMPA] A* + CMPA [A*-CMPA] (2 선) 이와 같은 평형상태에서 실제로 존재하는 화학종은 다섯 가지 (A, A*,CMPA 및 두개의 착물)이며, 광학분할은 이 다섯 가지 화학종 사 이의 상호 작용 및 다섯 가지의 화학종과 고정상 사이의 상호작용 등 여러 가지의 복합적인 메커니즘에 의하여 결정된다. 죽 광학분할은 1) 고정상에 의한 [A-CMPA 〕와 〔 A*-CMPA 回 흡착정도의 차이 , 2) A 와 CMPA 의 상호 작용및 A * 와 CMPA 의 상호작용의 차이, 3) 고정상에 의한 CMPA 의 흡착정도의 세 가지 요소가 서로 복합적으로 작용하여 이루어진다고 할 수 있다. 〔 A-CMPA 〕와 [ A * -CMPA 〕가 고정상에 흡착될 때 흡착성에 큰 차이가 있으면 이것은 고정상에 의하 여 공유결합성 부분 입체 이성질체가 분리되는 것과 비슷한 메커니즘 으로 분리된다고 생각할 수 있으며 A 와 CMPA 의 상호작용 및 A * 와 CMPA 의 상호작용에 차이가 있을 때는 강한 상호작용을 하는 광학 이성질체가 이동성을 따라 더 빨리 이동할 것이므로 더 빨리 분리되어 나을 것이다. 반면 고정상과 CMP A;,가 강한 상호작용을 할 때에는 이 것은 일종의 CSP 로 작용함으로써 CMPA 와 강한 상호작용을 하는 광학 이성질체보다는 CMPA와 약한 상호작용을 하는 광학 이성질체 가 더 빨리 분리되어 나올 것이다. 이들 세 가지 형태의 분리 메카니 즘이 독립적으로 이루어지거나 어느 하나의 메커니즘이 특별히 우세할
경우에는 용리순서와 광학 이성질체의 절대배열을 연관시킬 수 있으나 보통은 이둘 세 가지 형태의 분리 메커니즘이 복합적으로 이루어지므 로 용리순서와 광학 이성질체의 절대배열을 연관시키는 것은 극히 어 렵다고할수있다. CMPA 방법의 다른 문제점은 분리하는 동안 키랄이동상 첨가제가 계속 요구된다는 점이다. 따라서 분석이 목적이 아니라 다량의 라세미 체 를 광학분할하는 것이 목적이라면 CMPA 방법은 사용하기 곤란하 다. 또한 CMPA 방법에 있어서 컬럼에서 분리되어 나오는 것은 순수 한 형태의 광학 이성질체가 아니고 키랄이동성- 첨가제와의 혼합물(혹 은 일시적인 부분 입체 이성질 착물)이므로- 두 광학 이성질체에 대한 검 출기의 감응도가 달라서 두 피이크의 크기로부터 광학순도를 정확히 측정할 수 없을 뿐만 아니라 순수 광학 이성질체를 얻으려면 적절한 방법으로 키랄이동성 첨가제를 분리하여야 한다는· 문제점이 있다. 그러나 CSP 방법은, 키랄컬럼이 가격면에서 비키랄컬럼보다 비싸 며 현재 많은 종류의 CSP 들이 개발되어 있기는 하나 상품화가 되지 않은 CSP 인 경우 입체화학에 관련된 여러 문제점을 해결하려는 연구 자들이 쉽게 만들거나 구입하여 사용할 수 없다는 단점이 있음에도 불 구하고, CMPA 방법이 가지는 여러 문제점들울 가지고 있지 않다는 점에서 특히 선호되고 있다. 즉 CSP 방법으로 광학분할을 실시할 경 우 두 광학 이성질체가 직접분리되므로 검출기의 감응도에 차이가 없 으며 크로마토그램의 두 피이크 크기를 비교함으로써 두 광학 이성질 체의 존재비율을 정확하게 측정할 수 있다. 또한 CSP 방법에서는 키 랄고정상과 두 광학 이성질체 사이에 형성되는 일시적인 부분 입체 이 성질 착물의 안정성 차이에 의해서만 광학분할이 이루어지므로 광학분 할 메커니즘이 비교적 단순하다. 따라서 광학분할 메커니즘이 알려져 있지 않은 경우도 있지만 많은 경우에 정확한 광학분할의 메커니즘(혹 은 키랄성 인지 메커니즘)을 제시할 수 있다. 또한 CSP 방법에서는 소 량의 키랄 물질을 컬럼의 고형지지체로 사용할 수 있는 물질에 고정시
킴으로써 키랄고정상을 만들 수 있으며 키랄고정상으로 채워진 컬럼은 계속 사용할 수 있기 때문에 다량의 라세미체를 광학분할하거나 많은 수의 라세미체 시료를 광학분할할 때는 경제적인 이점도 크다. CSP 방법의 이와 같은 여러 장점 때문에 키랄고정상을 이용한 광학분할은 현재 정확하고 빠른 광학순도의 측정, 절대배열의 측정 , 광학적으로 순수한 광학 이성질체의 획득이라는 입체화학에 관련된 세 가지의 문 제점을 해결하는 데 있어서 거의 이상적인 방법으로 인식되고 있다. 6. 3. 2 키랄이동상 참가제법 (CMPA meth o d) 키랄이동상 첨가제법으로 가장 광범위하게 이용되며 많은 연구가 이 루어 진 분야는 리 간드 교환 크로마토그래 피 (Liga nd Ex c hang e Chroma t o gr a p h y : LEC) 에 기초를 둔 것으로서 키랄 리간드와 금속이 온 및 라세미체를 구성하는 두 개의 광학 이성질체 사이에 부분 입체 이성질 삼성분 착화합물의 형성 (ter nary com p lexa ti on) 을 통한 광학 분할이다 (84, 85, 87). 키랄 리간드 및 전이금속이온(보통 Cu2+ , N i 2+ , Co2+, Fe2+ , F e3+, Zn2+, Cd 2+ )을 함유하는 이동상을 보통의 비키랄컬럼 에 통과시켜 고정상과 이동상 사이에 평형을 유지한 후 분리하려는 라 세미체를 주입하면 키랄 리간드와 금속 사이에 형성된 이원 착화합물 의 키랄 리간드와 라세미체의 두 광학 이성질체 사이에 식 (2 - 10) 과 같 이 리간드 교환이 일어나 두 개의 부분- 입체 이성질 삼성분 착화합 물이 형성된다. [CL]n 〔 M]+ 肉 [CLJ n -1 [M] [피 + [C L] [CL]n 〔피+[언 [CL ]n 크 [M ] [S J + [C L] (2- 10 ) 위의 평형식에서 [CL] 은 키랄 리간드, [CL]n 〔 M] 은 이성분 착화합 물, [R 〕과 〔 S ]는 라세미체의 S- 이성질체와 R- 이성질체를 뜻하며
[CL J n - 1[M 託 R ] 혹은 園〕 n -1 〔 M] [ S 〕 는 삼성분 착화합물을 의미한다. n은 보통 2와 같거나 2 보다 큰 수이다. 형성된 두 개의 부분- 입체 이 성질 삼성분 착화합물은 그 자체에 안정성의 차이가 있을 것이며 고정상 과의 상호작용에 있어서도 차이가 있을 것이므로 앞의 6.3.1 절에서 기술한 여러 메커니즘에 따라 광학분할이 일어난다. 이때 키랄 리간드 는 아미노산이나 아미노산의 유도체와 같이 키랄중심을 가운데 두고서 금속과 킬레이트 (chela t e) 화합물을 용이하게 만들 수 있도록 두 개 혹은 두 개 이상의 작용기를 가전 광학활성 화합물이다. 주로 많이 사 용되는 키랄 리간드들은 광학활성 형태로 쉽게 구할 수 있는 아미노산 이나 아미노산 유도체이며 가끔 키랄 디아민 (88), 키랄 트리아민 (89), 타르타르산아미드 (90) 등이 키랄 리간드로 시용되기도 한다. 키랄 리 간드로서 적어도 두 개의 작용기를 가전 광학활성 화합물이 사용~다 는 것은 이 방법으로 광학분할이 가능한 라세미체 역시 키랄 리간드와 치환하여 금속과 킬레이트 화합물을 형성할 수 있도록 적어도 두 개의 작용기를 가져야 한다는 사실을 의미한다. 이와 같은 사실은 부분- 입 체 이성질 삼성분 착화합물의 형성을 통한 직접분리 방법의 커다란 한계 점이 되고 있다. 따라서 이 방법에 의하여 광학분할이 가능한 라세미 체들은 대부분 아미노산이나 이들의 유도체와 같이 두 개의 작용기를 가진 화합물들이며 (85), 이의에도 두 개의 작용기를 가전 a- 히드록시 카르복실산이나 (91, 92) /3-아미노알코올 (90), 디올 화합물 (93) 들이 광 학분할된 예가 보고되어 있다. 특히 광학분할시에 형광 검출기를 사용 함으로써 검출기의 감응도를 증진시키기 위하여 아미노산 라세미체는 단실 (dans y l) 유도체로서 광학분할된 예들도 보고되어 있다 (94, 95). 부분 입체 이성질 삼성분 착화합물을 형성함으로써 광학분할이 이루어 지는 CMPA 방법의 실제적인 크로마토 그램의 예는 그림 2-1~ 갇 다 (96). 그림 2-1~ 크로마토그램은 2 당량의 D-혹 은 L- 프롤린 (L -p ro li ne) 과 1 당량의 Cu (Il )를 포함하는 아세트산나트륨영의 완충용 액 (0. 05 N, pH 5.5) 을 이온교환수지가 채워진 컬럼에 통과시킨 후 똑
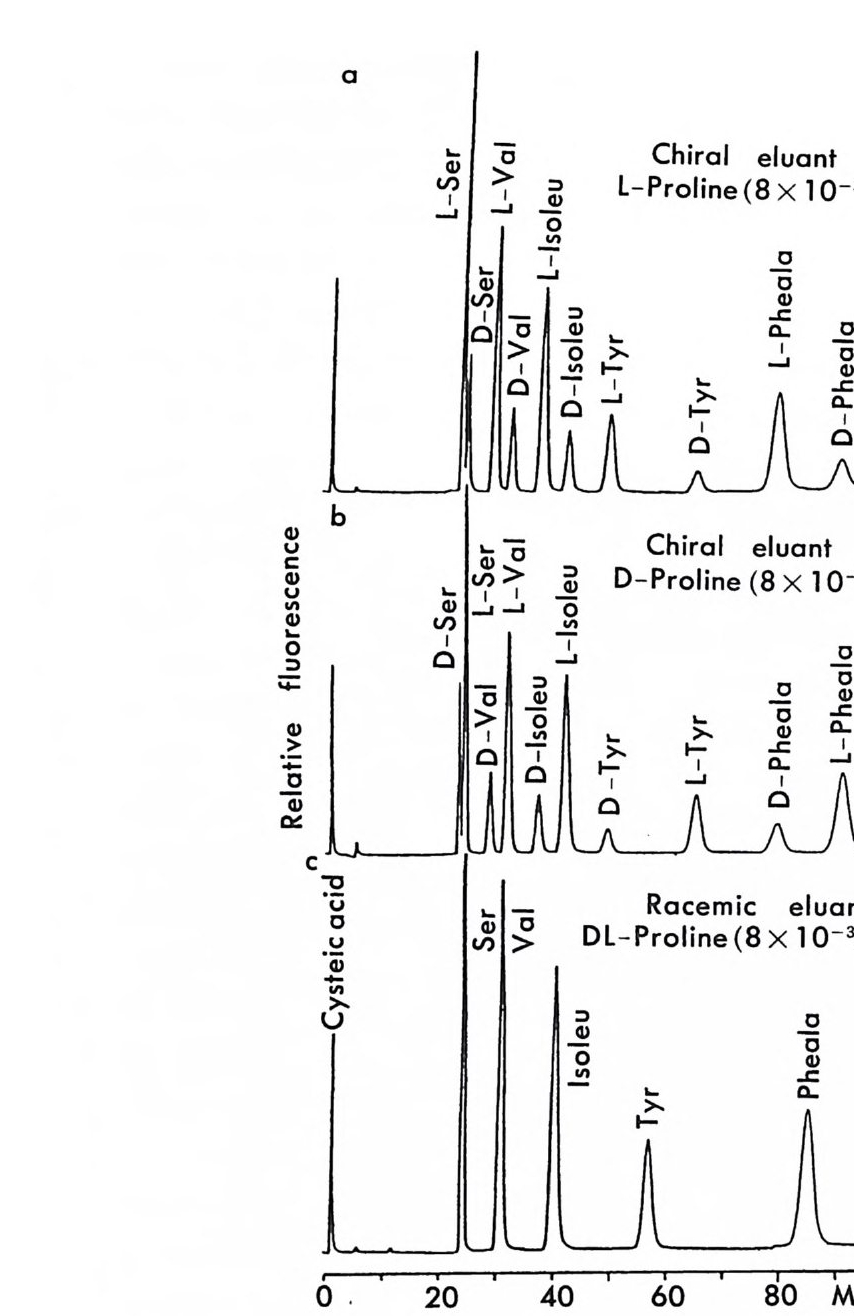 a
a
그림 2- 1 4 CMPA 방법에 의한 아미노산의 광학분할. CMPA: 프롤 린 -Cu (Il ), 그 의의 조건은 본문을 참조할 것 (참고문헌 : 96)
같은 양의 다섯 가지 아미노산 (D / L= 0 . 125 / 0.375) 을 녹안 시료용액 울 주입해서 얻은 것이다. 키랄 리간드로서 L- 프롤린과 D- 프롤린을 사용하였을 때 용리순서가 정확히 바뀌었음을 그립 2 - 14 에서 확인할 수 있을 뿐만 아니라 이동상 첨가제로 광학활성인 프롤린 대신에 라세 미 프롤린을 사용하고 다른 크로마토그래피 변수를 고정시켰을 때, 두 광학 이성질체에 해당하는 아미노산의 두 피이크가 정확히 중간 지점 에서 합치됨을 확인할 수 있다. 이와 같은 사실은 복잡한 크로마토그 램에서 각 피이크가 어느 이성질체에 해당하는가를 결정하는 데 유용 하게 사용될 수 있음울 시사해 주고 있다. 특히 반대의 절대배열을 가 진 광학 이성질체를 소량 포함하고 있는 광학활성 물질의 광학순도를 측정할 때는 소량의 광학 이성질체가 먼저 분리되어 나오게 함으로서 기준선 분리 (baselin e se p ara ti on) 가 가능해진다 (97). 이때 소량의 광 학 이성질체를 빨리 분리되어 나오게 하거나 니중에 분리되어 나오게 하는 것은 CMPA 방법에서는 다만 키랄이동상 첨가제의 절대배열만 R 에서 S 로 혹은 S 에서 R 로 바꾸면 되므로 디론 절대배열을 가진 광 학 이성질체를 극소량 포함하고 있는 광학활성 물질의 광학순도를 결 정하는 데 있거서 CMPA 방법은 특히 유용하다고 할 수 있다. 현재 국내에서도 L- 프롤린 혹은 L- 프로린 유도체와 같은 키랄리간 드와 Cu( Il )사이에 형성되는 키랄 착화합물을 CMPA 로 사용하여 라 세미 N- 단실아미노산을 광학분할하는 연구가 이루어지고 있으며 이 미 여러 편의 논문이 발표된 바있다 (98,99). 키랄 리간드와 전이금속 사이에 형성되는 키랄 착화합물을 키랄이동 상 첨가제로 사용하는 방법 의에도 라세미체와 함께 일시적인 부분 입 체 이성질체를 만들 수 있는 어떤 키랄 화합물도 키랄이동상 첨가제로 사용하여 광학분할에 응용할 수 있다. Hara 는 아미노산 유도체인 N -아세틸 _L- 발린-t유녁i아미드를 이동상 첨가제로 사용하였을 때 이 화합물은 라세미 N- 아세틸아미노 t숙녀i에스데르(1 00,101). N- 아세 틸아미노산 t냐 L 틸아미드(1 03), 혹은 N-(4- 니트로벤조일)아미노이소프
로필에스테르와(1 02), 함께 그림 2-15 와 같이 두 자리에서 수소결합을 하기 때문에 한 쌍의 부분- 입체 이성질 두 자리-킬레이트 이합체를 만 들고 이들은 안정성에 차이가 있기 때문에 실리카 겔 컬럼상에서 분리 가잘됨을확인하였다.
 CH3\\lY
CH3\\lY
그림 2-15 키랄이동상 첨가제인 N- 아세틸 -L- 발린 - t--¥-틸아미드와 라세미 아미노산 유도체 사이의 두 자리 수소결합 퀴닌 (qu in ine ) , 신코니딘 (ci nchon i d i ne) 과 같은 키 랄 아민들은 라세 미 카르복실산과 한 쌍의 부분- 입체 이성질 이온쌍 착화합물을 형성하 며 10- 캠퍼술폰산 (10-cam p horsul p hon ic aci d) 브나 N- 벤족시카르보 닐-글리실-프롤린 (N-benzoxy c arbony l- gl y c y lp r o li ne : ZGT) 깐과 갈 은 키랄 카르복실산들은 라세미 아민이나 라세미 아미노알코올(특히 f3 -차단제)등과 한 쌍의 부분 입체 이성질 이온쌍 착화합물을 형성한다.
 之뭉 °3H 0 마 Oi -NH ― CH2 정요。 H
之뭉 °3H 0 마 Oi -NH ― CH2 정요。 H
이들 한 쌍의 부분 입체 이성질 이온쌍 착화합물들은- 안정성에 차이가 있으며 고정상에 의한 흡착성질에 차이가 있기 때문에 LC 에 의하여 광학분할이 이루어진다. 이와 같이 라세미 화합물과 반응하여 한 쌍의 부분 입체 이성질 이온쌍 착화합물을 형성하는 키랄이동상 첨가제를 이용한 광학분할의 CMPA 방법은 Pe tt erson 에 의하여 많이 연구되 었다 (10 3- 10 5) . D - 글루구 ?一 人 쿠 구성된 시클로덱스트린 (c y clodex t r i n) 은 키랄공동 (chir a l cav ity)을 가지고 있기 때문에 라세미 화합물의 두 광학 이성 질체와 부분 입체 이성질 내포 화합물 (d i as t ereomer ic inc lusio n com- p ound) 을 가역적으로 형성한다. 따라서 시클로덱스트린을 키랄이동 상 첨가제로 사용할 경우에 라세미 화합물·과 시클로덱스트린 사이에 형성되는 한 쌍의 부분- 입체 이성질 내포 화합물은 안정성이나 혹은 고정상에 의한 흡착성에 차이가 있기 때문에 LC 에 의하여 분리가 가 능하다(1 06). 특히 시클로덱스트린을 키랄이동상 첨가제로 사용할 때 는 시료물질이 시클로덱스트린의 키랄공동에 끼워 들어가 내포 화합물 울 형성하므로 라세미 탄화수소 등과 같이 특별한 작용기가 없어서 다 른 방법으로는· 광학분할이 어려운 경우에도 광학분할이 가능하다는 특 칭이 있다.
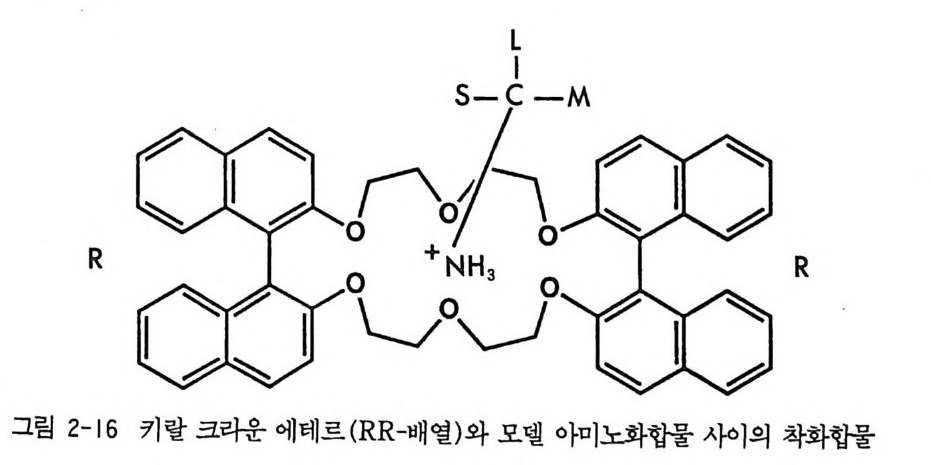 L_
L_
광학활성인 키랄 크리운 에테르 (crown e t her) 를 키랄이동상 첨가제 로 사용하면 손님분자(gu es t molecule) 인 라세미 아미노산 에스테르 등과 주인분자 (host molecule) 인 키 랄 크라운 에 데르 사이에 그림 2 -16 과 같은 부분 입체 이성질 착화합물이 가역적으로 형성되기 때문에 두 부분 입체 이성질 착화합물의 안정성 차이등에 의하여 보통의 비키 랄컬럼상에서 분리가 이루어진다(1 07). 키랄 크라운 에데르를 키랄이 동상 첨가제로 사용하여 라세미 화합물의 광학분할에 이용한 것이 아 마도 CMPA 방법에 의한 광학분할의 시초가 될 것이다. 광학활성인 2, 2, 2- 트리플르오르 -1-(10 - 메틸 -9 - 안트릴)에탄올 을 키 랄이동상 첨가제로 사용하였을 때는 라세미 화합물과 가역적으로 전하 이동 착화합물 (char g e tra nsfe r com p lex) 을 형성하며, 형성된 한 쌍의 전하이동 착화합물의 안정성 차이 및 실리카 겔에 의한 흡착성 차이동 에 의하여 광학분할이 이루어진다(1 08). (R)-2, 2, 2- 트리플르오르 - 1 - (10- 메틸 -9- 안트릴) 에탄올을 그림 2-17 에서 보는 바와 같이 메틸 2, 4- 디니트로페닐술폭시드 (me thy l 2, 4-d i n it ro p hen y lsul fo x i de) 와 전 하이동 착화합물 a 와 벼춘 형성한다. 착화합물 a 는 10- 메틸 -9- 안트릴 기와 2, 4- 디니트로페닐기가 감은 방향으로 나와 있어 강한 전하이동 상호작용을 하기 때문에 착화합물 隣} 비교하여 훨씬 더 안정하리라 고 생각할 수 있다. 실리카 겔에 의한 술폭시드의 흡착(두 광학 이성질 체가 똑같이 흡착됨)과 키랄이동상 첨가제 (카르바놀)와 술폭시드 사이 의 착화합물 형성이 식 (2-11) 과 같이 경쟁한다고 하면 키랄이동상 첨 가제와 강한 전하이동 상호작용을 함으로써 더 안정한 착화합물을 형 성하는 광학 이성질체가 더 빨리 실리카 겔에서 떨어져 나와 이동상으 로 이동하여 이동상 첨가제와 안정한 착화합물을 형성할 것이므로 먼 저 분리되어 나울 것을 쉽게 예측할 수 있다. 실리카(켈고-정상술)폭 시드 카르비(놀이-동상술)폭 시드 (2-11)
a 1군 0L ,,'-HH .. `.0, 02\N; 3:N02 CH3 b O/lcH\ OH` 2 CH3 ’ ’ IFlJl `3 그림이 상2-에17서 ,키 랄여이러동 상종 첨류가의제 인광 학키활랄 성카 르물비질늘들과 이메 틸키\겁 2랄,4이- 디동니0상트2 로첨페가닐제술로폭시 사드 의 두 광학 이성질체 사이에 형성된 전하이동 착화합물• a7} hll다 더 안정하다. 용되어 라세미 화합물들의 광학분할에 응용되고 있음을 살펴 보았다. 키랄이동상 첨가제로 사용가능한 모든 광학활성 물질들은 모두 적절한 방법으로 실리카 겔과 같은 컬럼의 고형지지체에 고정시킬 경우 키랄 고정상으로 사용될 수 있다. 실제로 키랄이동상 첨가제로 예를 든 위 의 모든 광학활성 물질들은 적절한 방법으로 고정상에 부착되어 키랄 고정상으로 사용되고 있으며 제 3 장부터 제惡}까지 이들 키랄고정상들 에 대한 자세한 기술이 있을 것이다. 6 . 3 . 3 키 랄고정상법 (CSP me th od) 과 세 점 상호작용 모델 간접분리 방법이나 직접분리 방법에서의 키랄이동상 첨가제법
(CMPA 방법)과 비교하여 키랄고정상법은 6.3.1 절에서 기술한 바와 같이 많은 장점을 가지고 있기 때문에 1980 년대에 들 어와서 키랄고정 상법에 의한 광학분할의 연구는 가히 폭발적으로 이루어졌다. 그 결과 로 많은 종류의 HPLC 용 키랄고정상들이 개발되었으며 이 분야에 대 한 총설논문들이 발표되었고 (2 , 39 , 109 ~ 114) 편집 단행본들이 출판된 바 있다 (40, 84, 85, 115, 116) . 이들 많은 키랄고정상들이 어떤 키랄성 인지 메커니즘 (ch i ral recog nition mechanis m ) 에 의하여 라세미 화합물을 광학분할하 는지 그 키랄성 인지 메커니즘을 이해하는 것은 간접분리 방법의 분리 메커 니즘 (6.2 . 2 참조)을 이해하는 것이 중요한 것과 마찬가지로 매우 중요 하다. 키랄고정상에 의한 키랄성 인지 메커니즘을 정확하게 이해함으 로서 주어전 라세미 화합물이 주어전 키랄고정상이 충전된 키랄컬럼상 에서 광학분할될 수 있는가 없는가를 예측할 수 있을 뿐만 아니라 용 리순서로부터 분리된 광학 이성질체의 절대배열을 결정할 수 있으며, 또한 주어진 라세미 화합물에 대하여 더 좋은 광학분할능을 보이며 여 러 종류의 라세미 화합물의 광학분할에 광범위하게 사용될 수 있는 새 로운 키랄고정상의 개발이 가능하기 때문이다. 그러나 키랄고정상에 의한 키랄성 인지 메커니즘을 이해하거나 밝히는 것은 그리 쉬운 일이 아니다. 키랄고정상에 의한 키랄성 인지 메커니즘은 키랄고정상에 따라 달라 지며 광학분할하려는 라세미 화합물의 구조에 따라 달라진다. 그러나 키랄고정상이 한 쌍의 광학 이성질체를 식별할 수 있기 위하여서는 키 랄고정상과 한 쌍의 광학 이성질체 중의 어느 하나의 광학 이성질체 사이에 적어도 세 점에서 동시 상호작용 (s i mul t aneous i n t era ci on) 이 있어야 하며 세 점에서의 상호작용 중 적어도 하나의 상호작용은 입체 선택적으로 이루어져야 한다는 매우 단순하며 일반적인 세 점 상호작 용 모델 (three po in t int e r acti on model) 이 키 랄고정상에 의한 광학분 할을 설명하는 데 아주 유용하게 사용되고 있다. 세 점 상호작용 모델
의 개념은 처음에는 효소반응이 입체 특이성 (s t ereos p ec ifi c ity)이라는 사실을 설명하기 위하여 도입되었다. 1933 년 Easson 등이 효소와 기 질 사이의 세 점 상호작용에 대하여 처음으로 기술하였으며 (117), 1948 년 Og s to n 5:. 효소반응의 입체 특이성을 설명하기 위하여 효소와 기 질 사이 의 반응에 대한 자물쇠와 열쇠관계 (rock and key relati on , 1890 년 E. F i sher 가 제안)의 변형으로서 효소와 기질의 세 작용기 사이 에 특징적인 결합이 있어야 한다고 제안하였다(1 18). 그러나 세 접 상 호작용 모델이 크로마토그래피에 의한 광학분할의 키랄성 인지 메커니 즘 설명에 처음 도입된 것은 1952 년 Dal g li esh 에 의해서이다 (119). 1951 년 Sonoh 등은 종이 크로마토그래피상에서 방향족 아미노산들의 광학 이성질체가 분리되는 것을 보고하였으며 (120), 이 연구의 확장으 로서 1952 년 Dal gli esh 는 방향족 아미노산을 디론 유도체로 만들거 나, 방향족기를 지방족기로 바꾸었을 때 종이 크로마토그래피상에서 광학분할이 이루어지지 않음을 확인하였다. 종이 크로마토그래피에 사용된 종이는 셀룰로오스의 구성성분인 글루코오스의 키랄성뿐만 아 니라 셀룰로오스 고분자의 나선성 때문에 키랄성을 가지는 키랄고정상 으로서 종이 키랄고정상과 두 광학 이성질체 · 사이의 입체 선택성을 설 명하기 위하여 Dal g li esh 는 처음으로 키랄고정상과 두 광학 이성질체 사이의 세 점 상호작용 모델을 제안하게 되었다. 그 후 키랄고정상에 의한 키랄성 인지를 설명하는 세 점 상호작용 모델은 Lochm ti ller 등 에 의하여 1975 년에 총설 논문에서 기술된 바 있으나(1 21), 1980 년대 에 들어서서 Pir k le 등에 의하여 광범위하게 이용되기 시작하였으며 022), P i rkle 과 저자 등에 의하여 새로운 키랄고정상의 개발에 성공 적으로 응용되기 시작하였다(1 23) . 키랄고정상의 키랄성 인지를 가장 간단하고 단순하게 설명하는 세 점 상호작용 모델은 그림 2-18 5!} 같다. 그림 2-18 에서 일반적인 (+)-광학 이성질체는 (+)-CSP 와 상호작 용이 가능한 상호작용 부위 (int e r acti on sit e) A', B', C' 를 가지고 있기
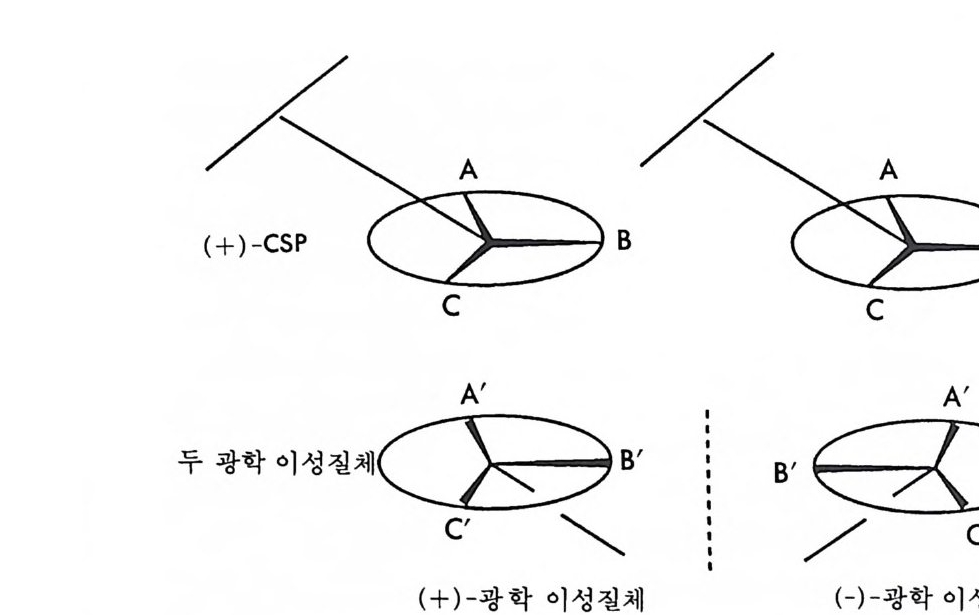 / /
/ /
그림 2-18 키랄고정상에 의한키랄성 인지의 세 접 상호작용모델 때문에 A, B, C 의 상호작용 부위를 가진 (+)-CSP 와 세 점에서의 상 호작용 죽 A-A', B-B', C-C' 의 상호작용울 한다. 그러나 ( + )-광학 이성질체의 거울상인 (-)-광학 이성질체는 입체적인 공간배열(절대배 열)의 차이 때문에 (+)-CSP 와 A-A' 상호작용과 c-c' 상호작용만이 가능하다. 이때, CSP 와 두 광학 이성질체 사이의 상호작용은 수소결 합, 정전기적 상호작용, 쌍극자켓戶f자 상호작용, 전하이동 상호작용 혹은 r-/ 상호작용 (charge tra nafe r int e r acti on ) 등과 감이 친화적 인 상호작용 (a tt rac ti ve i n t erac ti on) 뿐만 아니라 천수성 상호작용, 친지 질성 상호작용, 입체장애에 의한 상호작용 (re p uls i ve int e r acti on ) 등 모두 가능하다. 그림 2-18 에서 입체 배열의존 상호작용인 B-B' 의 상 호작용이 친화성인 경우 (+)-광학 이성질체가 (-)-광학 이성질체보 다 더 큰 친화력으로 CSP 와 상호작용하므로 CSP 상에 (+)-광학 이 성질체가 더 오래 머무르게 된다. 그러나 B-B' 상호작용이 입체장애 에 의한 상호작용일 경우에는 CSP 가 (+)-광학 이성질체와 상호작용
할 때 다큰 입체장애를 느낄 것이므로 ( - ) - 광학 이성질체가 키랄컬 럼에 더 오래 머무르게 된다. 따라서 분리의 메커니즘 죽 키랄성 인지 메커니즘을 정확하게 이해하게 될 때, 한 쌍의 광학 이성질체의 용리 순서를 예측할 수 있을 뿐만 아니라 용리순서로부터 광학 이성질체의 절대배열을 결정하는 것이 가능하게 된다. 그립 2 - 18 의 분자간 상호작용은 수소결합이나 정전기적 상호작용과 같이 접과 점 사이 의 상호작용인 단일점 상호작용 (sin g le -po in t i n t erac ti on) 과 전하이동 착물형성에 관여하는 상호작용이나 쌍극자 -쌍극자 겹침 (d ip ole-s t ack i n g)에 의한 상호작용과 같이 분자에 있는 점과 점 사이에서 상호작용이 일어나는 것이 아니라 면과 면 혹은 선 과 선 사이에서 상호작용이 일어나는 다중점 상호작용 (mul ti-p o i n t i n t erac ti on) 으로 나눌 수 있다. 특히 CSP 와 한 쌍의 광학 이성질체 사이에 씽극자켓드f자 겹침 상호작용이나 亢-T 상호작용과 같은 다중 점 상호작용이 있을 때는 이들 상호작용과 또 하나의 입체배열 의존 상호작용이 있기만 하면 두 광학 이성질체의 구별이 가능하게 된다. 이 경우에는 CSP 와 한 쌍의 광학 이성질체 사이에 두 종류의 상호작 용만이 작용하나 실제로는 한 쌍의 광학 이성질체를 구별하는떠 1 최소 한으로 필요한 세 점 상호작용보다 더 많은 상호작용이 존재하고 있다 고생각할수있다. Davankov 등은 키랄고정상의 키랄성 인지에 있어서 세 개의 동시 적인 상호작용이 항상 필요한 것은 아니며 키랄고정상과 두 광학 이성 질체 사이에 두 점에서의 상호작용이 있더라도 순간적인 부분· 입체 이 성질체를 구성하는 키랄고정상의 키랄 선택자와 광학 이성질체가 흡착 제 (고정상, 또는 키랄고정상 지지체)의 표면에 동시에 접촉한다면 그립 2-19 에서 보는 바와 같이 순간적인 두 개의 부분 입체 이성질체와 흡 착제 표면 사이의 상호작용에 차이가 있어서 한 쌍의 광학 이성질체가 구별된다고 하였다 (85, 124). 그러나 이와 같은 결론은, 흡착제의 표면 은 결국 키랄고정상의 일부로서 흡착제의 표면과 광학 이성질체 사이
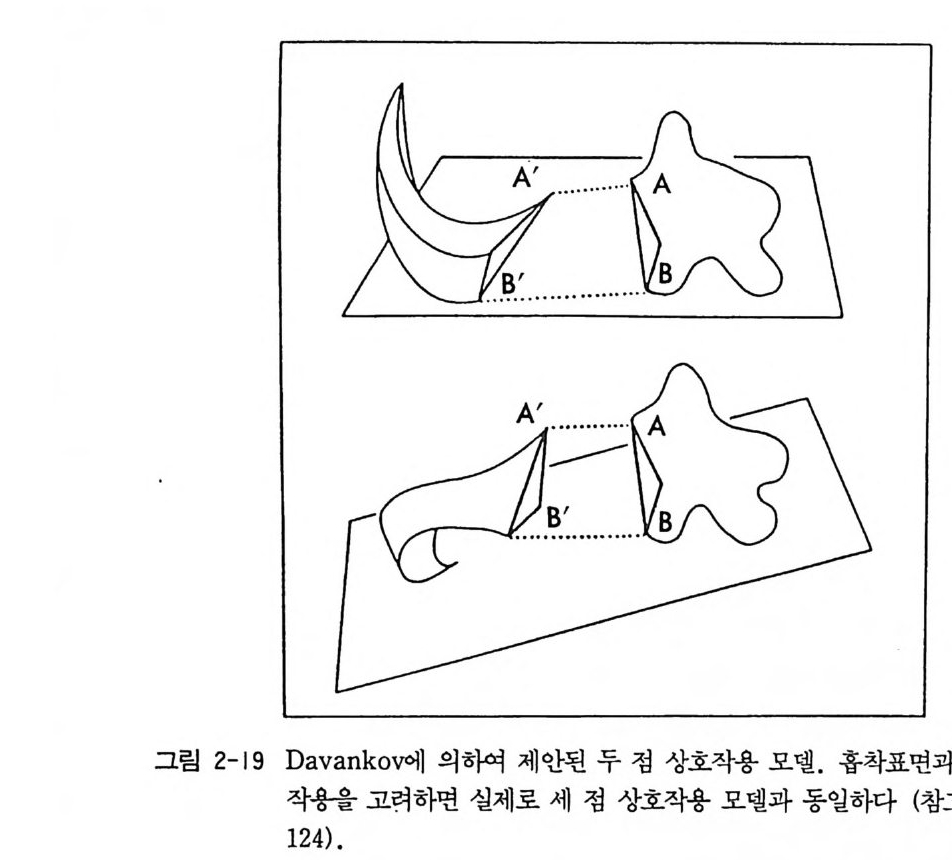 ..... ........ ..
..... ........ ..
의 상호작용 또한 키랄고정상과 광학 이성질체 사이의 상호작용으로 생각할 수 있다는 사시을 무시하였기 때문에 얻어전 것이다. 따라서 키랄고정상의 일부인 흡착제의 표면과 광학 이성질체 사이의 상호작용 울 제 碑] 분자간 상호작용으로 생각한다면, 특수한 경우에 두 점에서 의 동시 상호작용만으로도 키랄성 인지가 가능하다는 Davankov 의 생각은 세 점 상호작용 모델과 동일한 개념임을 알 수 있을 것이다• 실제로 키랄고정상과 두 광학 이성질체 사이에 어떤 종류의 상호작 용이 존재하여야 하며 상호작용을 하기 위하여 세 점은 공간적으로 어 떤 배열을 하여야 하는가 하는 문제 등을 정확히 밝힘으로서 정확한 키랄성 인지 메카니즘을 제시한다는 것은 극히 어렵다. 지금까지 키랄
고정상에 의한 라세미 화합물의 광학분할 메커니즘을 밝히려는 노력은 대부분 라세미 화합물이 광학분할되었을 때 두 광학 이성질체의 용리 순서, 라세미 화합물의 구조 변화에 따른 분리경향의 변화 및 키랄고 정상에 의한 광학분할은 세 점 상호작용 모델을 따른·다는 사실 등을 종합하여 키랄성 인지 메커니즘을 예측하려고 하는 것이었다. 그러나 최근 P i rkle 과 Pocha p sk y는 키랄고정상을 이루는 키랄 화합물과 라 세미 화합물 사이에 형성되는 부분 입체 이성질 착화합물의 1H NMR 스펙트럼을 관찰하여 부분 입체 이성질 착화합물을 형성하는 분자간의 NOE 효과 (Nuclear Overhauser E ff ec t)를 확인함으로써 부분 입체 이 성질 착화합물을 형성하는 분자간의 상호작용에 대한 직접적인 증거를 제시하여 세 점 상호작용 모델이 옮음을 밝힐 수 있었다(1 25, 126). 또 한 컴퓨터를 사용하여, 키랄고정상과 두 광학 이성질체가 상호작용하 여 두 개의 일시적인 부분 입체 이성질체를 만들 때의 결합에너지를 계산함으로써 결합에너지를 최소화하는 과정을 통하여 키랄고정상과 두 광학 이성질체 사이의 정확한 상호작용 부위를 밝힐 뿐만 아니라 키랄성 인지 메커니즘을 밝히려는 노력이 Lip k owi tz 등에 의하여 이 루어지고 있다(1 27 - 129). 아마도 가까운 장래에 키랄고정상에 의한 라 세미 화합물의 광학분할에 대한 정확한 키랄성 인지 메커니즘이 밝혀 지리라고예상된다. 지금까지 논의한 키랄고정상에 의한 키랄성 인지의 세 점 상호작용 모델은 아주 단순하고 간단한 모델로서 이 모델에 정확히 들어맞는 키 랄고정상은 그리 혼하지 않다. 아마도 Baczuck 에 의하여 개발된 키 랄고정상이 세 점 상호작용의 모델에 잘 들어맞는 최초의 키랄고정상 이 될 것이다. Baczuck은 l- 아르기닌을 기저로 한 키랄고정상을 만 들면 그림 2-20 에 보는 바와 같이 키랄고정상은 l- 디히드록시페닐알 라닌 (l-DOPA) 과 세 점에서의 동시적인 상호작용이 가능하나 d-DOPA 는 두 점에서의 상호작용만이 가능하므로 d i -DOPA 의 광 학분할이 가능할 것이라고 생각하였다. 실제로 Baczuck은 2, 4, 6- 트
 OH +H 2N—HNc_3― NN1O— 0HCC IC_ 니 c\_/ c
OH +H 2N—HNc_3― NN1O— 0HCC IC_ 니 c\_/ c
그림 2-20 Baczuck 의 아르기닌 키랄고정상과 DOPA 사이의 세 점 상호작용모델 리클로로-대칭성 -트리 아진 (2,4,6- t r i chloro-s y mme t r i c- t r i az i ne) 을 이 용하여 l- 아르기닌울 세파덱스 G-25 에 고정시킨다음 dl-DOPA를 광학분할한 결과 아주 잘 분리됨 (a = 1. 60) 을 확인하였다(1 30). 그러 나 예측과는 달리 용리순서에 있어서는 d-DOPA 가 컬럼상에 오래 머무름을 관찰하였다. dl-DOPA 의에도 l- 아르기닌 키랄고정상과 세 점 상호작용을 할 수 있는 티로신(tyr os i ne) 은 부분적으로 광학분 할됨이 확인되었으나 히드록시기를 가지고 있지 않은 페닐알라닌은 전 혀 광학분할되지 않았으며 이것은 키랄고정상과 광학분할하고자 하는 시료 사이의 세 점 상호작용이 중요함을 의미하고 있다. Baczuck 의 키랄고정상과는 달리 어떤 키랄고정상들은 키랄고정상 에 부착된 여러 키랄단위들이 서로 모여서 일종의 키랄공동 (ch i ral cav ity)을 형성하기 때문에 키랄인지가 이루어지거나 혹은 커다란 고 분자가 나선성을 가지기 때문에 고정상 전체가 근본적으로 삼처원적인 키랄환경을 제공하여 두 광학 이성질체의 구별이 이루어지는 경우가 있다. 이러한 경우에는 키랄고정상과 두 광학 이성질체 사이에 어떠한 상호작용이 키랄성 인지에 중요하게 작용하는가 하는 점을 밝히기가 아주 까다롭다. 이와 갇이 여러 키랄 단위가 모이거나 커다란 비대칭
성 고분자물질이 키랄고정상으로 작용하여 복합적으로 키랄성 인지가 이루어지는 것을 복합적 방식 (coop e rati ve manner) 에 의한 키랄성 인 지라고 할 수 있다. 반면 Baczuck 의 키랄고정상과 같이 키랄고정상 을 구성하는 개개의 키랄단위가 한 쌍의 광학 이성질체와 상호작용하 여 키랄성 인지가 이루어지는 경우를 독립적 방식 (ind ep e ndent manner) 에 의한 키랄성 인지라 할 수 있다 (39). 키랄고정상에 의한 키랄성 인지가 어떤 형태로 이루어지든지 한 쌍 의 광학 이성질체가 분리되는 것은 키랄고정상과 두 광학 이성질체 사 이에 형성된 한 쌍의 순간적인 부분· 입체 이성질체의 안정성 차이 때 문이며 이 안정성 차이와 두 광학 이성질체의 분리인자 a 와의 관계는 다음에 기술한 바와 같이 유도할 수 있다. 키랄고정상과 R- 광학 이성질체가 순간적인 CSP*R 부분 입체 이 성질체를 형성하고 키랄고정상과 s- 광학 이성질체가 순간적인 CSP* S 부분 입체 이성질체를 형성할 때 부분 입체 이성질체 형성에 대한 평형상수 및 부분 입체 이성질체의 안정성 관계는 식 (2-12) 와 같이 나 타낼수있다. CSP+R ~KR CSP*R, ~GR = -RTlnKR CSP+S ~Ks CSP*S, l:::.G s = —RT lnKs (2-12) 여기서 K 며 Ks 는 평형상수로서 고정상과 이동상 사이의 두 광학 이성질체의 분배계수(p ar titi on coe ffici en t)와 동일한 값이다. 6. GR 과 6. Gs 는 각각 순간적인 두 부분 입체 이성질체의 형성에 대한 자유에 너지 변화이며 순간적인 두 부분 입체 이성질체의 안정성과 같은 값이 댜 키랄고정상과 두 광학 이성질체가 위와 같이 한 쌍의 순간적인 부
분 입체 이성질체를 형성할 때 순간적인 두 부분 입체 이성질체의 안 정성 차이 !:::.!:::.G 는 식 (2-13) 과 같이 표현된다. 1::,1::,G = ( 6GR一 6Gs) = —R T In (KR/Ks) (2-13) 평형상수(혹은 분배계수) KR 과 Ks 는 키랄컬럼상에서 두 광학 이성 질체의 용량인자 kR' 및 ks’ 와 직접 비례하므로 6 . 2.1 절의 a 값에 대 한 정의와 연관시켜 순간적인 두 부분 입체 이성질체의 안정성 차이 6.6. G 는 식 (2-14) 와 같이 a 와 연관된다 (38). 6.6.G = -RTln(KR/Ks) = 一 RTln (kR'/ks') = —R TIna (2- 1 4) 따라서 순간적인 두 부분 입체 이성질체의 안정성 차이는 크로마토 그램으로부터 직접 측정이 가능한 분리인자 a 로부터 계산이 가능하 다. 현재는 HPLC 시스템의 발달로 인하여 순간적인 두 부분 입체 이 성질체의 안정성 차이 즉 66G가 40 J/m ol 정도로 작은 값일 경우 (a = 1. 01) 에도 HPLC 에 의한 분리가 가능하다. 그러나 분석이 목적 인 광학분할 (anal ytic al resolu ti on) 이 아니라 제조가 목적인 광학분할 (pre pa rati ve resolu ti on) 인 경우에는 a 값이 클수록 대량의 라세미 화 합물을 효율적으로 광학분합할 수 있다. 66G 가 특히 작은 값일 때 는 ,0,. G가 작아야 분해인자가 커져서 두 피이크의 구분이 쉬워진다. 큰 6G 값은 키랄고정상과 광학 이성질체 사이의 강한 상호작용을 의 미하며 상호작용이 강할수록 머무롬 시간이 길어지고 물질이동(혹은 질량이동 : mass t rans fe r) 의 효율이 떨어진다. 두 피이크의 분해능이 떨어지면 a 값이 작을 경우 두 피이크롤 구별하기가 어렵다. 결국 키 랄고정성을 개발하는 과정에서는 가능하면 6G 를 작게 하면서 ,0,.,0,.G 는 크게 하는 것이 바람직하다고 할 수 있다.
순간적인 두 부분 입체 이성질체의 안정성 차이 6 6. G 는 일시적인 두 부분 입체 이성질체의 형성에 대한 엔탈피 차이 6 6.H 및 엔트로 피 차이 스스 앉斗 식 (2 ~ 15) 와 같은 관계를 가진다. 66G = 66H —T6 6S (2-15) 일시적인 한 쌍의 부분- 입체 이성질체가 형성되는 과정에서 엔탈피 변화의 차이가 엔트로피 변화의 차이에 온도를 곱한 값보다 충분히 크 다면 6.6. G 는 주로 6 6. H 에 의존한다. 이때는 온도가 낮아짐에 따라 키랄고정상과 두 광학 이성질체 사이에 더 강한 상호작용이 있게 될 것이므로 크로마토그램의 피이크가 넓어지고 a 값은 증가한다. 대부분 의 경우에 키랄고정상에 의한 광학분할은 온도가 낮아짐에 따라 증가 하므로 (a 값이 증가) 6.6. H 에 의하여 지배된다고 할 수 있다. 그러나, 일시적인 한 쌍의 부분- 입체 이성질체의 형성에 대한 엔트로피 변화에 큰 차이가 있을 때 (죽 6 6. S 가 클 때 )는 T 6. 6. S 항이 6. 6. H 항 보다 커 지며 따라서 온도가 증가함에 따라 T 6. 6.~긱 절대값도 증가할 것이 므로 6.6. G 는 T 6.6. S 에 의하여 지배되고 온도가 증가함에 따라 a 값 도 증가할 것이다 (2). 키랄고정상에 의한 광학분할이 엔트로피 변화의 차이 ( 6 6S) 에 의하여 지배되는 예는 혼치 않으나 키랄고정상과 두 광 학 이성질체 사이에 일시적인 한 쌍의 부분 입체 이성질체를 형성하는 과정에서 참여하는 화학종의 수에 차이가 있기 때문에 엔트로피 변화 에 큰 차이가 있어서 광학분할이 엔트로피 변화의 차이 ( 6 6S) 에 의하 여 지배되는 경우가 보고된 바 있다 (131). Davankov는 l-N- 벤질프 롤린으로부터 유도된 키랄고정상을 이용하고 Cu(ll) 를 매개로 하여 N- 벤질프롤린을 광학분할할 때 (리간드 교환 크로마토그래피에 의한 광학 분할은 제懿낼 7 참조할 것) 온도 7} 증가함에 따라 a 값이 조금 증가함을 관찰하였다(1 31). 그림 2-21 에서 보는 것처럼 l, l- 착화합물은 축위치 에 용매분자인 물분자를 가지고 있으나 d, l- 착화합물은 벤질기의 입
 一l, l 一d, l
一l, l 一d, l
그림 2-21 l-N- 벤질프롤린 키랄고정상에서 N- 벤질프롤 린의 광학분할. /, /-착 화합물에 비교하여 d, I - 착 화합물이 적은 수의 리간드 를 가지고 있으 므로 엔트로피의 관점에서 볼 때 d, I- 착화합물의 형성이 더 쉽다. 체장애 때문에 축위치가 비어 있는 상태로 존재한다. 따라서 많은 리 간드 분지를 가지고 있는 l, l- 착화합물이 엔탈피의 관점에서 더 안정 하나 엔트로피 관점에서는 적은 수의 리간드를 가지는 d, l- 착화합물 이 더 형성되기 쉬우며 엔트로피 효과때문에 d, l- 착화합물이 l, l- 착 화합물보다 더 안정하다. 따라서 온도가 증가함에 따라 T 66 S 항이 더 중요하게 되고 a 값이 증가함을 예측할 수 있다. 참고문헌(제 2 장) 1. K. Mi sl ow, Intr o ducti on to Ste r eochemi stry, Benja m i n, New York, 1966. 2. W. H. Pir k le and T. C. Pochaps k y, in Advances in Chromato - gra ph y , vol 27, J. C. Gid d ing s, Ed., Marcel Dekker, New York, 1987, chap 3.
3. M. Raban and K. Mi sl ow, in Top ics in Ste r e o ch em i stry, vol 2, N. L. Allin g e r and E. L. Eli e!, Eds., W ile y -I nte r scie n ce, New York, 1967, pp. 199 -2 30. 4. J. Jac q u es, A. Collet and S. Wi el en, Enanti om ers, Racemate s , and Resoluti on s, W ile y -I nte r sci en ces, New York, 1981, chap . 2 5. K. K. Anderson, D. M. Gash, and J. D. Roberts o n, in Asym metr ic Sy n th e s is , vol 1, J. D. Morris o n, Ed., Academi c Press, New York, 1983, chap 4. 6. G. B. Kauff m an and R. D. My er s, J. Chem. Ed., 완 , 777 (1975). 7. H. E. Zaug g, J. Am. Chem. Soc., 끄, 2910 (1955). 8. I. Chib a ta , S. Yamada, M. Yamamoto and M. Wada, Jap a n Pate n t. 6,722,6 9 3(Nov. 8, 1967) ; Chem. Abs., 69, 36443 k. 9. A. Collet, M -J . Brie n ne and J. Jac q u es, Chem. Rev. 전Q, 215 (1980). 10. A. Lti ttri n g h aus and D. Berrer, Tetr a hedron. Lett ., No 10(1959). 11. M. B. Groen, H. Schadenberg and H. Wy nb erg, J. Org. Chem., 쁘 2797(1971). 12. K. S. Hay e s, W . D. Hounshell, P . Fin o cchia r o and K. Mi sl ow, J. Am. Chem. Soc., 쁘, 4152 (1977). 13. S. H. W iel en, in Top ics in Ste r eochemi stry, vol 6, N. L. Allin g e r and E. L. Eli e!, Eds., W iley -l n te r sci en ce, New York, 1971, pp. 107 -1 76. 14. B. T. Cho and W. S. Park, Bull. Kor. Chem. Soc., 브 , 618 (1989). 15. J. J ac q ue s, A. Collet and S. H. W iel en, Enanti om ers, Racemate s and Re so luti on s, Wi ley - l nte r sci en ce, New York, 1981, chap 7. 16. P. Newman, Optica l Resoluti on Procedures for Chemi ca l Com- po unds, vol 2, Op tica l Resoluti on Info r mati on Cente r , New York, 1981 . 17. P. Newman, Optica l Resoluti on Procedures for Chemi ca l Com- po unds, vol 1, Op tica l Resoluti on Inf or mati on Cente r , New York, 1981 . 18. J. Jac q ue s, A. Collet and S. H. W iel en, Enanti om ers, Racemate s and Resoluti on s, W iley -l n te r sci en ce, New York, 1981, chap 5.
19. D. G. Kais e r, G. J. Vavg ies sen, R. J. Reis c her and W. J. W echte r , J. Phann. Sci., 쁘 , 269 (19 76). 20. P. Newman, Opt ica l Re sol uti on Proced u res for Chem i ca l Com- po u nd s, v ol 3, Op tica l Resoluti on Info r mati on Cente r , N ew York, 1984. 21. P. Block , Jr . and M. S. Newman, Org. Sy n ., 샌, 120 (1968). 22. M. S. Newman, R. W. Wotr i n g , Jr., A. Pandit , and P. M. Cha- krabarti, J. O rg. Chem. , 완, 4293 (1966). 23. D. A. Ligh tn e r, D. T. Hefe lf in g e r, T. W. Powers, G . W. Frank, and K. N. Trueblood, J. A m. Chem. Soc., 완, 3492 (19 72). 24. A. C. Cop e, C. R. Ganell in, H. W. Joh nson, Jr., T. V. Van Auken, and H. J. S. Wi nk ler, J . Am. Chem. Soc., ~ . 3276 (19 63). 25. R. Arad-Yell in, B. S. Green, and M. Knossow, J. Am. Chem. Soc., 브~. 1157 (1980). 26. A. Collet, in Chir a l Sep a rati on s by HPLC ; Ap pli ca ti on s to Phar- maceuti ca l Comp ou nds, A. M. Krstu l ovic . Ed., Ell is Horw o od, Chic h este r , 1989, chap 4. 27. S. Yamagu c hi, in Asym metr ic S yn th e si s , vol 1, J. D. Morris o n, Ed., Academi c Press, New York, 1983, chap 7 . 28. W. H. Pir k le and D. J. Hoover, in Top ics in Ste r eo che m i stry, vol 13, N. L. Al ling e r, E. L. Eli el and S. H. Wi el en, Eds., J oh n W iley & Sons, New York, 1982, pp 263-331 . 29. G. R. Weis m an, in As ym metr ic Syn th e sis , vol 1, J. D. Morris o n, Ed., Academi c Press, New York, 1983, chap 8. 30. R. D. Sti pa novic , J. P. McCormi ck , E. 0. Schlemp e r, B. C. Ham- pe r, T. Shir n ny o zu and W . H. Pir k le, J. Org. Chem ., 템 2500 (1986). 31. W. H. Pir k le, M. R. Robert so n and M. H. Hy un , J . Org. Chem., 32. —4F9. ,’ J2. 4V 3i3 l la(1n9 i8, 4J)r.. , M. J. Costa n zo, R. R. Inners, M. S. Mutt er and D. E. McClure, J. Org. Chem., 템 3715 (1986). 33. M. J. Shap a ir o , A. E. Arch ina l, M. A. Jar ema, J. Org. Chem., 텐 5826 (19 89). 34. S. C. Benson, P. Cai, M . Colon, M. A. Haiz a . M. Tokles and J. K.
Sny d er, J Org. Chem., 완 , 5335 (19 88). 35. R. R. Fraser, in Asym metr ic Sy n th e sis , vol 1, J. D. Morris o n, Ed., Academi c Pres s, New York, 1983, chap 9. 36. G. R. Sulliv a n, in To p ics in Ste r eochemi stry, vol 10, N. L. Al ling er , E. L. Elie ! and S. H. Wi le n, Eds., Joh n Wi le y -I nte r sci en ce, New York, 1978, pp. 287-329. 37. M. D. McCreary, D. W. Lewi s, D. L. Wernic k and G. M. Whit es id e s, J. Am. Chem. Soc., 왠 1038 (1974). 38. L. R. Sny d er and J. J. Ki rk land, Intr o ducti on to Moder n Chromato - gra p hy , 2nd Ed., W ile y -I nte r scie n ce, New York, 1979. 39. W . H. Pir k le and J. Fin n , in Asym metr ic Sy n th e s is , vol 1, J. Morris o n, Ed., A cademi c Press, New York, 1983, chap 6. 40. R. W. Soute r , Chrromato g rap h ic Sep a rati on s of Ste r eois o mers, CRC Press, Boca Rato n , Florid a , 1985. 41. W. Lind ner, i n Chromato g r aph ic Chir a l Sep a rati on s, M. Zie f and L. J. Crane, Eds. , Marcel Dekker, New York, 1988. 42. J. Gal, LC. GC., ~. 106 (19 87). 43. J. Gal. in D1'U g Ste r eochemi stry, I. W. Wain e r and D. E. Dray e r, Eds., Marcel Dekker, New York, 1988, chap 4. 44. M. Ahnoff and S. Ein a rsson, in Chir a l Liq u id Chromato g r a ph y, W . J. Loug h , Ed., Blackie , Glasgo w and London, 1989, chap 4. 45. S. Pedrazzin i , W . Zanoboni- M uc iac ci a, C. Sacchi and A. For- gion e, J. Chromato g r., 브 ~. 214 (1987). 46. A. Sio u fi , D. Colussi, F. Marfi l a nd J. P. Dubois , J. Chromato g r., 브i, 131 (1987). 47. S. Gorog , H. Hereny i and M. Low, J. Chroma t o g r. 덴효, 417 (1986). 48. A. J. Hutt , S . Fournel and J. Caldwell, J. Chroma t o gr . 잔젼, 409 (1986). 49. J. Goto , N, Goto , T. Nambara, J. Chromato g r., 쁘언, 559 (1982). 50. J. Got o, M. Ito, S. Kats u ki, N. Sait o and T. Nambara, J. Liq. Chromato g r., 언 , 683 (1986). 51. R. Ty so n, Chem. & Ind., Feb . 15, 118 (2988).
52. W. H. Pir k le and M. S. Hoekstr a , J. Org. Chem. , 쁘 , 3904 (1974). 53. W. H. Pir k le and C. W. Boeder, J. Org. Chem., 샌 , 2091 (2978). 54. W. H. Pir k le and P. L. Rin a ldi, J. Org. Chem., 센 3803 (1978). 55. W. H. Pir k le and P. E. Adams, J. Org. Chem., ~ . 4111 (19 80). 56. W . H. Pir k le and P. E. Adams, J. Org. Chem., ~ . 4117 (19 80). 57. K. J. Mi ller , J. Gal and M. M. Ames, J. Chromato g r. , ~. 335 (1984). 58. W. H. Pir k le and J. R. Hauske, J. O rg. Chem. , 샌, 1839 (1977). 59. A. A. Gulaid , G. W. Houg h to n and A. R. Boobis , ]. Chromato g r., 318, 393 (1985). 60. W. H. Pir k le and R. W. Anderson, J. O rg. Chem., 쁘, 3901 (19 74). 61. W. H. Pir k le and J. R. Hauske, ]. Org. Chem., 쁘 , 2781 (1977). 62. E. ]. Corey and S.-i. Hashim oto , Tetr a hedron. Lett ., 쁘, 299 (1981). 63. N. Ni m ura, H. Og u ra and T. Ki no shit a, J. Chromato g r., 쁘왼, 375 (1980). 64. T. Ki no shit a, Y. Kasahara, and N. Ni m ura, ]. Chromato g r., 껀}, 77 (19 81). 65. J. Gal, J. Lig . Chromato g r., ~ , 673 (1986). 66. C. Banfi el d and M. Rowwald, J. P harm. Sci., 끄, 921 (19 83). 67. C. Banfi el d and M. Rowwald, J. Pharm. Sci., 챔 1392 (1984). 68. ]. M. Barksdale and C. R. Clark, J. Chromato g r. Sci., 챔 176 (1985). 69. H. Yag i, K. P. Vy as , M. Tada, D. R. Thakker and D. M. Jer in a , J. Org. Chem., 旦 , 1110 (1982). 70. W. H. Pir k le and J. R. Hauske, J. Org. Chem., 性 , 2436 (1977). 71. J. I. Seeman, C. G. Chavdaria n and H. V. Secor, J. Org. Chem., 텐 5419, (1985). 72. W. H. Pir k le and K. A. Sim mons, J. Org. Chem., 쁘 , 2520 (19 83). 73. D. J. Aberhart , J-A. Cott ing and H-J. Lin, Anal. B i ochem. 산i_!, 88 (1985). 74. W. H. Pir k le and J. R. Hauske, J. Org. Chem., 산, 801 (1976). 75. G. Helmchen, R. Ot t and K. Sauber, Tetr a hedron. Lett ., 3873 (1972).
76. G. Helmchen, H. Volte r and W . Schil le, Tetr a hedron. Lett ., 1417 (1977). 77. G. Helmchen, G. Ni ll. D. Flockerzi, W. Schule and M. S. K. Youssef, Ang e w. Chem. Int. Ed. Eng l., 恩 62 (1979). 78. M. Hudlic k y , Chem i stry of Or ga nic Fluor ine Comp o unds, Mac-mi llan , New York, 1961, p. 304. 79. W. H. Pir k le and P. E. Adams, J. Org. Chem. , 브 , 2169 (1979). 80. W . H. Pir k le and P. L. Rin a ldi, J. Org. Chem. , 브 , 1025 (1979). 81. W . H. Pir k le and C. W . Boeder, J . Org. Chem., 센 1950 (19 78). 82. S.G. Allenmark, Chromato g r aph ic Enanti os ep a rati on : Meth o ds an d Ap pli ca t i on s, Ellis Horwood, Chic h este r , 1 988. 83. C. Pett er son, A. Karlsson and C. Gi oe li, J. Chromato g r. , 쁘 !, 217 (1987). 84. A. M. Krstu l ovic , Ed., C hir a l Sep a rati on s by HPLC : Ap pica ti on s to Pharmace u ti c a l Comp o unds, Ell is Horwood, Ch ich este r , 1989, Part Il . 85. V. A. Dav a nkov, J. D . Navrati l and H. F. Waeto n . Lig a nd Ex ch ang e Chro m ato g r aph y , CRC Press, Boca Rato n , Florid a , 1988, chap 5. 86. V. A. Davankov and A. A. Kurga nov, Chromato g rap h ia , l 설, 339 (1980). 87. V. A. Davankov, A. A. Kurga nov and A. S. Bochkov, in Advances in Chromato g r aph y, vol 22 , J.C .Gi dd in g s , Ed., M arcel Dekker, New York, 1983, chap. 3. 88. A. A Kurga nov, V. A. Davankov, J. Chromato g r., 쁘 , 559(1981). 89. J.N . Lep a g e , W. Lind ner, G. Davie s , D.E. Seit z and B. L. Karge r, Anal. Chem. , 탄 , 433 (1979). 90. W. F. Lin d ner, I. Hirsc hbock, J. Lig. Chromato g r., ~ . 551 (1986). 91. R. Wernic k e, J. Chromato g r. Sc i., 챔 39 (19 85). 92. R. Horik a wa, H. Sakamoto and T. Tanim ura, J. Lig . Chromato g r., 안 537 (1986). 93. V. Forsman, J. Chromato g r., 쁘 , 217 (1984). 94. S. Lam, J. Chromato g r.' 천반, 485 (1982).
95. S. Lam and A. Karmen, J. Liq. Chromato g r., 언 , 291 (19 86). 96. P. E. Hare and E. Gi l- Av, Sc ien ce, 깐i, 1226 (1979). 97. J. A. Perry, J. D. Rate i k e and T. J. Szczerba, J. Chromato g r., 홍 57 (1987). 98. S. H. Lee, D. S. Oh and S. H. Bak, Bull. Kor. Chem. Soc., .!Q, 491 (1989). 99. S. H. Lee, D. S. Oh and B. E. Ki m , Bull. K or. Chem. Soc., ~. 341, (1988). 100. A. Dobashi and S. Hara, Tetr a hedron. Lett ., 센 1509 (1983). 101 . A. Dobashi and S. Hara. J. Chromato g r., 莖 11 (1983). 102. A. Dobashi and S. Hara, Anal. Chem. , 헨 1805 (1983). 103. C. Pett er son, in Chir a l Sep a rati o n s by HPLC ; Ap pli ca t i on s to Pharmaceuti ca l Comp ou nds, A. M. Krstu l ovic , Ed., Ellis Horw o od, Ch ich este r , 1989, chap 6. 104. G. Szepe si, in Chir a l Liq u id Chromato g r ap h y , W. J. Loug h , Ed., Blackie , Glasgo w and London, 1989, chap 11. 105. C. Pett er son and G. Schil l, i n Chro m ato g r aph i c Chir a l Sepa r ati on , M. Zie f and L. J. Crane, Eds., Marcel Dekker, New York, 1988, chap 11. 106. D. Sy b il sk a and J. Zukowski, in Chir a l Sepa r ati on s by HPLC ; Ap pli ca ti on s to Phannaceuti c a l Comp ou nds, A. M. Krstu l ovic , Ed., Ell is H orwood, Chic h este r , 1989, chap 7. 107. L. R. Sousa, D. H. Hoff m an, L. Kap la n and D. J. Cram, J. Am. Chem. Soc., 쁘, 7100(1974). 108. W. H. Pir k le and D. L. Sik k eng a , J. C hromato g r., ~ . 400 (1976). 109. W . H. Pir k le and T. C. Pochap s ky, Chem. Rev., 쁘 347 (1989). llO. W. H. Pir k le and G. S. Mahler, in Syn th e sis and Sepa r ati on Usin g Functio n al Polym ers, D. C. Sherrin g ton and P. Hodg e , Eds., Joh n Wi ley & Sons, 1 988, chap 9. lll. D . W. Armstr o ng , Anal. Chem., 헨 84A(1987). ll2. I. W . Wain e r and T. D. Doy le , LC Mag a zin e , 낌 88 (1984). ll3. S. Allenrnark, J. Bio c hem. Bio p h y . Meth d ., 언 , 1 (1984). ll4. 현명호, 《화학과 공업의 전보 》 , 갬 107 (1987).
115. W. J. Loug h , Ed., Chir a l Liq u id Chromato g r aph y , Blackie , Glas- go w and London, 1989. 116. M. Zie f and L. J. Crane, Eds., C hromato g r ap hi c Chir a l Se pa rati on s, Marcell Dekker, New York, 1988. 117. L. H. Easson and E. Ste d man, Bi oc hem . J., '!!_, 1257 (1933). 118. A. G. Og st e n , Natu re, 쁜 963 (19 48). 119. C. E. Dalgl i e s h, J. Chem. Soc., 3940 (1952). 120. M. Kota k e, T. Saken N. Nakamura, and S. Senoh, J. Am. Chem. Soc., 전, 2973 (19 51). 121 . C. H. Lochmi llle r and R. W. Soute r , J. Chromato g r.' 브집, 283 (1975). 122. W. H. Pi rk le, D. W. House and J. M. Fin n , J. Chromato g r., 쁘, 143 (19 80). 123. W. H. Pir k le, M. H. Hy u n and B. Bank, J. C hromato g r., 墨 585 (1984). 124. V. A. Davankov and A. A. Kurga nov, Chromato g rap h ia , 끄, 686 (1983). 125. W. H. Pir k le and T. C. Pochap sk y , J. A m. Chem. Soc., 쁘 , 5627 (19 86). 126. W . H. Pir k le and T. C. Pochap s ky, J. Am. Chem. Soc., 쁘, 5975 (1987). 127. K. B. Lip k owi tz, D. A. Demete r , C. A. Paris h and T. Darden, Anal. Chem., 헨 1731 (1987). 128. K. B. Lipk owi tz, S. Ante l l and B. Baker, J. Org. Chem., ~. 5449 (1989). 129. K. B. Lipk owi tz, D. A. Demete r , R. Zeg a rra, R. Lart er and T. Darden, J. Am. Chem. Soc., 墨 3446 (1988). 130. R. J. Baczuk, G. K. Landram, R. J. Dubois and H. C. Dehm, J. Chromato g r., 쁘, 351 (1971). 131 . A. A. Kurga nov, L. Ya. Zhuchkova and V. A. Davankov, J. Inorg. Nucl. Chem. , 셴 1081 (1978).
제 3 장 광학활성 천연물을 이용한 키랄고정상 1 서론 크로마토그래피를 이용하여 라세미 화합물을 직접 광학분할하려는 최초의 시도는 모두 광학활성 천연물을 이용한 것이었다. 광학활성 천 연물은 자연에서 쉽게 구할 수 있을 뿐만 아니라 가격이 저렴하다. 이 렇게 손쉽게 구할 수 있는 광학활성 천연물을- 적절한 방법으로 LC 컬 럼에 충전시킬 경우 이들은 키랄환경을 제공하기 때문에 키랄고정상으 로 작용하여 라세미 화합물을 광학분할할 수 있다. 키랄고정상으로- 혼 히 시도되어 온 광학활성 천연물은 녹말과 셀룰로오스와 같은 다당류 및 울 (wool), 실크, 수정 등이다 (1-3). 예를 들면, 1939 \! Henderson 3!} Rul 탁 d- 글루코스를 키랄고정상으로 사용하여 라세미 캠퍼 (cam- p hor) 유도체를 광학분할하는 데 성공하였고 (4), 1944년 Prelog 등은 Trage r 염기를 광학분할할 수 있었다 (5). 특히 광학활성 천연 화합물 들 중 감지녹말은 키랄고정상으로 사용되어 비아릴 (b i a ry l) 계통의 라 세미 화합물들을 광학분할할 수 있었다 (6, 7). 그러나 이들 천연화합 물들 죽 울, 실크, 다당류 등은 기계적인 성질(압축성 등)이 좋지 않
을 뿐만 아니라 극성이 높으며 다공성 구조이기 때문에 이들을 고정상 으로 사용할 경우에 물질이동이 효율적으로 이루어지지 않아 이들을 키랄고정상으로 사용하려는 많은 시도가 있었으나 크게 성공하지는 못 했다. 최근 Konrad 등은 광학활성 천연화합물들을 키랄고정상으로 사용하려는 연구에서 감자녹말은 키랄컬럼의 충전제로 어느 정도 유용 하나 울이나 실크, 셀룰로오스 등은 이들 자체로서는 키랄고정상으로 유용하지 않다는 결론을 얻었다 (8). 그러나 이들 광학활성 천연물들을 적당한 유도체로 만듦으로써 극성을 감소시킬 수 있을 뿐만 아니라 이 들 광학활성 자연물들을 기계적 성질이 우수하며 디공성인 실리카 겔 혹은 고분자 물질에 결합시키거나 흡착시킴으로써 HPLC 에 사용가능 한 키랄고정상들을 제조할 수 있다. 이들 중 특히 성공적으로 사용되 고 있는 HPLC 용 키랄고정상들은 셀룰로오스의 극성을 감소시킨 셀 룰로오스 유도체 키랄고정싱들과 시클로덱스트린이나 단백질 등을 실 리카 겔과 같은 고형지지체에 결합시킨 키랄고정상들이다. 2 셀룰로오스를 기저로 한 키랄고정상 2.1 셀룰로오스의 특성 및 역사적 배경 다당류인 셀룰로오스는 D- 글루코스가 /3-1, 4- 글루코시드 결합에 의 하여 이루어전 그림 3-1 과 같은 구조를 가전 천연 고분자 화합물이 다. 셀룰로오스는 분자량이 아주 큰 천연 고분자 화합물의 하나로서 셀룰로오스의 분자량은 보통 수만에서 수십만에 이르며 D- 글루코스 단위를 적어도 1500 개 이상 포함한다. 셀룰로오스는 셀룰로오스롤 구 성하는 D- 글루코스 단위가 광학활성이기 때문에 그 자체로서 광학활 성일 뿐만 아니라 셀룰로오스 사슬과 사슬 사이에 수많은 히드록시기 를 통하여 수소결합을 형성함으로써 사슬과 사슬둘이 모여 섬유형태의
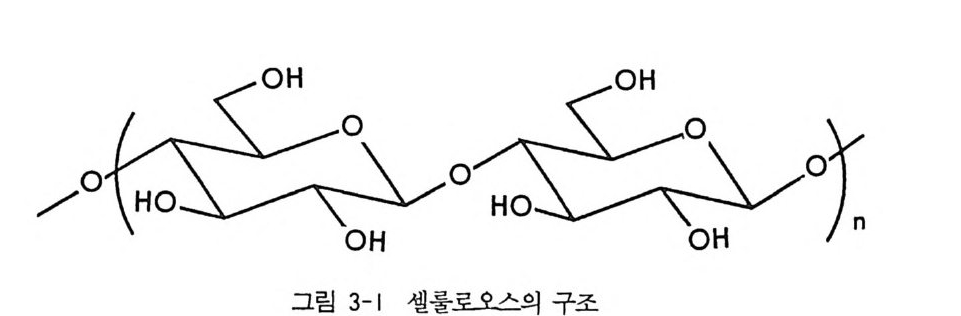 討
討
나선성 구조 를 가지기 때문에 셀룰로오스는 나선성에 의한 광학활성을 7}전 다. 광학활성 셀룰로오스겨춘 이용한 광학분할의 시초는 아마도 종이 크로 마토그레피상에서의 방향족 아미노산의 광학분할일 것이다(1, 2). 그 러나 자연상태의 셀룰로오스는 많은 히드록시기를 가지고 있기 때문에 극성이 클 뿐만 아니라 높은 압력하에서 수축되는 좋지 않은 기계적 성질 등으로 인하여 컬럼내에서의 원활한 물질이동에 문제가 있으며, 따라서 자연상태의 셀룰로오스를 키랄고정상으로 사용하여 라세미 화 합물을 광학분할하고자 하는 시도는 별로 성공적이지 못하였다 (3). 다 만 근래에 자연상태의 셀룰로오스 미세 입자를 액체 크로마토그래피의 컬럼충전제로 사용함으로써 DL- 트리프토판 (DL - t r ypt o p han) 이나 DL -디히드록시페닐알라닌 (DL-DOPA) 의 광학분할 등 성공적인 광학분 할의 예가 보고되고 있으며 (4) 이 의에도 리신 (lysi n e ) 등과 같은 극성 아미노산의 광학분할과 (5) 지방족 아미노산 N-2, 4- 디니트로페닐 유 도체의 광학분할 (6) 이 보고된 바 있다. 셀룰로오스의 크로마토그래피적 성질 및 광학 이성질체에 대한 입체 선택성을 증진시키려는 노력은 셀룰로오스의 히드록시기를 다른 작용 기 특히 에스데르기로 바꿈으로써 극성을 감소시킬 뿐만 아니라 키랄 고정상으로서의 셀룰로오스와 광학 이성질체 사이에 입체장애를 더 크 게 함으로써 광학 이성질체에 대한 키랄고정상의 입체 선택성을 증진 시키는 것이었다. 이러한 노력의 결과로서 1973 년 Hess~ Ha g el 에
의하여 미세결정형 셀룰로오스 트리아세테이트 (m i crocr y s t all i ne cellu-lose t r i ace t a t e : CTA-I) 가 키랄고정상으로 소개되었다 (7, 8) 그 후 Okamoto 등이 셀룰로오스 트리아세데이트롤 포함한 여러 종류의 셀 룰로오스 에스데르와 셀룰로오스 카르밤산 에스테르를 실리카 겔과 갇 은 고형지지체에 흡착시킴으로써 다양한 라세미 화합물들을 성공적으 로 광학분할할 수 있는 키랄고정상들을 개발할 수 있었다 (9).
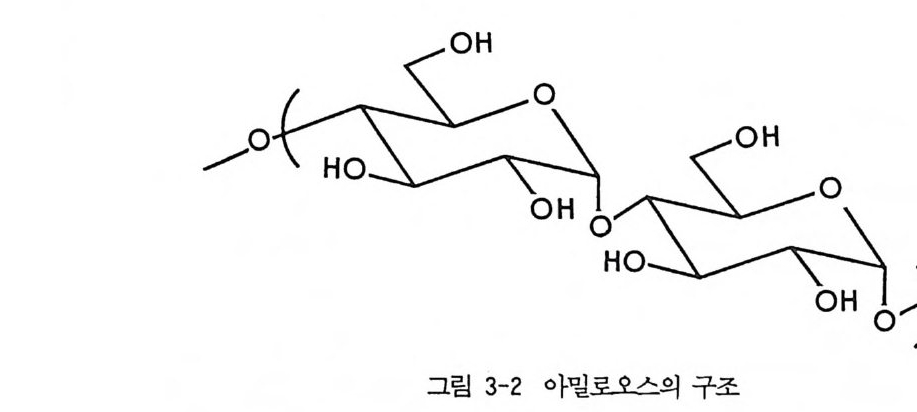 /
/
다당류로서 키랄고정상으로 사용된 천연화합물은 셀룰로오스 의에 도 셀룰로오스와 비슷한 구조를 가전 아밀로오스가 있다. 셀룰로오스 와는 달리 아밀로오스는 D- 글루코스가 a-1, 4- 글루코시드결합에 의하 여 이루어전 그림 3-2 와 같은 구조를 가진 천연화합물로서 아밀로오 스의 에스테르나 카르밤산 에스테르도 셀룰로오스의 에스데르나 카르 밤산 에스데르와 마찬가지로 여러 종류의 라세미 화합물을 광학분할하 는 데에 성공적으로 사용된 바 있다(1 0, 11) 2. 2 미세결정형 셀룰로오스 트리아세테이트 키랄고정상 1973 년 Hesse2 } Ha g el 에 의하여 키랄고정상으로. 처음 소개된 미 세 결정 형 셀룰로오스 트리 아세 데 이 트 (m i croc ry s t a lli ne cellulose
t r i ace t a t e:CTA-1) 는 (7) 현재 상업적으로 구할 수 있으며 (12) Hesse 와 Ha g el 의 방법에 따라 무수초산과 과염소산을 사용하여 미세결정 형 셀룰로오스겨춘 불균일상태에서 아세틸화함으로서 제조가· 가능하다 (그립 3-3) (13) .
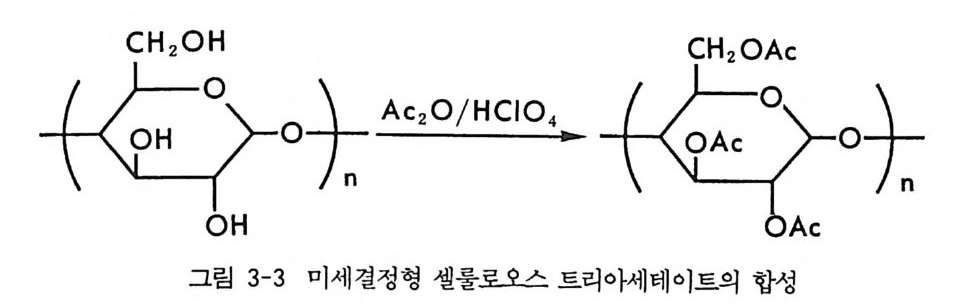 o}. Ac,0/HCIO, CH20Ac 나
o}. Ac,0/HCIO, CH20Ac 나
이와 같은 반응조건에서 광학 이성질체의 입체 선택성에 중요한 셀 룰로오스의 본래 구조는 그대로 유지되는 것으로 밝혀졌으며 (13 ) 미세 결정형 셀룰로오스 트리아세데이트를 키랄고정상으로 하는 라세미 화 합물의 광학분할은 아주 다양한 것으로 확인되었다 (3, 14-16). 그림 3-4 는 CTA-I 싱에서 광학분할이 가능한 라세미 화합물의 예들을 나타 낸 것이다. Tre g er 영기 (1) 가 광학분할될 뿐만 아니라 키랄면을 가전 화합물 (~) 의 광학분할, 큐물렌 (curnullene) 이나 비 아릴 (bia r y l) , 스피 란 (spi ra ne) 동 키랄축을 가전 화합물들 (4-1) 의 광학분할 및 C2 대칭축 울 가진 화합물들(§첼)의 광학분할 등 CTA-I 키랄고정상에서 광학분 할 가능한 화합물들은 아주 다양하다. 특히 CTA-I 키랄고정상에서 광학분할되는 라세미 화합물들의 대부 분이 방향족기를 가지고 있는 반면 스피로케탈 (s pi roke t al: 브,g)과 같이 방향족 화합물이 아닌 라세미 화합물들도 광학분할된다는 사실은 주목할 만하다. CTA-I 키랄고정상에서 광학분할되는 라세미 화합물 의 더 많은 예 및 광학분할의 조건은 참고문헌 (22) 에 자세히 요약되어 있으므로- 크게 참고가 될 것이다.
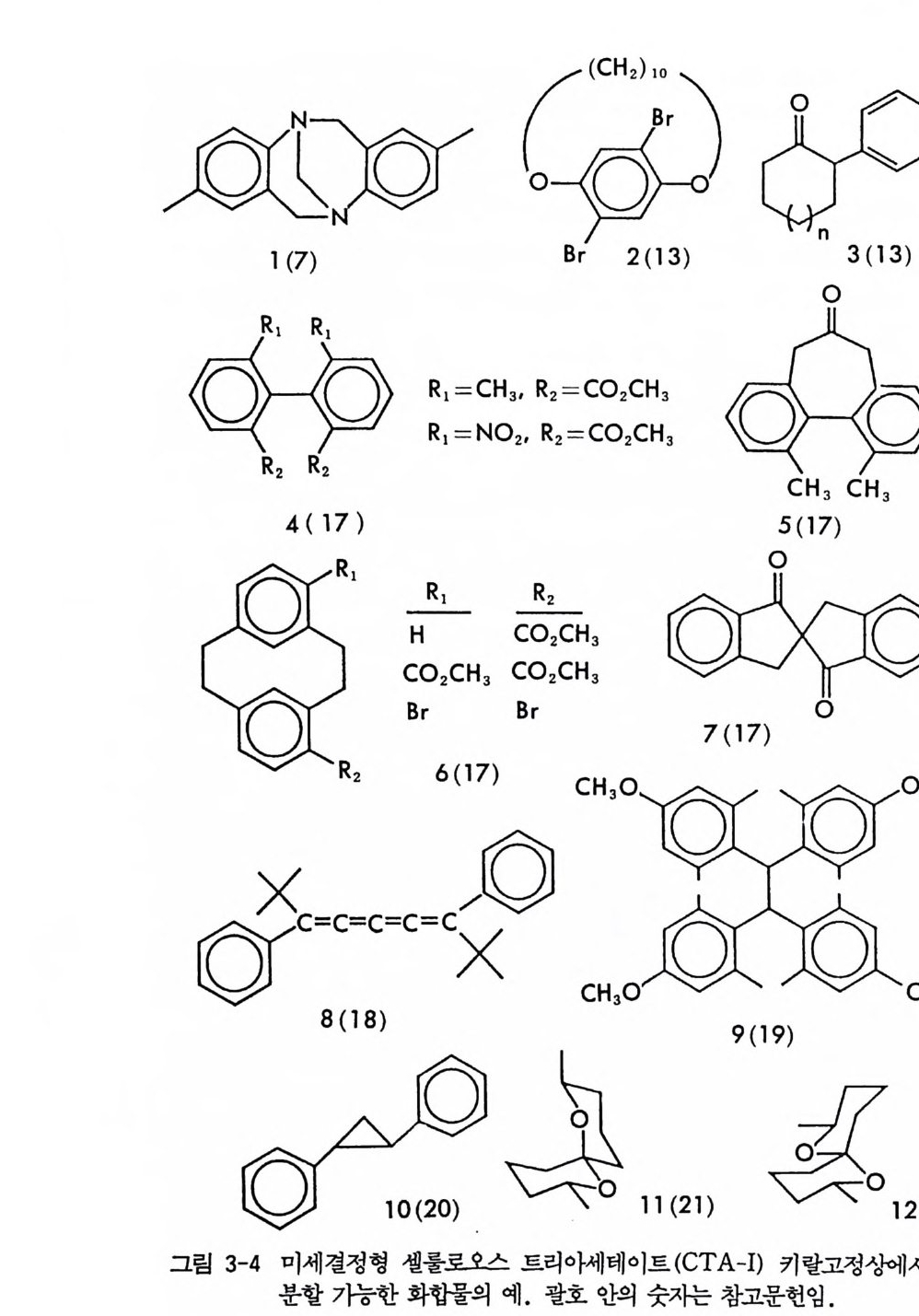 1 (7) Br 2(13) 3(13)
1 (7) Br 2(13) 3(13)
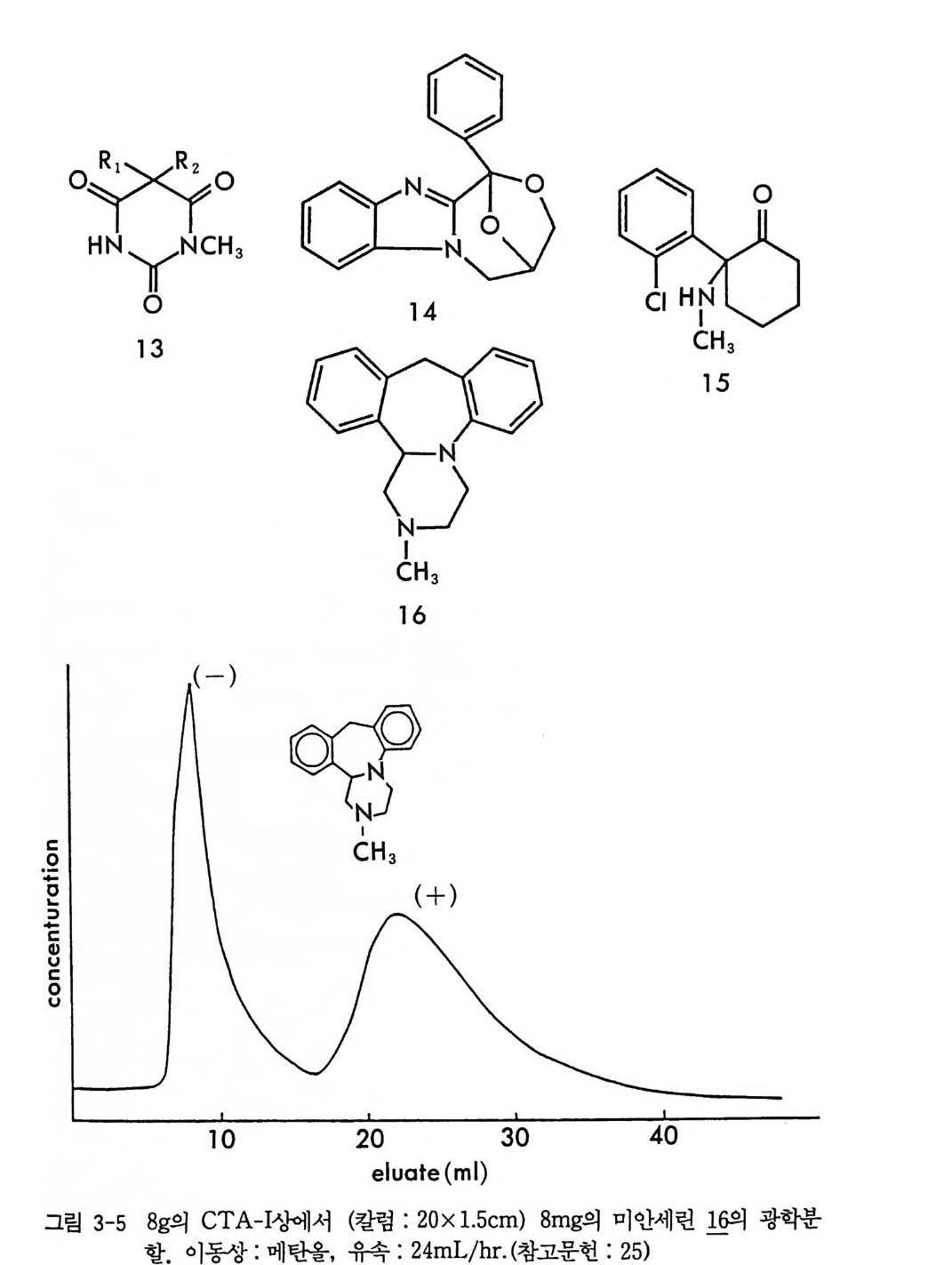 。二C :
。二C :
CTA-I 키랄고정상에서의 광학분할 중 홍미있는 것은 여러 종류의 라세미 의약품들의 광학분할이다. 여러 종류의 바아비탈산 유도체 (barbit ur at e) 묘이 제조규모(p re p ara ti ve scale) 로 광학분할되었을 뿐 만 아니라 (23, 24) 옥사파돌 (oxa p adol) ]A, 케타민 (ket am ine) ~. 미 안세린 (m i anser i n) 1§ 등이 제조규모로 완전히 광학분할되었으며 (25), /3 - 차단제의 옥사졸리돈 유도체가 광학분할되었다 (26), 그림 3- 志 미안세린 표辭] 제조규모 광학분할에 대한 크로마토그램을 제시 한 것이다 (25). CTA-I 키랄고정상에서 광학분할이 가능한 라세미 화합물들은 실로 다양하나, 라세미 화합물이 CTA-I 키랄고정상에서 광학분할되기 위 하여서는 어떤 구조적인 특칭을 가져야 하는가를 말하기는 아직 어렵 다. CTA-I 키랄고정상에 의한 광학분할의 메커니즘이 아직 확실히 밝혀지지 않았기 때문이다. 다만 CTA-I 을 용매에 놓인 후 다시 재결 정 하였을 때 얻어지는 더 안정한 미세결정형 셀룰로오스 트리아세테 이트인 CTA-II 를 \키랄 고정상으로 사용하여 광학분할하였을 때 그림 3-~ Trag e r 염기의 광학분할에서 보는 바와 같이 광학분할능 (a 값) 2 Hr 그림 3-6 CTA-1( 아래)과 CTA-11( 위) 키랄고정상에서 Tro g er 영기의 광학분 할 이동상 : 에탄올, 유속 : 0.2 m L/m in. (참고문헌 : 3)
이 감소할 뿐만 아니라 용리순서가 바뀐다는 사실로부터 (3) CTA-I 이 나 CTA - II 의 결정격자 구조가 광학 이성질체에 대한 입체 선택성에 중요한 역할을 할 것 이러~ 추측을 하고 있을 뿐이다. 죽 CTA-I 에 있어서 입체 선택성에 필요한 것은 셀룰로오스 트리아세데이트 분자의 특별한 작용기가 아니라 CTA-I 혹은 CTA-II 분자가 가지는 특별한 결정구조 때문에 만들어지는 키랄공동에 의하여 광학분할이 이루어전 다고 생각된다. 이 키릴공동의 크기가 제한되어 있고 특별한 모양을 한다면 라세미 화합물의 두 광학 이성질체 중 적어도 하나의 광학 이 성질체가 키랄공동에 적절한 방법으로 끼어들어가거나 혹은 키랄공동 과 적절한 방법으로 상호작용함으로써 광학분할이 가능하게 될 것이 다. 이것을 Hess 탁근 내포 크로마토그래피 (inc lusio n chromato g rap h y) 라고 명명하였다 ( 8) . 만일 CTA-I 를 용매에 녹인 후 다시 침전시킨다 면 CTA-I 이 가지고 있던 본래의 결정구조는 없어질 것이므로, 새로 얻어진 CTA 결정 (CTA - II) 에서의 광학분할의 경향이 바뀌리라는 것 을 쉽사리 예측할 수 있을 것이다. 최근에는 키랄 인덴(i ndene) 끄의 알킬 치환기 크기와 길이룰 바꿈 으로써 얻어지는 광학분할의 경향을 연구하여 CTA- I 키랄고정상에 의한 광학분할의 메커니즘을 규명하려는 시도가 있었으며 (27) 키랄인 돌화합물 陸의 구조를 체계적으로 변화시킬 때 얻어지는 광학분할의 경향(크로마토그램의 경향)을 컴퓨터를 이용하여 변수사이의 부분 최소 자승법 (pa rti al least squ are : PLS) 으로 연관시 킴으로써 CTA-I 키 랄
 二zR 三HR 3 。
二zR 三HR 3 。
고정상에서의 광학분할 메커니즘을 설명하고자 하였으나 (28) 광학분할 의 메커니즘을 알기에는 이론 단계이다. 그러나 회전장애 이성질체 (a t ro pi somer) 인 3-(2- 프로필페 닐 )-4- 메 틸 - 4- 티 아졸린 -2 귄븐 旦가 CTA-I 키랄고정상에서 광학분할될 때 주입하는 시료의 양에 따라 용 리순서의 역전현상(그림 3 - 7) 을 관찰할 수 있었던 것은 아주 홍미있는 일로서 두 개의 광학 이성질체가 키랄고정상에서 독립적인 머무름 메 카니즘을 갖고 있음을 시사해 주고 있다 (29). 즉 두 개의 광학 이성질 체의 머무름이 키랄고정상을 구성하는 미세결정형 트리아세틸셀룰로 오스의 서로 다른 부위에서 일어나고 있음을 시사해 주는 것으로 키랄 성 인지의 메커니즘을 규명함에 있어서 주목할 만한 실험 결과이다. 이와 같은 실험 결과는 R i zz i에 의하여 제안된, CTA-I 키랄고정상에 서 두 형태의 흡착 부위가 존재한다는 모델과도 일치하는 것이다 (30). 미세결정형 셀룰로오스: 트리아세데이트 키랄고정상에서의 광학분할
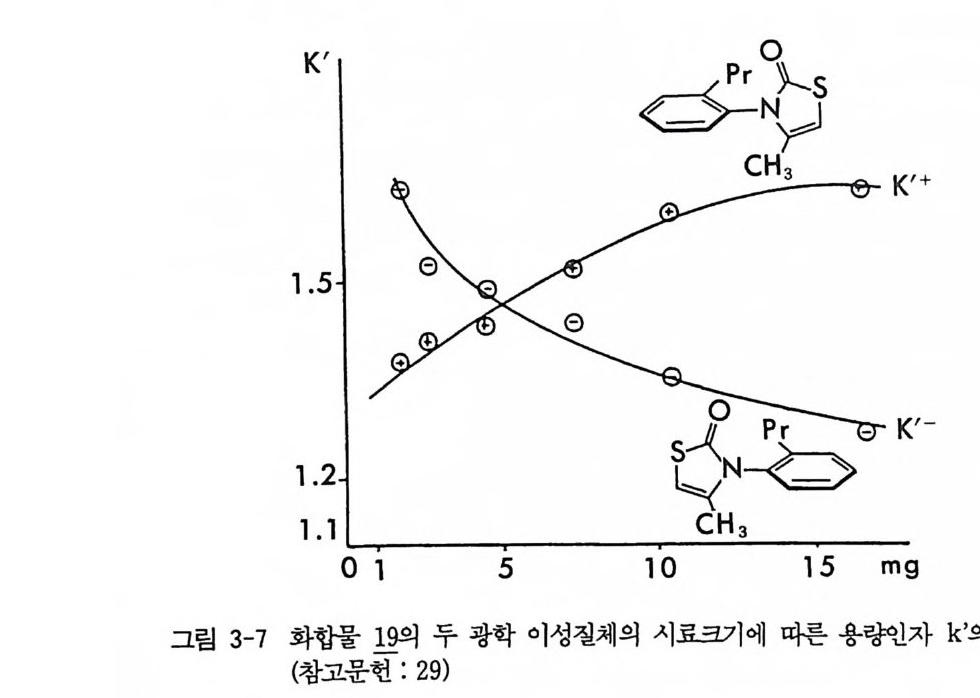 K' 三C :〉 K'+
K' 三C :〉 K'+
에 주로 사용되는 이동상은 에탄올과 물 (95: 5, v/v) 의 혼합용매이나 알코올, 에테르, 물-다른 종류 알코올의 혼합용매 혹은 방향족 탄화 수소 용매를 이동상으로 사용하여 광학분할이 성공적으로 수행된 예들 이 있다. 새로운 라세미 화합물의 광학분할을 시도할 때는 우선 에탄 울과 물의 혼합용매 (95 : 5, v / v) 를 이동상으로 사용하여 시험할 필요 가 있으며 광학분할이 되지 않을 때는 탄화수소 용매, 에데르, 방향족 탄화수소 용매나 이 들의 혼합용매를 사용해 볼 필요가 있다. 그러나 아세톤, THF, 아세토니트릴, 클로로포름, 디클로로메탄과 같은 용매 는 키랄고정상을 용해시키기 때문에 이동상으로 사용하여서는 안된 다. 물과의 혼합용매를 이동상으로 사용할 때 물의 함량이 70% 까지 는 괜찮은 것으로 확인되었으며 이온성 라세미 화합물의 광학분할에처 는 p H 를 변화시킬 수 있는 완충용액을 사용할 수 있다. 이때 p H 의 범위는 5-10 이 바람직하다 (31). 광학분할의 온도를 증가시킴에 따라 그림 3-8 에서 보는 바와 같이
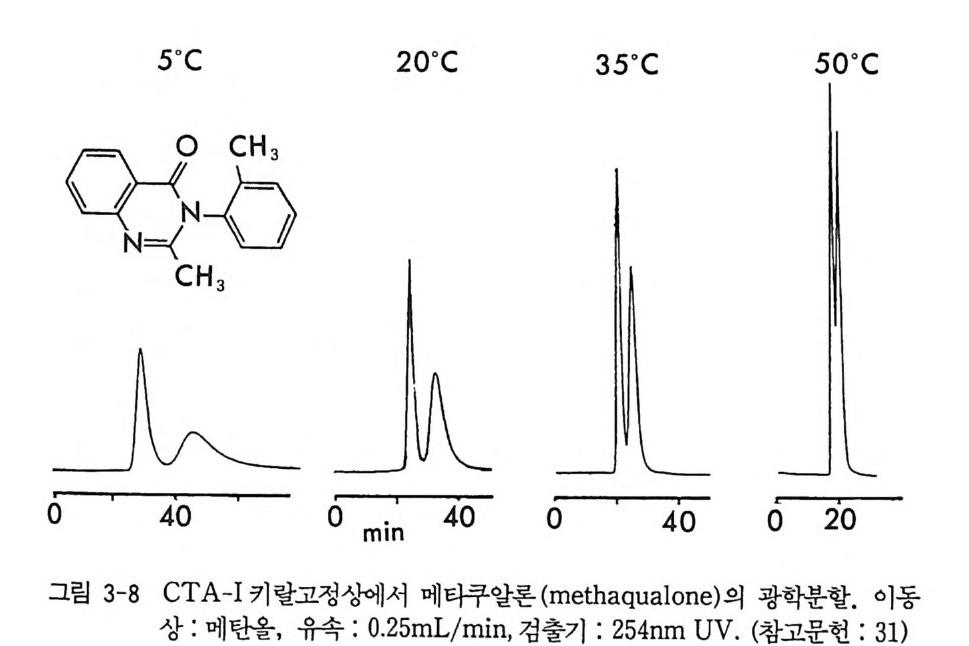 5°C 20°C 35·c 5o·c
5°C 20°C 35·c 5o·c
일반적으로 광학분할의 분해능이 크게 증가함을 알 수 있다 (31). 따라 서 온도를 높임에 따라 라세미화가 용이하게 되는 화합물을 제의하고 는 CTA-I 키랄고정상에서 광학분할할 때 높은 온도에서의 광학분할 은 좋은 분해능을 가전 크로마토그램을 얻기 위한 바람직한 방법이 될 것 0] 다. 2. 3 실리카 갤에 흡착된 셀룰로오스 유도체 키랄고정상 2.3.1 키랄고정상의 구조, 합성 및 응용 CTA-I 이 키랄고정상으로 처음 소개된 후 10 여 년이 지난 1984 년 Ichid a 등 (32) 과 Okamoto 등 (9, 33) 은 HPLC 용 키랄고정상으로는 아주 성공적인 새로운 형태의 셀룰로오스 유도체 키랄고정상들을 소개 하였다. 이들 새로운 형태의 셀룰로오스; 키랄고정상들은 크게 셀룰로 오스 트리에스테르를 실리카 겔에 흡착시킨 키랄고정상과 셀룰로오스 트리카르밤산 에스테르를 실리카 겔에 흡착시킨 키랄고정상으로 나눌 수 있다. 그동안 실리카 겔에 흡착된 셀룰로오스 유도체 키랄고정상들 이 많은 종류 합성되어 광학분할에 대한 연구가 실시되었으나 현재 많 이 사용되고 있는 대표적인 키랄고정상들은 그립 3-9 에 그 구조가 나 와 있는 키랄고정상들로써 키랄고정상 OA, OB, OJ , OK 는 셀룰로오 스 트리에스데르를 실리카 겔에 흡착하여 얻어진 것이며 키랄고정상 OC, OD, OF, OG 는 셀룰로 9-_ 트리카르밤산 에스데르를 실리카 겔에 흡착시켜 얻어진 것이다. 그림 3-9 의 키랄고정상들은 현재 모두 상업 적으로 구입이 가능한 키랄고정싱들이다(제 9 장 참조). 실리카 겔에 흡착된 셀룰로오스 유도체 키랄고정상의 일반적인 제조 과정은 비교적 간단한 편이다 (3, 9, 33, 34). 우선 셀룰로오스와 산염화 물 혹은 이소시아네이트를 일반적인 방법에 따라 반응시켜 셀룰로오스 트리에스데르나 셀룰로오스 트리카르밤산 애스데르를 얻는다. 이들 셀룰로오스 유도체의 생성은 원소분석이나, IR, NMR 을 사용하여 셀
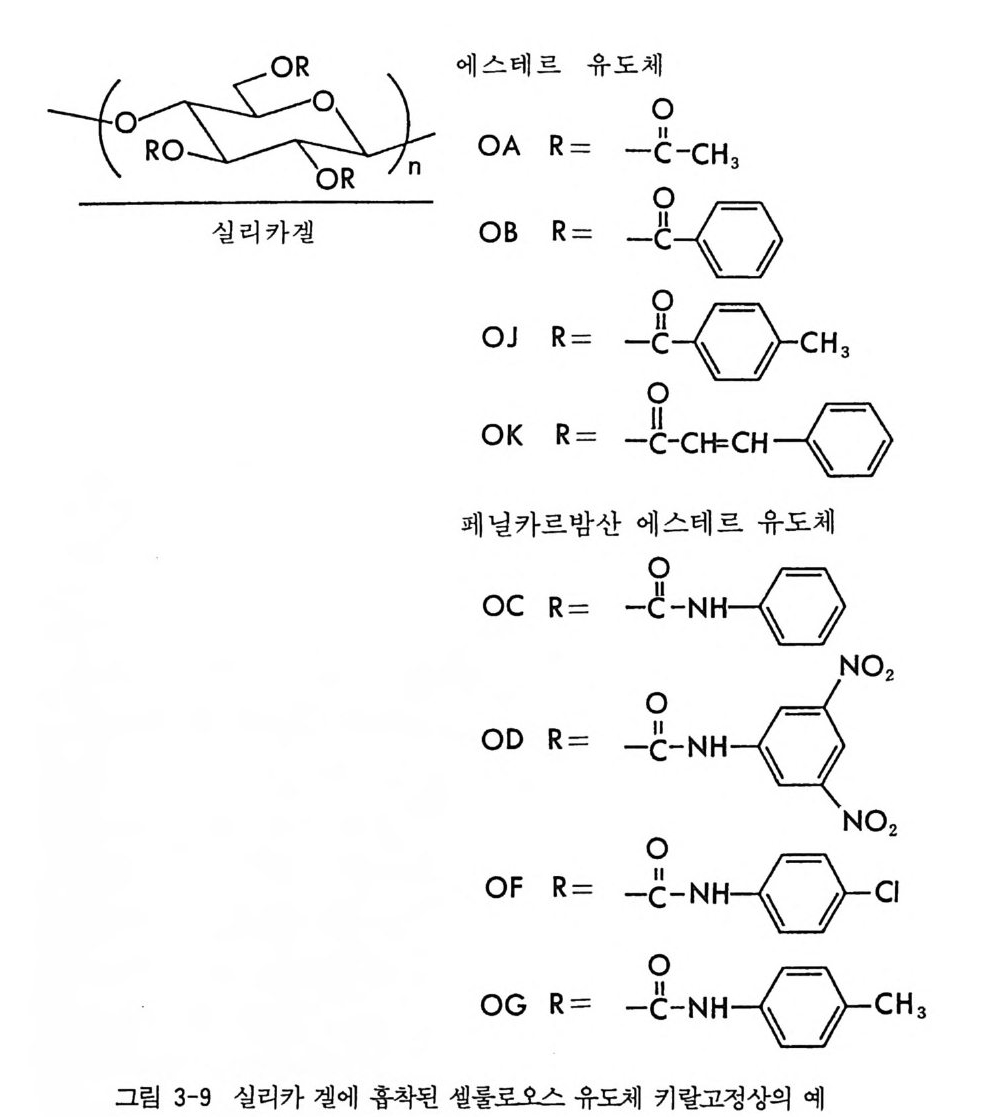 에스테르 유도체
에스테르 유도체
룰로오스의 히드록시기가 에스데르기나 카르밤산 에스테르기로 완전 히 바뀐 것으로 확인할 수 있다. 이렇게 합성된 셀룰로오스 유도체를 적절한 용매에 녹인 후, 이들을 과량의 3- 아미노프로필트리에톡시실 란이나 디페닐디메독시실란으로 미리 처리한 다공성 실리카 겔에 홉착 시키고 용매를 제거함으로써 실리카 겔에 흡착된 셀룰로오스 유도체
키랄고정상둘이 얻어진다. 이때 흡착되는 셀 룰로 오스 유도체의 무게 %는 대략 20% 정도인 것으로 확인되었다. 실리카 겔의 좋은 기계적 성질로 인하여 실리카 겔에 흡착된 셀룰로오스: 유도체 키랄고정싱들은 높은 압력에 견딜 뿐만 아니라 내구성이 좋기 때문에 보통의 슬러리 방법에 의하여 250 X 4.6mm 의 HPLC 컬럼에 충전되어 라세미 화합 물들의 광학분할에 응용된다. 실리카 겔에 흡착된 셀룰로오스 유도체 키랄고정상을 이용한 라세미 화합물의 광학분할은 아주 다양하다. 일반적으로 이들 키랄고정싱들 에 의하여 광학분할이 가능한 라세미 화합물들은 하나 이상의 방향족 고리를 가지거나, 카르보닐기, 술피닐 (sul fi n y l) 기, 시아노기, 니트로 기 등과 같은 7r:-결합작용기를 가지거나 혹은 히드록시기와 같은 극성 기를 가진 것들로서 그동안 실제로 광학분할되었던 많은 라세미 화합 물들이 참고문헌 (22) 에 자세히 요약되어 있다. 특히 광학분할이 잘 되는 라세미 화합물로는 방향족고리를 하나 이상 가지면서 다른 작용 기를 동시에 가지고 있는 화합물들이지만 방향족고리를 가지고 있지 않은 라세미 화합물들도 광학분할된 예가 많이 보고되어 있다. 표 3-1 의 광학분할 예에서 보는 바와 같이 (16 ) 라세미 화합물에 따라 여 러 키랄고정상에서의 분리인자 (a 값)의 값은 전부 다르기 때문에 일반 적으로 어느 화합물이 어느 키랄고정싱에서 가장 좋은 광학분할을 보 일 것인지를 정하기는 아주 어려운 실정이다. 표 3-1 의 여러 라세미 화합물들 중 특히 라세미 알코올의 경우 분자내에 방향족기를 가지고 있으면 직접 광학분할이 가능하나 분자내에 방향족기가 없을 때는 보 통 아세틸유도체나 벤조일유도체로 광학분할됨을 알 수 있다. 라세미 카르보닐화합물의 경우에도 방향족고리를 가지고 있지 않은 라세미 화 합물보다는 방향족고리를 가지고 있는 라세미 화합물의 경우에 더 좋 은 광학분할을 관찰할 수 있으며, 방향족고리를 가지고 있지 않은 라 세미 카르보닐 화합물의 광학분할에는 특히 키랄고정상 OA 가 유용함 울 확인할 수 있다. 표 3-1 에 예를 든 라세미 화합물 이의에도 키랄중
표 3-1 실리카 겔에 흡착된 셀룰로오스 유도체 키랄고정상 OA, OB, OC, OK 에서 여러 화합물들의 광학분할에 대한 분리인자 (a 값). 이동상 : 헥산 -2 - 프로판올 (9/1), 유 속 : 0.5mL/mi n. (참고문현 : 16) 화합물 OA OB oc OK o:::(Ph 1.22 1.47 1.32 1.15 Ph Trl:i ge r 영 기 1.31 1.00 1.32 2.82 Ph -C| H - CII - Ph 1.05 1.12 1.00 1.08 OH 0 1.07 1.47 1.14 1.26 ~Ph 江 CCOONNHHPPhh 1.13 2.0 6 1.25 1.52 다 61HH-CONH 2 1.08 1.00 1.00 1.00 0 건 Ph 1.39 1.17 1.07 門 1.31 1.04 1.00 1.00 같O° H 1.22 1.00 1.00 1.00 亨 1.00 1.31 1.25 1.36 갑 OH 1.00 1.61 1.12 1.00 ~군:0 H . R= COPh 11..000 0 11..363 3 11..000 0 11..000 0 R=H 1.00 1.18 1.00 1.00 R=COPh 1.00 1.00 1.00 1.00 R=H 1.00 1.00 1.00 1.00 성0 H 1.00 1.37 1.35 1.21
 cxfoH I 1.os 11 .16 11 .00 11. 1 4
cxfoH I 1.os 11 .16 11 .00 11. 1 4
심이 인이나 황인 라세미 화합물들도 셀룰로오스에스데르유도체 키랄 고정상이나 셀룰로오스카르밤산 에스데르 유도체 키랄고정상에서 광 학분할이 잘된다 (22) . 홍미있는 광학분할의 예는 키랄고정상 oc 상에서의 프로스타글란딘 합성의 전구체로 사용되는 4 - 히드록시 - 2 - 시클로펜테논 (4 - h y dro x y -2 -cyc l op e nte n one) 유도체들의 광학분할이다. 화합물 챕이나 (35) 화합 물낌이 (36) 키랄고정상 0C 상게서 광학분할되므로 프로스타글란딘 합 성에 응용이 가능할 것이다. 대표적인 크로마토그램은 그림 3-10 과 같다. 지금까지 예를 든 광학분할 가능한 라세미 화합물 이의에도 N- 벤질 옥시카르보닐 a- 아미노산에스데르(아미노산 유도체) 화합물들이 여러 키랄고정상들 중 특히 키랄고정상 oc 나 혹은 OD 상에서 광학분할이 잘됨이 보고되었다 (37) . 세스퀴테르펜류인 식물호르몬 아비시식산 (abs ci s ic a ci d:ABA) 은 키 랄고정상 OD 상에서 메틸에스테르의 형태로 광학분할됨이 보고되었으 나 (38) 라세미 카르복실산은 이동상에 소량의 산을 가할 경우에 유도
 。。
。。
그림 3-10 키랄고정상 0C 상거]서 4 - 히드록시 -2- 시클로펜테논유도체 20(a) 과 2l( b) 의 광학분할 a) 이동상 : 핵산 /2- 프로판올 (98/2), 귬출기 : 254nm UV. b) 이동상 : 핵산 /2- 프로판을 (85/15), 검출기 : 215 run UV. (참고문헌 : 35, 36) 체로 만들지 않고 직접 광학분할된다는 보고가 발표된 후 (39) 곧 핵산 -2- 프로판을-트리플르오르아세트산 (80 : 20 : 1) 을 이동상으로 사용하 여 키랄고정상 OD 상에서 광학분할되었다 (40). 그림 3-11 은 아비시식 산의 메틸에스테르와 산 자체를 키랄고정상 OD 상에서 광학분할한 크 로마토그램이다. 실리카 겔에 흡착된 셀룰로오스 유도체 키랄고정상들은 여러 종류의 라세미 화합물들의 광학분할에도 성공적으로 사용되었다. 진정제인 고리형이미드 계통의 화합물 죽 바아비탈산 유도체들 (barb it ura t es) 이 광학분할되었을 뿐만 아니라(1 6) 소영진통제인 2- 아릴프로피온산 계통
 CJH3 1CH3~ ?H f'c H~3c H -COOH
CJH3 1CH3~ ?H f'c H~3c H -COOH
그림 3-1 I (a) (RS)-ABA 메틸에스데르의 광학분할 컬럼 : OD, 이동상 : 핵산/ 2-프 로판올 (9 : 1) , 유속 : 0.5mL/mi n, 검출기 : 254nm UV. (b) (RS) -ABA 의 광학분할. 컬럼 : OD, 이동상 : 핵산 /2- 프로판을/트리풀르오 르아세트산 (80 : 20 : 1) , 유속 : 0.5 m L/mi n, 검출기 : 240nm UV. (참 고문헌 : 38, 40) 의 의약품들이 여러 종류의 유도체로서 (41) 혹은 유도체로 만들지 않 고 직접 광학분할되었다 (39, 42). /3-차단제로 사용되는 /3-아미노알코 올 계통의 라세미 의약품들은 특히 키랄고정상 0D 삼낭]서 광학분할이 잘되는 것으로 알려져 있다 (11,42,43). 특히 광학활성인 /3-차단제의 대량생산 전구체로 사용될 수 있는 D- 만니톨 (D-mann it ol) 에서부터 얻어진 광학활성 D 내 H 떳 l 술포닐에스데르 盤의 광학순도를 키랄고정 상 O B-를 이용하여 빠른 시간내 (錢小)에 결정할 수 있음이 확인된 것은 의약품의 비대칭 합성이라는 관점에서 홍미있는 일이다 (44). 이 의에도 항히스타민제 계통의 의약품, 아트로핀 계통의 의약품,
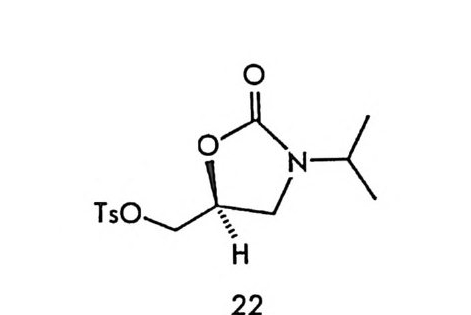 TsO< N- <
TsO< N- <
항고혈압작용제 의약품, 말라리아 치료제인 클로로퀸 (chloro qu i ne), 혈액응고 방지제인 와파린 (war fa r i n) 등 많은 종류의 의약품들이 광 학분할되었다 (11). 이들 의약품들은 보통 방향족 고리와 아미노기 혹 은 히드록시기, 카르복시기 등의 극성기를 가진 화합물들로서 방향족 고리를 가지면서 아미노기나 히드록시기를 가전 다른 라세미 의약품들 도 이들 키랄고정상에서 광학분할이 가능하리라는 것을 예측할 수 있 다. 그림 3 - 12 는 라세미 의약품들 중 소영전통제인 페노프로펜 ac) rOe fCH.c3 O OH b ) :::?HC5H2COCH3 。 10 mm. 20 0 10 mm. 20 30 40 그림 3-12 (a) 페노프로펜의 광학분할 컬럼 : OJ , 이 동상 : 핵산 /2- 프로판을/아 세트산 (9/1/0 . 05) , 유속 : l.O mL/mi n, 검출기 : 254run UV, (b) 와파 린의 광학분할, 컬럼 : OD, 이동상 : 핵산 /2-프 로판을-포름산 (80 : 20 : I). (참고문헌 : 11, 42).
(fe no p ro fe n) 과 혈액응고 방지제인 와파린의 광학분할에 대한 크로마 토그램을 제시한 것이다. 2. 3 .2 광학분할에 영향을 미치는 요인 및 광학분할 메커니즘에 대 한고찰 실리카 겔에 흡착된 셀룰로오스유도체 키랄고정상들의 광학분할에 영향울 미치는 요인들은 여러 가지가 있다. 똑같은 키랄고정상이라도 셀룰로오스 유도체의 물리적 성질이나 제조방법에 따라 광학분할 양상 이 달라지며 사용하는 이동상에 따라 광학분할 양상이 달라진다. 셀룰로오스 유도체의 여러 가지 물리적 요인 중 라세미 화합물의 광 학분할에 큰 영향을 미치는 것으로 알려져 있는 것은 셀룰로오스 유도 체의 분자량이다. Shib a ta 등은 일반적으로 고 분자량의 셀룰로오스 유도체 키랄고정상에서 광학분할이 잘 되는 라세미 화합물들은 - 저 분 자량의 셀룰로오스 유도체 키랄고정상에서 광학분할이 잘 안되며 반대 로 저분자량의 셀룰로오스 유도체 키랄고정상에서 광학분할이 잘되는 라세미 화합물들은 고 분자량이 셀룰로오스 유도체 키랄고정상에서는 광학분할이 잘 안되는 것으로 보고하였다 (3, 16) 트란스-스틸벤옥시드 나 Tra g er 염기 등의 광학분할은 키랄고정상의 분자량에 의하여 크게 영향을 받지 않는 것으로 알려져 있다. 셀룰로오스 유도체를 실리카 겔에 흡착시키는 과정에서 사용되는 용 매에 따라 키랄고정상에 의한 라세미 화합물의 광학분할은 달라진다. 표 3 - 2 에서 보는 바와 같이 흡착과정에서 사용한 용매에 따라 똑같은 형태의 키랄고정상이라도 a 값에 큰 차이를 보이고 있다 (16). 이와 같 온 사실은 셀룰로오스 유도체 키랄고정상이 라세미 화합물을 광학분할 할 때 적용되는 키랄성 인지의 메커니즘이 고정상-라세미 화합물간의 단순한 상호작용으로 이루어지는 것이 아니라 아직 밝혀지지 않은 복 잡한 메커니즘으로 이루어지고 있음울 시사해 준다. 실리카 겔에 흡착된 키랄고정상을 이용한 광학분할에 흔히 사용되는
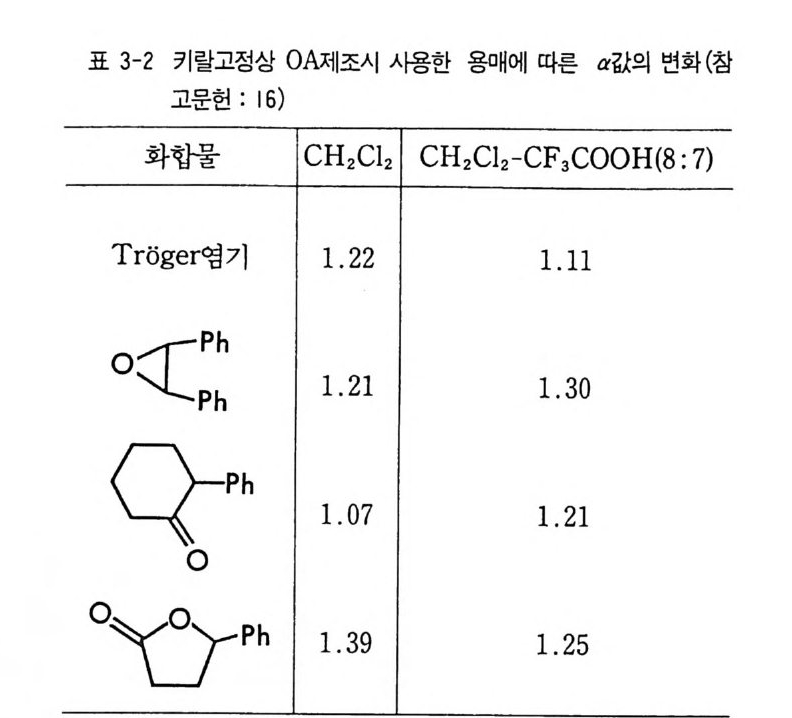 표 3-2 키랄고정상 OA제 조시 사용한 용매에 따른 a 값의 번화 (창
표 3-2 키랄고정상 OA제 조시 사용한 용매에 따른 a 값의 번화 (창
이동상으로는 핵산과 2- 프로판올의 혼합용매 (정상이동상) 및 에탄올 (혹은 메탄올)과 물의 혼합용매 (역상 이동상)등이 주로 사용되며 이동 상으로 사용되는 용매나 용매의 조성에 따라 라세미 화합물의 광학분 할이 크게 영향을 받는다. 대부분의 셀룰로오스 유도체 키랄고정상들 은 셀룰로오스 유도체가 실리카 겔에 흡착된 상태이므로 (셀룰로오스 유도체를 실리카 겔에 공유결합시킨 키랄고정상들도 보고되었으나 광학분할 의 정도는 실리카 겔에 셀룰로오스 유도체를 흡착시킨 키랄고정상에 비교하 여 디소 떨어지는 것으로 확인되었음 )(45) 실리카 겔에 흡착된 셀룰로오. 스 유도체를 용해시킬 수 있는 용매는 이동상으로 사용할 수 없다. 특 히 디클로로메탄이나 클로로포름과 같은 염소화된 용매들이나 아세 돈, THF, DMF, DMSO, 톨루엔, 아세토니토릴 등은 실리카 겔로부 터 셀룰로오스 유도체를 용해시키므로 사용할 수 없다. 최근에는 셀룰 로뚜 유도체 키랄고정상을 이용한 광학분할에 준임계 유체 크로마토
그래피 (SubFC) 기법을 사용한 예가 보고되고 있으며 이산화탄소와 2- 프로판울의 혼합용매를 이동상으로 사용하고 있다 (46). 라세미 화합물이 아미노기를 가지는 화합물인 경우 이동상에 소량의 디에틸아민을 가하면 피이크의 끌림이 없이 빠른- 시간내에 광학분할이 되는 것으로 알려져 있다 (43). 이동상에 소량의 아민이 존재하면 아미 노기를 가진 라세미 화합물이 키랄고정상에 흡착되고 탈착되는 속도가 빨라지기 때문이다. 라세미 카르복실산을 광학분할할 때에는 이동상 에 소량의 유기산울 가함으로써 a 값에 큰 영향을 줄 수 있을 뿐만 아 니라 머무름 시간에도 영향을 미친다. 표 3-3 에 의하면 이동상에 첨 가해 준 산의 종류에 따라 키랄고정상 OD 상에서 만델산의 광학분할 이 영향을 받고 있음을 확인할 수 있다 (39). 이동상으로 핵산 / 2- 프로 판을 (80 : 20) 의 혼합용매를 사용하거나 핵산/아세트산 (95 : 5) 의 혼합 용매를 사용하였을 때는 만델산이 컬럼에서 전혀 유출되지 않았으나 핵산 /2- 프로판올 (80 : 20) 의 혼합용매에 여러 종류의 유기산을 가하여 핵산 /2- 프로판을/유기산 (80 : 20 : 1) 의 혼합용매를 이동상으로 사용하 표 3-3 키랄고정상, . OD 상에서 만델산의 광학분할에 대한 이동상의 영향. 컬럼크기 : 25 x 0.46cm, 이동상 : 헥산 -2- 프로판올--tt (BO : 20 : I ) , 유속 재 .5mL/m i n, 온 도 : 25C (참고문헌 : 39) 산 k1, a’ Rs 없음 용리되지 않음 CH3COOH 용리되지 않음* CH3CH2COOH 2.0 2 1.37 。 CH3COOH 1.28 1.34 0.47 HCOOH 0. 74 1.37 1.19 CHCl2COOH 0.50 1.60 1.62 CC!a C OOH 0.55 1.66 1.74 CF3COOH 0.7 5 1.51 1.98 * 이동상 : 핵산 -CH3COOH (95/5)
면 표 3 - 3 에서 보는 바와 같이 좋은 광학분할을 확인할 수 있다. 일반 적으로 가하여 주는 유기산이 센 산일수록 광학분할이 더욱 효과적으 로 되어 a 값 및 R s 값이 증가함을 확인할 수 있다. 이동상에 산을 가 함으로써 만델산의 해리를 억제할 뿐만 아니라(만델산의 해리로 인하여 카르복실산이 음이온으로- 존재할 때 만델산은 고정상에 강하게 흡착될 것으 로 예상됨) 머무름 시간을 감소시키고, a 값이 영향을 받는 것으로 생 각할수있다. 키랄성 인지 메커니즘을 밝히는 일이 중요함에도 불구하고 실리카 겔에 흡착된 셀룰로오스 유도체 키랄고정상에서의 광학분할에 대한 키 랄성 인지 메커니즘은 아직 확실히 밝혀져 있지 않다. 그러나 실리카 겔에 흡착된 셀룰로오스 유도체 키랄성 키랄고정상에서의 광학분할양 상이 미세결정형 셀룰로오스 트리아세테이트 (CTA-1) 키랄고정상에서 의 광학분할 양상과는 다른 것으로 알려져 있다. 예를 들면 Trege r 염기를 CTA- I 키랄고정상에서 광학분할할 때는 (―)-이성질체가 먼 저 분리되어 나오나 실리카 겔에 흡착된 셀룰로오스 유도체 키랄고정 상에서는 ( + )-이성질체가 먼저 분리되어 나오는 것으로 밝혀졌다 (3, 9). 따라서 실리카 겔에 흡착된 셀룰로오스 유도체 키랄고정상에서의 광학분할은 CTA-I 의 결정성 때문예 생기는 키랄공동에 의한 광학분 할과는 다른 키랄성 인지 메커니즘에 의하여 일어난다고 생각할 수 있 다. 아직 확실히 밝혀지지는 않았지만 키랄고정상으로 작용하는 셀룰 로오스 유도체와 라세미 화합물 사이에 수소결합이나 (34, 37, 47, 48) TC -re 상호작용 및 쌍극자-쌍극자겹침 상호작용이 (48) 중요한 역할을 하 여 광학분할이 일어나고 있다는 몇 가지의 실험적 사실이 발표된 바 있다 . Okamoto 등은 실리카 겔에 흡착된 셀룰로오스 트리벤조산 에스데 르 키랄고정상 (OB) (47) 과 셀룰로오스 트리페닐카르밤산 에스테르 키 랄고정상 (OC) (34, 43) 의 페닐기에 여러 종류의 전자끌게 치환체나 전 자주게 치환체를 도입함으로서 (그림 3-13) 페닐기에 치환된 치환기의
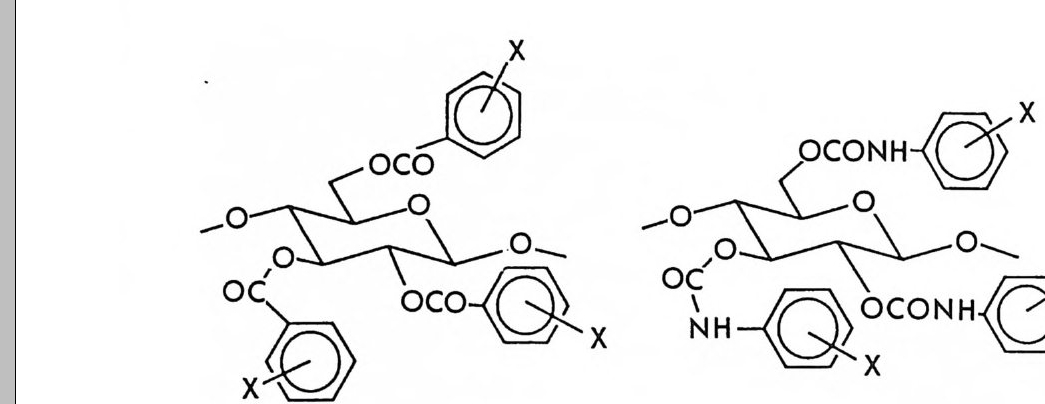 oct jJ ONHOX
oct jJ ONHOX
1. 3, 5-(0CH3) 2 7. 4-CF3 1. 4-0CH3 7. 4-Br 2. 4-0CH3 8. 3, 5-Cl2 2. 4-CH3 (OG) 8. 4-CF3 3. 4-C (CH3 ) 3 9. 3-CH3 3. 4-CH2CH3 9. 3,5-(C H3) 2 (0D) 546... 44H--(CFOH B3) ( OJ ) 111210... 233,,- C45H--((3CC HH33)) 22 456... 4H 4--(CFOl C(O) F) 111201... 333,,- 44 C- -HC(lJ C2 H 山 그림 3-13 페닐기에 여러 종류의 치환체가 치환된 셀룰로오스 트리벤조산 에스데 르 키랄고정상 및 셀룰로오스 트리페닐카르밤산 에스데르 키랄고정상 종류에 따라 광학분할이 큰 영향을 받음을 확인할 수 있었고 이와 같 은 사실은 페닐기에 치환되어 있는 치환체가 에스데르결합 혹은 카르 밤산 에스데르 결합의 수소결합(라세미 화합물과의)능력에 변화를 주기 때문에 광학분할에 영향을 미친다고 보고한 바 있다. 그림 3-14 는 셀룰로오스 트리페닐카르밤산 에스데르 키랄고정상의 페닐기에 여러 종류의 치환체를 치환하였을 때 치환체의 Hammet
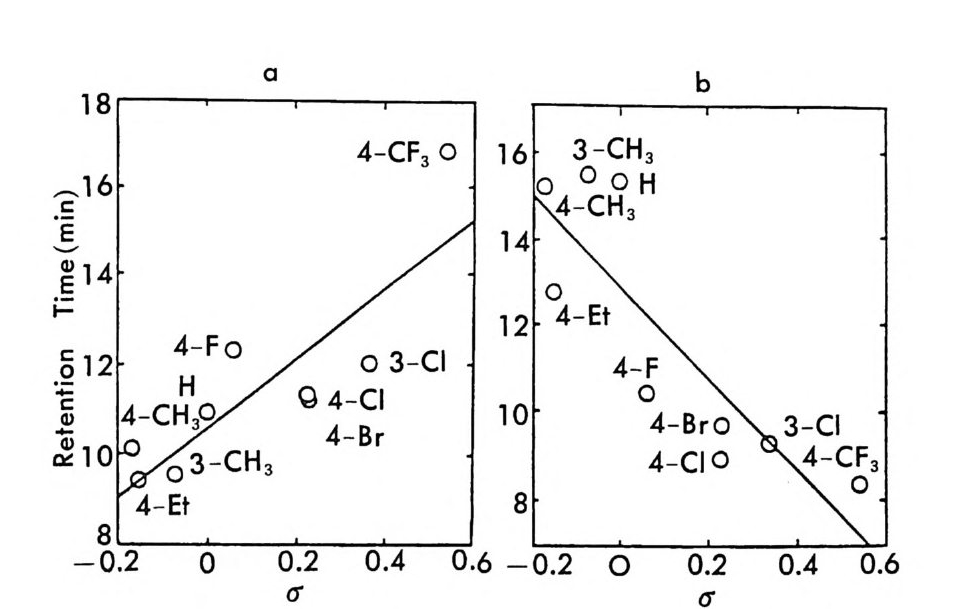 ;6l a /4/-CF 3] I 11164) 0o4 4--3oCE - CHto H3 3H b
;6l a /4/-CF 3] I 11164) 0o4 4--3oCE - CHto H3 3H b
그림 3-14 셀룰로오스 트리페닐카르밤산 에스테르 키랄고정상에서 페닐기의 치환 체 c 값과 a) 아세톤 및 b) 1-(9- 안트릴 )-2,2,2- 트리플르오르에탄을의 두 광학 이성질체 중 먼저 분리되어 나오는 광학 이성질체의 머무름 비 교도. (참고문헌 : 34)
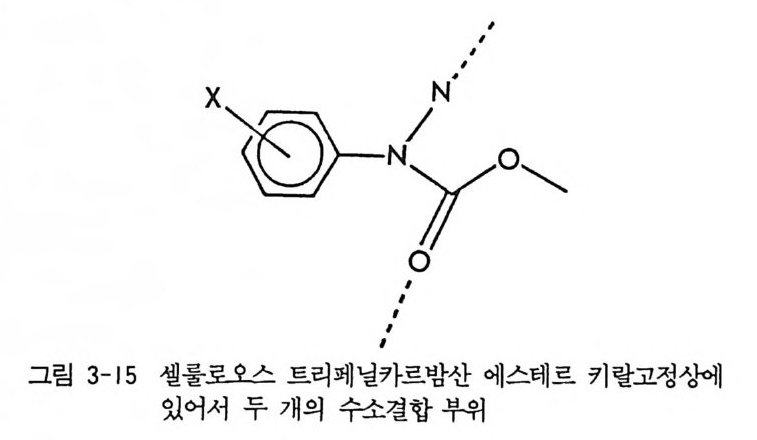 ,,,,
,,,,
셀룰로오스 페닐카르밤산 에스테르 유도체 키랄고정싱에 있어서 수 소결합에 참여할 수 있는 두 개의 부위 (그림 3-15) 죽 N-H 및 C=O 중 치환체 (X) 의 전자주기 효과가 증가함에 따라 C=O 산소의 전자 밀도가 증가하고 따라서 알코올과의 수소결합이 강하게 되어 알코올의
머무름이 증가한다. 반면 치환체의 전자끌기 효과가 증가하면 N-H 수소의 전자밀도가 감소하여 아세톤의 카르보닐기와 강한 수소결합을 형성하여 아세톤의 머무롬은 증가하게 됨을 예측할 수 있다. 이와 같 은 효과는 키랄고정상 OD 상에서 아미노산 유도체의 광학분할에서도 확인할 수 있다. 그림 3-1 6i본 N-(4 - 치환벤조일) 알라닌벤질에스테르 4- 치환체의 Hammet
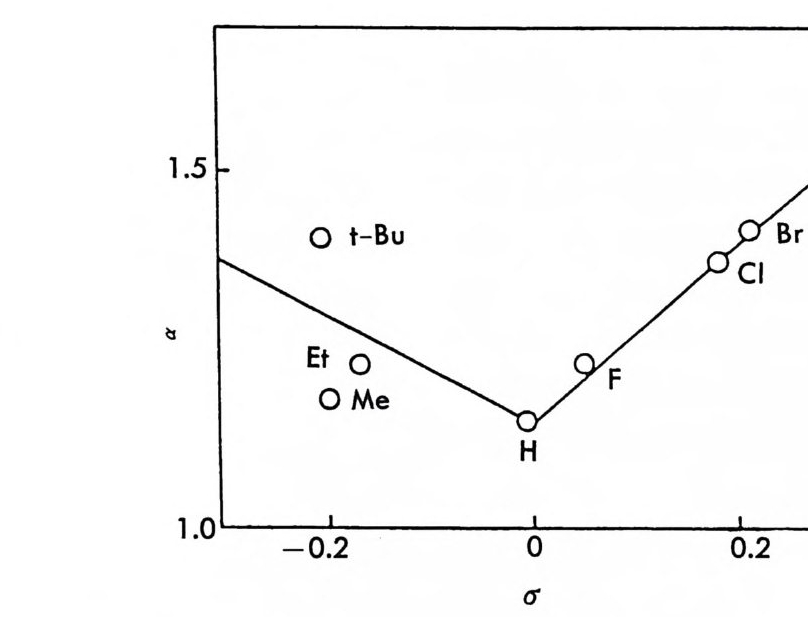 1.5
1.5
그림 3-16 키랄고정상 OD 상에서 N-(4- 치환벤조일)알라닌벤질에스데르의 광 회 분할에 대한 a 값 및 4- 치환체의 c 값과의 관계 (참고문헌 : 37)
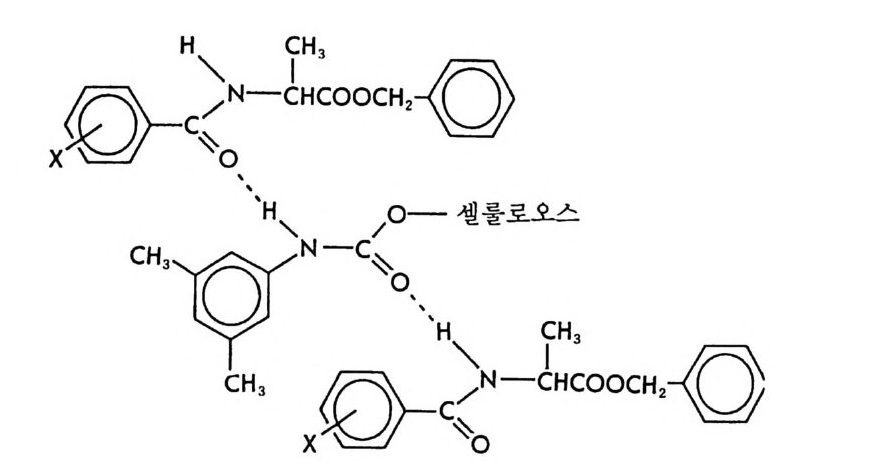 x 《》 _CH?, °N ―」C HH3C OOCH2 《》
x 《》 _CH?, °N ―」C HH3C OOCH2 《》
그림 3-17 키랄고정상 OD 와 N-(4- 치환벤조일)알라닌벤질에스테르의 수소결합상호작용 며, 4- 치환체의 전자끌기 효과가 커짐에 따라((J값 증가) N_ (4- 치환 벤조일)아미드에 있는 N-H 의 전자밀도가 감소하여 키랄고정상의 카 르보닐산소와 강한 수소결합을 하게 되어 그림 3-1~ 같은 결과를 얻을 수 있다고 생각할 수 있다. 한편 Wa i ner 등은 키랄고정상 OB 상에서 여러 종류 라세미 아민의 N- 벤조일유도체 盤과 라세미 카르복실산의 아닐리드 (ani l i de) 유도체 갬 및 器과 갬의 N-H 수소대신 알킬기가 치환된 화합물, 혹은 盤과 갬料 페닐기 대신 알킬기가 치환된 화합물 등 다양한 라세미 아미드 화합물들의 광학 분할을 연구함으로써 키랄고정상과 라세미 화합물 사 이의 수소결합이 광학분할에 중요한 역할을 할 뿐만 아니라 키랄고정 상의 페닐기와 라세미 화합물의 페닐기 사이의 7C-7C 상호작용 및 키랄 고정상과 라세미 화합물 사이의 쌍극자』射구자 상호작용이 중요한 역 할을 하고 있음울 시사하였다 (48). Wain e r 등은 또한 키랄고정상 O IY-J-ol l 서 썹혹은 갬롤 광학분할할 때 이동상으로서 핵산과 여러 종
CH3-{C H 2 )n-CNI HH-CCOHC 36 H5 CHa -( CH2 ln -CCI HO-NCHHC63 H 5 23 24 류의 알코올 혼합용매를 사용함으로써 사용한 알코올의 입체장애가 증 가함에 따라 표 3-4 에 보는 바와 같이 머무름 시간과 a 값이 모두 증 가함을 확인하였다 (49) . 이와 같은 실험결과는 라세미 화합물과 이동상중의 알코올이 키랄 고정상과의 수소결합에 대하여 서로 경쟁관계에 있음을 나타내는 것으 로서 알코올의 입체장애가 커질수록 경쟁력이 약화되어 분 석 성분과 키 랄고정상의 상호작용이 강화되므로 머무름 시간이 길어지며 알코올이 키 랄고정상의 키 랄공동주위 혹은 키랄공동에 있는 비키 랄 작용부위에 결합(수소결합)되어 키랄공동의 입체칭애 환경을 변화시키기 때문에 알코올의 구조에 따라 기질에 대한 입체 선택성 (a 값)에 영향을 미친 다고생각되고있다. 표 3-4 키랄고정상 O Wci백서 화합물 찬 (n = 3 및 4) 과 화합물 안 (n = 3 및 4) 의 광학 분할에 대한 이동상중의 알코올의 영향(이동상 : 0.67 M 알코올+헥산). 용량인 자는 먼저 분리된 광학 이성질체에 대한 값임 (참고문헌 : 50) . 화합물썽 (n=3) 화합물 썰 (n= 4) 화합물 낀 (n = 3) 화 합물 낀 (n = 4) 알코올 k' a’ k' a’ k' a’ k' a’ 메탄올 1.66 1.22 1.39 1.30 1.85 1.24 1.39 1.25 에탄올 2.00 1.48 1.67 1.53 2.1 0 1.50 1.65 1.52 1- 프로판을 2.1 6 1.61 1.83 1.61 2.15 1.56 1.71 1.62 2- 프로판을 3.7 0 1.62 3.5 7 1.81 3.6 2 1.66 2.91 1.93 2 부탄올 2.70 2.19 2.48 2.56 3.4 9 1.83 2.76 1.97 2- 펜탄올 3.91 3.21 3.56 3.46 4.80 2.1 1 4.03 2.01 2 - 헥산을 3.62 2.28 3.02 2.23 4.01 1.66 3.1 9 1.60 t부탄올 5.7 0 4.59 6.34 4.88 9.74 1.93 9.1 2 1.96
이와 같은 연구결과의 확장으로서 Wain e r 등은 최종적으로 키랄고 정상 OB 상에서 여러 종류 방향족 알코올의 광학분할에 대한 키랄성 인지 메커니즘을 제안하였다 (50). 즉 키랄고정상 0 8-'-o h게서 방향족 알 코올의 광학분할에 대한 키랄성 인지 메커니즘은 방향족 알코올의 히 드록시기와 키랄고정상의 에스테르카르보닐 산소 사이의 수소결합에 의한 부분 입체 이성질 착화합물의 형성, 형성된 부분 입체 이성질 착 화합물의 방향족기가 키랄고정상의 키랄공동에 끼어들어감으로써 얻 게되는 부분 입체 이성질 착화합물의 안정성 및 키랄공동에 방향족기 가 끼어들어갈 때 방향족 알코올의 두 광학 이성질체에 대한 입체장애 차이의 세 가지 복합적 요인에 의하여 결정된다고 제안되어 있다. 그 러나 이 세 가지 요인이 어떻게 작용하여 키랄성 인지가 일어나고 있 는가 하는 물음에 대한 구체적인 대답은 아직 없다. 이상에서 셀룰로오스 유도체 키랄고정상에 의한 광학분할에 키랄고 정상과 광학분할하고자 하는 라세미 화합물 사이의 수소결합 등이 중 요한 역할을 하고 있음을 확인하였으나(수소결합이 없는 경우에도 광학 분할이 가능한 예가 디수 있음 (22)) 아직 구체적인 키랄성 인지의 메커 니즘은 모르는 상태이다. 또한 지금까지 살펴온 바와 같이 각 키랄고 정상들마다 라세미 화합물에 대하여 다른 광학분할능을 보이기 때문에 어느 키랄고정상이 어떤 종류의 라세미 화합물의 광학분할에 특히 유 용한가를 이론적으로 결정하기는 아주 어렵다. 따라서 그동안 광학분 할이 시도된 바 없는 새로운 라세미 화합물을 광학분할하기 위하여 셀 룰로오스 유도체 키랄고정상을 사용하고자 할 때에는 그동안 광학분할 되었던 여러 종류의 라세미 화합물들이 어떤 키랄고정상에서 가장 잘 분리되었는가를 조사한 후 실제로 여러 종류의 키랄고정상을 사용하여 광학분할이 가장 좋은 키랄고정상을 선택하는 방법 의에는 없다. 이러 한 문제점에도 불구하고 방향족기와 극성 작용기를 동시에 가지고 있 는 라세미 화합물둘은 실리카 겔에 흡착된 셀룰로오스 유도체 키랄고 정상들 중의 어느 하나의 키랄고정상에서 광학분할되리라고 예측되기
때문에 셀룰로오스 유도체 키랄고정상들은 앞으로도 계속 여러 종류 라세미 화합물의 광학분할에 응용될 것으로 생각된다. 앞으로 더 정확 한 키랄성 인지의 메커니즘이 밝혀진다면 이 메커니즘은 키랄고정상의 광학분할능을 증진시키고 또 새로운 라세미 화합물의 광학분할 여부를 결정하는 데 유용하게 응용될 수 있을 것이다. 그러나 셀룰로오스 유 도체 키랄고정상의 특성 죽 여러 키랄단위들이 복합적으로 모여서 이 루어진 키랄고정상이라는 특성 때문에 명확한 키랄성 인지의 메커니즘 을 밝히기는 간단한 문제가 아니라는 것을 지적해 두고자 한다. 3 시클로덱스트린 키랄고정상 3 .1 시클로덱스트린의 구조 및 특성 시클로덱스트린 (cy cl odextr i n , CD) 은 여섯 개에서 열두 개까지의 D -(+)-글루코피라노오스(g luco pyr anose) 가 a-1, 4 - 글루코시드결합에 의하여 형성된 고리모양의 울리고당류 (o lig osacchar i de) 이다. CDo 사 존재하는 글루코피라노오스 단위의 수를 표시하기 위하여 그리이스 문 자가 사용된다. 죽 글루코피라노오스 단위가 여섯 개인 CD 는 a- C D, 일곱 개인 CD 는 [3- CD, 여덟 개인 CD 는 y- CD 등으로 표현되며 크 로마토그래피에 사용되는 CD 들은 보통 a, /3, y -CD 들로서 모두 상 업적으로구할수있다. 그림 3-l Si는 a-CD 의 구조를 나타낸 것이다. CD 분자의 물리적인 형태는 꼭대기를 잘라낸 원추형 모양으로써 a, /3, y -CD 의 구조적 특성 및 물리적 특성을 표 3-5 에 요약하였다. 꼭지롤 잘라낸 원추형 태 CD 분자의 윗부분店(넓은 부분)에는 글루코피라노오스단위의 C-2 위 치와 C-3 위치에 있는 2 차 히드록시기가 고정되어 있으며 특히 C-2 위치의 모든 Vl - 히드록시기는 시계회전 방향으로 배열되어 있다.
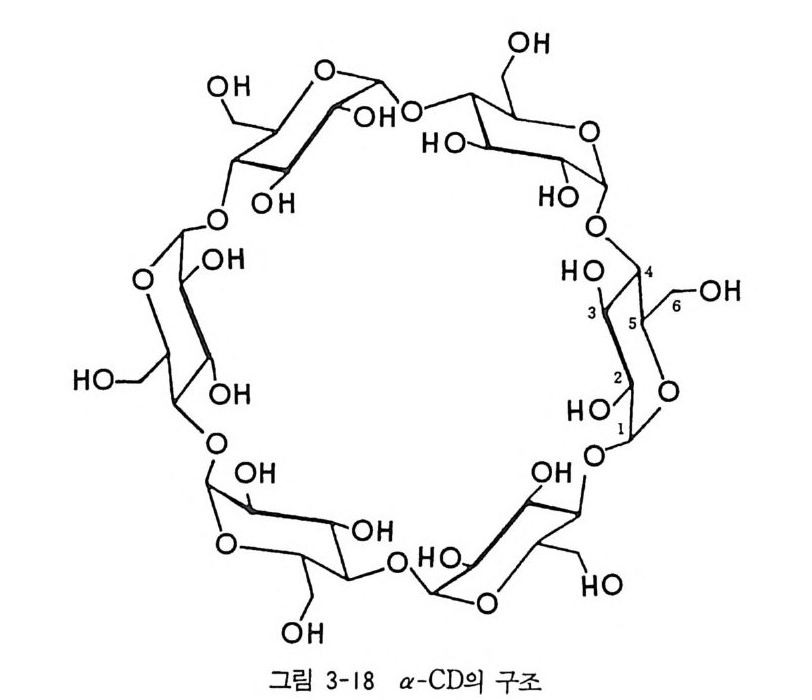 广 °H〈
广 °H〈
CD 분자의 아래부분(좁은 부분)에는 회전이 가능한, C-6 위치의 1 차 히드록시기가 있어서 CD 분자의 공동을 아랫쪽에서 가려막고 있다. CD 분자를 형성하는 글루코피라노오스단위 구조의 특성으로 인하여 CD 분자의 모든 히드록시기들은 분자의 외부로 향하고 있으며, CD 분 자의 공동내부는 C-H 에 의하여 형성된 두 개의 고리와 이 고리사이 에 글루코시드산소에 의해 형성된 고리로 이루어져 있다. 따라서 CD 분자의 공동내부는 소수성 (hy d rop h obic ) 이며 CD 분자의 공동입구는 히드록시기 때문에 친수성 (h ydro p h ili c) 이다. CD 분자를 구성하는 각 글루코피라노오스 단위가 광학활성인 키랄물질이므로 CD 분자는 광학 활성일 뿐만 아니라 CD 분자가 가지는 내부공동 또한 키랄공동이다. CD 분자의 특수한 구조적 특칭으로 인하여 CD 분자들은 CD 분자의 내부공동 크기에 적합한 많은 화합물들과 함께 내포 화합물(i nclus i on com p ound) 을 형성하며 특히 이들 화합물들이 라세미 화합물일 경우
표 3- 5 시클로덱스트린분자의 구조적 특 성 및 물리적 특 성 ( 이 표는 참고문헌 I 과 2 를 참 조하여 작성된 것임) 공동의 크기 ( A ) CD 글단위루의코 오스수 a b C 분자량 물25에°C ( 대g 한lO O용mL해- 도1) a-CD 6 5.7 13. 7 7.8 973 14.5 /3- CD 7 7.8 15.3 7.8 1135 1.8 y-C D 8 9.5 16.9 7.8 129 7 23. 2
 b
b
-C D 분자들은 부분 입체 이성질 착화합물 혹은 부분」 입체 이성질 내포 화합물을 형성한다. 이와 같은 특성으로 인하여 CD 분자들은 부분 입 체 이성질 내포 화합물의 재결정에 의한 광학분할(제 2 장 4 절 참조)에 이용되거나 액체 크로마토그래피에 의한 광학분할에서 키랄이동상 첨 가제 (제 2 장 6.3 . 2 철 참조)로 사용되어 왔을 뿐만 아니라 효소의 모델 화합물로서 여러 종류 반응의 촉매 혹은 비대칭 촉매로 사용되어 왔다 (3). 특히 CD 분자들을 키랄이동상 첨가제로 사용하여 액체 크로마토 그래피에 의한 라세미 화합물의 광학분할을 성공적으로 수행할 수 있 는 것은 이들 CD 분자들이 빠른 시간(확산지배 과정과 같은 속도)내에 가역적으로 입체 선택적인 내포 화합물을 형성할 뿐만 아니라 CD 분
자들은 넓은 p H 범위에 걸쳐 안정하며 독성이 없고 크로마토그래피에 사용되는 UV 검출기의 UV 범위내에서 빛을 흡수하지 않기 때문이다. 따라서 적절한 방법으로 CD 분자들을 실리카 겔과 같이 LC 고정상으 로 사용할 수 있는 고형지지체에 결합시킨다면 이들은- 키랄고정상으로 서 액체 크로마토그래피를 이용한 라세미 화합물의 광학분할에 사용될 수 있을 것이다. 3.2 시클로덱스트린 키랄고정상의 제법 및 특성 CD 분자를 키랄이동상 첨가제로 사용하여 액체 크로마토그래피상에 서 라세미 화합물을 광학분할하는 것이 많은 경우에 성공적이었지만, CD 분자들의 낮은 용해도로 인하여 이동상의 선택이 제한적이며 광학 분할시 CD 분자를 계속 공급해주어야 한다는 문제점들이 있다. 그러 나 CD 분자를 HPLC 컬럼의 충전제로 서용할 수 있는 고형지지체에 고정시켜 키랄고정상으로 이용할 경우에는 이러한 문제점들을 고려하 지 않아도된다. CD 분자를 기저로 한 키랄고정상으로는 CD 분자를 고분자화하여 겔로 만든 키랄고정상과 CD 분자를 교차결합시켜 얻어전 교차결합 CIHl 랄고정상 (4, 5, 6), 고분자화된 CD 분자를 직접 액체 크로마토그 래피의 컬럼충전제로 사용된 키랄고정상 등이 있으나 CD 고분자의 좋 지 않은 기계적 성질로 인하여 키랄컬럼충전제로서는 그리 성공적이 아니었다. 그러나 CD 분자를 실리카 겔에 고정시킴으로써 기계적 성 질이 좋은 키랄고정상을 제조하려는 노력이 많은 연구자들에 의하여 시도되었다. Fuji m ura 등은 실리카 겔과 3-((2 - 아미노에틸)아미노)프로필트리 메톡시실란을 톨루엔에서 환류함으로써 두 개의 아미노기를 가전 변형 된 실리카 겔을 얻을 수 있었고 이것을 1 차 히드록시기가 토실화된 a . 혹은 {3 -CD 와 반응시킴으로써 디아미노 형태의 C D.키랄고정상 l 을
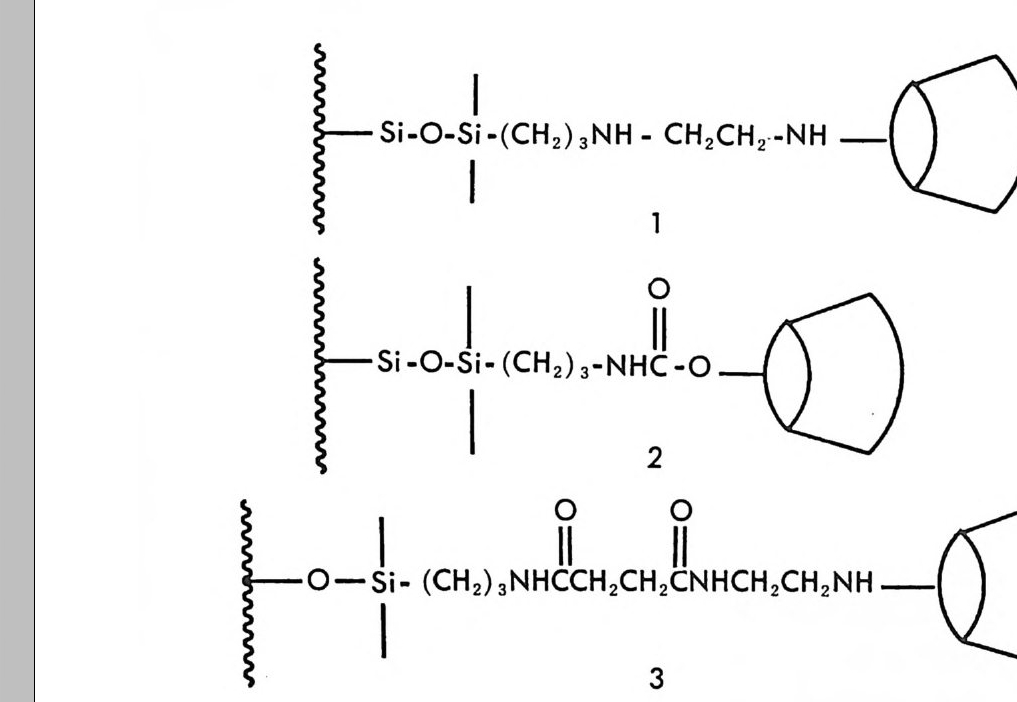
얻었으나 (7) 이 키랄고정상에서는 오로지 여러 종류의 방향족 구조 이 성질체의 분리만이 시도되었다. 그 후 Fu ji mura 등은 건조한 a 홈f-€:- [J -CD 와 3- 이소시아나토프로 필트리에톡시실란을 반응시켜 카르밤산 에스데르 형태의 트리에목시 실란 화합물을 만들고 이것을 건조한 피리딘 및 실리카 겔과 함께 환 류함으로써 카로밤산 에스테르결합을 가전 CD 키랄고정상 2 을 제조할 수 있었으며 CD 키랄고정상 2 에서 만델산 에스테르와 만델산의 페닐 기에여러치환체가치환된만델산에스테르를광학분할할수있었다 (8). Kawag uc hi 동은 3- 아미노프로필실리카 겔과 숙신산무수물 (sue- cinic anh y dr i de) 을 반응시켜 얻은 Su- 실리카 및 에틸렌디아민과 CD 를 반응시켜 얻은 일치환 a- 시클로덱스트린에틸렌디아민을 반응시켜 키랄고정상 g을 제조하였으나 방향족 화합물의 기하 이성질체 등을 분 리하는데 이용되었고 라세미 화합물의 광학분할에는이용되지 않았다 (9).
CD 키랄고정상 l, i, J은 모두 아미노기나 아미드기를 가지고 있 기 때문에 쉽게 가수분해될 뿐만 아니라 실리카 겔에 고정되는 CD 량 이 충분히 많지 못하며 존재하는 아미노기나 혹은 아미드기 등이 라세 미 화합물에 대한 입체 선택성에 영향을 미치는 문제 등을 내포하고 있다. 한편, CD 키랄고정상 l, ~. 앞虹다 더 광범위하게 사용되며 아미노 기나 아미드기를 포함하지 않는 CD 키랄고정상이 Armstr o ng 등에 의하여 개발되었다 00, 11). Arms t ron g은 3_ 글리시독시프로필실란과 실리카 겔울 반응시켜 3- 글리시독시프로필 실리카 겔울 얻은 후, 3- 글리시독시프로필 실리카 겔울 a-, /3절r-.g. y -CD 와 반응시킴으로 써 3- 글리시독시프로필 실리카 겔의 에폭시기가 CD 의 1 차 히드록시 기에 의하여 공격을 받아 에폭시고리가 열리면서 새로운 결합이 형성 되어 얻어진 CD 키랄고정상을 합성할 수 있었다(그림 3-19). Arms t ron g이 3 글리시독시프로필 실리카 겔과 여러 종류의 CD 를
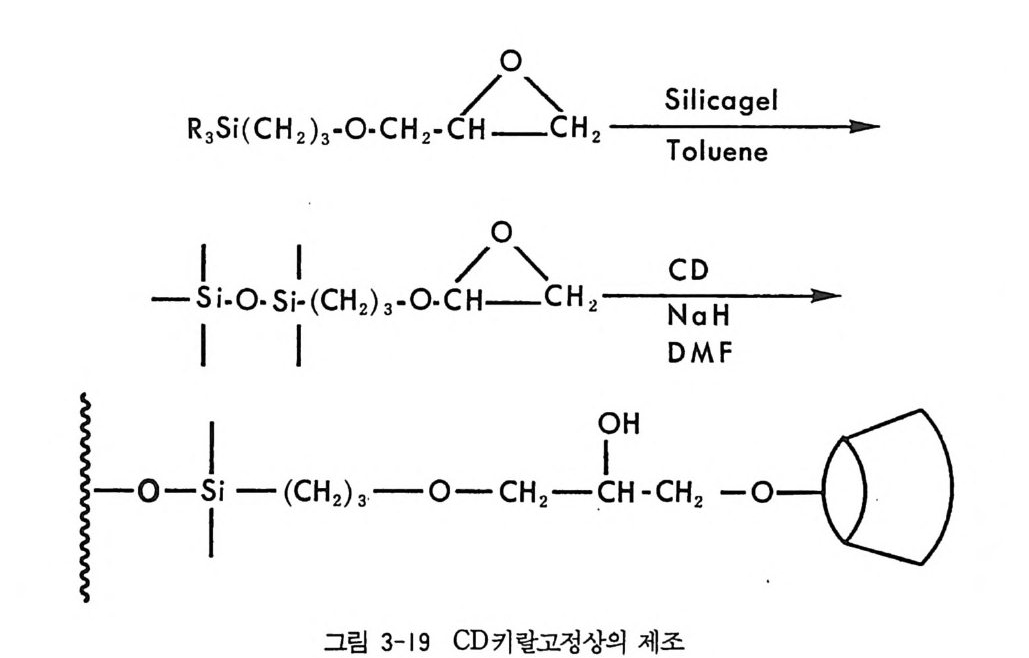 R3Si (C H2)3-0-CH2-C/H —°\CH 2 STio lliuc ae ng ee l
R3Si (C H2)3-0-CH2-C/H —°\CH 2 STio lliuc ae ng ee l
반응시켜 CD 키랄고정상들을 합성한 후 이들은 곧 상품화되었으며 현 재 상업적으로 구할 수 있는 CD 키랄고정상으로는 {3 - CD 를 기저로 한 키랄고정상인 Cy c lobnd I(/3 - CD 키 랄고 정상)과 모든 히드록시기가 완전히 아세틸화된 아세틸화 Cy c lobond I, y - C D-를 기저로 한 키랄 고정상인 Cy c lobond II(y - CD 키랄고정상), a - CD 를 기저로 한 키랄 고정상인 Cy c lobond III(a-CD 키랄고정상)와 모든 히드록시기가 완전 히 아세틸화된 아세틸화 Cy c lobond III 의 다섯 종류가 있다(제 9 장 참 조). 이들 다섯 종류의 CD 키랄고정상들은 결합된 CD 에 따라 표 3-6 과 같이 공동의 크기가 다르기 때문에 이들 C D:;기랄고정상들을 이용 하여 여러 종류의 다양하고 크기가 디른 라세마 화합물들을 광학분할 할수있을것이다. 표 3-6 상품화된 CD 키랄고정상 이름 형태 공동의 직경, A Cy cl obond I {3- CD 7.8 Cy cl obond Il y-C D 9.5 Cy cl obond III a-CD 5.7 Cy cl obond I Ac 아세틸화 {J- CD 7.8 Cy cl obond Ill Ac 아세틸화 a-CD 5.7 특히 완전 아세틸화된 /3 -CD 키랄고정상과 a-CD 키랄고정상은 이 들의 물리적 성질이나 착화합물 형성 등의 성질에 있어서 보통의 ¢ -C D? l 랄고정상이나 a - CD 키랄고정상괴는 현저히 다를 것이다. 예를 들면 아세틸화 CD 키랄고정상의 경우 보통의 CD 키랄고정상과 비교 하여 천수성이 감소하고 소수성이 훨씬 더 증가하게 될 것이며 CD 분 자의 공동입구에 있는 히드록시기의 수소결합능력에 현저한 변화가 있 게 된다. 따라서 라세미 화합물의 광학분할에 있어서도 아세틸화 CD
키랄고정상들은 보통의 CD 키랄고정상과는 다른 성질을 보이게 될 것 이다. 최근에는 /3 -CD 의 모든 히드록시기가 메틸화된 TM-/3- CD(hept ak is (2,3,6- t r i - O - me t hy l)- /3 -c yclodex t r i n) 를 옥타데실실리카 겔 컬럼 (ODS) 에 흡착시켜 일종의 키랄고정상으로 사용함으로써 바아비탈산 유도체와 만델산에스테르를 광학분할한 예가 보고된 바 있다 (12). 3.3 시클로덱스트린 키랄고정상을 이용한 광학분할의 예, 특성 및 키랄성 인지 메커니즘 CD 분자를 실리카 겔에 고정시킨 CD 키랄고정상을 이용한 광학분 할은 다양하다. CD 키랄고정상에서 분리가능한 라세미 화합물들은 대 부분 키랄중심탄소에 방향족기와 국성기를 함께 가진 화합물들이다. 이들 라세미 화합물들이 CD 키랄고정상에 의하여 광학분할되기 위하 여서는 키랄고정상을 이루는 CD 분자와 라세미 화합물 사이에 안정성 이 서로 다른 부분 입체 이성질 내포 화합물이 형성되어야 한다. 방향 족기를 가진 라세미 화합물이 CD 키랄고정상에서 광학분할될 때에는 라세미 화합물의 방향족기가 소수성인 CD 분자의 내부공동으로 끼어 들어가 가역적인 내포 화합물을 형성하는 것이 필수적이다. 방향족기 가 소수성인 CD 분자의 키랄공동에 끼어들어가 내포 화합물을 형성하 기 위하여서 는 이동상으로 물을 포함하는 용매 (역상)을 반드시 사용-하 여야 한다. 만일 정상 (normal p hase) 이동상을 사용하면 비극성인 용 매분자가 CD 분자의 키랄공동을 차지하므로 라세미 화합물과 내포 화 합물을 형성할 수 없으며 라세미 화합물은 오로지 CD 분자의 키랄공 동의부하고만 상호작용을 하게 되므로 일부 예의가 있기는 하지만 키 랄성 인지는 거의 일어나지 않는 것으로 알려져 있다 (14). 따라서 CD 키랄고정싱을 이용하여 광학분할할 경우에는 대부분 물과 적당한 유기 용매 (메탄올, 아세토니트릴, DMF, THF 등)를 적정한 비율로 혼합한
혼합용매를 이동상으로 사용한다. 보통은 물과 메탄올의 혼합용매가 많이 사용된다. 내포 착화합물이 형성될 때 CD 분자의 키랄공동 크기 및 라세미 화 합물의 크기 또한 아주 중요한 것으로 알려져 있다. 만일 라세미 화합 물의 크기 (특히 CD 분자의 키랄공동에 끼어들어 가는 방향족기의 크기)가 CD 분자의 키랄공동 크기보다 훨씬 크다면 내포 화합물은 형성될 수 없으며 라세미 화합물의 크기가 CD 분자의 키랄공동보다 훨씬 작으면 내포 착화합물이 형성되기는 하나 키랄공동내에서 키랄분자가 일정한 위치를 유지하지 못하고 자유로이 움직일 수 있으므로 키랄성 인지에 불리하게 된다. 그러나 서로의 크기가 맞아 꼭 맞는 내포 착화합물 (tigh t inc lusio n com p lex) 을 형성한 경우에는 키랄분자가 CD 분자의 키랄공동내에서 마음대로 움직일 수 없고 일정한 위치를 유지하기 때 문에 키랄성 인지에 더 유리하다고 할 수 있다. 예를 들면 나프탈렌기 는 a_CD 의 키랄공동에 끼어들어가기에는 너무 크나 /3 -CD 에는 쉽게 끼어들어가 그림 3-20 에 보는 바와 같이 가역적인 내포 화합물을 형 성할수있다. 방향족기를 가전 키랄물질의 방향족기가 CD 분자의 키랄공동으로
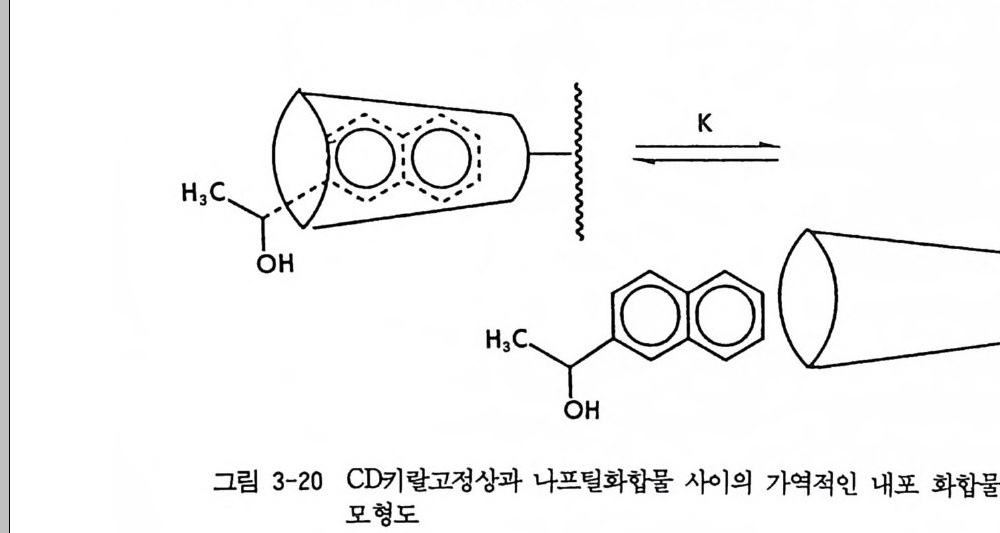 H3C 1~
H3C 1~
끼워들어가 내포 착화합물을 형성하는 것만으로는 키랄성 인지가 가능 하지 않다. 라세미 화합물의 방향족기가 CD 분자의 키랄공동에 끼어 들어가 가역적인 내포 착화합물을 형성하는 것과 동시에 라세미 화합 물의 키랄중심에 있는 극성기가 CD 키랄고정상 CD 분자의 키랄공동 입구에 일정한 방향을 유지하고 있는 C-2 히드록시기나 C-3 히드록시 기와 이성질체의 절대배열에 따라 다른 상호작용(대부분의 경우 수소결 합, 극히 일부분의 경우 입체반발 상호작용)을 함으로써 두 광학 이성질 체의 구별이 가능하게 된다(1 4). CD 키랄고정상에 의한 키랄성 인지의 예는 여러 종류의 라세미 의 약품들을 성공적으로 광학분할한 Arms t ron g의 연구에서 찾아볼 수 있다(1 5). Armstr o ng 등은 /J -CD 키랄고정상에서 메탄올과 물 혹은 아세토니트릴과 물의 혼합용매를 이동상으로 사용하여 표 3-7 에 제시 한 /J - 차단제, 진정제 등의 라세미 의약품들을- 광학분할할 수 있었다. 특히 이 연구에서는 /J - CD 키랄고정상에서 프로프라놀롤(p ro p ranolol) 의 광학분할시에 생성되리라고 예상되는 가장 안정한 두 개 의 부분 입체 이성질 내포 화합물의 컴퓨터 그림으로부터 키랄공동에 프로프라놀롤의 나프틸기가 똑같은 위치에 끼어들어가는 것 의에 프로 프라놀롤의 극성기와 /J -CD 분자의 키랄공동입구에 있는 히드록시기 사이의 상호작용에 의하여 키랄성 인지가 일어남을 분명히 밝혀주고 있다(1 5). 그립 3-21 의 컴퓨터 그림에 의하면 d - 프로프라놀롤과 l- 프 로프라놀롤의 나프틸기는 모두 CD 분자의 키랄공동에 똑같은 모양으 로 위치하고 있으며 d- 나 l- 프로프라놀롤의 키랄탄소에 붙어 있는 히 드록시기는 똑같이 CD 분자의 3- 히드록시기와 수소결합을 하고 있다. 그러나 d_ 프로프라놀롤의 ~}-ol-민과 CD 분자의 C-2 히드록시기 및 C-3 히드록시기 사이의 거리는 수소결합하기에 알맞는 3.3A 과 2.8 A 인 반면, d- 프로프라놀롤의 2 차아민과 CD 분자의 C-2 히드록시기 및 C-3 히드록시기와의 가장 가까운 거리는 3.8A 과 4 . 5A 이다. 따라서 d- 프로프라놀롤이 l- 프로프라눌롤에 비하여 CD 분자와의 상호작용이
표 3-7 /3 -CD 키랄고정상에서 광학분할된 라세미 의약품의 예 (참고문헌 : | 5) 의약품 /3-차단제 프로프라놀롤 (pr op r anolol) , 메토프롤롤 (meto p r olol) 항히스타민제 클로로페니 라민 (chlorop he nir a mi ne ) 칼슘~}단 X1] 베라파밀 (verap a mi l) , 니솔리디펜 (nis o lid i p e ne) , 니모디펜 (nim odip e ne) 이뇨제 클로르탈리돈 (chlorth a li do ne) 진정제 핵소바아비탈 (hexobarbit al) , 메포바아비탈 (mep o barbit al) , 메페니토인 (mep h eny toi n ) , 트리아졸린 (tri a z olin e ) , 펜숙시이미드 (ph en- suxim i de ) 항코르티코스테로 01 드제 아미노글루테티미드 (ami no g lu te t h im i de ) l:l l 스테로 01 드계소염제 케토프로펜 (keto p r ofe n ) 마취제 메타돈 (meth a done) 중추신경자극제 메 틸페니데 이트 (meth ylp h enid a te ) 더 좋아 키랄컬럼상에 더 오래 머무름을 쉽게 짐작할 수 있다. 이와 갇은 결론은 제 2 장에서 논의한 세 점 상호작용 모델과 유사하다고 할 수있다. 라세미 화합물의 방향족기가 CD 분자의 키랄공동에 끼어들어가 내 포 착화합물을 형성하기는 하나 라세미 화합물과 CD 분자 사이에 다 른 상호작용이 없어서 키랄성 인지가 일어나지 않는 경우는 라세미 의 약품인 와파린 (warf ar in ) 뜬 1 광학분할에서 볼 수 있다. {3 -CD 키랄고
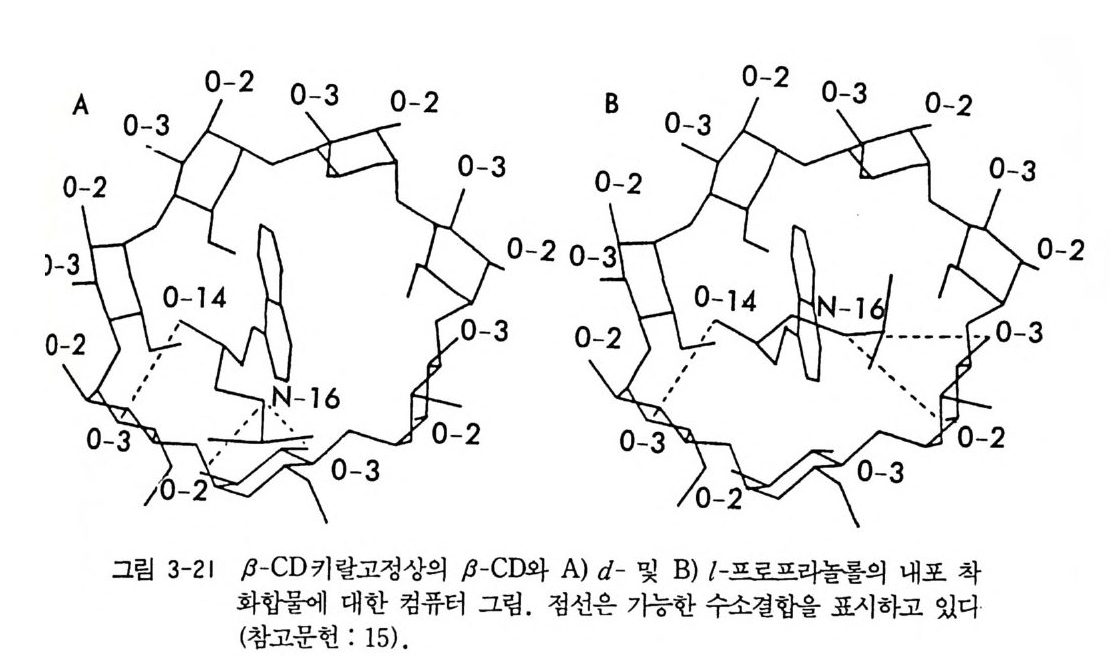 B 0, -2 0-3
B 0, -2 0-3
정싱에서 와파린은 광학분할되지 않음이 알려졌다. {J -CD 와 와파린에 의한 내포 착화합물의 컴퓨터 그립에 의하면, 와파린의 쿠마린고리에 있는 히드록시기와 곁사슬로 나와 있는 케론기가 헤미케탈을 형성한 상태로 CD 분자의 키랄공동에 끼어들어가나 키랄중심의 페닐기는 키 랄공동에서 너무 멀리 떨어져 있어서 키랄공동과 아무 상호작용도 할 수 없으므로 두 광학 이성질체의 광학분할이 불가능한 것으로 생각되 고 있다(1 5). 라세미 아미노산들은 대부분의 경우에 있어서 CD 분자의 키랄공동 에 꼭 끼어들어 맞기에는 너무 작기 때문에 CD 키랄고정상에서 광학 분할되지 않는다. 그러나 아미노산을 g-나프릴에스테르 §나 g-나프 틸아미드 § 혹은 단실 (dans y l) 유도체 1로 만들었을 때는 표 3-8 에 보 는 바와 같이 {J -CD 키탈고정상에서 광학분할이 잘되고 있다(1 0, 16). 표 3-8 에서 분명한 것은 아미노산의 카르복시기나 혹은 아미노기를 나프틸기의 유도체로 만들었을 때 광힉~ 잘되는 반면 N- 벤조일
 。 Yc5H 3
。 Yc5H 3
아르기닌 /3-나프틸아미드의 광학분할에서 보는 것처럼 카르복시기와 아미노기를 둘 다 다른 유도체 (하나는 나프틸기를 포함하는 유도체기)기 로 만들었을 때는 광학분할이 훨씬 감소한다는 것이다. 이것으로부터 라세미 화합물의 광학분할을 위하여서는 라세미 화합물은 /3 -CD 분자 의 키랄공동입구에 존재하는 C-2 히드록시기 혹은 C-3 히드록시기와 표 3-8 {J -CD 키랄고정상에서 아미노산 유도체들의 광학분할, 유속 : l.Om L/mi n 용리순 서 : 항상 D- 이성질체가 L --0|성질체보다 키랄고정상에 더 오래 머무름 (참고문 헌 : 10). 아미노산유도체 k1, k2, a’ 이동상(에탄올:물) 알라닌 /3_나프틸아미드 5.1 6.1 1.20 50 : 50 알라닌 /3-나프틸에스테르 1.0 1.8 1.80 50 : 50 단실 페닐알라닌 3.1 3.8 1.23 55 : 45 단실루이신 3.0 4.2 1.40 50 : 50 메티오인 /3-나프틸아미드 2.7 3.6 1.33 50 : 50 미N-드 벤 조일아르기닌 /3-나프틸이 28.5 30.4 1.07 50 : 50
수소결합을 할 수 있는 작용기를 가져야 광학분할이 잘될 수 있으리라 는 것을 짐작할 수 있다 (10) . /3 -CD 키랄고정상에서 나프틸기를 포함하는 유도체로 만든 아미노 산들이 광학분할되는 반면에 키랄공동의 크기가 훨씬 작은 a-CD 키 랄고정상에서는 방향족기를 가지는 아미노산들 예를 들면 트리프토판 (try p top h an) , 페닐알라닌, 티로신 (tyros in e ) 등이 유도체로 만들지 않고 직접 광학분할된다 (l7) . 그림 3-22 에서 보는 것처럼 6 국문르오르 트리프로판과 O- 메틸티로신은 트리프토판과 티로신에 비하여 머무름 시간이 증가하였을 뿐만 아니라 입체 선택성 (a 값) 및 분해능 (Rs 값)도 아주 좋아졌음을 알 수 있다. 이것은 아미노산의 방향족기 소수성이 증가함으로 인하여 방향족기가 a-CD 분자의 키릴공동으로- 끼어들어 갈 때 내포 화합물을 더 잘 만들기 때문이라고 예측할 수 있다. 방향족기를 가지는 여러 종류의 라세미 히단토인 (h y dan t o i n) 을 ¢ -CD 키랄고정상에서 광학분할한 Mag u ir ~ 연구결과는(1 8) Arm- s t ron g에 의한 연구결과와 마찬가지로 히단토인의 방향족기가 CD 분
 B C
B C
그림 3-22 a-CD 키랄고정싱에서 티로신 (A) ,페닐알라닌 (B) ,트리프토판 (C), 6~ 운 己 ?코 드리프토판 (D), 0- 메틸티로신(E펴 광학분할. 컬럼길이 : 25cm, 유속 : 0.5mL/mi n, 이동상 : 1% 의 트리에틸암모늄아세데이트 수용액 (pH , 5.1 ) (참고문헌 : 17)
자의 키랄공동에 끼어들어가 내포 착화합물을 형성함과 동시에 CD 분 자의 키랄공동입구의 히드록시기와 라세미 화합물의 극성기 사이의 상 호작용예 의하여 키랄성 인지가 이루어지고 있음을 보여주고 있다. 표 3-9 에서 보는 바와 같이 5- 위치의 알킬기 길이룰 증가시킴에 따라 a 표 3-9 /3 -CD 키랄고정상에서 라세미 히단토인의 광학분할(참고문현 : 18)
 X
X
R 화합R물' X k' a’ Rs 이동상(물/에탄을) H H H 0.67 1.09 0.2 90/10 CH3 H H 1.17 1.11 0.5 90/10 C2Hs H H 2.30* 1.21 2.0 90/10 n-C3 H1 H H 3.30 1.26 2.2 90/10 2-C3H1 H H 4.96 1.27 2.5 90/10 n-C ◄ H9 H H 4.40 1.14 1.1 90/10 C2Hs CH3 H 2.15* 1.47 4.2 90/10 C2Hs C2Hs H 2.18 1.26 2.2 90/10 C2Hs 2-C3H7 H 2.88 1.00 。 90/10 C2Hs H OH 2.87* 1.22 2.3 90/10 n-C3H1 H OH 5.98 1.31 3.6 90/10 * (R)-( -)-이성질체가 먼저 분리되어 나옴
값 및 머무롬 시간이 증가하나 5- 알킬기의 길이가 더 길어지면 (예를 들면 n -lj내) 머무롬 시간과 a 값이 감소함을 알 수 있다. 5- 알킬기의 길이가 길어짐에 따라 5- 알킬기와 /3 -CD 의 소수성 내부사이의 소수성 상호작용이 증가하여 머무롬 시간 및 a 값이 증가하는 것으로 예상되 나 5 - 알킬기가 너무 길어지면 분자모델에 의한 연구결과, 이것은 히 단토인과 /3 - CD 의 2 차 히드록시기 사이의 수소결합을· 방해하기 때문 에 오히려 머무름 시간과 a 값이 감소하는 것으로 생각되고 있다. 히 단토인 3 _ 위치의 알킬기 길이를 증가시키면 광학분할이 급격히 감소 하거나 광학분할이 전혀 이루어지지 않음을- 표 3-9 에서 알 수 있는데 이 때에도 분자모델의 연구에 의하여 3- 위치의 큰 일킬기가 히단토인 고리와 CD 분자사이의 수소결합을 방해하여 이러한 결과가 관찰되는 것으로 생각되고 있다. 특이한 것은 히단토안의 페닐기의 p-위치에 히드록시기가 치환되어 있는 경우에는 머무름 시간이 증가할 뿐만 아 니라 키랄 선택성 (a 값)도 증가하고 있음을 알 수 있는데 이것은 히단 토인의 페닐기의 p-위치에 히드록시기가 치환됨으로 해서 p-위치의 히드록시기와 /3 -CD 분자의 키랄공동 하단에 있는 일차 히드록시기가 수소결합을 형성하여 더 안정한 내포 착화합물을 형성하기 때문인 것 으로 (그립 3- 23 ) 생각된다.
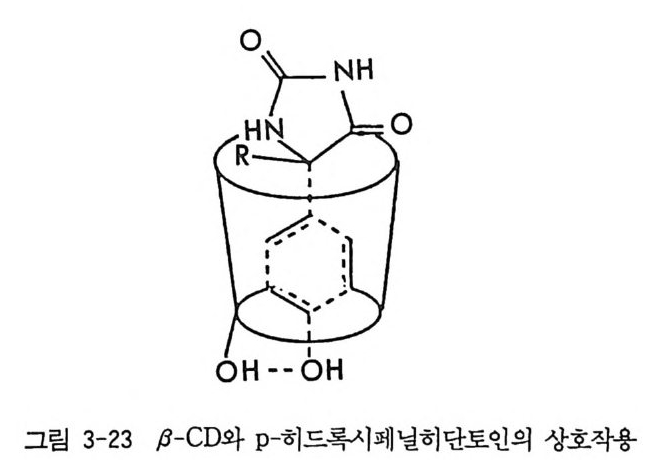 NH
NH
방향족기를 가지는 히단토인과 같은 디중고리 라세미 화합물의 광학 분할에 있어서 Armstr o ng 등은 키랄중심이 고리의 일부분인 화합물 들과 키랄중심이 비고리부분에 있는 화합물들로 나누어 광학분할의 양 상을 조사함으로서 CD 키랄고정상에서의 광학분할에 대한 라세미 화 합물 구조의 영향을 제안한 바 있다(1 9). 이 제안에 의하면 라세미 화 합물이 적어도 두 개 이상의 고리를 가지며 특히 이중 적어도 하나의 고리가 방향족고리일 때 CD 키랄고정상에서 광학분할이 잘되며 다중 고리 라세미 화합물의 키랄중심이 두 개의 방향족고리 사이에 있거나 혹은 하나의 방향족고리와 카르보닐기 사이에 있을 때 광학분할이 잘 된다. 특히 키랄중심이 이소퀴놀린화합물 §과 같이 접합 두 고리계의 일부가 될 때 광학분할이 아주 잘됨을 관찰할 수 있었다 (a = 2.3 4 , Rs = 3.0 ) (19).
 NHCH3
NHCH3
[3 -CD 키랄고정상에서 메탈로센 (meta l locene) 라세미 화합물의 광 학분할에 대한 Arms t ron g의 연구는 광학 이성질체의 구조에 조금만 변화가 있어도 [3 -CD 키랄고정상의 광학분할능에 크게 영향을 미치는 예들을 제시하여 주고 있다 (20). 그립 3-24는 /3 -CD 키랄고정상에서 메탈로센 라세미 화합물들이 광학분할된 예를 보여주는 크로마토~램 울나타낸것이다. 그립 3-24 에서 보는 메탈로센 화합물들이 광학분할이 잘되는 반면, (+)-a 페로세닐벤질알코올 (a- fe rrocen y l benzy lz lcohol) ~근 광학분 할되지 않는다. 아마도 키랄탄소에 붙어 있는 히드록시기가 입체칭애
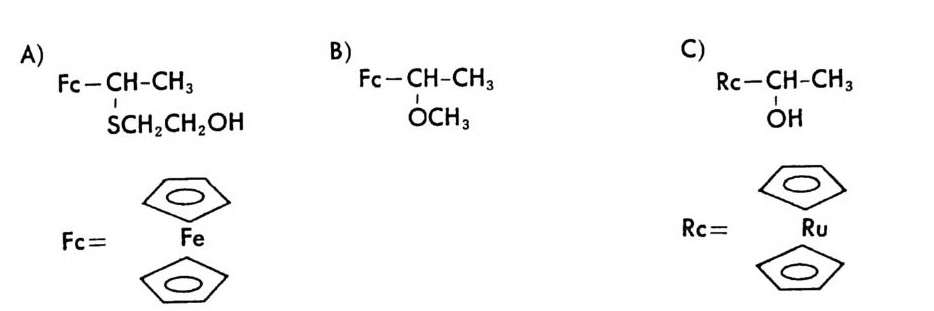 A) Fc-CSHC-HC2 HC 3H 20H B) Fe— CO’HC H-C3H 3 C) Rc-COHH- C H3
A) Fc-CSHC-HC2 HC 3H 20H B) Fe— CO’HC H-C3H 3 C) Rc-COHH- C H3
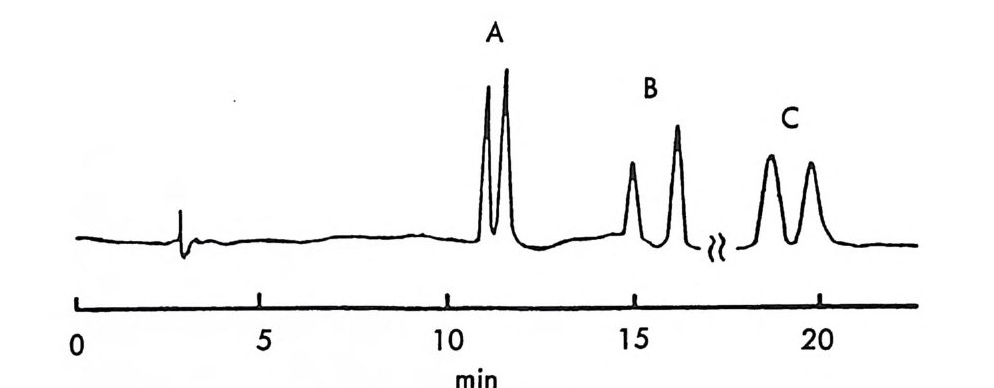 A
A
그림 3-24 /3 - CD 키랄고정상에서 메탈로센의 광학분할. 컬럼 : 25x 0 .4 c m, 이동 상 : 메탄올풍 H70 : 30) , 유속 : lmL/ m i n (참고문헌 : 20) 가 큰 페로센 (fe rrocene) 과 페닐기 사이에 숨어 있어서 CD 분자의 2 차 히드록시기와의 수소결합이 방해받기 때문인 것으로 생각된다. 그 러나 키랄탄소의 히드록시기를 티오에탄올로 치환하였을 때 (화합물 1Q) 키랄탄소에 있는 히드록시기가 주위의 입체장애가 큰 치환기 밖으 로 뻗어 나올 수 있어서 CD 분자의 2 차 히드록시기와 수소결합이 가 능하게 되어F 광e학?-분 할0이 일 어난다고 생각할 수 있다. CH-t -0 OH a=l.00 SCH2CH20H a=l .06 9 10
라세미 크라운 에데르의 광학분할에 있어서도 크라운· 에데르의 고리 크기에 따라서 또는 크라운 에데르 고리를 구성하는 원소의 종류에 따 라 /3 -CD 키랄고정상에서 서로 다른 양상의 광학분할을 보이고 있음 을 볼 때 (21), 광학 이성질체 구조의 조그마한 변화가 키랄성 인지에 큰 영향을 미친다는 사실을 다시 한번 확인할 수 있다. CD 키랄고정상에서의 광학분할은 지금까지 기술한 것 의에도 방향 족 카르복실산의 광학분할 (22, 23) ,니코틴 및 니코틴유사체의 광학분할 (24), 정신질환치료제로 쓰이는 라세미 의약품인 노미펜신 (nom ife n s i ne) 의 광학분할 등 (25) 다양하다. 특히 생물학적 시료에 존재하는 라세미 의약품인 데르부탈린 (t erbu t a li ne) 뵤을 /3 -CD 키랄고정상에서 직접 광학분할할 수 있었던 연구결과는 (26) 앞으로 라세미 의약품의 생체내 변화과정을 추적할 때 키랄고정상을 이용할 수 있다는 점에서 유의할 만하다. 또한 아세틸화된 /3 -CD 키랄고정상에서의 (土) - 노르 제스트렐 (nor g es t rel) Jl의 광학분할은 키랄성 인지 메커니즘의 관점 에서 홍미있는 연구결과이다 (27). (土)-노르제스트렐이 보통의 CD 키 랄고정상에서 광학분할이 되지 않는 것은 CD 키랄고정상과 (士)-노르
 HOH 戶O_아H c H 2 N H ' cIHHfCalH I3 C c。o H OH' , · C 三 CH
HOH 戶O_아H c H 2 N H ' cIHHfCalH I3 C c。o H OH' , · C 三 CH
제스트렐이 내포 착화합물을 형성하였을 때 노르제스트렐의 키랄중심 과 국성치환기가 CD 키랄공동의 입구에서 너무 멀리 떨어져 있어서 국성치환기가 CD 분자의 C-2 히드록시기 혹은 C-3 히드록시기와 수 소결합을 할 수 없으므로 광학분할이 되지 않으나 fJ _CD 분자를 아세 틸화하여 CD 분자의 수소결합부위를 확장하면 아세틸화 CD 분자와 노르제스트렐사이의 수소결합이 가능하여 광학분할이 되는 것으로 생 각되고 있다. CD 키랄고정상은 라세마 화합물의 광학분할에 이용될 수 있을 뿐만 아니라 부분 입체 이성질체의 분리, 구조 이성질체의 분리, 기하 이성 질체의 분리 등 다양한 종류의 이성질체 분리에 이용될 수 있다. 이들 이성 질체들의 분리의 유용성을 보이기 위하여 그림 3-25 에 프로스玉타 글란딘의 하나인 시스 _ 와 트란스 - 시프로스텐 (cip ro ste n e) 냐의 분리 예
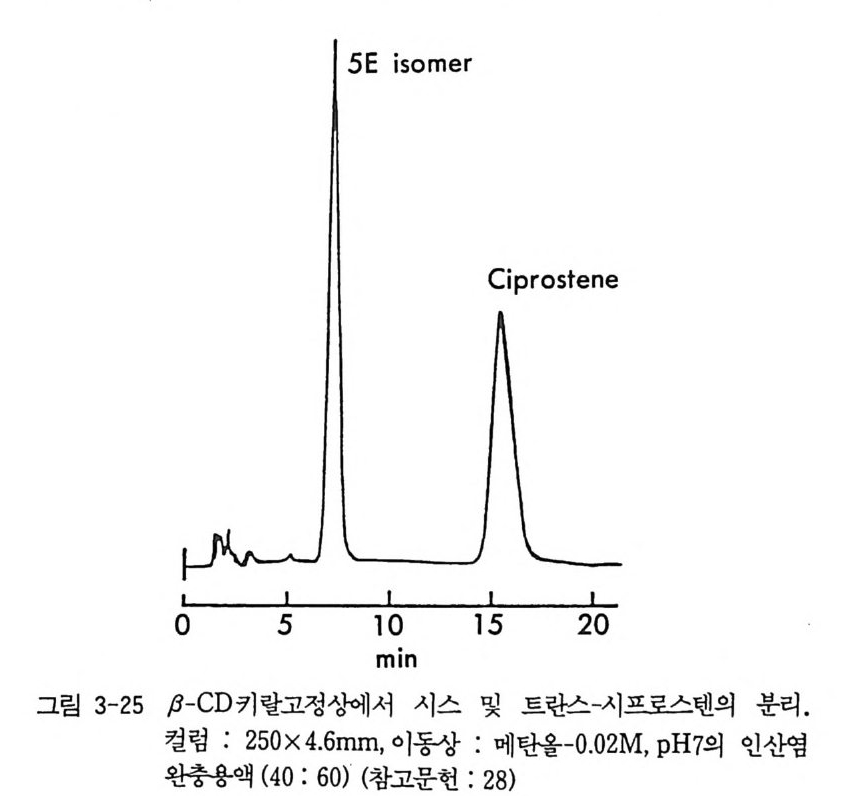 5E iso mer
5E iso mer
를 제시하였다 (28). 그림 3-25 에서 보는 것처럼 시스 및 트란스-이성 질체가 완전히 분리되는 것을 알 수 있으며 따라서 유기합성과정에서 생성되는 시스- 혹은 트란스-시프로스텐의 순도를 측정하는 데에도 CD 키랄고정상은 효과적으로 응용될 수 있을 것이다. CD 키랄고정싱을 이용한 라세미 화합물의 광학분할뿐만 아니라 기 하 이성질체의 광학분할, 부분 입체 이성질체의 광학분할, 스데로이 드 에피머의 광학분할 등 CD 키랄고정싱을 이용한 이성질체의 분리연 구들을 총망라한 Armstr o ng 등의 최근 총설논문을 참고하면 더 다양 한 응용의 예를 찾아 볼 수 있을 것이다 (29). 3.4 광학분할에 영향을 미치는 요인들 CD 키랄고정상에서 광학분할은 대부분 물과 적당한 유기용매의 혼 합물을 이동상으로 사용한 역상 (reverse p hase) 크로마토그래피에 의한 광학분할이다. CD 분자와 라세미 화합물사이에 형성된 내포 화합물은 CD 분자의 소수성인 키랄공동에 라세미 화합물이 끼어들어가서 생긴 것으로 이동성에 물이 많이 포함되면 많이 포함될수록 키랄공동에 끼 어들어간 화합물의 결합력 (소수성 결합)은 커지고, 따라서 보통의 역 상 크로마토그래피에서와 마찬가지로 머무름 시간은 증가하게 되며 이 동상의 조성에 따라 분리인자 a 값도 큰 영향을 받는다. 그립 3-2~ 본 물과 혼합되어 있는 유기용매인 메탄올의 조성에 따른 머무름 시간 (k'), 분리인자 (a), 분해인자 (Rs) 의 변화를 제시한 것이다(1 6). 따라 서 유기용매의 양을 적절히 조절함으로써 광학분할에 가장 적합한 이 동상의 조성을 결정할 수 있게 될 것이다. 물과 함께 사용 가능한 유기용매들 즉 DMSO, DMF, 아세토니트 랄 에탄올, 메탄을들 중 어느 유기용매를 물과 함께 사용하는 것이 광학분할에 가장 좋은가 하는 것을 결정하기는 어렵다. 그러나 대부분 의 발표된 연구에 의하면 물과 메탄올의 혼합용매를 이동상으로 사용
35% MeOH Co) 0일 。 그림 3-26 /3 -CDR— s키 =랄 고정7T상 ,m에. 'm서J 2 핵5 소바『言開鬪“巨0·%아 비H탈 (hex2ob Rarsb=i信 巨〈it1 a.l2) 의 H 광학분할. l0 10cm /3 -CD 컬럼, 유속 : l.Om L/mi n, 온도 : 2'l C , 이동상 : 메탄을/ 물(참고문헌 : 16) 하여 광학분할을 성공적으로 수행한 山예가 많이 있으므로 우선은 메탄 고올있다자과. 물할따이 라때 서혼에 합는C용 D우매 키선를랄 고물이정 동: 상메상을탄으 올로이 용=선昌하 5택0여 하 : 5는라0 인 세것 미이혼 합화일용합반매물적를을이 라광이고학동 분상할할으 수하로 사용하여 광학분할을 시도하고 머무름 시간 등을 조정하고자 할 때에 는 혼합용매의 조성을 조금씩 바꾸는 것이 바람직하다고 할 수 있다. 특히 아세토니트릴이나 에탄올은 메탄올에 비해 CD 분자의 공동에 대 한 친화성이 훨씬 좋은 것으로 알려져 있다 (27, 30). 이 때문에 메탄올 대신 아세토니트릴이나 에탄올을 유기용매로 사용할 경우에는 소량의 아세토니트릴이나 에탄올을 사용하여도 메탄올을 사용하였을 때와 같 은 효과가 있음이 알려져 있으며 (16) 머무름 시간을 감소시키거나 입
체 선택성 (a 값)을 증진시키기 위하여 아세토니트릴을 소량 첨가한 메 탄올이 유기용매로 사용되거나 혹은 메탄올 대신 아세토니트릴과 물을 적당한 조성으로 섞은 혼합용매를 광학분할의 이동상으로 사용하기도 한다(1 6). 이온화할 수 있는 작용기를 가진 라세미 화합물을 CD 키랄고정상에 서 광학분할할 때에는 이동상의 p H 를 변화시키거나 이동상에 염을 가함으로써 라세미 화합물과 CD 분자 사이의 상호작용(수소결합 등)을 변화시키고 따라서 입체 선택성을 중전시킬 수 있으며 머무름 시간을 조정할 수 있다. 라세미 카르복실산의 광학분할에서는 보통 인산염완 충용액과 아세토니트릴 혹은 메탄올을 적절히 혼합하여 p H 가 조정된 이동상을 사용한다 (22, 23). /3 -CD 키랄고정상에서 이동상의 p H 가 4.2 일 때 만델산은 광학분할되나 p H6 . 5 에서 광학분할현상이 완전히 사라지는 것은 이동상의 p H 가 CD 키랄고정상의 입체 선택성에 영향 울 미치고 있음을- 보여주는 예이다 (22). 이동상에 흔히 가미되는 염은 트리에틸암모늄아세테이트 (TEAA) 로서 부식성이 거의 없을 뿐만 아 니라 p H 가 조정된 완충용액으로 만들어져 다른 유기용매와 섞어 이 동상으로 사용된다 (17, 25) . 광학분할에 대한 이동상 pH 영향의 예는 니코틴유사체 화합물들의 광학분할에서도 찾아볼 수 있다. 니코틴유사체 화합물들은 보통 pH =7.l(TEAA 완충용액)에서 광학분할이 잘되나 p H 가 감소함에 따라 입체선택성 (a 값)이 감소하며 pH < 5 .0이 하에서는 광학분할이 전 혀 관찰되지 않음이 확인되었다 (24). 그림 3-27은 /3 -CD 키랄고정상 에서 (S,R) -노미펜신 브의 말레산염 ( (S,R)-nomi fen sin e hy d rog en malea t e) 의 광학분할에 대한 p H 의 영향을 나타낸 그래프로서 용량인 자 (k'), 분리인자 (a), 분해인자 (Rs) 가 p H에 영향을 받고 있음을 알 수 있다 (25). 이동상의 p H 를 조절할 때에는 실리카 겔과 CD 분자를 연결하는 공유결합부분을 보호하기 위하여 보통 p H 를 7.5 이하로 유 지하여야 하며 p H 가 3.0 이하일 때는 CD 분자가 분해될 수 있으므로
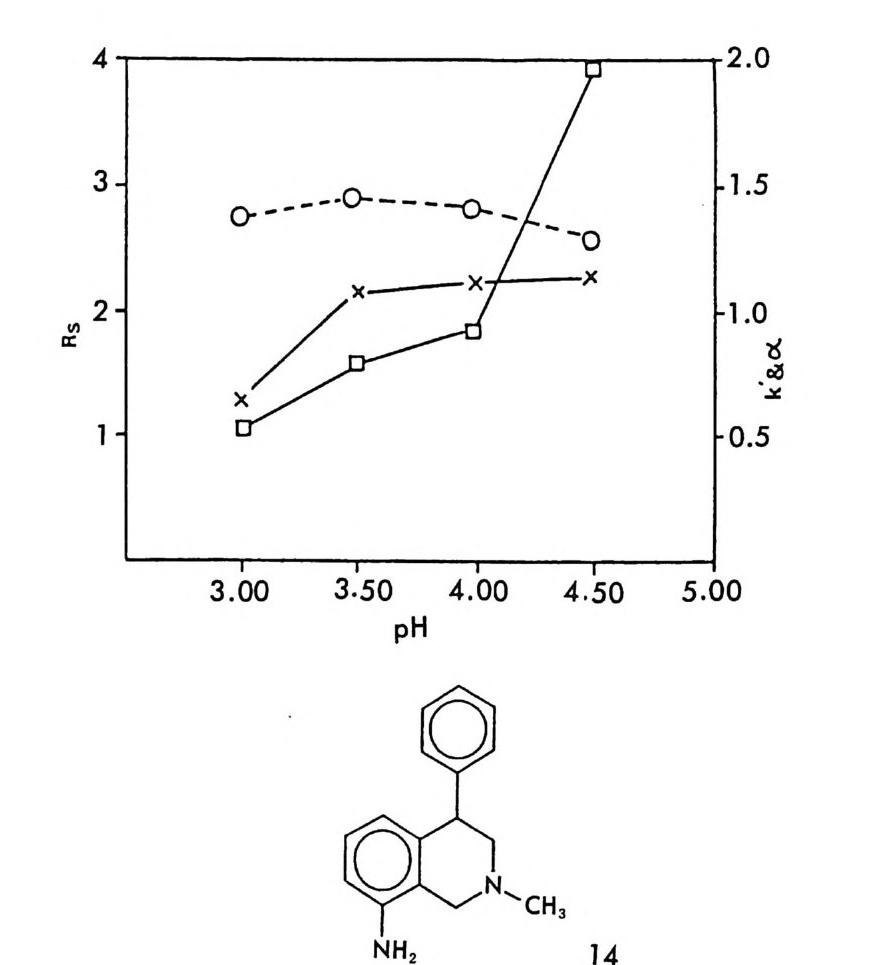 4 2.0
4 2.0
그림 3-27 /3 -CD 키랄고정상에서 (S, R)- 노미펜신 말레산염 (S, R)-nomi fen - sin e hy d rog en malea t e) 의 광학분할에 대한 p H 의 영향. 이동상 : TEAA완 충용액 (pH , 3-4 .5 ) , 20% 메탄을 유속 : 0.5 m L/m in, 온도 : 2 :r c, □ = 용량인자, 0= 분리인자, X= 분해인자(참고문헌 : 25) CD 키랄고정상을 이용할 때 사용 가능한 이동상의 p H 범위는 보통 3.0 〈p H 〈 7.5 이다 (25). 이동상으로 물과 유기용매의 혼합용매를 사용하는 것 의에 극히 일 부분이기는 하나 아세토니트릴을 이동상으로 하거나 핵산 : 2-프 로판 올의 혼합용액을 이동상으로 사용하는 정상 (normal p hase) 크로마토그
래피에 의한 CD 키랄고정상에서의 광학분할 예 들 이 보고되어 있다 (24 , 31 ). 유기용매를 이동상으로 사용할 때는 유기용매가 CD 분자의 . 키랄공동을 차지하기 때문에 역상 크로마토그래피에서 광학분할이 일 어나는 키랄성 인지 메커니즘과는 디른- 메커니즘으로 광학분할이 일어 난다고 예측된다. 예를 들면 물과 아세토니트릴 혼합용매 를 이동상으 로 사용하여 니코틴 유도체들을 광학분할할 때 아세토니트릴의 양을 증가시킴에 따라 용량인자와 분리인자가 점점 감소하다가 아세토니트 릴의 양이 60% 와 80% 사이에서 최소가 되며 아세토니트 릴 양이 80 %보다 증가함에 따라 다시 용량인자와 분리인자가 증가하 는 것을 그 림 3-28 에서 볼 수 있다(용리순서에는 변화 없음) (24 ) . 반면 아세토니 트릴 대신 메탄올을 물과의 혼합용매로 사용하였을 때는 메탄올의 조 성이 100% 에 이를 때까지 용량인자와 분리인자는 계속 감소함을 알
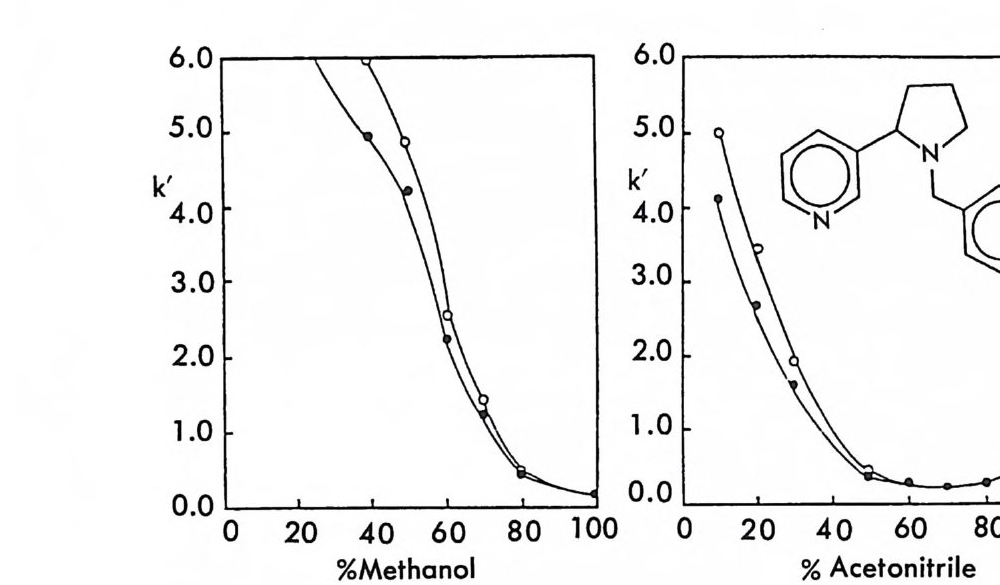 6.0 6.0
6.0 6.0
. 그림 3-28 /3 -CD 키랄고정상에서 라세미 N' -벤질노르니코틴 (N'-benzy l no mi co tin e) 의 광학분할에 대한 이동상의 영향. (0) : (R)-(+) - 이 성질체, (e) : (S)-( 一)-이성질체 , 컬럼 : 250 x 4.6mm, 유속: l. OmL/m in ( 참고문헌 : 24) •
수 있다 (24). 아세토니트릴을 사용한 경우 용량인자의 변화양상은 CD 분자에 의한 키랄성 인지 메커니즘이 변화하고 있음을 시사하여 주고 있다. 이때의 키랄성 인지 메커니즘은 용매가 CD 분자의 키랄공동을· 차지하므로 용질분자(라세미 화합물)가 내포 착화합물을- 형성하는 것 이 아니고 CD 분자의 히드록시기와 라세미 화합물의 극성기 사이의 입체 선택적인 상호작용에 의하여 키랄성 인지가 이루어진다고 예상될 뿐 아직 정확한 키랄성 인지의 메커니즘은 모르는 실정이다. 정상 크로마토그래피에 의한 CD 키랄고정상에서 광학분할의 특수한 예는 준임계 유체 크로마토그래피 (Subcriti ca l Fluid Liq u id Chromato - grap h y : SubFC) 에 의 한 포스핀옥시드 (ph osph in e oxid e ) 의 광학분할이 다 (31). 특히 최근 SubFC 에 의한 광학분할은 키랄고정상의 응용범위 를 확장하고 빠른 시간내에 광학활성 물질의 광학조성을 측정하는 새 로운 HPLO 키 분석수단을 제공한다는 의미에서 크게 관심을 끌고 있 다 (32). 준임계 상태의 CO2 와 메탄올의 혼합용액을 이동상으로 사용 하여 p-CD 키랄고정상에서 포스틴옥시드의 광학분할을 시도하였을 때 (SubFC) 그림 3_29 에서 보는 바와 같이 좋은 광학분할을 보여주고 있다 (31). 특이한 점은 핵산 : 2- 프로판을을 이동상으로 하는 정상 크 로마토그래피 (정상 LC) 의 광학분할과 비교하여 SubFC 에 의한 광학 분할의 결과가 더 좋은 뿐만 아니라 2- 나프틸포스핀옥시드 (2-na ph th yl pho sph ine ox i de) 와 O- 아니실포스핀옥시드 (0-a nis y l pho sph ine o- x i de) 의 용리순서가 뒤바뀌었다는 점이다. CO2 의 경우에는 다른 유기 용매와는 달리 크기가 작기 때문에 유동성이 좋아서 f3 _CD 의 키랄공 동에 들어가 있다고 하더라도 광학분할하고자 하는 라세미 화합물의 방향족기에 의하여 쉽게 치환되기 때문에 SubFC 에 의한 광학분할에 서는 실제로 내포 착화합물이 형성된다고 생각되며 따라서 2_ 나프릴 포스핀옥시드의 2- 나프틸기는 키랄공동으로 까어들어가 내포 착화합 물을 형성하기 때문에 머무름 시간이 커진다고 예측된다. 그러나 LC 에 의한 광학분할에서는 극성이 없는 핵산이 키랄공동을 차지하여 내
포 착화합물을 형성하기가 어렵기 때문에 2- 나프틸포스틴옥시드 는 그 림 3-29 에서 보는 바와 같이 컬럼에서 더 빨리 분리되어 나오며 광학 분할이 되지 않는다고 생각할 수 있다.
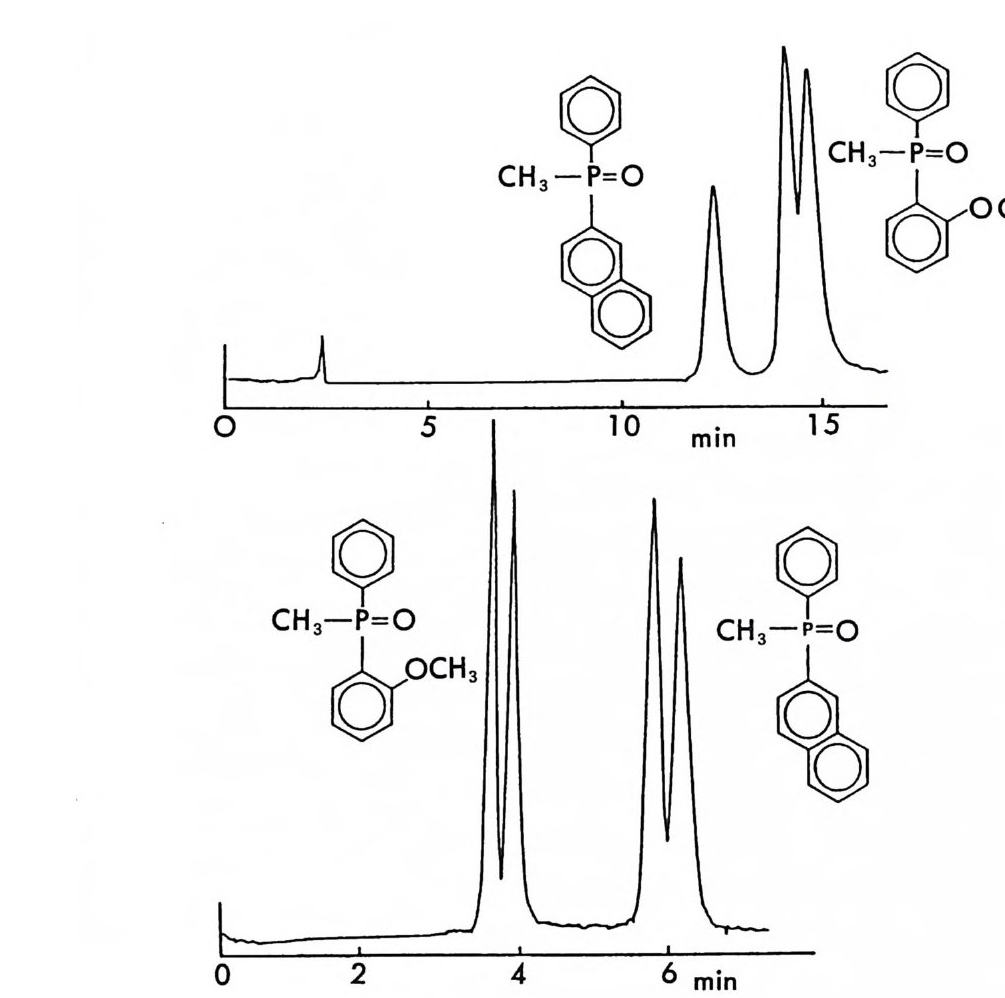 。I CH3I—: P= O 5’ IC H, —:Q P~l ’O0 m ·mc H 3 12f,5C H<3 00-P=: O
。I CH3I—: P= O 5’ IC H, —:Q P~l ’O0 m ·mc H 3 12f,5C H<3 00-P=: O
그림 3-29 /3 - CD 키랄고정상 (C y clobond I) 에서 2- 나프틸포스핀옥시드와 O- 아 니실포스틴옥시드의 광학분할. 위) LC, 이동상: 핵산-에탄올 (85:15, v/v) , 유속 lmL/m i n, 검출기 : 234 nmUV 아래 ) SubFC, 이동상 : CO 2-메 탄을 (92:8, w/w), 유속 : 4.5mL/m i n, 온도 : 25° C , 컬럼 평균 압력 : 150bar, 검출기 : 234nm UV (참고문헌 : 31)
CD 키랄고정상에서 광학분할을 실시할 때 온도의 영향은 다론 키랄 고정상에서의 광학분할과 비교하여 특히 크다. 내포 착화합물의 안정 성은 낮은 온도에서 크고 높은 온도에서는 낮아지기 때문이다. 내포 착화합물의 안정성이 온도가 낮을 수록 증가하는 것은 온도가 낮아짐 에 따라 용량인자가 증가하고 입체 선택성이 증가하는 것으로부터 알 수 있다. 그러나 온도가 너무 낮아지면 크로마토그램의 피이크의 폭이 넓어지고 머무롬 시간이 너무 길어지는 등의 문제가 있고 60-80°C 근 방에서는 CD 내포 착화합물이 완전히 해리되어 광학분할이 불가능할 수 있으므로 적절한 온도에서 광학분할을 시도할 필요가 있다. 일반적 으로 l8-31°C 온도 범위에서 광학분할을 실시하는 것이 적당하다. 온 도를 낮추면 일반적으로 분리인자도 증가하나 분해인자는 낮은 온도에 서 좋지 않은 물질이동의 영향으로 인하여 일반적으로 감소하는 경향 울 보인다(1 6). 그림 3-3 底든 시클로핵실페닐글리콜리산의 광학분할에
 T=57°C
T=57°C
대한 온도의 영향을 나타내는 크로마토그램을 제시한 것 이다 ( 22 ). CD 키랄고정상에서 광학분할할 때 이동상의 유속은 보통의 분석컬 럼 (250 X 4.6mm) 상에서 보통 0.5-2 . OmL / m i n 이다. 유속이 1.0 -2. OmL / m i n 일 때 피이크의 폭이 넓은 크로마토그램이 얻어지면 유속을 0.4mL/mi r@. 낮춤으로써 기준선분리 (baselin e se p ara ti on) 가 가능하 다. 표 3-1 0{든 25cm 의 /3 - CD 칼럼상에서 DDDD(tr a ns-a ,a'-(2, 2-dim eth y l -l, 3-dio x olane-4 , 5-diy l) bis (dip h eny lm eth a nol) ) 표의 광 학분할에 대한 이동상 유속의 영향을 나타낸 것이다(1 6). 유속의 영향 은 다른 라세미 화합물의 광학분할에서도 바슷한 양상을 보인다. 유속· 이 0 .4 0mL/m i~로 감소함에 따라 분리인자의 크기에는 큰 변화가 없으나 분해인자 (Rs) 는 크게 증가하고 있음을 표 3 - 10 에서 확인할 수
 HHsCs6C ~6H/O t > >1-5 << :.'. C!O (H'6C 6HHs5
HHsCs6C ~6H/O t > >1-5 << :.'. C!O (H'6C 6HHs5
표 3-10 /3- CD 키랄고정상에서 (+)-DDDD 가 광학분할될 때 이동 상 유속의 영향. 컬럼크기 : 250X4.6mm, 이동상 : 메탄올 울 (50 : 50, v/v) (참고문헌 : 16) k' 유속 (mL/m i n) + a’ Rs 1.00 0.58 0.71 1.22 0.8 5 0.80 0.57 0.71 1.24 0.92 0.60 0.56 0.70 1.24 0.97 0.40 0.55 0.69 1.25 1.12 0.30 0.5 6 0.70 1.26 1.22 0.20 0.53 0.66 1.24 1.20
있다. 광학분할에 대한 표 3-10 과 같은 유속의 영향은 내포 착화합물 의 특성으로서 아마도 유속이 클 때는 내포 착화합물을 형성하는 CD 분자로부터 용질분자가 떨어져 나오기가 어려워져 물질이동에 문제가 생기므로 이와 같은 현상이 관찰된다고 예측되고 있다. 이상에서 살펴본 바와 같이 CD 키랄고정상에 의한 광학분할에 영향 울 미치는 요인으로는 이동상의 상태 (조성, pH ), 온도, 이동상의 유 속 등 다양하기 때문에 주어전 라세미 화합물에 대한 최적의 광학분할 조건을 찾기 위해서는 모든 요인을 충분히 고려할 필요가 있다. 최근 에는 이들 요안 의에도 라세미 화합물을 녹이는 시료의 용매도 광학분 할에 영향을 미치는 것으로 알려지고 있다 (33). 따라서 가능하면 라세 미 화합물은 이동상으로 사용하는 용매에 녹여 CD 키랄 컬럼에 주입 될 필요가있다. CD 키랄고정상으로 충전된 HPLC 키랄컬럼의 과다사용으로 인한 컬럼효능의 감소현상은 CD 분자의 키랄공동에 비극성이 큰 불순~이 들어가 있기 때문이므로 컬럼을 통하여 메탄올이나 아세토니트릴 등을 계속 흘려주면 컬럼효능을 다시 증진시킬 수 있다. 오랜 시간 컬럼을 사용하지 않을 때에는 컬럼에 메탄올을 충전시켜두는 것이 바람직하다. 4 단백질을 기저로 한 키랄고정상 4.1 서론 단백질은 L- 아미노산의 펩티드결합에 의하여 이루어전 고분자물질 로서 특수한 삼처구조를 가지는 키랄 천연물이다. 특수한 삼차구조롤 가지는 키랄성으로 인하여 단백질은 다른 라세미 물질 특히 라세미 의 약품의 두 광학 이성질체와 입체 선택적인 상호작용을 한다는 사실이 얕거져 왔으며 최근까지 이에 관한 많은 연구가 이루어지고 있다. 예
를 들면 인체의 혈장에 존재하는 당단백질인 AGP(a,-a c i d g ly c o - p ro t e i n) 나 혈청알부민 (SA:serum album i n) 은 /3-차단제인 프로프라 놀롤 (p ro p ranolol) 의 두 광학 이성질체 중 하나의 광학 이성질체와 선 택적으로 상호작용을 한다는 사실이 연구보고 되었다(1). 라세미 의약 품과 같은 작은 분자들에 대한 단백질의 입체 선택성을 고려할 때 단 백질을 이용하여 라세미 화합물의 광학분할을 시도할 수 있다는 생각 울 할 수 있는 것은 당연한 귀결이다. 그 결과로서 AGP y-혹 은 소의 혈청알부민 (BSA:bovin e serum albumi n) 등을 HPLC 컬럼의 컬럼지 지체로 사용되는 실리카 겔에 고정시킨 키랄고정상들이 개발되어 여러 종류 라세미 화합물의 광학분할에 성공적으로 사용되고 있다. 특히 최근에는 특정한 라세미 화합물에 대하여서만 입체 선택성을 보이는 효소나 특정한 라세미 화합물 항원에 대한 항체를 적절한 HPLC 컬럼지지체에 고정시킴으로써 키랄고정성을· 제조하려는 노력들 이 시도되고 있다. 특정 라세미 화합물 항원에 대한 항체를 기저로한 키랄고정상은 일종의 주문자방식 (tai l or made) 의 키 랄고정상으로서 주 문자방식의 합성고분자 키랄고정상(제 4 장 참조)과 함께 홍미있는 키랄 고정상이 될 것이다. 4.2 AGP 키랄고정상 4.2.1 AGP 의 특성 및 AGP 키랄고정상의 제조 당단백질인 AGP (a1-aci d g l y co p ro t e i n) 는 분자량이 41, 000 인 고분 자물질로서 인체의 혈장 lOOmL 당 55-140m g이 존재하는 것으로 알 려져 있다. 이 당단백질은 181 개의 아미노산잔기와 디섯 개의 탄수화 물 단위로 이루어진 물질로서 탄수화물 부분에는 시알산 (s i a li c acid ) 이 포함된다. AG~ 분자는 1 다의 시알산을 포함할 뿐만 아니라 10-1 까 정도의 아스팔트산을 포함하며 AGP 를 구성하는 세린
(ser i ne) 도 카르복시기를 가진다. 반면 AGP 에 있어서 일차아미노기 는 13-1 헝 H 정도의 아르기닌 (ar gi n i ne) 이나 히스티딘 (h i s ti d i ne) 잔기 에 존재한다. 따라서 AGP 는 산성이 큰 당단백질로서 이 단백질의 등 전점 (iso electr i c p o i n t)은 인산염완충용액에서 2.7 인 것으로 알려져 있다 (2). AGP 를 실리카 겔에 고정시키려는 시도는 처음 Hermansson 에 의 하여 이루어졌으며 그 방법은 그림 3-31 과 같다 (3). 우선 실리카 겔울 3- 글리시독시프로필트리메목시실란과 반응시켜 반응성이 큰 에폭시드 고리 를 가진 실리카 겔을 얻은 후 이 실리카 겔과 AGP 를 완충용액에 서 반응시킴으로써 AG Pi-서의 자유 아미노기가 실리카 겔의 에폭시드 고리를 공격하여 AGP 가 공유결합에 의하여 실리카 겔에 고정된 AGP 키랄고정상을 얻을 수 있다. I /0\ -~i-O H + (CH30)3Si -( CH2)3·0·CH2-CH-CH2 __., I | | /0\ -~i-0 -~i - (CH2k0·CH2-CH·-CH2 + H2N·a-AGP ——— I I I I IH -~i -0-~ i -(CH2) 3-0·CH2-CH-CH2-NH• a-AGP I I 그림 3-31 a 1 -AGP 키랄고정상의 합성 이렇게 합성된 AGP 키랄고정상은 여러 종류의 라세미 화합물들의 광학분할에 성공적으로 사용되었다 (3, 4). 그러나 이 방법에 의하여 얻 어진 AGP 키랄고정상에서 실리카 겔에 고정된 단백질의 양이 실리카 겔 l g당 2.5m g에 불과할 뿐만 아니라 (3), p H 가 7.0 이상일 때 키랄
고정상의 안정성이 좋지 않기 때문에 새로운 방법에 의하여 AGP 키 랄고정상이 제조되었고 Enan ti oPac 이라는 상품명으로 상품화되어 있 다(제 9 장 참조). Enan ti oPac 에 대한 정확한 제조방법이 알려져 있지 는 않으나(특허로 되어 있고 (5), 논문에 정확히 밝히지 않았음) 대략의 과정은 다음과 같다. 우선 AGP 를 디에틸아미노에틸작용기를 가지고 있는 실리카 겔에 흡착시킨 후 과요오드산염으로 처리하여 AGP 의 탄 수화물 부분에 있는 알코올기를 알데히드기로 산화시킨다. 이렇게 되 면 아미노기 (AG Pt-사의 아미노기 및 디에틸아미노에틸 실리카 겔의 아미노 기)와 알데히드기 사이에 쉬프염기 (이민결합)가 형성되며 이것을 시아 노수소화붕소나트륨 (sodiu m cy a noborohy d rid e : NaCNBH 3 ) 로 환원함 으로써 단백질분자들 사이에 교차결합이 이루어지고 실리카 겔과 AGP 사이에 가수분해되기 힘든 공유결합을 가진 AGP 키랄고정상이 얻어진다. 이 방법에 의하여 고정된 단백질의 양은 실리카 겔 l g당 대략 180m g혹는 4.4X10- 열 정도인 것으로 알려지고 있다 (6). 4.2.2 AGP 키랄고정상을 이용한 광학분할의 특성 및 광학분할의 예 단백질 키랄고정상과 두 개의 광학 이성질체의 입체 선덱적인 상호 작용은 정전기적 상호작용과 소수성 상호작용에 의하여 주로 이루어지 며, 이의에 수소결합이나 전하이동 상호작용, 입체장애에 의한 상호 작용 등이 단백질 키랄고정상에 의한 키랄성 인지에 어떤 역할을 하고 있을 것으로 예상되고 있으나 아직 어떤 메커니즘에 의하여 서로 거울 상인 두 개의 광학 이성질체가 단백질 키랄고정상과 선택적으로 결합 하는가 하는 것은 밝혀져 있지 않다. 다만 단백질 키랄고정상과 두 개 의 광학 이성질체인 분석성분 사이에 정전기적 상호작용 혹은 이온쌍 상호작용이 중요하므로 이동상의 조성이나, 이동상의 pH , 이동상에 첨가된 이동상변형제 (mobil e p h ase modif ier ) 등이 단백질 키랄고정상 에 의한 라세미 화합물의 광학 분할에 있어서 입체 선택정의 크기 (a
값), 분석성분의 머무름 시간에 큰 영향을 미친디는 사실이 알려져 있 다. 이들의 영향은 다음 절 (4 . 2 . 3) 에서 논의하고 여기서는 어떤 종류 의 라세미 화합물들이 AGP 키랄고정상에서 광학분할되며 광학분할의 특성은 무엇인가에 대하여 살펴보고자 한다. AGP 키랄고정상에서 광학분할이 가능한 라세미 화합물은 크게 세 종류로 나눔 수 있다. 즉 아미노기를 가전 영기성 화합물로서 양이온 성 (이온쌍 을 형성할 때 양이온성임)인 라세미 화합물코} 흔히 카르복시기 를 가지는 산성 화합물로서 음이온성 (이온쌍을 형성할 때 음이온성임 )인
표 3-11 AGP 키랄고정상에서 광학분할 가능한 영기성 라세미 의약품의 예 라세미 의약품 참고문헌 g-아미노알코올 계통의 라세미 의약품 알프레놀롤 (alpr e nolol) 7* 에페드린 (ep h edrin e ) 8, 9 라베탈롤 (lab eta l ol) 8, 9 메토프롤 롤 (meto p ro lol) 8, 9, 10 나돌 롤 (nadolol) 8, 9 옥스프레놀롤 (oxp re nolol) 7* , 8 핀돌 몰 (pind olol) 7* 유사에 페드린 (ps eudoep h edrin e ) 8 터부탈린 (ter buta l i ne ) 8 항읍t린성 의약품 (an ti -chol i ner gi c drug s) 아트로핀 (atr o p ine ) 8, 9 클리디니움 (clid i n i u m ) 8 호모아트로핀 (homoatr o p ine ) 8 메펜솔레 이트 (mep e nsolatl e ) 3, 4, 9 메틸아트로핀 (meth y la tr o p ine ) 8 메 틸호모아트로핀 (meth y lh omatr o p ine ) 8, 9 옥시팬사이클이민 (oxy ph ency lim i ne ) 3, 8 트리디핵세틸 (tri d i h e xeth y 1) 8 트로피카미드 (tro p ica mi de ) 4
마취제 계동의 라세미 의약품 부피비카인 (bup ivi c a in e ) 4, 8 코카인 (cocain e ) 8 디페로돈 (dip e rodone) 8 케타민 (keta m i ne ) 4 메피비카인 (mep ivi c a in e ) 4, 8 진통제 계동의 라세미 의약품 부토파놀 (buto r ph anol) 8 메터돈 (meth a done) 8 펜타조신 (pe nta z oc ine ) 4 페니 라미돌 (ph eny ram i do l) 4 프로폭시펜 (pro p o xy ph ene) 8 항히스타민 계통의 라세미 의약품 브로모페 닐하이드라민 (bromop h eny lh y d rami ne ) 8 브로모페니라민 (bromop h enir a mi ne ) 8 클로로페니 라민 (chlorop h en ira mi ne ) 4, 9 카르비녹사민 (carbin o xami ne ) 8 디 메 틴덴 (dim eth ind ene) 8 독실아민 (doxy la mi ne ) 8, 9 프로메타전 (pro meth a zin e ) 3, 4, 8 기타염기성 의약품 아미노글루테티미드 (ami no g lu th e th im i de ) 11 시클로펜톨레 이트 (cyc l op e nto l ate ) 8, 9 디소피라미드 (diso p yram i de ) 3, 4, 8, 12, 13 도부타민 (dobuta m i ne ) 9 메토르판 (meth o rp h an) 8, 9 메틸펜디데 이트 (meth ylp e nd ida te ) 8, 9 펜메트라전 (ph enmetr a zin e ) 8, 9 페녹시벤자민 (ph enoxy b enzami ne ) 8 테트라히드라졸린 (tet r a hy d razoli ne ) 9 토카이니드 (toc ain ide ) 8, 9 베 라파밀 (verap a mi l) 9
*옥사졸리돈 (oxazo li done) 유도체의 광학분할
라세미 화합물 및 중성인 라세미 화합물의 세 종류이다. 많은 종류의 라세미 의약품들 중 아미노기를 가지는 염기성 라세미 화합물들은 AGP 키랄고정상에서 광학분할이 잘된다고 보고되었다. 표 3-11 에는 그동안 AGP 키랄고정상에서 광학분할된 염기성 라세미 의약품들의 예를 요약하여 놓았다. 이들 라세미 의약품들에 대한 AGP 키랄고정상의 입체 선택성은 라세미 화합물의 구조와 밀접한 관 계를 가지는 것으로 알려져 있다. 예를 들면 메토프롤롤 (me t o p rolol) 과 관련된 /3 - 아미노알코올을 광학분할할 때 암모늄이온(메토프롤롤이 광학분할과정에서 이온쌍을 형성함으로써 생성됨)과 수소결합부위 (메토프 롤운]서는 히드록시기)와의 거리가 광학분할능에 영향을 미치며 암모 늄이온 주위의 입체장애 또한 광학분할에 영향을 미친다 (8). 표 3-12 표 3-12 AGP 키랄고정상에서 메토프롤롤 (me t o p rolol) 계의 라세 미 화합물의 광학분할에 대한 구조의 영향. 이동상 : 0.02 M 인산염완충용액내에 0 . 001M 의 브롬화 테트라프로필암모늄 염, pH 7.0, 컬럼 : Enan ti oPac( 참고문헌 : 8) CH30CH2CH2- 0-OCH2晶y H -( CH2) n-NHR1 R1 n a’ 2-pr op yl 1 1.49 2-pr o p yl 2 1.14 2-pr op yl 3 1.00 t-B uty l 1 1.73 n-pr op yl 1 1.23 eth y l 1 1.12 meth yl 1 1.0 H 1 1.17
 H 。
H 。
에서 보는 바와 같이 히드록시기와 암모늄이온을 형성하는 아미노기사 이의 메틸렌기의 수가 l 에서 邸녁 1 증가함에 따라 a 값은 1. 49 에서 1.0~ 변화하는 사실을 알 수 있으며 아미노기에 붙어 있는 R 기 의 입체장애가 감소함에 따라 a 값 또한 감소함을 알 수 있다. AGP 키랄고정상의 입체 선택성에 대한 8- 아미노알코올의 아미노기에 붙어 있는 입체장애의 영향이나 아미노기 및 히드록시기 사이의 거리의 영 향은 터부탈린(t erbu t al i ne) l 과 메타프로테레놀 (me t a p ro t erenol) i 의 광학분할 및 (8) 최근에 연구보고된 아미노알코올 글리콜산에스데르 (gly c oli c e s t er) 의 광학분할에서도 (14) 찾아볼 수 있다. 터부탈린 l 과 메타프로테레놀 2 는 아미노기에 t유 L 틸기와 이소프로필기가 붙어 있 다는 사실이외에는 똑같은 구조를 가지고 있으나 터부탈린은 AGP 키 랄고정싱에서 광학분할이 되는 반면 (a= l.22 ), 메타프로데레늘은 광 학분할되지 않음이 보고되었다 (8). 항준民核! 의약품 (an ti -cho li ner gi c dru g s) 으로 쓰이는 시클로핵실 (3- 티에닐)굴리콜산에스데르의 광학분할에 있어서도 그립 3-32 에서 보 는 바와 같이 비대칭 탄소와 아미노기 사이의 거리 및 아미노기에 치 환되어 있는 알킬기의 크기에 따라 광학분할의 정도가 달라짐을 알 수 있다 (14). AGP 키랄고정상에서 산성 라세미 화합물들의 광학분할연구는 주로 진통소영제 계통의 의약품인 2- 아릴프로피온산 (2-ar y l p ro pi on ic aci d) 의 광학분할과 에토토인 (eth o to i n ) , 핵소바아비탈 (hexobarbit al) 등과
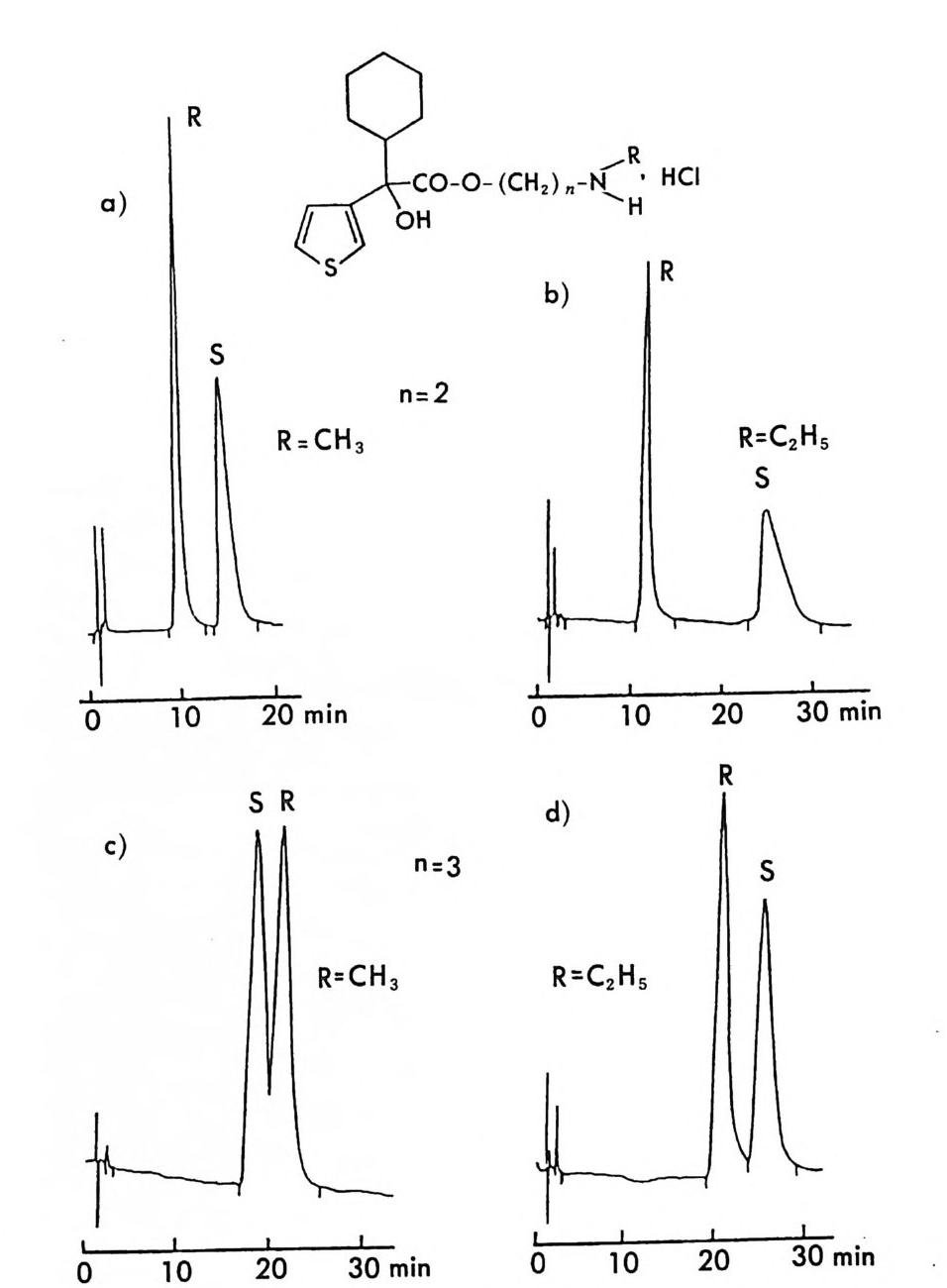 R
R
그림 3-32 의판A 을G광P (학키a, 분랄b 할고: 1정 0%상이, 동c(, 상Cdh : :i 6 r8%aml) -MA( Gv나P/v트)) 에 륨(서인참 산고아문염미헌완노 충:알 1용4코)액 올 (p글H 리, 콜7 . 0산)에+2스 - 프테르로
같이 산성 양성지를 가지고 있는 라세미 의약품들의 광학분할에서 찾 아볼 수 있다 (9 , 12). AGP 키랄고정상에서 광학분할된 산성 라세미 화합물들의 예를 표 3-13 에 요약하였으며 대표적인 크로마토그램의 예를 그림 3-33 에 제시하였다(1 2). 표 3-13 의 여러 라세미 카르복실 산들 중 2- 페닐부티르산 (2- p hen y lbu tyri c ac i d) 은 카르복시기와 페닐 기 이의에 다른 상호작용기를 가지고 있지 않음에도 이 화합물의 두 광학 이성질체에 대한 AGP 키랄고정상의 입체 선택성은 아주 좋음 (a = l. 97) 이 알려져 있다. 반면에 같은 조건에서 2- 페닐프로피온산 과 3 - 페닐부티르산의 두 광학 이성질체에 대한 AGP 키랄고정상의 입 체 선택성은 상대적으로 훨씬 낮은데 (각각 ct = 1.15, ct = I . 20) 아마 도 키랄중심에 치환된 알킬기의 길이 및 키랄중심과 카르복시기 사이 의 거리가 입체 선택성에 영향을 미치고 있는 것으로 짐작된다 (9). 갇 은 광학분할 조건에서 2- 페녹시프로피온산의 두 광학 이성질체에 대 한 AGP 키랄고정상의 높은 입체 선택성 (a = 1. 99) 은 페녹시기의 수 표 3-13 AGP 키랄고정상에서 광학분할 가능한 산성 라세미 의약품들의 예 라세미화합물 참고문헌 페아노부프프로로펜펜 ((ifbnuo pp rro o ffee nn )) 99121991129919219 케토프로펜 (keto p ro fe n ) 나프록센 (nap ro xen) 2 페닐平키르산 (2-ph eny lb uty ric aci d) 3 페닐뚜라르산 (3-ph eny lb uty ric ac id) 2-페 학巴로피온산 (2-ph enoxy pr op ion ic aci d) 2 페닐프로피온산 (2-ph eny lp r o p ion ic aci d) 에토토인 (eth o to i n ) 핵소바아비탈 (hexobarbit al) 밴드로플루메티아지드 (bendrofl um eth i a z id e )
 O) b)
O) b)
그림 3-33 AGP 키랄고정상 (Enan ti oPac) 에서 a) 이부프로펜 및 b) 2 - 페녹시프 로피온산의 광학분할• 이동상 : 인산영완충용액 (pH 7.0)+0.5% 2 - 프 로판을 +a) 9.8 m M N, N~ 디메틸옥틸아민 +b) 4.9mM N, N- 디메틸옥틸 아민, 유속' : 0.9mL/mi n (참고문헌 : 12) 소받게 능력이 증진됐기 때문인 것으로 예측된다 (9). 산성 라세미 화 합물의 하나인 식물호르몬 아부식산 (abs ci s ic ac id) ~은 카르복시기와 키랄탄소가 많이 떨어져 있으나 AGP 키랄고정상에서 광학분할이 가 능한 것으로 보고되어 있다(1 5). 지금까지 살펴본 염기성 라세미 화합물들과 산성 라세미 화합물들은 모두 이온성 화합물들이나 이들 라세미 화합물들을 이온성이 아닌 유 도체로 바꾸었을 때 AGP 키랄고정상에 의한 입체 선택성이 증가하는 몇몇 예들이 보고되어 있다. 여러 종류의 /3-차단제 (아미노알코올)들을
。 3 C`H3C | OOH R-O-CH2CO!!° ir 4—· .CNIH -2C - :H.:/_ CCHH33
포스젠(p hos g ene) 과 반응시켜 옥사졸리돈 (oxazol i done) 유도체 4 로 만 들었을 때 표 3-14 에 요약된 바와 같이 이온성인 화합물로서 광학분할 될 때 보다도 입체 선택성 (a 값)이 크게 증진되는 것을 알 수 있다 (6, 7). 메토프롤롤 (me t o p rolol) 의 옥사졸리돈 유도체는 이동상변형제로 10% 의 2- 프로판을을 사용하였을 때 광학분할되지 않으나 이동상변형 제를 사용하지 않았을 때는 a = 1. 9 5£.서 광학분할이 잘됨을 알 수 있다 (4.2.3 참조). 만델산은 AGP 키랄고정상에서 광학분할되지 않으나 만델산의 메틸 에스데르나 에틸에스테르는 표 3-15 에서 보는 바와 같이 광학분할되 고 있다. 특히 만델산을 만델산 에틸에스데르로 바꾸었을 때 머무름 시간 뿐만 아니라 입체 선택성이 크게 증가한 것은 키랄고정상과 광학 표 3-14 AGP 키랄고정상에서 衍由제 및 p-차단제의 옥사졸리돈유도체의 광학분할 비교. 컬럼 : 실리카 겔에 흡착된 AGP (A : 150mg /g, B : 180-mg /g) , 이동상 : 인산염 완충용액 0 . 02M, pH 7.0+A) 10% 2- 프로판을 B) O. lM NaCl+ B% 에탄을 (참고문헌 : 6) (A) /3-차단제 (B ) 옥사졸리 돈유도체 /3-차단제 kI, a’ kI' a’ 알프레놀롤 (alpr e nolol) 9.0 1.57 메토프롤롤 (meto p ro lol) 6.1 1.64 3.0 l.95* 옥푸레놀롤 (oxp re nolol) 11.2 1.25 28 1.64 핀돌롤 (pind olol) 4.6 1.65 프로프라놀롤(p ro p ranolol) 27 1.12 27 5,7 *변형제 사용하지 않은것
표 3-15 AGP 키랄고정상에서 만델산 및 만델산에스테르의 광학분 핼 컬럼 : AGP 키랄고정상 (150m g/g실리카겔). 이동 상 : 인산염완충용액 (pH 7.0 : µ = 0.0 2 ) + 2% 2 - 프로판을 (참고문현 : 7) 시료 | kl, a’ 만델산 1.13 1.00 만델산 메틸에스테르 1.04 1.27 만델산 에틸에스데르 1.47 1.93
분할하고자 하는 분석성분 사이에 소수성 상호작용이 중요하게 작용하 고 있음을 시사하고 있다고 할 수 있다. 이온성 라세미 화합물들을 비 이온성 유도체로 바꾸었을 때 입체 선택성의 증가에 대한 몇 가지의 예에도 불구하고 아직 이들에 대한 정확한 키랄성 인지의 메커니즘은 규명되지 않은 상태이다. 이온성이 아닌 라세미 화합물들에 대한 광학분할의 예는 이온성 라 세미 화합물을 비이온성 유도체로 만든 후의 광학분할의에도 몇 가지 의 예를 찾아 볼 수 있다. 그 중 홍미있는 사실은, 세포독성을 나타내 는 것으로 알려진 a- 메틸렌 -T 부티로락톤 (a-me t hy lene- y -bu tyr olac t one) 류의 화합물 5 는 R 과 R2 의 종류에 따라 a 값이 1. 23 에서 부터 1. 74 까지 좋은 입체 선택성을 보이며 a- 메틸렌 -r -Jf티로락탐 (a -meth y le ne-r-buty rol acta m ) 류의 화합물 敬든 R 과 R2 의 종류에 따라 짝이 1. 09 에서부터 4.70 까지 다양한 입체 선택성을 보인다는 사실 이다 (16). AGP 키랄고:정상:에서< 키랄O중 심이 황인 라R2세< 미 화합물의O 광 학분할이
최근 보고된 바 있다. 알벤다졸 (albendazole:ABZ) 과 펜벤다졸 (fen bendazole :FBZ) 은 벤즈이미다졸 계통의 구충제로서 이들 의약품 들을 경 구두여 하면 이 들은 술폭시 드 (sulfo x id e ) 인 SOABZ J_과 SOFBZ ~ 산화되며 술폭시드 화합물 1 과 §이 실지로 구충작용을 하는 것으로 알려져 있다 07). SOABZ J_과 SOFBZ §.f는 키랄중심이 황인 키랄 술폭시드화합물로서 아무런 이동상변형제도 사용하지 않고
 』亡 :\NH 『 CH3
』亡 :\NH 『 CH3
AGP 키랄고정싱에서 광학분할이 잘되는 것으로 보고되었다 (18). 그 립 3-34 는 AGP 키랄고정상에서 SOFBZ .9-] 광학분할에 대한 크로마 토그램을 제시한 것이다.
 。 10 20 mi n
。 10 20 mi n
AGP 키랄고정상을 이용하여 라세미 의약품들의 인체내 대사과정을 밝히려는 연구는 특히 최근 라세미 의약품들을 규제하려는 움직임에 비추어 (제 1 장 참조) 홍미있는 일이다. 디소피라미드 (d i so py ram i de: DP) 는 항부정맥 의약품으로서 라세미 형태로 사용되고 있다. 디소피 라미드는 인체내에서 모노 -N- 데이소프로필디소피라미드 (mono -N -deis o p ro p yld is o p yra mi de : MND) 로 전환되는데 (그림 3-34) 인체내에 서 DP 의 두 광학 이성질체 존재량과 MND 의 두 광학 이성질체의 존 재량을 AGP 키랄고정상을 이용하여 쉽게 측정할 수 있으며 경구두여 후 시간에 따른 존재량의 변화를 측정한 예는 그림 3-3~ 같다(1 9, 20).
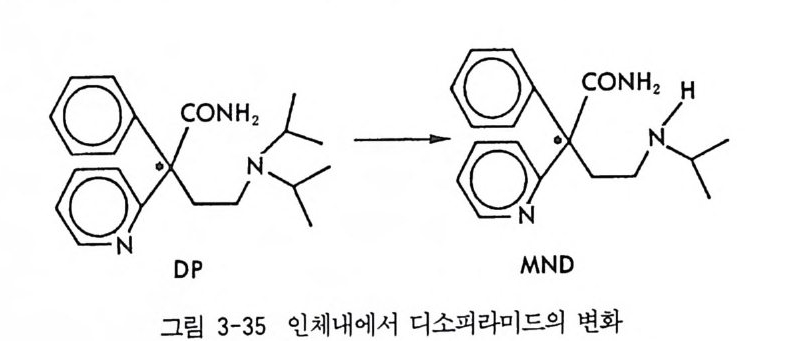 CONH2 H
CONH2 H
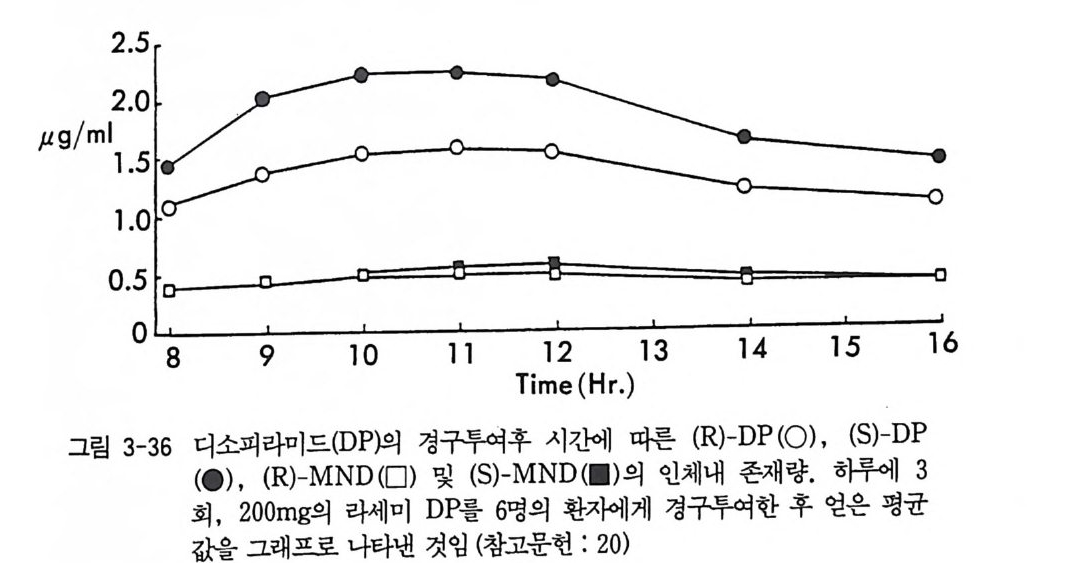 2.5
2.5
국소마취제인 부피바카인 (bup iva cain e ) 안나 기관지 천식에 이용되 는 살부타몰 (salbu t amol) 1Q을 인체에 두여하였을 때 혈액 혹은 소변 에 존재하는 이둘 라세미 의약품들의 두 광학 이성질체 양을 AGP 키 랄고정상을 이용하여 직접 분석한 예들이 있다 (21, 22). 이와 같이 인
 [~_NH SNeHo 9CH s:CHHs CH2 0 H: g-CH2 N H -l:H3
[~_NH SNeHo 9CH s:CHHs CH2 0 H: g-CH2 N H -l:H3
체내에서의 라세미 의약품의 두 광학 이성질체의 존재량 변화와 대사 물질의 존재량을 쉽게 측정할 수 있기 때문에 키랄고정상을 이용하면 여러 라세미 의약품들이나 광학활성 의약품들의 인체내 약리작용에 대 한 연구 및 약리학적 인 속도론 연구 (ph armacokin e ti cs) 에 크게 도움 이 될 것으로 기대되고 있다. 4.2.3 AGP 키랄고정상에 의한 광학분할에 영향을 마치는 요인들 AGP 키랄고정상에 의한 라세미 화합물의 광학분할에 대하여 현재 키랄성 인지 메커니즘이 밝혀져 있지는 않다. 그러나 AGP 키랄고정 상에서 이온성 라세미 화합물들이 광학분할될 때 키랄고정상과 라세미 화합물사이에 이온-이온 상호작용 및 소수성 상호작용 등이 중요한 역 할을 하고 있는 것으로 생각되고 있다. 따라서 AGP 키랄고정상에 의 한 광학분할은 이동상으로 주로 사용되는 인산염완충용액에 첨가되는 이동상변형제 (mobil e p h ase mod ifi er) 의 종류 및 양, 이동상의 pH , 인산영완충용액에 있어서 인산염의 농도 등에 의하여 큰 영향을 받는 다. 이동상에 첨가되어 라세미 화합물의 두 광학 이성질체의 컬럼내 머
표 3-16 AGP 키랄고정상에 의한 광학분할시에 이동상에 첨가되는 유기번형제 비이온성 변형제 1 가 알코올 : l- 프로판을, 2- 프로판을, 에탄을 디올 : 에틸렌글리콜 (e t h y lene g l y lcol) , 프로 필렌글리콜 (pro p yle ne gly c ol) , 1, 2 국 L 탄디올 아미노기 : 6- 아 미 노 핵 산 산 (6-ami n ohexan- oic a ci d) , /3-알라닌, 루이신 (아미노산은 적절한 조건하에서 비이온성임 ) 양이온성변형제 3 차아민 : N, N- 디메틸옥틸아민 (DMOA) , N, N -디메틸에틸아민 4 차암모늄영 : 브롬화 데트라프로필암모늄(t e t ra p ro pyla mmoniu m bromi de ) , 브롬화 테트 라부틸암모늄 (tet r a buty la mmoniu m bro- mi de ) 아마노산 : 1, 2- 디 아미 노부탄산 (1, 2-dia m i no - buty ric aci d) 음이온성변형제 카르복실산 : 옥탄산 (oc t ano i c ac id) , 부티 르산 (buty ric aci d) 데칸산 (decano i c aci d) 아미노산 아스팔트산 (aspa rt ic ac id) 술팜산 : 시클로핵 실술팜산 (cy c lohexy ls ulfa m i c aci d)
무름과 입체 선택성에 영향을 미치는 이동상변형제는 표 3-16 에 보는 바와 같이 비이온성 변형제, 양이온성 변형제 및 음이온성 변형제의 세 종류로 구분할 수 있으며 (23) 이들은 이동상에 첨가되어 AGP 키 랄고정상에서의 두 광학 이성질체의 머무름 시간을 조절할 뿐만 아니 라 두 광학 이성질체에 대한 입체 선택성을 증진시킨다. 비이온성 변형제 특히 알코올 변형제를 이동상에 첨가하였을 때는 일반적으로 라세미 화합물의 두 광학 이성질체 머무름 시간이 감소할 뿐만 아니라 입체 선택성도 감소하는 것으로 알려져 있다. AGP 키랄
고정상에서의 광학분할에 대한 비이온성 변형제의 영향 예를 그림 3-37 에 제시하였다 00) • AGP 키랄고정상에 의한 라세미 화합물들의 광학분할은 이온-이온상호작용과 함께 AGP 분자와 라세미 화합물분 자 사이의 소수성 상호작용도 중요하게 작용하기 때문에 이동상에 비 이온성 유기용매를 가함으로써 AGP 분자와 라세미 화합물분자 사이의 소수성 상호작용을 감소시켜 머무름 시간을 감소시킬 뿐만 아니라 입 체 선택성도 감소시키는 것으로 예측되고 있다 (3, 4, 8, 9). 양이온성 변형제나 혹은 음이온성 변형제를 이동상에 첨가하였을 때, AGP 키랄고정상에 의한 라세미 화합물들의 광학분할은 라세미 화합물들의 구조, 사용한 변형제의 종류 혹은 이동상의 pH 등에 따 라 아주 복잡한 양상을 나타내므로 현재 완전히 이해되고 있는 상태는 아니다. 그러나 일반적으로 양이온성 변형제를 이동상에 첨가하였을
2.5 ak1,R. 5 1· 8a 27 k,R · 2.0 1 2b。 1.7 1.5 1.6 1.j· 5 1.0 1.5 I 1.0 LL.._ 。 0.5 1.0 1.5 1.0 1.5 2.0 2.5 % 2-Prop a nol %Aceto n it ril e 그림 3-37 AGP 키랄고정상 (Enan ti oPac) 에서 메토프롤롤 (me t o p rolol) 의 광학 분O) 한+에 a) z대...,프 한로 비판을이,온 성b) 아변세형토제의니 트영릴향 .( 참이고동문상헌 : : 인10산) .염 완충용액 (pH 7.
때 양이온성 라세미 화합물의 머무름 시간은 감소하고 음이온성 라세 미 화합물의 머무름 시간은 증가하며 음이온성 변형제를 이동상에 첨 가하였을 때는 이와 반대의 영향을 미치는 것으로 알려지고 있다. 이 와 같은 사실은 키랄고정상을 구성하는 AGP 분자의 이온-이온상호작 용부위에 대하여 같은 전하를 가진 이동상변형제와 라세미 화합물 사 이에 서로 경쟁이 있기 때문인 것으로 예상된다. 양이온성 변형제를 이동상에 첨가하였을 때 음이온성 라세미 화합물의 광학분할에 대한 입체 선택성 (ct값)은 일반적으로 증가한다. 그러나 음이온성 변형제를
 K'
K'
그림 3-38 AGP 키랄고정상에서 양이온성 라세미 화합물들이 광학분할될 때 이 동상에 첨가되는 DMOA 의 영향. D= (-)-프로메타전, .= (+) -(S프) -로메메피타J:진1,硏 인 O, &= =(S) ( -R)디 -소메피피라바미카드인,. •이 동= 상(R ): -2%디 소2피-라 프미로드판,을 /!::인:,. =산 염완충용액 (v/v) (pH 7 .1 5)+DMOA, 유속 0.5 m L/m in (참고문헌 : 4).
가하였을 때는 일반적으로 a 값이 아주 작거나 광학분할이 전혀 일어 나지 않는 것으로 알려져 있다. 반면 양이온성 라세미 화합물의 입체 선택성에 대한 이동상변형제의 영향은 훨씬 다양한 것으로 알려지고 있다 (23). 그림 3-38 은 AGP 키랄고정상에서 양이온성 광학 이성질체들의 머 무름 시간에 대한 양이온성 변형제인 N, N- 디메틸옥틸아민 (DMOA) 의 영향을 그래프로 나타낸 것이다 (4) . 일반적으로_ DMOA 의 농도가 커질수록 머무름 시간(용량인자 k' )이 감소하는 것으로 관찰되고 있 다. 한편 그림 3-3 9i:근 음이온성 광학 이성질체에 대한 양이온성 변형제 의 영향을 나타낸 그래프로서 양이온성 변형제인 DMOA 의 농도가 증가함에 따라 머무름 시간이 증가할 뿐만 아니라 입체 선택성 (a 값)
k' a’ 100 5 4 3 50 2 1 1 5 10 L 1 5 10 mM DMOA mM DMOA 그림 3-39 AGP 키랄고정상 (Ena ti oPac) 에서 음이온성 라세미 화합물이 광학분 할될 때 양이온성 변형제인 DMOA 의 영향. 나프록센( □), 이부프로 올펜 +(0D)M. O이A 동. 유상속 : :인 0산.9영m완L/충m용i n 액 ( 참(p고H 문 7헌.0 ): +1 20) . 50% (v/v) 2-프 로판
도 크게 증가하고 있음을 확인할 수 있다(1 2). 특히 나프록센 (na p roxen) 의 경우 DMOA 의 농도가 증가함에 따라 a 값이 1. 1 에서 4. 辱 크게 증가하였는데 이것은 이 과정에서 (+)-이성질체에 대한 k' 값은 크게 증가하는 반면 (-)-이성질체에 대한 k' 값은 감소한 결과 로서 이와 같은 사실은 AGP 키랄고정상과 나프록센의 두 광학 이성 질체가 서로 상호작용할 때 같은 메커니즘에 의하여 상호작용이 이루 어지는 것이 아니고 디른 메커니즘에 의하여 상호작용이 이루어지고 있음울 시사해 주고 있다. 양이온성 라세미 화합물의 입체 선택성에 대한 이동상변형제의 영향 의 다양성은 비슷한 구조를 가진 아트로핀 (a t ro pi ne), 메틸아트로핀
a’ 3.8 3·4 39 2· 218 J.4 I _. l.Ot . 三一소\? A B C D E O.J ~ M O.O lM 0.003M 0.0 0 1M O.OlM 그림 3-40 AGP 키랄고정상에서 아트로핀( ... ), 메틸아트로핀(♦ ), 메틸호모아트 로핀 (v')의 광학분할에 대한 이동상변형제의 영향. 이동상 : 인산염완 충용액 (pH 7.0) +이동상변형제 A : 2_ 프로탄을, B : 2, 4- 디아미노부 탄산, C : 브롬화 테트라프로필암모늄, D : 옥틸 술폰산, E : 옥탄산 (참고문헌 : 9)
(meth y la tr o p ine ) 및 메 틸호모아트로핀 (meth y lh omatr o p ine ) 의 광학분 할에 대한 여러 종류의 이동상변형제의 영향을 그래프로 나타낸 그림 3-4@ 로부터 자명하다 (9). 아트로핀과 메틸호모아트로핀의 구조의 차이점은 두 가지에 불과하다. 메틸호모아트로핀의 경우 3 차 아민기 대신 4 차 암모늄염을 가지고 있디는- 사실과 아트로핀의 경우 키랄탄소 와 히드록시기 사이에 메틸렌기가 있다는 것으로서 구조에 있어서의 조그마한 차이점이 입체 선택성에 있어서는 아주 큰 영향을 미친디는 사실이 홍미롭다고 할 수 있다. 특히 메틸호모아트로핀과 아트로핀의 광학분할에 대한 옥탄산 (oc t ano i c a ci d: 음이온성 변형제)변형제의 영향 은 완전히 반대라는 사실이 홍미롭다. g - 아미노알코올인 에페드린 (eph edrin e ) 과 유사에페드린 (p seudoe p hedr i ne) 은 서로 부분 입체 이 성질 관계에 있는 화합물로서 입체구조는 다르나 화학구조는 동등한 화합물인 양이온성 라세미 화합물이다. 그러나 AGP 키랄고정상에서 의 광학분할에 대한 이동상변형제의 영향은 그립 3-41 에서 보는 바와 같이 완전히 반대임을 관찰할 수 있다 (9). AGP 키랄고정상에서의 광학분할에 대한 이동상의 pH 영향 또한 고려하여야 할 중요한 사항이다. 보통 실리카 겔울 컬럼의 고형지지체 로 사용하는 키랄고정상에 있어서 적합한 p H의 범위는 3.0 - 7. 誌巳1 이 p H 의 범위 밖에서는 키랄고정상 손상을 입울 우려가 있다 (9). 양이온성 라세미 화합물들을 광학분할할 경우 3.0-7.5 범위내에서 이동상의 p H 를 낮추면 일반적으로 머무름 시간 (k') 도 감소한다. 그러 나 음이온성 라세미 화합물들을 광학분할할 경우에는 p H 를 낮춤으로 써 머무름 시간 (k') 이 증가한다 (9, 12). p H 를 낮춤으로써 키랄고정상 울 구성하는 AGP 분자가 가지고 있는 아미노기에 양성자가 첨가되고 따라서 음이온성 라세미 화합물.(보통 카르복실산음이온을 가짐)과의 정 전기적 인력 혹은 이온-이온상호작용이 증가되어 머무름 시간이 증가 하는 것으로 예상된다. 반면 p H 를 높임에 따라 AGP 분자가 가지는 카르복시기는 더욱 카르복실산음이온으로 될 것이고 따라서 양이온성
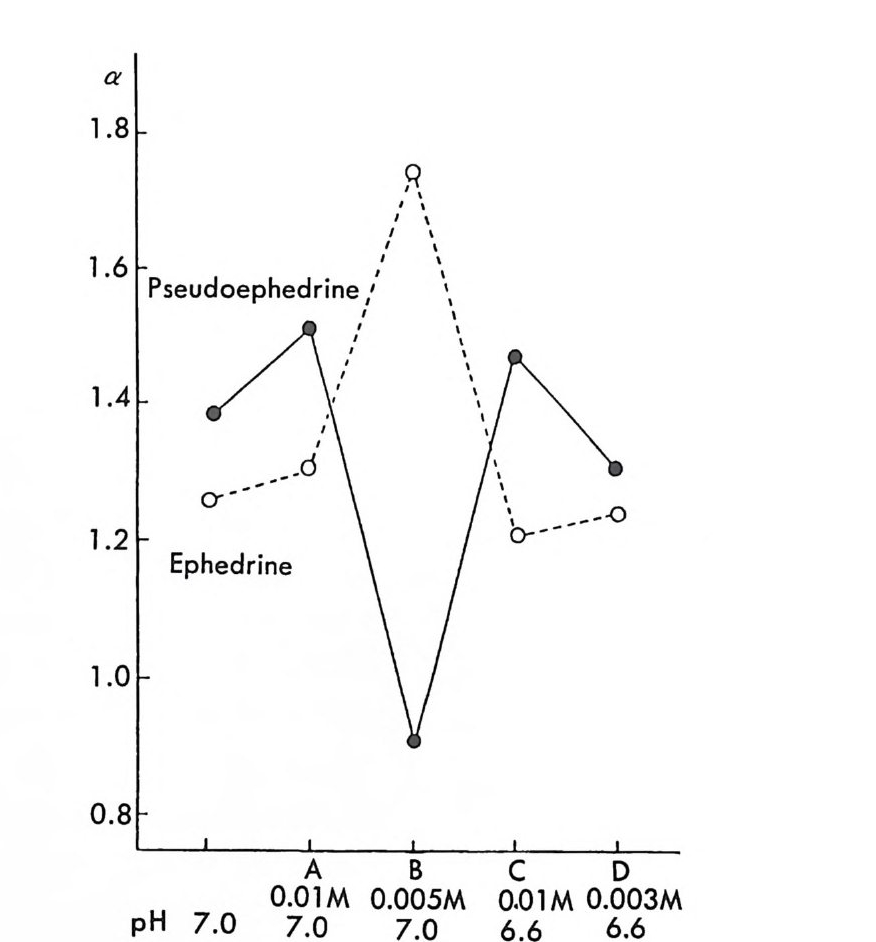 a’
a’
그림 3-41 AGP 키랄고정싱에서 에페드린과 유사에페드린의 광학분할에 대한 이 동상변형제의 영향. 이동상 : 인산영완충용액 (pH 7.0 ) + 이동상 변형제 A : 6- 아미노핵산산, B : 옥탄산, C : 아스팔트산, D : 브롬화 테트라 프로필암모늄 (참고문헌 : 9)
라세미 화합물(보통 아민화합물)과의 정전기적 상호작용이 증가하여 머 무름 시간이 증가할 것으로 예상된다. 이와 같은 사실은 p H의 변화 에 따른 양이온성 라세미 화합물과 음이온성 라세미 화합물의 광학분 할 변화를 나타낸 표 3-17 에 잘 나타나 있다. p H 에 따른 입체 선택성 (a 값)의 변화는 라세미 화합물의 종류에 따
표 3-17 AGP 키랄고정상에서 광학분할에 대한 p H 의 영향. 이동상 : 0.02M 인산염완충 용액+번형제(참고문헌 : 9) 라세미 화합물 변형제 pH 시(c클y c로l o펜p e톨 nt레o l 이at트e ) (do독x실y l a0 m떤i n e ) kI' a’ kI' a’ 브롬화 데트라부틸암모늄 7.0 18 1. 70 9.5 1.23 (0.003M) 6.0 4.8 2.09 6.1 1.37 NaCl (O.lM) + 7.5 14 1.96 9.8 1.15 2- 프로판올 (0.33M) 6.5 4.8 1.79 6.2 1.16 2- 페닐부탄산 2- 페닐프로피온산 kI' a( kI' a’ 브롬화테트라프로필암모늄 6.6 5.3 1.70 2.6 1.15 (0.003M) 6.1 10.8 1.65 4.5 1.16
라 달라질 뿐만 아니라 이동상에 첨가된 변형제에 따라서도 달라전다. 표 3-17 에서 보는 바와 같이 시클로펜톨레이트 (c y clo p en t ola t e) 나 독 실아민 (dox y lam i ne) 의 경우 0.003M 의 브롬화 데트라부틸암모늄을 첨가하여 광학분할할 때는 p H 를 7.0 에서 6. 但녁i 낮추면 a 값은 증 가하나 O.lM 의 NaCl 과 0.33M 의 2- 프로판을을 이동상에 첨가하여 광학분할할 때는 p H 가 7.5 에서 6. 또냐i 변할 때 a 값이 감소하거나 변화가 거의 없음을 알 수 있다. AGP 키랄고정상에서 라세미 화합물들이 광학분할될 때 이동상의 이온세기 (ion ic s t ren gt h) 는 분석성분의 머무름 시간에 분명히 영향을
미칠 것으로 예측되나 그 효과는 키랄고정상의 AGP 분자와 분석성분 사이의 상호작용이 정전기적 상호작용인가 혹은 소수성 상호작용인가 에 따라 다소 복잡하리라고 예상된다. AGP 키랄고정상에서 광학분할 에 미치는 이동상의 이온세기 영향에 대한 체계적인 연구결과는· 아직 없는 실정이다. 다만 인산염완충용액을 이동상으로 사용할 경우에 대 부분의 광학분할에서 인산염완충용액의 농도는 0 . 02M 이며 이동상에 간혹 NaCl 을 첨가하여 이동상의 이온세기를 증가시켜 광학분할을 성 공적으로 수행한 예들이 보그 L 되어 있다 (8). AGP 키랄고정상에서 광학분할에 대한 온도의 영향은 다른 키랄고 정상에서의 광학분할에 대한 온도의 영향과 마찬가지로서 온도가 증가 하면 머무름 시간이 감소할 뿐만 아니라 입체 선택성 (a 값)도 감소한 다 (12). 그러나 그 정도는 라세미 화합물에 따라 달라진다. 마지막으로, AGP 키랄고정상에 의한 광학분할에 영향을 미치는 요 인으로서 실리카 겔에 고정된 AGP 분자의 양을 고려할 필요가 있다. 실리카 겔에 고정되는 AGP 의 양이 증가함에 따라 일반적으로 머무름 시간은 증가하나 입체 선택성은 실리카 겔 l g당 80m g정도의 AGP 분 자가 고정될 때 최대가 됨이 알려져 있으며 (4), AGP 키랄고정상의 제조과정에서 유념하여야 할 사항이라고 할 수 있다. 4.3 BSA 키랄고정상 4. 3 .1 BSA 의 특성 및 BSA 키랄고정상의 제조 소혈청 알부민 (Bovin e Serum Albumi n : BSA) 은 분자량이 66210 인 구상단백질로서 581 개의 아미노산으로 이루어진 폴리펩티드사슬 하나 로 구성되어 있다. BSA 를 구성하는 하나의 펩티드사슬은 구성성분인 시스틴 (c y s t e i ne) 사이에 17 개의 분자내 디술파이드 가교(di sul phi de br i d g e) 를 형성함으로써 9 개의 이중곡선 (double loo p)으로 되어 있다 (24) BSA 은 등전점이 4. 7 로서 상대적으로 산성인 단백질이며 pH 7.0
에서 알짜전하는 ― 18 이다. 또한 유기용매인 벤젠의 물에 대한 용해 도가 순수한 물보다는 BSA 이 녹아 있는 수용액에서 더 크다는 사실 은 BSA 분자가 소수성이라는 사실을 시사해 주고 있다 (2 5 ). 이와 같 은 성질을 가전 BSA 는 전하를 띠지 않은 소수성 화합물과 상호작용 할 수 있을 뿐만 아니라 음이온성 화합물과 상호작용할 수 있음이 알 려져 있다. 그러나 큰 소수성 기를 가지고 있지 않은 양이온성 화합물 과는 상호작용을 하지 않음이 알려져 있다. BSA 분자가 라세미 화합 물과 상호작용할 때 입체 선택성은 맨처음 L - 트리프토판 (L - t r ypt o p han) 이 D-트 리프토판보다 혈청단백질과 훨씬 강한 상호작용을 한다 는 사실에서부터 알려지기 시작하였으며 (26), 그 후 BSA 이 친화 크 로마토그래피 (aff ini t y chroma t o gr a ph y)에 응용되어 광학분할에 사용 된 것은 1973 년 S t ewar t와 Doher ty가 BSA 를 세파로스 (seph arose) 에 고정시켜 D, L- 트리프토판을 광학분할한 것이었다 (27). 그 후 La g ercran t z 는 세파로스에 고정된 BSA 혹은 디른 종류의 혈청알부 민을 키랄고정상으로 사용하여 트리프토판과 와파린 (war fa r i n) 의 광 학분할을 시도하였다 (28, 29). 1982 년에 이르러 Allenmark 는 스웨덴 Pharma ci a 사의 활성화 CH-Seph arose 4B 와 에폭시활성화 Sep h ar-ose 6B 에 BSA 를 고정시켜 , 그림 3-42 에 있는 구조를 가졌다고 예상
HIN \ -NH c/ o` B s A OH 十 o y o~O~NH-BSA OH 그림 3-42 BSA-세 파로스 키랄고정상의 예상구조. (위) : CH-Seph arose 4B 로 부터 유도된 키랄고정상, (아래) : 에폭시활성화 Seph arose 6B 로부터 유도된키랄고정상
되는 BSA 세파로스 키랄고정상을 제조하여 ( 3 0) 방향족 아미노산과 라세미 술폭시드, N- 벤조일아미노산 등의 광학분할에 성공적으로 옹 용하였다 (31). 그러나 세파로스의 좋지 않은 기계적 성질로 인하여 BSA 세파로스 키랄고정상을 직접 HPLC 시스템에 응용할 수는 없었 고, BSA- 세파로스 키랄고정상을 유리컬럼에 충전하고 이 유리컬럼을 HPLC 시스템에 연결하여 광학분할을 실시하였기 때문에 큰 a 값으로 인하여 광학 이성질체들의 기준선 분리가 가능하였으나 컬럼의 효율 (특히 분해능)은 그다지 좋은 편이 아니었다. 1983 년 Allenmark 는 최초로 BSA를 실리카 겔에 화학결합시켜 BSA- 실리카 겔 키랄고정상을 제조하고 흔히 사용되는 HPL C-용 스 텐레스 컬럼에 충전하여 HPLC 시스템을 이용한 라세미 화합물의 광 학분할에 성공적으로 응용하였다 (32). 그러나 BSA- 실리카 겔 키랄고 정상의 정확한 제조방법은 보고되지 않았으며 1983 년부터 Resolvosil (제 9 장 참조)이라는 상품명으로 상품화되어 있다. Allenmark 에 의하여 제조된 BSA- 실리카 겔 키랄고정상 의에 Erlandsson 등은 BSA 를 실리카 겔에 흡착시켜 키랄고정성을 제조하 였고 (33) , Rog e rs 등은 클루타르알데히드 (g lu t araldeh y de) 를 교차결 합 시약으로 사용하여 4- 아미노부틸-실리카 겔 (4- 아미노부틸디메틸매독 시실란과 실리카 겔을 반응시켜 얻은 것)에 BSA를 결합시킨 BSA_,).J 리 카 겔 키랄고정상을 제조하였다 (34). 4.3.2 BSA 키랄고정상을 이용한 광학분할의 특성 및 예 BSA 키랄고정상에서 광학분할이 가능한 라세미 화합물들은 보통 중성 혹은 음이온성 화합물들이고 양이온성 라세미 화합물들은 몇 가 지의 예를 제외하고는 광학분할이 잘되지 않는다. BSA 키랄고정성에 서 광학분할이 광범위하게 연구된 음이온성 라세미 화합물은 아미노산 이다. 여러 종류의 라세미 아미노산들이 N- 벤조일유도체 旦 (31, 32), N- p-니트로페닐유도체 !Z_( 35), N-2, 4- 디니트로페닐유도체 묘
(35, 36), 나프토일 (na p h th o y l) 유도체 11(35)' 프탈이미드(p h th al imi de) 유도체 ~(37), N- 벤젠술포닐 (benzenesul fo n y l) 유도체 브 (37), 혹 은 단실유도체 끄 (36) 로서 광학분할이 가능하다. 그림 3-43 은 트레오닌(t hreon i ne) 의 프탈이미드 유도체와 세린 (se i ne) 의 N- 벤젠술포닐 유도체의 광학분할에 대한 크로마토그램을 제시한 것으로서 좋은 광학분할을 확인할 수 있을 뿐만 아니라 (37) 표 3-18 에서 보는 바와 같이 아미노산 결사술의 소수성에 따라 머무롬 시 간과 입체 선택성에 큰 차이가 있음을 확인할 수 있다 (36). 이들 아미 노산 유도체들이 BSA 키랄고정상에서 광학분할될 때 두 광학 이성질
 。갱0 -NH- J H- 갱0 OH O , N -0- NH- 『 -[OH
。갱0 -NH- J H- 갱0 OH O , N -0- NH- 『 -[OH
체의 용리순서는 유도체의 종류 및 아미노산의 종류에 따라 다르다는 사실이 확인되고 있다. 그립 3 - 43 에서처럼 프탈이미드 유도체 및 N -벤젠술포닐 유도체의 경우 D- 이성질체가 먼저 분리되어 나오나 표 3-1~ 모든 N-2, 4- 디니트로페닐아미노산의 광학분할에서는 L- 이
성질체가 먼저 분리되어 나온다. 마이크로컬럼내에서 BSA 분자를 실 리카 겔에 흡착시켜 얻은 BSA 키랄고정상을 이용한 비교적 최근의 광학분할연구에서는 표 3-19 에서 보는 바와 같이 유도체기의 종류 및 아미노산의 종류에 따라 용리순서가 달라짐을 확인할 수 있다 (38). BSA 키랄고정상과 광학분할하고자 하는 라세미 분석성분사이의 소수 성 상호작용, 정전기적 상호작용, 전하이동 상호 작용 등이 용리순서 를 결정하는 것으로 추측되나 아직 정확한 키랄성 인지 메커니즘은 밝 혀져 있지 않다. 최근에는 BSA 키랄고정상에서 구조가 아미노산의 유도체라고 생각
D— L— D— 。 10 min 0 10 20 min 30 그림 3-43 BSA 키랄고정상에서 a) 트레오닌 N- 프탈이미드 유도체와 b) 세린 N -벤젠술포닐 유도체의 광학분할. a) 이동상:인산영완충용액 (50mM) +2 . 0% 1- 프로판을 (pH 7.10) , b) 이동상 : 인산염완충용액 (50mM) pH : 5.78, 유속 : l.Om L/mi n, 검출기 : 225run UV (참고문 헌 : 37)
표 3-18 BSA 키랄고정상에서 아미노산 N-3, 5- 디니트로페닐 유도체들의 광학분할. 이 동상 : 인산염완충용액 (50mM) + 2% 1 - 프로판올 (pH 6.8 4 ) 유속 : l.5 m L/mi n (참고문헌 : 36) 아미노산 kI' k2' a’ 세린 (serin e ) 2.99 3.46 1.16 알라닌 (alanin e ) 20.5 26.8 1.30 메티오닌 (meth i o n in e ) 19. 8 80 4.00 아스팔트산 (asp a rtic ac id) 10.5 75.5 7.21 글루탐산 (glu ta m i c acid ) 11 .1 13.5 1. 21 페 닐글리 신 (ph eny lg l y c ine ) 36.2 93.6 2.59 페 닐알라닌 (ph eny la lanin e ) 44. 8 64.3 1.44 티로신 (tyros in e ) 29.9 38. 8 1.30 표 3-19 마이크로 컬럼 AGP 키랄고정상에서 아미노산 유도체의 광학분할에 대한 용리순 서. 이동상 : 0.05M 인산염완충용액(p H 7.0)+2% 2- 프로판올(참고문헌 : 38) >0 \ H.>. R Ar N COOH H Ar R= IC H3 R = CH2IlC H2CH3 R = CH2CIll H (CH3) 2 (a) 벤조일 (Benzoy l) L< D D< L D< L (b) m- 니트로벤조일 L< D D< L D< L (c) p-니트로벤조일 L
할 수 있는 류코보린 (leu covorin ) 旦이 광학분할된 바 있다 (39) . 트리 프토판이나 5- 히드록시트리프토판, 키누레닌 (k yn ure ni ne) 과 같은 방 향족아미노산의 경우에는 유도체로 만들지 않고 BSA 키랄고정상에서 광학분할이 가능하다 (30).
BSA 키랄고정상에서 광학분할이 가능한 라세미 아민화합물(양이온 성 화합물)의 예는 극히 드물다. 국소마취제로 쓰이는 프릴로카인 (pr il oc ain ) 旦의 경우에는 인산염완충용액 (pH = 8. 9, 50mM) 을 이동 상으로 사용하여 광학분할이 어느 정도 잘되는 것으로 (a = 1. 60) 보고 되어 있다 (40).
 广°H HCNNHlN3IcNHI:OH: Hl1二9 _ c 。 。 H
广°H HCNNHlN3IcNHI:OH: Hl1二9 _ c 。 。 H
BSA 키랄고정상에서 광학분할이 가능한중성 라세미 화합물둘은다 양하다. 키랄황화합물인 술폭시드화합물낌과 술폭시이민화합물낌이 광학분할되며 (35, 40) 혈액응고 방지제 (혹은 혈전증치료제)로 쓰이는 와 파린 (war fa r i n) 과 같은 쿠마린 유도체 盤 (40), 전정제로 쓰이는 옥사 제팜 (oxaze p am) 과 같은 벤조디아제핀 (benzodia z ep in) 유도체 盤 (40), 바아비탈산 유도체 (barbit ur at e) 겜 (41) , 벤조인 ~(35, 39 42) , a- 메 틸렌 - y-부티로락돈 器 등이(1 6) BSA 키랄고정상에서 광학분할됨이 보고되었다. 특히 (+)-벤조인의 경우에는 그림 3-44 에서 보는 바와 갇이 글루타르알데히드에 의하여 교차결합된 BSA 이 실리카 겔에 물
 H3C 仁OCH3 3 f : R dcb,,, RRR== =C CB(HrC (HC3H )J 3 2 CC6HH3/\/S 4~N 。 -C-NH-Ar
H3C 仁OCH3 3 f : R dcb,,, RRR== =C CB(HrC (HC3H )J 3 2 CC6HH3/\/S 4~N 。 -C-NH-Ar
그림 3-44 BSA 키랄고정상(글루타르알데히드교차결합 BSA- J리카 겔)에서 (+)-벤조인의 광학분할. 칼럼 : 150x4.6mm, 이동상 : 인산염완충용액 (50mM, pH 8.0) +2% 1- 프로판을, 유속 : l.O mL/mi n, 검출기 : 247run UV (참고문헌 : 42)
리적으로 고정된 키랄고정상에서 a 값이 2 . 7~ 냐i 광학분할이 아주 잘 이루어지고 있음을 확인할 수 있다 (42). 이들 여러 중성 라세미 화힙물들의 광학분할은 치환기의 종류에 따 라 입체 선택성 (a 값)및 머무름 시간 (k') 에 큰 차이가 있음이 알려져 있다. 예를 들면 의약품으로 중요한 키랄 황화합물인 술폭시드화합물 챕의 광학분할은 표 3 - 20 에서 보는 바와 같이 5- 위치의 치환기에 따 라 a 값 및 머무름 시간에 큰 차이가 있음을 확인할 수 있으며 (40) 벤 표 3-20 BSA 키랄고정상에서 술폭시드화합물 찬의 광학분할. 이동상 : 50mM 인산염완 충용액 + 2% I -프로판올, pH 6.0 ( 참고문헌 : 40) 화합물 kI' k2, a’ 20a 11. 7 26.9 2.3 20b 20. 9 57.4 2.7 20c 7.3 47.4 6.5 20d 10. 4 78.5 7.5 조디아제핀 유도체 盤 (40) 이나 바아비탈산 유도체 겜 (41) 의 광학분할 에 있어서도 치환기에 따라 광학분할의 특성이 달라진다는 사실이 확 인되었다 . 그러나 광학분할 특성이 치환기에 따라 어떻게 달라지는지 현재로서는 정확한 메커니즘을 모르는 실정이다. 다만 표 3-20 에 보 는 바와. 같이 술폭시드화합물낌의 방향족기에 붙어 있는 치환기의 종 류에 따라 광학분할특성이 크게 변화하는 사실로부터 BSA 키랄고정 상의 BSA 분자와 라세미 화합물 사이의 정전기적 상호작용, 소수성 상호작용, 수소결합 상호작용의에 전하이동 상호작용도 중요한 역할 을 하고 있다고 추측할 수 있다.
4. 3 .3 BSA 키랄고정상에서의 광학분할에 영향을 미치는 요인들 BSA 키랄고정상에서의 광학분할에 사용되는 표준이동상은 1- 프로 판을을 소량 함유하고 있는 인산염완충용액 (p H 범위 4.5-8 . 0) 으로서 광학분할은 이동상의 pH , 이동상으로 사용되는 완충용액의 인산염농 도, 사용되는 이동상변형제의 종류 및 양에 따라 크게 영향을 받는다 는사실이 알려져 있다. 일반적으로 아미노산의 NH 기가 아실기나 혹은 아릴기로 치환된 라세미 아미노산의 유도체들은 이동상의 p H 를 증가시킴에 따라 그립 3-45 에서 보는 바와 같이 머무름 시간이 감소할 뿐만 아니라 입체 선
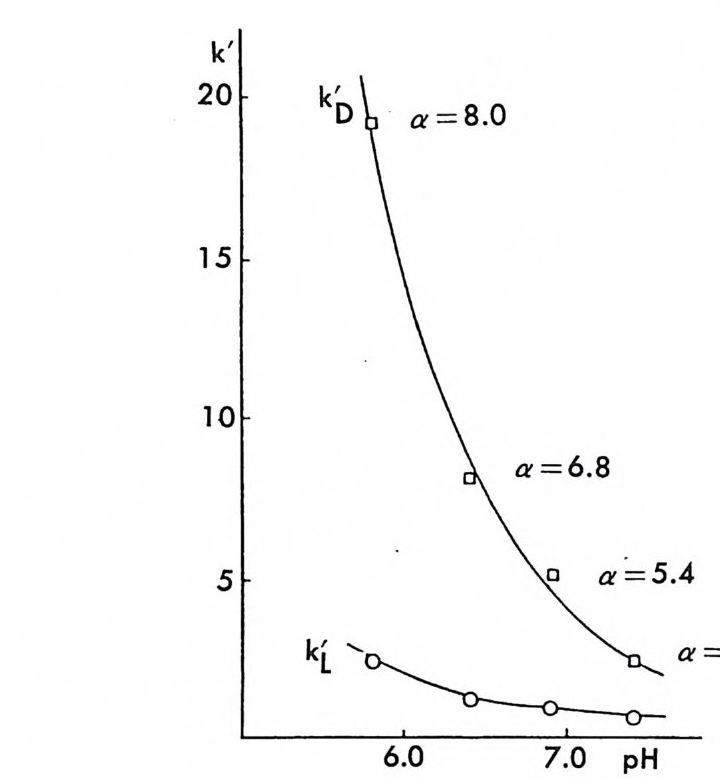 k'
k'
그림 3-45 BSA 키랄고정상에서 아스팔트산 N-2,4- 디니트로페닐유도체의 광학 분할에 대한 p H 의 영향. 이동상: 인산염완충용액 (50mM)+2% 1-프 로판을(참고문헌 : 36)
태성도 감소한다 (36). 이동상의 p H 가 증가하면 BSA 의 음하전수가 증가하게 되고 따라서 아미노기가 치환된 아미노산이나 혹은 다른 라 세미 카르복실산(음이온성 분석성분)사이의 정전기적 상호작용이 감소 하게 되어 머무롬 시간이 감소하는 것으로 생각되고 있다 (35). 그러나
 50 .£+0 a 5k0' Ir 24b la
50 .£+0 a 5k0' Ir 24b la
그림 3-46 BSA 키랄고정상에서 바아비탈산유도체 249 -l 광학분할에 대한 p H의 영향. 이동상 : 50mM 인산염완충용액 +6 굼 1- 프로판을. ki' (O ), kz' (6.) , a ( □) (참고문헌 : 41)
중성 라세미 화합물의 광학분할에 대한 이동상의 p H 영향은 다소 복 잡하다. 바아비탈산유도체 (barbit ur ate ) 갬 의 광학분할에 대한 이동상 의 pH 영향(그림 3-46) 에서 보는 바와 같이 치환기의 조그마한 변화 에 의해서 p H에 따른 머무롬 시간의 변화 및 입체 선택성의 변화가 아주 다른 양상을 보이고 있다 (41). 광학분할에 대한, 이와 같은 pH 의 영향은 아직 설명하지 못하고 있다. 다만 겜 a 의 경우 산성인 N -H 기를 가지고 있으므로_ p H 가 증가함에 따라 머무름 시간이 감소하 는 것은 음이온성 라세미 화합물의 광학분할에 대한 p H 의 영향과 비 • 슷한 경향을 보이는 것으로 생각할 수 있다. 특히 씬 C 와 겜 d 의 광학 분할에서는 R1 치환체가 페닐기인가 벤조일기인가에 따라 전혀 다른 p H의 영향을 보이고 있으며 p H에 따라 머무름 시간의 극대 혹은 극 소를 관찰할 수 있는 것은 p H 가 변함에 따라 광학분할 메커니즘이 달 라지고 있음을 짐작하게 한다. 카르복시기를 가지는 라세미 화합물의 광학분할에 있어서는 이동상 으로 사용하는 인산염완충용액의 인산영농도를 증가시켜 이동상의 이 온세기를 크게 하면 분석성분의 머무름 시간이 감소한다 (35, 40). 이동 상의 이온세기를 증가시킴으로써 키랄고정상의 BSA 분자와 분석성분 사이의 쿨룽인력 혹은 정전기적 인력이 감소하기 때문에 이러한 현상 이 관찰된다고 생각된다. 그러나 BSA 키랄고정상에서 N- 벤조일페닐 알라닌을 광학분할할 때는 그림 3-47 에서 보는 바와 갇이 완충용액의 농도를 lOOmM 이상으로 증가시키면 머무름 시간이 계속 감소하나 200mM이 지나서는 다시 머무름 시간이 증가한다 (35). 완충용액의 농 도가 lOOmM 에 이를 때까지는 키랄고정상의 BSA 분자와 분석 성분 사이의 정전기적 인력이 감소하여 머무름 시간이 감소하나 완충용액의 농도가 200mM 이상으로 증가하면 키랄고정상의 BSA 분자와 분석성 분 사이의 소수성 상호작용이 중요한 역할을 하게 되어 머무름 시간이 증가하는 것으로 예측되고 있다. N- 나프토일아미노산 (N-naph th o y la mi no ac ids ) 등과 같이 소수성
 k'
k'
그림 3-47 ~SA 키랄고정상에서 N- 벤조일페닐알라닌 (0), N- 벤조일알라닌 (O) , N- 벤조일세린 ( 스 )의 광학분할에 대한 완충용액 농도의 영향 (참고문헌 : 35)
이 큰 화합물들은 BSA 키랄고정상과 강한 소수성 상호작용을 하여 키랄고정상에 오래 머무른다. 소수성 상호작용에 의한 분석성분의 머 무름 시간은 이동상에 흔히 1- 프로판을과 같은 알칸을 (alkanol) 을
0-8% 사이의 적절한 비율로 첨가하여 이동상변형제로 사용함으로써 조정이 가능하다 (35, 40). 일반적으로 이동상에 첨가하는 알칸을의 양 울 증가시킴에 따라 키랄고정상의 BSA 분자와 분석성분 사이의 소수 성 상호작용이 약화되어 (BSA 분자와의 소수성 상호작용에 대하여 일컨올 과 분석성분이 경쟁관계에 있게 되므로) 그립 3-48 에서 보는 바와 같이 라세미 분석성분의 머무름 시간이 감소할 뿐만 아니라 입체 선택성 (a 값)도 감소하며 사용하는 알칸을의 소수성이 커지면 이와 같은 효과는 더 크게 나타난다 (43) .
 k'
k'
그림 3-48 BSA 키랄고정상 (Resolvos il)에서 N- 벤조일알라닌의 광학분할에 대 한 알코올 이동상변형제의 영향. 이동상 : 인산염완충용액 (50mM, pH 7.0) +이동상변형제, 에탄올 (O), . l- 프로판을(t:::,,), 1 -4~안 □) (참 고문헌 : 43)
이동상변형제로서 트리클로로아세트산과 같은 유기산을 사용하였을 때도 알칸을을 이동상변형제로 사용하였을 때와 마찬가지로 분석성분 의 머무롬 시간이 감소하였을 뿐만 아니라 입체 선택성이 감소하는 것
이 확인되었다 (39). 특히 트리클로로아세트산 등의 유기산은 BSA 분 쟈게 결합되어 있는 와파린 (war fa r i n) 을 치환한다는 사실이 보고된 것울 고려할 때 (39) BSA 키랄고정상에서의 와파린의 광학분할에 대 한 트리클로로아세트산의 영향은 크리라고 짐작할 수 있다. 실제로 BSA 키랄고정상에서 와파린을 광학분할할 때 트리클로로아세트산 (5.0mM) 을 이동상변형제로 사용하면 와파린의 두 광학 이성질체의 머무름 시간은 거의 50% 감소하고 입체 선택성은 단지 4% 만이 감소 하는 사실이 확인되었다 (39). 적절한 이동상변형제를 사용하여 광학분 할함으로써 입체 선택성에는 큰 손실없이 머무롬 시간을 크게 단축시 킬 수 있다는 것은 광학활성 물질의 광학순도를 결정할 때 분석시간을 크게 줄일 수 있다는 점에 있어서 큰 효과를 기대할 만하다고 하겠다. 4.4 기타 단백질을 기저로 한 키랄고정상 4.4.1 오보유코이드 키랄고정상 오보뮤코이드 (ovomuco i d) 는 계란의 흰자에서 쉽게 분리해낼 수 있 는 일종의 당단백질(g l yco p ro t e i n) 이다. Mi w a 등은 아미노프로필 실 리카 겔 (am i no p ro py ls i l i ca g el) 과 탄산 N, N- 디숙신이미딜 (N, N -dis u cci ni m i dy l carbona t e) 을 반응시키고 여기에 오보뮤코이드를 포 함하는 완충용액 (pH = 6.8) 을 가함으로써 넓은 p H 범위에서 안정한 오보뮤코이드 키랄고정상을 제조할 수 있었다 (44). 이 키랄고정상은 소수성인 분석성분과 강한 소수성 상호작용을 하는 것으로 알려져 있 으며 따라서 여러 종류의 방향족 라세미 화합물들의 광학활성에 적절 하게 이용될 수 있다 (45). 오보뮤코이드 키랄고정상에서 광학활성이 보고된 라세미 화합물들은 소영전통제로 쓰이는 플루르바프로펜 (flur bip ro fe n ) , 이부프로펜 (ibu p rpfen ) , 케토프로펜 (keto p ro fe n ) 및 항콜린성 의약품인 프로글루미드(p ro g lum i de) 낍과 같은 음이온성 라 세미 화합물들이며 오보뮤코이드 키랄고정상에서 광학활성이 보고된
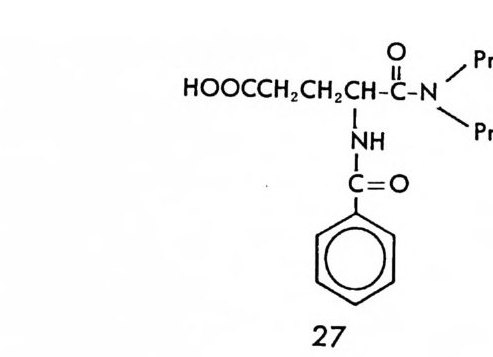 O P 『 O
O P 『 O
양이온성 라세미 화합물들은 클로르페니라민 (chlorph enir a mi ne ) 갬, 클로르프레날린 (chlorpr e nali ne ) 盤, 핀돌롤(pi ndolol) 述, 톨페리손 (tol pe ris o ne) 석 등이 있다. 오보뮤코이드 키 랄고정상에서 광학활성된 라세미 화합물들 중 중성 라세미 화합물로 생각되는 화합물에는 골격 극이완제로 사용되는 클로르페네신카르밤산 에스데르 (chlor p henes i carbamate ) 盤가 있다. 오보뮤코이드 키랄고정상에서의 광학활성에 대한 전형적인 크로마토그램은 그림 3-49 와 같다.
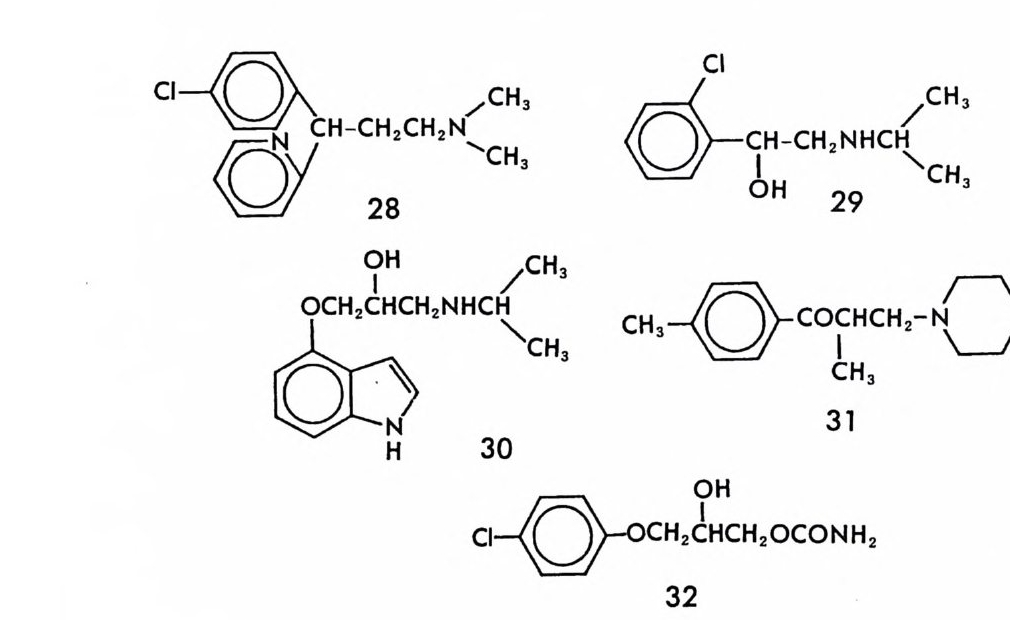 Cl< 仁>C H-C2 H82 CH2N`/CCHH33 d-rH-C\ :Hc(::
Cl< 仁>C H-C2 H82 CH2N`/CCHH33 d-rH-C\ :Hc(::
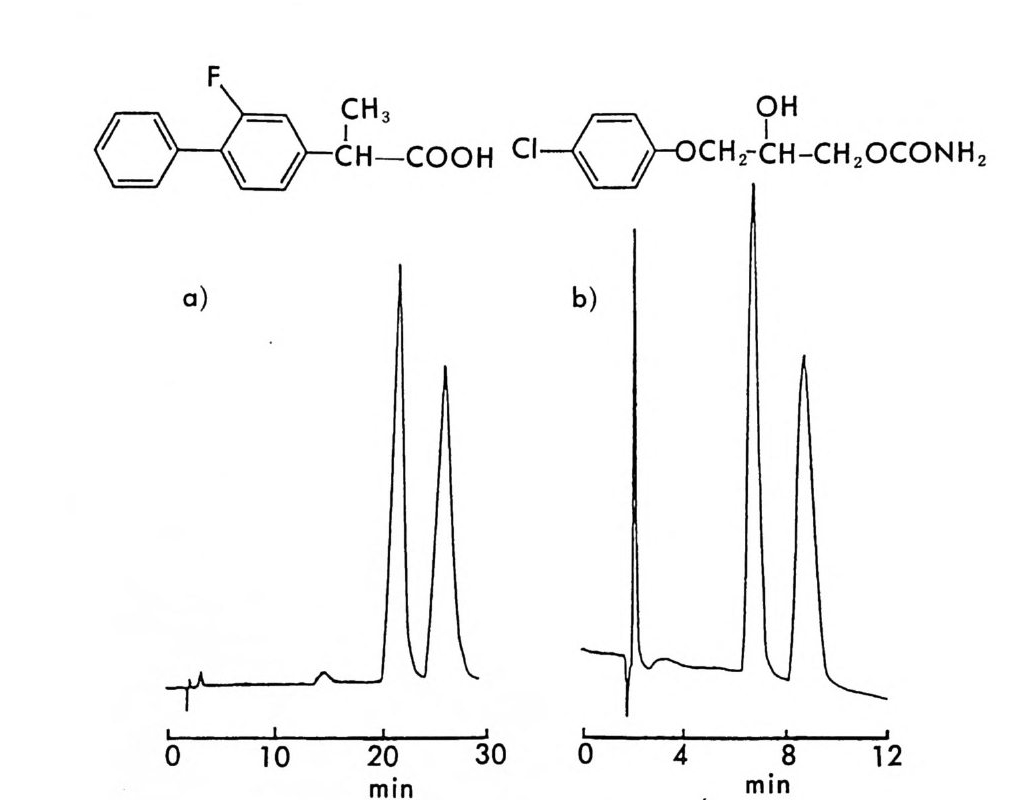 二F :HH二 cooH c1- Q-ocH ,-i :_CH ,OCONH,
二F :HH二 cooH c1- Q-ocH ,-i :_CH ,OCONH,
그림 3-49 오보뮤코이드 키 랄고정상에서의 광학분할. a) 플루르비프로펜 (flur bi. pro fe n ), 이동상 : 20mM 칼륨인산염완충용액 (pH 5 . 5)+5mM 옥틸술 폰산나트륨, 검출기 : 220nm UV, 유속 : l.Om L/mi n b) 클로르페네신 (chlorph enesin ) . 이동상 : 20mM 이수소인산칼륨완충용액 (pH 5.5 ) , 검출기 : 220nm UV, 유속 : l.Om L/ m i n (참고문헌 : 46)
AGP 키랄고정상과 BSA 키랄고정상에서의 광학분할과 마찬가지로 오보뮤코이드 키랄고정상에서의 광학분할은 이동상의 pH 및 이동상 변형제에 의하여 영향을 받는다. 음이온성 라세미 화합물의 광학분할 에서 이동상의 p H 를 증가시키면 머무롬 시간과 입체 선택성이 감소 하고 반면 양이온성 라세미 화합물의 광학분할에서 이동상의 p H 를 증가시키면 머무름 시간은 증가하나 입체 선택성은 라세미 화합물의 구조에 따라 증가하기도 하고 감소하기도 한다 (45). 표 3-21 에서 보는
표 3-21 오보유코이드 키랄고정상에서의 광학분할에 대한 p H 의 영향. 이동상 : 20mM 인산칼륨완충 g액 (참고문헌 : 45) 화합물 pH kI' k2, a’ 플루르비프로펜 (flur bip ro fe n ) 5.0 13.12 16. 73 1.28 5.5 10. 95 13.8 9 1.27 6.0 7.28 9.00 1.24 케토프로펜 (keto p ro fe n ) 5.0 12. 41 14 .97 1.2 1 5.5 9.55 11. 60 1.2 1 6.0 6.15 7.2 8 1.1 8 클로르페니 라민 (chlorph enir a mi ne ) 5.5 3.20 7.2 8 2.28 6.0 9.50 29.83 3.1 4 핀돌롤 (pind olol) . 5.5 0.46 0.68 1.48 6.0 1.31 1.88 1.4 4
바와 같이 음이온성 라세미 화합물인 플루르비프로펜의 광학분할에서 이동상의 p H 를 5.0 에서 5.5 , 6. 虹냐i 증가시킴에 따라 k1 ’ 값 과 k2, 값이 감소하며 a 값도 감소함을 확인할 수 있다. 그러나 머무름 시간 의 큰 변화에 비교하여 a 값의 변화는 그다지 큰 편이 아니다. 반면 양이온성 라세미 화합물인 클로르페니라민 광학분할에 있어서 p H 를 5.5 에서 6. 但도료 증가시키면 k1’ 과 k1’ 값이 크게 증가하며 a 값도 증 가한다. 그러나 양이온성 라세미 화합물인 핀돌롤의 광학분할에 있어 서는 p H 를 5.5 에서 6. 虹냐i 증가시켰을 때 k1 ’ 과 k2’ 값은 증가하는 반면 a 값은 감소함을 알 수 있다. 오보뮤코이드 키랄고정상에서 광학분할에 대한 이동상변형제의 영 향은 다른 종류의 단백질 키랄고정상에서의 광학분할과 대동소이한 것 으로 확인되고 있다 (45) . 4.4.2 a- 키모트립신을 기저로 한 ACHT 키랄고정상 a- 키모트립신 (a-ch ym o t r yp s i n:ACHT) 은 췌장에서 분비되는 소화
효소로서 페닐알라닌(p he), 티로신(ty ros i ne , tyr), 트리프토판(trp)과 같은 방향족 아미노산 L- 이성질체의 카르복시기를 포함하는 결합의 위치에서 펩티드사슬을 입체 선택적으로 가수 분해한다. 따라서 a- 키 모트립신을 컬럼 고형지지체에 고정시켜 키랄고정상으로 사용할 경우 에 페닐알라닌, 티로신, 트리프토판과 같은 방향족 아미노산들의 두 광학 이성질체를 구분할 수 있을 뿐만 아니라 페닐알라닌, 티로신, 트 리프토판을 포함하는 펩티드(특히 디펩티드)들의 광학 이성질체들을 구별해 낼 수 있을 것으로 기대된다. Wain e r 등은 공유결합된 글루타르알데히드를 포함하는 천수성고분 자 -실 리카 겔 (J. T. Baker 제품)을 인산염완충용액 (0.l M , pH 7.0) 으로 씻은 후 이것을 a - 키모트립신이 용해되어 있는 보론산나트륨용액 (0. lM, pH 8.7) 에 첨가하여 5 - 6 C 에서 적어도 1 8-'-]간 보관하고 1 8-'-]간 후 고체상을 분리해내어 인산염완충용액 (0. lM , pH 7.0) 으로 씻어줌으 로써 ACHT 키랄고정상을 제조할 수 있었다. 이때 고정상 l g당 고정 된 a- 키 모트립신의 양은 50 - 53m g인 것으로 계산되었다 (46, 47). 이 와 같이 제조된 ACHT - 키랄고정성을 이용하여 Wain e r 등은 표 3-22 에 보는 바와 같이 티로신과 트리프토판뿐만 아니라 페닐알라닌, 티로신, 트리프토판을 포함하는 여러 종류 디펩티드들의 광학 이성질 체들을 광학분할할 수 있었다 (47). 특히 ACHT- 키랄고정상을 N - 토 실 -L 페 닐 알 라 닌 클로로메 틸 케 톤 (N-to s yl - L-ph eny la lan ine chlor-ometh y l keto n e : TPCK) 으로 처리함으로써 효소의 활성화자리를 차단 하여 (48) 얻어전 비활성 ACHT- 키랄고정상에서도 활성 ACHT- 키랄 고정상에서와 마찬가지로 표 3-22 에서 보는 바와 같이 아미노산과 디 펩티들의 광학분할이 확인되었다 (47). 이와 같은 사실은 ACHT- 키랄 고정싱에 의한 광학분할에 있어서 ACHT 분자와 디펩티드 혹은 아미 노산의 상호작용은 ACHT 분자의 활성화자리에서 일어날 뿐만 아니 라 ACHT 분자와 아미노산 혹은 디펩티드 사이의 소수성 상호작용도 광학분할에 중요하게 작용하고 있음을 시사해 주고 있다. 예를 들면
표 3-22 ACHT- 키랄고정상 및 비 활 성 ACHT - 키랄고정상에서의 광 학 분 할. 컬럼 : 100x4.6mm, 이동상 : 인산영완충용액 (0.1 2 3M , pH 6.0 ) 유속 : 0 . 30mL/m i n( 참 고문헌 : 47) ACHT - 키랄고정상 비활성 ACHT - 키랄고정상 분석성분 k' a’ k' a’ D-Ty r 0.3 4 1.03 0.36 1.00 L-Ty r 0.3 5 0.3 6 L-Ty p 1.00 1.16 1.02 1.03 D-Ty p 1.16 0.9 9 L- Le u- D -Phe 0.3 9 1.00 0.5 5 1.07 D-Leu-L-Phe 0.39 0.59 L-Leu- L - P he 0.41 1.05 0.5 9 1.05 D- L eu- D -Phe 0.43 0.6 2 L-Leu-D-Ty r 0.4 7 1.00 0.4 4 1.02 D- L eu-L-Ty r 0.47 0.45 L-Leu-L-Ty r 0.6 9 1.00 0.53 1.00 D-Leu-D-Ty r 0.69 0.5 4 L-Ty p- D-Leu 1.05 1.04 1.1 8 1.02 D- T y p- L-Leu 1.09 1.20 D- T yp-D-Leu 1.91 1.02 1.5 7 1.01 L-Ty p- L-Leu 1.95 1.59
ACHT- 키랄고정상과 비활성 ACHT- 키랄고정상에서의 Leu-Ph@ l- Leu-T y r 의 광학분할에 대한 용량인자 k' 를 비교할 경우 표 3-22 에서 보는 바와 같이 Leu-Ph 타곤 비활성 ACHT- 키랄고정상에서 더 큰 용 량인자값을 보이나 Leu-T y r 은 활성 ACHT- 키랄고정상에서 용량인 자값이 더 크다. Ph 얹논 T y r 보다 소수성이 더 큰 반면, T yr은 ACHT 에 대하여 더 큰 친화성을 보이기 때문에 활성 ACHT- 키랄고 정상에서는 분석성분과 ACHT 분자 사이의 효소활성화자리에서의 상
호작용이 더 중요하여 Leu - T y r 의 경우 Leu-Ph 안i다 더 큰 k' 값을 보이나, 비활성 ACHT- 키랄고정상에서는 소수성 상호작용이 중요하 게 되어 Leu-Phe 이 더 큰 k' 값을 보이는 것으로- 예측할 수 있다 (47). ACHT 키랄고정상에서 아미노산 및 디펩티드의 광학분할에 대한 입체 선택성은 표 3 - 22 에서 보는 바와 같이 그다지 좋은 편이 아니 다. 그러나 ACHT- 키랄고정상은 특정한 종류의 라세미 화합물에 대 하여서만 입체 선택성을 나타내는 효소를 이용하여 특정한 라세미 화 합물의 광학분할에 유용하게 응용할 수 있는 키랄고정성을- 제조할 수 있는 가능성을 보인 좋은 예가 될 것이다.
참고문헌(제 3 장, I) 1. S. V. Rog o zhin and V. A. Davankov, Russ. Chem. Rev. 선 r, 565 (19 68). 2. D. R. Buss and T. Vermeulen, Ind. and Eng. Chem., 헨 12(1968). 3. C. H. Lochmi ille r and R. W. Soute r , J. Chromato g r., 墨 283 (1975). 4. G. M. Henderson and H. G. Rule, J. Chem. Soc., 1568 (1939). 5. V. Prelog and P. W iel and, Hel. Chem. Acta . , 끄 , 1127 (1944). 6. H. Hess, G. Burge r and H. Musso, Ang e w. Chem. Int. Ed. Eng l., 끄 . 612 (1978). 7. R. K. Hayn es, H. Hess and H. Musso, Chem. Ber. 맨 r, 3733 (1974). 8. G. Konrad and H. Musso, Lieb ig s Ann. Chem., 1956 (1986). (제 3 장, 2) 1. M. Kota k e, T. Saken N. Nakamura, and S. Senoh, J. Am. Chem. Soc., 전 , 2973 (1951).
2. C. E. Dalgl ies h, J. Chem. Soc., 3940 (19 52). 3. T. Sh iba ta , I. Okamoto , K. Ishii , J. Liq. Chromato g r. 안 313 (19 86). 4. G. Gti bi t z, W. Jel lenz and J. Schonleber, J. High Res. Chromato g r . Commun., 던, 31 (1980). 5. T. Fukuhara, M. Isoy a ma, A. Shim ada, M. ltoh and S. Yuasa. J. Chromato g r., 쁘?, 562 (1987). 6. T. Fukuhara, M. ltoh , M. Isoy a ma, A. Shim ada, and S. Yuasa. J. Chromato g r., 쁘산, 325 (19 86). 7. G. Hesse and R. Hag e l, Chromato g rap h ia , ~. 277 (19 73). 8. G. Hesse and R. Hag e l, Chromato g rap h ia , ~. 62 (1976). 9. Y. Okamoto , M. Kawashim a, K. Yamamoto and K. Hata d a, Chem. Lett ., 739 (1984). 10. Y. Okamoto , R. Aburata n i, T. Fukumoto and K. Hata d a, Chem. Lett ., 1857(1987). 11. Y. Okamato , R. Aburata n i, K. Hata n o and K. Hata d a, J. Liq. Chromato g r., 브, 2147 (19 88). 12. E. Merck, Darmsta d t, Germany . 13. G. Hesse and R. Hage l, Jus tu s Lieb ig s Ann. Chem., 996 (19 76). 14. G. Blaschke, Ang e w. Chem. Int. Ed. Eng l., 브 13 (1980). 15. W. H. Pir k le and B. C. Hamp er , in Prepa r ati ve Liq u id Chromato g r a - phy , B. A. Bidl i ng me y e r Ed., Elsevie r , Amste r dam, 1987, chap 7. 16. A. Ich ida and T. Sh iba ta , in Chromato g r a p hi c Chir a l Se pa rati on s, M. Zie f and L. J. Crane, Eds., Marcel Dekker, New York, 1988, chap 9. 17. K. Schlog l and M. W idh alm, Chem. Ber., 墨 3042 (19 82). 18. K. Berts c h and J. C. Joc h im s, Tetr a hedron. Lett ., 4 379 (19 77). 19. K. Schlog l, W. Weis s enste i n e r and M. W idh alm, J. Org. Chem., 旦, 5025 (1982). 20. K. R. Lind ner and A. Mannschreck. J. Chromato g r., 뿌, 308 (1980). 21. R. Isaksson, T. Lil jefo r s and P. Rein h oldsson, J. Chem. Soc. Chem. Comm., 137 (1984). 22. T. Sh iba ta , K. Mori and Y. Okamoto , in Chir a l Se pa rati on s by HPLC : Ap pli c a ti on s to Pharmaceuti ca l Comp ou nds, A. M. Krstu l ov ic, Ed., Ell is H orwood, Ch ich este r , 1989, chap fa.
23. P. Newman, Optica l Re solu ti on Proc e d u res for Chem i ca l Com- pou n ds, vol 2, Op tica l Resoluti on Info r mati on Cente r , N ew York, 1981, Part Il , pp. 1000- 10 06. 24. G. Blaschke, H. -P , Kraft and H. Markg raf , C hem. Ber., 壁 3611 (1983). 25. G. Blaschke, J. L ig. Chromato g r., 언 , 341 (1986). 26. R. Isaksson and B. Lamm, J. Chromato g r., 혼 , 436 (1986). 27. A. Husseniu s , R . Isaksson and 0. Mats s on, J . Chromato g r., 堅 155 (19 87). 28. P. Erlandsson, R. Isaksson, I. Ni lss on and S. Wold. J. Chromato g r., 쁘 , 364 (1989). 29. C. Roussel, J.- L. Ste i n , F. Beauvais and A. C. Esip s oi, J. Chromato g r. , 쁘 , 95 (19 89). 30. A. M. Ri zz i, J . C hroma t ogr . 산~ . 71 (19 89). 31. R. Isaksson, P. Erlandsson, L. Hansson, A. Holmberg and S. Berner, J. Chromato g r., 쁘~. 257 (1990). 32. A. Ichid a , T. Shib a ta , I. Okamoto , Y. Yuki, H . Nami ko shi and Y. Tog a , Chromato g rap h ia , 브 , 280 (1984 ). 33. Y. Okamoto , M. Kawashim a and K. Hata d a, J . Am. Chem. Soc. 산§ 5357 (1984 ). 34. Y. Okamot o, M. Kawashim a and K. Hata d a, J. Chromato g r., 쁘침, 173 (19 86). 35. Y. Okamoto , R. Aburata n i, M. Kawashim a, K. Hata d a and N. Okamura, Chem. Lett ., 1767 (19 86). 3367.. LY. . MOi lklearm aontdo , H .Y B. usKha, iJd . a C, hrRo. maAtbo ug rra.,t 뽀a n i ; , 3a3n7d ( 19K89. ). Hata d a, J. Chromato g r, 旦 I , 367 (1989). 38. I. D. Rail ton , J. Chroma t o gr.신笠, 371 (1987). 39. Y. Okamoto , R. Aburata n i, Y. Kaid a and K. Hata d a, Chem. Lett ., 1125 (1988). 40. Y. Okamoto , R. Aburata n i, K. Hata d a, J. Chromato g r:, 쁘, 454 (1988). 41. D. M. McDanie l , B. G. Snid e r, J. Chroma t o gr.맨산, 123 (1987).
42. Ap plica ti on Guid e for Chir a l Columm Selecti on , 1989, Daic e l Chem- ica l Industr i e s , LTD. 43. Y. Okamoto , M. Kawashim a, R. Aburata n i, K. Hata d a, T. Ni- shiy a ma and M. Masuda, Chem. Lett ., 1237 (19 86). 44. A. M. Krstu l ovic , G. Rossey, J.-P . Porzie m sky , D. Long and I. Chekroun, J. Chromato g r., 브~. 461 (1987). 45. Y. Okamoto , R. Aburata n i, S.-i, Mi ur a and K. Hata d a, J. Liq. Chromato g r., 팸 1613 (1987). 46. P. Macaudie r e, M. Caude, R. Rosset and A. Tambute , J. Chromato g r. S c i., '!]_, 583 (1989). 47. Y. Okamoto , R. Aburata n i and K. Hata d a, J. Chromato g r., 홍 95 (1987). 48. I. W. Wain e r, and M. C. Alembik , J. Chromato g r.' 호 , 85 (19 86). 49. I. W. Wain e r, M .C. Alembik and E. Smi th, J. Chromato g r., 쁘 , 65 (1987). 50. I. W. Wain e r, R. M. Sti ffin and T. Shib a ta , J. Chromato g r., 虫 139 (19 87). (제 3 장, 3) 1. T. J. Ward and D. W. Armstr o ng , in Chromato g r aph i c Chir a l Sep a rati on s, M. Zie f and L. J. Crane, Eds., Marcel Dekker, New York, 1988, chap 5. 2. D. Sy b il sk a and J. Zukowski, in Chir a l Se pa rati on s by HPLC : Ap pli ca ti on s to Pharmaceuti ca l Comp ou nds, A. M. Krstu l ovic , Ed., El lis H orwood, Chic h este r , 1989, chap 7. 3. M. L. Bender, and M. Komi ya ma, Cy c lodextr in Chemi stry, Sp ri n g e r -Verlag, Berlin , 1978. 4. A. Harada, M. Furue and S.-I. Nozakura, J. Polym . Sci., 브, 189 (1978).
5. B. Zsadon, L. Decsei, M. Szil a si, F. Ti. id i:i s and J. Szejt li, J. Chromato g r. , 壅 127 (19 83). 6. B. Zsadon, M. Szil a si, L. Decsei, A. Uj ha zy and J. Szejt li, J. Chromato g r., 쁘~. 428 (1986). 7. K. Fuji m ura, T. Ueda and T. Ando, Anal, C hem., 완 , 446 (1983). 8. K. Fuji m ura, M. Ki tag a wa, H. Takay a nag i and T. Ando, J. L iq . Chromato g r., 언 , 607 (19 86). 9. Y. Kawag uc hi, M . Tanaka, M. Nakae, K. Funazo and T. Shono, Anal. Chem., 완 , 1852 (19 83). 10. D. W. Armstr o ng and W. DeMond, J. Chromato g r. Sci., 쁘, 411 (1984). 11. D. W . Armstr o ng , U. S. Pate n t, No 4, 539, 399, Sep. 3, 1985. 12. J. Zukowski and D. Sy b il s ka, J. Boja r ski and J. Szejt li, J. Chromato g r., 쁘, 381 (1988). 13. J. Zukowski and R. Nowakowski, J. Liq. Chromato g r., 프, 1545 (1989). 14. T. J. Ward and D. W. Armstr o ng , J . Liq. chromato g r., ~ . 407, (19 86). 15. D. W. Armstr o ng , T. J. Ward, R. D. Amstr o ng and T. E. Beesley , Sc ien ce, 썬~ . 1132 (19 86). 16. W. L. Hi nz e, T. E. Rie h l, D. W . Armstr o ng , W. DeMond, A. Alak and T. Ward, Anal. Chem., 몬 , 237 (19 85). 17. D. W . Armstr o ng , X. Yang , S. M. Han and R. A. Meng e s, Anal. Chem. , 5 9, 2594 (1987). 18. J. H. Mag uire , J. Chromato g r., 흰 , 453 (1987). 19. S. M . Han, Y. I. Han and D. W. Armstr o ng , J . Chromato g r., 뜨, 376 (1988). 20. D. W. Armstr o ng , W. DeMond and B. P. Czech, Anal. Chem., 몬 , 481 (1985). 21. D. W. Armstr o ng , T. J. Ward, A. Czech, B. P. Czech and R. A. Barts c h, J. Org. Chem., 텐, 5556 (1985). 22. K. G. Feit sm a, J. Bosman, B. F. H. Drenth and R. A. De Zeeuw, J. Chromato g r., 호 , 59 (1985). 23. K. G. Feit sm a, B. F. H. Drenth and R. A. De Zeeuw, J. Chromato g r., 쁘?, 447 (1987).
24. J. L Seeman, H. V. Secor, D. W . Armstr o in g , K. D. Ti m mons and T. J. Ward, Anal. Chem., 쁘, 2120 (1988). 25. H. Y. Aboul-Enein , M. R. Islam and S. A. Bakr, J. Liq. Chromato g r., 11, 1485 (1988). 26. L.-E . Edholm, C. Lind berg, J. Paulson and A. Walhag e n, J. Chromato g r., Biom . Ap pl., 썬산, 61 (1988). 27. L. Coventr y , in Chir a l Liq u id Chromato g r ap hy , W. J. Loug h , Ed., Blackie , Glasgo w and London, 1989, chap 8. 28. B. G. Snid e r, J . Chroma t o gr.선완, 548 (19 86). 29. S. M. Han and D. w. Armstr o ng , in Chir a l Sep a r ati on s by HPLC : Ap pli ca ti on s to Phannaceu ti ca l Comp ou n ds, A. M. Krstu l ov ic , Ed., Ellis Horwood, Chic h este r , 1989, chap 10. 30. Y. Mats u i, K. Mochid a , Bull. C hem. Soc. Jpn ., ~ . 2808 (1979). 31. P. Macaudie r e, M. Caude, R. Rosset and A. Tambute , J. Chromato g r., 쁘 135 (1987). 32. P. Macaud ier e, M. Caude, R. Rosset and A. Tambute , J. Chromato g r. Sc i., 낀, 583 (1989). 33. T. D. W ilso n, J. Chroma t o gr . 신~. 31 (1988). (제 3 장, 4) 1. U. K. Walle, T. Walle, S. A . Bai and L. S. Olanoff , Clin . Pharmacol. Therap ., ~ . 718 (1983). 2. K. Schmi d, in the Plasma Prote i n s , vol 1, F. W. Putn a m, Ed., Academi c Press, New York, 1975, chap. 4. 3. J. H ermansson, J. Chromato g r. 쁘언, 71 (1983). 4. J. Hermansson, J. Chromato g r., 쁘천, 67 (1984). 5. J. Hermansson, Eur. Pat. Ap pl. No. 84850169, 8, Pub!. No. EPO 128886 AZ. 6. J. H ermansson and G. Sch ill, in Chromato g r ph ic Chir a l Se pa rati on s,
M. Zie f and L. J. Crane, Eds., M arcell Dekker, New York, 1988, c hap 10. 7. J. Hermansson, J. Chromato g r., 쁘~. 379 (1985). 8. G. Schil l, I. W . Wain e r and S. A . Barkan, J . L iq . Chromato g r., 언 , 641 (1986). 9. G. Schil l, I . W. Wain e r and S. A. Barkan, J. Chromato g r .. ~ , 73 (1986). 10. K. Balmer, B.- A . Persson and G. Schil l, J. Chromato g r., 巴 107 (19 89). 11. H. Y. Aboul-E nein and M. R. Islam, J. Chromato g r, Sc i., 쁘 , 616 (19 88). 12. J. Hermansson and M. Erik s son, J. liq . Chromato g r., 언 , 621 (1986). 13. P. L. Corre, D. Gi ba ssie r , P . Sado and R. L. Verge , J. C hromato g r., 쁘, 211 (19 88). 14. M. Lien ne, M. Caude, R. Rosset and A. Tambute , J. Chromato g r., 쁘, 406 (1989 ). 15. H. M. Nonhebel, J . Chromato g r., 쁘 , 374 (1987). 16. M. Lien ne, M. Caude, R. Rosset and A. Tambute , J. Chromato g r., 쁘 , 55 (19 88). 17. S. E. Marrin e r, D . L. Morris, B. Dic k son and J. A . Bog a n. Eur. J. Clic . Pharmacol. , 쁘 , 705 (1986). 18. M. Lie n ne, M. Caude, R. Rosset and A. Tambute , J. Chromato g r., 472, 265 (1989). 19. J. Hermansson, M. Erik s son and 0. Ny qu is t , J. Chromato g r., 쁘; , 321 (19 84). 20. P. L. Corre, D. Gi ba ssie r , P. Sado and R. L. Verge , J. Chromato g r., 쁘, 211 (1988). 21. E. D. J. L ee, S. B. Ang and T. L. Lee, J . Chromato g r., 쁘, 203 (19 87). 22. Y. K. Tan and S. J. Soldin , J. Chromato g r., ~. 187(1987). 23. I. W. Wain e r, in Chir a l Liq u id Chromato g r aph y , W. J. L oug h , Ed., Blackie , Glasgo w and London, 1989, chap 7. 24. T. Pete r s, Jr., in the Plasma Prote i n s , vol 1, F. W. Putn a m, Ed., Academi c Press, New York, chap. 3.
25. A. Rey, J. A. Rey n olds, H. Polet and J. Ste i n h ardt, B io c hem., §_, 2606 (19 66). 26. R. H. McMenamy and J. L. Oncley, J. Bio l . Chem. , 혹 1436 (19 58). 27. K. K. Ste w art a nd R. F. Doherty , Proc. Natl . Acad. Sci. U . S. A., 잰 2850 (1973). 28. C. Lag e rcrantz , T. Larsson and H. Karlsson, Anal. Bi oc hem., 왠 352 (1979). 29. C. Lag e rcrantz , T. Larsson and I. Denfo r s, Comp . Bio c hem. Phys i o l . [C] , 쁘, 375 (1981). 30. S. Allenrnark, B. Bomg ren and H. Boren, J. Chromato g r., 쁘 I, 473 (1982). 31. S. Allenrnark, and B. Bomg ren , J. Chromato g r., 쁘 ~. 297 (1982). 32. S. Allenrnark, B. Bomg re n and H. Boren, J. Chromato g r. , 쁘산, 63 (1983). 33. (P1.9 E86r)la. n dsson, L. Hansson and R. Isakkson, J. Chromato g r., 370’, 4 75 34. M. Aubel and L. B. Rog e rs, J. C hromato g r., 쁘, 415 (1987). 35. S. Allenrnark, B. Bomg ren and H. Boren, J. Chromato g r., 완 § , 617 (1984). 36. S. Allenrnark and S. Andersson, J. Chromato g r., 쁘 ~. 231 (1986). 37. B. Bomg ren and S. Allenrnark, J. Liq. Chromato g r., 언 , 667 (1986). 38. J. Vi nd evog el , J . V. Di jck and M. Verzele, J. C hromato g r., 션 T, 297 (1988). 39. I. W. Wain e r and Y.-Q . Chu, J. Chromato g r., ~. 316 (19 88). 40. S. Allenrnark, J. Liq. Chromato g r., 언, 425 (1986). 41. S. Allenrnark, S. Andersson and J. Boja r ski, J . Chromato g r., 쁘 479 (1988). 42. R. A. Thomp so n, S. Andersson and S. Allenrnark, J. Chromato g r., ~ . 263 (1989). 43. S. Allenmark, in Chir a l Sep a rati on s by HPLC : Ap pli ca ti on s to Pharmaceuti ca l Comp ou nds A. M. Krstu lovic , Ed., Ell is H orwood, Ch ich este r , 1989, chap 11. 44. T. Mi w a, M. Ich ika wa, M. Tsuno, T. Hatt or i, T. Miya kawa, M.
Kay a no and Y. Mi ya ke, Chem. Pharm. Bull. (Toky o) , 완, 682 (19 87). 45. T. Mi w a, T . Mi ya kawa, M. Kay a no and Y. Mi ya ke, J. Chromato g r., 쁘, 316 (19 87). 46. I. W. Wain e r, P . Jad aud, G. R. Schonbaum, S. V. Kakodkar and M. • P. Henry, Chromato g rap h ia , 空, 903 (19 88). 47. P. Jad aud and I. W . Wain e r, J . Chromato g r., 쁘 165 (1989). 48. D. M. Blow, Acc. Chem. Res., 언 , 145 (1976).
제 4 장 광학활성 합성고분자를 이용한 키랄고정상 1 서론 광학활성 천연고분자(광학활성 천연물)을 키랄고정상으로 사용하여 라세미 화합물의 광학분할이 가능한 것과 마찬가지로 광학활성인 합성 고분자를 키랄고정상으로 사용하면 여러 종류의 라세미 화합물을 광학 분할할 수 있을 것으로 쉽게 예상할 수 있다. 광학활성 천연고분자들 은 그 종류가 한정적인 반면 합성방법이나 사용·하는 모노머에 따라 다 양한 종류의 광학활성 합성고분자를 합성할 수 있으므로 사용목적에 따라 다양한 종류의 합성고분자 키랄고정상을 제조하여 라세미 화합물 의 광학분할에 시용할 수 있을 것으로 예상할 수 있다. 그러나 현재 광학활성 합성고분자를 기저로한 키랄고정상의 종류는 그다지 다양한 편이 아니다. 광학활성 합성고분자는 크게 고분자의 나선성때문에 키랄성인 것, 키랄모노머를 출발물질로 하여 중합반응을 실시하였기 때문에 모노머 의 키랄성으로 인하여 합성된 고분자가 키랄성인 것, 합성고분자 구조 에 키랄공동이 존재하기 때문에 키랄성인 것의 세 가지로 분류할 수
있으며 모두 그 자체로서 혹은 실리카 겔에 흡착시키거나 실리카 겔에 공유결합시켜 키랄고정상으로 사용할 수 있다. 2 광학활성인 나선성 고분자를 기저로 한 키랄고정상 2.1 광학활성인 나선성 고분자의 합성 및 광학활성인 나선성 고분자를 기저로 한 키랄고정상의 제조 구조의 나선성때문에 광학활성인 고분자의 합성은 1979 년 Okamo t o 에 의하여 처음 보고되었다 (1). 트리페닐메틸메타크릴레이 트(t rip heny lme t h y l me t hacry la t e:TrMA) 를 광학활성 키랄음이온 개 시제인 (―)-스파르타인-¥-틸리튬 착물 (s p ar t e i ne - Bu Li -com p lex) 존 재 하에 ― 7 S' C 에서 톨루엔을 용매로 하여 음이온 중합하면 비대칭 유 발 중합반응에 의하여 이소탁틱 비닐 폴리머 (iso ta c ti c vin y l po lym e r) 인 (+)구문리트리페닐메틸메타크릴레이트 (PTrMA) 가 얻어진다(그림 4-1). (+)-PTrMA는 순수하게 한쪽 방향만의 나선성을 가진 고분자 물질로서 나선성때문에 광학활성을 나타내는 최초의 합성고분자이다.
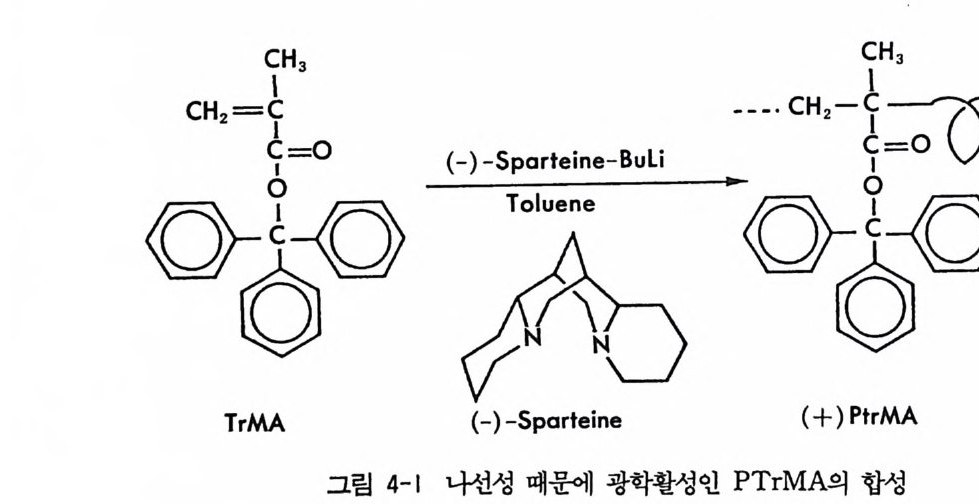 언 3 언
언 3 언
비대칭 유발 중합반응의 음이온개시제로서 (+) - (2S,3S) -디메목시 -1, 4 비스(디메틸아미노)부탄 - 리튬아미드칙물 (DDB-L i ami de com- p lex) 을 사용하였을 때는 (-)-스파르타인유 L 틸리튬 착물을 사용하였 울 때와 마찬가지로 광학활성인 ( + )-PTrMA 가 얻어지며 (+)-DDB l 과 마찬가지로 (— )-DDB- 리튬아미드 착물을 음이온 개시제로 시용 하여 중합반응하면 (-)-PTrMA 도 같은 방법으로 합성할 수 있다 (2).
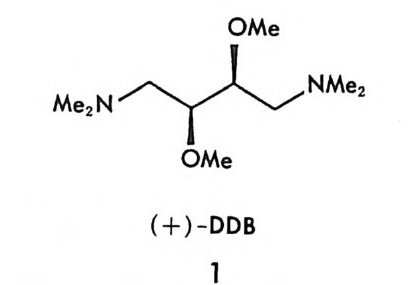 OMe
OMe
결정성이며 불용성인 (+)-PTrMA 는 그 자체로서 라세미 화합물 의 광학분할에 응용-할 수 있는 키랄고정상으로 사용할 수 있다. 실제 로 1. 3 g의 (+)-PTrMA를 20-44µm 로 잘게 분쇄한 후 250X4.6 mm( 내경)인 HPL C-용 컬럼에 충전하여 여러 종류 라세미 화학물들의 광학분할에 성공적으로 사용된 바 있다 (3,4,5). 그러나 고분자입자의 부서지기 쉬운 성질때문에 분쇄한 (+)-PTrMA 로 충전된 키랄컬럼 의 내구성은 제한되어 있다. 분쇄한 (+)-PTrMA 로 충전된 키랄컬럼의 내구성에 대한 단점을 보완하는 방법으로, 유기용매에 가용성인 저분자량의 (+)-PTrMA 롤 합성하고 이것을 다공성인 실리카 겔에 흡착시킴으로써 내구성이 좋은 키랄고정상을 제조하여 라세미 화학물들의 광학분할에 응용하고 있다 (6). 이때 키랄고정상을 제조하기 위하여 사용되는 저분자량의 (+)-PTrMA는 (-)-스파르타인우틸리튬 착물 대신 (+)-6- 벤질스 파르타인유 L 틸리듐 착물을 음이온 개시제로 사용하여 합성된 것이다.
THF 에 녹인 저분자량의 (+)-PTrMA 가 흡착되는 실리카 겔은 트 리에틸아민 존재하에서 톨루엔을 용매로 사용하여 입자 크기가 lOµm 인 다공성 실리카 겔 (Lic h rosph er SI 1000, Merck 제)과 디클로로페닐 실란을 환류시켜 얻은 것으로서 이렇게 변형된 실리카 겔울 저분자량 의 (+)-PTrMA 의 THF용 액과 함께 처리함으로써 (+)-PTrMA 키랄고정상이 얻어진다. 이렇게 제조된 (+)-PTrMA 키랄고정상은 실리카 겔의 좋은 기계적 성질로 인하여 좋은 내구성을 보이는 것으로 확인되었으며 현재는 Chir a lpa k OT(+) 라는 상품명으로 공급되고 있다 (제 9 장 참조). 그러나 (+)-PTrMA 키랄고정상의 단점은 메탄을과 같은 용매를 이동상으로 사용할 경우에 (+)-PTrMA 분자의 에스데르결합이 서서 히 가수분해되어 메틸트리페닐 메틸에데르를 형성하기 때문에 (+) -PTrMA 키랄고정상이 손상을 받는다는 것이다. 따라서 알코올 계통 의 용매를 이동상으로 사용할 경우에는 PTrMA 분자의 가수분해 속 도를 줄이기 위하여 낮은 온도에서 크로마토그래피를 실시하여 광학분 할할 필요가 있으며 컬럼을 보관할 때 사용하는 용매로는 알코올 계통 의 용매를 사용하지 말아야 한다. 디페닐 -2- 피리딜메틸메타크릴레이 트 (D2P y MA) 나 디페닐 -4- 피리딜메틸메타크릴레이트 (D4P y MA) 인 경 우에는 TrM A:보다 알코올에 의한 가수분해속도가 훨씬 늦은 것으로 알려져 있다. 따라서 D2P y MA 나 D4P y MA 를 출발물질로 하여 키랄 음이온 개시제 존재 하에 비대칭유발 음이온 중합반응을 하면 (+) -PTrMA 와 비슷한 광학활성 고분자 PD2Py M A 2 와 PD4Py M A J이 얻어지며, (+)-PTrMA 키랄고정상의 제조방법을 이용하여 2 혹은 3 을 기저로 한 키랄고정상이 제조된다 (7,8). 2 혹은 3 을 기저로 한 키 랄고정상은 알코올에 의한 가수분해 속도가 (+)-PTrMA를 기저로 한 키랄고정상보다 20 배 정도 느리므로 메탄올을 이동상으로 사용하 여 컬럼에 아무 손상 없이 실온에서 사용가능한 것으로 알려져 있다. 실제로 PD2P y MA를 실리카 겔에 흡착시켜 얻어진 키랄고정상은
HPLC 용 스데인레스강 컬럼에 충전되어 Chir a lpa k OP(+) 라는 상 품명으로 공급되고 있다(제 9 장 참조).
 Cl H3 C1H 3
Cl H3 C1H 3
다공성 실리카 겔에 흡착된 (+)-PTrMA 키랄고정상 혹은 (+) -PD2Py M A 키랄고정상을 사용하여 광학분할할 때에는 방향족 탄화 수소계의 용매나 클로로포름, THF 와 같은 용매를 이동상으로 사용 할 수 없다. 왜냐하면 실리카 겔에 흡착된 고분자가 용매에 용해되기 때문이다. 그러나 최근에는 (+)-PTrMA 를 실리카 겔에 공유결합시 킨 키랄고정상의 제조에 관한 연구가 보고되었으며 이 공유결합 (+) -PTrMA 키랄고정상은 클로로포름이나 혹은 THF 에 용해될 수 있는 광학활성 물질이나 나선성 고분자의 분석에 유용하게 이용될 수 있다 (9). 2.2 광학활성인 나선성 고분자 키랄고정상에서의 광학분할 예 1980 년, 분쇄한 (+)-PTrMA를 기저로 한 키랄고정상이 처음 제 조되어 라세미 화합물의 광학분할에 사용되었고 이어서 1981 년 다공 성 실리카 갤에 흡착된 (+)-PTrMA 키랄고정상이 제조되어 라세미 화합물의 광학분할에 사용되어 두 키랄고정상 모두 라세미 화합물의
학분할에 응용할 수 있음이 확인되었다. 표 4 - 1 과 그림 4 - 2 의 대표적 인 크로마토그램에서 보는 바와 같이 두 키랄고정상에서 치환된 1, 1' -비나프틸 (b i na p h t hy l) 계통의 라세미 화합물, 라세미 디카르복실산 유 도체, 라세미 디올 유도체, 라세미 에폭시 화합물 등의 두 광학 이성 질체가 잘 분리되고 있을 뿐만 아니라 이의에 키랄성 3 차아민 화합물 인 Tra g er 염기, 핵사헬리센 (hexahe li cene), 방향족 라세미 알코올
표 4-1 (+)-PTrMA 를 기저로 한 두 종류 키랄고정상에서의 광학분할 예. 이동상 : 메 탄올, 유속 0 . 50mL/m i n( 참고문헌 : 6) 라세미 화합물 R (+실)리-P 카 T rCMSAP - 분쇄 (+)-P TrMACSP a’ Rs a’ Rs 핥oac(COCOOCRRO R NOOHHPh P h 261...332 177 231...384 503 231...512340 210...347 98 OCH3 1.73 0.9 3 1.65 1.39 Br 1.41 1.06 1.00 。 :COR OPh 1.22 0. 4 1.29 1.50 OCOR Ph 2.16 4.26 1.45 1.68 COR NHPH 1.78 3.0 3 1.18 0.6 * R CONHPh 1.24 0.5 1.18 0.6 · <(CONHPh Ph 1.44 1.35 1.46 0.98 0~Ph H 1.22 0.87 1.00 。 . R Ph 5.2 1 5.30 2.1 7 2.39 *유속 0.20mL/m in
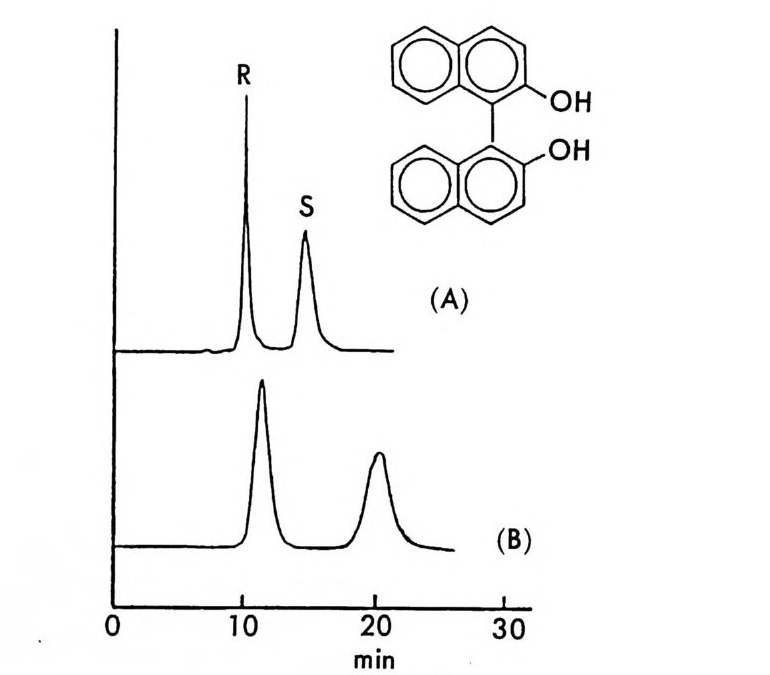 R s (A) 二二
R s (A) 二二
그림 4-2 분쇄된 (+) - PTrMA 키랄고정상 (A) 과 (+)-PTrMA 실리카 겔 키 랄고정상 (B) 에서의 비나프톨 (b i na p h th ol) 의 광학분할. 이동상: 메탄 울, 유속 : 0.7 2 mL/m in ( A) , 0.50mL/mi n (B) (참고문헌 : 10)
등이 광학분할된다 (4 , 5,6,10). (+)-PTrMA 키랄고정싱에서 광학분 할되는 이들 라세미 화합물의 특징은 모두 방향족기를 가지고 있거나 혹은 방향족기의 유도체들이라는 것이다. 특히 24 탄을과 같은 라세 미 화합물은 (+)-PTrMA 키랄고정상에서 광학분할되지 않으나 이 알코올울 벤조산에스테르 혹은 3, 5- 디클로로페닐벤조산에스테르 유도 체로 만들었을 때 광학분할이 잘된다는 사실이 보고되었으며 (11) 자일 로스 (xy lo se) 혹은 아라비노스 (arabin o se) 를 데트라벤조산에스데르 유 도체로 만들었을 때 그림 4-3 에 보는 바와 같이 잘 분리되고 있다(I O, 12). 따라서 (+)-PTrMA 를 기저로 한 키랄고정상에서의 광학분할에 는 키랄고정상과 광학분할하고자 하는 라세미 화합물 사이의 7t-7t상 호작용이 중요한 역할을 하고 있음을 추측할 수 있다.
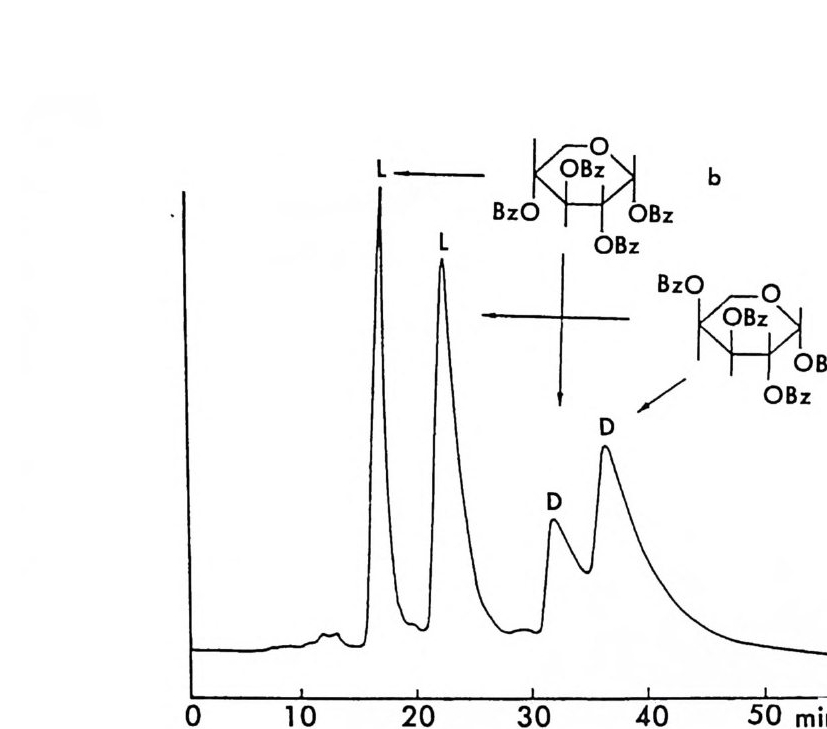 L —Bz0멱 겨 0 Bz b
L —Bz0멱 겨 0 Bz b
그림 4-3 (+)-PTrMA 질리카 겔 키랄고정상에서 아라비노스 (a) 와 자일로스 (b) 테트라벤조산에스테르 유도체의 광학분할. 이동상 : 메탄을, 유속 : 0.5mL/mi n, 온도 : lSC (참고문헌 : 12) 표 4-1 과 그림 4-2 에 제시된 광학분할의 예에서 보는 바와 같이 (+)-PTrMA- 실리카 겔 키랄고정상이 분쇄한 (+)- P TrMA 키랄고 정상보다 일반적으로 더 큰 분리인자 (a 값)와 더 좋은 분해인자 (Rs 값)를 보임으로써 두 키랄고정상 중 (+)-PTrMA- 실리카 겔 키랄고 정상이 더 좋은 광학분할능을 보이는 키랄고정상으로 판명되었을 뿐만 아니라 기계적 성질이 우수하여 내구성이 좋기 때문에 1981 년 이후 (+)-PTrMA 키랄고정상을 이용한 광학분할의 연구는 대부분 (+) - PTrMA 질리카 겔 키랄고정상을 이용하여 이루어졌다. (+)-PTrMA 질리카 겔 키랄고정상을 이용한 라세미 화합물들의 HPLC 광학분할은 아주 다양하다. (+)-PTrMA 자체가 나선성인 광 학활상 물질이므로 나선성때문에 키랄성인 라세미 화합물들이 잘 광학 분할될 뿐만 아니라 Co(acac)3, Cr(acac)3, Al(acac)3 와 갇은 키랄
유기금속 화합물(1 3, 1 4) 과 키랄중심이 인이나 황인 라세미 화합물들도 (15 ) 광학분할이 잘되는 것으로 보고되었다. 특히 키랄중심이 인인 화 합물들 중 살충제 로 사용되 는 EPN (o-ety l o- (4 - n it ro p h eny l) ph eny lp h onoth io n ate ) 1 및 살리티온 (sa lit h i on) ;i 등도 광학분할된 다(1 5). (+ ) -PTrMA 키랄고정상에서 그동안 광학분할된 많은 종류 의 라세미 화합물들의 자세한 목록은- 참고문헌 16 에 수록되어 있다.
 Ph-s~ -0- 0-NO, 二0....;〈S OCH3
Ph-s~ -0- 0-NO, 二0....;〈S OCH3
이들 중 입체화학적으로- 홍미있는 라세미 화합물들은 그림 4-4 와 갇 으며 비극성 라세미 화합물들로서 키랄축이나 키랄면을 가지고 있기 때문에 키랄성이거나 혹은 나선성때문에 키랄성인 화합물들이다. ( + )-PTrMA - 실리카 겔 키랄고정상은 생물학적으로 중요한 라세 마 화합물들의 광학분할에도 응용되었다. 예를 들면 항생제의 합성중 간체인 화합물 §의 광학분할에 ( + )-PTrMA - 실리카 겔 키랄고정상 이 사용되었고 47 H 의 이성질체가 가능한 화합물 1 을 (+)- P TrMA 질리카 겔 키랄고정상에서 광학분할할 때 세 개의 피이크로 분리됨이 확인되었을 뿐만 아니라 a- 토코페롤 8 이 아세트산에스데르 유도체로 서 ( + ) - PTrMA- 실리카 겔 키랄고정상에서 분리되는 것으로 확인되 었다 (10,1 2 ). 이 의에도 무공해 살충제로 최근 많은 관심의 대상이 되 고 있는 피레쓰로이드(py re t hro i d) 계 살충제의 일종인 페노트린 (ph enoth r in ) 2 의 시스 및 트란스 이성질체의 두 광학 이성질체가 광 학분할될 뿐만 아니라 (25) 키랄중심이 하나 더 존재하는 피레쓰로이드 계 화합물인 광학활성 델타메트린 (delta m eth rin) 꼬과 이것의 광학 이성질체가 광학분할된다 (26). 의약품으로는 항협심증제인 닐바디핀
 i:? H: 二M 3e 。0 \o 〉 N 广Cl Cl c)3
i:? H: 二M 3e 。0 \o 〉 N 广Cl Cl c)3
그림 4-4 (+)-PTrMA 키랄고정상에서 광학분할된 화합물들의 예. 괄호안의 숫 자는 참고문헌임 (이 그립은 참고문헌 1~ 참조한 것임 ) .
(nilv ad ipine ) ll 의 두 광학 이성질체가 혈장에서 추출되어 (+) -PTrMA 키랄고정상 (C hi ral p ak OT) 에서 메탄올-물 (95 : 5) 의 혼합용 매를 이동상으로 사용하여 광학분할되는 것으로 보고되어 있다 (27). 최근에는 세포독성을 갖고 있는 것으로 알려진 a- 메틸렌가―부티로락 톤 화합물 묘의 광학분할이 보고된 바 있다 (28).
 >
>
2.3 광학활성인 나선성 고분자 키랄고정상에서의 광학분할 특 성 및조건 (+)-PTrMA 키랄고정상에서 많은 라세미 화합물들이 광학분할되 었지만 아직 정확한 키랄성 인지 메커니즘은 밝혀져 있지 않다. 그동 안 연구된 광학분할의 결과로부터 다만 몇 가지의 단편적인 키랄성인 지의 가능성을 추측하고 있을 따름이다. (+)-PTrMA- 실리카 겔 키랄고정상을 이용하여 HPLC 상에서 라 세미 화합물을 광학분할할 때 주로 사용되는 이동상은 메탄올이며 이 의에 핵산 -2- 프로판올의 혼합용매, 아세토니트릴, 핵산 등이 사용될 수 있다. 특히 극성용매인 메탄을을- 이동상으로 사용할 경우에 좋은 광학분할을 확인할 수 있으며 (+)-PD2PYMA- 실리카 겔 키랄고정상 (Chir a lpa k OT) 에서의 광학분할에서는 메탄올 이동상에 물을 20% 까 지 가함에 따라 분석성분의 머무롬 시간이 증가할 뿐만 아니라 두 광 학 이성질체에 대한 입체 선택성이 증가한다 (25). 이와 같은 사실은 분석성분의 소수성기와 키랄고정상의 소수성부분 특히 트리페닐메틸 기 사이의 소수성 상호작용이 키랄성 인지에 중요하게 작용하고 있음 울 추측하게 한다. 키 랄고정상과 분석성분 사이의 소수성 상호작용의 중요성때문에 특별한 작용기가 없어서 다른 종류의 키랄고정상에서는 광학분할이 잘되지 않는 라세미 화합물들(그림 4- 伴] 라세미 화합물들) 이 (+)-PTrMA- 실리카 겔 키랄고정상에서는 광학분할이 가능한 것 으로예상되고있다. (+)-PTrMA- 실리카 겔 키랄고정상을 이용한 광학분할에 있어서 특별히 이동상으로 사용할 수 없는 용매는 톨루엔과 같은 방향족탄화 수소, CH2Cl2, CHC13, 아세톤, THF, DMSO 등으로서 이둘 용매 둘은 2.1 절에서 기술한 바와 같이 실리카 겔에 흡착된 (+)-PTrMA 분자를 용해시켜 키랄고정상의 수명을 단축시키기 때문이다. 메탄올 울 이동상으로 사용할 때에는 2.1 절에서 기술한 바와 같이 (+)
-PTrMA 분자의 가수분해를 주의할 필요가 있으며 따라서 가능한 낮 은 온도에서 크로마토그래피를 실시할 필요가 있다. 보통 5°C 이하에 서는 메탄올에 의한 ( +)- PTrMA 의 가수분해속도가 거의 무시할 수 있을 정도라는 사실이 알려져 있다 (12). (+)-PTrMA- 실리카 겔 키랄고정상에서 라세미 화합물들의 광학 분할은 실리카 겔에 흡착되는 (+)-PTrMA 의 양에 의해서도 영향을 받는 것으로 알려져 있다 (27) . 그림 4-5 에서 보는 바와 같이 흡착되는 (+ ) 一 PTrMA 의 양이 증가함에 따라 2 , 2' -디히드록시 -1, 1' -디나프틸 의 머무롬 부피 (머무름 시간과 동일함)의 차이는 계속 증가하나 트란스 -1, 2- 시클로부탄디카르복실산 디알닐리드의 경우에는 머무름 부피의 차이에 극대점이 있고 흡착되는 (+)-PTrMA 의 양이 더욱 증가하면
 (-E3 二O0 HH
(-E3 二O0 HH
그림 4-5 (+)-PTrMA 실리카겔 고정상에서의 광학분할(머무름 부피의 차이 : 세로축)에 대한 (+)-PTrMA/ 실리카 겔의 무게비의 영향(참고문헌 : 10)
머무름 부피의 차이는 오히려 감소하고 있다. 즉 광학분할하고자 하는 라세미 화합물의 구조에 따라서 많은 양의 ( + )-PTrMA 이 흡착된 키 랄고정상에서 광학분할이 찰되는 라세미 화합물들이 있는 반면 소량의 (+)-PTrM 回 흡착된 키랄고정상에서 광학분할이 잘되는 라세미 화 합물들이 있다. 실리카 겔에 흡착되는 (+)-PTrMA 의 양이 소량일 때는 각 (+)-PTrMA 분자 하나하나가 키랄성 인지에 관여하나 흡착 되는 (+)-PTrMA 의 양이 많아지면 각각의 나선성 고분자가 밀집하 게 되고 따라서 나선성 고분자 시술 사이에 새로운 키랄공간을 형성하 여 각각의 나선성 고분자가 나타내는 키랄성 인지와~ 다른 키랄성 인
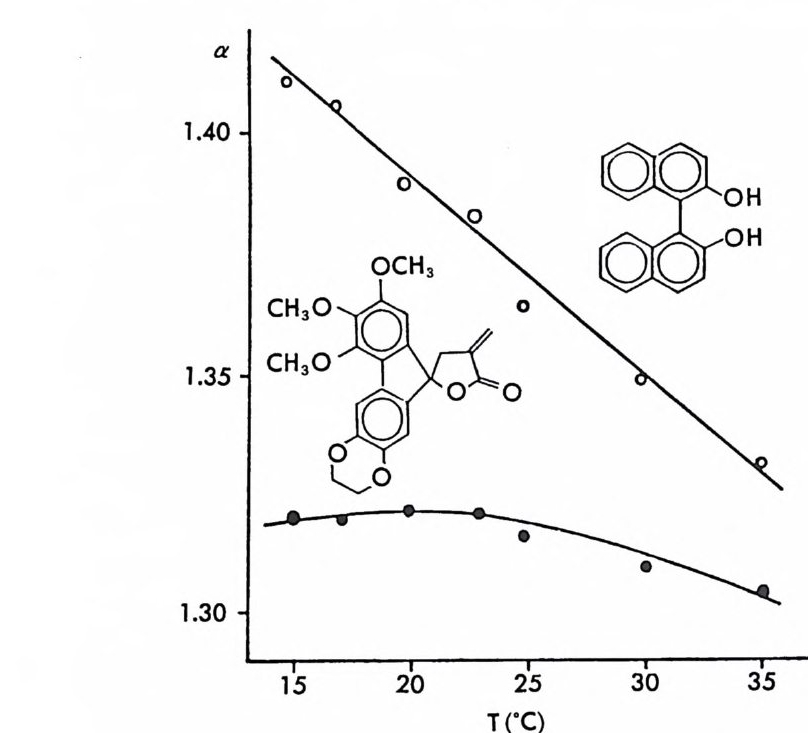 a(
a(
그림 4-6 학(분+)할-에P Tr대M한A 질온리도카의 겔영 (향C.h i r이al p동 a상k :O이T)산 화키탄랄소고-정메싱탄에올서 ( 9S2u:b8,F C w광/ w) , 유속 : 5mL/m in, 온도 : (jC , 기 압 : 140bar, 검출기 : 229nmUV (참고문헌 : 28)
지를 나타내기 때문에 이와 같은 현상이 관찰된다고 추측하고 있다. 라세미 화합물의 구조에 따라 다른 키랄성 인지 메커니즘에 의하여 광 학분할되는 것으로 생각할 수 있는 다른 예는 이산화탄소와 메탄올을 혼합한 준임계 유체를 이동상으로 사용하는- 준임계 유체 크로마토그래 피 (SubFC) 에서의 광학분할에 대한 온도의 영향을 나타내는 그립 4-6 의 그래프에서도 확인할 수 있다 ( 2 8). 온도의 변화에 따라 다른 구조 를 가진 두 화합물의 a 의 값이 다른 양상으로 변화하는 것은 이 두 화합물이 다른 키랄성 인지 메커니즘에 의하여 광학분할이 되고 있음 울 시사하고 있다. 그러나 아직 정확한 키랄성 인지 메커니즘을 논의 하기에는 이른 단계이다. (+)-PTrMA 실리카 겔 키랄고정상에서의 광학분할 특성 중 홍미 있는 사실은 C2 축과 두 개의 방향족기를 포함하는 라세미 화합물의 두 광학 이성질체 중, C2 급현] 수칙한 방향에서 분자를 바리볼- 때 방 향족기에 대하여 P - 나선성을 가지는 이성질체가 항상 키랄컬럼에 오 래 머무른다는 사실이다(그림 4-7 ) (4,10, 12 ). (+) - PTrMA 분자는
 N< HO
N< HO
그림 4-7 (+)-PTrMA 질리카 겔 키랄고정상에서 오래 머무르는 광학 이성질체 (C2 축 및 두 개의 방향족기를 가침 )의 구조
(+)-PTrMA 분자와 아주 강한 상호작용을 하며 반대의 나선성울 가 진 (― )-PTrMA 와는 약한 상호작용을 한다는 사실과 비교하여, P -나선성을 가진 이성질체들이 (+)-PTrMA- 실리카 겔 키랄고정상에 오래 머물기 때문에 (+)-PTrMA 분자의 나선성은 P 라고 예측할 수 있다 (4). 이것은 절대배열을 알기가 힘든 고분지물질의 절대배열을 결 정하는 데에 키랄고정상 기법이 응용될 수 있다는 점에서 중요한 의미 가 있다고 하겠다. 또한 P- 나선성을 가지는 이성질체는 (+) -PTrMA 실리카 겔 키랄고정상에 항상 오래 머무른다는 사실로부터 그림 4-7 에 제시한 화합물들과 비슷한 구조를 가전 라세미 화합물들 의 두 광학 이성질체의 절대배열은 용리순서로부터 예측이 가~노할 뿐 만 아니라, 나선성을 가지는 라세미 화합물들이 나선성 키랄고분자 키 랄고정성에서 항상 일정한 용리순서를 보인다는 사실로부터 키랄고정 상을 이루는 고분자의 나선성과 분석성분의 나선성 사이의 상호작용이 키랄성 인지에 중요한 역할을 하고 있음을 예측할 수 있다. (+)-PTrMA- 실리카 겔 키랄고정상의 키랄성 인지에 있어서 (+) -PTrMA분 자의 트리페닐메틸기에 있는 세 개의 페닐기의 구조 또한 중요한 역할을 한다는 실험적인 증거가 있다. 광학활성인 이소탁틱고 분자 L狩 } H를 기저로 한 키랄고정상이 (+)-PTrMA- 실리카 겔 키 랄고정상이 광학분할할 수 있는 많은 종류의 라세미 화합물들에 대하 여 아무런 입체 선택성도 보이지 않는다는 사실이 확인되었다 (10). (+)-PTrMA 분자는 덩치가 큰 트리페닐메틸기의 존재로 인하여 고 정된 나선성 구조를 가지기 때문에 좋은 광학분할능을 보인다고 생각 할 수 있으며 또한 트리페닐메틸기의 큰 소수성으로 인하여 소수성인 라세미 화합물과 강한 소수성 상호작용을 할 수 있고 트리페닐메틸기 의 트리페닐기가 프로펠라 구조를 가지기 때문에 여러 라세미 화합물 둘의 광학분할이 가능한 것으로- 생각된다. (+)-PTrMA 키랄고정상 울 구성하는 (+)-PTrMA 분자의 트리페닐메틸기는 왼쪽 방향 프로 펠라 구조 및 오른쪽 방향 프로펠라 구조가 가능하다. 그러나 (+)
(패 )를 仁광학c분H 2 할 1할》《귓〔3_0clI_ J H C 때 JHc二두 광 학 이성질체 仁중 A C-H 亡H이2성_-1 5tC〔질4 H _ =H체 C』O가H 2 n 키 < 랄컬>럼에 -PTrMA- 실리카 겔 키랄고정상에서 트리스(아세틸아세토나토 )M 더 오래 머무르는 사실을 확인하였으며 A- 아성질체의 분자모델과 트 리페닐메틸기의 분 자모델을 조사한 결과 A -이성질체와 왼쪽 방향 프 로펠라구조의 트리페닐메틸기가 서로 완전히 겹쳐지는(프로펠라의 날개 끼리) 안정한 상호작용을 하고 있음을 확인할 수 있었다(1 3). 따라서 (+)-PTrMA- 실리카 겔 키랄고정상을 구성하는 (+)-PTrMA분 자 의 트리페닐메틸기는 그림 4 정과 같이 왼쪽 방향 프로펠라 구조를 하 /?\ <汀O/M> \/
그림 4-8 (+)-PTrMA 분자의 트리페닐메틸기 구조 및 4- 및 A- 트리스(아세틸 아세토나토 )M 에)의 구조( A 및 4 의 명명법은 무기화합물의 IUPAC 명명규칙을 참조할 것). (참고문헌 : 13)
고 있다고 생각할 수 있으며 트리페닐메틸기의 이러한 프로펠라 구조 는 라세미 화합물의 광학분할에 중요한 역할을 하고 있다고 생각할 수 있다. 3 광학활성 폴리아미드를 기저로 한 키랄고정상 3.1 키랄고정상의 제조 광학활성 폴리아미드는 광학활성인 모노머 죽 광학활성 아크릴아미 드나 광학활성 메타크릴아미드를 라디칼 중합함으로써 얻을 수 있다. 그림 4-9 에서 보는 바와 같이 염화 아크릴로일 (acryl o y l chlor i de) 과 (S) -페닐알라닌에틸에스데르를 반응시켜 광학활성인 아크릴아미드를 쉽게 얻을 수 있고 (S)-1- 페닐에틸아민 혹은 (S)-1- 시클로핵실에틸 아민과 염화 메타크릴로일 (meth a cry lo y l chlor i de) 을 반응시켜 광학활 성인 메타크릴아미드를 쉽게 얻을 수 있다. 이렇게 합성된 광학활성 아크릴아미드나 광학활성메타크릴아미드는 교차결합 시약인 에틸렌디 울디아크릴레이트 (1,2-e thy lened i old i acry la t e)1 존재하에서 라디칼 개 시제인 1 몰 %의 아조이소부티로니트릴 (azo i sobu ty ron it r i le:AIBN) 을 사용하여 라디칼 중합하면 공중합체인 흰색의 구술형 폴리아미드를 거 의 정량적인 수율로 얻을 수 있다(1 ,2). 이 고분자는 교차결합되어 있 기 때문에 용매에 불용성일 뿐만 아니라 기계적으로 안정하여 일정한 입자 크기의 구술형 고분자만을 모아 컬럼에 충전할 경우 라세미 화합 물의 광학분할에 사용할 수 있다. 그러나 위와 같이 제조된 고분자 키 랄고정상은 톨루엔, 디옥산(di oxane), THF 등과 같은 유기용매 (이 동상)에서 팽윤하기 때문에 저압 크로마토그래피에 사용할 때에는 아 무 문제점이 없으나 고압하에서는 고분자 입자들이 압축될 뿐만 아니 라 고분자의 팽윤정도가 용매에 따라 달라지기 때문에 고압 액체 크로
 C6HsCH2 —k~HH2-C OOC2H5 CTHo2 l=u CenHe- C OCI C6H5CH2 -CNIH H--C
C6HsCH2 —k~HH2-C OOC2H5 CTHo2 l=u CenHe- C OCI C6H5CH2 -CNIH H--C
마토그래피 (HPLC) 나 경사용리 (grad ie n t eluent) 크로마토그래피에 사용하기에는 부적절하다. 그러나 최근에는 폴리아미드 키랄고정상의 이와 같은 문제점을 보완하기 위하여 폴리아미드를 기계적 성질이 우 수한 실리카 겔에 결합시킴으로써 HPLC 를 이용한 라세미 화합물의 광학분할에 적절하게 이용할 수 있는 키랄고정상이 새로이 게발되었다
(2,3). 실리카 겔에 결합된 광학활성 폴리아미드 키랄고정상의 제조는 세 가지 방법으로 가능하다. 상업적으로 구할 수 있는, 디올기를 가지고 있는 실리카 겔 (Lich rosorb D i ol) 과 아크릴산 혹은 메타크릴산을 에스데르화 반응하 여 아크릴레이트 혹은 메타크릴레이트 고정상을 얻은 후 이것울 앞서 합성한 광학활성 아크릴아미드 혹은 메타크릴아미드의 용액과 섞어 라 디칼 중합반응을 진행시키면 광학활성인 모노머들은 실리카 겔에 에스 데르기를 통하여 결합된 아크릴 혹은 메타크릴기과 함께 공중합반~卜이 일어나 공중합체를 형성함으로써 결국은 광학활성인 폴리아미드가 실 리카 갤에 공유결합된 키랄고정상 (CSP 5) 이 얻어전다(그립 4- 1 0). 이
 -~I-IlC H2COI HH -COH1 H2 C에H스2 =테C르H-화C반OO응H ―「I1iS합 - cH 2 cH'0—cHCH_2oHT ='C0,H -, ccC - I-I
-~I-IlC H2COI HH -COH1 H2 C에H스2 =테C르H-화C반OO응H ―「I1iS합 - cH 2 cH'0—cHCH_2oHT ='C0,H -, ccC - I-I
때 교차결합 시약은 필요없으며 실리카 겔에 결합된 아크릴 혹은 메타 크릴기와 공중합반응을 하지 않고 광학활성인 모노머끼리 중합된 폴리 아미드는 톨루엔에 용해하므로 단순한 거름과정을 통하여 쉽게 제거된 다. 이렇게 얻은 폴리아미드 - 실리카 겔 키랄고정상은 무게로 10-20% 의 폴리아미드를 포함하는 것으로 밝혀졌다 (3). Lic h rosorb D i ol 에 (S) - N - 아크릴로일페닐알라닌에틸에스데르 (N -ac ryl o y lp h eny la lanin e eth y l es t er) 와 같은 광학활성 모노머를 흡착시 킨 후 중합반응을- 진행시키면 광학활성 아크릴아미드_실리카 겔 키랄 고정상이 얻어전다. 아직 정확한 합성 메커니즘은 모르나, 일단 광학 활성 모노머와 라디칼 개시제가 실리카 겔에 흡착된 후 가열하면(보통 톨루엔 서스펜션 상태) 다공성인 실리카 겔의 표면(혹은공동)에서 중합 반응이 일어나 합성된 고분자가 실리카 겔 공동에 갇히게 되므로 고분 자가 실리카 겔에 붙잡혀 있는 폴리아미드-실리카 겔 키랄고정상 (CSP §_)이 만들어지는 것으로 추측하고 있다(그림 4-11). 이렇게 제 조된 폴리아미드-실리카 겔 키랄고정상의 고분자 물질은 n- 핵산, 톨 루엔, 디옥산, 2- 프로판을과 같은 유기용매에 의해서 실리카 겔로부 터 씻겨나오지 않는 것으로 알려져 있다 (2,3). 그러나 실리카 겔에 흡착된 광학활성 모노머를 중합하여 얻은 폴리 아미드-실리카 겔 키랄고정상 (CSP §_)은 실리카 겔 표면에 실라놀 (s i lanol) 기가 많이 존재하기 때문에 이 실라놀기와 광학분할하고자 하 는 라세미 화합물 사이에 비입체 선택적인 수소결합 상호작용이 존재 하게 되고, 라세미 화합물의 두 광학 이성질체가 입체 선택성과는 상 관없이 키랄고정상에 너무 오래 머무르게 된다. 이러한 문제점을 보완 하기 위하여 위에서 얻은 폴리아미드-실리카 겔울 디메틸옥틸클로로 실란 (d i me t h y loc ty l chloros il ane) 과 반응시키면 실리카 겔 표면의 실 라놀기가 적절히 제거된 폴리아미드-실리카 겔 키랄고정상 (CSPl) 이 얻어전다(그립 4-11). 특히 N- 아크릴로일 -(S) -페닐알라닌에틸에스데 르를 실리카 겔에 흡착시켜 중합하고 실리카 겔의 실라놀기를 제거하
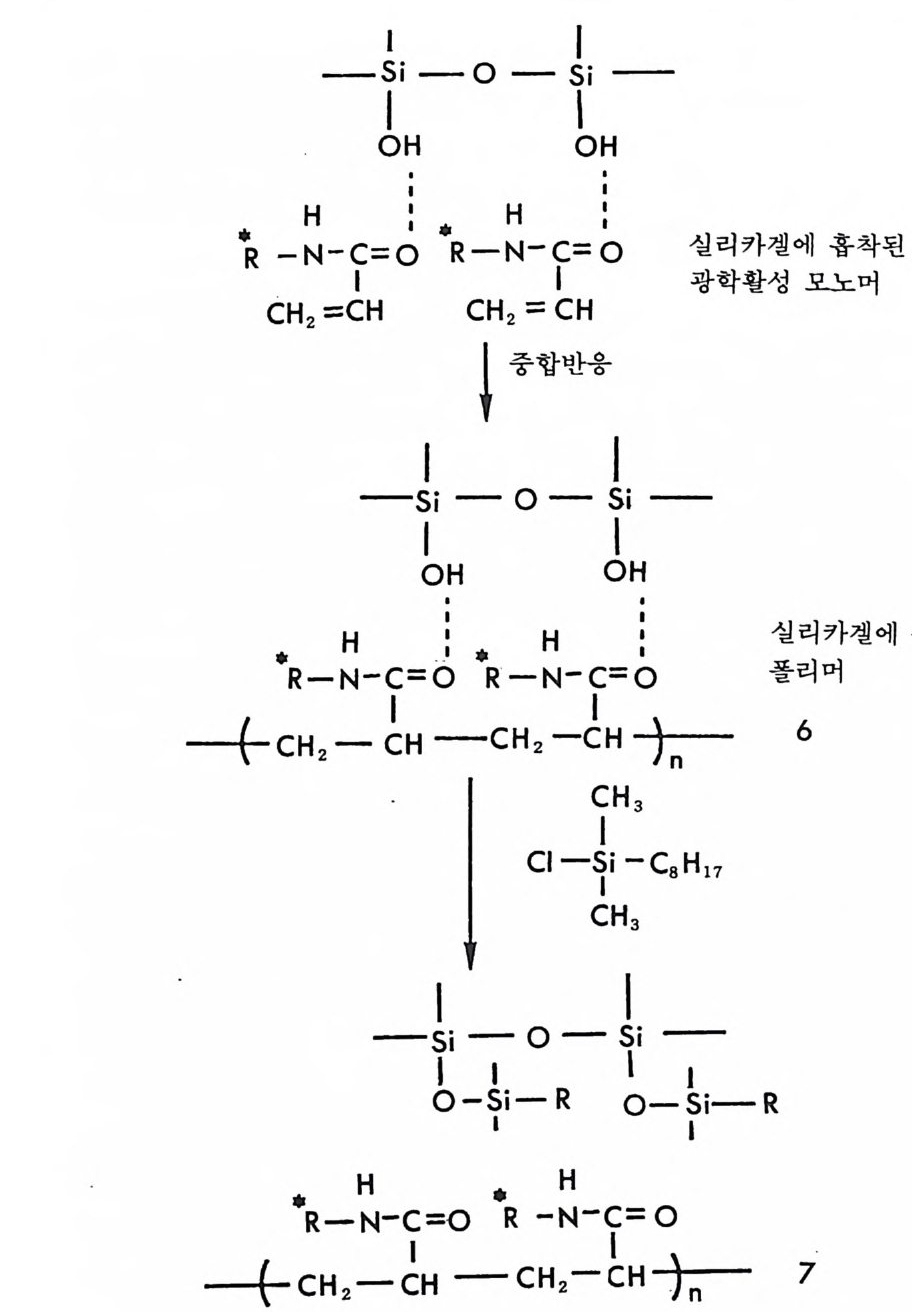 -SlI? i- 0 一 SLI i ? ―
-SlI? i- 0 一 SLI i ? ―
그림 4-11 폴리아미드수i리카 겔 키랄고정상 (CSP_§ 및 CSP'!_) 의 제조과정
여 얻어진 CSP Z 형태의 키랄고정상은 Chir a S p her 라는 상품명으로 상품화되어 있다 (제 9 장 참조) . 3.2 폴리아미드 키랄고정상에서의 광학분할 및 특성 입자크기가 50-lOOµm 정도인, 교차결합된 광학활성 폴리아미드 키 랄고정상 (CSPl,I, J)을 저압 펌프를 사용하여 유리컬럼에 충전한 후 저압 액체 크로마토그래피를 실시하면 여러 종류의 라세미 의약품들이 광학분할된다(1, 4). 이뇨제인 클로르탈리돈 (chlor t hal i done) ~. 530m g이 톨루엔과 디옥 산의 1 : 1 혼합용매를 이동상으로 하여 페닐알라닌에틸에스데르를 포 함하는 폴리아미드 키랄고정상 (CSP .!_) 250 g이 충전된 컬럼상에서 그 림 4-12 와 같이 정량적인 수율로 완전히 광학분할된다 (1,5). 특히
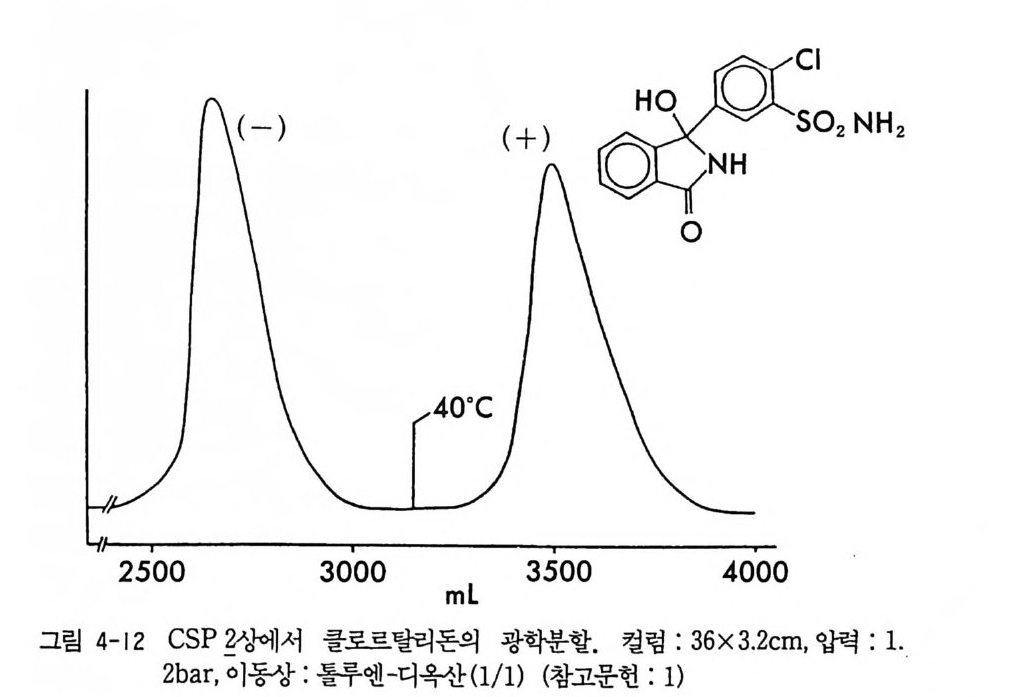 Cl
Cl
(+)-이성질체의 머무름 시간이 너무 길기 때문에 광학분할에 필요한 시간을 절약하고 이동상의 사용량을 줄이기 위하여 (-)-이성질체가 완전히 분리되어 나온 후 컬럼온도를 40°C 로 올려 크로마토그래피를 실시한 사실이 인상적이다. 전정제로 알려져 있으나 태아기형을 유발시키는 부작용때문에 의약 품 시장에서 판매금지된 탈리도미드(t hal i dom i de) 는 폴리아미드 키랄 고정상인 CSP ~ 상에서 완전히 광학분할된다. 65 g의 폴리아미드 키 랄고정상 (CSP 집)이 충전된 80x2.3cm 의 컬럼상에서 벤젠과 디옥산 의 4 : 1 혼합용매를 이동상으로 사용하여 500m g의 탈리도미드가 단 한번의 크로마토그래피에 의하여 완전히 광학분할되며 이렇게 분리된 광학 이성질체를 사용하여 태아기형을 유발시키는 부작용을- 시험한 결 과 오로지 (S)-(-) -탈리도미드만이 태아기형에 관계있다는- 사실이 확인되었다(1). 클로르탈리돈과는 다른 종류의 이뇨제인 벤조티아디아전 (benzo t h iad ia z in e ) 계통의 라세미 의약품들도 폴리아미드 키랄고정상인 CSP 2 상에서 광학분할된다 (6). 예를 들면 펜플루티자이드(p e nf lu ti z i de) ~ 와 벤드로플르메티아자이드 (bendro fl ume t h i az i de) lQ의 경우 톨루엔 과 디옥산의 1 기 혼합용매를 이동상으로 하여 CSP 2 상에서 완전한
 H2NC0F&3二' -TA( N5H: y N HC H 2(CH2)4HC2H N3 0F2 s3 亡C~gNH yr H C H2Cs Hs
H2NC0F&3二' -TA( N5H: y N HC H 2(CH2)4HC2H N3 0F2 s3 亡C~gNH yr H C H2Cs Hs
기준선 광학분할은 관찰되지 않으나 그립 4-13a 에 보는 바와 같이 어 느 정도 광학분할됨을 확인할 수 있다. 특히 베메티자이드 (beme t izi d e ) 旦의 경우에는 곁사슬에 존재하는 두번째의 키랄중심때문에 실 제로 네 개의 입체 이성질체가 가능하나 그림 4-13b 에 보는 바와 같 이 네 개의 입체 이성질체 죽 두 개의 광학 이성질체쌍이 분리되고 있 음을 알 수 있다 (6).
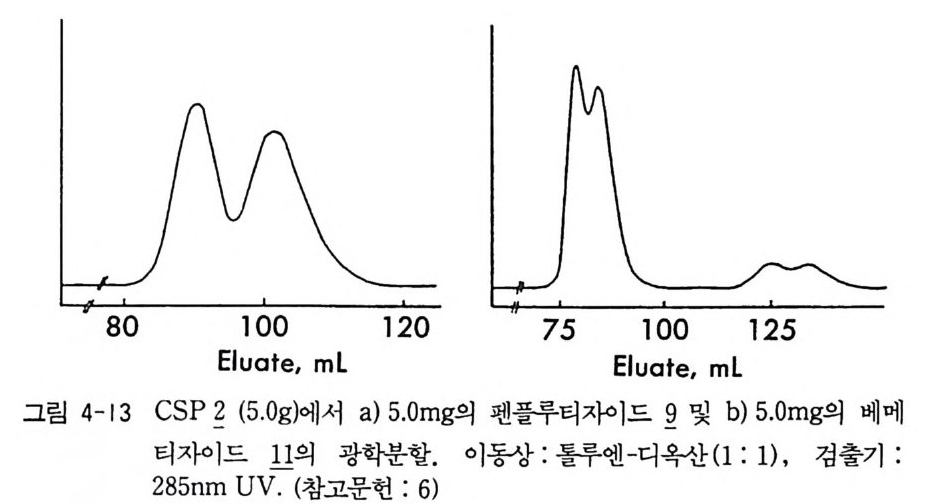 80 100 120 75 100 125
80 100 120 75 100 125
옥사자포스포린 (oxazaph osph orin e ) 골격을 가전 항암제들은 키랄 중심이 인원자인 키랄 화합물이다. 이들 항암제 중의 하나인 이포스파 미드(ifo s fa m i de) 표와 2- 데클로로에틸이포스파미드 (2-dechloroe th yl ifo s fa m i de ) 묘은 그림 4-14- 9.} 같이 폴리아미드 키랄고정상인 CSP l 에서 광학분할된다 (7). 2- 데클로로에틸이포스파미드의 경우 자유아 미노기와 키랄고정상 사이의 수소결합이 머무름 시간 및 입체 선택성 울 증가시키는 것으로 예측된다. 키랄중심이 황원자인 라세미 화합물 술포디이미드 (sul fodi m i de)H 는 폴리아미드 키랄고정상인 CSP ~에서 톨루엔과 디옥산의 1 : 1 혼 합용매를 이동상으로 하여 광학분할이 찰되나 이동상에 메탄올을 조금
첨가하면(톨루엔 / 디옥산 / 메탄올 = 47 : 47 : 6) 머무롬 시간이 훨씬 짧아 짐을 그림 4-15 에서 확인할 수 있다(1) . 이동상 중의 메탄올과 분석성
CH2CH2 C I CH2CH2 C I CA\/P< O l2 c\P/\o 13 o· 'NHCH2CH2CI I '---0 NH2 20 m30l 40 20 50 ml 60 70 그립 4-14 a) CSP 1( 5.0 g)ol]서 a) 5 . 0 g의 이포스파미드 및 b) 5.0m g의 2- 데클로 로이포스과미드의 광학분할 컬럼 29.S x l.Oc m, 이동상 : 톨루엔-디옥 산(1 : 1) (참고문헌 : 7) o; PN f /ss시 f>o \l4 b 250 a 50 70 90 ml 그림 4-15 a) CSP ~ (6.2 g제서 15m g의 술포디이미드 브의 광학분할 컬럼 : 24xl .Oc m, 이동상 : 톨루엔/디옥산/메탄올= 47 : 47 : 6 및 b) CSP .!_(5.0 g제서 2 . 0m g의 술포디 이미드의 광학분할 컬럼 : 29.5 x l.Oc m, 이 동상 : 톨루엔/디옥산 (1 : 1) (참고문헌 : 1)
분이 키랄고정상과의 수소결합에 대하여 서로 경쟁관계에 있으며 또한 이동상 중의 메탄올과 분석성분 사이의 수소결합에 의하여 분석성분의 머무름 시간이 감소한다고 생각할 수 있다. 이와 같은 실험결과는 키 랄고정상과 분석성분 사이의 수소결합이 키랄성 인지에 중요한 역할을 하고 있음을 시사해 주는 것이다. 근육이완제로 사용되는 클로르메지논 (chlormezanon) ~裝근 폴리아 미드 키랄고정상인 CSP l 에서 광학분할되지 않으나 N- 데메틸클로르 메자논 (N - deme t h y lchlormezanon) 1§梵 7 광학분할된다(그립 4-16). N 대메틸클로르메자논의 경우 N-H 기의 산성 수소와 키랄고정상 사 이의 수소결합이 머무름 시간을 증가시킬 뿐 아니라 입체 선택성을 증 가시키는 것으로 예측되고 있다. 지금까지 예를 든 라세미 화합물들 이의에도 폴리아미드 키랄고정상
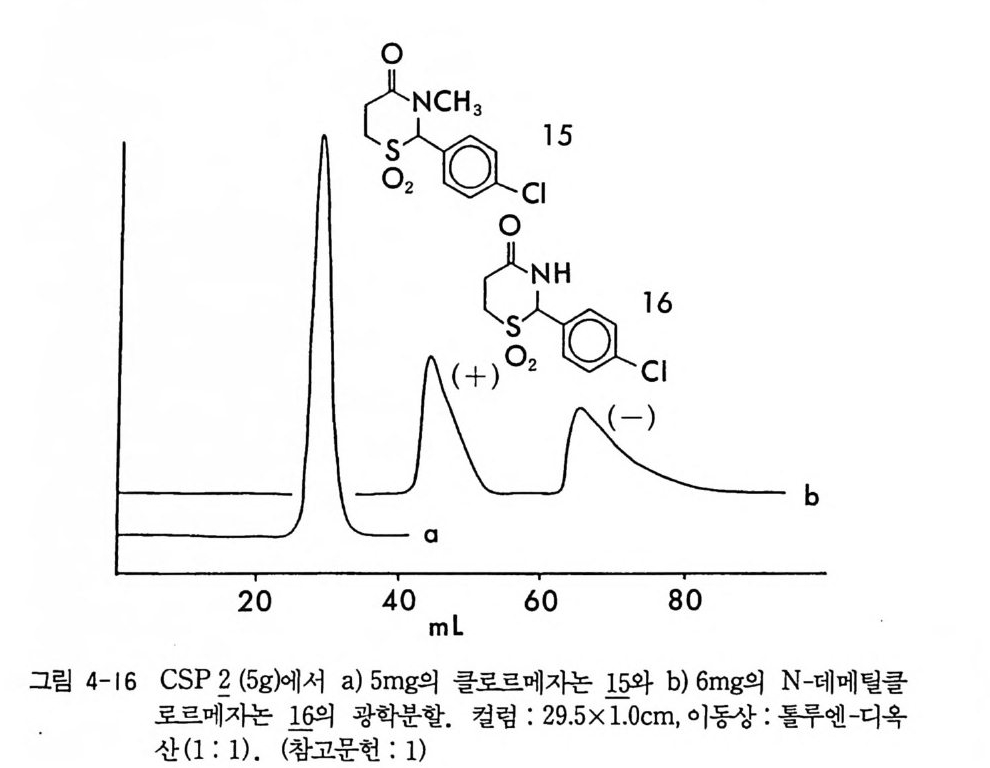 m〔〔i。l 0 {NHlc15s 二0)l C6_ b
m〔〔i。l 0 {NHlc15s 二0)l C6_ b
에서 광학분할이 가능한 라세미 화합물에는 옥사제팜 (oxaze p am) 끄, 옥사졸람 (oxazolam) 뵤 케타졸람 (ke t azolam) ~' 향응고제인 아세토 쿠마롤 (acenocoumarol) 챕 등이 있다.
 Cl 二 50H c / >H >~O N `
Cl 二 50H c / >H >~O N `
광학활성인 폴리아미드를 기저로 한 키랄고정상 죽 CSP l, ~, J에서 광학분할이 가능한 모든 라세미 화합물들은 광학활성 폴리아미드-실 리카 겔 키랄고정상에서도 모두 광학분할된다. 광학활성 폴리아미드 국J리카 겔 키랄고정상은 스데인레스스틸 컬럼에 충전되어 HPLC 에 응용되기 때문에 저압 크로마토그래피에 의한 광학분할에 비교하여 좋 은 피아크의 모양을 기대할 수 있을 뿐만 아니라 많은 경우에 있어서 기준선 분리에 의한 광학분할이 가능하다. 그림 4-17 은 폴라이미드수 l 리카 겔 키랄고정상인 CSP .Q 및 CSP §_
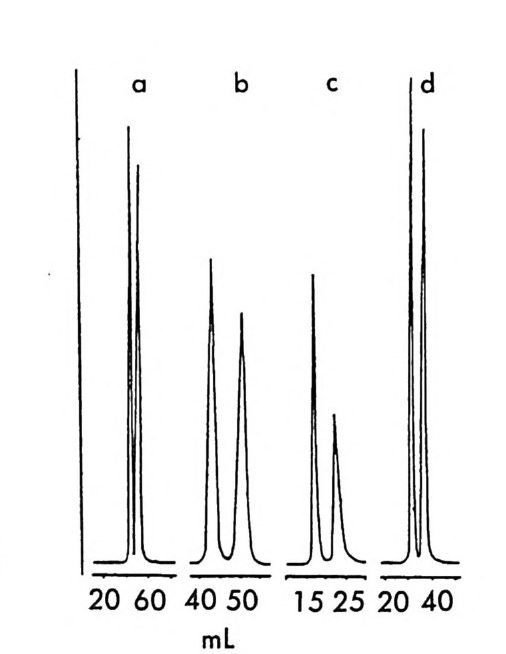 a b c d
a b c d
그림 4-17 a) CSP ~ (페닐알라닌 에틸에스테르)에서 펜플루티자이드 g b) CSP ~ (페닐에틸아민)에서 N- 데메틸클로르메자논 拉 c) CSP ~ (시클로핵실에틸아민)에서 탈리도미드 d) CSP § (페닐알라닌에틸에스데르)에서 클로르탈리돈 院] 광학분할. 유속 (mL/mi n) , 이동상의 조성 (n- 핵산/디옥산) : a) 1.5, 53 : 47, b) 1. 0, 65 : 35 c) 1.0, 7.5 : 25 d) 0.7, 55:45 (참고문헌 : 3)
에서 여러 종류의 라세미 의약품들의 광학분할에 대한 크로마토그램을 나타낸 것이다. 어느 경우에나 크로마토그램은 좋은 피이크 모양을 보 일 뿐만 아니라 모두 기준선 분리가 이루어지고 있음을 확인할 수 있 다 (3). 이뇨제인 펜플루티자이드 針근 그립 4-13a 에서 보는 바와 같이 폴리 아미드 키랄고정상인 CSP2 에서 기준선 분리가 이루어지지 않으며 벤드로플르메티아자이드 L條 든 펜플루티자이드 9보 다 광학분할이 더 잘 되지 않는 것으로 알려져 있으나(1) 갇은 라세미 화합물을 광학활 성 폴리아미드수 l 리카 겔 키랄고정상인 CSP l( 페닐알라닌 에틸에스테 르)에서 HPLC 를 이용하여 광학분할할 경우 그림 4-18 :i!} 같이 완전
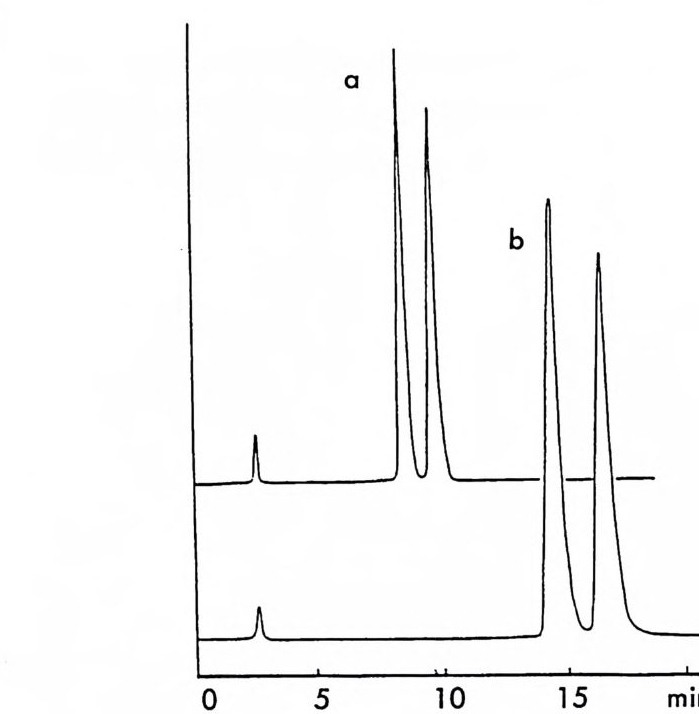 a
a
그림 4-18 CSP'!_( 페닐알라닌에틸에스데르)에서 a) 15µ g의 펜플루티자이드 g 및 b) 15µ g의 벤드로플루메티아자이드 L伊 ] 광학분할. 컬럼 : 250x 4 mm, 이동상 : n- 핵산/디옥산 /2- 프로판을 (65 : 34 : 1) , 유속 : lmL/ m i n, 압 력 : 55bar, 검출기 : UV 254run (참고문헌 : 2)
한 기준선 분리가 이루어짐을 확인할 수 있다 (2). 옥사졸람 上읽끈 네 개의 입체 이성질체가 가능하나 폴리아미드-실리 카 겔 키랄고정상인 CSP 1( 페닐알라닌에틸에스테르)에서 그립 4-19 와 같이 네 개의 입체 이성질체가 모두 분리되고 있다. 최근에는 상품화되어 있는 키랄고정상 Chir a S p her 를 이용하여 HPLO0네 서 4 홈冷 5- 위치가 알킬기로 치환된 라세미 y-혹 은 8 -락돈(쌀 혹은 盤)을 광학분할한 바 있으며 치환된 알킬기의 길이 및 고리의 크기가 입체 선택성 (a 값)에 미치는 영향에 대한 연구결과가 보고된 바 있다 (8).
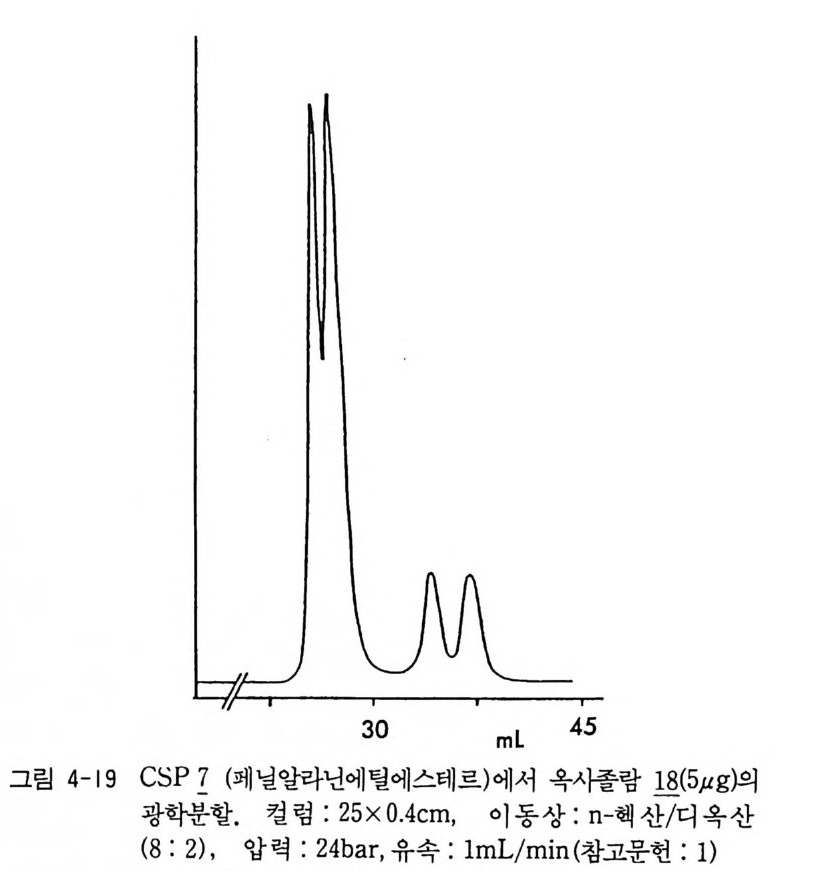 30 ml 45
30 ml 45
그립 4-20 에 보는 바와 같이 R7] 의 길이가 길어짐에 따라 a 값이 증가하며 Y- 락톤 낍이 0- 락톤 盤보다 광학분할이 더 잘 됨을 알 수 있다. 이와 같은 결。과 는 분석성분의 크기 혹은 구\조가 입체 선택성에 영향을 미치는 예로서 Y- 락본의 경우 오각형고리가 거의 평면 (hal f 21 22
a’ 1.4 ,,0-·--. o··· .. o 1.3 1.2 O- 1.1 1.1 。 2 3 4 n 5 6 7 8 9 그림 4-20 CSP J_( Chir a S p her) 에서 락돈 씩 ( 0 ) 과 盤 ( □ )의 광학 분 할에 대한 양기 사슬길이 (n) 의 영향. 이동상 : t냐 L 밀 메 필 에테르 - 핵산 (5 : 95), 유속 : l.2m L/mi n. ( 참고문헌 : 8)
-p lanar) 을 아루기 때문에 두 광학 이성질체의 구분이 용이하며 R7] 의 길이가 길어지면 H 및 R 기의 구분이 쉬워지기 때문에 광학분할이 더 잘 되는 것으로 생각할 수 있다. 광학활성인 폴리아미드를 기저로 한 폴리아미드 키랄고정상 및 폴리 아미드_)..J리카 겔 키랄고정상에서의 광학분할은 지금까지 기술한 바 와 갇이 많은 종류의 라세미 의약품에 걸쳐 다양하나 아직 광학분할에 대한 정확한 키랄성 인지 메커니즘을 모르고 있는 실정이다 . 그러나 라세미 화합물들에 대한 폴리아미드 키랄고정상의 광학분할능력은 고 분자가 만들어지는 방법에 따라 아주 달라지기 때문에 광학활성 폴리 아미드를 기저로 한 키랄고정상의 광학분할능은 고분자에 결합된, 키 랄중심을 가지고 있는 개개의 광학활성 치환기 (예를 들면 페닐알라닌에 틸에스테르, 페닐에틸아민 등)나 분석성분과의 입체 선택적인 상호작용 에 의하여 결정되는 것이 아니라 고분자의 3 차 구조에 의해서 결정된 다고 예측되고 있다 (4). 예를 들면 광학활성 모노머를 만든 후 라디칼 중합에 의하여 얻어진 폴리아미드 키랄고정상 (CSP i. ~ 혹은 4) 에서
 n C6H5-~N*HH -2C Ha + -f- cH2_~C& l: . 七0 .. C-sfH csHH2-C •’N― H- -CcI H=C H片o3
n C6H5-~N*HH -2C Ha + -f- cH2_~C& l: . 七0 .. C-sfH csHH2-C •’N― H- -CcI H=C H片o3
라세미 만델산이나 라세미 만델산아미드는 광학분할이 잘되나 교차결 합된 폴리아크릴산염화물(p ol y acr yli c aci d chlor i de) 에 (S)-1- 페닐에 틸아민을 반응시켜 얻은 광학활성 폴리아미드 키랄고정상 盤은 (그립 4-21) CSP 19-} 같은 조성을 가지면서도 라세미 만델산을 거의 광학분 할하지 못하는 것으로 밝혀졌다. 따라서 커다란 광학활성기를 가지고 있는 아미드 모노머가 라디칼 중합하여 CSP z_,~ 혹은 4 가 만들어지 는 과정에서 커다란 광학활성기의 존재로 인하여 각 고분자사슬 사이 에 특정한 키릴공동을- 가전 삼치원적인 고분자가 합성되는 것으로 생 각할 수 있다. 결국 라세미 화합물들이 광학활성인 폴리아미드를 기저 로 한 키랄고정상에서 광학분할되는 것은 라세미 화합물의 두 광학 이 성질체가 키랄고정상의 키랄공동에 끼어들어갈 때 입체 선택성이 있기 때문이라고 생각되며 특별히 고분자 사술의 CO - NH 기와 라세미 화합 물 사이의 수소결합 혹은 쌍극자짱戶구자 상호작용이 라세미 화합물의 두 광학 이성질체가 폴리아미드의 키랄공동에 끼어들어가게 되는 중요 한 친화적인 상호작용이라고 생각할 수 있다. 따라서 폴리아미드를 기 저로 하는 키랄고정상에서 광학분할이 가능한 라세미 화합물 혹은 의 약품들은 대부분 아미드기, 이미드기 혹은 아미노기와 같은 극성기를 가전 국성물질임을 쉽게 예측할 수 있을 것이다. 이러한 예측이 가능함에도 불구하고 폴리아미드를 기저로 한 키랄고 정상에서의 광학분할은 아주 복잡하여 어떤 고정상에서 어떤 라세미체 가 어떻게 광학분할될 것인가를 예측하는 것은 아주 어렵다. 예를 들 면 앞에서 본 바와 같이 클로르탈리돈針논 CSP 2 상에서 완전히 광학
분할되나 CSP .4에서는 전혀 광학분할되지 않는 반면 탈리도미드는 CSP .4에서 완전히 광학분할되고 CSP Z 에서는 광학분할되지 않는다. 이와 같은 광학분할의 특성은 현재 전혀 설명되지 못하고 있는 실정이 다. 4 키 랄공동을 가진 고분자 키 랄고정상(주문자방식의 키랄고 정상) 적당한 키랄형판 (ch i ral tem p la te ) 분자와 결합하고 있는 모노머를 출발물질로 사용하여 고분자를 합성한 후 키랄형판 분자를 제거하면 키랄형판분자가 차지하고 있던 공간과똑같은 키랄공간 혹은 키랄공동 울 가전 고분자가 합성될 수 있을 것으로 예상된다. 따라서 이와 같은 키랄공동을 가진 고분자를 키랄고정상으로 사용하면 키랄형판 분자로 사용하였던 물질의 라세미 형태를 쉽게 두 개의 순수한 광학 이성질체 로 광학분할할 수 있을 것이다. 왜냐하면 키랄형판 분자와 똑같은 키 랄성 혹은 입체구조를 가전 광학 이성질체는 키랄고정상의 키랄공동에 찰 맞춰 끼어들어갈 수 있는 반면 키랄형판 분자의 키랄성과 반대의 키랄성을 가전 광학 이성질체는 키랄공동에 끼어들어갈 수 없어서 두 개의 광학 이성질체가 구별되기 때문이다. 이것은 마치 항원에 대하여 형성된 항체가 항원과만 선택적으로 상호작용하는 원리나 혹은 생체내 의 효소가 특정한 기질 (subs t ra t e) 에 대하여서만 활성을 나타내는 것 과같은이치이다. 키랄형판 분자의 존재 하에서 카랄공동을 가전 키랄고분자가 형성되 는 과정에 대한 모델을 그림 4-22 에 나타내었다(1). 그림 4-22 에서 보는 것처럼 적절한 작용기를 가지고 있고 중합이 가능한 비닐유도체 와 키랄형판 분자가 적절한 결합을 형성하고 있을 경우, 교차결합된
 틀〈一
틀〈一
그림 4-22 키 랄 형판 분자 를 이용하여 키랄공동을 가진 고분자 키랄고정싱을· 합성 하는 과정. 키 랄 공동 고분자의 키랄공동은 세 개의 상호작용부위 (• ’ A · ) 를 가진다(참고문헌 : 1).
고분자를 합성할 수 있는 적절한 반응조건하에서 중합반응을 실시하면 키랄형판 분자를 내부에 포함하는 고분자가 합성된다• 고분자가 합성 된 후에 적절한 방법으로 키랄형판 분자를 제거하면 고분자 내부에 키 랄형판분자의 입체구조와 똑같은 모양을 가지며 적절한 부위에서 키랄 형판 분자와 상호작용이 가능한 작용기를 가전 키랄공동이 형성된다. 이렇게 합성된 키랄공동을 가전 고분자의 키랄공동에는 키랄형판분자 로 사용되었던 광학 이성질체는 끼어들어갈 수 있으나 반대 절대배열 을 가진 광학이성질체는 키랄공동에 끼어들어갈 수 없을 뿐만 아니라 상호작용부위 (그림 4- 2 2 에서 세 자리 상호작용)의 위치도 달라져서 키랄 공동내의 상호작용부위와 적절한 상호작용을 할 수 없게 된다. 따라서 합성된 키랄공동 고분자를 키랄고정상으로 사용할 경우에 형판화합물 의 라세미체를 광학분할할 수 있을 것으로 기대된다. 그러나 키랄형판 분자를 이용하여 합성된 키랄공동 고분자를 기저로 한 키랄고정상은
여러 종류의 다양한 라세미 화합물들의 광학분할에 응용될 수 없으며 오로지 형판으로 사용되었던 형판화합물 라세미체의 광학분할에만 특 히 유용하다는 단점이 있다. 따라서 다른 라세미 화합물을 광학분할하 고자 할 때에는 이 라세미 화합물의 두 광학 이성질체 중 하나의 순수 한 광학 이성질체를 키랄형판분자로 사용하여 이 광학 이성질체가 적 절하게 들어맞는 키랄공동을 가전 고분자를 새로 합성하고 이것을 키 랄고정상으로 사용하여야 할 것이다. 이와 같은 특성을 고려하여 Wul ff는 이와 같은 키랄고정상을 주문자방식 (Ta i lor-made) 의 키랄고 정상이라고 호칭한 바 있다 (2). 적절한 키랄형판 분자를 사용하여 키랄공동을 가진 고분자를 합성하 고 이것을 키랄고정상으로 사용하여 형판화합물의 라세미체를 광학분 할한 예는 그다지 많은 편은 아니나 주로 Wul ff에 의하여 연구되었다 (1, 2). 여기서는 키랄형판 분자를 사용하여 합성된 키랄공동고분자 중 입체 선택성이 좋은 것으로- 알려진 고분자를 선택하여 이들의 합성, 이들에 의한 입체 선택성 및 HPLC 용 키랄고정상으로서의 가능성에 대하여 살펴보고자 한다.
 <> <>
<> <>
키릴공동을 가진 키랄고정상을 제조하기 위하여 Wul f秒卜 주로 사용 한 키랄모노머는 4- 비닐벤젠봉산 (4-v i n y lbenzene boric a ci d) 과 페닐 -a-D- 만노피라노시드(p hen y 1-a-D-manno pyr anos i de) 가 두 개의 에 스테르결합에 의하여 결합된 화합물 止료써, 여기서 키랄형판분자는 페닐 -a - D- 만노피라노시드 2 이다(1, 4). 두 개의 작용기 (주로 비닐기)를 가전 다량의 교차결합시약과 함께 모노머 l 을 비활성용매 중에서 라디칼 중합을 실시하면 내부표면적이
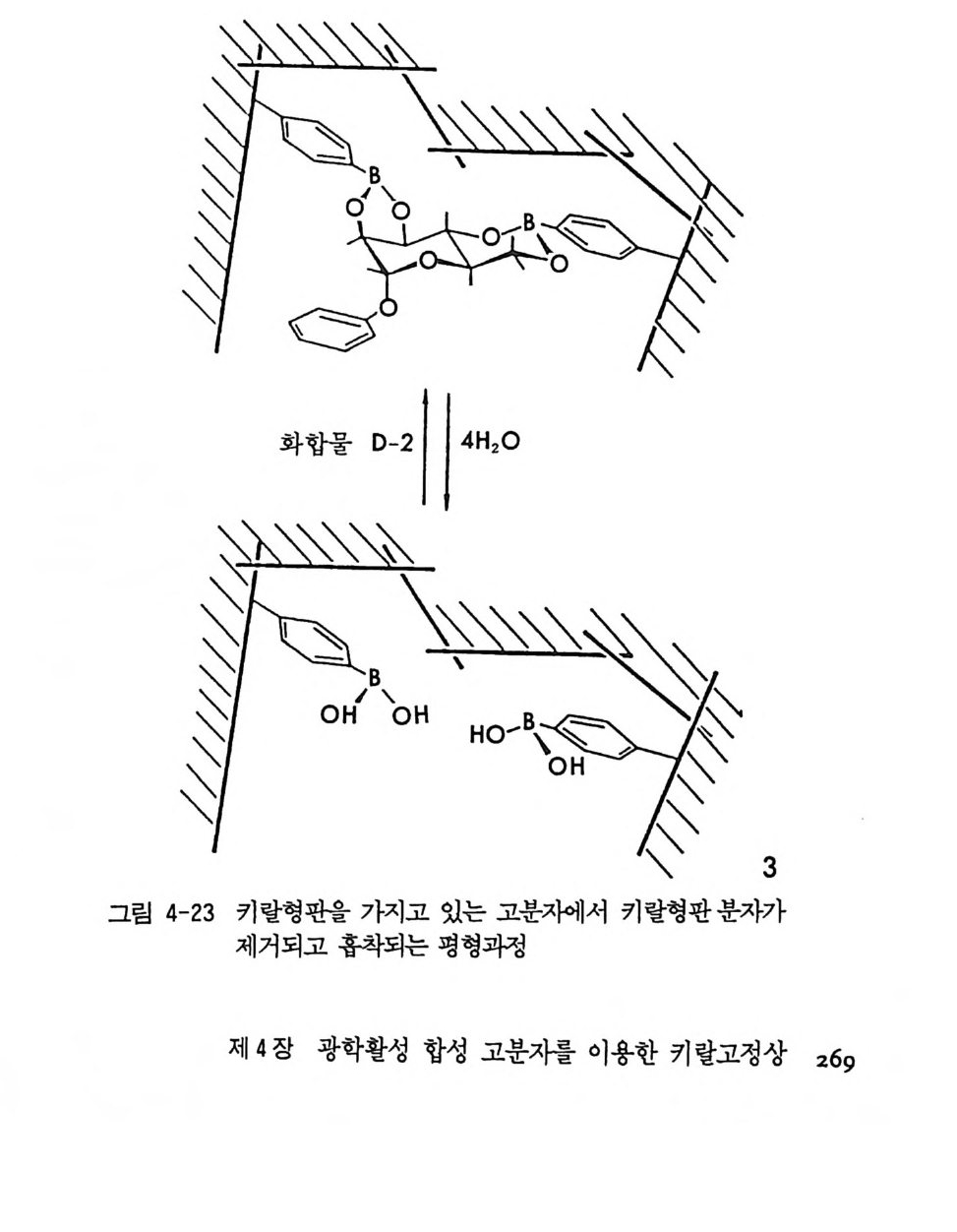 〔`〕}\\
〔`〕}\\
아주 큰 다공성인 고분자가 얻어지며 이 고분자를 물 혹은 알코올로 처리하면 그림 4-23 에서 보는 것처럼 형판분자의 40-90% 가 떨어져 나와 자유키랄공동을 형성하게 된다. 이렇게 얻은 키랄공동을 가전 고 분자를 평형조건에서 형판화합물의 라세미체 (페닐 -a-D, L- 만노피라노 시드)로 처리하였을 때 키랄형판으로 사용되었던 광학 이성질체가 선 택적으로 고분자에 흡착된다는 사실이 확인되었으며, 이때 고분자와 용액상에 존재하는 D- 이성질체와 L- 이성질체의 비, 죽 분배계수 혹 은 분리인자 a 는 고분자가 만들어지는 조건에 따라 달라지나 최적조 건에서 만들어진 고분자의 경우 a = 3 . 65 인 것으로 확인되었다(1). 이 와 같은 결과는 키랄형판 분자 존재 하에서 고분자를 합성한 후 키랄 형판 분자를 제거하였을 때 고분자가 키랄형판과 똑같은 모양의 키랄 공동을 유지할 뿐만 아니라 키랄형판 분자와 작용하는 고분자 작용기 의 키랄공동내 위치 (그림 4 - 23 에서는 봉산의 OH기 위치) 또한 키랄형 판 분자와 다시 결합할 수 있는 적절한 위치에 그대로 유지되고 있음 울시사해 주고있다. 키랄형판 존재하에서 고분지를 합성한 후 키랄형판을 제거하였을 때 고분자가 키랄형판과 같은 모양의 키랄공동을 유지하는- 능력은 고분자 를 합성할 때 사용하는 교차결합 시약의 종류 및 사용하는 교차결합 시약의 양에 아주 크게 영향을 받는다. 왜냐하면 키랄형판을 제거한 후 키랄형판과 똑갇은 형태의 키랄공동을 유지하기 위해서는 고분자의 안정성이 특히 중요하며 이 안정성은 교차결합의 정도에 의하여 결정 되기 때문이다. 그림 4-24 는 교차결합 시약인 p-디바닐벤젠, 에틸렌 글리콜디메타크릴레 이트 (ety h leneg ly c oldim eth a cryl a te ) , 부틸렌디올 디메타크릴레이트 (bu ty lene dioldi me t hac ry la t e) 존재 하에서 모노머 l 을 라디칼 중합하고 키랄형판을 제거하여 키랄공동을 가전 고분자 3 을 합성하였을 때 형판화합물의 두 광학 이성질체에 대한 흡착분배계수 a 와 교차결합 정도의 관계를 그래프로 나타낸 것이다. 교차결합의 정도가 10% 미만에서는 입체 선택성을 전혀 보이지 않
 400 a-value
400 a-value
그림 4-24 키 랄공동고분자 쁜 l 교차결합 정도 및 입체 선택성과의 관계 . 에칠렌글리콜디메타크릴레이트( ■), 부틸렌디올디메타크릴레 이트(•), p-디비닐벤젠(~) (참고문헌 : 1)
으며 교차결합의 정도가 증가함에 따라 입체 선택성이 크게 증가하고 있음을 알 수 있다. 특히 교차결합 시약이 에틸렌글리콜디메타크릴레 이트인 경우에는 교차결합 정도가 50% 까지는 a 값이 1.50 정도로 증 가하다가 교차결합 정도가 66.7% 로 증가하면 a 값이 3.04 로 급격히 증가하고 교차결합의 정도가 95% 에서는 a 값이 3.66 임을 알 수 있 다. 그러나 디비닐벤젠울 교차결합 시약으로 사용한 경우에는 교차결 합의 정도가 크게 증가하여도 입체 선택성은 그렇게 크게 증가하고 있 지 않다. 표 4-2 는 교차결합 시약을 달리하여 교차결합의 정도가 다 른 키랄공동고분자 3 을 합성하였을 때 이들 고분자의 물리적 성질을 나타낸 것으로서 표 4-2 로부터 그립 4-2~ 결과를 쉽게 짐작할 수 있다 (2). 키랄형판 라세미체의 두 광학 이성질체에 대한 고분자의 입체 선택
표 4:.2 교차결합 시약을 달리하여 얻은 키랄공동 고분자 Pl 물리적 성질 (참고문현 : 2) 고분又} 교차결합 함께사용한 교차결합 내부표면적 형판분리 팽윤성 분리인자 시약 모노머 정도(%) (m2/g) 정도(%) a’ P1 P-DVB sty ren e 71 661 42. 2 1.36 1.67 sty ren e 95 825 41 .3 1.27 1.70 P2 EGDM MMA 63 169 89. 5 1.87 3.0 4 없음 95 383 81 .7 1.69 3.66 p - DVB : p-디바닐벤젠, EGDM : 에 틸 렌글리콜디메타크 릴 레이 트, MMA : 메 틸메타크릴레이트
성은 교차결합 정도가 증가함으로 갖게 되는 키랄공동의 안정성에도 관계되지만 고분자의 유연성도 중요한 역할을 한다. 고분자의 유연성 이 결핍되면 키랄형판 분자가 고분자에서부터 쉽게 떨어져 니올· 수 없 을 뿐만 아니라 가역적으로 다시 쉽게 결합하기도 어려워진다. 고분자 의 유연성 척도는 고분자의 팽윤성 (swellab ility)이다. 즉 팽윤성이 클 수록 유연성이 크다고 할 수 있다. 표 4_2 에서 보는 바와 같이 p -DV 賤 교차결합 시약으로 사용하여 얻은 교차결합 정도가 95% 인 고분자의 괭윤성은 1. 27 인 반면 EGDM 을 교차결합 시약으로 사용하 여 얻은 교차결합 정도가 95% 인 고분자의 팽윤성은 1. 69 이다. 따라 서 EGDM 을 교차결합 시약으로 사용하여 합성된 고분자 P2 의 유연 성은 p -DV B-을 교차결합 시약으로 사용하여 얻은 고분자 P1 의 유연 성보다 훨씬 크며 그 결과로서 키랄형판분자가 고분자에서 떨어져 나 온 정도는 고분자 P1 의 경우 4 1. 3% 인 반면 고분자 P2 의 경우에는 8 1. 7% 이다. 결국 고분자 P2 는 훨씬 많은 키랄공동을 가지며 유연성 도 크기 때문에 라세미 형판화합물에 대하여 고분자 P1 보다 훨씬 좋 은 입체 선택성을 보인다고 생각할 수 있다. Fuji i 등은 D, L- 벤질발린에 대하여 거의 100% 의 입체 선택성을
 x /0< 00 H ( ClRy, c2R、e’' 0 -X h l 'n e2 D ·Ia m. .n.. 0X:P :N O\H5Ix
x /0< 00 H ( ClRy, c2R、e’' 0 -X h l 'n e2 D ·Ia m. .n.. 0X:P :N O\H5Ix
보이는 키랄공동고분자의 합성을 보고한 바 있다 (5). 4-( p-비닐벤질 옥시)살리실알데히드와 (1 R, 2R)-1,2- 디아미노시클로핵산의 번응에 의 하여 생긴 쉬프염기리간드 _1-2} Co(OAc)2 및 N- 벤질 _D- 발린의 반응 에 의하여 형성된 6 배위 코발트착물 5 를 출발물질로 사용하여 스티렌 및 디비닐벤첸과 함께 아조이소부티로니트릴 (AIBN) 개시제 존재 하 에 공중합하면 녹색의 고분자 칙물§이 얻어진다(그림 4-25). 이 녹색 의 고분자착물을 3M HCLl 료 처리하면 키랄형판 분자인 N- 벤질 -D -발린이 제거되어, 유기용매에 녹지 않는 갈색의 키랄공동 고분자 착 물 1 이 얻어진다. 고분자 착물 1 을 과량의 라세미 N- 벤질발린으로 처리하면 몇 시간내에 녹색의 고분자 착물 §이 얻어지며 이것을 다시 3M HCL 로 처리하면 99.5% 의 광학순도를 가진 N- 벤질J)-발린이 회수됨을 확인할 수 있고 분배계수 a 는 무려 6.82 임이 확인되었다. 그러나 쉬프염기 4를 스티렌 및 디비닐벤젠과 함께 공중합하여 얻은 고분자를 CoCl2 와 반응시켜 고분자 착물을 만들었을 때 이 고분자 착 물의 입체 선택성은 고분자 착물 1 보다 현저히 감소할 뿐만 아니라 고 분자가 아닌 Co- 쉬프염기 4 착물의 입체 선택성 또한 고분자 착물 1 보다 현저히 감소하고 있음을 표 4-3 에서 확인할 수 있다 (5). 이 결과 는, 키랄형판 분자의 존재 하에서 고분자를 합성한 후 키랄형판 분자 롤 제거하여 얻은 키랄공동 고분자의 입체 선택성에 대하여 키랄형판
표 4-3 N- 벤젤 -D- 발린을 키랄형판으로 사용하여 합성된 고분자 1, 키랄형판없이 합성 된 갇은 형태의 고분자 및 쉬프염기 산 -Co 착물의 입체 선택성 (참고문헌 : 5) 착물 kc. G0(kJ /m ol) 고분자착물 1 682 16.2 비키랄형판고분자 18.5 7.2 트란스 -[Co(4) (H20 넓 Cl 24.8 8.0 °K= {[Co(D - a. a.)][L-a.a.-H ]/{ [Co(L-a. a)]/ [ D-a.a . -H] }
분자가 중요한 역할을 하고 있음을 시사해 주는 좋은 예라고 할 수 있 다. 이 의에도 키랄형판 분자 존재하에 고분자를 합성한 후 특정한 키랄 공동을 만들어 광학 이성질체 분리에 응용하고자 하는 예는 디수· 있다 (6-10). 예를 들면 Anderson 등은 p-비닐벤조산과 페닐알라닌에틸에 스테르 사이에 형성된 염을 출발물질로 사용하여 디비닐벤젠과 함께 공중합반응을 하고 페닐알라닌에틸에스데르를 제거하여 키랄공동을 가전 고분자물 합성하였으며 (6) Necker 동은 프롤린의 비닐벤질에스 데르유도체를 스티렌 및 디비닐벤젠과 함께 공중합반응을- 하고 프롤린 을 제거함으로써 키랄공동을 가전 고분자를 합성하였고 (7) 이들은 키 랄형판 라세미체에 대하여 어느 정도의 입체 선택성을 보임이 확인되 었다. 키랄형판 분자를 이용하여 합성된 키랄공동 고분자는· 라세미 형판화 합물에 대하여 좋은 입체 선택성을 나타내기 때문에 일정한 입자크기 를 가지는 이돌 고분자입자들을 HPLC 컬럼에 충전하면 HPLC 에 의 한 라세미 화합물(특히 형판화합물)의 광학분할에 이용될 수 있을 것이 다. 키랄공동 고분자 1 은 N- 벤질 -D, L- 발린에 대하여 아주 좋은 입 체 선택성을 보이나 이 고분자를 이용한 HPLC 광학분할은 현재까지 시도된 바 있다. 아마도 아미노산과 키랄공동 고분자 1 사이의 리간드 교환반응이 너무 늦게 일어나므로 HPLC 에 응용될 수 없는 것으로 예측된다. 키 랄공동 고분자 3 을 이용한 HPLC 광학분할은 Wulff 등에 의하여 많이 연구되어 있으나 그렇게 성공적인 것은 아니었다(1, 2,11). 키랄 형판과 결합하고 있는 모노머 l 을 13.2% 포함하는 고분자를 합성한 후 키랄형판 분지를 제거한 키랄공동 고분자 3 을 키랄고정상으로 사용 하여 라세미 형판화협물 (D, L-.?_) 을 광학분할한 대표적인 크로마토그 램은 그림 4-26 와 같다(1). 그림 4-26 에서 보는 것처럼 피이크가 넓 고 긴 끌림을 가지고 있어서 두 광학 이성질체가 완전하게 분리되고
s t a 『t
그림 4-26 키랄공동고분자 g울 기저로 한 키랄고정상에서 ( 土 ) -2 의 광학분할 이 동상 : 아세토니트릴 /NH3 수용액 (25%)/ H 2 0( 91 : 4 : 5) .. 온도 : 실온 (참고문헌 : 1)
있지 않다. 키랄공동 고분자 3 을 기저로 한 키랄고정상에서 HPLC 에 의한 라세미 화합물의 광학분할은 크로마토그래피를 하는 동안 키랄공 동 고분자와 분석성분 사이에 공유결합이 형성되고 끊어지는 과정이 입체 선택적으로 계속 일어나서 이루어지나 이 과정이 너무 늦기 때문 에 피이크의 모양이 아주 넓으며 피이크가 길다란 끌립현상을- 보이는 것으로 예상된다. 따라서 피이크의 넓어짐과 끌림을 완화시키는 것은 키랄공동을 가진 고분자 키랄고정상과 분석성분 사이에 가역적인 공유 결합형성을 빠르게 일어나도록 하는 것으로서 Wul ff는 적절한 이동상 울 선택하거나 크로마토그래피를 수행하는 온도를 높임으로써 분석성 분의 머무름 시간을 조절하여 크로마토그래피에 의한 광학분할의 최적 조건을 찾고자 노력하였다(1). Wulff 등은 키랄형판 화합물인 D-~ 와 이것의 광학 이성질체인 L -2 에 대한 키랄공동 고분자 뿐 1 결합상수를 여러 용매내에서 측정한 결과 키랄공동 고분자 3 과 키랄형판 화합물 2 사이의 에스테르화 반응 의 평형에 가장 좋은 용매는 메탄울과 아세토니트릴임을 확인하였고 여기에 물을 첨가하면 가수분해에 의하여 평형은 키랄공동 고분자 뿐} 키랄형판화합물 2 쪽으로 이동하여 결합상수는 감소한다는 사실을 확
인하였다. 또한 용매에 암모니아나 피페리딘과 같은 질소원자를 가전 염기를 소량 첨가하면 B-N 결합의 형성으로 인하여 가수분해에 더 안 정한 에스데르결합을 형성하기 때문에 결합상수는 증가하는 것을 확인 하였다(표 4-4 ) . 이와 같은 결합상수에 대한 실험 및 크로마토그래피 에 의한 실험 등의 결과, 이동상으로서는 그림 4-26 에서 보는 바와 같이 4% 의 암모니아수용액 (25% NH 3 ) 과 6% 물을 첨가한 아세토니 트릴 혼합용매가 광학분할에 가장 좋은 것으로 확인되었다 (1). 키랄공동을 가진 키랄고분자 3 을 기저로 한 키랄고정상에서의 광학 분할은 크로마토그래피를 실시하는 온도에 크게 영향을 받는다. 키랄 고분자 3 을 HPLC 컬럼에 충전하고 여기에 (D, L)-2 를 주입하였을 때 D-2 는 키랄공동내에 존재하는 두 개의 봉산과 두 자리에서 봉산에 스데르 결합을 형성하는 동시에 키랄공동의 모양에 정확하게 들어맞아 고정상에 머무른다. 반면 L - 2 분자의 입체구조와 키랄공동내에 존재하 는 두 개의 봉산의 위치가 두 자리에서의 붕산에스테르결합을 형성하 기에 적합하지 않기 때문에 L-2 분자는 키랄고정상과 한 자리에서만 봉산에스데르결합을 형성하는 것으로 · 생각할 수 있다. 온도가 증가함 에 따라 봉산에스테르결합의 가수분해속도는 봉산에스데르결합의 형
표 4~4 키랄공동 고분자 笠에 대한 D-~ 및 L 겔의 결합상수(참고문헌 : I) 결합상수 용매 L-2 D- 2 a’ MeOH 0.37 1.31 3.54 MeOH, 0.1 % H20 0.1 7 0.3 9 2.31 MeOH, 2% NH 기체 0.46 0.7 8 1.68 CH3CN 5.4 8 20. 28 3.72 CH3CN, 5% H20 0.1 0.2 6 2.60 CH3CN, 4% H20, 1% NH3* 0.31 0.47 1.54 *25% NH3 수용액
성속도보다 빨라진다. 따라서 온도가 증가함에 따라 분석성분의 머무 름 시간은 계속 감소하며 또한 봉산에스테르결합이 형성되고 끊어지는 속도(가수분해속도)가 증가하기 때문에 컬럼내 분석성분의 물질이동이 원활해져서 피이크의 모양이 좁아진다. 이와 같은 효과는” 키랄공동고 분자 3 과 한 자리에서만 봉산에스데르결합을 형성하는 L-2 의 경우 크 게 나타나나 D-Z 의 경우에는 D-2 가 키랄공동내 두 자리에서 붕산에 스테르결합을 형성할 뿐만 아니라 D-2 의 입체구조와 일치하는 키랄공 동에 위치하기 때문에 특별히 안정하여 온도에 의한 효과가 L- 塾i다 는 훨씬 적게 나타난다. 그림 4-27 은 키랄공동고분자 3 을 기저로 한
 L
L
그림 4-27 2온키m도랄L 공:/ m8동(iJ n ' C고, ,양 분격이자 동: g8상울0 b :기 a아r저, 세a로 토= 니하 3트.는37릴 키( +참랄1고0고문%정헌 H상 :2에 10서2+) 5%(士 )N-I碑- 1]3 , 광유학분속할 :. o .
키랄고정상(키랄모노머 l 을 13. 2% 포함하는 고분자로부터 제조된 것)에 서 라세미 화합물 2 의 광학분할을 80° C 에서 실시한 크로마토그램이다 (2, 12). L - 이성질체에 대한 피이크가 좁고 날카로운 반면 D- 이성질체 에 대한 피이크는 80 ° C 에서도 상대적으로 넓으며 긴 끌림을 가지고 있 으나 기준선 분리에 의한 광학분할을 확인할 수 있다 (12). 기준선 분 리에 의한 광학분할로 인하여 이동상의 유속을 증가시키더라도 분해인 자에 큰 영향없이 광학분할할 수 있기 때문에 광학분할에 필요한 분석 시간은 훨씬 감소한다. 결론적으로 적절한 키랄형판 분자 존재 하에서 고분자를 합성한 후 키랄형판 분자를 제거하여 얻은 키랄공동을 가진 고분지를- 키랄고정상 으로 이용하여 라세미 형판화합물의 두 광학 이성질체를 HPLC 상에 서 광학분할하고자 하는 착상은 아주 좋으나 아직 크게 성공적이지 못 하다. 키랄형판 분자와 고분자 사이의 가역적인 상호작용(대부분의 경 우 공유결합의 형성)이 늦은· 속도로 이루어지지 때문에 물질이동에 문 제가 있게 되어 크로마토그램의 피이크가 넓어지고 길게 끌리는 현상 이 문제점으로 지적된다. 이동상을 조절하고 광학분할의 온도롤 높임 으로써 이러한 문제점을 어느 정도 해결할 수 있으나, 더 많은 연구에 의하여 키랄형판 분자와 고분자 사이의 가역적인 상호작용이 U 바른 속 도로 이루어지는 키랄형판 분자들을· 새로 찾아야 할 필요가 있다. 특 히 키랄형판분자를 이용하여 얻은 키랄공동 고분자 키랄고정상은 오로 지 하나의 라세미 화합물(형판 라세미 화합물)의 광학분할에만 응용될 수 있다는 점이 큰 단점으로 지적되고 있다. 그러나 한 가지 라세미 화합물만을 광학분할하고자 하는 목적에는 훌륭하게 응용~ 수 있을 것으로예상된다.
참고문헌 (제 4 장, 2) l. Y. Okamoto , K. Suzuki, K. Ohta , K. Hata d a and H. Yuki, J. Am. Chem. Soc., 쁘, 4763 (1979). 2. Y. Okamoto . H . Shohi and H. Yuki, J. Polym . Sc i. Polym . Lett . Ed., _21,, 601 (1983). 3. H. Yuki. Y. Okamoto and I. Okamato , J. Am. Chem. Soc., 브 2, 6356 (1980). 4. Y. Okamoto , I. Okamoto and H. Yuki, Chem. Lett ., 835 (19 81). 5. Y. Okamoto and H. Yuki, i n As ym metr ic R eacti on s and Proces ses in Chemi stry, E. L. Elie l , Ed., ACS Sy m p o siu m Serie s 185, Americ a n Chemi ca l Soc iet y , Washin g ton D. C. 1982, pp 280-282. 6. Y. Okamoto , S. Honda, I. Okamoto , H. Yuki, S. Murata , R. Noy o ri and H. Takay a, J. Am. Chem. Soc., 쁘, 6971 (1981). 7. Y. Okamoto , H. Mohri, M . Ishik u wa, K. Hata d a and H. Y uki, J. Polym . Sci. Polym . Sy mp., 꼬 125 (1986). 8. • Y. Okamoto , M. Ishi ku ra, K. Hata d a and H. Yuki, P olym . J., 拉, 851 (1983). 9. Y. Okamoto , H. Mohri , M. Nakamura and K. Hata d a, Ni ppo n Kag a ku Kais h i, 4 35 (1987). 10. Y. Okamoto and K. Hata d a, J. Liq. Chromato g r., 언, 369 (1986). 11. Y. Okamoto , S. Honda, K. Hata d a and H. Yuki, B ull. Chem. Soc. Jpn ., ~ . 3053 (1985). 12. Y. Okamoto and K. Hata d a, In Chromato g 1 'a ph ic Chir a l Se pa ra- tion s, M. Zie f and L. J. Crane, Eds., Marcel Dekker, New York, 1988, chap 8. 13. Y. Okamoto , S. Honda, E. Yashim a and H. Yuki, Chem. Lett ., 1221 (19 83). 14. Y. Okamoto , E. Yashim a and K. Hata d a, J. Chem. Soc. Chem. Commun., 1051 (1984). 15. Y. Okamoto , S. Honda, K. Hata d a, I. Okamoto , Y. Tog a and S. Kobay as hi , Bull. Chem. Soc. Jpn ., ~, 1681 (19 84). 16. Y. Okamoto and K. Hata d a, in Chir a l Sepa r ati on s by HPLC :
Ap pli ca ti on s to Pharmq ce u ti ca l Comp ou nds, A. M. Krstu l ovic , Ed., Ell is H orwood, Chic h este r , 1989, chap 12. 17. R. Noy o ri, N . Sano. S. Murata , Y. Okamoto , H. Yuki and T. Ito, Tetr a r~d r on, Lett ., 겜 2969 (1982). 18. M. Nakazaki, K. Yamamoto , T. Ikeda, T. Ki tsu ki and Y. Okamoto , J. Chem. Soc. Chem. Commun., 7 87 (1983). 19. Y. Okamoto , E. Yashim a. K . Hata d a and K. Mi slo w, J. O rg. Chem., 49, 557 (1984). 20. Y. Okamoto , S. Honda. H. Yuki, H. Nakamura, Y. Iitak a and T. Nozoe, Chem. Lett ., 1149 (1984). 21. N. Harada, J . Iwabuchi, Y. Yokoda, H . Uda, Y. Okamoto , H. Yuki, and Y. Kawada, J. Chem. Soc. Perkin . Trans. I, 1 845 (1985). 22. W. Ki ss ener and F. Vog tle , Ang ew . Chem. Int. Ed. Eng l., 샘 222 (1985). 23. K.-H . Duchene and F. Vog tle , Ang ew . Chem. Int. Ed. Eng l. , 샘 885 (1985). 24. K. Yamamoto , K. Noda and Y. Okamoto , J. Chem. Soc. Chem. Commun., 1421 (19 85). 25. D. M. Joh ns, in Chir a l Liq u id Chromato g r a ph y , W . J. Loug h, Ed., Blackie , Glasgo w and London, 1989, chap 10. 26. C. Mein a rd and P. Bruneau, J. Chromato g r., 壁 169 (1988). 27. Y. Tokuma, T. Fuji wa ra and H. Nog uc hi, J. P harm. Sc i., 7§., 310 (1987). 28. P. Macaudie r e, M. Caude, R. Rosset and A. Tambute , J. Chromato g r. S c i., 꼬 583 (1989). 29. Y. Okamoto , S. Honda, K. Hata d a and H. Yuki, J. Chromato g r., 완Q, 127 (1985). (제 4 장, 3) 1. G. Blaschke, J. Liq . Chromato g r., 안 341 (1986). 2. G. Blaschke, in Chromato g r a ph i c Chir a l Sep a rati on s, M. Zie f and L.
J. Crane, Eds., M arcel Dekker, New York, 1988, chap 7. 3. G. Blaschke, W. Broker and W. Fraenkel, Ang e w. Chem. Int. Ed. Eng l., ~. 830 (19 86). 4. G. Blaschke, Ang e w. Chem. Int. Ed. Eng l., 브, 13 (19 80). 5. G. Blaschke and H. Markg raf , Chem. Ber. , 墨 2031 (1980). 6. G. Blaschke and J. Maib a um, J. Phara. Sci., 젼, 438 (1985). 7. G. Blaschke and J. Maib a um, J. Chromato g r., 훅 329 (19 86). 8. M. Huff er and P. Schreie r , J. Chromato g r., 堅 137 (1989). (제 4 장, 4) 1. G. Wulff, H.-G. Pol and·M . Mi na rik , J. Liq . Chromato g r., ~ . 385 (19 86). 2. G. Wulff and M. Mi na rik , in Chromato g r ph ic Chir a l Sepa r ati on s, M. Zie f and L. J. Crane, Eds., Marcel Dekker, New York, 1988, chap 2. 3. G. Wulff, W. Vespe r, R . Grobe-E in s ler and A. Sarhan, Makromol. Cham., 墨 2799 (1977). 4. G. Wulf f, in Polym er Reag e nts and Cata l ys t s ,W . T. Ford, Ed., ACS Sy mpo siu m Serie s , 쁘, 186 (1986), chap 9. 5. Y. Fuji i, K. Mats u ta n i and K. Ki tuc hi, J. Chem. Soc. Chem. Com- mum., 415 (1985). 6. L. Andersson, B. Sellergr en and K. Mosbach, Tetr a hedron. Lett ., 7. 썹J. D 5a2r1n1e n(1 9a8n4d) .D . C. Neckers, T etr a hedron. Lett ., n, 1913 (1980). 8. K. J. Shea, E. A. Thomp s on, S. D. Pandey and P. S. Beauchamp , J. Am. Chem. Soc., ~. 3149 (1980). 9. J. Darnen .an d D. C. Neckers, J. Org. Chem 述 1382 (1980). 10. R. Arshady and K. Mosbach, Makromol. Chem., 혹 687 (1981). 11. G. Wulff and W. Vespe r, J. Chromato g r., 壁 171 (1978). 12. G. Wulff and M. Mi na rik , J. High Resolut. Chromato g r. Chro- mato g r. Commun., ~' 607 (1986).
제 5 장 리간드교환키랄고정상 1 서론 리 간드 교환 크로마토그레피 (Liga nd Exchang e Chromato g rap h y : LEC) 는 착물을 형성하고 있는 금속이온과 분리하고자 하는 리간드 혼 합물 사이에 리간드 교환에 의하여 새로운 금속 착물이 선택적으로(혹 은 다른 속도로) 형성되고 또 깨어지는 과정을 거쳐 혼합물을 분리하는 방법으로서 1961 년 He lffe r i ch 에 의하여 처음으로 보고되었다 (1). 만 일 착물을 형성하고 있는 리간드가 광학활성인 키랄리간드이면 광학활 성 금속이온 착물은 다른 리간드로 작용할 수 있는 라세미 혼합물과 입체 선택적으로 리간드 교환을 할 수 있어서 두 개의 광학 이성질체 를 구분할 수 있을 것으로 예상할 수 있다. 광학활성 리간드와 금속이온 사이에 형성된 착물과 라세미 화합물 사이의 입체 선택적인 리간드 교환방법을 이용한 광학분할 방법 특히 크로마토그래피에 의한 광학분할 방법은 크게 두 가지로 나눌 수 있 다. 한 가지 방법은 광학활성 리간드와 금속이온 사이에 형성된 광학 활성 착물을 크로마토그래피 이동상에 첨가하여 비키랄고정싱에서 라
세 미 화합물을 광학분할하는 키 랄이 동상첨 가제 법 (Chir a l Mobil e Phase Addit ive :CMPA meth o d, 제 2 장 참조)이며, 다른 방법은 광학활 성 리간드와 금속이온 사이에 형성된 광학활성 착물을 액체 크로마토 그래피의 고정상으로 사용할 수 있는 고형지지체에 고정시켜 라세미 화합물을 광학분할하는 키랄고정상법 (CSPme t hod) 이다. 두 방법 모 두 리간드 교환 크로마토그래피를 이용한 광학분할에 유용하게 사용될 수 있으며 모두 장·단점을 가지고 있다. CMPA 방법에 대하여서는 이미 제 2 장에서 기술하였으므로 여기서는 리간드 교환 키랄고정상을 이용한 광학분할에 대하여 기술하고자 한다. 리간드 교환 키랄고정상이 라세미 화합물의 광학분할에 처음 이용된 것은 1968 년 Ro g ozh i n 과 Davankov 에 의해서이다 (2). Ro g ozh i n 과 Davankov는 광학활성인 L- 프롤린을 교차결합된 폴리스티렌에 고정 시킨 후 암모니아수용액 중의 황산구리로 처리하여 리간드 교환 키랄 고정상 l 을 제조할 수 있었다. 개개의 구리이온은 고분자에 결합된 두 개의 L- 프롤린 분자와 이5성분 \(b i n_a r ey〔: u) 6H착H尸 : 물을 형성한다. 키랄고정상 l
울 컬럼에 충전한 후, 예를 들어 라세미 아미노산의 수용액을 주입하 면, 이 아미노산과 고정상에 부착되어 있는 L- 프롤린 사이에 리간드 교환이 일어나 다음과 같이 가역적으로 삼성분(t ernar y) 착물이 형성 된다.
RPro -Cu -P roR + HA;: ::! RPro -Cu-A+ H ProR 여기서 HA 는 아미노산, F 百균은 고정상에 고정된 L- 프롤린 음 이온을 의미한다. 라세미 아미노산의 두 광학 이성질체가 리간드 교환 에 의하여 각각 삼성분 착물을 형성할 때 두 개의 삼성분 착물은 서로 부분 입체 이성질체의 관계에 있으며 안정성에 차이가 있게 된다. 따 라서 서로 부분 입체 이성질체의 관계에 있는 두 개의 삼성분 착물- 중 더 안정한 착물을 형성하는 광학 이성질체는 키랄고정상에 오래 머무 르며 상대적으로 불안정한 착물을 형성하는 광학 이성질체는 키랄고정 싱에 머무르는 시간이 짧아 두 개의 광학 이성질체가 구분된다. 광학활성 착물을- 고형지지체에 고정시켜 키랄고정상으로 이용할 수 있기 위하여 중요한 사항은 키랄고정상을 이루는 광학활성 착물과 광 학분할하고자 하는 라세미 화합물 사이에 입체 선택적이며, 빠르고 가 역적인 리간드 교환반응이 가능하여야 한다는 것이다. 가역적인 리간 드 교환반응에 의한 착물의 형성은 고정된 리간드의 종류, 금속이온의 종류, 이동상(완충용액의 농도, pH, 금속이온의 농도, 온도) 및 고형 지지체의 종류에 따라 달라진다. 빠르고 가역적인 리간드 교환반응이 가능한 금속이온둘은 보통 Cu(Il ), Zn( Il ), Ni (Il ), Cd( Il )과 같은 2 가의 전이금속 양이온들이며 특히 리간드 교환 키랄고정상을 이용한 광학분할에 주로 시용되어 온 금속이온은 Cu (Il)이다. 반면 Co(il l), Cr(III), P t(N)과 같은 금속 양이온들에 의하여 만들어전 착물들은 아주 안정하기 때문에 일반적으로 이들 금속 양이온들을 금속이온과 리간드 사이의 빠른 리간드 교환반응을 이용하여 혼합물을 분리하는 리간드 교환 크로마토그래피에 사용하기에는 적당하지 않다. 리간드 교환 키랄고정상을 이용한 HPLC 광학분할은 Davankov 등의 최근 저서 (3) 『 리간드 교환 크로마토그래피 (Lig a nd Exchang e Chroma to g rap hy ) 』 에 자세히 기술되어 있으며 리간드 교환 키 랄고정상 울 이용한 광학분할의 좋은 참고문헌이 될 것이다. 여기서는 리간드
교환 키랄고정상의 세 가지 · 형태 즉 키랄 리간드 화합물을 고분자에 고정시킨 키랄고정상, 키랄 리간드 화합물을 실리카 겔에 고정시킨 키 랄고정상 및 키랄 리간드 화합물을 실리카 겔에 코팅시키거나 혹은 홉 착시킨 키랄고정상에 대하여 이들 키랄고정상들의 합성, 광학분할의 특성, 키랄성 인지 메커니즘 등에 대하여 기술하게 될 것이다. 2 리간드 교환 키랄고정상의 제조, 광학분할의 특성, 광학 분할메커니즘 2.1 키랄 리간드 화합물을 고분자에 고정시킨 키랄고정상 키랄 리간드를 고분지에 고정시킨 키랄고정상의 대부분은 광학활성 아미노산울 아미노기를 통하여 폴리스티렌 혹은 폴리아크릴아미드에 공유결합시킨 것들이다. 여러 종류의 광학활성 아미노산들 중 특히 고 리형 아미노산인 L- 프롤린 (p ra li ne) 이나 L- 히드록시프롤린을 고분자 에 고정시킨 키랄고정상이 Cu (Il)와 착물을 형성한 후 라세미 아미노 산들의 광학분할에 성공적으로 사용되었다.
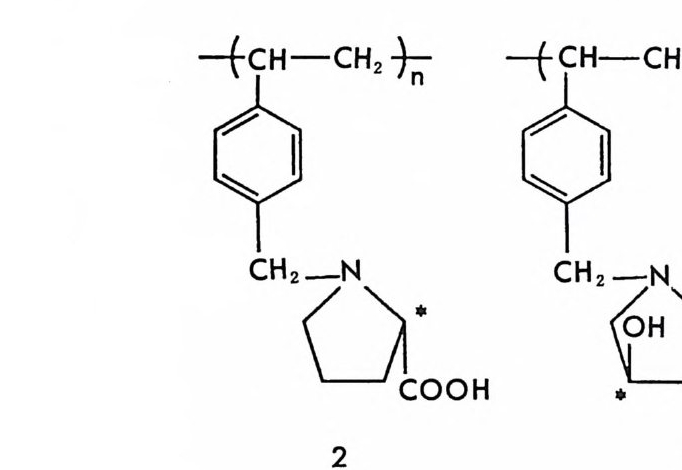 CH2:* CH2 臼·
CH2:* CH2 臼·
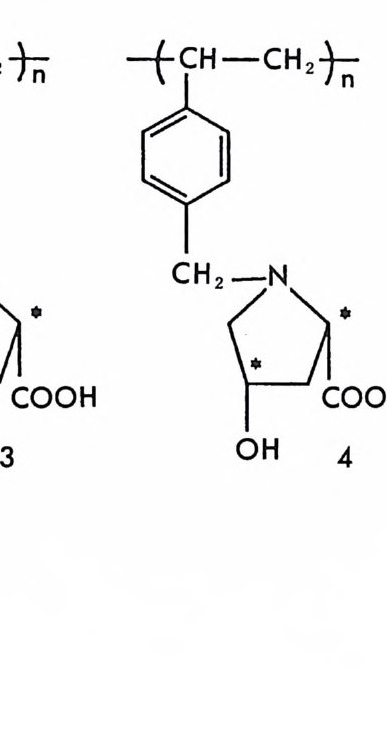 c H :广。H
c H :广。H
L- 프롤린을 기저로 한 리간드 교환 키랄고정상은 교차결합된 폴리 스티렌수지의 페닐기의 파라위치에 클로로메틸화 반응을 한 후 이것을 L- 프롤린과 반응시켜 L _ 프롤린을 고분자에 고정시키고 고분자에 고 정된 L- 프롤린에 Cu(Il )이온을 처리하여 제조할 수 있다 (2,3,4,5). 이렇게 제조된 세 종류의 리간드 교환 키랄고정상 2, g 및 싣는 여러 종류의 라세미 아미노산 및 Cu (Il)이온과 함께 입체 선택적으로 삼성 분 착물을 형성함으로써 라세미 아미노산의 광학분할에 성공적으로 사 용되었으며 용리순서에 있어서 몇 가지의 중요한 특성을 보이고 있다. 이들 세 종류의 리간드 교환 키랄고정상 중 L- 히드록시프롤린 키랄고 정상 3 에서의 광학분할에 대한 크로마토그램의 예는 그림 5-1 과 같으 며 (3,4) 세 종류의 키랄고정상에서의 여러 라세미 아미노산들의 광학 분할은 표 5 - 1 에 요약되어 있다 (6,7,8).
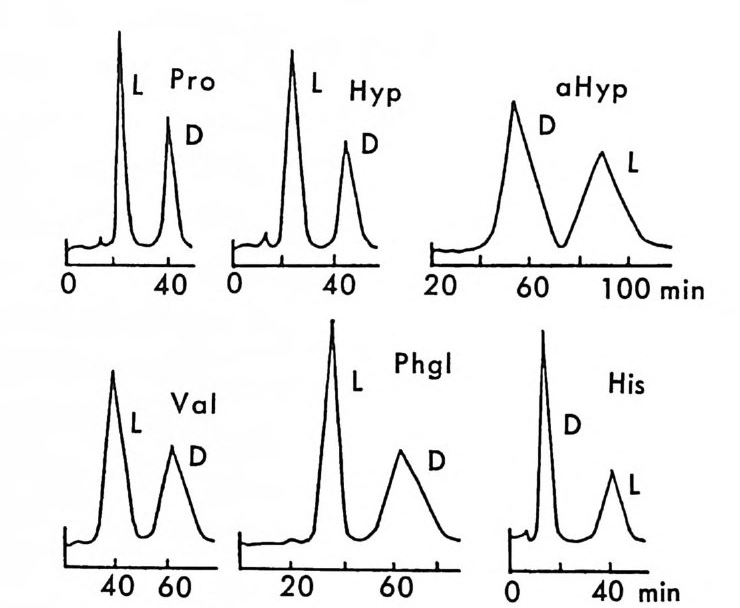 L Pro
L Pro
그림 5-1 키랄고정상 연 (3.86 mmole/g, 입자크기 50µm, Cu(Il )함량 : 30-65%) 에서 @回 라세미 아미노산의 광학분할. 컬럼 : 14X 0.8cm, 이동상 : 0. 05-l. OM NH.OH, 유속 : 13-25mL/hr, 검출기 : 206run UV (참고문헌 : 3)
표 5-1 키랄고정상 2, 3, 4 (금속이온 : Cu(Il ) 이온)에서 라세미 아미노산의 광학분할 에 대한 분리인자 a = ko'/kL' 아미노산 CSP2 CSP3 CSP 4 알라닌 (Ala) 1.08 1.04 1.04 발린 (Va l) 1.29 1.61 1.58 루이신 (Leu) 1.27 1.70 1. 54 트레오닌 (Thr) 1.38 1.52 1.48 페 닐글리신 (Phe g l y) 1.67 2. 22 1. 78 프롤린 (Pro) 4.0 5 3.9 5 1.84 히드록시프롤린 (hPro) 3.8 5 3.17 1.63 알로히드록시프롤린 (aH yp) 0. 43 0.6 1 1.48 히스티딘 (H i s) 0.37 0.36 1.32 。比팔트산 (As p} 0.91 1.00 0.8 1 글루탐산 (Glu) 0.62 0.8 2 0.69
표 5-1 에서 보는 것처럼 대부분의 라세미 아마노산들의 광학분할에 서는 D- 이성질체가 L 이성질체보다 컬럼에 더 오래 머무른다. 키랄 고정상의 리간드와 Cu(Il) 이온 및 이동상중의 리간드 화합물 사이에 형성되는 삼성분 착물은 보통 두 아미노산의 질소원자와 산소원자를 트란스-위치에 둔 평면 4 배위착물로서, L- 프롤린 혹은 L- 히드록시프 롤린 키랄고정상 災} 뿐] 경우 평면 4 배위 착물 축방향의 두 개 배위 자리 중 하나는 벤질기가 가로 막고 있고 다론 하나의 배위자리는 이 동상의 물 혹은 암모니아 분자가 약하게 배위되어 있는 것으로 생각되 고 있다. 따라서 이동상중의 D- 아미노산과 L_ 아미노산이 키랄고정상 과 함께 삼성분 착물을 형성할 때 삼성분 착물의 구조는 그립 5_2 와 같다고 할 수 있다. L- 아미노산과 키랄고정상의 삼성분 착물(그립 5 -2a) 의 경우 아미노산의 a_ 알킬치환체와 4 배위착물의 축방향에 배위 되어 있는 물분자 사이의 입체장애로 인하여 그다지 안정한 구조를 가 지고 있는 것으로 생각할 수 없으나 D 一아미노산과 키랄고정상과의 삼
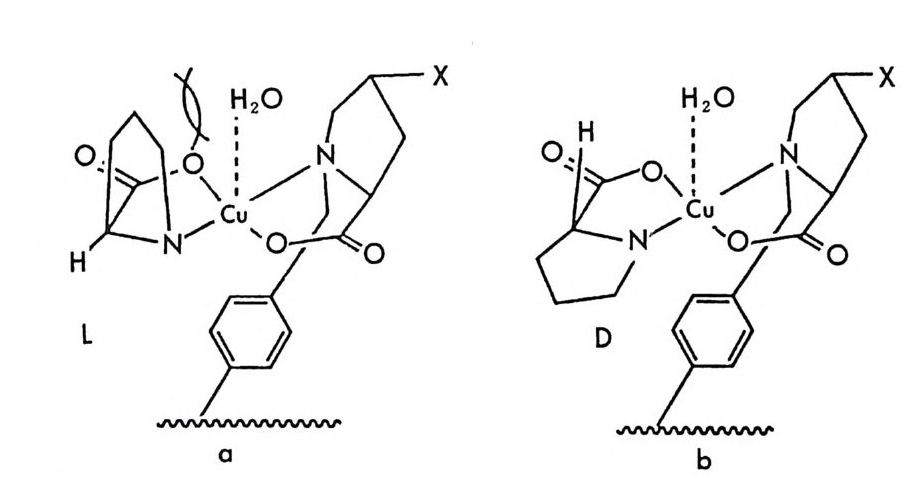 a b
a b
그림 5-2 키 랄 고정상 2 (X= H) 혹은 3 (X = OH) 과 D, L- 아미노산 사이에 형 성된 삼성분-착물의 구조. b 가〕낸다 상대적으로 더 안정하다.
성분 착물(그립 5-2b) 에 있어서는 아미노산의 a- 알킬치환체가 소수성 인 폴리스티렌 지지체 혹은 벤질치환체로 향하고 있어서 서로간에 소 수성 상호작용이 작용하므로 역상 크로마토그래피에 있어서는 상대적 으로 더 안정하여 D- 아미노산이 컬럼에 더 오래 머무르게 된다고 생 각할 수 있다. 그러나 표 5-1 에서 보는 바와 같이 알로히드록시프롤 린 (aH yp), 히스티딘(Hi s), 글루탐산 (Glu), 아스팔트산 (As p)과 같은 세 자리 리간드인 아미노산의 경우에는 L- 아미노산이 키랄컬럼에 더 오래 머무른다. 이들 세 자리 리간드인 아미노산들의 두 광학 이성질 체중 L- 이성질체가 리간드 교환 키랄고정상 컬럼에 더 오래 머무르는 이유는 그림 5-3 에 보는 바와 같이 L - 아미노산의 경우 세번째의 리간 드가 착물의 축방향 배위자리를 차지하여 안정하게 되는 반면 D- 아미 노산의 경우 아미노산의 세번째 리간드와 키랄고정상의 벤질치환체 사 이의 입체장애로 인하여 상대적으로 불안정하게 되기 때문이다. L- 알로히드록시프톨린을 기저로한 리간드 교환 키랄고정상 心에서 여러 라세미 아미노산들의 광학분할에 대한 용리순서는 표 5-1 에서 보는 바와 같이 키랄고정상 2 및 g에서의 광학분할에 대한 용리순서와
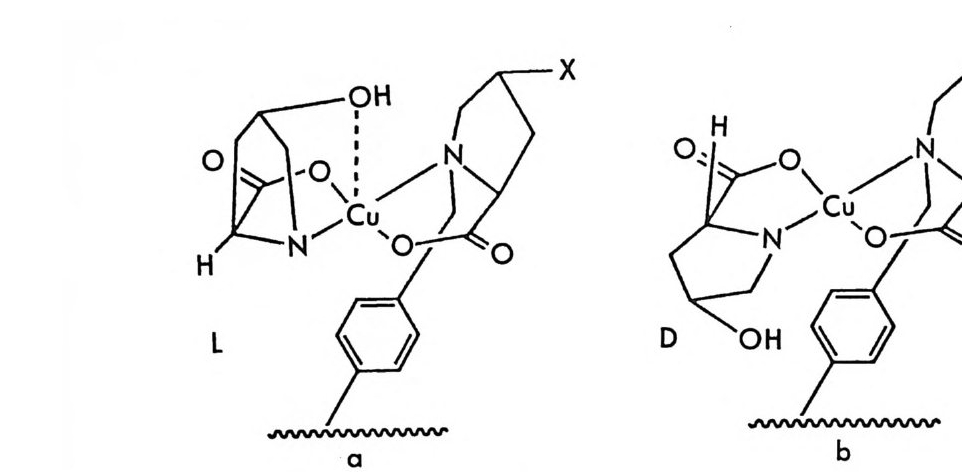 b
b
그림 5-3 키랄고정상 2 (X = H) 혹은 3 (X = OH) 과 세 자리 리간드인 D, L -히드록시프흘린 사이의 착물형강. a.7}-M 다 상대적으로 더 안정하다.
비슷하나 알로히드록시프롤린 및 히스티딘과 같은 세 자리 리간드인 아미노산의 광학분할에 대한 용리순서는 키랄고정상 2 와 g에서의 용 리순서와는 달리 보통의 두 자리 리간드인 아미노산의 광학분할에 대 한 용리순서와 같다. L- 알로히드록시프롤린을 기저로 한 리간드 교환 키랄고정상 뜬] 경우 그림 5-4 에서 보는 바와 같이 L- 알로히드록시프 롤린 고정상중의 히드록시기가 착물의 축방향 배위자리를 차지하게 됨 으로 세 자리 리간드인 아미노산의 세번째 리간드가 배위할 자리가 없 어진다. 따라서 세 자리 리간드인 아미노산이나 두 자리 리간드인 아 미노산 모두 L- 이성질체보다는 D- 이성질체가 더 안정한 착물을 형성 하여 컬럼상에 오래 머무른다고 예상할 수 있다. 그러나 L - 아스팔트 산이나 L- 글루탐산인 경우에는 착물의 축방향에 배위하고 있는 고정 된 리간드의 히드록시기와 아미노산의 a- 알킬기의 끝에 있는 카르복 시기 사이의 수소결합으로 인하여 안정한 착물을 형성하기 때문에 표 5-1 과 같이 L- 이성질체가 컬럼에 더 오래 머무르는 것으로 Davankov 등은 설명하였다 (9). 이와 같은 키랄성 인지 메커니즘을 기초로 하여 리간드 교환 키랄고
그립 5-4 키 랄 고정상 ~ D, L - 아미노산 사이의 삼성분 착물형성 . D - 이성질체가 더 안정한 차물 을 형성한다.
정상의 입체 선택성을 증진시키려는 시도가 있었다. 그림 5 - 2 에서 브 는 것처럼 키랄고정상 2 혹은 꼰斗 D- 아미노산 사이에 형성된 삼성분 착물의 안정성은 아미노산의 a - 알킬 치환체와 고정상의 벤질치환체 사이의 소수성 상호작용이 중요하게 작용하는 것으로 인식되었다. 따 라서 이 소수성 상호작용을 증진시킬 수 있다면 두 삼성분 착물사이의 안정성 차이는 더 크게 되어 더 좋은 입체 선택성을 관찰할 수 있을 것이다. 이와 같은 생각에 따라 Zolot a rev 등은 L- 히드록시프롤린을 기저로 한 리간드 교환 키랄고정상 §를 제조하였다(1 0). 폴리스티렌수 지를 SnC14 존재 하에서 할로겐화알킬과 반응시켜 (Frie d el-C raf ts
 ab RR== (CH03) 3C —-
ab RR== (CH03) 3C —-
alky la ti on ) 페닐기에 알킬기가 치환된 폴리스티렌수지 를 얻은 후 키 랄고정상 2 정 및 산롤 제조하는 방법과 마찬가지로 클로로메틸화 반 옹, L- 히드록시프롤린과의 반응, 구리이온의 처리과정을 거쳐 키랄고 정상 §가 제조되었다. 그림 5 - 5 에서 보는 바와 같이 키랄고정상 院] 경우 고정상의 벤질기에 붙어 있는 알킬기의 영향으로 인하여 D- 아미 노산은 고정상과 더 강한 소수성 상호작용을 할 수 있으며 D - 아미노
 一a 一b
一a 一b
그림 5-5 키랄고정상 ~ D, L - 아미노산 사이의 삼성분 착물형성 . b 가 탸 L 다 더 안정하다.
산에 대한 입체 선택성이 증진되는 것이 확인되었다. 그러나 아미노산 의 a- 알킬 치환체에 친수성기를 가전 아미노산들 죽 세린 (ser), 트레 오닌(thr) 등의 경우에는 키랄고정상 §에서 입체 선택성이 다소 감소 하였다. 아마도 a- 알킬 치환체와 고정상의 R.7] 사이의 소수성 상호 작용보다는 아미노산의 a- 알킬 치환체에 존재하는 천수성기때문에 입 체장애가 작용하여 입체 선택성이 감소하는 것으로 예측된다. 폴리스티렌수지와 같은 소수성 고형지지체를 사용한 리간드 교환 키 랄고정상에서 라세미 아미노산을 광학분할할 때는 일반적으로 D - 아미 노산이 컬럼에 오래 머무르는 반면, 폴리아크릴아미드나 폴리메타크
릴레이트와 같은 친수성 고형지지체를 사용·한 리간드 교환 키랄고정상 에서 라세미 아미노산을 광학분할할 때는 일반적으로 L- 아미노산이 컬럼에 오래 머무르는 것으로 관찰되었다. Lefe b vre 등은 교차결합 된 폴리아클릴아미드에 L- 프롤린을 고정시켜 키랄고정상 §울 제조하 였고 (11) Davankov 등은 폴리 2,3- 에폭시프로필메타크릴레이트에 L- 히드록시프롤린 메틸에스테르를 결합시키고 메틸에스테르기를 가수 분해하여 리간드 교환 키랄고정상 1 을 제조하였다 (12). 이들 리간드 》二HII_CCH』 O— CH2— c0HO ― H62古C IIclCH 晶=-― 3CO H C —H 27 C 一H2순 — ]N * * COOH
교환 키랄고정상에 의하여 형성되는 삼성분 착물에 있어서 고형지지체 중의 -C = O 기 혹은 -0-H 는 그림 5-6 에서 보는 바와 같이 평면에 위치하는 4 배위착물의 두 개 축방향배위자리 중 고형지지체쪽인 아래 배위자리를 차지한다. 따라서 착물을 형성하는 D- 아미노산의 a 유알킬 치환체는 고형지지체와 소수성 상호작용을 할 수 없으며 오히려 D- 아 미노산의 a- 알킬 치환체와 고형지지체의 카르보닐기, 혹은 히드록시 기 사이의 입체장애 때문에 D- 아미노산과 키랄고정상 사이에 형성된 삼성분 착물은 불안정하고 L- 아미노산과 키랄고정상 사이에 형성된 삼성분 착물이 더 안정하여 L- 아미노산이 컬럼에 오래 머무르리라고 예상할 수 있다. 이와 같은 예상은 L- 프롤린의 유도체인 화합물 g을
(D: /a 〉N CHH-CH2 二 \ 三: 3COH2~
그림 5-6 키랄고정상 전 (a) 혹은 I(b) 및 아미노산 사이의 삼성분 착물의 구조
합성하고 발린 혹은 페닐알라닌과 삼성분 착물을 만들었을 때 L- 발린 혹은 L 페닐알라닌을 포함하는 부분 입체 이성질체 착물이 D- 발린 혹은 D- 페닐알라닌울 포함하는 착물보다 더 안정하디는 사실이 실험 적으로 밝혀짐으로써 (13) (그림 5-7) 그 타당성이 입증되 었을 뿐만 아 니라 리간드 교환 키랄고정상 §에 의한 라세미 아미노산의 광학분할은 아미노산의 a- 알킬치환체의 크기가 분리인자 a 와 밀접한 관계에 있 다는 사실이 실험적으로 밝혀짐으로써 그 타당성이 입증되었다(1 4).
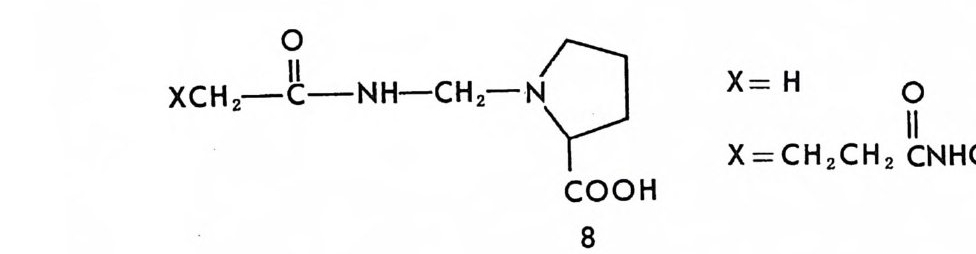 XCH,_[ -N H— CH 2-N: X= H 問
XCH,_[ -N H— CH 2-N: X= H 問
죽 아미노산의 a- 알킬치환체위 크기가 증가함에 따라 Ala (a = 1. 00) , 아미노부티르산 (a = 1.25 ) , n-Val (a = 1.30 ) , Pheala (a = 2. 00) , Ty r (a = 2.0 0 ) , Trp ( a = 2 .4 5) 순으로 a 값이 증가할 뿐만 아니
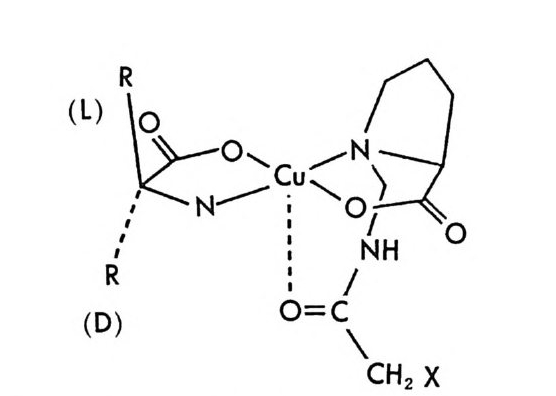 (DR) O=CI\
(DR) O=CI\
그림 5-7 화합물 院} 아미노산 사이의 삼성분 착물 화합물 距斗 L - 아미노산이 더 안정한 착물을 형성한다.
라 특히 a- 알킬치환체가 가지를 가지고 있을 때 다음에 보는 바와 · 같 이 효과적으로 광학분할됨이 확인되었다. Val(a = 1.7 5), Isoleu(a = 1.5 5), Ser(a = 1.85 ), Thr(a = 1.90 ) 아미노산의 a- 알킬치환체 크기가 증가함에 따라 D- 이성질체의 경 우 지유롭게 회전할 수 있는 a- 알킬치환체와 착물의 축방향 아래 배 취자리를 차지하고 있는 고형지지체의 카르보닐기 사이에 입체장애가 증가하여 착물의 안정성이 감소함으로 a 값이 증가하리라는· 것을 쉽게 짐작할 수 있다. 그러나 리간드 교환 키랄고정상 § 혹은 1 에서 라세미 프롤린을 광학분할할 때는 다른 아미노산과는 달리 D- 아미노산이 컬 럼에 오래 머무르는 사실이 확인되었다. 프롤린의 a- 알킬치환체는 아 미노기와 고리를 형성하기 때문에 다른 아미노산의 a- 알킬치환체와는 달리 자유롭게 회전할 수 없으므로 D- 프롤린과 함께 형성된 삼성분 착물의 경우 입체장애를 크게 느끼지 못하여 D- 이성질체가 컬럼에 더 오래 머무르는 것으로 설명되고 있다 (5). 리간드 교환 키랄고정상 § 혹은 1 에 의한 키랄성 인지 메커니즘에 근거하여 Qu iv o ron 등은 키랄고정상 § 혹은 1 의 광학분할능을 증진
시킬 목적으로 축방향의 배위자리에서 구리이온과 더 강한 결합을 할 뿐만 아니라 입체장애 효과를 더 크게 나타낼 수 있는 피리딘치환체를 가전 리간드 교환 키탈고정상 읽춘 제조하였다 0 5 ). 2- 메틸 -5- 비닐피 리딘 Lg t 출발물질로 사용하여 세 단계를 거쳐 5- 비닐 2-(N-L- 프롤 리노나토에틸)에틸피리딘 旦울 얻은 후 AIBN 을 개시제로 사용하여 라디칼 중합을 실시하고 가수분해함으로써 총 다섯 단계 를 거쳐 합성
二<.:9 一 》C IHzc0 。 H CH3 言I • COOC , H, cH10 11 된구5- 리8 과키이zN_ 랄온 갇고이과정 고강상하정 判게상t 의 결리 합피간할리드 딘 뿐교기 환아가 니 키착라랄물 고의피정 리축상딘방으고향로리 에배사 위용의자할한리 경큰를우 차입에지체 하장그여애림 효과 때문에 키랄고정상 왔즌 키랄고정상 § 혹은 1 보다 더 좋은 광학분 할능을 보일 것으로 예측할 수 있다. 예측한 바와 마찬가지로 리간드 교환 키랄고정상 얽근 여러 종류 라 세미 아미노산의 광학분할에 성공적이었으며 크로마토그램의 예는 그 립 5-9 와 같다. 그림 5-9 에서 프롤린을 제의하고는 L- 아미노산이 항 상 컬럼에 오래 머무르는 사실을 확인할 수 있다(1 6). 또한 키랄고정 상 §에서의 광학분할과 키랄고정상 9 에서의 광학분할을 비교함으로써
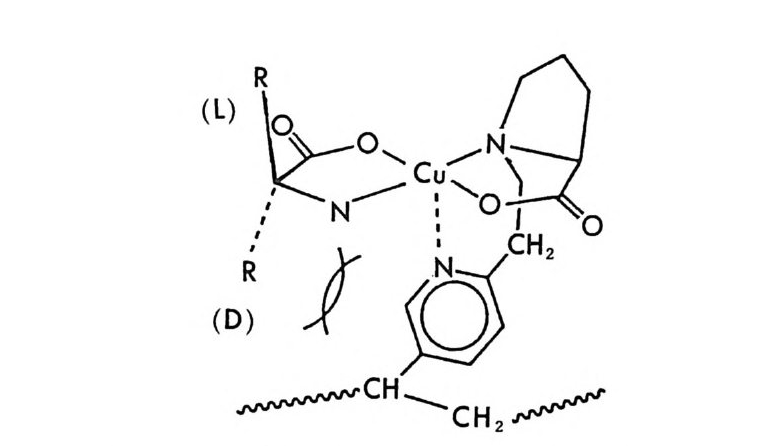 (D
(D
그림 5-8 키랄고정상 呼와 아미노산 사이의 삼성분 착물
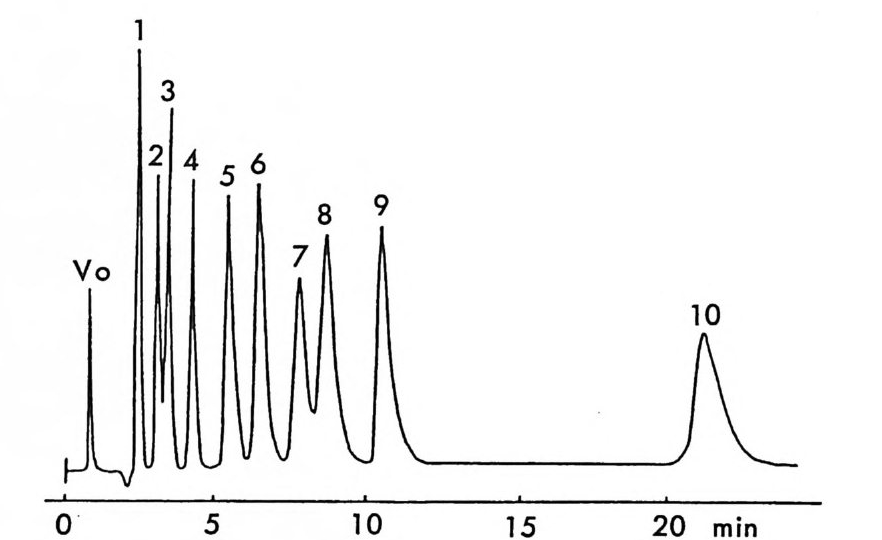 3
3
그림 5-9 럼[키N 랄:H 1고.C5정xI] 상0 =.4 6 09 m.0 m0(2,실 N리,이 유카 동 속겔 상 에: 2 : mH흡L2착0/된m/C inH안, 3온에C도N서 (:3아 20o미/7C0노 ,) 산 ;의 아〔 N 미H광 노 학』 분산=할 0.: .4l .N 컬D ; -ph eala ; 2. L-ph eala ; 3. L- 피페콜산 ; 4. D- 피페콜산 ; 5. D-tr p ; 6. D -tyr ; 7. L-tr p ; 8. L-ty r ; 9. L -pr o ; 10. D-pr o (참고문헌 : 16)
키랄고정상 釣에서의 광학분할이 키랄고정상 §에서의 광학분할보다 훨 씬 우수함을 확인할 수 있었다 (17).
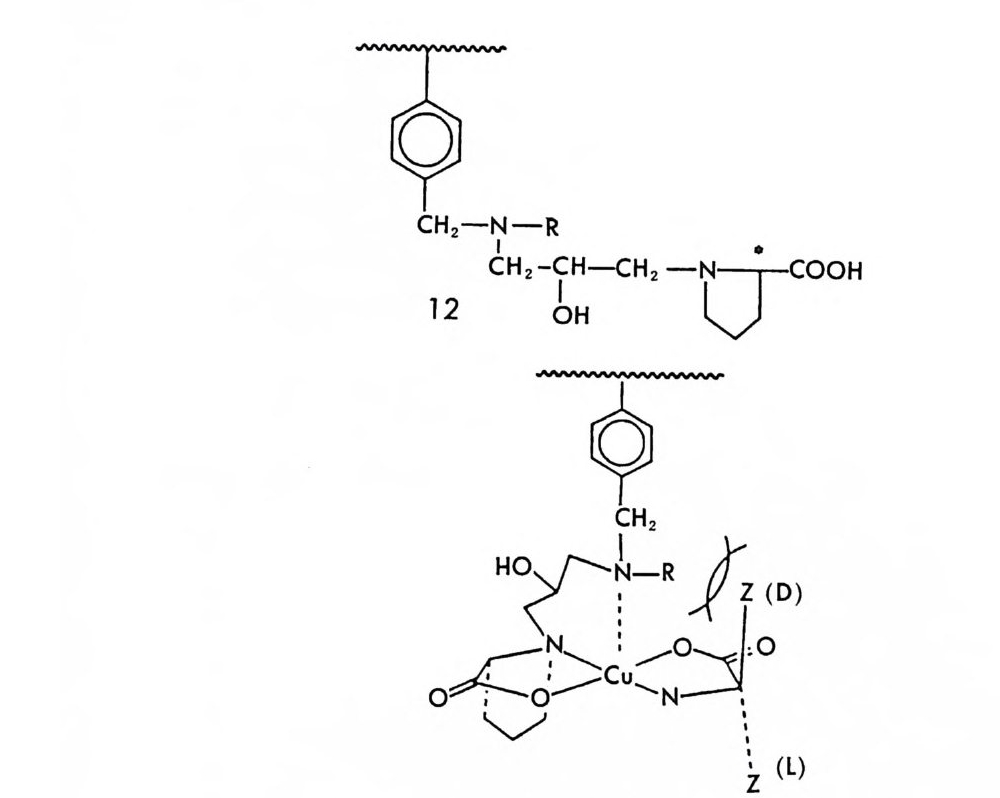 : C1H22 _ C선H _2R占-C H H- CH 2 -N汀~CO OH
: C1H22 _ C선H _2R占-C H H- CH 2 -N汀~CO OH
그림 5-10 키랄고정상 L疾 } 아미노산의 삼성분 칙물 L- 아미노산이 더 안정한 착물을 형성한다.
최근에는 키랄고정상 묘를 합성하여 키랄고정상의 R7] 가 아미노산 의 광학분할에 영향을 미치고 있음이 보고된 바 있다(1 8). 그립 5-10 과 같은 삼성분 착물의 구조를 고려할 때 L- 아미노산이 키랄고정상에 오래 머무를 것은 예상할 수 있으며 이 예상은 실제의 용리순서와 일 치함이 확인되었다. 2.2 키랄 리간드화합물을 실리카 겔에 고정시킨 키랄고정상 폴리스티렌, 폴리아크릴아미드 혹은 폴리메타크릴레이트가 고형지 지체인 리간드 교환 키랄고정상을 이용한 라세미 아미노산의 광학분할
이 성공적이었으나 고분자 를 고형지지체로 하는 이 들 키랄고정상둘은 높은 압력 하에서 안정하지 않기 때문에 HPLC 에 직접 응용하기 위 하겨 §, 1 혹은 9 와 같은 고분자 를 실리카 겔에 흡착시켜 키랄고정상 으로 사용하기도 하였다(1 6,17). 그러나 광학활성 아미노산이나 혹은 리간드분자로 사용할 수 있는 광학활성 화합물을 실리카 갤에 직접 공 유결합시킬 수 있다면 이들은 리간드 교환 키랄고정상으로서 HPLC 시스템에 응용되어 라세미 화합물의 광학분할에 유용하게 쓰일 수 있 울것이다• Cubit z 등은 5µm 의 실리카 겔과 3 - 글리시독시프로필메톡시실란을 반응시켜 에폭시 실리카 겔울 얻은 후 이것을 아미노산의 나트륨 염과 반응시켜 아미노산이 실리카 겔에 고정된 키랄고정상 묘을 합성할 수 있었다(그립 5- 11 ) (19 ).
 -SI i-O H + (CH10) 1Si — (CH2 ) 1 -0-CH2- C H/_0_:\C H2
-SI i-O H + (CH10) 1Si — (CH2 ) 1 -0-CH2- C H/_0_:\C H2
그림 5-1 | 키랄고정상 브의 합성
이들 키랄고정상들에 의한 광학분할에 있어서, 특히 고정된 리간드 가 L- 프롤린과 같은 고리형 아미노산인 경우, 그립 5 - 12 에서 보는 바 와 같이 아미노산의 D- 이성질체와 착물의 축방향 배위자리를 차지하 는 고형지지체의 히드록시기 사이의 입체장애로 인하여 착물 5-12~ 큰 착물 5-12~다 상대적으로 불안정하다 (20). 따라서 리간드 교환 키
 -s\I-o -slI-( CaH , ) (0> 一 sI! -o:- s!I- (CHb, ) ;'0>
-s\I-o -slI-( CaH , ) (0> 一 sI! -o:- s!I- (CHb, ) ;'0>
그림 5-12 키랄고정상 13(L- 프롤린리간드)과 아미노산(p heala) 사이의 삼성분 착물. 착물 a;,~ b 보다 더 안정하다.
랄고정상 묘에서는 프롤린의 광학분할만 제의하고는 거의 항상 L- 아 미노산이 키랄컬럼에 더 오래 머무르는 것으로 확인되었다(1 9,20). Guib i t z 등은 L- 프롤린이의에도 L- 히드록시프롤린, L- 아제티딘카 르복실 산 (azet idi ne carhoxy lic acid ) _li, L- 피 페 콜산 (pipe coli c ac id) L譯 실리카 겔H에 고d정시켜 이둘 리간드 교환己 키랄고0 정0H상 에 의한 광학 COOH
분할을 조사하였는데 특히 L- 피페콜산이 실리카 겔에 고정된 키랄고 정상 묘이 라세미 아미노산에 대하여 좋은 입체 선택성을 보임이 확인 되었다 (19, 20). 이들 리간드 교환 키랄고정상 표에 의한 광학분할은 라세미 아미노산의 광학분할에만 국한되지 않고 여러 종류의 a- 히드 록시카르복실산의 광학분할에도 응용되었을 뿐만 아니라(그립 5-13a)
(20, 21) 갑상선호르몬의 광학분할 (그림 5-13b) (20, 22) 및 아미노산의 단실유도체 (dansyl deriv a ti ve ) , 디펩티드인 D, L-Leu-D, L-Leu 의 네 개 입체 이성질체의 광학분할(그림 5-13c) 등에 응용되었다 (20). Yuki 등은 에폭시실리카 겔과 (lR, 2S) 쵸r-g. (lS, 2S)-2- 카르복시 메틸아미노 -1,2- 디페닐에탄올의 나트륨 염을 반응시켜 리간드 교환 키탈고정상 拉울 제조하였다(그립 5-14). (23,24) 이렇게 제조된 리간
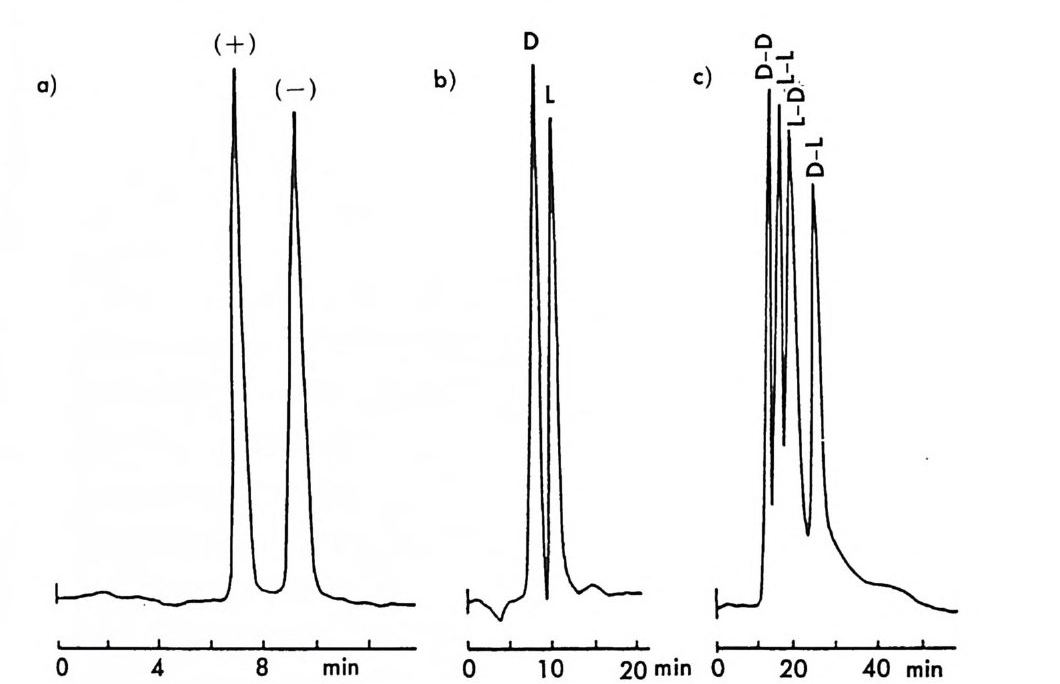 a) (+) (一) b) D L c) qo _,d-, -,,1 ,
a) (+) (一) b) D L c) qo _,d-, -,,1 ,
그림 5-13 a) 키랄고정상 旦 (리간드 : A- 히드록시프롤린)에서 D, L- 만델산의 광 학분할 컬 럼 : 25x0.4 6 cm, 이 동상 : 10-•M Cu( Il )SO., 유속 : 2mL/mi n, 온도 : 20'C , 검출기 : 223nm UV (참고문헌 : 21) b) 키 랄 고정상 13 (리간드 : L- 프롤린)에서 D, L- 티록신(tyr ox i ne) 의 광학분 할(65 : 컬35급) + O: 5. lxm0M .4 6 Ccmu ,( I이l )동 , 상p :H O .=l M 8 .암5, 모유늄 속아 세: l테m이L트/m-i 아n, 세검토 출니 트기 릴: 254nm UV( 참고문헌 : 22) C) 키랄고정상 13( 리간드 : L- 히드록시프롤 린)에서 DL-Leu-DL-Le 떠 분리. 이동~: 10-sM Cu(Il )SO., 유 속 : lmL/mi n, 검출기 : 223nm UV. (참고문헌 : 20)
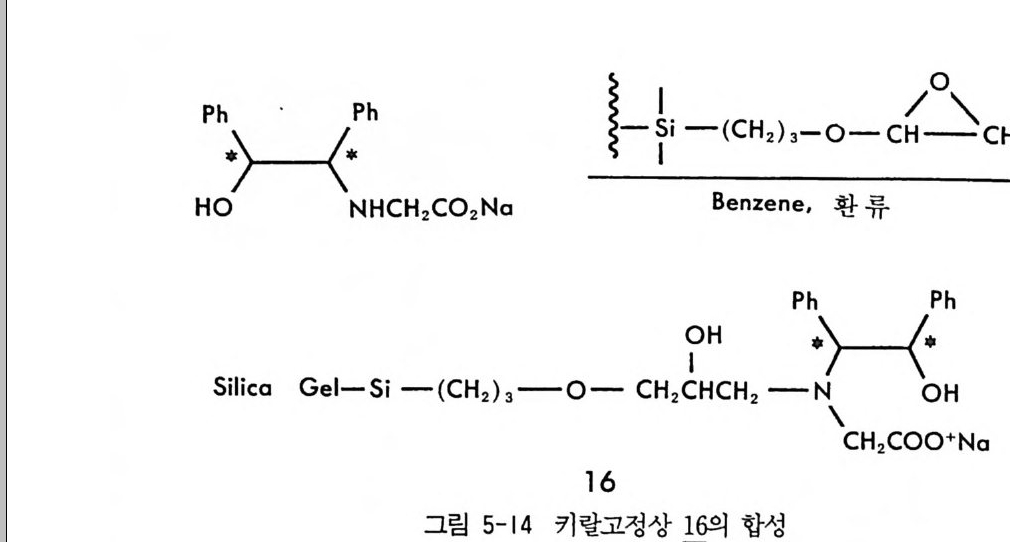 Ph* 曰• *P h l-fi -( CH2)3-O— C~ ~CH,
Ph* 曰• *P h l-fi -( CH2)3-O— C~ ~CH,
드 교환 키랄고정상 L얹 본 여러 종류의 라세미 아미노산뿐만 아니라 a -히드록시카르복실산, 라세미 아미노산 유도체둘의 광학분할에도 성 공적으로 응용되었다 (24). (lR, 2S)- 이성질체로부터 유도된 키랄고정상 L倍 경우 대부분의 라세미 아미노산을 광학분할하는 반면 DL- 세린 (ser) , DL-트 레오닌 (thr) , DL- 아스팔트산, DL- 아스파라전 (aspa r-ag ine ) 및 DL- 히스티딘을 광학분할할 수 없으나 (1S, 2S) -이성질체로 부터 유도된 키랄고정상 L統 ~ DL- 글루타민(g lu t am i ne), DL- t유느이 신, DL- 티로신(tyr os i ne) 을 광학분할할 수 있을 뿐만 아니라 (lR, 2S) -이성질체로부터 유도된 키랄고정상에서 광학분할되지 않는 DL -세란 DL- 트레오닌, DL- 아스팔트산, DL- 아스파라진을 광학분할할 수 있음이 확인되었다. 따라서 서로 부분 입체 이성질체 관계에 있는 (1S, 2S)- 및 (IR, 2S) -이성질체롤 기저로 한 두 종류의 키랄고정상 표 은 라세미 아미노산의 광학분할에 있어서 서로 보완적인 관계에 있다 고할수있다. Davc}. n kov 등은 (Et 0 ) 3S i -X-L- 프롤린 혹은 (Et 0 ) 3S i -X-L- 히드 록시프롤린을 실리카 겔과 반응시키거나 혹은 실리카 겔과 3 풍문로로 프로필트리에독시실란을 반응시켜 얻어진 변형된 실리카 겔울 요오드
켜 -키0랄-0 \고s/정0ER상=。 -tx_끄 H ! 一 울' HN 합 성 二_g할 수 R 있17었 다 (adb2c)5))) X,XXX2===6=,--- C-2((CC7CHH)HH.22 22-~ )많)3s0--은 라-세CH미2 -아 미노 나트륨 (Nal) 존재하에서 L- 프롤린 혹은 L- 히드록시프롤린과 반응시 산들이 키랄고정상 끄상에서 구리 (n) 이온과의 삼성분 착물 형성과정 을 통하여 광학분할된다는 사실이 확인되 었다. 특히 Pheala, Trp, Leu, Ileu, T y r 과 같이 큰 소수성기를 가진 아미노산들의 머무름 시간 은 키랄고정상의 X 기의 소수성이 커질수록 증가한다는 사실이 확인되 었으며 따라서 리간드 교환 키랄고정상과 아미노산 사이의 소수성 상 호작용이 광학분할에 중요한 역할을 하고 있다는 사실이 확인되었다. 특히 Ala, Val, Ileu, Leu, Pheala 과 같이 a- 위치에 단순한 알킬기를 가전 a- 아미노산들을 리간드 교환 키랄고정상 끄 P 에서 광학분할할 때 D- 아미노산이 컬럼에 오래 머무를 뿐만 아니라 일반적으로 아미노 산의 a- 위치의 알킬기 크기가 증가할수록 입체 선택성 (a 값)도 증가 한다는 사실이 확안되었다 (26). 그림 5-1~ 같은 삼성분 착물의 형성 울 가정할 때 아미노산의 a- 알킬기와 고정상 연결사슬의 소수성 상호 작용 때문 고정상과 D- 아미노산 사이에 형성된 삼성분 착물이 더 안 정하리라고 예측되며 a- 알킬기의 길이가 증가할수록 소수성 상호작용 이 더 크게 작용하여 더 좋은 입체 선택성을 보이는 것으로 생각된다• Karge r 등은 긴 알킬사슬을 통하여 아미노산울 실리카 겔에 결합 시킨 리간드 교환 키랄고정상을 제조할 때, 아미노산-알킬사슬 사이 에 단순한 알킬기를 가진 알킬사슬을 도입함으로써 실리카 겔에 결합 되는 아미노산의 농도를 조정할 수 있었을 뿐만 아니라 이들 키랄고정
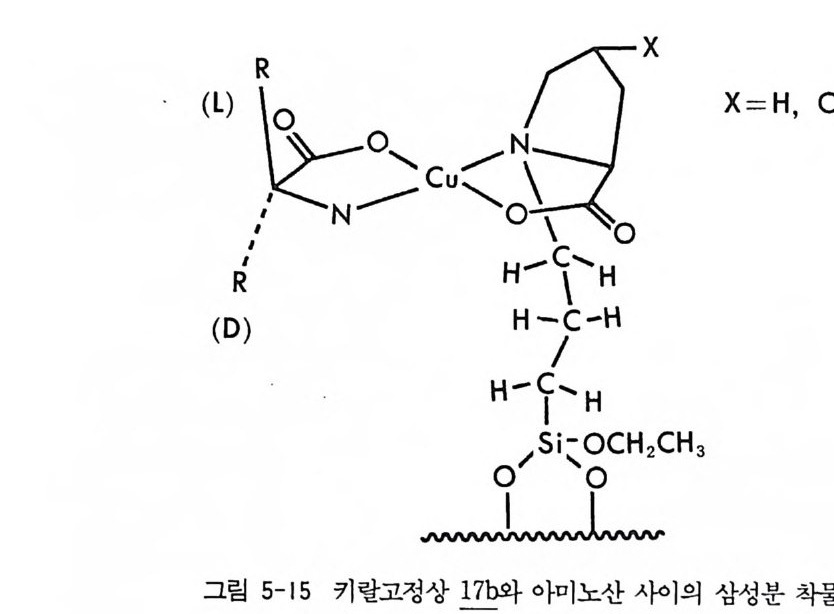 X=H, OH
X=H, OH
상에 의한 입체 선택성을 크게 증진시킬 수 있었다 (28,29). Karge r 등은 운데센산 (undeceno i c a ci d) 과 발린 t-1f-t일에스데르를 반응시켜 아미드를 얻은 후 디메틸클로로실란과 함께 히드로실릴화 반응을 하여 아미노산에 모노둘툐뚝꾹 l 릴기가 도입된 화합물 陸울 합성할 수 있었 다. 화합물 표울 실리카 겔에 공유결합시킨 후 반응하지 않은 실라놀 기를 제거하고 (end cap ping ) t숙녀i에스데르기를 가수분해하여 제거 하면 실리카 겔 단위표면적당 아미노산의 농도가 큰 리간드 교환 키랄 고정상 1Q( C-CSP 브 :C= concen t ra t ed) 가 제조된다(그립 5-1 6). 한편 화합물 L院 } 알킬기 (R) 의 탄소수가 C4 , C10 혹은 C20 인 알킬 디메틸모노클로로실란 [R(CH3)2S i Cl 〕울 적절한 비율로 혼합하여 실리 카 겔에 결합시킨 후 아미노산의 t유녁i에스테르기를 제거하면 알킬사 슬-아미노산 사이에 다른 알킬기가 끼어들어감으로써 실리카 겔 단위 표면적당 아미노산의 농도가 낮은 리간드 교환 키랄고정상 1Q( D-CSP 19:D= dilu t ed) 가 합성된다(그림 5-1 7). 이와 같이 제조된 키랄고정상 D-CSP 브는 실리카 겔 단위표면적당
H2C =CH-(CH2)8-COOH + H2N - CCIH H (C-HC 3O) 2O -t- B u 132) )) THSFHiMl Ai2cD P, a tS 실 Cg l es온 l lc― 『 —o e—I3Is 「CHZ3SJiH _ C 「〉1 OIII 5 |CCHH3)O0c0IIcIccc_CH- NH J_c卜 H H-Nuo) HHOY° _ H2 c H_ ( H.IH8 _t_B(C_uHL3: H C0H(c CISi H (CH3) 2 0 B C02 C-CS_P 19 그림 5-.1 6 HCM-CDSSP =1 9h 의e x합am성eHC과th 정y l(d TisE i lAa z =a n삼e)풀 르오르 아세트산, 활성리간드의 농도가 0 . 2µmo l/ m 낀J 반면 C-CSP~ 인 경우에는 실 리카 겔 단위표면적당 활성리간드의 농도가 2.5µmo l/ m 히다. 따라서 C-CSP 브로 충전된 150x 4.6mm 스데인레스스틸 컬럼내에서 착물형 성에 이용되는 Cu (Il)의 량은 l40µmo 부서 D-CSP 브로 충전된 컬 럼내에서 착물형성에 이용되는 Cu (Il)량, 3.6µmol 의 거의 40 배에 해 당한다. 그러나 표 5-2 에서 보는 바와 같이 아미노산의 단실유도체를 광학분할할 때 C10-D-CSP 登가 C-CSP 브에 비하여 더 오랜 머무롬 시간을 보일 뿐만 아니라 더 좋은 입체 선택성을 보인다는 사실은 아 주 홍미있는 일이다 (28). C10 인 알킬기에 의하여 뭄혀진 C10-D-CSP 旦에서의 아미노산 단실유도체의 광학분할 예 (그림 5-18) 에서 보는 바
TH3 2) HMDS 18 + -OCl S-Si t( iHC- H3( C 山 H (2C)H 92)- 10CCH O3 NH —CH- C OOH CH (CH 山 1) Sil ic a ge l, pyri d i d e , 실 온 I 3)TFA, 실온 -OSi (CH3) 2 (CH2) 9CH3 -0Si (CHa)2 (CH2)9(H3 -OSi ( CH3) 2 (CH2) 9CHa CI H (CH3)2 -OSi (CH3) 2 (CH2) 10CONH-CH-COOH ·-O Si ( CH3) 2( CH2) 9(H3 C10-D-CSP 19 그림 5-17 C,0-D-CSP 19 의 제조과정 (HMDS, TEA 는 그림 5-16 참조) 궁
』 n-E iq』_sd us.vn'0a 1-a '>usv-0 na1'0 0. 뇨1 dsv-1 -1 O 5 10 15 20 min 그림 5-18 C10-D-CSP ~앙논]서 아미노산단실유도체의 광학분할. 이동상 : 10-•MCu(Il ), 30% 아세토니트릴, 0.035M 아세트산, pH 5.5 (+ NH3) , 온도 : 55' C (참고문헌 : 28)표 5-2 키랄고정상 브에서 아미노산 단실유도체의 광학분할. 이동상 : 10-• M Cu(Il ), 30%, 아세토니트릴, 0 . 035M 아세트산, pH 5 . 5(+NH3) ,은도 : 55C (참고문헌 : 28) DAS-AA k' C- C SP 19 a’ kC'1 0-D-CSP 1a9’ D 4.90 Ala 1.57 분리안됨 2.3 4 L 11. 45 D 4.32 Val 1.80 분리안됨 2.7 5 L 11.88 D 7.2 5 Leu 3.3 1 분리안됨 3.4 4 L 24.95 D 1.46 3.69 Ser 1.15 2.1 7 L 1.68 8.0 0 D 1.29 3.0 1 Thr 1.16 3.3 8 L 1.49 10. 20 D 1.30 4.69 Asn 1.44 3.3 3 L 1.87 15. 64 D 1.61 1.65 Asp 1.60 2.70 L 2.5 8 4.46 D 0.7 9 1.05 Glu 1.19 1.47 L 0.9 4 1.54
와 갇이 크로마토그램의 피이크 모양 또한 아주 좋음을 확인할 수 있 다 (28). 이와 같은 결과는 그림 5-19 에서 보는 바와 같이 C-CSP 브의 경우
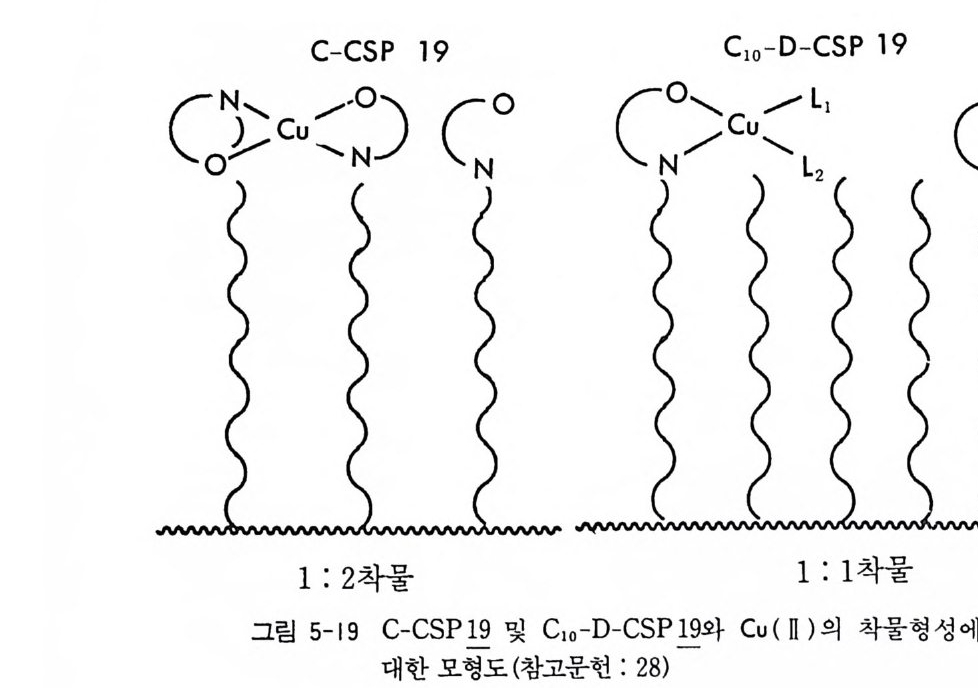 C-CSP 19 C10-D-CSP 19
C-CSP 19 C10-D-CSP 19
Cu( Il )과 실리카 갤에 결합된 아미노산 리간드 사이에 1 : 2 칙물형성 이 가능한 반면 C10-D-CSP 브의 경우에는 1 : 1 착물형성만이 가능 하다는 사실로써 설명이 되어 있다 (28). C - CSP 핀의 경우 1 : 2 착 물을 형성하기 때문에 아미노산의 단실유도체와 구리이온 및 고정된 리간드 사이의 착물형성이 어려운 반면 1 : 1 착물을 형성하는 C10-D _CSP 브의 경우에는 아미노산의 단실유도체와 구리이온 및 고정된 리간드 사이의 착물형성이 용이하므로 C10-D-CSP 브는 아미노산의 단실유도체에 대하여 더 오랜 머무름 시간을 보일 뿐만 아니라 더 좋 은 입체 선택성을 보임을 쉽게 짐작할 수 있다. 발린 대신 L - 프롤린이 결합된 C20-D-CSP ~를 이용하여 Karge r 등은 여러 종류 아미노알코올의 살리실알데히드 쉬프염기유도체 챕을 성공적으로 광학분할할 수 있었다 (29) . 특히 알라니놀 (alanin o l : 2 -am i no p ro p an-1-ol) 의 쉬프염기와 페닐에틸아민의 쉬프염기는 광학분
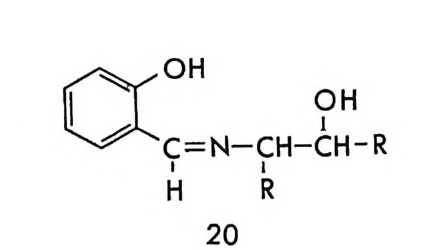 :::N_FH- 『 HH-R
:::N_FH- 『 HH-R
할되지 않는 반면 1- 아미노프로판 -2-¾(l - am i no p ro p an - 2 - ol) 은 광학 분할된다는 사실 등으로부터 고정된 키랄리간드급몽f이온 - 쉬프염기의 삼성분 칙물은 가능한 두 가지 구조 중 쉬프염기가 세 자리 리간드로 작용하는 그립 5 - 20 죤와 같은 구조를 가질 것으로 제안되었다 (29). 왜 냐하면 그림 5-20 Q와 같은 구조일 경우 페닐에틸아민 쉬프염기의 광 학분할이 가능하여야 할 뿐만 아니라 알라니놀의 경우 키랄중심과 이 민기의 질소사이 거리가 l - 아미노프로판 -2~ 탸 L 다 더 가까워서 더 잘 광학분할될 것으로 . 예상되나 실제로는 반대현상이 관찰되었으며 이와
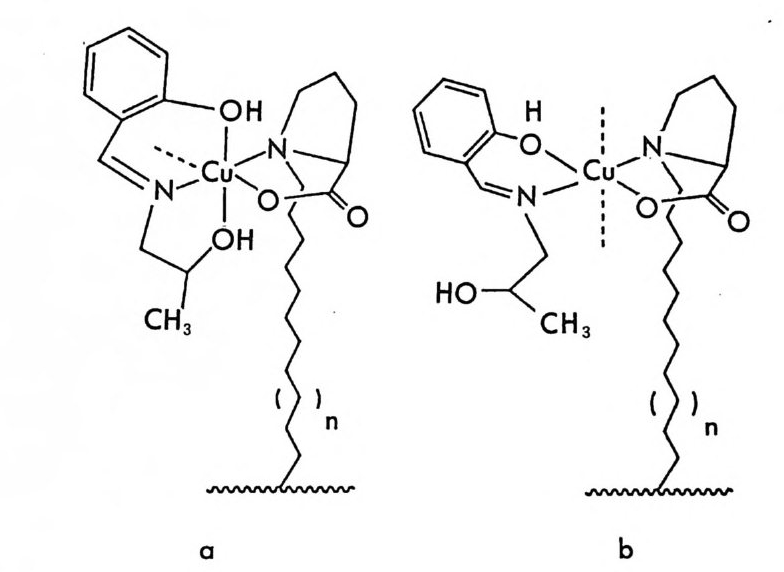 :HH0ONIIII\c/uY. 'III
:HH0ONIIII\c/uY. 'III
그림 5-20 C 2 0 - D-CSP19( 리간드 : L- 프롤린)와 아미노알코올의 살리실알데히드 쉬프염기 사이의 삼성분 착물 구조
같은 현상은 그림 5-20 브와 같이 쉬프염기가 세 자리 리간드로 작용하 는 착물의 구조로부터 설명이 가능하기 때문이다. 키랄 리간드물질을 실리카 겔에 직접 공유결합시킨 리간드 교환 키 랄고정상들이 높은 온도나 낮은 pH 혹은 높은 p H에 불안정한 것을 보완하기 위하여 Davankov 등은 고분자에 키랄 리간드물질이 결합 되어 있고 이 고분자가 실리카 겔에 결합되어 있는 키랄고정상들을 - 제 조하였다 (30). 죽 스티렌과 메틸비닐디메목시실란 공중합체의 페닐기 파라위치에 크로로메틸화 반응을 한 후 이 고분지를 실리카 겔 (Lic h rosorb S i -100) 과 반응시켜 고분자가 결합된 실리카 겔을 얻고 요오드나트륨 존재 하에서 이 실리카 겔과 L - 프롤 린메틸에스데르를 반응시키고 가수분해함으로써 키랄고정상 낌이 합성되었다. 또한 폴 리스티렌을 클로로 메틸화 반응한 후 소량의 3- 아미노프로필트리에폭 시실란과 반응시켜 트리에독시실릴기가 일부 치환된 고분자를- 얻고 이 것을 실리카 겔과 반응시킨 후 반응하지 않고 남아 있는 일부분의 클 로로메틸기를 L- 히드록시프롤린메틸에스데르로 치환한 후 가수분해함 으로써 키랄고정상 盤가 합성되었다. 이들 키랄고정상 낌과 끄는 여
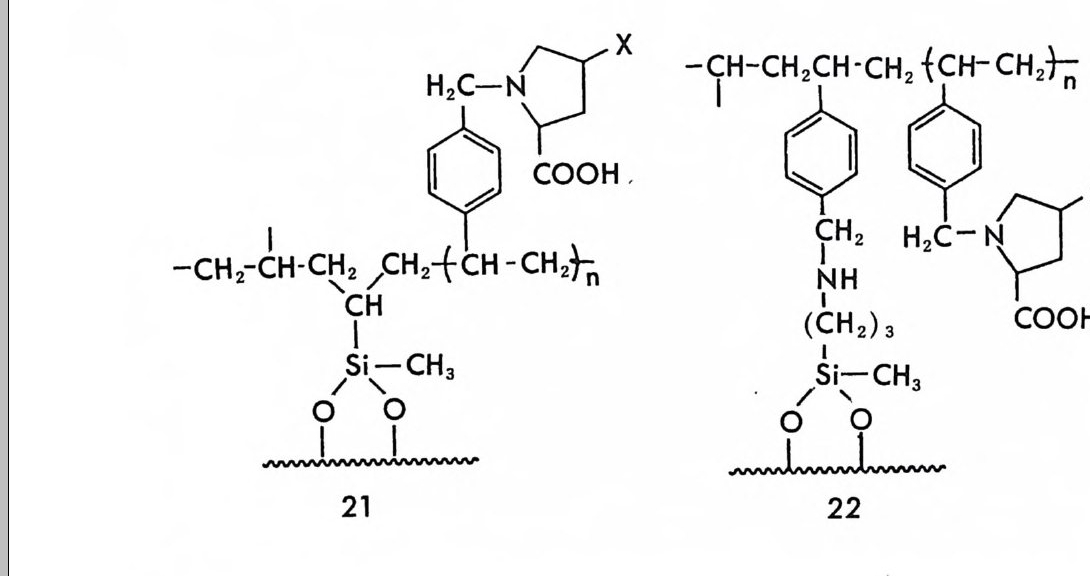 -CH,- 〉 H-C0I 선 / 2\ S1i6\ _ 0-C IC HH:232 :\--C 二H2 九 2 〈〉2E1仁〈2 (e/Hxo0
-CH,- 〉 H-C0I 선 / 2\ S1i6\ _ 0-C IC HH:232 :\--C 二H2 九 2 〈〉2E1仁〈2 (e/Hxo0
러 종류 아미노산의 광학분할에 성공적이었으며 실리카 겔에 폴리스티 렌이 결합되어 있으므로 소수성이 아주 커서 Trp, Pheala, Leu, Ty r 과 같은 소수성인 아미노산들의 머무름 시간이 특히 길다는 사실이 확 인되었다 (30). 2.3 키랄 리간드 화합물을 실리카 겔에 흡착시킨 키랄고정상 리간드 교환 키랄고정상들 중 특히 홍미있는 형태는 키랄 리간드물 질을 고분자나 실리카 겔과 같은 컬럼의 고형지지체에 공유결합시키지 않고 역상 크로마토그래피용 고정상인 RP-C8 고정상 혹은 RP-C1a 고정상과 긴 알킬기를 가지고 있는 키랄 리간드 사이에 소수성 상호작 용에 의하여 리간드가 고정상에 흡착된 키랄고정상들이다. 키랄 리간 드가 고분자 고형지지체에 공유결합된 키랄고정상이나 실리카 겔에 공 유결합된 키랄고정상과 비교하여 키랄리간드가 역상_고정상에 흡착됨 으로써 얻어진 키랄고정상은 상업적으로 구할 수 있는 역상 HPLC 컬 럼을 이용하여 쉽게 제조할 수 있다는 점에서 특히 매력적이다. 그러 나 이들 키랄고정상에서 사용가능한 이동상은 흡착된 키랄리간드가 이 동상에 끌려 컬럼에서 빠져나오지 않고 역상-고정상과 키랄 리간드 사 이의 소수성 상호작용이 충분히 커서 키랄리간드가 역상_고정상에 홉 착된 상태로 유지되도록 충분히 극성을 갖고 있어야 한다는 제한점이 있다. Davankov 등은 N- 헵틸 -L- 히드록시프롤린 (C1-L-Hy p) , N- 데실 -L- 히드로시프롤린 (C10-L-Hy p) , N_ 헥사데실 -L- 히드록시프롤린 (C16-L-Hy p) 혹은 N- 데실 -L- 히스티딘 (C16-L-H i s) 을 메탄올 혹은 메탄올과 물의 혼합용매에 녹여 RP-C! 죠 L 정상으로 충전되어 있는 HPLC 컬럼을 통하여 흘려보낸 후 아세트산구리(Il)의 메탄올/물 혼 합용매를 홀려줌으로써 N- 알킬 -L- 히드록시프롤린 혹은 N_ 알킬 -L 히
스티딘이 RP-C1 s.:i1.정상에 흡착된 키랄고정상을 제조할 수 있었다 (31). 이들 키랄고정상들은 여러 종류 라세미 아미노산의 광학분할에 아주 성공적이었으며 대표적인 크로마토그램은 그림 5 - 21 과 같다. 이들 키랄고정상에 의한 라세미 아미노산의 광학분할 특징은 그림 5-21 에서 보는 바와 같이 D- 아미노산이 L- 아미노산보다 항상 컬럼에
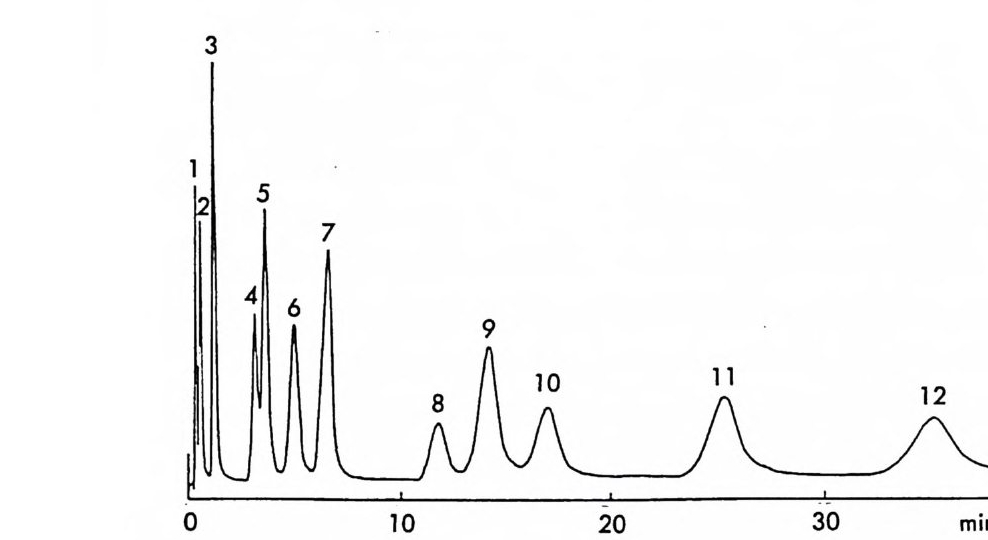 3
3
그림 5-21 Lich rosorb RP-C18 에 흡착된 C16-L-H yp키랄고정상에서 라세미 아 미노산들의 광학분할 컬럼 : 100x4.2mm, 이동상 : 메탄을/물(1 5/ 85) , 10-•M CuAc2, pH 5.0, 유속 : 2mL/mi n, 온도 : 2(J'C , 검출기 : 254nm UV ; l L-Ala, 2 D-Ala, 3 L-Nval, 4 L-Leu, 5 L-Nleu, 6 D -Nval, 7 L-Pheala, 8 D-Leu, 9 L-Trp , 10 D-Nleu, 11 D-Pheala, 12 D-Trp . (참고문헌 : 31)
더 오래 머무르며 아미노산의 a- 알킬기의 길이가 증가할수록 Ala
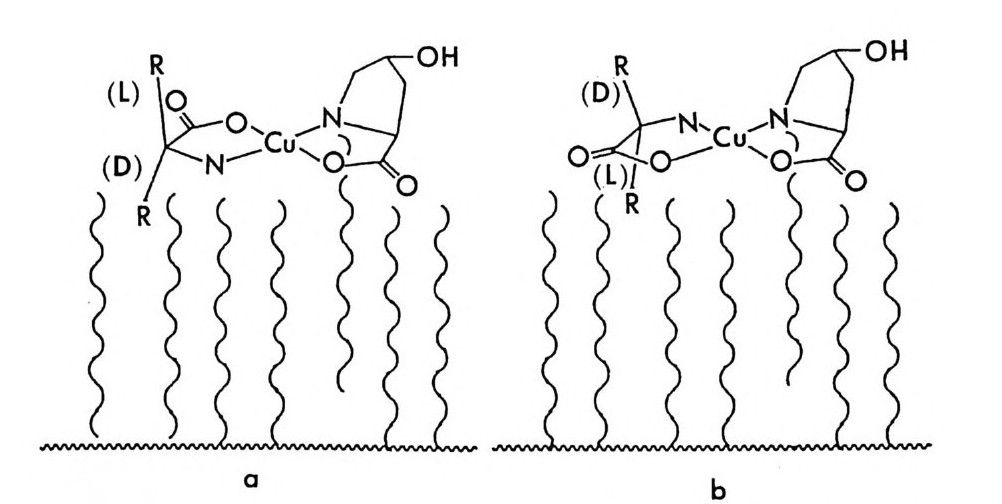 R /UH R /\ ...,H
R /UH R /\ ...,H
그림 5-22 RP-C1a 고정상에 흡착된 C10-L-H yp키랄고정상에 의한 삼성분 착물 형성의 모델. a7} b 보다 더 안정하다.
분할에 아주 중요하게 영향을 미친다는 사실을 시사해 주는 것으로서 이들 키랄고정상에 의한 라세미 아미노산의 광학분할 모델은 그림 5-22 과 같다. 키랄고정상은 N- 알킬 -L-HYI 펴 N- 알킬기가 RP-C1a 고정상의 옥 타데실사슬둘 사이로 소수성 상호작용에 의하여 끼어들어가 형성되 며, 이동상중의 D- 아미노산, Cu(II) 및 흡착된 키랄리간드에 의하여 형성된 삼성분 착물에 있어서 이동리간드안 D_ 아미노산의 a- 알킬기 는 고정상의 옥타데실사슬로 향하여 소수성 상호작용을· 하기 때문에 특히 안정화된다(그림 5-22. 인. 이동상중의 이동리간드인 L_ 아미노산 도 Cu(II) 및 흡착된 키랄리간드 사이에 삼성분 시스-착물을 형성하 여 (그립 5-22~) L- 아미노산의 a- 알킬기가 고정상의 옥타데실사술 사 이로 끼어들어감으로 소수성 상호작용을 예상할 수 있다. 그러나 리간 드 교환 키랄고정상을 이용한 라세미 아미노산의 광학분할에 대한 많 은 연구결과 시스-착물보다는 트란스-착물이 훨씬 더 안정한 것으로 알려져 있다 (31). 따라서 5-22 Q.!키 착물에 비교하여 5-22~ 형태의 착
물이 훨씬 안정하다고 생각할 수 있어 키랄리간드가 역상 - 고정상에 홉 착된 키랄고정상에서의 광학분할 특징을 자명하게 이해할 수 있다. N- 알킬 -L-H yp이 RP-C1s 고정상에 흡착된 리간드 교환 키 랄고정 상은 라세미 아미노산의 광학분할에만 응용되지 않고 최근에는 노르셰 페드린 (nore p hedr i ne) 과 노르에페드린유사체의 광학분할에도 성공적 으로 응용되었다 (32) . N, N- 디옥틸 -L- 알라닌을 RP-C1s 고정상에 흡착시킨 리간드 교환 키랄고정상은 소수성인 라세미 아미노산뿐만 아니라 친수성인 라세미 아미노산의 두 광학 이성질체에 대해서도 좋은 입체 선택성을 보인다 (33). 특히 N, N- 디옥릴 -L- 알라닌을 RP-C1s 고정상에 흡착시킨 키 랄고정상에서의 광학분할 중 흥미있는 것은 이소루이신 (i soleu ci ne) 의 네 가지 입체 이성질체가 아주 깨끗이 분리된다는 사실이다 (33). 이 의에 N, N- 디옥틸 - L 혹은 D- 알라닌이 RP-C1 S.::고정상에 흡착된 키 랄 고정상은 아미노산의 N- 아세틸유도체 및 N- 카르보벤족시 (N -carbobenzox y :BZ) 유도체의 광학분할에도 응용되었고 글리신 -DL 우이신 (g l y c y l-DL-leuc i ne) 과 같은 디펩티드의 광학분할 및 DL- 만 델산, DL- 젖산, DL- 타르타르산과 같은 a- 히드록시카르복실산의 광 학분할에도 성공적으로 응용되었다 (34). 3 리간드 교환 키랄고정상에서의 광학분할에 대한 이동상 의 조성, pH 및 온도의 영향 리간드 교환 키랄고정상에 의한 라세미 화합물의 광학분할은 이동상 중의 이동 리간드와 금속이온, 고정상의 키랄 리간드 사이에 삼성분 착물이 가역적으로 형성될 때 두 개의 가능한 부분- 입체 이성질 착물 의 안정성 차이에 의하여 결정된다. 두 개의 부분- 입체 이성질 착물의 안정성이 크면 머무름 시간이 길어지며 두 개의 부분 입체 이성질 착
물의 안정성에 큰 차이가 있으면 좋은 입체 선택성을 관찰할 수 있게 된다. 착물의 안정성은 이동상의 조성뿐만 아니라 이동상의 pH 및 온도에 의해서도크게 영향을받는다. 예를 들어서 이동상에 녹아있는 Cu(Il) 이온의 농도를 증가시키면 키랄고정상의 키랄리간드와 이동상의 이동 리간드 사이에 리간드교환 이 빨라져서 머무름 시간이 감소할 뿐만 아니라 온도를 높여주면 또한 리간드 교환이 빨라지기 때문에 머무름 시간이 감소할 것을 쉽게 예측 할 수 있다 (3 3 ). 그러나 입체 선택성에 대한 이동상중 Cu(Il )이온농 도의 영향 및 온도의 영향은 그리 큰 편이 아니다. 이동상의 p H 를 증가시키면(보통 p H4-6 . 5 범위에서)일반적으로 리간 드의 머무름 시간이 증가할 뿐만 아니라 입체 선택성도 증가한다. p H 를 증가시킴으로써 이동상 중의 리간드인 아미노산은 양성자를 잃 어서 더 안정한 삼성분 착물을 형성할 수 있기 때문이다. 리간드 교환
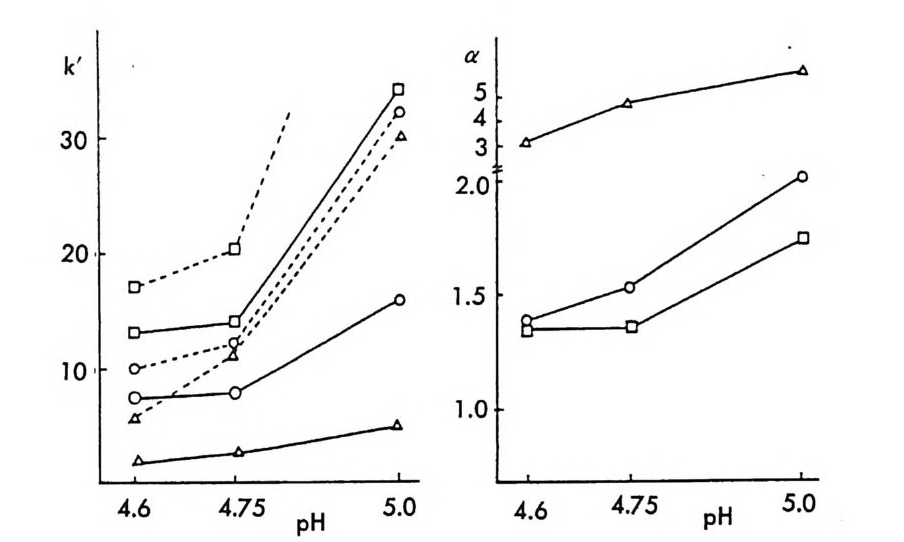 2kO,, ’ 3o· 고’’’ . .’ / ',,, ' ,, ' ,,’,, ’ ‘ , 1]2..50 l/
2kO,, ’ 3o· 고’’’ . .’ / ',,, ' ,, ' ,,’,, ’ ‘ , 1]2..50 l/
그림 5-23 키랄고정상 22 에서 Ty r, Leu, Pr~ 광학분할에 대한 p H 의 영향. 이동상 : O.OlM 암모늄아세테이트, 유속 : 3mL/mi n. kL' (一), k 。’ (…), Ty r(O ), Leu(O), Pro(6. ) (참고문헌 : 30)
키랄고정상 盤에서 p H 를 증가시키면서 Ty r, Leu, Pro 을 광학분할하 였을 때 머무름 시간의 증가와 입체 선택성의 증가를 그림 5 - 23 에서 확인할수있다 . 이 동상 이 물과 메탄올, 에탄올, 아세토니트릴과 같은 유기용매의 혼합물일 경우에는 물과 유기용매의 조성에 따라 머무릅- 시간 및 입체 선택성이 달라진다 . 특히 이동상중의 리간드 및 Cu(Il) 이온과 고정 리 간드 사 이 에 형성된 삼성분 착물의 안정성에 소수성 상호작용이 중 요하게 작용할 경우에 이동상중의 유기용매 조성비가 커질수 록 소수성
표 5-3 RP-C,s 고정상 에 서 N- 데실 -L - H yp 이 흡착된 키랄고정상에서 광학분 할 에 대한 이동상의 영향 . 이동상 : 아세토니트릴/물, 10- •M CuAc2, 온도 : 20'C (참고문 헌 : 31) 30/70(v/v ) 20/ 80 (v/ v) 15/ 85 (v/ v) 10/ 90 (v/ v) 아미노산 k' a’ k' a’ k' a’ k' a’ 1.03 1.44 1.63 2. 45 Val 1.24 1.57 1.72 2.0 9 1.27 2.26 2.80 5 .12 1.84 2. 57 3.5 9 Ty r 1.29 1.17 1.28 1.50 2.16 3.2 9 5. 39 1.90 2.7 5 3.81 Met 1.41 1.28 1.37 1.50 2.43 3.7 6 5.72 1.72 3.0 0 4.73 7.4 5 Nleu 1.37 1.77 1.97 2.1 7 2.3 5 5.3 0 9.3 3 16.1 2.10 4.3 7 7.46 13.3 Pheala 1.31 1.63 1.75 1.95 2.76 7.1 3 13.1 26.0 2.78 6.9 9 14. 4 27. 6 Trp 1.15 1.38 1.45 1.48 3.2 0 9.65 21 .0 40.7
상호작용을 약하게 하기 때문에 머무름 시간을 단축시킬 뿐만 아니라 경우에 따라서는 입체 선택성을 감소시키기도 한다. 표 5-3 은 N - 데실 - L - H yp이 RP-C1 s.:i:대상에 흡착된 키 랄고정상에 서 여러 조성의 아세토니트릴과 물의 혼합용매를 이동상으로 사용하여 여러 종류의 아미노산을 광학분할한 것을 요약한 것으로서 이동상에 있어서 유기용매인 아세토니트릴의 양이 증가함에 따라 머무름 시간이 감소할 뿐만 아니라 입체 선택성도 감소하는 예를 제시한 것이다 (31). 물과 유기용매의 혼합용매를 이동상으로 사용할 때에는 혼합용매의 혼합조성에 의해서 머무름 시간이나 입체 선택성이 달라지지만 사용-하 는 유기용매의 종류에 따라서도 머무름 시간이나 입체 선택성이 달라 전다. 일반적으로 국성이 낮은 유기용매를 물과 혼합하여 이동상으로 사용할 경우에 머무름 시간이나 입체 선택성에 더 큰 영향을 미치게 된다. 왜냐하면 이동상 중의 리간드와 고정상 사이의 소수성 상호작용 이 더 크게 영향을 받기 때문이다. 그러나 지금까지 살펴본 이동상의 조성, pH , 광학분할 온도 등이 라세미 화합물의 광학분할에 미치는 영향은 실로 다양하기 때문에 일반적으로 리간드 교환 키랄고정상에서 라세미 화합물의 광학분할에 대한 최적 HPLC 조건을 찾아내는 것은 그리 간단한 일이 아니며 키랄고정상에 따라 또는 광학분할하고자 하 는 라세미 분석성분의 구조에 따라 최적 HPLC 조건이 달라질 수 있 디는 사실을 주의할 필요가 있다.
참고문헌(제 5 장) 1. F. Helff er ic h , Natu r e (London) , 쁘, 1001 (19 61). 2. S. V . Rog o zhin and V. A. Davankov, German Pate n t, 1,932,190, Jan . 8, 1970, Ap pl. USSR, Jul y 1, 1968, Chem. Abstr ., 뜨, 90875c (1970). 3. V. A. Davankov, J. D . Navrati l and H. F. Walte n , Liga nd Exchang e Chromato g rap h y, CRC Press, Boca Rato n , Florid a , 1988.
4. V. A. Davankov, in Adva n ces in Chr01nato g ra p hy , vol. 18, J. C. Gid d in g s, Ed., Marcel Dekker, New York, 1980, chap 4. 5. V. A. Davankov, A. A. Kurga nov and A. S. Bochkov, Adv. in Advan ce s in Chromato g r aph y , vol. 22, J. C . Gi dd in g s , Ed., M arcel Dekker, New York, 1983, chp . 3. 6. V. A. Davankov and Yu. A. Zolota r ev, J. Chromato g r ., 쁘 , 295 (1978). 7. V. A. Davankov and Yu. A. Zolota r ev, J. Chromato g r., 쁘 , 285 (1978). 8. V. A. Davankov and Yu. A. Zolota r ev, J. Chromato g r ., .!-i턴, 303 (1978). 9. V. A. Davankov, Yu. A. Zolota r ev and A. A. kurga nov, J. Liq . Chromato g r., ~. 1191 (1979). 10. Yu. A. Zolota r ev and N. F. My as oedov, J. Chromato g r., 264, 377 11. (B1.9 8L3e).f e b vre, R . Audebert and C. Qu iv o rori , J . Liq. Chromato g r., !, 761 (1978). 12. I. A. Yamskov, B. B. Berezin , V. A. Davankov, Yu. A. Zolota r ev, I. N. Dosta v alov and N. F. My a soedov, J. Chromato g r., 217, 539 (1981). 13. D. Muller, J. Jo zefo n vic z and M. A. Peti t, J . Inorg. Nucl. Chem., 샌, 1083 (1980). 14. J. Boue, R. Audebert and C. Qu iv o ron, J. Chromato g r ., 쁘, 185 (1981). 15. D. Charmot, R. Audebert and C. Qu iv o ron, J. Polym . Sc i., : Poly. Lett . Ed., 낀, 59 (1986). 16. D. Charmot, R. Audebert and C. Qu iv o ron, J. Liq. Chromato g r., 한 1769 (1985). 17. D. Charmot, R. Audebert and C. Qu iv o ron, J. Liq. Chromato g r., 한 1753 (1985). 18. R. Jin and B. He, J. L iq. Chromato g r., 프, 501 (1989). 19. G. Gubit z, F. Juffma nn and W . Jel lenz, Chromato g rap h ia , 쁘, 103 (1982).
20. G. Gobit z, J. Liq . Chromato g r., 언 , 519 (1986). 21. G. Gubit z a nd S. Mi he llye s , C hromato g rap h ia , ~ . 257 (19 84). 22. G. Gobit z a nd F. Juf f m ann, J. Chromato g r., 쁘 , 391 (19 87). 23. Y. Yuki, K . Saig o , K. Tachib a na and M. Haseg a wa, Chem. Lett ., 1347 (19 86). 24. Y. Yuki, K . Saig o , H. Ki m oto , K. Tachib a na and M. Haseg a wa, J. Chromato g r., 4 00, 65 (19 87). 25. K. K. Ung e r, A . A. Kurga nov and V. A. Dav a nkov, Ang e w. Chem. Int. Ed. Eng l., 안 , 930 (19 82). 26. P. Roumelio t i s, K. K. Ung e r, A . A. Kurga nov, V. A. Davankov, J. Chromato g r., 쁘 ~ . 51 (19 83). 27. P. Poumelio t i s, A. A. Kurga nov and V. A. Davankov, J. Chromato g r., 쁘, 439 (19 83). 28. B. Feib u sh, M. J. Cohen and B. L. Karge r, J. C hroma t o gr . 연多 3 (1983). 29. L. R. Gelbe r, B. L. Karge r, J . L. Neumey e r and B. Feib u sh, J. Am. Chem. Soc., 쁘 , 7729 (1984). 30. A. A. Kurga nov, A. B. Tevlin and V. A. Davankov, J. Chromato g r., 쁘, 223 (19 83). 31. V. A. Davankov, A. S. Bochkov, A. A. Kurga nov, P. Roumeli ot i s and K. K. Ung er , Chromato g rap h ia , 프, 677 (1980). 32. S. Yamazaki, T . Takeuchi and T. Tan im ura, J. Liq. Chromato g r., 12, 2239 (1989). 33. H. Ki ni w a, Y. Baba, T. Ishid a and H. Kato h , J. C hromato g r., 쁘 l, 397 (1989). 34. H. Kato h , T. Ishid a , Y. Baba and H. Kiniw a, J. Chromato g r., 4 73, 241 (19 89).
제 6 장 크라운 에테르 키랄고정상 1 서론 196 저 Pederson 에 의하여 처음 소개된 크라운 에테르는 (l) l 혹 은 2 와 같은 커다란 고리형의 폴리에테르 화합물로서 특정한 크기의 공동을 가질 뿐만 아니라 폴리에데르 고리에 산소원자가 존재하기 때 문에 폴리에테르 고리의 산소원자가 리간드로 작용하여 많은 금속이온 들과 착물을 형성하며 (2,3) 여러 종류의 암모늄 양이온돌과 세 개의 N-H•·… ·0 형태의 결합에 의하여 착물을 형성한다는 사실이 알려져 있다 (4). 크라운 에데르와 금속이온 혹은 유기 암모늄이온이 크라운 에데르의 공동개 끼워들어가서 착물을 형성하기 때문에 크라운 에데르를 보통 주인분자 (host molecule) 라 하고 양이온을 손님분자 (gue st molecule) 라 한다. 형성되는 착물의 안정성은 주인분자인 크라운 에데르에 있어 서 리간드로 작용하는 산소원자의 수와 배열에 관계될 뿐만 아니라 크 라운 에테르가 가지고 있는 공동의 크기에 의해서도 결정되나 같은 종 류의 크라운 에테르에 대하여서는 손님분자인 금속 양이온의 크기, 금
 oc°0(~ o@o ✓ l。O Xn) A nio n ex°0(~ O8'O J l°0 Xn )A nio n
oc°0(~ o@o ✓ l。O Xn) A nio n ex°0(~ O8'O J l°0 Xn )A nio n
속 양이온의 전자밀도 등에 의해서 결정되며 암모늄 양이온의 경우에 는 크라운 에테르의 산소원자와 암묘늄 양이온 사이에 형성되는 N-H …… 0 형태의 수소결합 수 및 암모늄양이온 주위에 있는 다른 치환체 들의 크기와 형태에 의하여 영향을 받는다. 결국 크라운 에테르와 여 러 종류의 금속 양이온 혹은 유기 암모늄 양이온 사이에 형성되는 착 물들은 모두 다른 안정성을 가지며 따라서 크라운 에데르는 여러 종류 의 금속 양이온돌을 구별할 수 있을 뿐만 아니라 유기 암모늄 양이온 들도구별할수있다. 크러운 에테르를 적절한 방법으로 HPLC 컬럼의 충전제로 사용할 수 있는 고형지지체에 결합시키거나 혹은 이동상에 크라운 에데르를 첨가하여 크로마토그래피를 실시할 경우 크라운 에테르와 금속 양이온 들 혹은 유기 암모늄 양이온들 사이에 형성되는 착물의 안정성 차이 때문에 금속 양이온들과 유기 암모늄 양이온들~ 분리할 수 있음을 쉽 게 예상할 수 있다. 실제로 많은 종류의 금속 양이온둘이 크라운 에테 르가 결합된 크라운 에테르고정상이나 혹은 크라운 에데르를 첨가한 이동상을 사용하여 분리되었을 뿐만 아니라 인체의 뇨 혹은 혈장중에 존재하는 암피실린 (amp icillin) , 세파렉신 (ce p halex i n) 과 같은 /3-락 탐계의 항생제 등과 (5) 아미노산을 포함한 많은 아미노 화합물들을 분 리할수 있었다 (6). 크라운 에테르에 적절한 키랄기를 도입하거나 혹은 키랄장벽 (chir a l
barr i er) 을 도입하면 키랄성을 가진 광학활성 크라운 에데르를 합성할 수 있다 (7). 예를 들면 Cram 등은 크라운 에데르에 키랄성인 비나프 틸 (b i na p h t hy l) 기를 도입하여 집과 伊} 같은 키랄 크라운 에테르를 합 성할 수 있었으며 (8, 9, 10) 바페난트릴기 (b ip henan t hry I) 가 도입된 키 랄 크라운 에테르 ~(11) 와 탄수화물기가 도입된 키랄 크라운 에데르 §(12) 의 합성 등 여러 종류의 키랄 크라운 에테르의 합성에 대한 연구 가 보고되어 있다 (6,7). 이들 키랄 크라운 에테르는 아미노산과 같이 아미노기를 가지고 있는 라세미 화합물의 암모늄염과 서로 안정성이
 A B A
A B A
다른 두 개의 부분 입체 이성질 착물을 형성하며 이때 안정성 차이는 키랄 크라운 에데르의 구조와 손님분자인 라세미 아미노 화합물의 구 조에 따라 크게 달라진다는 사실이 많이 연구되었다 (6, 13-16). 따라서 키랄 크라운 에데르를 크로마토그래피의 컬럼에 사용할 수 있는 고형 지지체에 적절히 결합시켜 크라운 에테르 키랄고정상을 만들거나 혹은 키랄 크라운 에데르를 이동상에 첨가하여 (CMPA 방법) 크로마토그래 피를 실시하면 아미노기를 가진 라세미 화합물을 광학분할할 수 있을 것으로예상할수있다. 실제로 키랄 크라운 에테르 쓰를 클로로포름이나 CH 2 Cl 2 에 첨가한 용액을 이동상으로 사용하여 여러 종류의 라세미 아미노산을 광학분할 하는 CMPA 방법이 Cram 에 의하여 연구보고된 바 있다 (17,18). 키 랄 크라운 에데르가 고정상으로 사용될 수 있는 고형지지체에 결합된 크러운 에데르 키랄고정상에서의 광학분할에 대한 연구는 광학분할이 가능한 라세미 화합물이 아미노기를 포함하는 라세미 화합물에 제한되 는 등의 이유로 인하여 다양한 편은 아니다. 키랄 크라운 에데르를 실 리카 갤에 결합시킨 키랄고정상에서의 광학분할 및(1 8,19) 키랄 크라 운 에테르를 교차결합된 폴리스티렌 p-디비닐벤젠수지에 결합시킨 키 랄고정상에서의 광학분할 (20,21), 키랄 크러운 에데르를 RP-C1 s.JI.정 상에 흡착시킨 키랄고정상에서의 광학분할의 예가 (22) 보고되어 있다. 2 크라운 에데르 키랄고정상의 제조 및 광학분할의 특성 Cram 등은 키랄 크라운 에데르 ~(RR_ 쁘邊 브롬화반응하여 6- 위치에 U 粹의 브롬원자가 치환된 키랄 크라운 에테르를 얻은 후 부 틸리튬울 가하여 저어주고 디메틸디클로로실란과 반응시켜 6- 위치에 때의 디메틸클로로실릴기가 치환된 키랄 크러운 에테르를 얻을 수 있 었다. 이어서 디메틸클로로실릴기가 치환된 키랄 크라운 에테르를 실
리카 겔과 반응시킨 후 메탄올로 처리하여 반응하지 않은 디메틸클로 로실릴기를 모두 디메틸메톡시실릴기로 바꾸고, 최종적으로 변형된 실리카 겔을 트리메틸클로로실란으로 처리하여 실리카 겔의 반응하지 않은 실라놀기 (― S i OH) 를 제거함으로써 키랄 크라운 에데르笠가 실 리카 겔에 공유결합된 키랄고정상 1 을 얻을 수 있었다(1 8,19). 그림 6 - 1 에 있는 키랄고정상 ?의 구조는 키랄 크라운 에테르의 네 개 나프 틸기 중 단지 하나의 나프틸기를 통하여 실리카 겔에 공유결합된 모양 을 나타낸 구조이나 실제로는 하나 이상의 자리에서 실리카 겔에 결합 될 수 있다. 이와 같이 제조된 키랄고정상 1 은 4.65% 의 탄소(무게 %)를 포함하는 것으로 확인되었다. 키랄고정상 1 에서의 광학분할 예 는 그다지 다양한 편이 아니다. 페닐에틸아민, 페닐글리신의 메틸에 스테르 혹은 이소프로필에스테르, 트리프로판의 메틸에스테르, 페닐 알라닌 및 발린의 메틸에스테르가 HCI 염 혹은 HPF6 염으로 광학분할 된 것이 보고되어 있다 (18,19). 이들 라세미 화합물들 중 발린과 페닐 알라닌 혹은 p-히드록시페닐알라닌의 메틸에스테르를 제의하고는 모 두 D- 이성잘체가 컬럼에 오래 머무르며 페닐에틸아민의 경우에는 S -이성질체가 컬럼에 더 오래 머무르는 것으로 확인되었다. 크라운 에
CH3300[--SS&fii 3 .....,.. ~ CH3 (RR)-(D) 착물 : 아미노산 (RR)-(S) 착물 : 페닐에틸아민 7 그림 6 키 키랄고정상 7 의 구조 및 세 접 결합모델. L= Ar, S= H, M= 알킬 혹 은메독시카교보닐.
데르의 고리에 있는 산소원자와 손님분자의 아민영기 사이에 세 자리 에서의 수소결합에 의하여 착물이 형성될 때 손님분자의 키랄중심에 붙어 있는 세 개의 치환기 즉 입체적으로 커다란 L- 기, 중간크기의 M- 기, 제일 작은 S- 기가 그림 6 ― l 과 같이 배열될 때 안정한 착물을 형성하여 컬럼에 오래 머무르는 것으로 예측할 수 있다. 즉 입체장애 를 크게 받는 L- 기가 입체장애가 가장 작은 부위에 위치하며 입체장 애를 가장 작게 받는 S- 기가 입체장애가 가장 큰 나프틸기 를 향하고 있어서 그림 6-1 의 형태를 가진 착물은 S- 기와 M- 기가 바뀐 부분 입 체 이성질 착물보다 더 안정할 것으로 기대된다. 그러나 가장 큰 기인 L- 기가 페닐기나 인돌기보다 작은 벤질기 혹은 이소프로필기일 때는 그림 6-2 에서처럼 크라운 에데르고리의 산소원자와 손님분자인 아민 염 사이의 세 자리 수소결합 의에 전자밀도가 큰 크라운 에데르고리의 산소원자와 손님분자인 아미노산에스데르의 카르보닐탄소(이웃 산소원 자의 전자끌기 효과 때문에 전기양성상태임) 사이에 제伊] 상호작용이 작 용하여 그림 6-2 의 착물이 더 안정하기 때문에 용리순서가 바뀌는 것 으로 Cram 은 제안하였다 (18, 19). 키랄고정상 1 에서의 광학분할에 대 한 대표적인 크로마토그램은 그림 6- 狩} 같으며 클로로포름을 이동상 으로 사용하여 광학분할할 때 페닐알라닌 메틸에스데르 HCI 염의 두
 H3
H3
그림 6-2 키랄고정상 1 과 L 페닐알라닌 메틸에스데르 사이에 형성된 안정한 착 물의구조
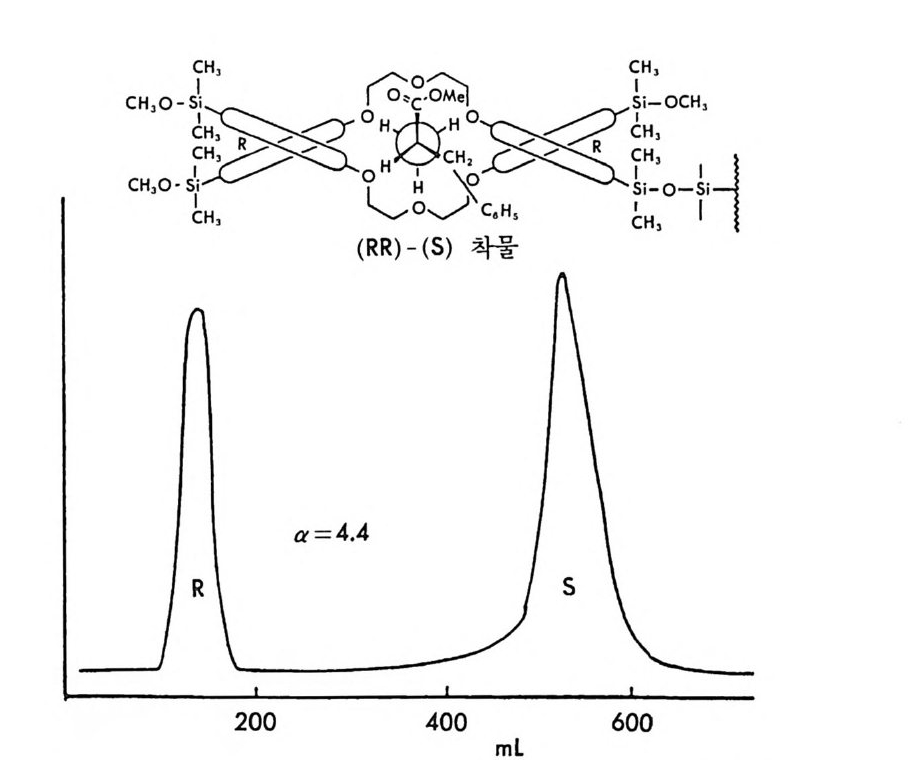 CHHO0CSH , 二_ _ _ 三CH,, 0\l
CHHO0CSH , 二_ _ _ 三CH,, 0\l
그림 6-3 키랄고정상 7 에서 페닐알라닌메틸에스테르 HCI 염의 광학분할. 이동상 : CHCIJ ( 참고문헌 : 18, 19)
광학 이성질체가 아주 잘 분리되고 있음을- 확인할 수 있다(분리인자, a = 4.4). Cram 등은 (R)-2,2' -디히드록시 -3,3' -디메틸 -1,1' 비나프틸울 브 롬화 반응하여 (R)-6,6' -디브로모 -3,3' -디메틸 -2,2- 디히드록시 -1, 1- 비나프틸을 얻은 후 (R)-2 , 2' -비스- (1, 4- 디옥사 -6- 토실옥시핵실) -1,1' -비나프틸과 함께 THF-KOH 용액내에서 가열함으로써 키랄 크라운 에데르 쁘 (RR- 쁘)를 얻을 수 있었다 (20). RR 첼런을 n~ 틸 리튬으로 처리하여 에틸렌옥시드를 가하면 ~ 쁘 (6%), ~(60 %)의 세 종류 키랄 크라운 에데르가 혼합물로 얻어지는데 이중 ~를 알루미나 컬럼을 통하여 분리해낸 후 이것을 p-위치가 클로로메틸화
된 교차결합 폴리스티렌디비닐벤젠수지에 결합시킴으로써 키랄 크라 운 에데르 RR_ 한가 고분자에 공유결합된 크리운 에데르 키랄고정상 g가 합성되었다 (20,21). 키랄고정상 읽큰 키랄고정상 7 과 마찬가지로 아미노산의 에스데르를 광학분할하는 데 응용되었을 뿐만 아니라 아미노산 자체의 광학분할에 도 성공적으로 응용되었다 (20,21). 그림 6 - 4 는 키랄고정상 9 상에서 페닐글리신의 과염소산염을 광학분할할 때 얻어진 대표적인 크로마토 그램을 나타낸 것이다. Cram 은 키랄고정상 9 에서의 광학분할에 대하 여 예비논문과 실험 전과정을 자세히 기술한 논문을 발표하였으나 (20, 21), 두 논문에 있어서 페닐알라닌 및 페닐알라닌유사체의 광학분할에 대한 용리순서가 일치하고 있지 않다. 실험 전과정을 자세히 논술한
 B c('ol
B c('ol
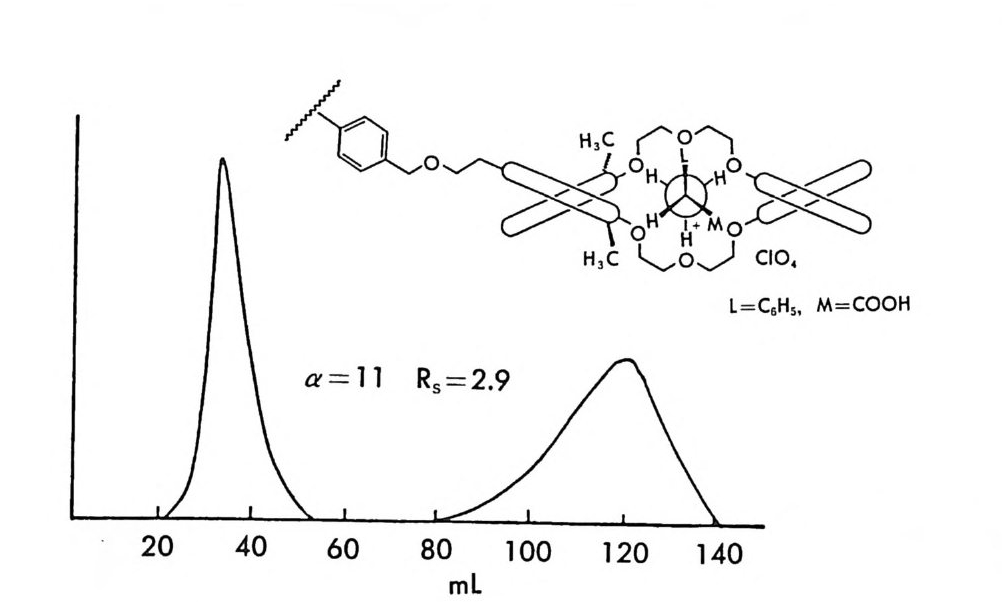 /:三玉。〈
/:三玉。〈
그림 6-4 키랄고정상 距i l 서 페닐글리신의 광학분할 및 안정한 착물의 구조. 이동상 : CH3CN-CHCI3 (10/90) (참고문헌 : 20)
논문에 의하면 (21) 광학분할을 시도한 모든 아미노산 및 아미노산에스 데르에 있어서 항상 D- 이성질체가 칼럼에 더 오래 머무르는 것으로 발표되었다 (21). 이와 같은 용리순서는 D- 아미노산의 키랄중심에 붙 어 있는 세 종류의 치환체 죽 입체장애가 큰 페닐, 벤질, 혹은 알킬기 와 같은 L- 기, 메틸에스데르기 혹은 카르복시기와 같은 중간정도 크 기의 M- 기, 입체장애가 가장 작은 H- 기가 그림 6- 伊} 같이 배열되 어 L- 기가 입체장애가 가장 작은 공간을 차지하고 H- 기는 입체장애 가 가장 큰 나프틸기를 향하고 있어서 안정한 착물을 형성한다고 가정 함으로써 설명이 가능하다(그립 6-1 의 착물의 구조와 같은 구조임). L -이성질체의 경우에는 그림 6-4 에서 H 기와 M 기가 바뀐 형태의 착물 울 형성하나 이와 같은 착물에 있어서 M 기는 입체장애가 큰 메틸나프 틸기를 향하게 되므로 D- 이성질체와의 착물보디는 불안정하게 될 것 으로생각되고있다. 최근에는 Shin b o 등이 키랄 크라운 에테르 꼬울 RP-C1s 고정상에
흡착시킨 역상 크로마토그래피용 크라운 에데르 키랄고정상을 노 제조하 였다 (22). 죽 메탄올-물의 혼합용매에 키랄 크러운 에테르 lQ울 용해 한 후 이 용액을 상업적으로 구할 수 있는 RP-C18 컬럼을 통하여 홀 려중으로써 RP-C1a 에 흡착된 크라운 에데르 키랄고정상이 쉽게 제조 되었다. 이때 키랄 크러운 에테르가 RP-C18 고정상에 효과적으로 홉
 (>
(>
착되도록 하기 위하여 Shin b o 등은 메탄을-물 혼합용매의 메탄올 양 울 80% 에서 45% 까지 단계적으로 감소시키면서 흡착과정을 실시하였 다. 이렇게 제조된 키랄고정상은 Crown Pak 이리는· 상품명으로 상품 화되어 있으며 (제 9 장 참조) 표 6-1 에서 보는 바와 같이 과염소산수용 액을 이동상으로 사용하여 많은 종류의 라세미 아미노산 및 페닐에틸 아민을 광학분할하는 데 성공적으로 사용되었다. 그립 6- 武근 크라운 에데르 旦이 RP-C1s 고정상에 흡착된 키랄고정상에서 세 종류의 라 세미 아미노산이 완전히 광학분할되는 것을 보여주는 대표적인 크로마 토그램으로서 세 종류의 아미노산이 완전히 기준선에서 광학분할될 뿐 만 아니라 피이크의 모양 또한 아주 좋음을 확인할 수 있다. 아미노산의 두 광학 이성질체 용리순서는 그립 6-5 와 갇이 키랄 크 라운 에테르의 절대배열이 S 일 때는 L- 이성질체가 컬럼에 더 오래 머 무르며 키랄 크라운 에테르의 철대배열이 R 일 때는 표 6-1 에서 보는 바와 같이 어느 경우에나 D- 이성질체가 컬럼에 더 오래 머무르는 것
표 6-1 RP-C1 sJ.!.정상에 흡착된 R- 잰 키랄고정상에서의 광학분할. 컬럼 : 125x4mm, 흡착된 R-10 의 량 : 58mg , 이 동상 : 10 -2 M 과염소산용액, 유속 : 0.5 m L/mi n, 검 출기 : 200run 혹은 254run UV (참고문헌 : 22) 18'C zc 라세미 화합물 kL' kD, a’ Rs kL, kD, a’ Rs 알라닌 (Ala) 0.2 4 0.4 8 2.02 1.44 0.37 1.02 2.72 3.70 발린 (Val) 0.8 9 1.01 1.14 0.6 6 1.06 1.31 1.24 1.38 루이신 (Leu) 3.1 1 5.1 4 1.65 5.7 3 3.82 8.37 2.19 8.7 1 이소루이신 (lsoleu) 2.44 2.88 1.18 1.62 2.8 2 3.70 1.31 2.4 8 (알al로so °lle 소u)루 0] 신 2.3 5 2.60 1.10 0.90 2.75 3.2 0 1.16 1.39 프롤린 (Pro) 0.30 0.3 0 1.00 0.4 1 0.41 1.00 페 닐알라닌 (Pheala) 9.25 11. 50 1.24 2.75 14.27 19.87 1.39 4.46 페닐글리신 (Phe g l y) 2.0 0 9.1 7 4.5 8 13. 66 2.83 25.71 9.07 17.06 시스틴 (C y s) 0.24 0.4 3 1.81 0.8 8 0.3 4 0.89 2.60 2.7 4 아스팔트산 (As p) 0.14 0.2 8 2.06 0.72 0.32 0.7 0 2.18 1.77 글루탐산 (Glu) 0.3 2 1.18 3.73 4.94 0.58 3.54 6.08 10. 73 리신 (L y s) 0.51 0.6 3 1.22 0.61 1.38 1.85 1.35 1.82 페닐에틸아민 10.36 6.5 0 1.59 2.02 25. 05 18.05 1.39 3.4 4
이 확안되었다. 이와 갇은 용리순서는 Cram 키랄고정상에서의 용리 순서와 일치하는 결과이다. 표 6-1 에 있는 페닐에틸아민에 대한 용리 순서는 아미노산의 용리순서와 일치하는 결과로서 다만 절대배열을 결 정하는 Cahn-In g old-Prelo g의 명명규칙때문에 페닐에틸아민의 경우 L- 이성질체가 더 오래 머무르는 것으로 표현된 것 뿐이다. 표 6-1 에서 보는 것처럼 낮은 온도에서 두 광학 이성질체의 머무름 시간이 더 클 뿐만 아니라 입체 선택성 (a 값) 또한 더 크다. 이와 같 은 사실은 낮은 온도에서 라세미 아미노화합물과 키랄 크라운 에데르 사이에 더 강한 착물을 형성하고 있음을 시사해 주는 것으로서 키랄 크라운 에데르-아미노산 착물의 안정성에 대한 온도의 영향을 조사한
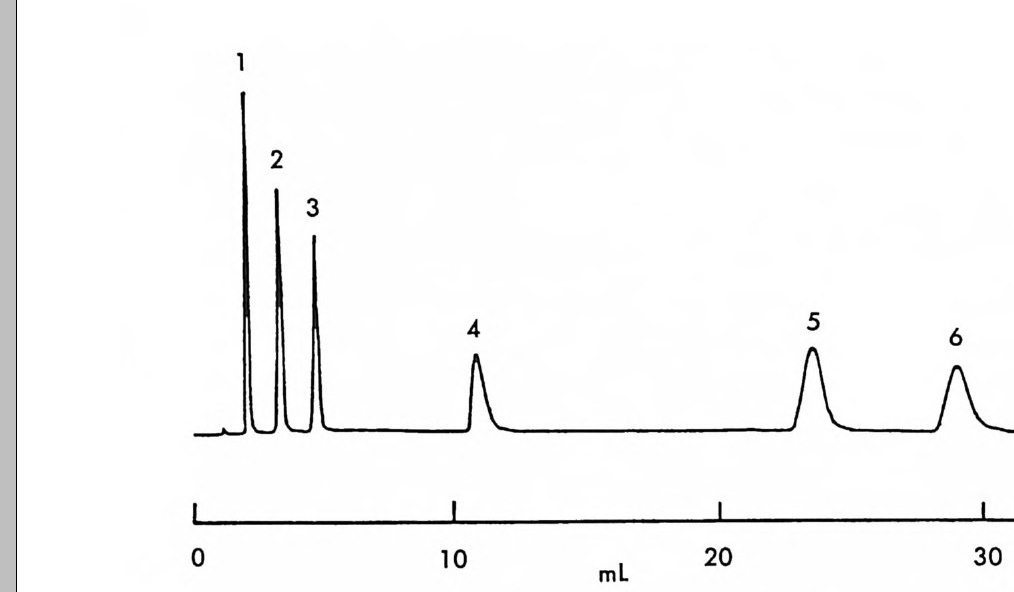 2
2
그림 6-5 RP-C18 고정상에 흡착된 S - 브 키랄고정상에서의 광학분할. 컬럼 : 125x 4mm, 흡착된 S-10 의 량 : 49mg , 이동상 : 10- 2M 과염소산용액 . 검출기 : 200nm UV, 온玉 : ?C . 1 = D- A rg, 2 = L- A m, 3 = D-Met. 4 = L-Met, 5 = D-Ty r, 6=L-Ty r. (참고문헌 : 22)
Cram 의 연구결과와 일치하는 내용이다 (15, 16). RP-C1s 고정성에 흡착된 키랄 크라운 에테르 꼬상에서 광학분할에 영향을 미치는 다른 주요한 요인은 흡착된 크라운 에테르의 양과 이동 상에 첨가되는 산의 농도 및 산의 종류이다. 일반적으로 흡착되는 키 랄 크라운 에데르의 양을 증가시킬 경우 표 6-2 와 같이 a 값이 증가함 울 확인할 수 있으며 표 6- 邸녁격 L 터는 이동상중의 산(과영소산)의 농 도를 증가시킴에 따라 분석성분의 머무름 시간이 증가할 뿐만 아니라 분리인자인 a 값도 다소 증가하며 이동상중의 산으로는 HCl
표 6-2 RP - C 교정상에 흡착되는 키랄 크라운 에테르 L碑 앙에 따른 분리인 자 (a) 의 번화. 이동상 : 10 -2 M 과염소산용액, 유속 : 0 . 5mL/ mi n, 검 출기 : 200run 혹은 254runUV, 온도: 1 8' C , 컬럼 : 125x4mm( 참고문헌 : 22) a, 흡착된 크라운 에테르 1~ 양 (m g) 아미노산 17(R) 39(S) 49(S) 58(R) 알라닌 (Ala) 1.36 1.60 1.65 2.0 3 루 0] 신 (Leu) 1.28 1.47 1.53 1.65 페 닐알라닌 (Pheala) 1.11 1.18 1.20 1.24 메티오닌 (Me t) 1.48 1.80 1.85 2.06 페 닐글리신 (Phe g!y) 2.6 8 3.6 3 3.97 4.5 8 글루탐산 (Glu) 1.90 2.65 2.8 9 3.7 3 티로신 (T y r) I. IO 1.15 1.16 1.21 아르기닌 (Ar g) 1.30 1.43 1.51 1.75
표 6-3 RP-C1 교정상에 흡착된 키랄 크라운 에테르 쁘 . (58m g~백서 페닐글리신 및 메 티오닌의 광학분할에 대한 이동상중의 산의 영향. 유속 : 0.5 m L/mi n, 온도 : I B'C , 검출가 : 200run 혹은 254run UV. (참고문헌 : 22) 페닐글리신 메티오닌 이동상 kL, kD’ a’ kL' kD, a’ • 3x 10- sM HCIO, 0.78 3.0 3 3.8 9 0.98 1.76 1.80 10 - ◄ M HC104 0.8 7 3.38 3.8 7 0.9 9 1.81 1.83 3 x 10 - ◄ M HCl04 0.9 2 3.62 3.95 1.05 1.91 1.83 10-3M HCl04 0.99 4.08 4.12 1.10 2.08 1.89 3x 1Q- 3M HCIO, 1.24 5.2 9 4.2 6 1.28 2.51 1.96 10-2M HC104 1.98 9.1 0 4.60 1.75 3.6 2 2.0 7 10-2 M HN03 1.37 5.82 4.25 1.23 2.4 1 1.96 10-2M HCl 1.34 5.70 4.24 1.16 2.28 1.96 5x l0~ 3M H2 SO . 1.17 4.92 4.1 9 1.02 2.00 1.96
A+ + x- ; == AX A 나 U+x - == ACX 여기서 A+ 는 양성자화된 아미노산 (A) 을 나타내며 X - 는 이동상중의 무기산 음이온, t는 고정성에 흡착된 키랄 크라운 에테르, AX 는 A+ 와 X 자이의 이온쌍, 天己표는 A+, C 및 x - 사이의 착물을 의미 하고 문자의 위에 그은 선은 고정상을 의미한다• 이중 KCX의 형성 은 입체 선택성에 직접 관련이 있으며 이온쌍인 XX 의 형성은 두 이 성질체의 머무름 시간을 증진시키나 입체 선택성은 감소시킬 것이다 (KX는 아미노산 염이 소수성 상호작용 등에 의하여 고정상에 머 물 러 있는 상태를 의미한다). C 의 농도를 중전시키면(죽 흡 착되 는 키 랄 크라운 에 테르 브의 양을 많게 하면) 가역적으로 많은 KCX가 형성될 것이므로 분리인자 a 값이 증가할 것을 쉽게 예측할 수 있다. 이동상중의 산의 농도를 증가시켜 X 리 양을 많게 하면 XX 가 증가할 뿐만 아니라 雲의 양도 증가하게 되어 분석성분의 머무름 시간이 증가할 뿐만 아니라 입체 선택성도 증가할 것으로 예측된다. 특히 고정상에 AX 가 머무는 것은 오로지 친지질성 (혹은 소수성) 상호작용에 의하여 이루어 지는 것이므로 X- 의 친지질성 혹은 소수성이 증가하면 머무름은 더욱 증가하며 (23) 따라서 표 6 - 3 에서 보는 바와 같이 X_ 의 친지질성의 순 서 죽 Cl- 〈 N03- 〈 Cl04 _ 순에 따라 머무름 시간이 증가함을 예측할 수 있고 이것은 실리카 겔에 흡착된 크라운 에테르 고정상에서 알카리 금 속이온을 분리할 때 음이온에 따른 머무름 시간의 변화와 일치하는 내 용이다 (23). 참고문헌(제 6 장) 1. C. J. Pederson, J. Am. Chem. Soc., 89, 2495 (1967).
2. C. J. Pederson and H. K. Frendorff , Ang e w. Chem. Int. Ed. E ng l. , 브 16 (1972). 3. R. M. Izatt R. E. Terry, B. L. Hay mo re, L . D. Hanson, N. K. Dalley , A. G. Avondet and J. J. Chris t e n sen, J. A m. Chem. Soc., 쁘 , 7620 (1976). 4. R. M. Izatt , J. D. Lamb, N. E. Izatt , B . E. Rossit er , Jr., J. J. Chris t e n -sen and B. L. Hay m ore, J. Am. Chem. Soc., 쁘, 6273 (1979). 5. T. Nakag a wa, A. Shib u kawa and T. Uno, J. Chromato g r., 쁘 , 695 (1982). 6. A. Shib u kawa and T. Nakag a wa, in Chir a l Se pa rati on s by HPLC : Ap pli ca t i on s to Pluin n aceu ti ca l Comp o unds, A. M . Krstu lovic , Ed., Ell is Horwood, Chic h este r , 1989. chap 16. 7. J. F. Sto ddart, in To pi c s in Ste r e o c he m i stry, vol17, E. L. Eli el and S. H. W ile n, Eds., Joh n W iley and Sons, New York, 1987, pp 207- 28 8. 8. E. P. Ky b a, M . G., Sie g e l, L . R. Sousa, G . D. Y. Sog a h and D. J. Cram, J. Am. Chem. Soc., 쁘 , 2691 (1973). 9. E. P. Ky b a, G. W. Gokel, F. de Jon g , K. Kog a , L. R. Sousa, M. G. Sie g e l, L . Kap la n, G. D. Y. Sog a h and D. J. Cram, J. Org. Chem., 쁘, 4173 (19 77). 10. S. C. Peacock, D. M. Walba, F. C. A. Gaeta , R. C. Helge son and D. J. Cram, J. Am. Chem. Soc., 쁘~ . 2043 (1980). 11. K. Yamamoto , H. Fukushim a, Y. Okamoto . K. Hata d a and M. Nakazaki, J. Chem. Soc. Chem. Commun., 1111 (1984). 12. D. Gehin , P. D. Cesare and B. Gross, J . Org. Chem.' 탄, 1_90 6 (1986). 13. R. C. Helge son, J. M. Ti m ko, P. Moreau, S. C. Peacock, J. M. May e r and D. J. Cram, J. Am. Chem. Soc., 쁘 , 6762 (1974). 14. E. B. Ky b a, K . Kog a , L. S. Sousa, M. G. Sie g e l and D. J. Cram, J. Am. Chem. Soc., 쁘, 2692 (1973). 15. E. P. Ky b a, J. M. Ti m ko, L. J. Kap la n, F. de Jon g , G. W. Gokel and 16. DS.. CJ. . CPreaamco, cJk. ,A Lm. .A C. Dheomm. e Sieo rc ,. ,F 쁘~~. A4.5 5G5 a(e1t9a 7, 8R). . C. Helge son, J. M. 17. TL.i mR .k Soo aunsda , DD. .J .H C. rHamof,f mJ. aAnm, L. .C Kheamp l.a Sno, c D,,. J1 .0 0C, r .8 a1m90, J(1. 9A78m) .. Chem.
18. SLo cR.., S96o, u7s1a0,0 G (.1 D97. 4Y).. Sog a h, D. H. Hoff m an and D. J. Cram, J. Arn. Chem. Soc., 100, 4569 (1978). 19. G. D. Y. Sog a h and D. J. C ram, J. Am. Chem. Soc., 안 , 1259 (1975). 20. G. D. Y. Sog a h and D. J. Cram, J. Am. Chem. Soc 기 쁘 , 3038 (1976). 21. G. D. Y. Sog a h and D. J. Cram, J. Arn. Chem. Soc 나안, 3035 (1979). 22. T. Shin b o, T. Yamag uc hi, K. Ni sh im ura and M. Sug iur a, J. Chromato g r., 쁘흔, 145 (1987). 23. T. Iwachid o , H. Nait o, F. Samukawa, K. Ishim aru and K. Toei, Bull. Chem. Soc. Jpn ., 쁘, 1475 (1986).
제 7 장 전하이동 착물형성 키랄고정상 1 서론 키랄고정상과 광학분할하고자 하는 라세미 화합물 사이의 상호작용 에는 수소결합, 정전기적 상호작용, 쌍극자-쌍극자 상호작용, 소수성 혹은 천지질성 상호작용, 입체장애 등이 있으나 키랄고정상과 광학분 할하고자 하는 라세미 화합물 사이의 亢-亢상호작용(혹은 전하이동 상 호작용)이 여러 다른 상호작용과 함께 아주 중요한 역할을 하여 광학 분할이 이루어질 때, 이 키랄고정상울 특히 전하이동 착물형성 키랄고 정상이라고 지칭하여 이 부분에서 다루고자 한다. 전하이동 착물형성에 대한 분자궤도함수 이론의 간단한 모델은 그림 7-1 과 같다. 그림 에 서 보는 바와 같이 r- 전자주게 (D 분 7 자)의 HOMO 에너지 준위와 亢-전자받게 (A 분자)의 LUMO 에너지 준위가 비슷할 경우에 D 옵!자와 A 나손자가 접근하면 HOMO-LUM(), 상호작 용에 의하여 안정한 D-A 착물이 형성된다 0). D-A 착물의 HOMO 에 서부터 LUMO 에로의 전자전이에 필요한 에너지는 D 웁!자와 A 읍본자 의 전자전이에 필요한 에너지보다 작기 때문에 보통 D-A 착물의 UV
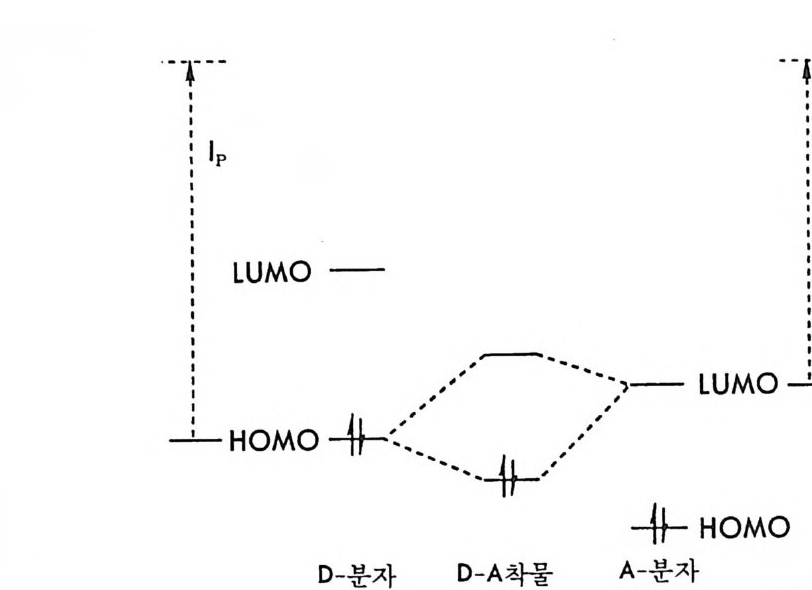 ·--.- -- --r-
·--.- -- --r-
그림 1-| 전하이동 착물형성에 대한 분자궤도함수 이 론 의 간 단한 모 델
-Vi s 스펙트럼에 의하면 D 옵 F 자나 A 옵 F 자의 UV-Vi s 스펙트럼보다 간 파장 쪽에서 새로운 UV-Vi s 흡수가 관찰된다. D 윤 F 자의 HOMO 에너지 준위는 D 윤 F 자의 이온화 에너지 (ion iz a ti ot i on po te n ti al : I 사에 해당하며 A분 자의 LUMO 에너지 준위는 A 분자의 전자 친화도 (electr o n a ffi n ity : E A ) 에 해당하므로 D-A 착물의 안정성은 결국 D 분자의 I p와 A 분자의 EA 에 의존하는 값으로써 D 웁 F 자의 HOMO 와 A 분자의 LUMO 의 에너지 차이가 작을수록 더 커진다. 보통은 방향족 탄화수소가 전자주게 분자 혹은 전자받게 분자로 작용하는데 , 서로 접하는 방향족 고리의 수가 증가하면 HOMO 의 에너지 준위가 증가할 뿐만 아니라 방향족 고리에 치환된 알킬기, 알콕시기 등의 전 자를 내어주는 치환기 또한 방향족 고리의 HOMO 에너지를 증가시키 므로 전자롤 내어주는 치환기가 붙어 있는 다중고리 방향족 화합물은 전하이동 착물형성에 있어서 좋은 D--{ 손자로 작용한다. 반면, 방향족 고리에 치환되어 있는 —N 02, -Cl, —C N 등과 같은 전자를 끄는 치 환기는 방향족 고리의 LUMO 에너지 준위를 낮추기 때문에 전자를
끄는 치환기가 붙어 있는 방향족 고리는 전하이동 착물형성에 있어서 좋은 A 분자로 작용한다(전자밀도가 큰 방향족 탄화수소를 r- 염기성 분 자라 하고 전자밀도가 상대적으로- 낮은 방향족 탄화수소를 7C- 산성 분자라 하기도 한다). T- 전지주게와 T- 전자받게 사이에 형성되는 전하이동 착물의 형성 엔탈피는 보통 몰당 수 kcal 에 불과하기 때문에 착물형성속도 및 해 리속도는 아주 빠르다. 따라서 r- 전자주게 혹은 T- 전자받게로 작용 하는 방향족 탄화수소를 액체 크로마토그래피의 고정상으로 사용할 수 있는 고형지지체 (보통 실리카 겔)에 결합시켜 고정상으로 사용할 경우 에 고정상과 여러 종류의 방향족 탄화수소 사아에 형성되는 전하이동 착물들은 안정성이 모두 다를 것이므로 이 고정상은 여러 종류의 방향 족 탄화수소를 쉽게 분리할 수 있음을 예상할 수 있다 (2,3). 실제로 l,2,g 및 伊} 같은 고정상들이 여러 종류의 방향족 탄화수소 혼합물
 02N N02 -NS|0I 2i- ( CNH022) 3Hl°2NON <二 'N (NCIO H022) 3 N 02 卜ilc士士 1~
02N N02 -NS|0I 2i- ( CNH022) 3Hl°2NON <二 'N (NCIO H022) 3 N 02 卜ilc士士 1~
을 분리하는 데 이용되었다 (4). r- 전자주게 혹은 r- 전자받게인 방향족 탄화수소가 컬럼의 고형지 지체에 결합된 액체 크로마토그래피용 고정싱을- 이용하여 여러 종류의 방향족 탄화수소 혼합물을 분리할 수 있다는 개념은 GII-Av 등에 의 하여 라세미 화합물의 광학분할에도 응용되었다 (3,5). (R)-2-(2, 4, 5, 7 대트라니트로 -9 플루오레닐리 덴아미노옥시 )프로피온산 (TAPA) 웁 헥사헬리센의 두 광학 이성질체와 안정성이 다른 두 개의 부분 입 체 이성질 7(-7(착물을 형성하며 이들의 결정화에 의하여 헥사헬리센 의 두 광학 이성질체를 분리할 수 있다는 사실이 Newman 등에 의하 여 (6) 보고된 것을 기초로 하여 Gi l- Av 등은 R-( 一 )-TAPA 를 실리 카 겔에 결합시킴으로써 (R)-(-)-TAPA 키랄고정상 §울 제조하였 고 여러 종류의 헬리센 광학~ 성공적으로 응용되었다(그림 7-2).
 H쿨 OII
H쿨 OII
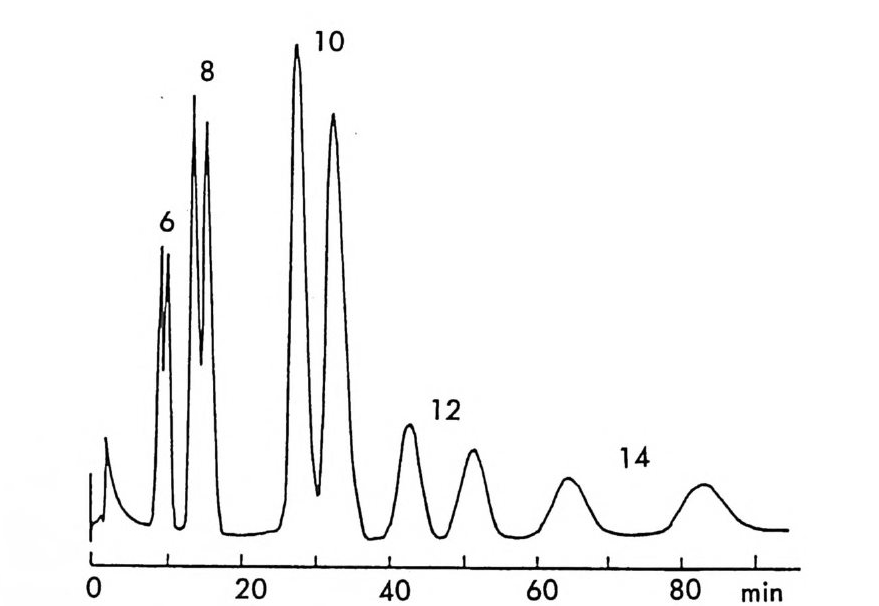 8 10
8 10
그림 7-2 키랄고정상 §에서 여러 종류 헬리센의 광학분할. 6, 8, 10, 12, 14 는 각 각 6-, 8-, 10- , 12- 및 14- 헬리센을 의미함. P-(+)- 헬리센의 머무름 시간이 더 김 . 이동상 : CH2CI 균시클로핵산 (25/75) , 검출기 : 280nm UV( 참고문헌 : 5)
(R)-TAPA 키랄고정상 § 의에도 헬리센의 광학분할에 사용된 전 하이동 착물형성 키랄고정상으로는 광학활성인 비나프틸 -2,2' -디올을 기저로 한 키랄고정상 1 과 N- 디니트로페닐알라닌울 기저로 한 키랄 고정상 § 등이 보고된 바 있다 (3,7). 그러나 이들 키랄고정상은 헬리 센 계통 화합물의 광학분할에만 응용되었을 뿐이다.
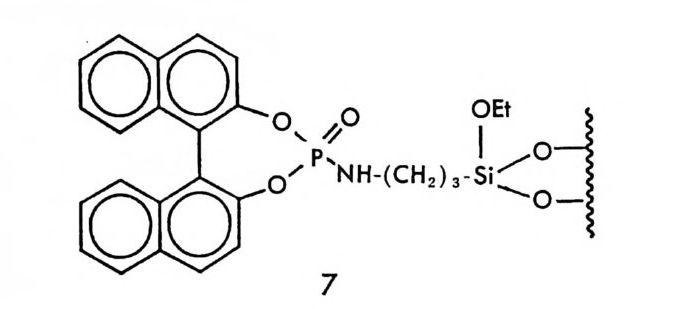 0`/O7E/O 페cH 2 빼 . 13 SoiE /-tl 00
0`/O7E/O 페cH 2 빼 . 13 SoiE /-tl 00
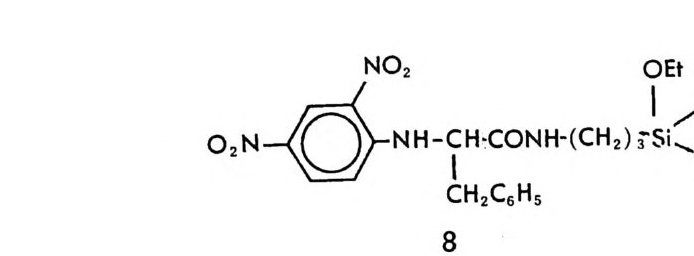 °2N 二N0 N2H - f HCONH-(CH2) ;O\Eit <:\
°2N 二N0 N2H - f HCONH-(CH2) ;O\Eit <:\
위에 제시한 키랄고정상들이 전하이동 착물형성 키랄고정상의 효시 이기는 하나 전하이동 착물형성 키랄고정상의 제조 및 이들 키랄고정 상게서의 광학분할에 대하여 체계적인 연구를 시작함으로써 1980 년대 액체 크로마토그래피에 의한 광학분할의 연구에 기폭제의 역할을 하였 을 뿐만 아니라 액체 크로마토그래피에 의한 광학분할의 연구에 지대 한 공헌을 한 과학자는 미국 일리노이대학의 W. H. Pir k le 교수이다. 따라서 전하이동 착물형성 키랄고정상을 특별히 P i rkle- 형 키랄고정 상이라고 지칭하기도 한다. P i rkle- 형 키랄고정상은 T - 전 자주게 (7C -염기성기) 혹은 亢-전자받게 (7C- 산성기)로 사용할 수 있는 키랄화합물 울 실리카 겔에 결합시킨 키랄고정상으로서 광학분할하고자 하는 라세 미 화합물과 입체 선택적으로 T_r 상호작용, 수소결합, 쌍극자켓戶f 자 상호작용, 입체장애에 의한 상호작용을 함으로써 두 개의 광학 이 성질체를 구별하나 이들 여러 상호작용 중 특히 江_亢상호작용이 필수 적인 것으로 인식되고 있다. 다른 종류의 키랄고정상들이 대부분 시행착오의 방법에 의하여 제조 된 반면 P i rkl e{본 철저하게 이미 얻어진 실험결과와 키랄성 인지 메 커니즘을 근거로 하여 새로운 형태의 키랄고정상을 합리적 방법으로 고안하였다. 제 7 장에서는 P i rkle 이 여러 종류의 키랄고정상을· 어떻 게 개발하였는가 하는 점에 중접을 두어 기술하면서 이들 키랄고정상 둘의 특칭과 키랄성 인지의 메커니즘 및 유사한 종류의 다른 키랄고정 상들에 대하여 기술하게 될 것이다. P i rkle- 형 키랄고정상에 대하여 서는 그동안 다수의 총설논문들이 발표되었으므로 이들 논문들이 P i rkle- 형 키랄고정상을 이해하는 데 많은 도움이 될 것이다 (8, 9, 10).
2 카르비놀 (carb i no l) 키랄고정상 Pir k le 동은 NMR 을 이용하여 광학활성 물질의 광학순도 및 절대 배열을 결정하고자 하는 지속적인 연구의 결과로서 NMR 키랄 용매 화 시약 (ch i ral solvati ng ag e nt : CSA) 인 카르비놀 l 을 개발할 수 있 었다(1).
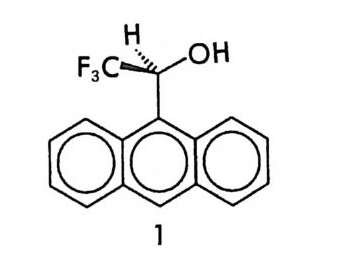
NMR 키랄 용매화 시약인 카르비놀 l 은 여러 종류의 광학활성 물 질의 광학순도 및 절대배열을 결정하는 데 성공적으로 응용되었을 뿐 만 아니라 카르비놀 l 을 이동상에 첨가하여 액체 크로마토그래피에 웅 용함으로써 (CMPA 방법) T- 산성기를 가전 라세미 화합물의 광학분 할에도 성공적으로 응용되었다(제 2 장 참조). NMR 용매화 시약으로 서 카르비늘 l 의 역할 및 키랄 이동상 첨가제로서 카르비놀 l의 역할 에 대하여 Pir k le 등은 정확한 메커니즘을 규명할 수 있었기 때문에 당연한 귀결로서 Pir k le 등은 카르비놀 l 을 적절한 방법으로 실리카 겔에 결합시켜 키랄고정상을 제조하면 이 키랄고정상을 이용하여 여러 종류의 라세미 화합물들을 광학분할할 수 있을 것으로 기대하였다. 특 히 카르비늘 l 은 강한 1t-염기성기인 9_ 안트릴기를 가지고 있기 때문 에 카르비놀 l 을 기저로 한 키랄고정상에서는 카르비놀 1 과 1(-1(상호 작용을 할 수 있는 1t-산성기를 가전 라세미 화합물들이 광학분할될 수 있을 것으로 기대하였다. 카르비놀 l 을 기저로한 카르비놀 키랄고정상 2 는 그림 7- 碑] 과정
Br2 니( )I( )I( )I A CS2 CH3 CH2Br 仁〉 1,E- t (CH2) 3-SH 戶 [°0:s:(E;H2) 3-s-C H2 2
그림 7-3 카르비늘 키랄고정상 2 의 제조과정
에 의하여 제조되었으며 (2), 이와 같이 제조된 키랄고정상 2 는 예상한 바와 마찬가지로 여러 종류의 r- 산성기를 가전 라세미 화합물들의 광 학분할에 성공적으로 응용되었다. r- 산성기를 가지고 있는 라세미 술 폭시드 화합물이 광학분할되었을 뿐만 아니라(그립 7-4 및 표 7- 1 ) 라 세미 알코올, 라세미 아민, 아미노산의 에스테르, a- 히드록시카르복 실산의 에스데르, 라세미 티올, 라세미 아미노알코올의 3, 5- 디니트로 벤조일 유도체 g이 광학분할된다는 사실이 확인되었다(표 7- 1 ) (2,3). 화합물 笠기 3 , 5- 디니트로벤조일기는 강한 7r:-산성기로 작용하여 키 랄고정상 ~ 9- 안트릴기와 亢-亢착물형성에 도움을 줄 뿐만 아니라 3,5- 디니트로벤조일기의 C=O 기는 키랄고정상과 수소결합을 할 수 있는 염기성 부위로 작용하며 화합물 ~ B 로 표시한 염기성 부위 또 한 키랄고정상의 산성 부위와 수소결합을 할 수 있기 때문에 키랄고정 상 2 에 의한 광학분할의 메커니즘은 그림 7-59 .} 같이 설명할 수 있 다 . 화합물 g은 그 구조에서 보는 바와 같이 -C=O 기와 키랄중심 의 -C ― H 가 서로 가리워져 있을 때 가장 안정한 형태 (confo rm a-
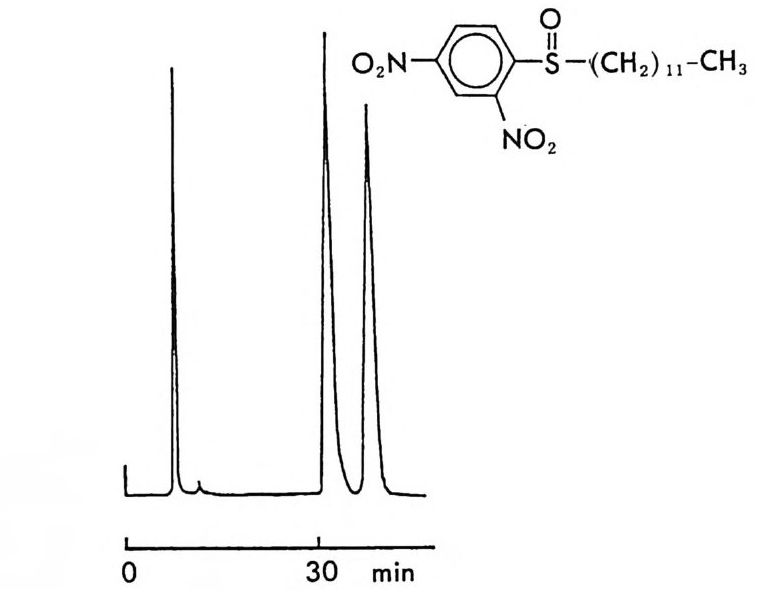 02N 를? _(CH2) II-CH3
02N 를? _(CH2) II-CH3
그림 7-4 카르비늘 키랄고정상 2 에서 n - 도데실 2,4- 디니트로페닐술폭시드의 광학분할. 이동상 : 핵산 -2- 프로판을 (4 / 1). (참고문헌 : 2)
ti on) 를 갖는다고 가정하면 (R) ―空} 키랄고정상 2 사이에는 그림 7 -5 전에서 보는 바와 같이 7l 국상호작용과 두 위치에서의 수소결합에 의하여 안정한 부분 입체 이성질 착물을 형성한다고 생각할 수 있다. (S) -洋키랄중심탄소 C ―Q결합사이의 회전에 의하여 (R)-3 과 마
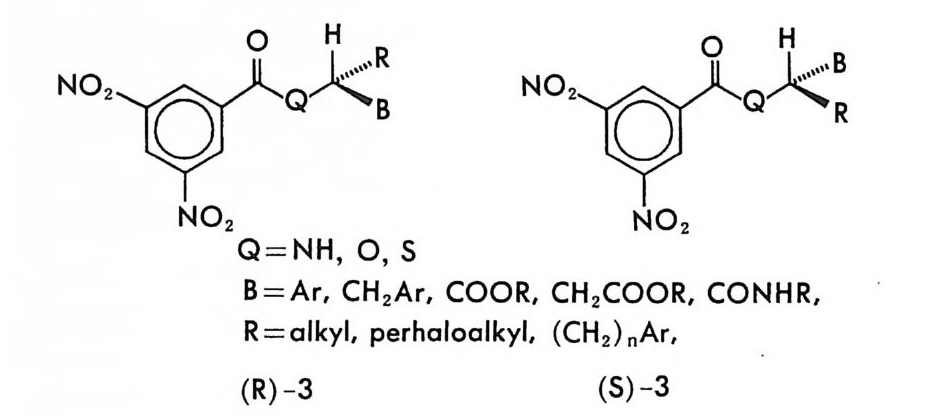 N02< Q\: N 。2 0¢NQo ZH」 BR
N02< Q\: N 。2 0¢NQo ZH」 BR
표 7-1 카르비놀 키랄고정상 2 에서 라세미 화합물의 광학분할. 이 동상 : 2- 프로판올-핵산 (20/80) (참고문헌 : 2) 。 7e-A c id; . s11 : R R 1r:- acid a’ k1' meth yl 2, 4-(N02) 2 CsH3 1.12 13.05 n-buty l 2, 4-(N 02) 2 CsH3 1.19 6.32 n-octy l 2, 4-(N02) 2 CsH3 1.24 4.44 n-dodecy l 2, 4-( N02) 2 CsHJ 1.26 3.3 3 n-dodecy l 4-N02C6H, 1.09 2.78 3 Q B R a’ k1' NH C6Hs CH3 1.19 3.0 1 NH C02CHs CH3 1.08 4.1 6 NH C02CHa CH2C6Hs 1.10 4.0 0 NH CONHC4H9 CH2C6Hs 1.56 1.22 NH CONHC4Hg CH2CH(CH3)2 1.65 3.76 NH CONHC4H9 C6Hs 1.78 0.7 5 。 C6Hs CH3 1.08 1.56 s。 CC06H2Cs H3 CCHH33 11..0075 31..34 79
찬가지로 그립 7-5~ 에서 보는 바와 같은 부분 입체 이성질 착물을 형 성하나 키랄중심에 있는 입체정애가 큰 R7] 가 -C = O 기와 가리위전 위치에 있기 때문에 그 안정성은 그림 7-5~ 착물의 안정성에 비교하 여 상대적으로 낮다. 따라서 키랄고정상 災} 안정한 착물을- 형성하는 (R)- 梵 컬럼에 오래 머무르고 (S)- 發든 먼저 분리되어 나올 것을 쉽게 예측할수있다.
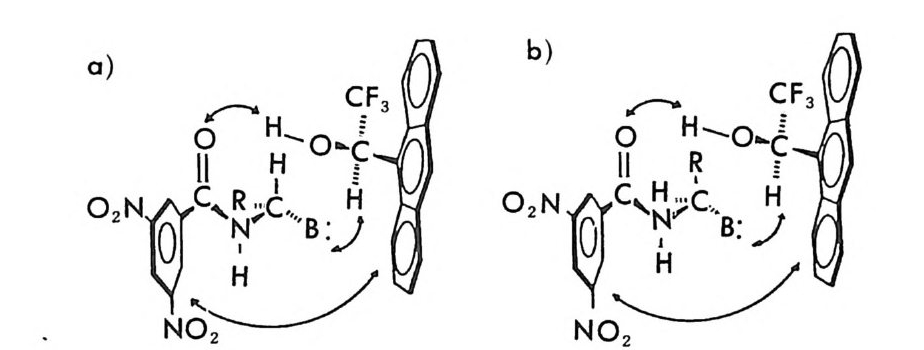 a)
a)
그림 7-5 키 랄고 정상 단 에 서 呼} 광학분할에 대한 키랄성 인지 메커니즘의 모델
그림 7 - 5 와 같은 키랄성 인지 메커니즘은 두 광학 이성질체의 용리 순서와 B 로 표시한 치환기의 염기성 세기를 바꾸었을 때 광학분할에 미치는 영향 등을 체계적으로 조사함으로써 도출된 결과이다. 그립 7-5 의 키랄성 인지 메커니즘의 특징은 키랄고정상과 화합물 g 사이의 7C - 7C 상호작용 및 두 위치에서의 수소결합을 고려한 것이므로 Dal g l i esh 의 세 점 상호작용 모델(제 2 장 참조)에 잘 들어 맞는 경우 라고 할수 있다. 아미노산의 3, 5- 디니트로벤조일유도체는 자유 카르복시기를 가지고 있기 때문에 정상이동상을 사용하여서는 키랄고정상 2 에서 광학분할 이 어려우나 역상 이동상을 사용하여 광학분할이 가능하다는 사실이 알려져 있다 (3). 3 7C- 산성 키랄고정상 3.1 키랄성 인지의 상호성 개념 여러 종류의 전하이동 착물형성 키랄고정상을 고안하고 개발하는 데 있어서 P i rkl e-,'.본 키랄성 인지의 상호성 개념 (reci pro c ity concep tion
ot chir a l reco gniti on) 을 아주 유용하고 적절하게 이용하였다. 키랄성 인지의 상호성 개념은 키랄성의 인지에는 상호성이 있다는 개념으로서 순수한 광학 이성질체 A 가 라세미 화합물 B 의 두 광학 이성질체를 구별할 수 있다면 역으로 순수한 광학 이성질체 B 는 라세미 화힙물 A 의 두 광학 이성질체를 구별할 수 있다는 것이다(엄밀한 의미에서 키 랄성 인지의 상호성 개념이 완전히 맞는 것은 아님 (1)). 따라서 순수한 광학 이성질체 (+)-A 로부터 얻어진 키랄고정상 CSP- ( + ) - A 가 라 세미 화합물 (土 )-B 를 광학분할할 수 있다면 역으로 순수한 광학 이 성질체 (+)-B 혹은 (― )-B 로부터 유도된 키랄고정상 CSP-(+ ) -B 혹은 CSP ―(-)-困즌 라세미 화합물 ( 士 ) - A 를 광학분할할 수 있을 것이다 (2). P i rkl 送는 이와 같은 키랄성 인지의 상호성 개념을 이용하 여 여러 종류의 새로운 키랄고정상들을 성공적으로 개발할 수 있었다. 그러나 광학적으로 순수한 A 혹은 B 를 실리카 겔에 고정시킬 때 고 정방법에 따라 키랄성 인지의 상호성 개념이 엄밀하게 성립한다고 단 언할 수 없다는 사실을 유의할 필요가 있다. 3.2 페닐글리신 -DNB 및 루이신 -DNB 키랄고정상의 제조 및 광학분할의 특성 카르비늘 키랄고정상 (2 절의 키랄고정상 2) 에서 특히 광학분할이 잘되 고 있는 아미노산 유도체들은 페닐글리신아미드의 3, 5- 디니트로벤조 일 (DNB) 유도체와 루이신아미드의 3, 5- 디니트로벤조일 (DNB) 유도체 이다 (2 절의 표 7-1 참조). 따라서 키랄성인지의 상호성 개념에 따라 페 닐글리 신의 DNB 유도체 (PGDNB) 혹은 루이 신의 DNB 유도체 (LeuDNB) 를 아미드 결합을 통하여 실리카 겔에 결합시킬 경우 이들 은 라세미 카르비늘을 광학분할할 수 있는 제 양1 ] 대 키랄고정상(카르비 놀 키랄고정상을 제 1 세대 키랄고정상이라 할 때 아미노산 DNB 에서 유도 된 키랄고정상을 키랄성 인지의 상호성 개념에 따라 카르비놀 키랄고정상에 서부터 유래하였다고 생각하여 제 筑]대 키랄고정상이라 지칭한다 (3) )으로-
사용할 수 있을 것울 쉽게 예상할 수 있다. 광학적으로 순수한 형태로 쉽게 구할 수 있는 페닐글리신이나 루이 신은 염화 3, 5- 디니트로벤조일 (3, 5-din i t ro benzoy l chlor i de) 과 반응 시켜 아미노산의 DNB 유도체를 만든 후 짝지음 시약인 N- 에독시카 르보닐 -2_ 에독시 -1, 2 工]히드로퀴놀린 (EED Q)존재 하에서 아미노프로 필 실리카 겔과 반응시키면 PGDNB 혹은 LeuDNB 가 아미드 결합에 의하여 실리카 겔에 결합된 키랄고정상 l(PGDNB CSP) 혹은 키랄고 정상 .?_(LeuDNB CSP) 가 얻어진다(그림 7-6) (4).
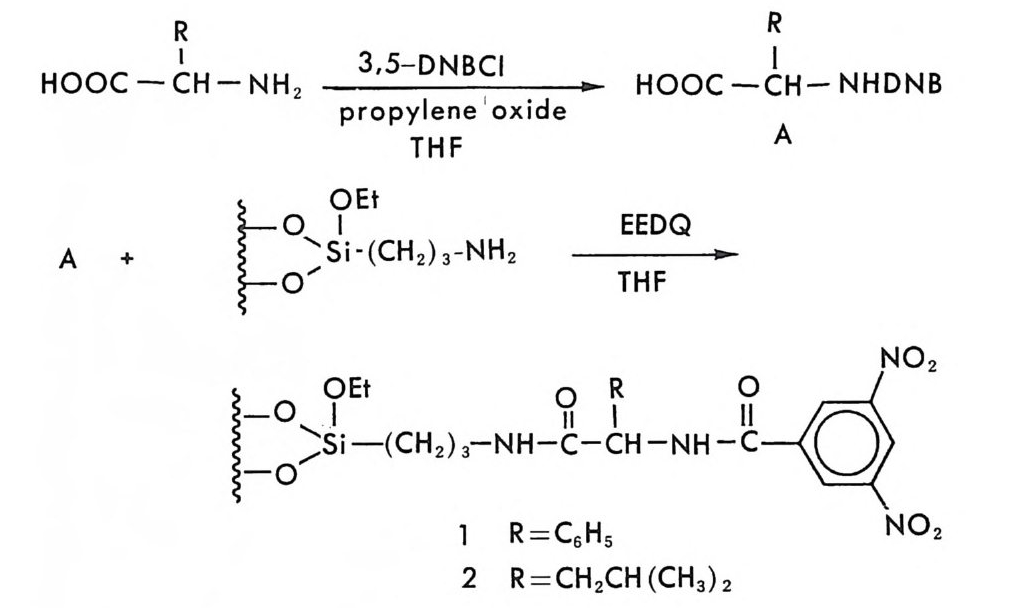 HOOC-CRI H -NH2 p3r o,5p 一yT DlHe NFnB eC 'Io x id e HOOC-CRIA H -N HDNB
HOOC-CRI H -NH2 p3r o,5p 一yT DlHe NFnB eC 'Io x id e HOOC-CRIA H -N HDNB
그림 7-6 아미노산 -DNB 키랄고정상 i 및 突] 제조
키랄고정상 l 과 2는 아미노산 -DNB 가 공유결합에 의하여 실리카 겔에 결합된 키랄고정상인 반면 아미노산 -DNB 유도체와 아미노프로 필 실리카 겔을 짝지음 시약없이 단순히 섞어줌으로써 아미노프로필 실리카 겔의 아미노기와 아미노산 유도체의 카르복시기 사이에 이온결
합기 형성되므로 이온결합 키랄고정상 g 혹은 산가 쉽게 제조된다 (2). 특히 아미노프로필 실리카 겔이 채워진 HPLC 컬럼을 통하여 아미노 산의 DNB 유도체가 녹아 있는 THF 용액을 흘려준 후 순수한 THF 용액을 홀려주면 HPLC 컬럼내에서 이온결합 키랄고정상 설 혹은 산가 아주 쉽게 생성된다(i n sit u ge nerat ion ) (2 ) . 이온결합 키랄고정상 g 혹은 蘇는 n 책산에 20% 의 2 - 프로판을이 혼합되어 있는 혼합용매보다 극성이 더 큰 이동상을 사용하지 않는 한 이온결합이 깨어져서 아미노 산 -DNB 가 컬럼에서 분리되어 나오는 일이 없다는 사실이 실험적으 로확인되었다.
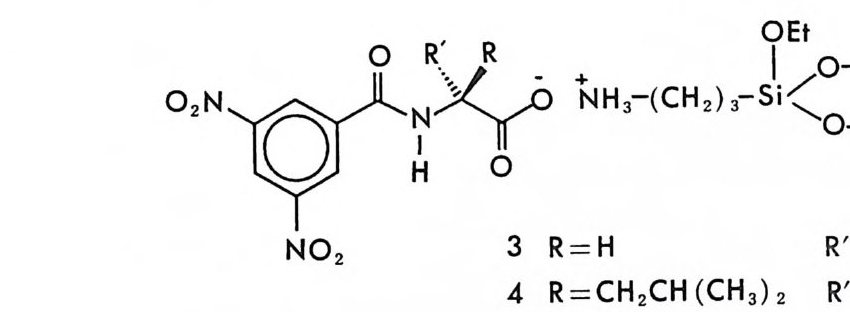 02N` 스즈/R.: 《R 타 (CH2) 3-OS\E
02N` 스즈/R.: 《R 타 (CH2) 3-OS\E
이렇게 제조된 아미노산 -DNB 키랄고정상둘은 광학분할하고자 하 는 라세미 화합물과의 T_ 元착물형성에 필요한 강한 亢-산성기 의에 수 소결합 자리 및 쌍극자 등을 가지고 있기 때문에 (그림 7- 7 ) 여러 종류 의 다양한 라세미 화합물들을 광학분할할 수 있을 것으로 기대된다. 아미노산 -DNB 키랄고정상에서 광학분할이 가능한 것으로 보고된 라세미 화합물들은 키랄성 인지의 상호성 개념에 의하여 광학분할될 것으로 기대된 2,2,2- 트리플르오르 -1-(9- 안트릴)에탄올 (2 절의 키랄 고정상 劉 모체 물질)을 비롯하여 아주 다양하다. 현재까지 개발된 여 러 종류의 키랄고정상 중 가장 광범위한 종류의 라세미 화합물들을 광 학분할할 수 있는 키랄고정상은 아마도 아미노산 _DNB 키랄고정상일 것 0] 다.
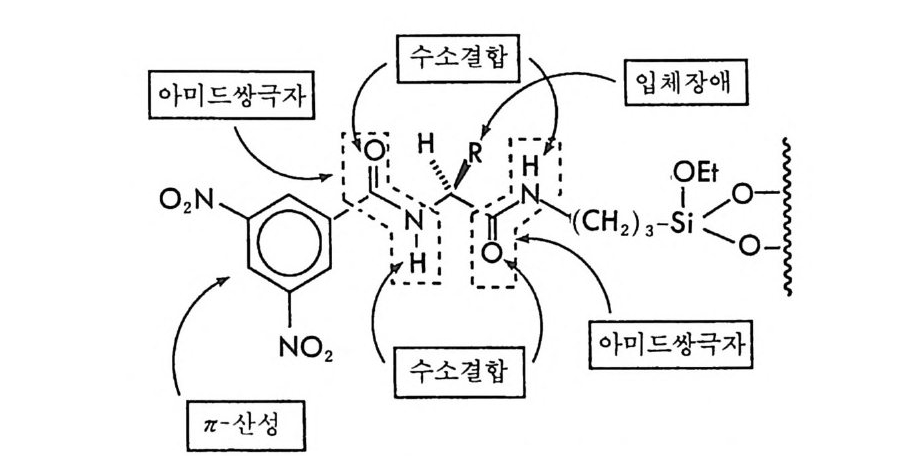 言
言
그림 1-1 아미노산 -DNB 키랄고정상의 가능한 상호작용 부위
아미노산 -DNB 키랄고정상에서 광학분할이 가능한 라세미 화합물 둘은 크게 두 가지로 분류할 수 있다. 즉 분자내에 7t:-영기성기로 사 용될 수 있는 방향족기를 가지고 있는 라세미 화합물들과 7t:-염기성기 를 가지고 있지 않은 라세미 화합물의 두 종류이다. T- 염기성기를 가 지고 있지 않은 라세미 화합물인 경우에는 r- 염기성기를 도입하여야 할 필요가 있으며 분자내에 7t:-염기성기를 가지고 있는 라세미 화합물 인 경우에도 키랄고정상과의 상호작용이 효과적이지 못하여 아미노산 -D NB 키랄고정상에서 광학분할되지 않을 경우에는 다른 7t:_염기성기 를 분자내에 도입할 필요가 있다. 7t:-염기성 유도체로 만들지 않고서 아미노산 -DNB 키랄고정상 l, 2, g 혹은 간에서 직접 광학분할이 가능한 라세미 화합물들의 예는 그 림 7-8 과 같다. 그림 7-8 의 화합물들은 모두 키랄중심이나 혹은 키랄 중심에서 가까운 위치에 T- 염기성기로 작용할 수 있는 방향족기를 가 지고 있으며 그 의에 키랄고정상과 수소결합, 혹은 쌍극자켓戶f자 상 호작용을 할 수 있는 작용기들 예를 들면 히드록시기, 카르보닐기, 아 미노기, 술폭시기, ― P=O 기 등을 가지고 있는 화합물들이다. 그림 7-8 에 있는 라세미 화합물들 이의에도 7t:-염기성기로 작용할 수 있는
 알코올
알코올
 아미노산유도체 2 - 카로복시 인돌린 인화합물〈
아미노산유도체 2 - 카로복시 인돌린 인화합물〈
 락탐A ~1 JH A :〕 工H R(Ar) 아비탈산A유도:체~ 1: 0
락탐A ~1 JH A :〕 工H R(Ar) 아비탈산A유도:체~ 1: 0
그립 7-8 아미노산 -DNB 키랄고정상 1, 2, 3 혹은 산에서 광학분할된 라세미 화 합물들의 예. 괄호안은참고문헌임.
방향족기와 함께 아미노산 - DNB 키랄고정상들과 적절한 상호작용을 할 수 있는 작용기를 가진 라세미 화합물들은 모두 아미노산 -DNB 키 랄고정상에서 광학분할될 것으로 기대된다. 그림 7- 陰] 대부분 화합물들은 직접 광학분할이 가능하나 자유 아 미노기나 자유 카르복시기를 가지고 있는 경우에는 키랄고정상의 지지 체인 실리카 겔과의 강한 상호작용으로 인하여 머무롬 시간이 너무 길 거나 혹은 크로마토그램의 피이크의 모양이 너무 넓어져서 분석의 목 적으로 적합하지 않기 때문에 자유 아미노기나 자유 카르복시기를 적 당한 유도체로 변화시켜줄 필요가 있다. 예를 들면 /3_차단제인 아미 노알코올 ?의 경우 N - 라우로일 (l auro y l) 유도체로 광학분할이 되거나
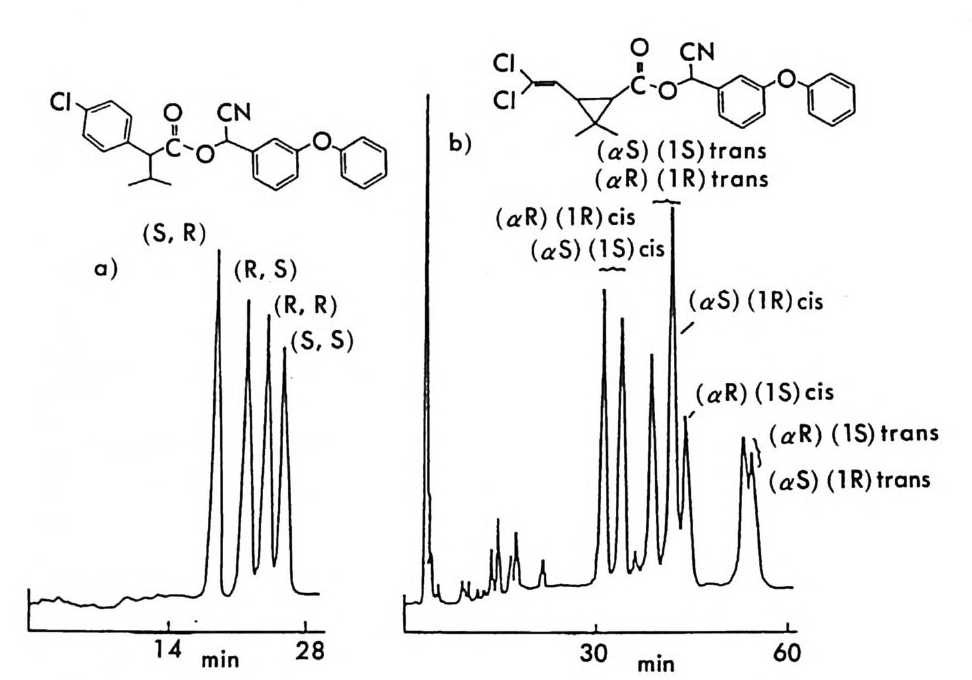 Cl~운 `o 도 °0 b) CCl l~, 군(.;.S°) ` (1SC0)Nt r~a ns0 -0
Cl~운 `o 도 °0 b) CCl l~, 군(.;.S°) ` (1SC0)Nt r~a ns0 -0
그림 7-9 키랄고정상 %。버]서 피레트로이드계인 a) 펜발러레이트 및 b) 사이퍼 메트린의 광학분할 a) 이동상 : 2- 프로판을-핵산 (0 .1/ 99 . 9) , 유속 : 2mL/mi n, b) 이동상 : 2- 프로판을-핵산 (0 .1/99 . 9) , 유속 : lmL/m in (참고문헌 : 32)
혹는 옥사졸리돈유도체 썬으로 광학분할이 가능하다. a - 아릴알킬아 민은 N- 아실유도체 (Y = 알킬)나 카르밤산에스데르유도체 (Y = -0 -R) 혹은 요소유도체 (Y = -NH-R) 끄로서 광학분할이 가능하며 암페타민 의 유사체화합물들은 N- 아실유도체 쌩로서 광학분할이 되고 있다. 반면 자유 카르복시기를 가지고 있는 소영진통제 계통의 의약품인 a - 아릴프로피온산 亞는 에스데르유도체, 일차아미드, 이차아미드 혹은 삼차아미드 유도체로서 광학분할이 이루어지고 있다. 항말라리아 의 약품인 할로판트린 (halo fa n t r i ne) l§의 경우 자유 아미노기 를 다른 유 도체로 변화함이 없이 광학분할되고 있으나 이 경우에는 컬럼내의 머 무름 시간을 조정하기 위하여 소량의 트리에틸아민을 이동상에 첨가하 여 광학분할이 이루어지고 있다 (3 1). 그림 7- 邸] 예들 이의에도 광학분할의 예는 다양하다. 키랄중심이 하나 이상인 라세미 화합물의 경우에는 가능한 입체 이성질체가 모두 분리되는 경우가 많다. 예를 들면 무공해 농약으로- 알려진 피레트로이 드(pyr o t hro i d) 계의 펜발러레이트(f envalera t e) 는 . 엄 n 의 키랄중심을 가지고 있어서 네 개의 입체 이성질체가 가능하나 그립 7 - 9 틴에서 보 는 바와 같이 네 개의 입체 이성질체가 모두 분리되며 세 개의 키랄중 심을 가지고 있는 사이퍼메트린 (c yp erme t hr i n) 의 경우 앙 n 의 입체 이 성질체가 가능하나 그림 7-9~ 에서 보는 바와 같이 두 개의 피이크만 겹칠 뿐 잘 분리되고 있음을 확인할 수 있다 (32) 항말라리아제인 프리마퀸(p r i ma q u i ne) 의 대사물인 바페닐 아합체는 그림 7-10 에서 보는 바와 같이 두 개의 키랄중심을 가지고 있을 뿐만 아니라 비페닐 이합체에 의한 키랄성이 있기 때문에 모두 8 개의 입체 이성질체로 존재한다. 가능한 앙 n 의 입체 이성질체들 중 두 개의 피이 크만이 겹치고 결국 U 粹의 피이크로 잘 분리되고 있음을 그립 7-10 에 서 확인할 수 있다 (33). T - 염기성기로 사용될 수 있는 방향족기를 가지고 있지 않기 때문에 아미노산 -DNB 키랄고정상에서 광학분할되기 위하여 r- 염기성 유도
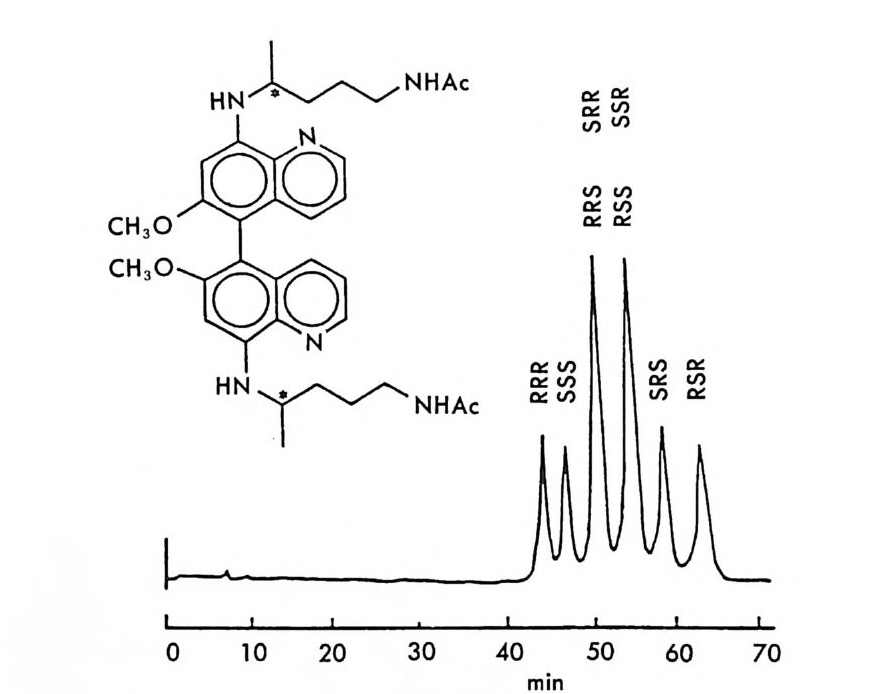 HN~NHAc HHSH SS
HN~NHAc HHSH SS
그림 7-10 키랄고정상 1 상에서 프리마퀸의 대사물인 비페닐이합체의 광학분할. 컬럼 : 250X 4 .6mm, 이동상 : 핵산 -2- 프로판을-아세토니트릴 (850/105/ 75, v/v/v) , 유속 : lmL/ m i n, 검출기 : 254nm UV (참고문헌 : 33)
체로 만들어야 하거나 혹은 T- 염기성기를 가지고 있으나 아미노산 -DNB 키랄고정상에서 광학분할될 때 크로마토그램의 모양을 좋게 하 거나 혹은 입체 선택성을 증진시키기 위하여 亢-염기성 유도체로 만들 어야 하는 라세미 화합물들의 예는 그림 7-11 과 같다. 키랄중심에 아 미노기를 가지고 있거나 혹은 키랄중심에 가까운 위치에 아미노기를 가지고 있는 라세미 화합물들은 보통 N-1- 나프토일 (nap h th o y l) 혹은 N-2- 나프토일 유도체로서 광학분할되거나 나프틸카르밤산 에스데르 유도체 , 나프틸요소 유도체로서 광학분할이 가능하다. 이들 아미노화 합물들은 벤조일유도체로서도 광학분할이 가능하나 일반적으로 나프 틸기를 가전 유도체로 만들 때 나프틸기의 높은 亢-염기성으로 인하여
 R1-CNI HH--8(CC- H A 2r) -R2 R 2 -0- CH,
R1-CNI HH--8(CC- H A 2r) -R2 R 2 -0- CH,
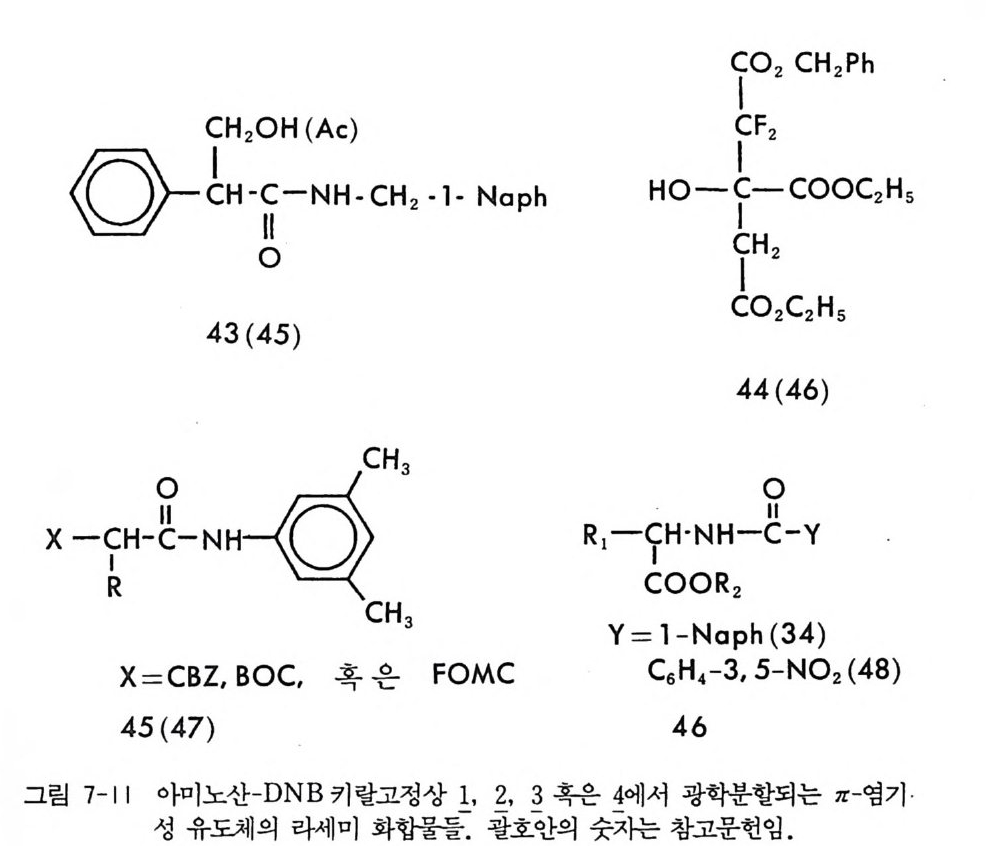 Q-bCHH2-밍0~H -N (AHc-)C H, -1-Nop h HO— Ctc七IIO H—22 CCO HO2 PCh2 Hs
Q-bCHH2-밍0~H -N (AHc-)C H, -1-Nop h HO— Ctc七IIO H—22 CCO HO2 PCh2 Hs
광학분할이 더 찰되는 것으로 알려져 있다• /3-차단제로 쓰이는 아미 노알코올 계통의 화합물 역시 아미노기를 가지고 있기 때문에 아미노 기의 1- 나프틸요소 유도체, 2- 나프틸카르밤산 에스데르 유도체, 1- 나 프토일 유도체 등으로 광학분할이 가능하다(그립 7-11). /3-아미노알코올을 아릴알데히드와 반응시키면 옥사졸리돈 유도체 보이 얻어지나 시스-와 트란스-이성질체가 가능하다 (4 1. 42). 그러나 시스-이성질체가 재결정 과정에서 선택적으로 얻어지는 사실이 확인 되었으며 이 시스-이성질체는 페닐글리신 -DNB 키랄고정상 g에서 두 광학 이성질체로 광학분할된다. 예를 들면 에페드린과 2- 메독시 -1-나 프트알데히드의 반응에서 얻어전 c i s 감누사졸리돈 유도체는 키랄고정상
g에서 광학분할이 잘되는 것으로 알려져 있다 (a = 1.36 ) (42). 소영진통제로 사용되는 2 - 아릴프로피온산이나 트로픽산(t ro pi c a ci d) 은 카르복시기를 r- 염기성이 큰 방향족기 를 가지고 있는 아릴아 민과 반응시켜 아미드유도체로 만들면 (그림 7 - 11 의 샌 및 션) 이들 유 도체들은 아미노산 - DNB 키랄고정상에서 광학분할이 가능·하다. 키랄 카르복실산들(샌 혹은 선)은 보통 1- 나프틸메틸아미드, 벤 질 아미드, p-알킬 혹은 p - 알콕시아닐리드유도체로 만둘면 아미노산 - DNB 키 랄 고정싱에서 광학분할이 잘 되나 최근에는 이들 소영진통제 계통의 화 합물들을 3, 5- 디메틸아닐리드유도체나 혹은 l - 나프틸아미 드 유도체로 만들면 입체 선택성이 오히려 증가한다는 사실이 저자의 연구실에서 확인되었다 (49). 아미노산은 아미노 화합물로 취급할 수도 있고 카르복실산 화합물로 취급할 수도 있다. 따라서 아미노산에스테르는 l - 나프토일 유도체로 만듦으로써 아미노산 -DNB 키랄고정상에서 광학분할이 가능하며 (그림 7-11 의 뻔 아미노산의 N-CBZ (benzoy lo xy carbony l) , N- B OC (ter t -buty lo xy carbony l) 혹은 N-FMOC (9-fl uo reny lm eth o xy c arbony l) 화합물들은 3,5- 디메틸아닐리드유도체로서 광학분할이 가능하다(그립
 47C H3 CH3 (CH2) l2CsE0 Ht0B 2— -5x0C》\。?4 80CNH HH 2了 N H。 MEet 。니广거°49 H
47C H3 CH3 (CH2) l2CsE0 Ht0B 2— -5x0C》\。?4 80CNH HH 2了 N H。 MEet 。니广거°49 H
7-11 의 1§_) . 한 가지 홍미있는 사실은 루이신 메틸에스테르의 DNB 유도체가 페닐글리신 - DNB 키랄고정상 l 에서 광학분할된디는- 것이 다. 아미노산에스데르의 DNB 유도체는 강한 T- 산성기를 가지고 있 음에도 불구하고 r- 산성 키랄고정상에서 광학분할이 가능하다는 사실 로부터 키랄고정상과 분석성분 사이에 7(-7(상호작용이 아닌 수소결합 이나 씽극자 - 쌍극자 상호작용이 카랄성 인지에 중요한 작용을 하고 있 음을 예측할 수 있다 (48). 이와 같은 예측은 亢-염기성기로 작용할 수 있는 방향족기를 전혀 가지고 있지 않은 라세미 화합물 중에도 아미노 산 - DNB 키랄고정상에서 광학분할이 가능한 것이 있다는 사실로부터 도 확인이 가능하다. T- 염기성기로 작용할 수 있는 방향족기를 가지 고 있지 않으나 아미노산 ― DNB 키랄고정상에서 광학분할이 가~錢} 라 세미 화합물에는 헥소바아비탈 旦, 세코바아바탈 怪, 에토숙시미드 (eth o suxim i de ) 산 등의 화합물과(1 5) 키랄 지방산에폭시드 텐의 에 스데르 혹은 아미드유도체 등이 있다 (50). 3.3 키랄성 인지의 메커니즘 페닐글리신 -DNB 혹은 루이신 -DNB 키랄고정상에서의 광학분할은 키랄고정상과 광학분할하고자 하는· 라세미 화합물 사이의 7C 급·상호작 용 및 수소결합, 쌍극자-쌍극자 상호작용, 입체장애에 의한 상호작용 등이 입체 선택적으로 작용하여 이루어진다고 생각된다. 그러나 아미 노산 -DNB 키랄고정상에는 그림 7-7 에서 본 바와 같이 7C- 산성기 아 의에 수소결합이 가능한 부위 및 씽국자켓匡f자 상호작용부위가 여럿 이 있으며 광학분할하고자 하는 라세미 화합물에도 7C- 염기성기 이의 에 수소결합부위나 씽극자 등을 여럿 가지고 있기 때문에 구체적으로 키랄고정상과 분석성분 사이에 어떤 상호작용에 의하여 광학분할이 이 루어지는가 하는 점을 밝히는 것은 그리 간단한 문제가 아니다. 실제 로는 광학분할하고자 하는 라세미 화합물의 구조에 조금의 변화가 있
더라도 키랄성 인지의 메커니즘이 달라질 수 있기 때문에 키랄성 인지 메카니즘을 논의할 때에는 많은 실험 데이타 를 근거로 하여 조심스럽 게 접근할 필요가 있다. 특히 Pir k le 등은 광학분할하고자 하는 라세 미 화합물의 구조를 변화시킴으로써 이에 따른 입체 선택성의 변화, 머무롬 시간의 변화, 용리순서의 변화를 관찰하여 관찰된 결과에 부합 되는 합리적인 키랄성 인지의 메커니즘을 제시하였고 아직 광학분할된 일이 없는 라세미 화합물의 두 광학 이성질체의 용리순서, 입체 선택 성의 정도 등을 예측할 수 있었다. 3.2 절에서 광학분할이 가능한것으로 알려진 많은 라세미 화합물에 대하여 키랄성 인지의 메커니즘이 제안되어 있으나 여기서는 몇 가지
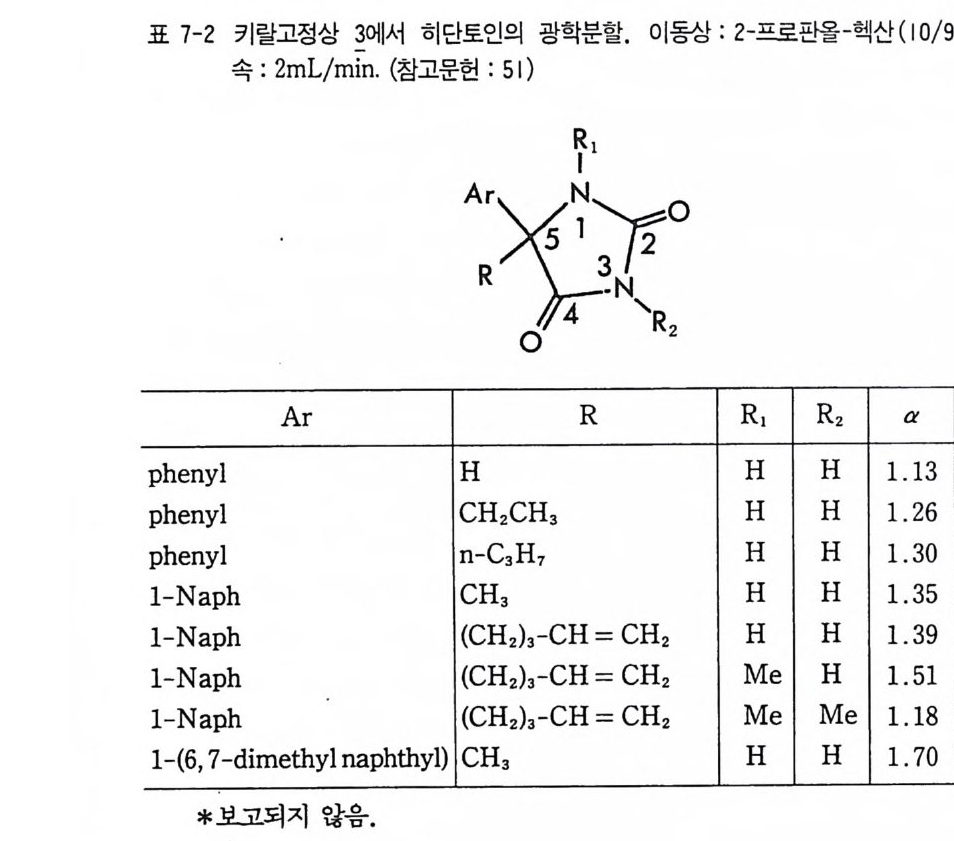 표 7-2 속키 랄: 2고m정L상/m i3 n 에. 서( 참히고단문헌토 인: 5의1 ) 광학분할. 이동상 : 2- 프로판올-헥산 ( 10/90) , 유
표 7-2 속키 랄: 2고m정L상/m i3 n 에. 서( 참히고단문헌토 인: 5의1 ) 광학분할. 이동상 : 2- 프로판올-헥산 ( 10/90) , 유
의 예를 선택하여 키랄성 인지의 메커니즘을 어떻게 규명할 수 있는가 에 대하여 중점적으로 논의하고자 한다• 표 7-2 는 페닐글리신 - DNB 키랄고정상 g 에서 히단토인의 광학분할 에 대한 실험결과를 요약하여 놓은 것이다 (51). 표 7-2 에 의하면 히단 토인 방향족기의 T - 염기성이 증가함에 따라 a 값이 증가할 뿐만 아니 라 R 기의 크기가 커질수록 a 값이 증가하고 있음을- 확인할 수 있다. 한편 1 - 위치와 3 - 위치의 N-H 수소 대신 메틸기로 치환하면 입체 선 태성이 크게 떨어지나 1 - 위치의 N-H 수소만을 메틸기로 치환하면 입 체 선택성은 오히려 증가한다는 사실로부터 3- 위치의 N-H 수소는 키 랄고정상과의 수소결합에 참여하고 있다고 가정할 수 있다. 이러한 실 험적 사실에 부합되는 키랄성 인지의 메커니즘은 그림 7-12 와 같다. 그림 7-12 는 (S) -히단토인 방향족기와 키랄고정상 DNB 사이의 7C-7C 상호작용, (S) -히단토인 4- 위치의 카르보닐산소와 키랄고정상 아미드 기의 N- H 수소 사이의 수소결합, 히단토인 3- 위치의 N-H 수소와 키 凡 H \\l/:`o oEIst/§ 그림 7-1。22 N페 ’ 닐글리>신 -DNY。B 키IN랄고 YH정상 ~i\p갭h (S)H -3 +히N단 토c인H 2 사 ) 3이 一 의 부분0 `입o체 이 성질 착물. 그립은 히단토인이- 키랄고정상의 후면으로 접근하여 키랄 고정상과 상호작용을 하고 있음을 나타내고 있다. R 이 클수록 히단토 인은 키랄고정상과 착물을 형성하기에 알맞은 형태를 가진다.
랄고정상의 카르복시 음이온(혹은 공유결합 키 랄고 정성에서는 연 결 아미 드기의 카르보닐 산소) 사이의 수소결합에 의하여 형성되는 순간적인 부분 입체 이성질 착물을 나타낸 것이다. (R) -히단토인의 경우에는 그림 7-12 와 같은 세 자리에서 상호작용이 불가능하거나 힘들기 때문 에 결국 히단토인의 광학분할시 컬럼에 더 오래 머무르는 광학 이성질 체는 그림 7-12 와 같이 키랄고정상과 안정한 부분 입체 이성질 착물 울 형성하는 (S) 홉]단토인으로 예상할 수 있으며 실제로 절대배열이 알려진 히단토인의 머무름 시간과 비교하여 이와 같은 예상의 전위여 부를 판가름할 수 있다. 그림 7-12 와 같은 키랄성 인지의 메커니즘에 서 주의하여야 할 점은 히단토인이 키랄고정상과 상호작용할 때 (면과 면의 상호작용 : fac e-to - fa c e int e r acti on ) 키랄고정상의 양면 (지면의 전 면과 후면) 중 앞면은 덩치가 큰 페닐기가 가로 막고 있을 뿐만 아니라 키랄고정상의 카르복시 음이온과의 수소결합을· 위하여 히단토인은 오 로지 키랄고정상의 후면으로만 접근하여 착물을 형성한다는 것이다. 이때 키랄고정상의 형태는 분자내 수소결합 등으로 인하여 그림 7-12 에 그려진 형태 그대로 유지된다고 생각되고 있다. 그림 7-12 와 같은 형태의 키랄성 인지 메커니즘은 亢-염기성기로 작 용할 수 있는 방향족기와 수소결합 주게로 작용할 수 있는 부위 및 수 소결합 받게로 작용할 수 있는 부위를 가진 라세미 화합물들의 광학분 할에 적용할 수 있다. 그림 7-1 3,'.본 히단토인 등을 포함한 N_ 아릴아 미노에스데르, 2- 카르복시인돌린 등 모두 비슷한 키랄성 인지 메카니 즘에 의하여 광학분할이 되는 것으로 예측되는 라세미 화합물들의 亢 - 염기성기, 수소결합 받게 부위 및 수소결합 주게 부위를 표시한 것이 다. 한편, 프탈리드(p h t ha li de) 계통의 화합물과 같이 T(-염기성기와 수 소결합 받게로 작용할 수 있는 작용기는 가지고 있으나 수소결합 주게 로 작용할 수 있는 작용기를 가지고 있지 않은 라세미 화합물들의 광 학분할에 있어서는 키랄고정상과 분석성분 사이의 상호작용에 T-T(
 2 2
2 2
그림 7-13 1 : 7r- 염기성기. 2 : 수소결합 받게. 3 : 수소결합 주게를 가지고 있어 서 아미노산 -DNB 키랄고정상에서 광학분할이 가능한 라세미 화합물 둘의 예
상호작용과 수소결합 의에 입체반발 상호작용이 주요한 역할을 한다. 그림 7 - 14 는 (R) -페닐글리신 -DNB 키랄고정상 l 에서 1- 나프틸기가 치환된 프탈리드의 광학분할에 대한 키랄성 인지 메카니즘을 제시한 것이다(1 7). 프탈리드의 1_ 나프틸기와 키랄고정상 DNB 사이의 亢-亢 상호작용과 키랄고정상 DNB 아미드의 N-H 수소와 프탈리드의 카르 보닐 산소 사이의 수소결합 이의에 프탈리드의 벤조고리 (benzo rin g) 가 키랄고정상의 커다란 페닐기와 상대적으로 작은 카르복시아미드기 룰 구별하기 때문에 프탈리드는 입체반발이 작은 카르복시아미드쪽으 로 접근이 용이하여 프탈리드의 두 광학 이성질체가 구분되며 결국 (R) -이성질체가 컬럼싱에 더 오래 머무르게 된다. 프탈리드처럼 키랄
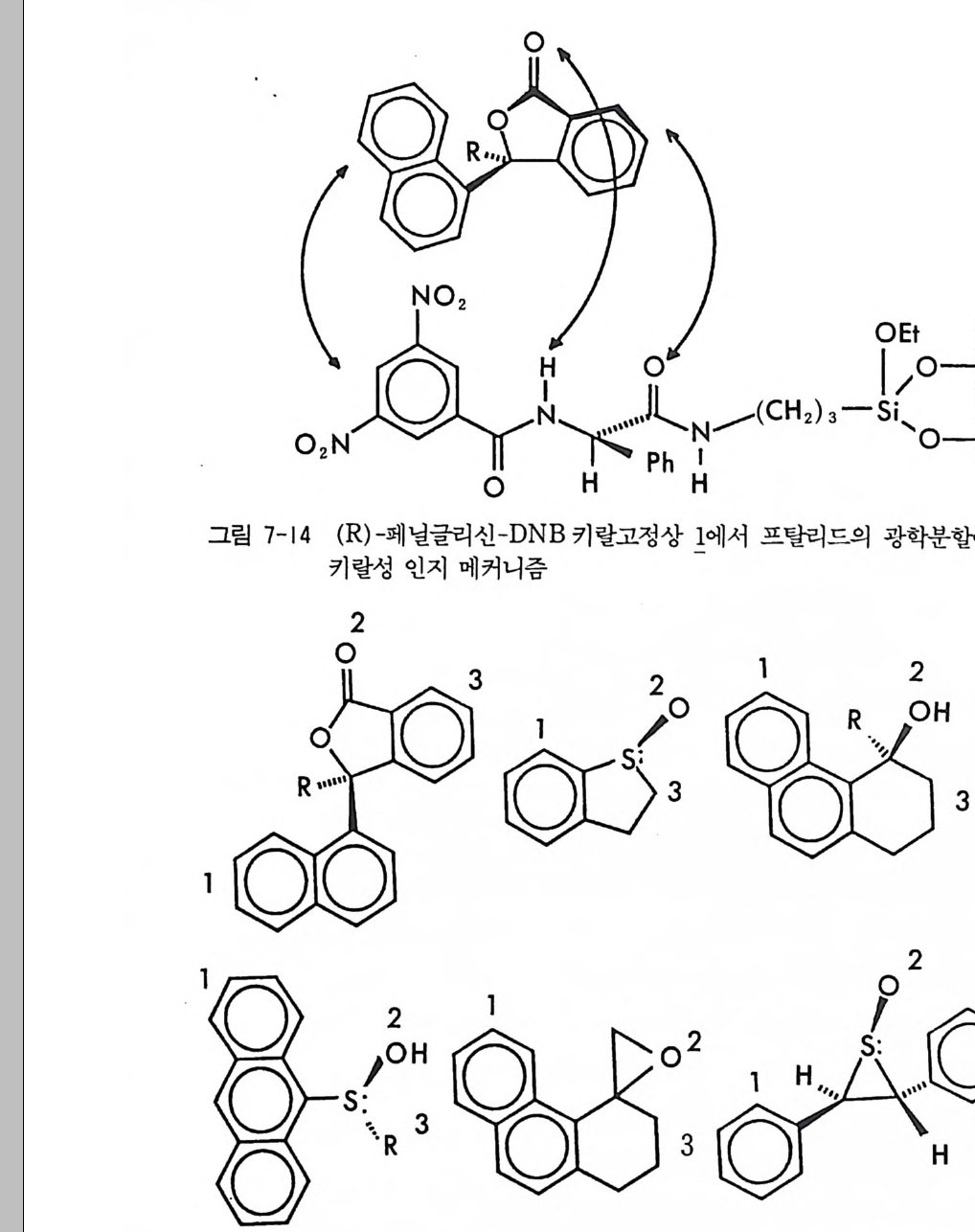 02N 0 H Ph ~
02N 0 H Ph ~
그림 7-'15 1 : 1r- 염기성기, 2 : 수소결합 받게, 3 : 입체장애 역할기를 가지고 있 물어서들 의아예미 노산 -DNB 키랄고정상에서 광학분할이 가능한 라세미 화합
고정상과 광학분할하고자 하는 라세미 화합물 사이의 입체반발 작용이 입체 선택성에 중요한 역할을 하는` 화합물의 예 및 각 부위의 특성은 그림 7-15 .2} 같다. a- 아릴알킬아민 (a-aryl a lky la mi ne ) N- 아실유도체들의 광학분할에 대한 키랄성 인지의 메커니즘은 지금까지 논의된 키랄성 인지의 메커 니즘과는 다르다. 표 7-3 에 의하면 ¢·아릴기의 T- 염기성이 증가하고 알킬기인 R 기의 크기가 커짐에 따라 입체 선택성은 증가하는 반면 N _아실기의 길이가 길어짐에 따라 입체 선택성은 감소하고 있다 (22). 용리순서는 (R) 一페닐글리신 -DNB 키랄고정상 l 에서 항상 (S) -이성 질체가 컬럼에 더 오래 머무론다. 이와 같은 관찰결과에 가장 부합되
표 7-3 (R) -페닐굴리신 -DNB 키랄고정상 l 에서 N- 아실 a- 아릴알킬아민의 광학분할. 이동상 : 2- 프로판올-핵산 ( I 0/90) , 유속 : 2mL/mi n, 용리순서 : 오래 머무르는 이 성질체 (참고문헌 : 22) Ar-CfH -NH-fC -Y Ar R Y a’ kI’ 용리순서 6, 7-(CH1pa-h)N2 e-anlyp- N hl aph CCCHHH333 nnnnCnCCC-----HHH2CCCCCH33&3133u 1sHHHH H171223 33 112111111.........42758437 1010023568 1653658340.........021749773 sssssssss i-C 3H7 CH3 2.40 4.1 s n-C11H23 1.39 1.6 s
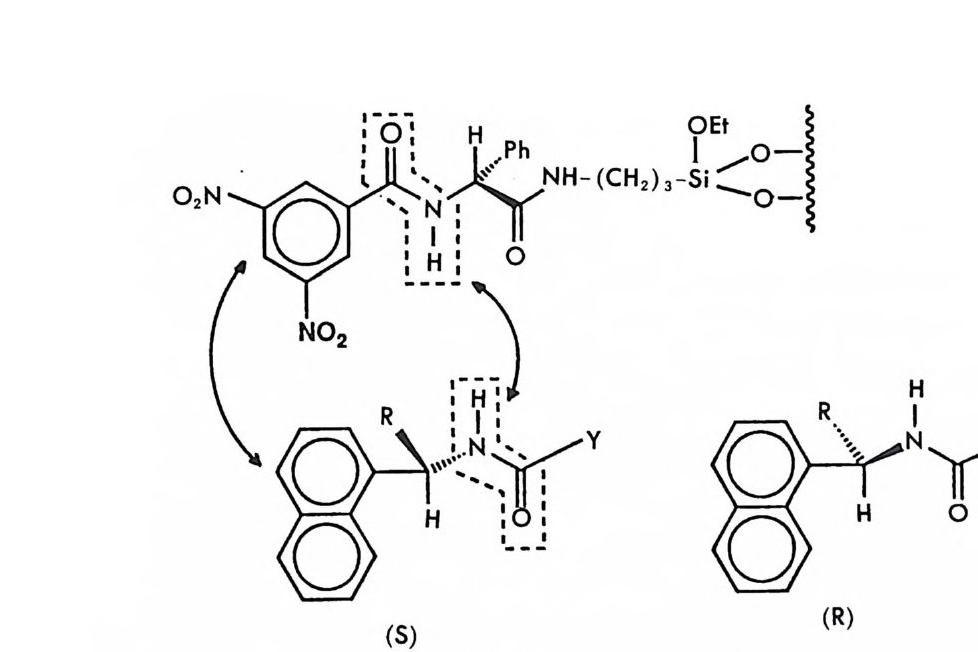 。2 N 闇〔 H IH홉 2 3 oEl t/0- I 了Y
。2 N 闇〔 H IH홉 2 3 oEl t/0- I 了Y
그림 7-16 (R) -페닐글리신 -DNB 키랄고정상 1 에서 N- 아실 a- 아릴알킬아민의 광학분할에 대한 키랄성 인지 메커니좋
는 키랄성 인지의 메커니즘은 모델을 이용한 연구 등의 결과 그림 7-1 詞 같을 것으로 제안되었다 (22). 그림 7-16 에서 N- 아실 a_ 아릴알킬아민의 형태 (con fo rma ti on) 는 R- 기의 입체장애 및 나프틸기의 7- 위치 주변수소 (p er i -hy dro g en) 의 입체장애 효과로 인하여 자유회전이 제한된 것으로 예측되는 구조이 다. N- 아실 a- 아릴알킬아민은 키랄고정싱에 접근할 때 키랄고정상 페닐기의 입체칭애가 없는 지면위로 접근하여 카랄고정상과 상호작용 하리라고 예상된다. 이와 같은 예상하에 키랄고정상과 N- 아실 a- 아 릴알킬아민은 亢 _T 상호작용 및 키랄고정상의 DNB 아미드쌍극자와 분석성분의 N- 아실아미드 쌍극자 사이의 쌍극자켓》 구자겹침에 의해 서 부분 입체 이성질 착물이 형성되며 (S) _이성질체의 경우에는 R 기 가 키랄·고 정상과의 접근면 반대방향에 위치하므로 (R) -이성질체와 비 교하여 더 안정한 부분 입체 이성질 착물을 형성할 것으로 생각되고
이것은 (S) - 이성질체가 컬럼상에 더 오래 머무르는 사실과 부합된다. a- 아릴알킬아민의 a- 아릴기의 7C - 염기성기를 증가시킴에 따라 키랄고 정상과 더 강한 7C-7C 상호작용이 가능하며 키랄중심에 위치한 R- 기의 크기가 커지면 그림 7-l ~ a - 아릴알킬아민의 형태가 더욱 고정될 것 이므로 R - 기의 크기가 커짐에 따라 입체 선택성이 증가할 것을 예측 할 수 있다. N - 아실기의 알킬기인 Y 는 키랄고정상의 연결가지 (connecti ng arm) 와 거의 평형하게 실리카 겔로 향하고 있다. 따라서 Y 의 길이가 증가함에 따라 Y 와 키랄고정상 연결가지 사이의 입체장 애 및 실리카 겔 표면과의 입체장애가 증가할 것이므로 키랄고정상과 N - 아실 a - 아릴알킬아민 사이에 형성된 부분 입체 이성질체 착물의 안정성은 감소하여 입체 선택성이 떨어질 것으로 생각할 수 있다. 따 라서 그립 7 - 16 에 제안된 키랄성 인지의 메커니즘은 광학분할에 대한 표 7 - 3 의 결과를 가장 적합하게 설명하고 있다고 할 수 있다. 7C- 영기 성기를 가지고 있지 않기 때문에 7C- 영기성 유도체로 만들어
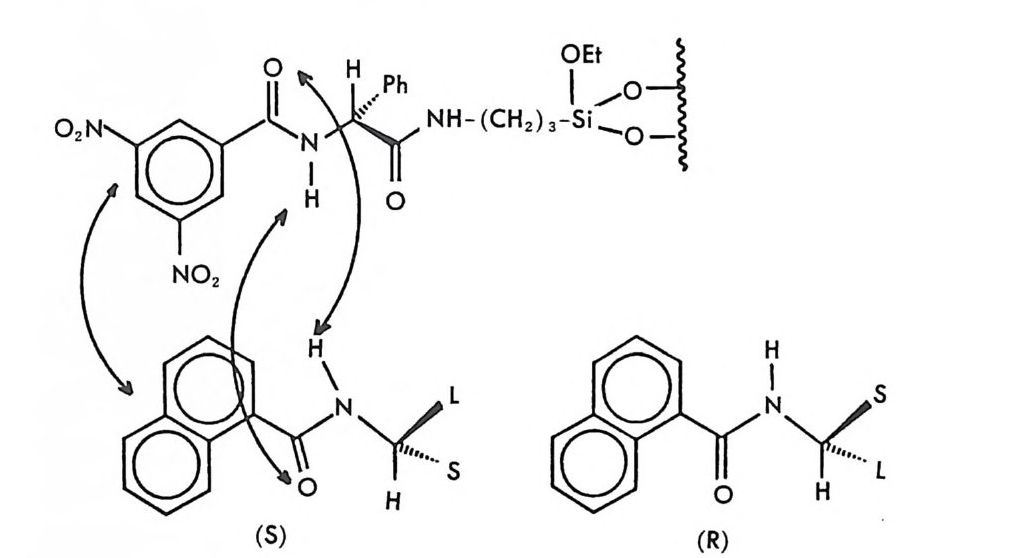 O, N , ~ ./。l l • ••, Ph NH- (CH ,) ,-':니
O, N , ~ ./。l l • ••, Ph NH- (CH ,) ,-':니
그림 7-17 (R) -페닐글리신 -DNB 키랄고정상에서 지방족 아민 1- 나프토일 유도 체의 광학분할에 대한 키랄성 인지 메커니즘
아미노산 -DNB 키랄고정상에서 광학분할된 라세미 화합물들에 대한 광학분할의 메커니즘도 비슷하게 설명할 수 있다. 예를 들어 지방족 아민의 l- 나프토일 유도체를 아미노산 - DNB 키랄고정상에서 광학분 할 경우에 그립 7-17 에서 보는 바와 같이 7C- 7 C 상호작용 및 두 위치에 서의 수소결합(혹은 비껴 서 있는 쌍극자끼리의 쌍극자-씽극자 겹침)의에 (S) -이성질체의 경우 입체장애가 작은 S- 기가 키랄고정상방향으로 향 하여 입체반발을 덜 받기 때문에 (S) ― 이성질체가 더 안정한 부분 입 체 이성질 착물을 형성하여 두 광학 이성질체가 구분된다 (34).
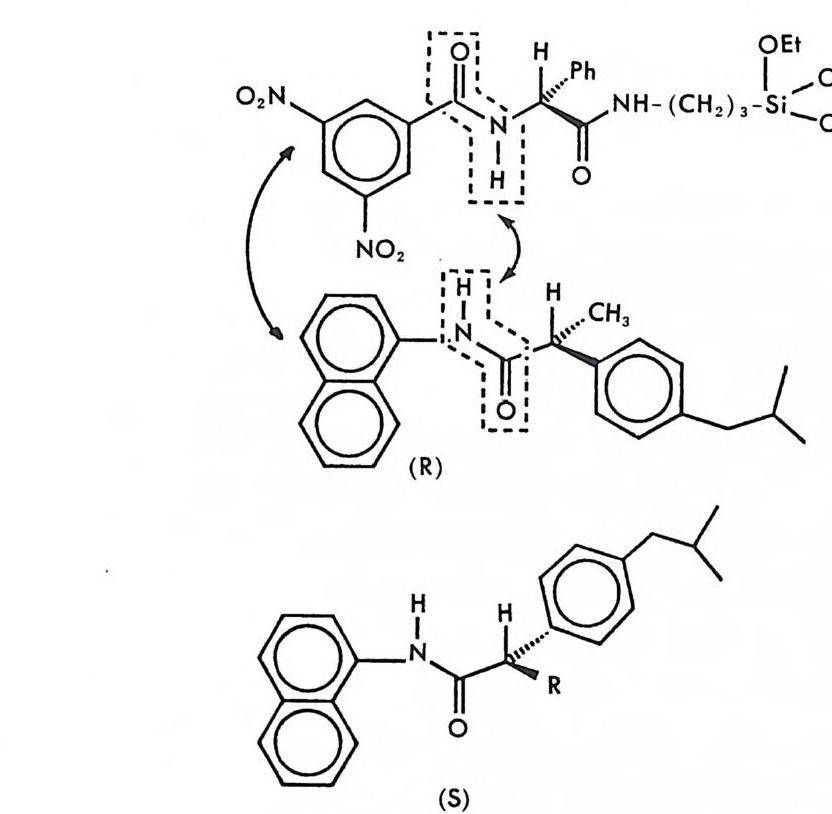 \0二 I
\0二 I
그림 7-18 (R) -페닐글리신 -DNB 키랄고정성에서 이부프로펜 1- 나프틸아미드 유도체의 광학분할에 대한 키랄성 인지 메커니즘
a- 아릴프로피온산의 광학분할에 대하여서는 그동안 여러 가지의 키 랄성 인지 메커니즘이 제안되었으나(1 8, 19, 43 , 44 ) 그림 7-1~ 쌍극자 - 쌍극자 겹침메커니즘이 a - 아릴프로피온산 유도체들의 광학분할에 대 한 실험결과를 가장 적절히 설명할 수 있는 것으로 생각된다 (20,49). 그림 7- 1 ~ 씽국자-쌍극자 겹침 메커니즘에 의하면 (R) -이성질체가 (S) - 이성질체보다 더 안정한 착물을 형성하므로 키랄고정상에 더 오 래 머무르게 된다.
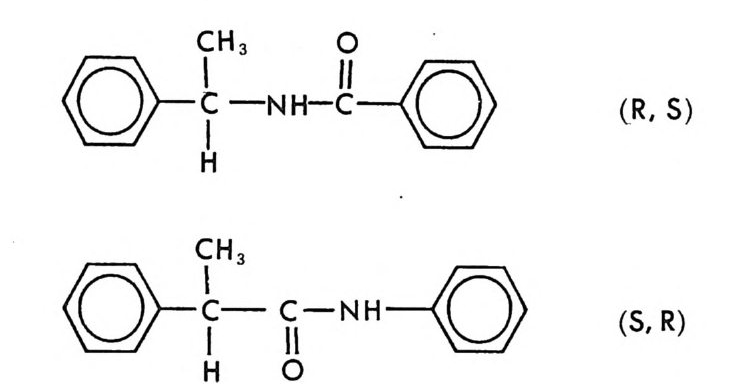 CH3 0
CH3 0
그림 7-19 (R) -페닐글리신 -DNB- 키랄고정상에서 아민 유도체 및 카르복~산유 도체의 광학분할에 대한 용리순서의 처이. (R,S) 는 (R) -이성질체가 먼 저 분리되어 나옴을 의미함.
Wain e r 등은 페닐에틸아민의 벤조일 유도체와 a- 페닐프로피온산 의 아닐리드 유도체를 광학분할할 때 용리순서가 그림 7-19 와 같이 서로 반대가 되는 것은 분석성분의 아미드 쌍극자와 키랄고정상의 아 미드 쌍극자 사이의 상호작용에는 방향성이 있기 때문이라고 생각하였 으며 정확한 메커니즘을 제시하지 못하였다 (35). 그러나 아민유도체의 광학분할에 대한 그림 7-17 의 키랄성 인지 메커니즘 및 a- 아릴프로피 온산 유도체의 광학분할에 대한 그림 7-1~ 키랄성 인지 메커니즘을 이용하면 그립 7-19 의 두 화합물의 광학분할에 대한 용리순서의 차이 점울 쉽게 설명할 수 있을 것이다 .
3.4 (R) -페닐글리선 -DNB 키랄고정상에 의한 제조목적의 광학 분할 (R) 페닐글리신 -DNB 키랄고정상이나 (S) 유머신 - DNB 키랄고정 상은 제조과정이 쉬울 뿐만 아니라 (R) -페닐글리신 혹은 (S) 추유이신 은 상업적으로 쉽게 구할 수 있기 때문에 이들 키랄고정상들을 다량으 로 제조하는 것은 그리 어려운 일이 아니다. 따라서 이들 키탈고정상 에서 광학분할이 가능한 모든 라세미체들은 많은 양의 키랄고정상이 충진된 용량이 큰 컬럼을 이용할 경우(보통 HPLC 시 스 템 대신 MPLC 시스템을 사용함) 한 번에 많은 양의 라세미 화합물 들을 광학분할할 수
 (a) c,~)
(a) c,~)
그림 7-20 (R) 대닐글리신 -DNB 키랄고정상 2 에서 라세미 벤조디아제피논의 a) 분석용 광학분할 및 b) 제조용 광학분할• a) 이동상 : 2- 프로판올-핵 산 (10 /90) , 유속 : 2mL/mi n, b) 이동상 : 2- 프로판을-핵산 (5/95) , 유속 : 5mL/mi n. (참고문헌 : 16)
있으며 따라서 짧은 시간내에 광학적으로 순수한 두 광학 이성질체를 동시에 얻을 수 있다. 이론적으로, 분석용 컬럼에서 광학분할이 가능 한 모든 라세미 화합물들은 제조용의 큰 컬럼을 이용하여 광학분할할 수 있으나 광학분할을 효과적으로 하기 위하여서는 a 값이 클수록 제 조목적의 광학분할이 용이해전다는 사실에 유의할 필요가 있다. 예를 들면 그림 7-20 혼는 250 x 4.6mm 의 컬럼에 충전된 (R) -페닐글리신 -D NB 키랄고정상 1 에서 라세미 벤조디아제피논의 분석용 광학분할을 나타낸 것으로서 a 값이 충분히 크기 때문에 (a = 1. 93) 그림 7-20 !2에 서 보는 바와 같아 크기가 250 x 10mm 인 컬럼에 충전된 키랄고정상 1 에서 104m g의 라세미 벤조디아제피논이 거의 완전한 기준선 분리에 의하여 광학분할됨을 확인할 수 있다(1 6). 표 7-4 는 그동안 제조목적으로 광학분할이 이루어진 광학분할의 예 를 제시한 것이고 그림 7 - 21 은 표 7- 유] 화합물 중 히단토인의 제조
 H 0 (R)
H 0 (R)
그림 7-21 (R) -페닐글리신 -DNB 키랄고정상 笠에서 히단토인의 제조목적 광학분 할 컬럼크기 : 2x30in , 시 료크기 : 1.2g , 이동상 : 2- 프로판을-핵산(1 9 / 90) , 유속 : 60mL/mi n, 검출기 : 254nm UV (참고문헌 : 51)
표 7-4 (R) -패닐글리신 -DNB 키랄고정상 3 에서 제조목적의 광학분 할 라세미 화합물 크시기료 a’ 컬럼크기’ 맡이동:상(로핵산 I 麟 。 Me RR == C( CHH3 , ). C H=CH, I14 ..0103gg 21..06 08 z2m i.n.. XX 33 00 mi. n.. I 102 5532 RR ==C (CHHJ 2 h CH= C H2 I 11..60 00 gg 11..5375 22 mi.n.. xX 33 0Q im. n.. I 1100 I 5511,, 5522 l.OOg 1.86 Z in. X 30 in. I 5 I 52, 53 吳 CFaCHOH R=H 1.00 g 1.46 2in . x30i n. 2 53 R= CH3 l.OO g 1.47 Zin . X30i n. 5 52, 53 R = CH3 2 . 00g 1. 47 2 in. X3 0 in. 5 52, 53 : R
二H_c` 、 NR H ?C ( CH2 ) 8 CH = CH2 RR == 시CH클(로CH핵3실)2 1j 73 .. 0O0Ogg 1I 22 .. 2137 I1 22 iinn.. xx 3300 !inn.. 21 5353 。 N )_CH_C3 O (CH2 ) 。 CH = CH2 I 100g I I 6 in. x 4 ft. I 10 I 54 H II 。 OH CCH0H33 0 :OCH, (:IH3 l700mg l I lOmmx 50cm I 10 I 55 Si + CI H3
목적 광학분할에 대한 크로마토그램의 예를 제시한 것이다. 표 7-4 에 제시한 제조목적의 광학분할 중 특히 홍미있는 광학분할은 라세미 N -(2- 나프틸)알라닌 에스데르의 광학분할로서 싱품·화되어 있는 (R)- 페 닐글리신 -DNB 키랄고정상 13k g를 4 ft x6 i nch( l. 2m x 15.2cm) 인 컬 럼에 충전하여 한 번에 100 g의 라세미 화합물을 광학분할할 수 있음 울 확인할 수 있다. 이 예에서 보는 바와 같이 용량이 큰 컬럼을 사용` 하여 다량의 라세미 화합물을 한 번에 광학분할할 수 있기 때문에 이 와 같은 제조목적의 광학분할은 앞으로 라세미 의약품의 광학분할에 유용하게 쓰일 수 있을 것으로 기대된다. 페닐글리신 -DNB 키랄고정상이나 루이신 -DNB 키랄고정상은 제조
과정이 아주 쉽기 때문에 플래쉬 컬럼 크로마토그래피 (flas h column chroma t o g ra p h y)에 의한 광학분할에도 응용이 가능하다. 예를 들면, 58µm 의 실리카 겔에 (R) -페닐글리신 -DNB 가 고정된 키랄고정상 120 g을 150x40mm 의 플래쉬 컬럼에 충전하여 플래쉬 크로마토그래 피를 실시함으로써 200m g의 벤조디아제피논이 광학분할된 바 있다 (56) . 3.5 다른 종류의 r 국 R 성 키랄고정상 페닐글리신 -DNB 혹은 루이신 -DNB 를 기저로 한 키랄고정상 이의 에 다른 아미노산들의 DNB 유도체들을 실리카 겔에 고정시켜 키랄고 정상으로 이용한 예들을 다수 찾아볼 수 있다. (R)-N- (펜타플루오르벤조일)페닐글리신을 아미노프로필 실리카 겔 에 이온결합시킨 키랄고정상 인은 (R) -페닐글리신 -DNB 키랄고정상 劉 DNB 대신 펜타플루오르벤조일기가 r- 산성기로 작용하는 키랄고 정상으로서 (R) -페닐글리신 -DNB 키랄고정상 g에 의한 라세미 화합 물의 광학분할과 비교하여 크게 중전된 것은 없으나 질소를 포함하는 라세미 화합물을 광학분할할 경우에는 키랄고정상 앞i디는 키랄고정 상 원에서 광학분할이 다소 증진된 것으로 확인되었다 (57). (R)-N-(3 , 5- 디니트로벤조일 )-1- 나프틸글리신을 아미노프로필 실
 巨- E(t C H2)3_N 』- r-NH 『* F
巨- E(t C H2)3_N 』- r-NH 『* F
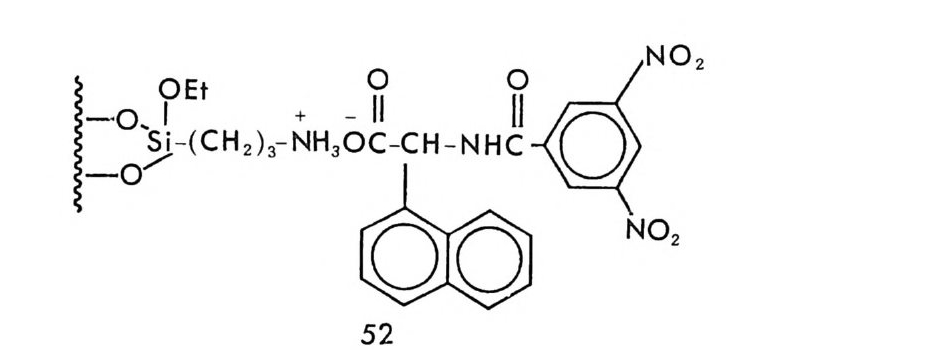 Q soi/_ Et`C H( 2 ) 3 NH 3 。 〉NO2 O2
Q soi/_ Et`C H( 2 ) 3 NH 3 。 〉NO2 O2
리카 겔에 이온결합시킨 키랄고정상 盤는 페닐글리신 -DNB 키랄고정 상 합의 페닐기대신 덩치가 큰 나프틸기를 키랄중심에 가지고 있기 때 문에 3.3 절에서 논의한 키랄고정상의 형태 (con fo rma ti on) 를 더 견고 히 유지할 것이므로 페닐글리신 -DNB 키랄고정상 얗i다 더 좋은 입체 선택성을 보일 것으로 기대된다 (29). 실제로 많은 종류의 라세미 화합 물들의 광학분할에 있어서 나프딜글리신 -DNB 키랄고정상 쌀가 페닐 글리신 -DNB 키랄고정상 g보다 더 좋은 입체 선택성을 보이는 것으로 확인되었다 (58). 그림 7-22 는 키랄고정상 g과 키랄고정상 盤상에서 펜프로파트린 (f en p ro p a t hr i n) 의 광학분할을 비교한 것으로써 키 랄고정 상 g상에서 두 광학 이성질체가 어느 정도 겹쳐 분리되는 반면 키랄고 정상 迎상에서는 완전한 기준선 분리가 이루어지고 있음을- 확인할 수 있다. 티로신(tyr os i ne)-DN B-를 실리카 겔에 결합시킨 키랄고정상 th io -DNBTy r -E 완 혹은 th io -DNBTy r -A ~는 아미노산의 카르복시 기를 통하여 실리카 겔에 결합된 다른 아미노산 _DNB 키랄고정상들과 는 달리 티로신의 p-히드록시기를 통하여 실리카 겔에 결합된 키랄고 정상으로서 최근 Caude 등에 의하여 연구보고되었다 (48,58). 특히 (S)-th i o -DNBTy r -A 완는 키 랄화합물인 t유 L 틸 N- 아릴술피나모일 에스테르(t er t -bu ty l N-aryl s ulph ina moy l es t er) 의 광학분할에 있어서 (R) -페닐글리산 DNB 키랄고정상 l 보다 월등한 입체 선택성을 보이 는 것으로 확인되었다. 전형적인 크로마토그램의 예는 그림 7-23 과
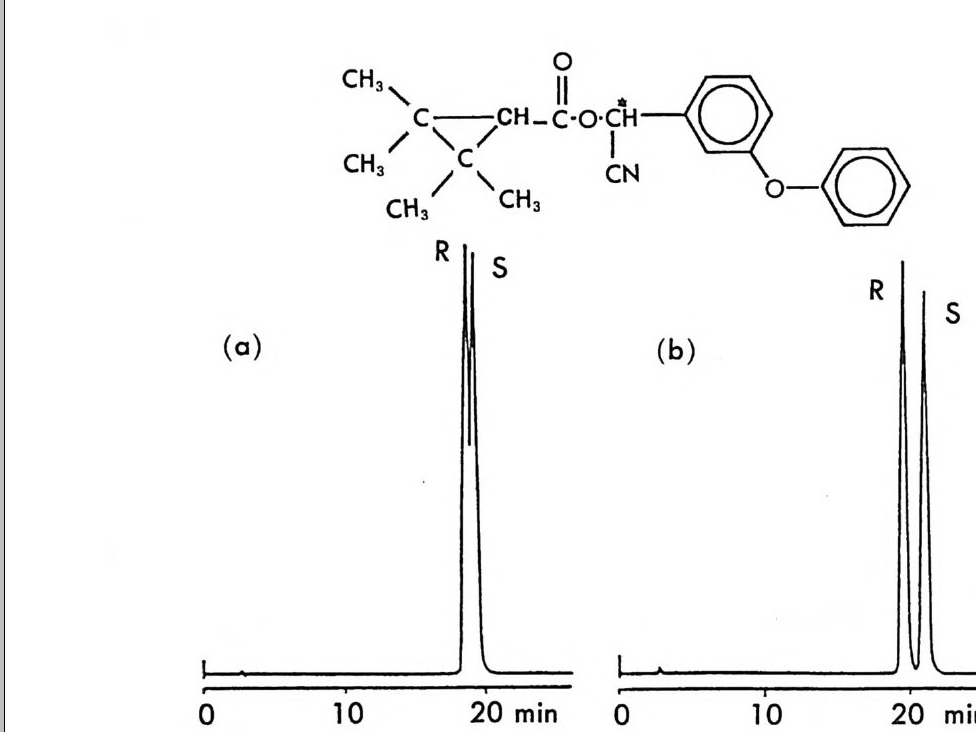 CCHH33 C'/-CH. 3 /: CC H\ C-HC3 。· 0 ·fC-*NH - QO o_ ( 0)
CCHH33 C'/-CH. 3 /: CC H\ C-HC3 。· 0 ·fC-*NH - QO o_ ( 0)
그림 7-22 a) 키랄고정상 ~ b) 키랄고정상 쁘에서 펜프로파트린의 광학분할. 이동상 : 핵산 -1, 2- 디클로로에탄-에 탄올 (500 : 20 : 0.1), 유속 : lmL/mi n (참고문헌 : 29)
같다. 그립 7-23 에서 보는 바와 같이 N-( p-트리플루오르메틸페닐)술 피나모일의 t냐맨에스데르는 (R) -페닐글리신 -DNB 키랄고정상 l 에 서 전혀 광학분할되지 않는 반면 (S)-th io -DNBTy r- A ~상에서는 입체 선택성이 아주 좋음을 알 수 있다 (a= 1. 97). 그림 7-23 에서 보 게 되는 입체 선택성의 큰 차이는 아미노산 -DNB 가 실리카 겔에 결 합될 때 연결가지 (connec ting arm) 의 방향성과 밀접한 관계를 가질 것으로 생각된다. (R) -페닐글리신 -DNB 키랄고정상 l 과 술피나모일 에스테르가 亢국상호작용 및 수소결합 혹은 쌍극자켓戶f자 상호작용 에 의하여 서로 접근할 때 분석성분의 t-lj녀i에스테르기는 키랄고정상 의 연결가지 혹은 실리카 겔 표면과 심한 입체장애를 일으킬 것으로
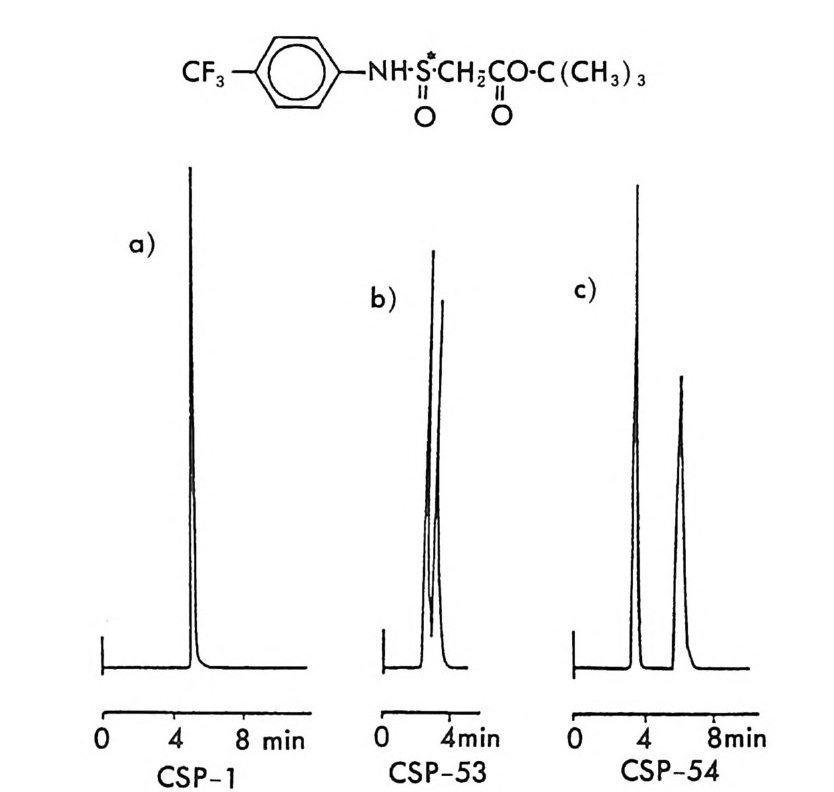 CF3 -0- NH 휴· ·CH 유 ?·C (CH3) 3
CF3 -0- NH 휴· ·CH 유 ?·C (CH3) 3
그림 7-23 키랄고정상 1, 53, 및 54 에서의 광학분할의 비교. a) 이동상 : 핵산 쿨로후璃(8 0/' 20)' 유족 : 2mL/mi n, 검출기 : 260nm UV. b) c) 이동상 : 핵산죽문로로포름 (50/50) , 유속 : 2mL/mi n, 검출기 : 260nm UV (참고문헌 : 59) .
예상되는 반면 (S)- t h i o-DNBT yr -A 키랄고정상 완에서는 연결가지 가 다른 방향으로 향하고 있으므로 이와 같은 입체장애를 받지 않을 것으로 예상된다. 따라서 키랄고정상 왼는 N- 아릴술피나모일에스데 르와 더 강한 상호작용을 하여 더 좋은 입체 선택성을 보이는 것으로 생각할 수 있다. 또한 (S)- thi o-DNBT y r-E 키랄고정상 왑에 비하여 (S)-th io -DNBTy r- A 키랄고정상 완는 분석성분과의 수소결합에 참여할 수 있는 아미드 -N-H 기를 하나 더 가지고 있으므로 분석성분
과 더t 강00 `한/O 상Eclt$호H 2 작}3용-Ss을-)1,,( c -t 하OH 2h 여) 3 -더NO T 좋은 > r 입체 c 선H 2 택 CH성1C을CoOHIJ II IO보NH 이C 는區 것으로 생 각할수있다. 仁:\\卞 S·- (CHD 2)B 3 -Yo 를E 5 3 C H2\HN.HN:〔 :Q:g二:: (S)-th i o - DNBTy r- A 54
아미노산 -DNB 유도체를 기저로 한 키랄고정상 이의에도 7r:-산성 키랄고정상들은 다양하다. 술폭시드 (sul fo x i de) 혹은 셀렌옥시드 (selenox i de) 의 광학분할에 응용된 키랄고정상 쭙는 (R, R)- 1, 2- 디아 미노시클로핵산과 Y- 글리시독시프로필 실리카 겔울 반웅시킨 후 염화 3, 5- 디니트로벤조일 (DNBCl) 을 처리하여 제조되었다 (59).
 \二 `,,,.1 iE -
\二 `,,,.1 iE -
키랄고정상 쳅은 f3 - 아미노산 - DNB 를 실리카겔에 공유결합시켜 얻 은 키랄고정상으로서 아주 성공적인 亢-산성 키랄고정상 중의 하나이 다 (60). P i rkl 퓰든 f3-락탐 완을 p냇 H 떳!술폰산 존재 하에서 10 쉽든데 센 -1 국춘(l O-undecen-1-ol) 과 반응시켜 f3-아미노에스데르를 얻은 후 이 에스데르를 DNB 유도체로 만들고 亢-염기성 키랄고정상을 이용하 여 광학분할한 후 히드로실릴화 반응 및 실리카 겔과의 결합 반응을 통하여 키랄고정상 원을 합성할 수 있었다.
 三: 二 N 〈:jh5t 7 _:0(CH2) 『\ o 」
三: 二 N 〈:jh5t 7 _:0(CH2) 『\ o 」
키랄고정상 인은 페닐글리신 -DNB 키랄고정상 l 에 비하여 크기가 큰 이웃한 t숙녀 l 기 및 페닐기 때문에 더 고정된 형태를 가지게 되며 따라서 프탈리드 헤데르고리 아민의 1- 나프토일 유도체, O- 아실페닐 알킬카르비놀 등의 광학분할에 있어서 페닐글리신 -DNB 키랄고정상 보다 훨씬 좋은 입체 선택성을 보임이 확인되었다 (60). 그림 7-24 는 키랄고정상 연에서 a-(1- 안트릴)에틸아민 N- 아실유도체의 광학분할 에 대한 크로마토그램을 제시한 것이다. 키랄고정상 연에 존재하는 두번째의 키랄중심 (t부될기가 결합된 키 랄중심)은 키랄성 인지 메커니즘에 특별한 작용을 하지 않고 다만 분 석성분과의 중요한 상호작용 부위로 작용할 수 있는 연결에스데르기의 방향을 조절하는 역할을 하고 있울 것으로 생각되고 있다 (60). 키랄고정상 완과 완는 최근 저자 등에 의하여 제조된 키랄고정상으 로서 N-(3,5- 디니트로벤조일 )-(1S, 2R) -노르에페드린을 실리카 겔에 공유결합시킨 것이다 (61,60). 노르에페드린은 히드록시가 결합된 탄소
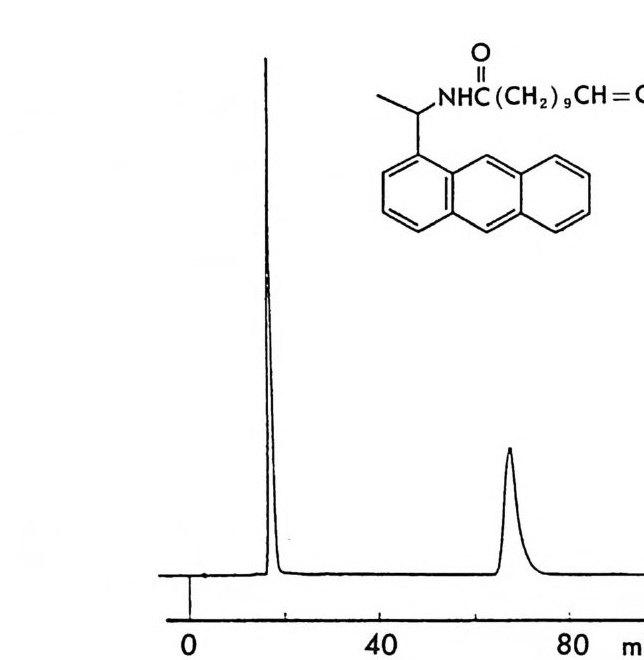 NH?C ( CH2) 9CH = CH2
NH?C ( CH2) 9CH = CH2
그림 7-24 키랄고정상 57 에서 a-(1- 안트릴)에틸아민 N- 아실유도체익 광학분할. 。듐장 : 2- 프로판을-핵산 (20/80) ,유속 : lmL/mi n (참고문헌 : 60)
와 아미노기가 결합된 탄소사이의 회전에 대한 낮은 에너지 장벽 때문 에 그 형태가 유연한 것으로 알려져 있으며 따라서 (1R, 2R) -노르에 페드린을 기저로 하여 제조된 키랄고정상 왕과 완 또한 유연한 형태 롤 가질 것으로 생각된다. 분자내 수소결합때문에 고정된 형태를 갖는 것으로 생각되어지는 페닐글리신 -DNB 키랄고정상 l 보다 더 고정된 형태를 가지는 것으로 생각되는 키랄고정상 笠이 많은 경우의 광학분 할에 있어서 페닐글리신 -DNB 키랄고정상 l 보다 더 좋은 입체 선택성 울 보이는 반면 유연한 형태를 가지는 것으로 생각되는 키랄고정상 완 과 완는 라세미 화합물의 형태 (co nf orma ti on) 에 따라 페닐글리신 -DNB 키랄고정상 1 과는 다른 입체 선태성을 보인다는 사실이 확인되 었다. 그림 7-2 닭근 페닐글리신 -DNB 키랄고정상 l 과 키랄고정상 햄
 ..-o...O NI HIc C H_2 ) 3 oSEi t- /o\;o'b' h; i ',t 0II OI Et~ O-
..-o...O NI HIc C H_2 ) 3 oSEi t- /o\;o'b' h; i ',t 0II OI Et~ O-
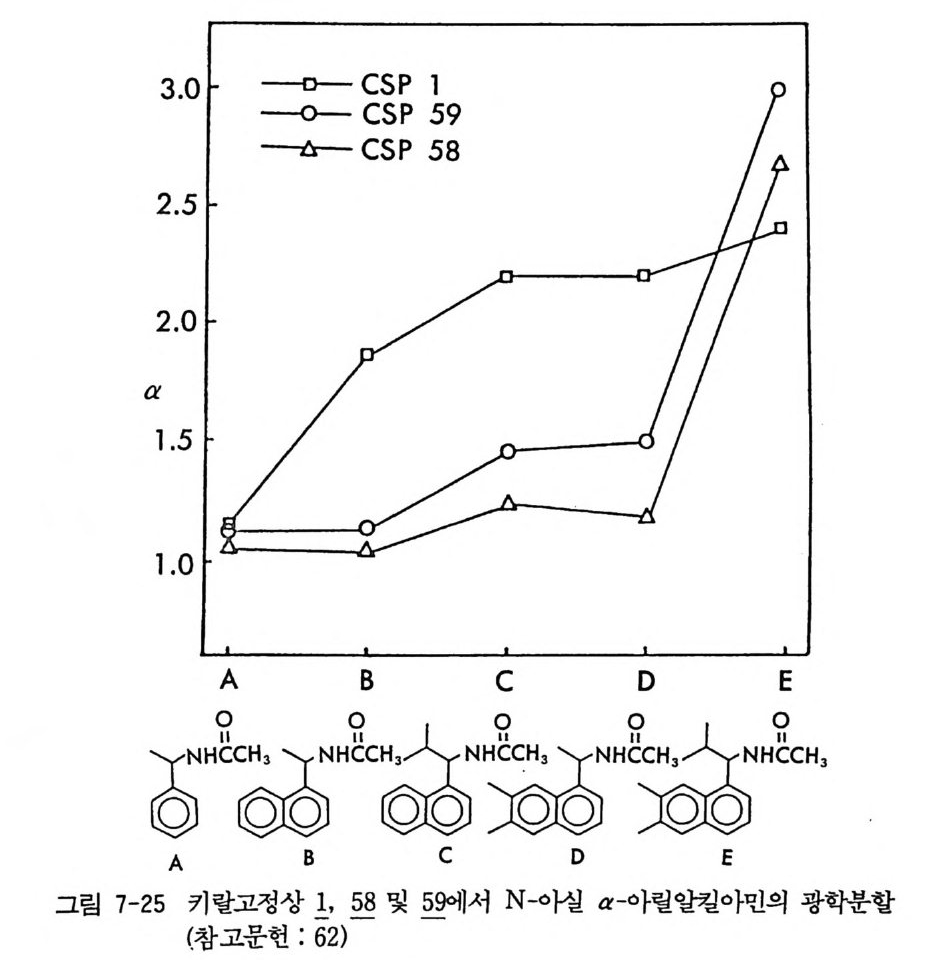 3.0t- 조 CSP 1
3.0t- 조 CSP 1
및 완상에서 N~ 아실 a- 아릴알킬아민의 광학분할에 대한 a 값을 도시 한 것으로써 다소 유연한 형태 (con fo rma ti on) 를 가지는 라세미 화합 물들은 고정된 형태를 가전 키랄고정상 l 에서 광학분할이 더 잘되는 반면 나프틸기의 주변수소(p er i -h y dro g en) 와 이소프로필기의 큰 입체 장애때문에 고정된 형태를 가진다고 생각되는 라세미 화합물 E 는 유 연한 형태를 가진다고 예상되는 키랄고정상 완과 쳄상에서 더 좋은 광학분할을 나타내고 있다 (62). 이와 같은 사실은 키랄고정상과 라세 미 화합물 사이의 세 점 상호작용(제 2 장 참조)에 미치는 키랄고정상의 형태 및 라세미 화합물 형태의 영향을 고려함으로써 설명이 가능하다. 죽 고정된 형태를 가지는 키랄고정상은 세 점 상호작용에 필요한 세 개의 상호작용 부위를 고정된 위치에 가지고 있기 때문에 키랄고정상 의 세 개 상호작용 부위에 대응하는 세 개의 상호작용 부위를 정확한 위치에 갖고 있는 라세미 화합물은 키랄고정상과의 상호작용에 별 문 제가 없으나(이와 같은 경우는 극히 제한적인) 세 점 상호작용 부위를 정확한 위치에 갖고 있지 않는 라세미 화합물은 키랄고정상과 세 점에 서 상호작용하기 위하여 그 형태를 변형하여야 할 필요가 있다. 따라 서 유연한 형태를 가전 라세미 화합물은 쉽게 형태를 변형시킬 수 있 기 때문에 고정된 형태를 가진 키랄고정상에서 광학분할이 잘될 것으 로 생각할 수 있다. 마찬가지로 유연한 형태를 가진 키랄고정상은 쉽 게 형태를 변형시킬 수 있어서 고정된 형태의 라세미 화합물과 세 점 에서 상호작용할 수 있다. 따라서 결론적으로 고정된 형태의 키랄고정 상은 유연한 형태의 라세미 화합물 광학분할에 유리하며 유연한 형태 의 키랄고정상은 고정된 형태의 라세미 화합물 광학분할에 유리하리라 고 생각할 수 있다 (62). 그러나 이 결론은 극히 논리적인 것으로서 키 랄고정상과 라세미 화합물의 형태 이의에 키랄고정상과 라세미 화합물 사이에 상호작용하는 힘의 크기, 입체장애때문에 생기는 두 분자(키랄 고정상 및 라세미 화합물)의 접근의 용이성 등 많은 요스계} 고려하여 입 체 선택성을 논의할 필요가 있다.
3.6 광학분할에 대한 이동상의 영향 아미노산 -DNB 키랄고정상 혹은 7C - 산성 키랄고정상에서 광학분할 에 주로 사용되는 이동상은 n_ 핵산과 2- 프로판올 (0 . 1-20% )을 적절히 섞어 극성을 조절한 혼합용매이다. 핵산과 2- 프로판올의 혼합용매 대 신 핵산과 다른 용매의 혼합용매를 사용하여 7C- 산성 키랄고정상에서 광학분할한 예는 디수 있으나 (63) 혼합용매 중의 2- 프로판을 대신 수 소결합 양성자 받게로서의 능력이 2- 프로판올과는 디른” 알코올이나 혹은 수소결합의 양성자 주게로서 작용하는 용매 혹은 강한 쌍극자 역 할을 하는 용매를 사용함으로써 키랄고정성의 입체 선택성 뿐만 아니 라 분석성분의 머무름 시간에 대한 이들 용매들의 영향을 자세히 연구 한 것은 Z i e f와 Tambute 등이다 (27,64). 이동상의 조성에 따른 입체 선택성 및 머무름 시간의 변화는 키랄고 정상과 이동상 사이 상호작용 및 키랄고정상과 분석성분 사이 상호작 용의 경쟁에 의하여 결정된다고 할 수 있다. 표 7- 았근 여러 종류의 알 코올과 클로로포름 및 디클로로에탄올 핵산과 적절히 혼합하여 (R) 페닐글리신 - DNB 키랄고정상 l 에서 메틸 (9 대난트릴)페닐포스핀옥 시드의 머무름 시간 (k2 ' ) 을 거의 일정하게 유지하였을 때의 a 값 및 이 동상의 조성을 나타낸 것으로서 입체 선택성이나 머무름 시간은 이동 상의 극성에 거의 무관하고 입체 선택성은 사용한 용매의 구조와 밀접 한 관계를 가지고 있음을 나타내고 있다. 이동상의 알코올은 그림 7 -26~ 에서 보는 바와 같이 키랄고정상과 두 점에서 수소결합을 형성하 며 (알코올은 수소결합 양성자 받게 및 양성자 주게의 역할을 동시에 할 수 있다) 알코올 R- 기의 크기가 클수록 입체장애로 인하여 고정상과의 상호작용의 세기는 약해지고 따라서 분석성분은 그만큼 쉽게 키랄고정 상과 결합하고 있는 알코올을 치환하여 그림 7-26~ 같은 착물을 형 성한다. 따라서 똑같은 조성의 핵산과 알코올의 혼합용매를 이동상으 로 사용할 때 알코올의 R 기가 커질수록 분석성분의 머무름 시간은
표 7-5 (R) -페닐글리신 -DNB 키랄고정상 l 에서 메틸 (9- 페난트릴) 페닐포스핀옥시드의 광학분할에 대한 이동상의 영향(참고문현 : 27) 이동상(핵산+용매) 핵산중의 터용매% P'* kz ' a’ Xe* eth a nol 0.52 10 0.52 10. 6 1.37 n-buta n ol 0.59 18 0.78 9.3 1.39 2-pr o p a nol 0.55 20 0.86 10.6 1.45 t-b uta n ol 0.56 50 2.10 9.9 1.47 Xd* CHCl3 0.41 55 2.30 10. 6 1.50 Xn* 1, 2-dic h loroeth a ne 0.49 85 2.9 9 9.9 1.33 * p' :용매의 극성인자, x •. xd, Xn 은 용매의 선택성 파라미터로서 각각 수소 결합 양성자 받게, 수소결합 양성자 주게, 쌍극자로서의 성질을 나타내 는 값이다. 따라서 알코올은 양성자 받게、 클로로포롬은 양성자 주게, 1, 2 쿨로로에탄은 쌍극자로서의 성질을 나타낸다고 할 수 있다. 자세한 정의는 참고문헌 63 및 6 톄卜 참조할것.
길어지고 입체선택성은 증가하게 된다. 표 7-5 에 의하면 똑같은 머무 롬 시간을 유지하기 위하여 사용하여야 할 t국냐묘匡의 농도는 에탄올 에 비하여 무려 5 배나 증가하고 있음을 알 수 있다 (27). 한편 수소결 합 양성자 주게로만 작용하는 클로로포름은 그립 7-26 ~와 같이 키랄 고정상과 한 점에서 수소결합을 하며 분석성분에 의하여 쉽게 치환이 가능하므로 키랄고정상과 분석성분 사이에 상대적으로 강한 상호작용 울 예측할 수 있다. 따라서 키랄고정상에 더 오래 머무르는 이성질체 의 머무름 시간 (k2' )을 표 7-5 에서 보는 바와 같이 10 . 終녁 1 유지하기 위하여 에탄을/핵산 혼합용매의 조성은 10/90 인 반면 클로로포름/헥 산 혼합용매의 조성은 55/45 까지 클로로포름이 양이 증가하고 있음을 확인할수있다.
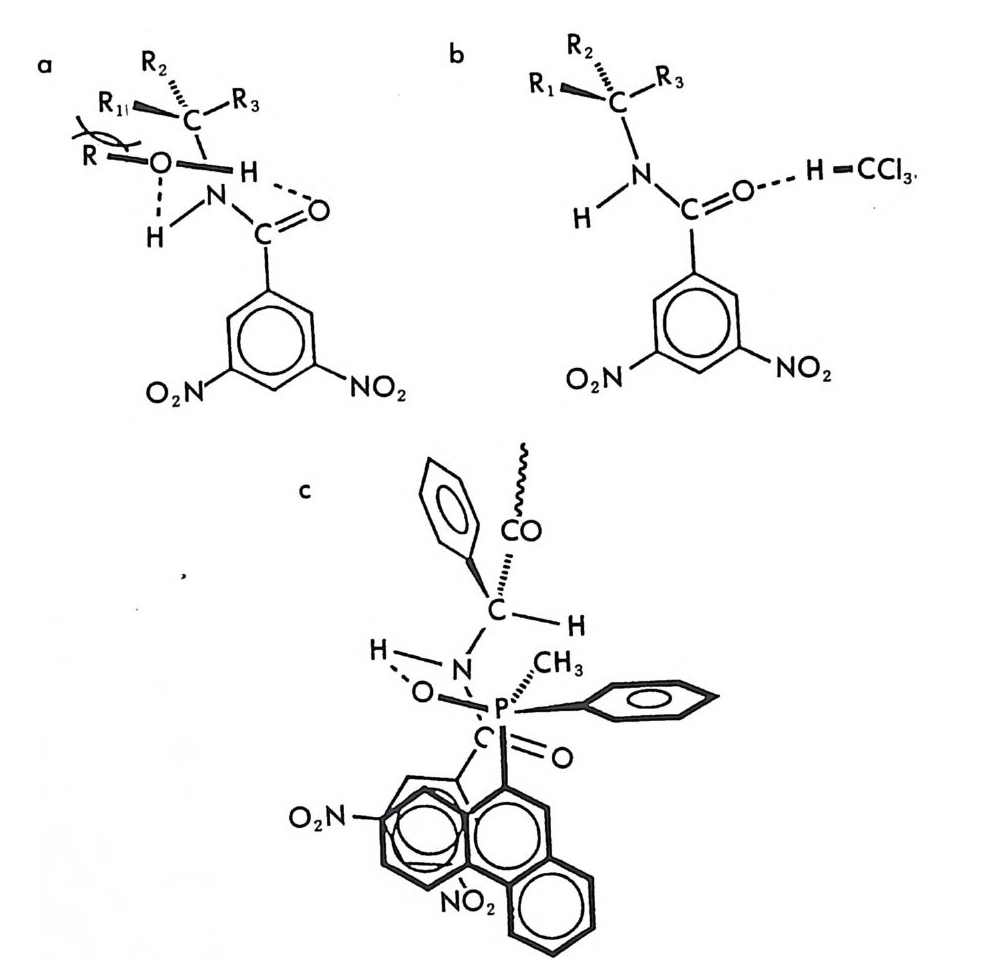 a R11 -R. .2 .. :\:.,,,.,R3 b R1~R2C ,.,.,R3
a R11 -R. .2 .. :\:.,,,.,R3 b R1~R2C ,.,.,R3
그림 7-26 키랄고정상 1 과 a) 알코올 b) 클로로포름 c) (S) -메틸 (9- 페난트릴) 페닐포스핀옥_ 시 드의 상호작용 모델 (참고문헌 : 27)
수소결합의 양성자 받게로 작용하는 알코올과 양성자 주게로 작용하 는 클로로포름 용매가 광학분할에 미치는 영향은 (S)-th io- DNBTy r -A 키랄고정상 완에서 티로신의 DNB 유도체를 광학분할할 때 이동 상의 조성에 따라 용리순서가 달라지는 실험결과(그림 7-27) 에서부터 국명하게 확인할 수 있으나 (48) 이 경우에 있어서 이동상에 따른 키랄 성 인지 메커니즘의 변화 이유는 아직 확실하지 않다. 다만 입체 선택
k,20 (S)C H2 =C HCH2 0 - 0—-C H02 -C = CH - NNH H•n.- DC N,HB9 (S) 15 0\8 \12 .mi n\ .( s,I , 4 8 (I 5 \ \ 二 •• 。 。 20 40 60 80 100 A : Hexane 85 %B B : Hexane 35 Eth a nol 15 Chlorofo rm 65 그림 7-27 키랄고정상 왼에서 티로신 -DNB 유도체의 광학분할에 대한 이동상의 영향 . 유속 : 2mL/m in, 온도 : 25 C , 검출기 : 254nm UV, (S) -이성질 체의 k'( ■), (R)- 이성질체의 k'(•) (참고문헌 : 48)
성에 반대의 영향을 미치는 경쟁관계에 있는 키랄성 인지 메커니즘이 두 개 있다고 생각하고 있을 따름이다. (S)- th io -DNBTy r - A 키랄고정상 완에서 N - ( p - 메록시페닐)술피 나모일에 스 데르의 광학분할은 그립 7-28 에서 보는 바와 같이 이동상 의 조성에 따라 용리순서에는 변화가 없으나 그립 7-27 과 마찬가지로 삼성분 용매의 조성에 따라 머무름 시간이 최저점을 보이는 것을 확인 할 수 있으며 입체 선택성은 계속 증가하고 있음을 확인할 수 있다 (58). 그림 7 - 28 과 같은 실험결과는 두 가지 사실 죽 1) 분석성분인 술피나모일에 스 테르는 에탄올 / 핵산 혼합용매보다는 클로로포름에 훨
 k8cH3 7。 6.릅5 H4'sNIIO c3 H2 2cOC ca c-'H -3 `b '3‘
k8cH3 7。 6.릅5 H4'sNIIO c3 H2 2cOC ca c-'H -3 `b '3‘
그림 7-28 키랄고정상 완에서 N- 아릴술피나모일에스데르의 광학분할에 대한 이 동상의 영향, 유속 : 2mL/mi n, 온도 : 실온 , 검출기 : 260run UV, k1 ' (A) , k/ (0) , a (e) (참고문헌 : 58)
씬 큰 용해도를 보이며 2) 에탄올은 키랄고정상에 강하게 흡착되어 있 어 클로로포름 분자에 의하여 쉽게 치환이 일어나지 않는다는 사실을 고려함으로써 설명이 가능하다. 핵산-에탄올의 이성분 혼합용매에 소 량의 클로로포름을 가하면 클로로포름때문에 분석성분의 용해도가 크 게 증가하여 이동상을 따라 분석성분은 쉽게 이동할 수 있고 따라서 k' 값은 감소하며 이것은 그림 7-28 의 왼쪽부분의 k' 값의 감소에 해당 한다. 반면 핵산-클로로포름의 이성분 혼합용매에 소량의 에탄올을 가하면(그림 7-2~ 오른쪽) 그 즉시 에탄올은 키랄고정상과 결합하고 있는 클로로포름을 치환하여 키랄고정상과 분석성분의 상호작용은 더 욱 어려워지므로 k' 값은 감소한다(그림 7-28 의 오른쪽 부분). 결과적으 로 삼성분 혼합용매의 조성에 따른 k' 값은 그림 7 - 28( 혹은 그림 7-27) 과 같이 최소점을 보이게 된다. 한편 a 값은 그림 7-27 의 예와는 달리 클로로포름의 조성이 증가함에 따라 계속 증가하여 B 조성의 용액을 95% 가하였을 때 최대 값을 보이나 그 이유는 확실하지 않다. 역상 (reverse ph ase) 이동상을 이용한 광학분할은 공유결합 키랄고 정상에서 가능하나 실제의 예는 그리 혼한 편이 아니다. 특별히 역상 이동싱을 이용하여 광학분할하여야 하는 경우는 분석성분의 용해도등 에 문제가 있을 때이다. 최근에는 초임계 유체 크로마토그래피 (SFC) 혹은 준임계 유체 크로마토그래피 (SubFC) 를 이용한 광학분할이 시도 되고 있다. SFC나 SubFC 울 이용하여 광학분할할 경우에 이동상으 로서는 이산화탄소와 알코올, 클로로포름 혹은 물의 혼합용매를 사용 한다. SFC 혹은 SubFO 긱 장점으로는 짧은 시간 동안에 분리 혹은 분석이 가능하며 따라서 이동상의 사용량을 줄일 수 있어 경제적이며 G@ 검출기와 유사한 종류의 검출기를 사용할 수 있다는 이점이 있 다. SFC 혹은 SubFC를 이용하여 키랄 고정상에서 라세미 화합물을 광학분할한 최초의 예는 페닐쿨리신 -DNB 키랄고정상 l 에서 포스핀 옥시드의 광학분할이다 (66). SFC 혹은 SubFC 를 이용하여 라세미 화 합물을 광학분할할 경우에 입체 선택성이나 분해능 (resolu ti on) 은 상
 Rs
Rs
그림 7-29 키 랄고 정상 l 에서 단위시간당 분해능에 대한 LC 및 SubFO 긱 비교. 시료 : N - (2 - 나프 틸 )-2- 아마노헵탄. LC( □ ) : 이동상 : 핵산 -2 - 프로판 올 (95 : 5) , 온도 : 35° C , 검 출 기 : 234mn UV. 유속 : 0.7 - 3 . 5 m L/mi n, SubFC(. ) : 이동상 : C0 2 -2- 프로판을 (95 : 5), 컬럼압력 : 230bar, 온도 : 25°C , 검 출 기 : 234mn 유속 : 2-7 m L/ m i n (참고문헌 : 36)
황] 따라 다소 차이가 있으나 HPLC 에 의한 광학분할의 결과와 비 교하여 비슷한 것으로 확인되고 있다. 그러나 단위시간에 대한 분해능 은 SFC 혹은 SubFO 긱 경우에 월등하게 증가된 것으로 확인되고 있 다 (그림 7-29) (36, 48) .
4 7C- 염기성인 a-<>} 릴알킬아민 키랄고정상 4.1 a -O r 릴알킬아민 키랄고정상의 디자인 아미노산 -DNB 키랄고정상에서 광학분할이 잘되는 라세미 화합물 의 두 광학 이성질체 중 어느 하나의 광학 이성질체를 적절한 방법으
로 실리카 겔에 고정시켜 새로운 키랄고정상을 만 들 경우 이 키랄고정 상은 키랄성 인지의 상호성 개념에 따라 아미노산 DNB 유도체의 광 학분할에 성공적으로 응용될 수 있을 것이다. 따라서 아미노산 -DNB 키랄고정성에서 광학분할이 잘되는 모든 화합물은 모두 제 3 세대 상호 성 키랄고정상(아미노산 -DNB 키랄고정상을 제 2 세대 상호성 키랄고정상 이라 할 때 아미노산 -DNB 키랄고정상으로부터 유래된 키랄고정싱을· 제 3 세대 상호성 키랄고정상이라 지칭함)의 후보이나 아미노산 - DNB 키랄고 정상에서 광학분할이 잘되는 화합물 중 어느 것을 선택하여 새로운 제 양1 ] 대 상호성 키랄고정상으로 이용할 것인가를 결정할 때는 여러 가지 요소들울 고려하여야 할 필요가 있다. 우선, 아미노산 - DNB 키랄고 정상에서 광학분할할 때 큰 a 값을 보이는 화합물을 제 3 세대 상호성 키랄고정상의 후보로 선택할 필요가 있으며 이 화합물은 쉽게 합성이 가능할 뿐만 아니라 광학적으로 순수한 형태로 쉽게 얻을 수 있어야 한다. 다른 한 가지 고려하여야 할 점은 이 화합물을 실리카 겔에 고 정시킬 때 고정방법이 키랄성 인지에 영향을 미치지 않아야 한디는· 것 01 다. 제 양 1] 대 상호성 키랄고정상으로서의 한 가지 후보는 N- 아실 a - 아 릴알킬아민이다. 이 화합물은 3.3 철의 표 7-3 에서 보는 바와 같이 광 학분할이 잘될 뿐만 아니라 a - 아릴기의 종류와 키랄중심에 결합되어 있는 알킬기의 크기를 조절함으로써 광학분할능 (a 값)을 증가시킬 수 있기 때문에 아미노산 -DNB 키랄고정상에서 광학분할이 잘 되는 a -아릴알킬아민을 실리카 겔에 고정시킬 경우에 아미노산의 DNB 유 도체를 비롯하여 라세미 아민이나 라세미 아미노알코올의 r - 산성 유 도체를 광학분할할 수 있을 것이다. N- 아실 a- 아릴알킬아민을 기저 로 한 제 양 1] 대 상호성 키랄고정상을 디자인하는 과정에서는 이 키랄 고정상이 여러 라세미 화합물들에 대하여 좋은 광학분할능을 보이도록 하기 위하여 다음과 갇은 사실들은 고려할 필요가 있다. 첫째로 a- 아 릴기는 1-나 프틸기를 사용하여야 주변수소(p er i -h y dro g en) 에 기인하
 HI
HI
는 고정된 형태 (con fo rma ti on) 를 유지할 수 있으며 둘째 a- 아릴기의 T- 염기성을 증진시키기 위하여 l- 나프틸기에 메틸기를 도입할 필요가 있으며 셋째 키랄중심에 있는 알킬기는 충분히 커서 키랄고정상의 형 태 (con fo rma ti on) 를 고정시킬 수 있어야 하며 넷째 a- 아릴알킬아민 울 실리카 겔에 연결할 때 연결가지 (connecti ng arm) 의 길이는 충분 히 길고 비극성이어서 광학분할시에 실리카 겔 표면이나 연결가지가 키랄고정상의 키랄부분과 분석성분 사이의 상호작용을 방해하지 않아 야 한다는 것이다(1). 이와 같은 네 가지 조건에 맞추어 디자인된 키 랄고정상의 구조는 l 과 갇다. 키랄고정상 1 은 키랄성 인지의 논리에 따라 합리적으로 디자인된 최초의 키랄고정상이라고 생각해도 무방할 것 0] 다. 4.2 a -0t릴알킬아민을 기저로 한 제 앙1 ] 대 상호성 키랄고정상 의 제조, 개발 및 광학분할의 특성 키랄성 인지의 논리에 따라 합리적으로 디자인된 키랄고정상 l 은 2, 3- 디메틸나프탈렌을 출발물질로 사용하여 그립 7-30 3!} 같이 제조되 었다 (1, 2). 2, 3 一디메틸나프탈렌의 Frie d el-Craft s 알 킬화 반응, 환원 성 아민화 반응, 영화 운데세노일 (undecenoy l chlor i de) 과의 반응, 아 미노산 _DNB 키랄고정상에서의 광학분할, 히드로실릴화 반응 및 실 리카 겔과의 처리과정을 거쳐서 키랄고정상 l 이 합성되었으며 키랄고
 O 二 b ..
O 二 b ..
그림 7-30 키랄고정상 1 및 鮮] 제조. (a) Isobuty ryl chlori de , AICl3, CH2 CClH2,2 =실 C온H,(C -H(b2))n NC글 O CCNIB, Htr3i e, t h yCla Hm3iC ne0 , 2CNHH2 ◄ 야, CH실온J O. H (,d 환) 키류,랄 고정(c) 상에서의 광학분할, (e) HSi( O C2Hs)3, H2Pt C ls, (g) Sµm 실리카 겔, benzene, 환류.
정상의 연결가지의 길이가 광학분할에 미치는 영향을 관찰하기 위하여 똑같은 방법으로 키랄고정상 2 가 합성되었다. 이렇게 합성된 키랄고 정상 l 과 2는 강한 r- 염기성기를 가지고 있기 때문에 T- 산성 라세미 화합물들을 광학분할할 수 있을 것으로 예상된다. 실제로 키랄고정상 l은 아미노산에스데르 혹은 아미노산아미드
DNB 유도체의 광학분할에 아주 성공적이었으며 대표적인 크로마토그 램은 그림 7 - 31 와 같다. 아미노산에스테르나 아미노산아미드의 DNB 유도체의 광학분할 이의에도 특히 홍미룰 끄는 광학분할은 a- 아릴알 킬아민 DNB 유도체의 광학분할이다. N-3,5- 디니트로벤조일 a- 아
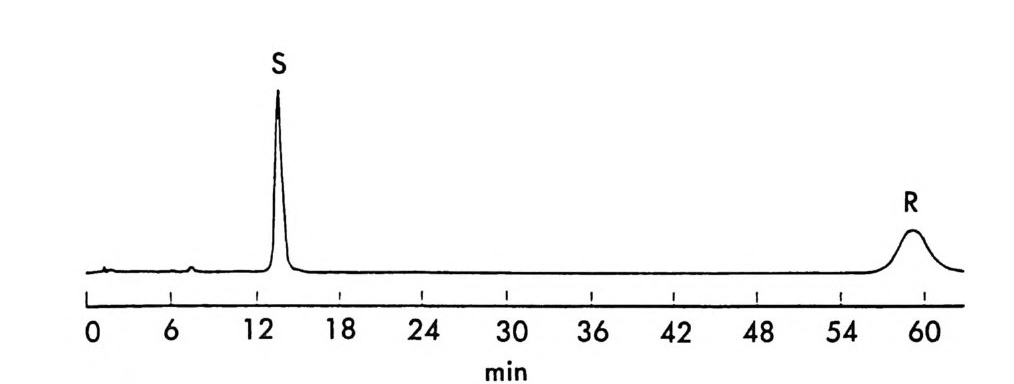 s
s
그림 7-31 키랄고정상 1 에서 페닐알라닌메틸에스테르 DNB 유도체의 광학분할. 이동상 : 2 - 프로판을 _ 핵산 (20 / 80), 유속 : 2mL/mi n, 검출기 : 254nm UV. (참고문헌 : 1)
릴알킬아민을 광학분할할 경우에 키랄중심에 있는 알킬기의 길이가 증 가함에 따라 그림 7-32 에서 보는 바와 같이 키랄고정상 2 에서 a 값은 점점 감소하다가 결국에는 용리순서가 바뀐다는 사실이 확인되었다 (2). 키랄고정상 2 에서 용리순서의 뒤바뀜은 서로 역으로 작용하는 두 개의 키랄성 인지 메커니즘이 서로 경쟁관계에 있다는 사실울 강하게 암시해 주고 있다. 그림 7-33 에 키랄고정상 l 혹은 2 에서의 광학분할을 합리적으로 설 명할 수 있는 서로 경쟁관계에 있는 두 개의 키랄성 인지 메커니즘이 제안되어 있으며 이와 같은 키랄성 인지 메커니즘은 그림 7-349 .} 같 이 단순화한 모형도로서 나타낼 수 있다. 그림 7-33 및 7-3~ 쌍극 자겹침 과정은 (R) -이성질체를 선택적으로 키랄고정상에 머물게 하는 키 랄성 인지 메커니즘으로서 키 랄고정상과 (R) -이성질체 사이에 1[-1[
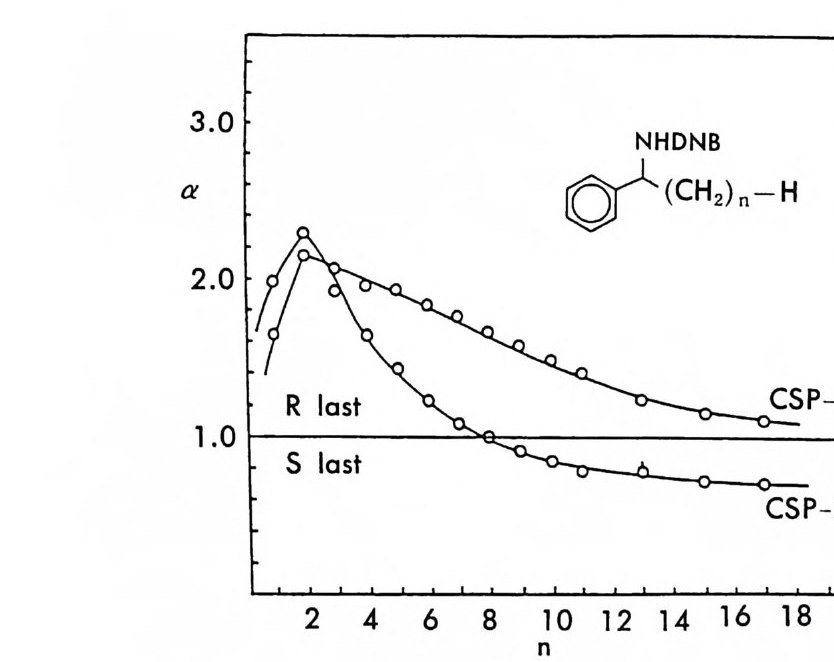 3.0 NHDNB
3.0 NHDNB
그림 7-32 키랄고정상 1 및 2 에서 a- 페닐알킬아민 DNB 유도체의 광학분할. 이 동상 : 2- 프로판을-핵산 (20/80)
상호작용과 · 아미드쌍극자-아미드쌍극자의 겹침에 의하여 키랄고정상 과 (R) -이성질체가 상호작용하고 있으며 (R) -이성질체의 키랄중심에 있는 알킬기 (R, 그립 7-33 에서는 메틸기이며 일반적으로 페닐기보다 입체 적으로 작다고 가정됨)는 키랄고정상의 두 연결가지 사이로 끼어들어가 는형상이다. 반면, 그림 7-3~ 그림 7-3~ 수소결합 과정은 (S) -이성질체를 선택적으로 키랄고정상에 머물게 하는 키랄성 인지 메카니즘으로서 키 랄고정상과 (S) -이성질체 사이의 亢-元상호작용 및 두 위치에서의 수 소결합에 의한 상호작용을 나타내고 있다. 이때 (S) -이성질체의 키랄 중심에 있는 입체장애가 작은 알킬기는 키랄고정상의 이소프로필기와 거의 평행한 방향으로 향하고 있으며 키랄고정상의 연결가지와는 전혀 방향이 다르다. (R) -이성질체와 (S) -이성질체를 각각 선택적으로 키
|o 기 凡H。 \e `/ 0 \(SCI)/i H- =0O2 ) 0En t H
。H父-N 있H냐 ~ PCH3h / 10 02 N 쌍극자겹침 과정 수소결합 과정 그립 7-33 키 랄고 정상 1 혹은 2 에서 a - 페닐알킬아민 DNB 유도체의 광학분할에 대한 두 개의_ 경쟁 대커니즘 -----CJ 그림 7-34 키랄고정상 l 혹은 혼에서 a- 아릴알킬아민 DNB 유도체의 광학분할에 대한 두 개의 경쟁 메커니즘 모형도 . (좌)쌍극자겹침 과정, (우)수소 결합과정랄고정상에 머무르게 하는 두 개의 메커니즘이 서로 경쟁한다면 더 안 정한 착물을 형성하는 메커니즘이 우세하게 될 것이며 만일 두 메커니 즘에 의하여 형성되는 착물의 안정성이 동일하다면 광학분할은 관찰되 지 않을 것이다. 쌍극자겹침 과정이 우세할 경우 (R) -이성질체는 (S) -이성질체보다 키랄고정상에 더 오래 머무르며 정확한 이유는 확실치 않으나 실지로 (R) -이성질체가 키랄고정상 l 에 더 오래 머무르는 사 실이 관찰되었으므로 쌍극자겹침 과정이 더 우세하다고 할 수 있다. 그러나 분석성분의 알킬기 길이가 증가함에 따라 (R) -이성질체의 알 킬기가 키랄고정상의 연결가지 사이로 끼어들어가는 것이 접점 어려워 지기 때문에 쌍극자겹침 과정은 점점 불리하게 되는 반면 수소결합 과 정은 분석성분의 알킬기 길이에 의하여 큰 영향을 받지 않는다. 결국 분석성분의 알킬기 길이가 길어짐에 따라 두 과정에 의한 착물의 안정 성 차이가 점점 감소하게 되고 따라서 a 값도 그림 7-32 에 보는 바와 같이 점점 감소한다. 연결가지의 길이가 짧은 키랄고정상 2 에서는 분 석성분의 알킬기가 길어지면 (R) _이성질체의 알킬기가 키랄고정상의 연결가지 사이로 끼어들어가기가 더 어려워질 뿐만 아니라 알킬기의 길이가 조금만 길어져도 금방 실리카 겔 표면과 입체장애 등을 받게 되므로 쌍극자겹침 과정은 더욱 불리하게 되고 그림 7-32 에서 보는 ’ 바와 같이 알킬기의 길이가 옥틸기 (n = 8) 로 길어졌을 때 두 경쟁과정 에 의하여 형성된 착물의 안정성은 같아져서 광학분할이 되지 않는 것 으로 생각할 수 있다. 알킬기의 길이가 더욱 길어지면 (n> 8) 이제는 수소결합 과정에 의한 착물이 더 안정하게 되어 결국은 용리순서의 뒤 바뀜을 관찰할 수 있게 된다. 그립 7-32 에서 n = 2 일때 a 값의 극대점 은 분석성분에 있어서 형태 (co nf orma ti on) 의 고정성 증가로 인한 효 과로생각되고있다. 이와 같은 경쟁관계에 있는 두 개의 키랄성 인지 메커니즘을 기본으 로 하여 키랄고정상 g과 4 에서 a- 아릴알킬아민 DNB 유도체의 광학 분할을 합리적으로 예측할 수 있다. 키랄고정상 꾼斗 ,싣 는 키랄아민의
 CH3
CH3
키랄중심에 있는 알킬기를 통하여 키랄아민을 실리카 겔에 결합시킨 것으로써 결합가지의 방향이 키랄고정상 l 과 2 의 결합가지 방향과는 다르다. 따라서 그림 7 一 35Sl 모형도에서 보는 바와 같이 두 개의 경 쟁 메커니즘 중, 쌍극자겹침 과정에서는 분석성분의 알킬기가 더 이상 키랄고정상의 결합가지 사이로 끼어들어가지 않고 반대로 수소결합 과 정에서는 분석성분의 알킬기가 키랄고정상의 결합가지 사이로 끼어들 어간다. 따라서 분석성분의 알킬기 길이가 길어침에 따라 본래 우세한
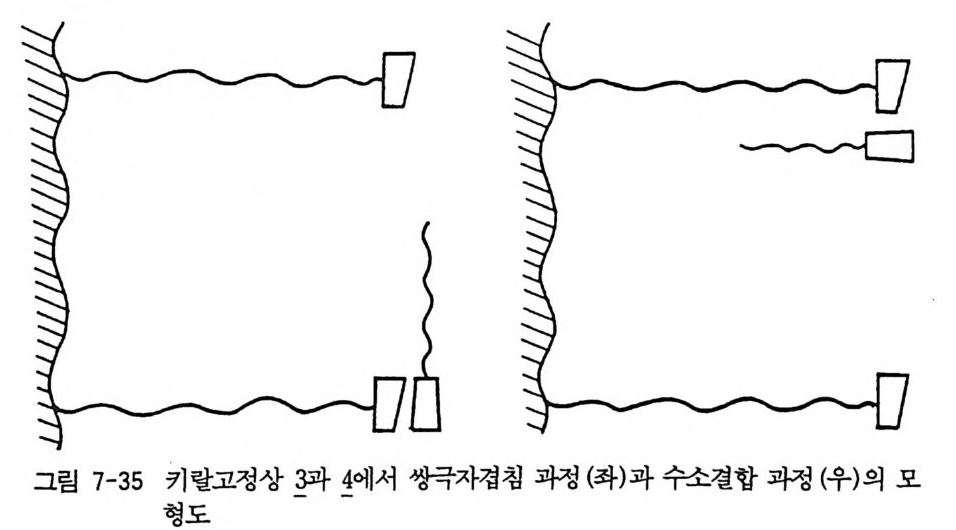 그림 7-35 키랄고정상 定} 산에서 쌍극자겹침 과정 (좌)과 수소결합 과정 (우)의 모
그림 7-35 키랄고정상 定} 산에서 쌍극자겹침 과정 (좌)과 수소결합 과정 (우)의 모
쌍극자겹침 과정은 분석성분의 알킬기 길이에 별로 영향을 받지 않는 반면, 수소결합 과정은 점점 불리하게 되므로 두 과정에 의하여 형성 되는 착물의 안정성에는 점점 큰 차이가 생겨서 a 값은 계속 증가하리 라고 예측할 수 있으며 이러한 경향은 결합가지의 길이가 짧은 키랄고 정상 g에서 더 크게 나타날 것이다. 그림 7-3& g-네 개의 키랄고정상 L 2, g 및 4 에서 일련의 p - 메톡시 페닐알킬아민 DNB 유도체의 광학분할에 대한 a 값을 도시한 것으로 써 분석성분의 알킬기 길이가 증가함에 따른 a 값의 변화가 두 개의
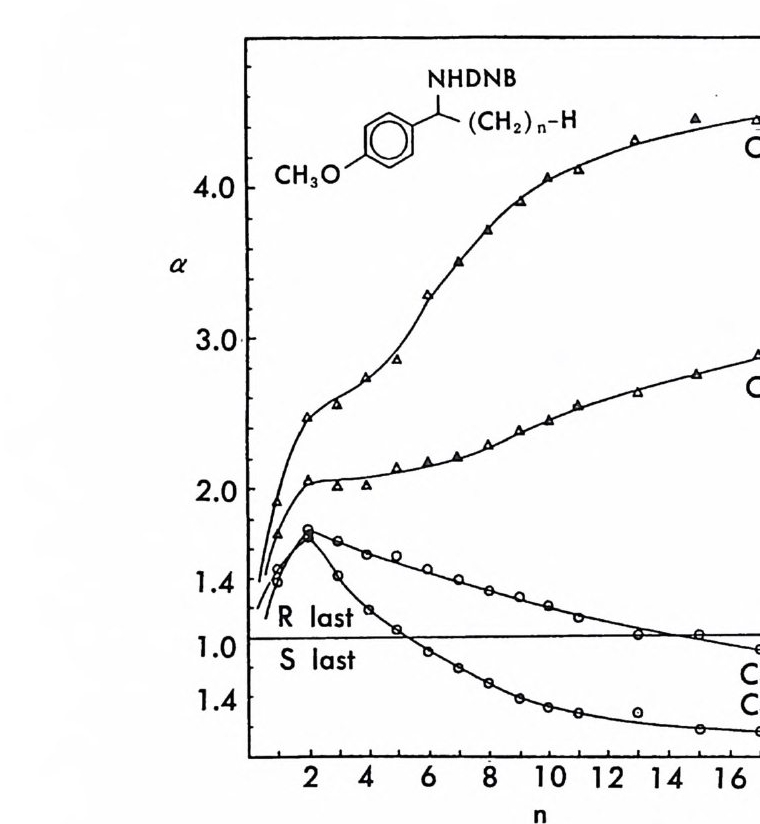 NHDNB
NHDNB
그림 7-'-36 키랄고정상 1, 2, 3 및 4 에서 p-메목시페닐알킬아민 DNB 유도체의 광학분할. 。「동장 : 5- 프로광을-핵산 (20/80) (참고문헌 : 2)
경쟁관계에 있는 키랄성 인지 메커니즘으로 예 측한 바와 정확히 일치 함을확인할수 있다. 한편, 아미노산에스데르 혹은 아미노산아미드 DNB 유도체를 광학 분할하는 과정에서도 에스테르 혹은 아미드의 알킬기 길이를 증가시킴 에 따라 a 값에 연속적인 변화가 관찰되나 그 경향은 a- 아릴알킬아민 DNB 유도체의 광학분할에서 관찰된 a 값 변화의 경향과는 다르다. 죽 아미노산에스데르 DNB 유도체 혹은 아미노산아미드 DNB 유도체 의 에스데르알킬기 혹은 아미드알킬기의 길이룰 증가시킴에 따라 키랄 고정상 l 과 2 에서 a 값은 계속 중가하는」 반면 (연결가지가 짧은 키랄고정 상 2 에서 a 값은 더 크게 증가함) 키 랄고정상 꾼斗 걷에서는 a 값이 계속 감소한다. 아미노산에스테르나 아미노산아미드 DNB 유도체의 광학분 할에 있어서도 쌍극자겹침 과정과 수소결합 과정의 두 경쟁과정이 관 여하는 것으로 생각되고 있으며 이 경우에는 확실한 실험적 증거는 없 으나 아마도 그립 7-37 의 수소결합 과정에서 보는 것처럼 분석성분의
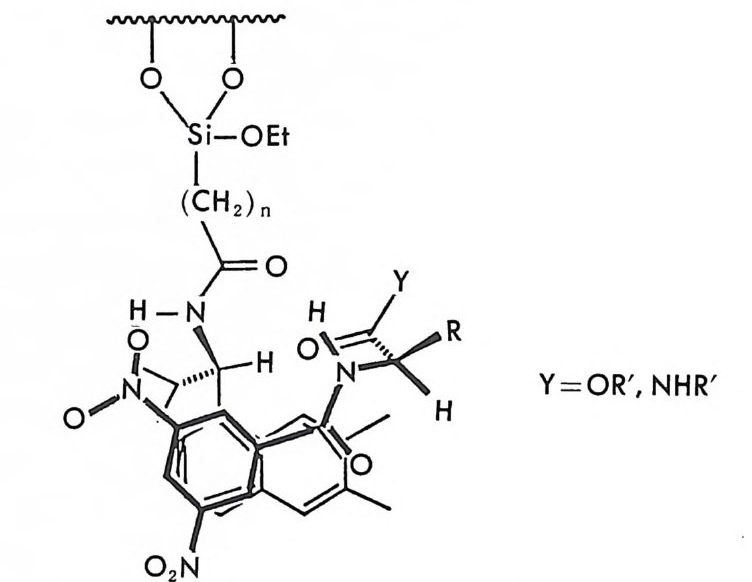 三0 \(_I O0 Et
三0 \(_I O0 Et
그림 7-37 키랄고정상 l 혹은 2 에서 아미노산에스데르 DNB 유도체 혹은 아미노산아너드 DNB 유도체의 광학분할에 대한 수소결합 과정
카르보닐 산소와 키랄고정상의 키랄중심에 있는 수소(바로 이웃의 전기 음성도가 큰 질소와 방향족기로 인하여 어느 정도 산성을 가질 것으로- 추측 됨) 사이의 추가적인 수소결합 때문에 수소결합 과정이 쌍극자겹침 과 정보다 우세한 과정이라고 생각할 수 있다. 따라서 키랄고정상 l 과 2 에서는 아미노산에스테르 DNB 혹은 아미노산아미드 DNB의 에스테 르알킬기 혹은 아미드알킬기의 길이가 증가함에 따라 본래 열세인 쌍 극자겹침 과정이 더욱 더 위축되는 반면 본래 우세한 수소결합 과정은 알킬기의 길이에 의하여 별로 영향을 받지 않으므로 두 과정 사이의 에너지 차이는 점점 증가하여 a 값은 계속 증가한다. 반면 키랄고정상 훔 산에서는 아미노산에스테르 DNB 혹은 아미노산아미드 DNB의 에 스데르알킬기 혹은 아미드알킬기 길이가 길어짐에 따라 본래 우세한 수소결합 과정에서 에스테르 알킬기 혹은 아미드 알킬기가 키랄고정상 의 결합가지 사이로 끼어들어가기가 점점 어려워지므로 두 과정 사이 의 에너지 차이는 점점 감소하며 따라서 a 값은 계속 감소한다. 지금까지 기술한 바와 같이 키랄고정상 l,£ 겔 과 간에서 키랄아민 DNB 유도체나 아미노산에스데르 혹은 아미노산아미드 DNB 유도체 의 광학분할은 결국 경쟁관계에 있는 두 메커니즘의 균형에 의하여 결 정되며 이 균형을 적절히 조절함으로써 광학분할을 조절할 수 있다는 생각을 할 수 있다. 키랄고정상 l 혹은 2 와 같은 키랄고정상은 키랄중 심에 있는 알킬기 (키랄고정상 l 과 2 에서는 이소프로필기임 )의 크기를 조 절함으로써 두 경쟁 메커니즘의 균형점을 조절할 수 있을 것으로 생각 된다. 죽 키랄고정상 l 혹은 空] 키랄중심에 있는 이소프로필기보다 더 큰 알킬기를 가지고 있는 키랄고정상 §에 있어서 두 경쟁 메커니즘 중 수소결합 과정은 더 압박을 받게 될 것이므로(키랄고정상의 키랄중 심에 있는 알킬기의 크기가 커지면 그림 7-3~ 수소결합과정이나 그립 7-37 의 수소결합과정에서 보는 것처럼 이 알킬기와 a- 아릴알킬아민 DNB 의 알킬기나 혹은 아미노산에스테르 DN& 긱 에스테르알킬기사이의 입체장 애가 증가함) a- 아릴알킬아민 DNB 유도체와 같이 쌍국자겹침 과정이
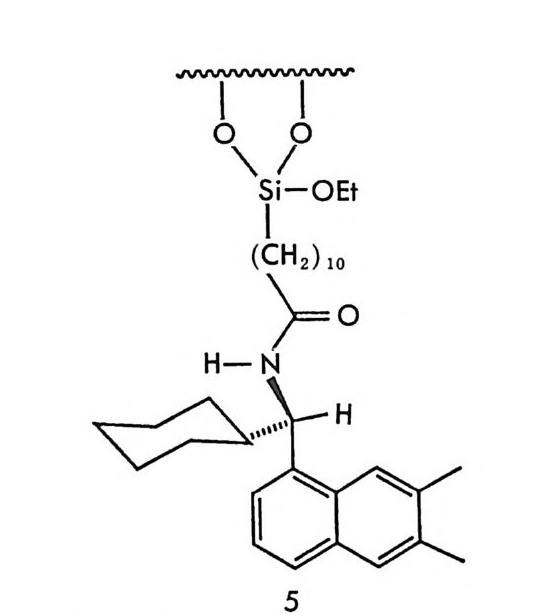 |
|
우세한 라세미 화합물들은 키랄고정상 l 보다 키랄고정상 울에서 광학 분할이 더 잘될 것이고 반면 아미노산에스테르나 아미노산아미드 DNB 유도체와 같이 수소결합 과정이 우세한 라세미 화합물들은 키랄 고정상 향比다 키랄고정상 l 에서 광학분할이 더 잘될 것으로 예상할 수 있으며 이러한 예상은 사실과 일치함이 확인되었다 (3). 한편, 키랄고정상 伊} 같은 형태의 키랄고정상에 있어서 N- 아실기 롤 아세틸기보다 훨씬 큰 기로 대체하면 두 경쟁 메커니즘 중 쌍극자 겹침 과정이 크게 압박받게 될 것이므로 본래 수소결합 과정이 우세한 것으로 생각되는 아미노산에스테르 혹은 아미노산아미드의 DNB 유도 체들은 N- 아실기가 피발로일(pi valo y l) 기인 키랄고정상 §에서 광학분 할이 잘될 것으로 예측된다. 실제로 이들 라세미 화합물둘은 키랄고정 상 §에서 광학분할이 아주 잘되는 것으로 확인되었으며 전형적인 크로 마토그램은 그림 7-38 와 같다 (2, 3) . 특히 그림 7-389 .J 크로마토그램은 길이가 5cm 에 불과한 짧은 키 랄 컬럼 (50mmx4.6mm) 을 사용하여 얻은 것으로서 a 값은 무려 8.29 에 이르고있다.
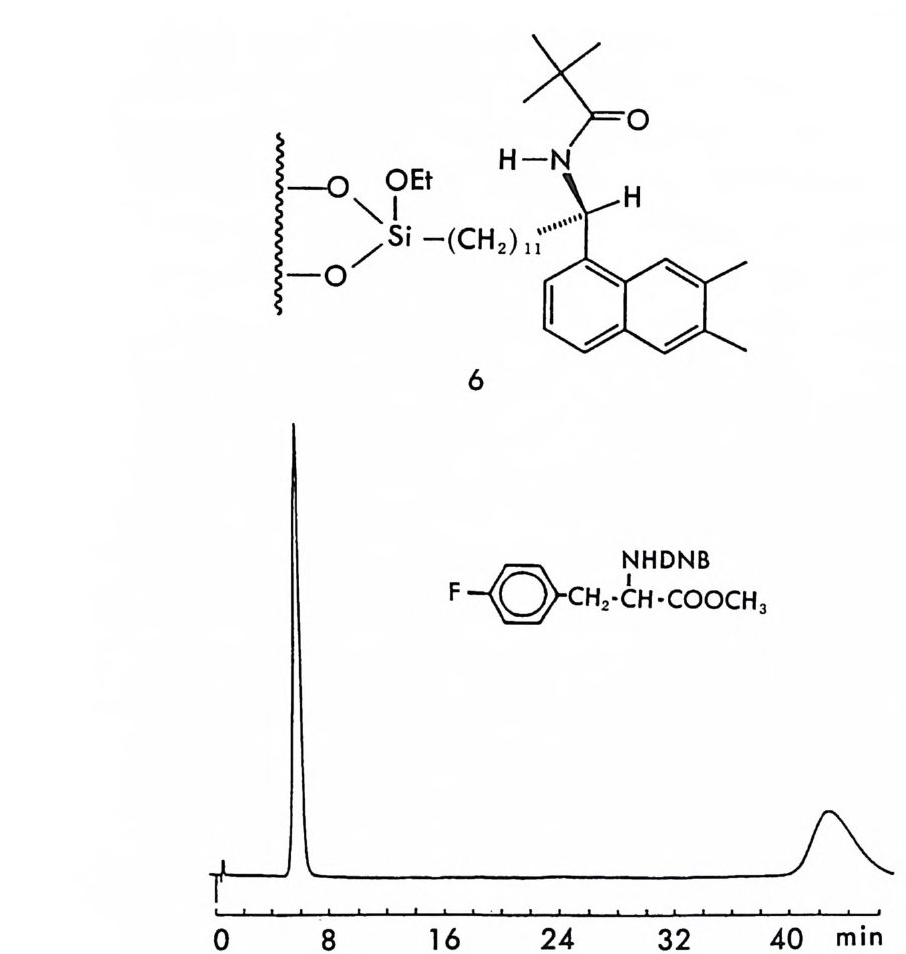 —一-O0 \l Q E t H 一\。H
—一-O0 \l Q E t H 一\。H
그림 7-38 키랄고정상 6 에서 4 급툐프녁녀]닐알라닌메틸에스데르 DNB 유도체의 광학분할. 컬럼 : 4.6x 50mm, 유속 : 2mL/mi n, 검출기 : 254nm UV. 이동상 : 2- 프로판을-핵산 (20/80) (참고문헌 : 3)
그림 7-3 떠 모형도에 의하면 키랄성 인지의 두 경쟁메커니즘이 옳 은 경우에 키랄고정상의 연결가지 사이의 거리가 쌍극자겹침과정에 영 향을 미칠 것으로 예상할 수 있다. 키랄고정상 l 의 경우 실리카 겔에 결합되는 키랄아민의 양을 많이 하면 연결가지 사이의 거리는 감소하 며 따라서 쌍극자겹침 과정에 있어서 분석성분 분자가 키랄고정상의
연결가지 사이로 끼어 들 기 는 그만큼 어려워져서 씽국지겹침 과정은 압 박을 받을 것이며 반면 실리카 겔 에 결합되는 키랄아민의 양을 줄일 경우에 연결가지 사이의 거리는 길어져서 그림 7-34 의 쌍극자겹침과 정은 그만큼 용이하게 될 것이다. 실리카 겔에 결합되는 아민의 양이 각각 0.4 m mole/g , 0.2 m mole/g , O . lmmole / g인 세 가지의 키랄고정 상 l 에서 ¢페닐알킬아민 -DNB 의 광학분할은 그림 7 - 39 와 같다 ( 4 ). d 페닐알킬아민 DNB 의 알킬기 길이가 짧울 때는 세 가지 키랄고정 상에서의 광학분할에 큰 차이점이 없으나 알킬기 길이가 길어짐에 따 라 가장 많은 양의 키랄아민이 실리카 겔에 결합된 키랄고정상에서 a 값은 가장 작을 뿐만 아니라 급격히 감소하고 있다. 많은 양의 키랄아 민이 실리카 겔에 결합된 키랄고정상의 경우 위에서 언급한 바와 같이 연결가지 사이의 거리가 짧기 때문에 분석성분의 알킬기 길이가 길어
NHDNB 2.0 二 (CH 2 )n-H a’ 1.5 1.0 ' 2 4 6 8 10 12 14 16 18 n 그림 7-39 결합된 아민의 양이 다론 키랄고정상 .!_( □ : O.lmmole/g, b,. : 0. 2mmole/g, O : 0 .4 mmole / g)에서 a- 페닐알킬아민 DNB 유도체의 광학분할(참고문헌 : 4)
짐에 따라 이 알킬기는 쌍극자겹침 과정에서 키랄고정상의 결합가지 사이로 끼어들어가기가 힘들어져 쌍극자겹침 과정이 크게 압박을 받아 a 값은 급격히 감소하게 되는 것이다. 그러나 두 경쟁 메커니즘 중 수 소결합 과정이 우세한 아미노산에스데르 DNB 유도체의 광학분할은 그림 7-40 에서 보는 바와 같이 가장 많은 양의 키랄아민이 실리카 겔 에 결합된 키랄고정상 l 에서 가장 좋으며 에스데르의 알킬기가 증가함 에 따라 a 값은 중가하고 있다.
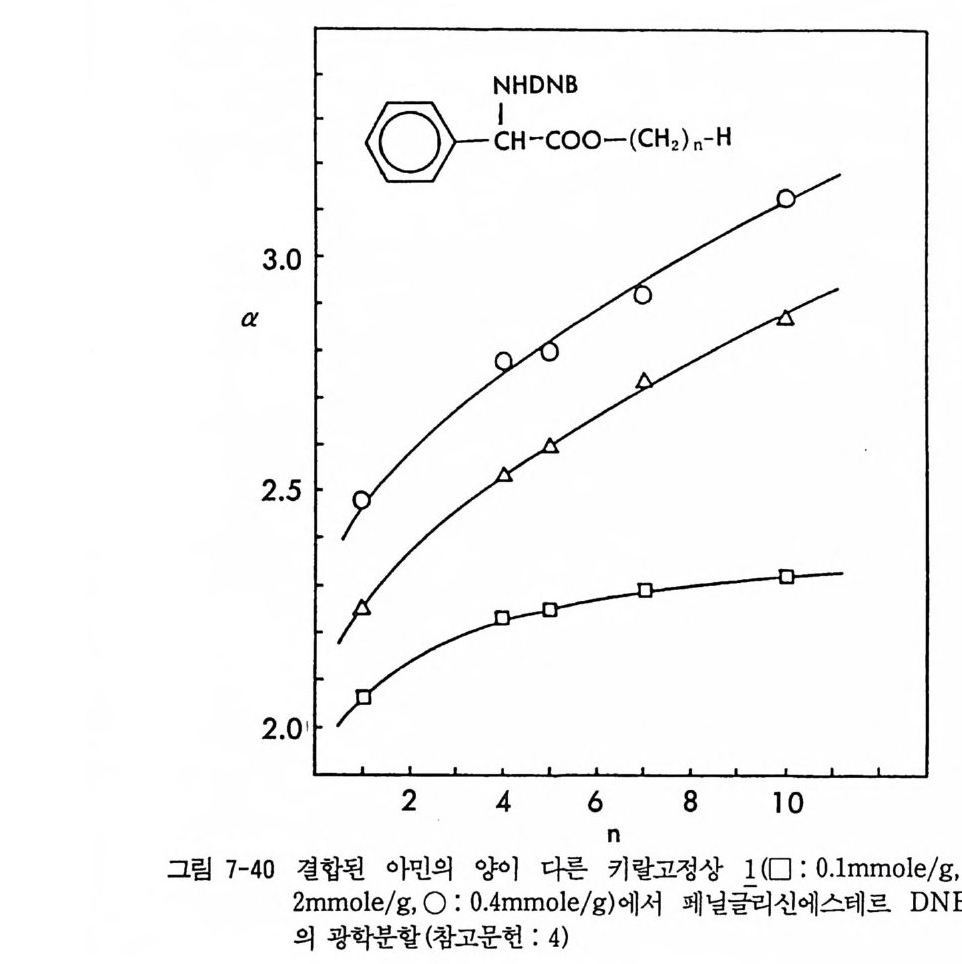 NHDNB
NHDNB
키랄고정상 1 의 짧은 연결가지 사이 거리가 광학분할에 미치는 영향 은 키랄아민을 실리카 겔에 연결하는 연결가지 사이에 n 감臼틸기나 혹 은 n 유 L 틸기와 같은 알킬기를 도입하더라도 관찰할 수 있다 (4). 그림 7-41 의 모형도와 같이 키랄고정상의 연결가지 사이에 알킬기를 도입 하면 연결가지 사이의 거리가 짧은 경우와 마찬가지로 쌍극자 과정에 서 분석성분의 알킬기가 연결가지 사이로 끼어들어가기가 힘들어져서 많은 양의 아민을 실리카 겔에 결합시킨 키랄고정상과 같이 행동할 것 으로 예상할 수 있다. 실제로 키랄고정상의 연결가지 사이에 n 감뎀i 기를 도입한 키랄고정상 l 은 아미노산에스테르 혹은 아미노산아미드 DNB 유도체의 광학분할 및 a - 아릴알킬아민 DNB 유도체의 광학분할 에 있어서 상대적으로 많은 양의 키랄아민이 실리카 겔에 결합된 키랄 고정상 l 과 같은 경향을 보인다는 사실이 확인되었다 (4). 경쟁관계에 있는 두 개의 키랄성 인지 메커니즘에 의하여 합리적으 로 개발된 키랄고정상 _l_- 6 이의에도 키랄고정상 l 과는 형태
그림 7-41 n- 알킬기가 키랄고정상의 연결가지 사이에 도입된 키랄고정상에서 쌍극자겹침 과정의 모형
(con fo rma ti on) 의 고정성이 다르리라고 예상되는 키랄고정상 7 및 상 업적으로 쉽게 구할 수 있는 키랄아민인 a-(1- 나프틸)에틸아민에서 부터 유도된 키랄고정상 §이 키랄고정상 l 의 제조방법과 비슷한 방법 으로 제조되어 광학분할에 응용되었다 (3). 특히 키랄고정상 §은 상업 적으로 쉽게 구할 수 있는 키랄아민을 출발물질로 하기 때문에 아미드 합성 과정, 히드로실릴화 과정, 실리카 겔에의 고정 과정인 3 단계 과 정에 의하여 쉽게 제조될 수 있디는- 것이 큰 장점이라고 할 수 있다. 지금까지 기술한 a- 아릴알킬아민을 기저로 한 키랄고정성들을- 이용 하여 광학분할이 가능한 라세미 화합물들은 a- 아릴알킬아민 DNB 유 도체와 아미노산에스데르 DNB 유도체 및 아미노산아미드 DNB 유도 체 이외에도 다양하다. 다만 이들 키랄고정상들이 강한 r- 염기성기를 가지고 있는 r- 염기성 키랄고정상들이기 때문에 이들 키랄고정상에서 광학분할이 가능하도록 라세미 화합물들은 대부분 강한 T- 산성기 유 도체인 3, 5- 디니트로벤조일 유도체나 혹은 3, 5- 디니트로아닐리드 유 도체로 만들어야 한다는 제한점이 있다. 아미노알코올 라세미체는 N-DNB 유도체나 혹은 아미노기 및 히드
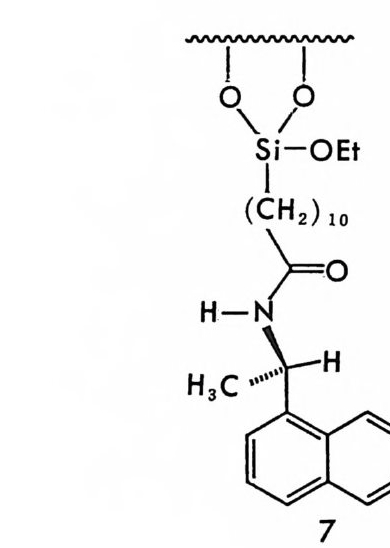 0I \ SI/i - 0O I Et
0I \ SI/i - 0O I Et
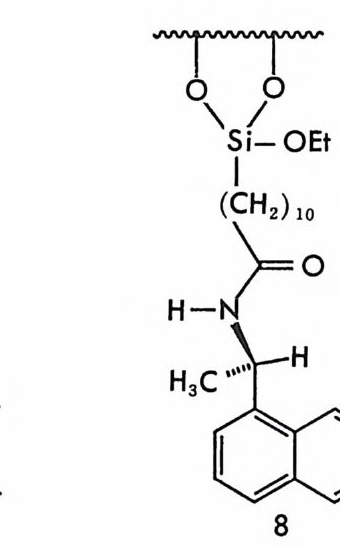 H0-\ >SI Ii- 0O EOt
H0-\ >SI Ii- 0O EOt
록시기의 이 DNB 유도체로서 광학분할이 가능하며 라세미 알코올이 나 라세미 디올은 DNB 유도체로서 광학분할이 가능하다(1 , 2). 라세 미 알코홀이나 라세미 디올의 경우 DNB 유도체 9 로서 광학분할이 가 능하기는 하나 이들은 키랄고정상과의 수소결합에 참여할 수 있는 양 성자 주게를 가지고 있지 않기 때문에 광학분할이 크게 효과적이지는 못하며 라세미 알코올이나 라세미 디올을 3, 5- 디니트로페닐이소시아 네이트와 반응시켜 얻은 카르밤산 에스테르유도체 L따 냐i서 광학분할 이 훨씬 더 잘된다는 사실이 확인되었다 (5,6). 라세미 알코울이
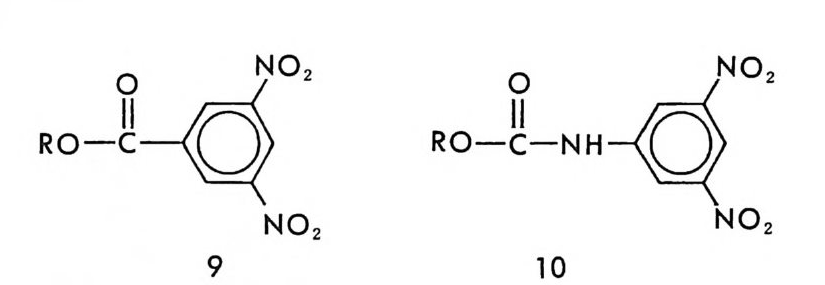 。 OIIc RO ―빌。 _NH -QN 02
。 OIIc RO ―빌。 _NH -QN 02
3,5- 디니트로페닐카르밤산 에스데르유도체로 광학분할이 잘되므로 히 드록시카르복실산에스테르의 3,5- 디니트로페닐카르밤산 에스테르유도 체 또한 a- 아릴알킬아만 키랄고정상에서 광학분할이 찰될 것으로 기 대된다 (7,8). 그림 7-42 는 락트산에틸에스데르의 3, 5- 디니트로페닐카 르밤산 에스데르유도체를 키랄고정상 l 에서 광학분할하였을 때의 전 형적인 크로마토그램이다. a- 히드록시카르복실산에스데르의 3,5- 디 니트로페닐카르밤산 에스데르유도체를 키랄고정상 l 에서 광학분할할 때에도 에스테르의 알킬기 길이를 증가시키면서 조사한 a 값의 변화로 부터 경쟁관계에 있는 두 개의 키랄성 인지 메커니즘과정이 키랄성 인 지에 관여하는 것으로 제안되었다 (8). 라세미 아민의 경우에도 라세미 알코올과 마찬가지로 3, 5- 디니트로 벤조일 (DNB) 유도체 혹은 3,5- 디니트로페닐우레이드 유도체(라세미 알코올의 경우 3, 5- 디니트로페닐카르밤산 에스테르유도체와 동일함)로서
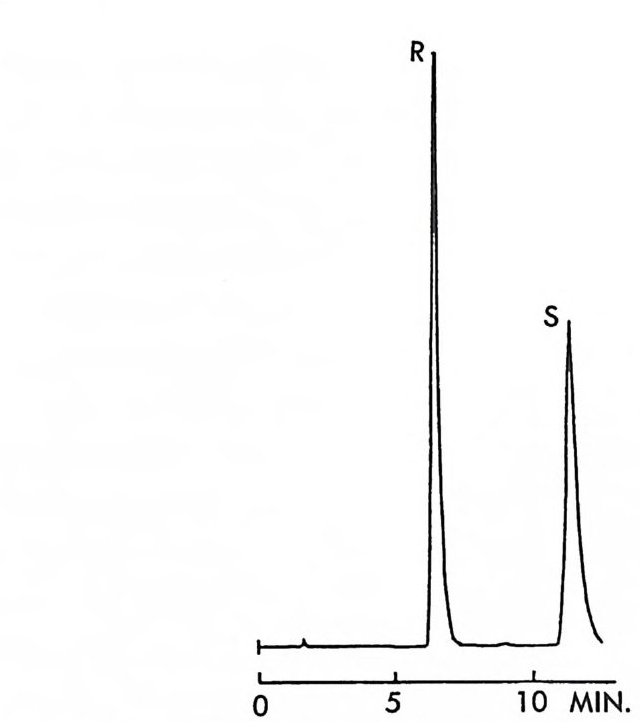 R
R
그림 7-42 키랄고정상 !에서 락트산 에틸에스데르의 3, 5- 디니트로페닐카르밤산 2에m스L테/m르i n의, 검출광기학 분: 2할54 nm이 U동V상 ( :참 2고- 프문헌로 판: 7을) -핵산 (10 / 90), 유속 :
광학분할이 가능하다. 특히 라세미 일차아민의 경우에는 3, 5- 디니트 로페닐우레이드 유도체로서 광학분할되는 것보다 DN B-유도체로서 광 학분할이 잘되는 반면 라세미 이차아민이나 라세미 고리형 이차아민의 경우에는 DN B-유도체보다는 3, 5- 디니트로페닐우레이드유도체로서 광 학분할이 찰되는 것으로 확인되었다 (9). 이와 같은 사실은 일차아민의 3,5- 디니트로페닐우레이드 유도체가 입체 선택성과는 관계없는 수소 결합 양성자 주게를 여분으로 가지고 있으며 반면 아차아민 혹은 고리 형 이차아민의 DNB 유도체는 수소결합 양성자 주게를 전혀 가지고 있지 않다는 사실을 고려하면 이해가 가능한 실험결과라고 할 수 있 다. /3-아미노산은 /3-아미노산 n 감댑 l 에스데르의 DNB 유도체로 키랄
고정상 l 에서 광학분할이 잘된다(1 0). 또한 항생제로 많이 쓰이는 /3 -락탐 계통의 화합물을 n 감댑i알코올로 처리하여 /3-아미노에스테르를 만든 후 염화 3, 5- 디니트로벤조일과 반응시키면 결국 /3-아미노산 n 옥틸에스테르의 DNB 유도체가 되기 때문에 이것은 특히 키랄고정상 l 과 §에서 광학분할이 잘되는 것으로· 보고된 바 있다 (11, 12) . 특히 광 학활성인 /3-락탐은 항생제의 합성에 중요하기 때문에 비대칭유발반응 에 의한 합성이 많이 연구되고 있으며 이 과정에서 합성된 /3-락탐의 광학활성을 측정하는 데 키랄고정상이 크게 유용한 것으로 확인되었다 (11, 12) . 두 개의 라세미 아미노산의 펩티드결합에 의하여 얻어전 디펩티드는 다의 입체 이성질체로 존재한다. 디펩티드의 C- 말단을 에스테르로 바꾸고 N- 말단을 DNB 유도체로 바꾸었을 때 디펩티드의 네 개 입체 이성질체는 키랄고정상 l 혹은 §에서 완전히 분리되는 것으로 확인되 었다 (13 , 14) (그립 7-43) .
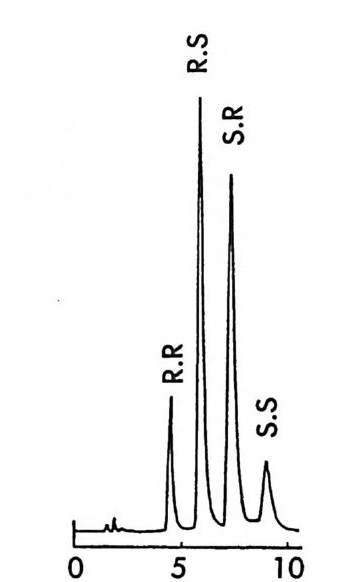 s·~
s·~
그림 7-43 키랄고정상 l( 철대배열 : S) 에서 N-DNB-va l-I eu-OCH3 의 광학분 2할54 nm이 U동V상 ( :참 2고- 프문로헌 판: 을14-) 핵산 (10/90), 유속 : 2mL/m in, 검출기 :
카르보닐기의 g위치에 키랄중심을 가진 라세미 카르보닐화합물은 다른 키랄고정상에서도 광학분할된 예가 혼치 않으나 카르보닐화협물 의 옥심유도체를 3, 5- 디니트로페닐이소시아네이트와 반웅시켜 얻은 유도체 브은 키랄고정상 l 에서 광학분할된다 (15). 카르보닐화합물들 중 시스 및 트란스김呂]이 만들어지는 경우에는 그림 7-44 에서 보는 바와 같이 시스감呂] 및 트란스깊呂]의 3, 5- 디니트로페닐카르밤산 에 스데르유도체가 모두 광학분할되고 있음을 확인할 수 있다 (15).
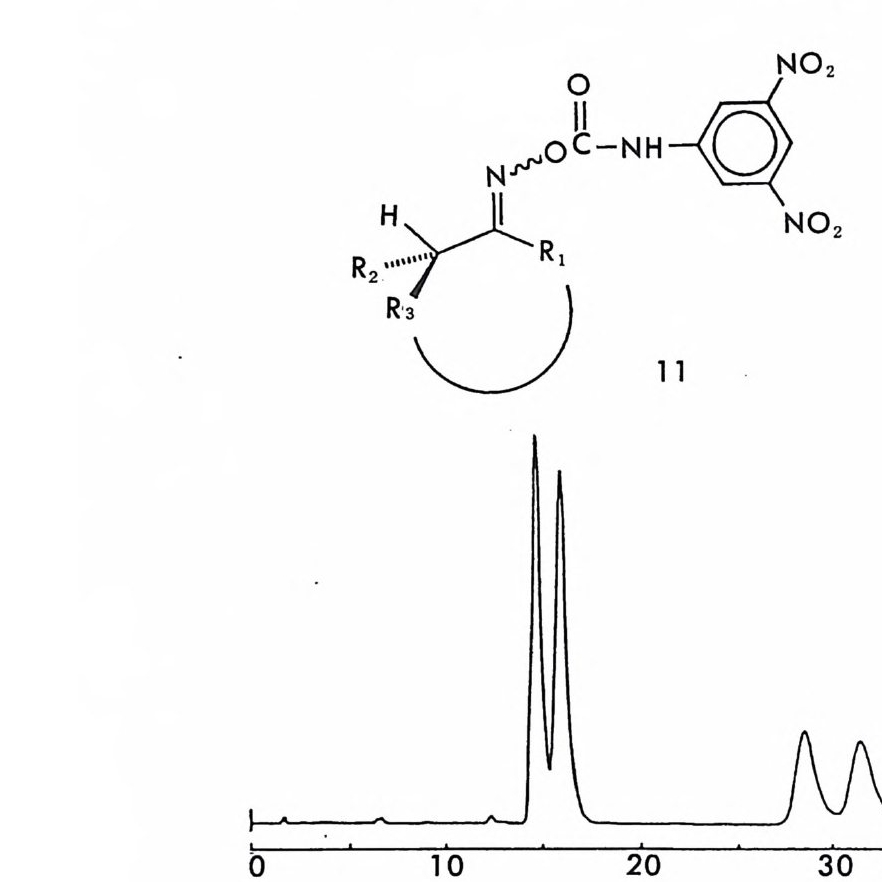 ro 。도 -NH -qN0 2
ro 。도 -NH -qN0 2
그림 7-44 키랄고정상 1 에서 2- 페닐부티로페논케톡심 3, 5- 디니트로페닐카르밤산 에스테르유토체의 광학분할 이동상 : 2- 프로판을-핵산 (10/90) , 유 속 : 2mL/m in, 검출기 : 254mn UV. (참고문헌 : 15)
소영진통제로 쓰이는 라세미 a- 아릴프로피온산들은 3, 5- 디니트로 페닐아닐리드 유도체로 광학분할이 가능하다(1 6). 그림 7-4 5-i근 소영
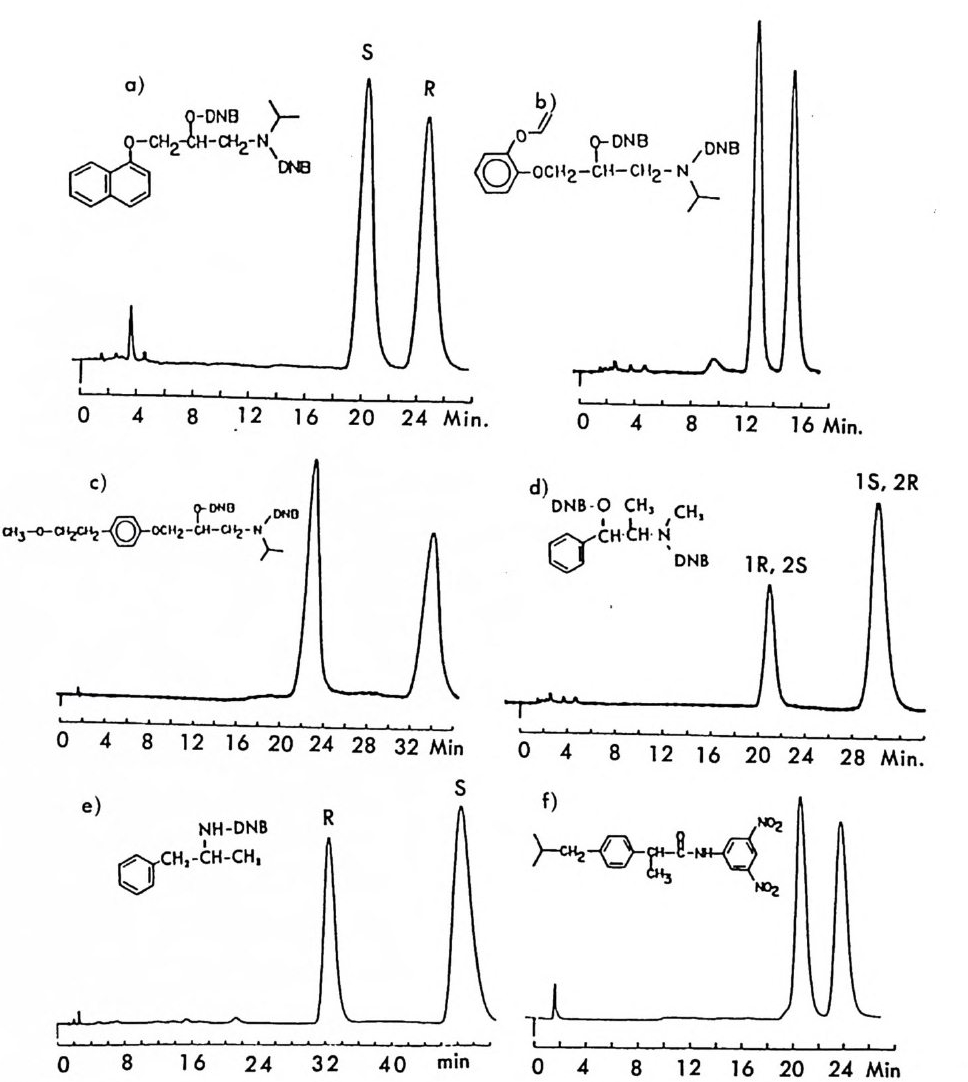 :CH2&°:&-N 「呼 s R 0@yoc l 2 G CONBI 룔 I N OI」 N B
:CH2&°:&-N 「呼 s R 0@yoc l 2 G CONBI 룔 I N OI」 N B
그림 7-45 a- 아릴알킬아민을 기저로 한 키랄고정상 6 에서 라세미 의약품 3, 5- 디 니트로벤조일 (DNB) 유도체들의 광학분안. a) 프로프라놀룰 b) 옥숙: 프레놀룰 c) 메토프롤롤, d) 에페드린, e) 암페타민, f)이부프로펜의 3,5- 디니트로아닐리드 유도체. 이동상: 핵산 /2- 프로판을 (80/20), 검 출기 : 254nm UV. (참고문헌 : 3)
전통제로 쓰이는 이부프로펜의 3, 5- 디니트로페닐아닐리드 유도체 광 학분할 뿐만 아니라 /3-차단제인 프로프라놀롤(p rop ranolol) 유도체의 광학분할 등 중요한 생리활성을 나타내는 의약품들의 광학분할에 대한 크로마토그램을 제시한 것이다. 여러 종류의 라세미 의약품들이 a- 아 릴알킬아민을 기저로 한 키랄고정상에서 광학분할이 가능하기 때문에 키랄고정상을 이용하면 라세미 의약품들의 인체내 변화과정 죽 광학활 성의 변화과정을 쉽게 추적할 수 있을 것으로 기대된다. 지금까지 살펴본 a- 아릴알킬아민을 기저로 한 키랄고정상에서의 광 학분할은 이동상으로서 대부분 핵산과 2- 프로판올의 혼합용매 를 사용 하여 실시된 것이나 역상 이동상을 사용한 광학분할 또한 가능하다. 예를 들면 아미노산의 DNB 유도체는 자유카르복시기를 가지고 있기 때문에 분석성분이 고정상에 너무 강하게 흡착되어 정상 이동상으로는 광학분할이 어려우며 물과 메탄올의 혼합용매에 NaHC03 을 소량 첨 가한 역상 이동상을 사용하여 광학분할이 가능하다(1. 17). 역상 이동 싱을 사용하여 광학분할하는 경우에도 분석성분과 키랄고정상 사이에 친지질성 상호작용이 중요하게 작용하는 경쟁관계에 있는 두 개의 키 랄성 인지 메커니즘에 의하여 키랄성 인지가 일어난다는 사실이 제안 되었다 (17). a- 아릴알킬아민을 기저로 한 키랄고정상 l 이나 키랄고정상 坪} 변 형된 형태로서 세 가지 다론 종류의 키랄고정상이 개발되어 있다. 키 랄고정상 g, 묘 혹은 브는 키랄아민이 우레아결합에 의하여 실리카 겔에 고정된 것이다 (18). 특히 키랄고정상 묘는 이소시아네이트 표를 3- 아미노프로필트리에톡시실란과 반응시킨 후 실리카 겔에 고정시켜 얻어지기도 하나 이소시아네이트 표의 반응성이 아주 크므로, 3- 아미 노프로필 실리카 겔이 충전된 상업용 컬럼에 이소시아네이트 L禁 ] CH2Cl2 용액을 재순환조작을 통하여 1 시간 정도 계속 흘려중으로써 HPLC 컬럼내에서 자동적으로 키랄고정상 표가 생성되도록 할 수 있 다(1 8). 키랄고정상 묘는 a-(1- 나프틸)에틸아민을 3- 시아나토프로필
 >H0HI -\ -(NNCS.II. /.. H) i -). 02.OI = - ) - EHn0 t 丁HH-\- N(NSI>Ii나 - O 2=)E3o t
>H0HI -\ -(NNCS.II. /.. H) i -). 02.OI = - ) - EHn0 t 丁HH-\- N(NSI>Ii나 - O 2=)E3o t
트리에독시실란과 반응시킨 후 실리카 겔과 함께 처리하는 간단한 과 정을 거쳐서 합성된 것으로써 (3) 상품화되어 있으며 (제 9 장 참조) Oi 등에 의해서도 연구 보고된 바 있다(1 9).
 O=C=N
O=C=N
우레아결합 키랄고정상들에 있어서 우레아결합은 아미드결합 키랄 고정상에 있어서 아미드결합보다 형태적으로 (co n£ orma ti onall y) 더 고정되어 있을 뿐만 아니라 이동상중의 2- 프로판을에 의하여 더 많이 용매화되어 있을 것으로 기대되기 때문에 경쟁관계에 있는 두 개의 키
랄성 인지 메커니즘 과정 죽 쌍국자겹침 과정과 수소결합 과정 중 쌍 극자겹침 과정에 크게 영향을 미치리라고 생각할 수 있다. 따라서 쌍 극자겹침 과정이 우세한 a- 아릴알킬아민 DNB 유도체를 우레아결합 키랄고정상에서 광학분할할 때에는 아미드결합 키랄고정상에서의 광 학분할과 비교하여 a 값이 작을 것으로 생각된다• 그러나 수소결합 과 정이 우세한 아미노산에스데르 DNB 유도체를 우레아결합 키랄고정상 에서 광학분할할 때에는 아미드결합 키랄고정상에서의 광학분할과 비 교하여 훨씬 더 좋은 광학분할을 기대할 수 있다. 그립 7-4 6{본 아미 노산에스테르 DNB 유도체의 광학분할을 도시한 것으로써 우레아결합 키랄고정상에서 훨씬 더 좋은 광학분할을 확인할 수 있다.
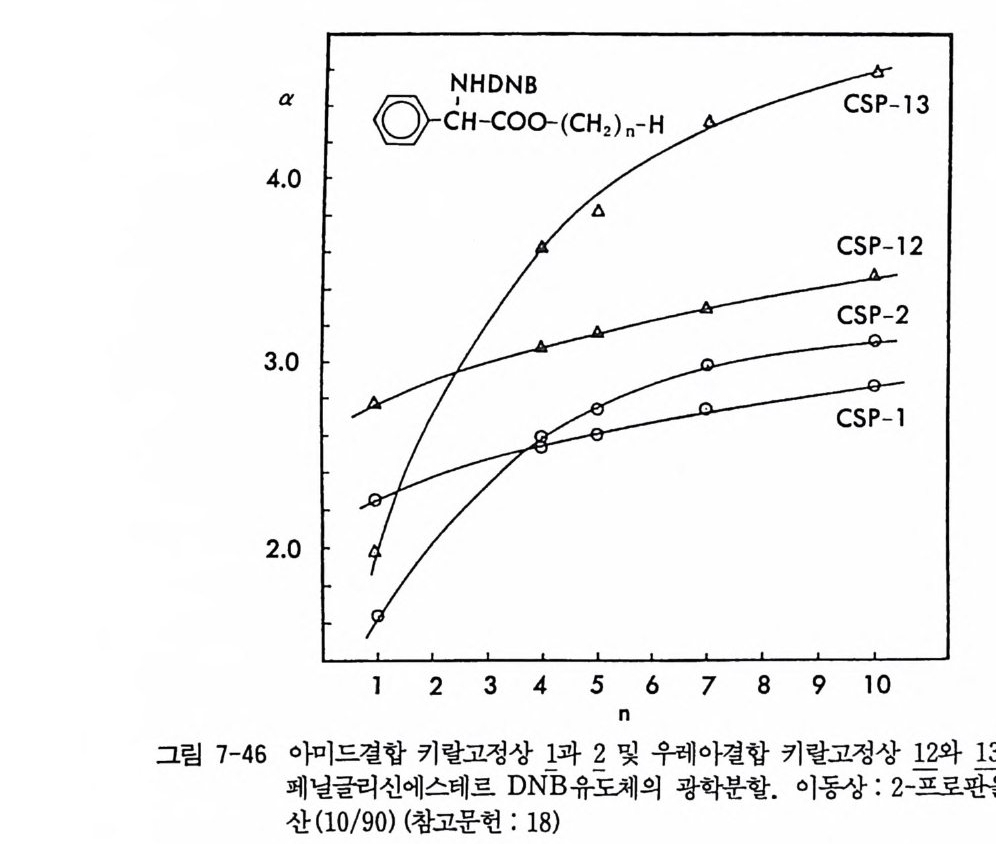 a’ 다 C선H H -DcN OB O - (CH 2 )0-H
a’ 다 C선H H -DcN OB O - (CH 2 )0-H
키랄고정상 伊} 같은 형태의 키랄고정상에서 N- 아세틸기를 N- 3, 5 디니트로벤조일로 치환한 키랄고정상 Lg 폰 같은 키랄고정상내에 강 한 r- 염기성기와 강한 T- 산성기를 동시에 가지고 있기 때문에 r - 염 기성 라세미 화합물들도 키랄고정상 L硏 J-ol l 서 광학분할이 가능한 것 으로 확인된 바 있다 (3 , 20). 또한 키탈고정상 伊 1 N_ 아세틸기 대신 (S) - N - (a 페닐)부탄오일기를 가전 키랄고정상 끄은 두번째 키랄중
 。
。
심에 있는 페닐기의 큰 입체장애로 안하여 쌍극자겹침 과정에 크게 영 향을 미칠 것으로 예상되나 현재 a- 히드록시카르복시산에스데르 3, 5- 디니트로페닐카르밤산 에스테르의 광학분할에 있어서 키랄고정상 l 보다 훨씬 좋은 광학분할능을 나타낸다는 사실이 보고되었을 뿐 (8) 앞 으로 더 많은 연구가 진행될 것으로 기대된다. 4.3 a· 아릴알컬아민을 기저로 한 T- 염기성 키랄고정상의 다른 예 P i rkl 랴 저자에 의하여 개발된 a- 아릴알킬아민 키랄고정상 의에
여러 종류의 a- 아릴알킬아민 키랄고정싱들이 시행착오의 방법으로 개 발되었다. a- 아릴알킬아민을 기저로한 키랄고정상을 ` 제조하는 데 있 어서 문제점은 광학적으로 순수한 a- 아릴알킬아민울 어떻게 얻을 수 있는가 하는 것이기 때문에 대부분의 키랄고정상들은- 상업적으로 쉽게 구할 수 있는 a-(1 - 나프틸)에틸아민이나 a- 페닐에틸아민을 기저로 하여 합성되었다. 그동안 a- 아릴알킬아민을 기저로 한 키랄고정상의 제조과정에 대하여 한편의 특허가 발표되었으며 (21) 그림 7-47 에 요약 된 키랄고정싱들이 연구보고된 바 있다. 이들 키랄고정상들은 모두 1C -영기성 키랄고정상들이므로 4.2 절에서 논의된 모든 라세미 화합물들 의 r- 산성 유도체들이 정도의 차이는 있으나 이들 키랄고정상에서 광 학분할될 것으로 생각된다. 그립 7-47 의 키랄고정상 챕은 a-(1- 나프 틸)에틸아민 및 발린을 기저로 한 키랄중심을 둘 가전 키랄고정상으로 서 T- 산성기 유도체로 만든 라세미 화합물의 광학분할에 있어서 키랄 중광학심분을할 하능나을만 보가일지 고뿐 만있 는아 키니랄라고 펜정프상로 旦파 트혹린은 (f旦 en에 p r o비 p a하 t h여r i n)더 겜 ,좋 은s -3307( 쁘), S-3308( 깐), 알레트롤론 (alle t hrolone) ('l]_), 알코올 라세미 체와 에스테르 라세미체들을 T- 산성 유도체로 만들지 않고 직접 광학 분할할 수 있는 것으로 보고되었다 (24) . 꼬깥 1 의 라세미 화합물들은 4.2 절의 키랄고정상들을 이용하여서도 직접 광학분할될 수 있을 것으 로 기대되나 아직 보고된 예는 없다. 그림 7-47 의 키랄고정상 盤은 키랄고정상 캡과 마찬가지로 두 개의 키랄중심을 가진 키랄고정상으로서 절대배열이 (R/R) 인 키랄고정상 과 (R/S) 인 키랄고정상의 두 종류 키랄고정상이 합성되었다 (29). (R/ R) 키랄고정상 챙과 (R/S) 키랄고정상 갬은 라세미 화합물의 광학분할 에 대하여 큰 차이점을 보이지는 않으나 이동상의 조성에 따라 광학분 할의 변화는 아주 큰 것으로 확인되었다. 죽 아미노산에스테르의 DNB 유도체를 광학분할할 때 이동상으로서 핵산 -2-프 로판올 혼합용 매를 사용하면 (R/R) -키랄고정상 盤과 (R/S) -키랄고정상 盤에서의
 「。〉: E〔 (CH2)3-N1H8::2:C0NH-:i 3:
「。〉: E〔 (CH2)3-N1H8::2:C0NH-:i 3:
그림 7-47 a-아 릴알킬아민을 기저로 한 키랄고정상들의 다른 예. 괄호내의 숫 자는참고문헌임.
 Acoo- 『~ 00 -0 \:,,,:HH-\\33 CH3
Acoo- 『~ 00 -0 \:,,,:HH-\\33 CH3
광학분할 사이에 큰 차이가 없으나 디클로로메탄과 핵산의 혼합용매를 이동상으로 사용하면 (R/S) -키랄고정상 썽에서는 헥산 -2- 프로판올 혼합용매를 이동상으로 사용할 때와 비슷하게 광학분할되나 (R/R) -키랄고정상 盤에서는 전혀 광학분할이 되지 않는다. 그러나 이러한 광학분할의 결과는 아직 합리적으로 설명되고 있지 않다. 그림 7-47 의 키랄고정상 낌은 아미노프로필 실리카 겔이 충전된 HPLC 컬럼에 디숙신이미도카르보네이트 (d i suc ci n i m i do carbonate : DSC) 의 아세토니트릴 용액과 펜타에틸렌핵사민 (pe nta e th y le nehex- am i ne) 의 아세토니트릴 용액을 차례로 흘려주어 여러 개의 아미노기 를 가진 실리카 겔 컬럼을 얻은 후 여기에 광학활성인 (R)- 혹은 (S) 숙신이미도 1-(a- 나프틸)에틸카바메이트의 아세토니트릴 용액을 홀 려주어 제조된 것으로서 (25) 아마도 단위 실리카 겔 표면적당 많은 수 의 1-(a-나 프틸) 에틸아민이 실리카 갤에 결합되어 있을 것으로 예측 된다. 키랄고정상 쌀상에서 광학분할이 시도된 라세미 화합물들은 아 미노산의 p-브로모페닐카르바밀유도체로서 역상이동상 (0.15M 나트륨
아세데이트(p H = 5) -아세토니트릴 : 30/70) 을 사용-하여 광학분할이 가능 하나 a 값은 그리 큰 편이 아니었다. 5 a --0}릴알킬아민을 기저로 하지 않은 7l_ 영기성 키랄고정 상 5.1 제 3 세대 亢_염기성 키랄고정상의 제조 및 광학분할의 특성 키랄성 인지의 상호성 개념에 따라, 아미노산 -DNB 키랄고정상으 로부터 유래되어 제조된 제 ~ ➔ l 대 키랄고정상들은 4.2 절의 a- 아릴알 킬아민을 기저로 한 키랄고정싱들 이의에도 여러 종류가 개발되어 라 세미 화합물들의 광학분할에 응용되었다. 3 . 3 절에서 기술된 바와 같이 히단토인 화합물들은 페닐글리신 -DNB 키랄고정상에서 광학분할이 잘되며(1 ,2) 합성이 용이하기 때문 에 페닐글리신 -DNB 키랄고정상에서 특히 광학분할이 잘되는 히단토 인을 선택하여 적절히 실리카 겔에 고정하면 제 양1 ] 대 상호성 키랄고 정상으로 사용할 수 있을 것이다 (2). 특히 히단토인 화합물들은 해당 하는 케톤 화합물로부터 쉽게 합성될 뿐만 아니라 제조용의 페닐글리 신 -DNB 키랄컬럼을 이용하여 쉽게 광학분할될 수 있기 때문에 (3) 광 학활성 히단토안을 기저로 한 키랄고정상을 제조하는 것은 그렇게 큰 문제가 아니다. 그림 7-4~ 세 종류 히단토인 키랄고정상 l,~ 및 2 은 5- 위치에 亢-염기성기로 작용할 수 있는 나프틸기 및 끝에 이중결 합을 가전 알킬기를 가지고 있는 광학활성 히단토인의 히드로실릴화 반응과 실리카 겔에의 고정과정을 거쳐서 합성되었다 (4). 이들 키랄고정상들은 아미노산 DNB 유도체의 광학분할(이동상은 역상 : 메탄올풍t -KHC03) 뿐만 아니라 아미노산에스데르 DNB 유도 체, 아미노산아미드 DNB 유도체 등 아미노산 유도체들의 광학분할 및 아미노알코올 DNB유도체, 아민 DNB 유도체, 아미노인산에스테
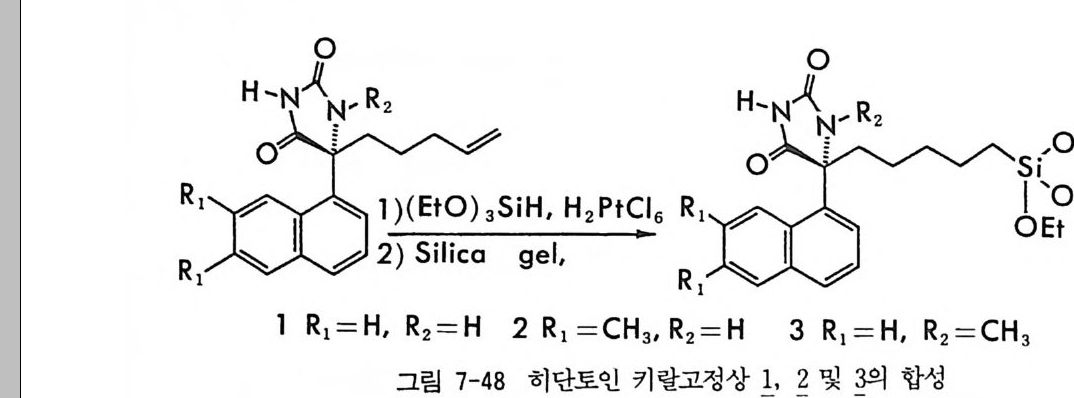 。
。
르 DNB 유도체들의 광학분할에도 성공적이었다 (4). 특히 히단토인 키랄고정상 2 는 히단토인 키랄고정상 l 에 비하여 T- 염기성이 더 큰 방향족기를 가지고 있어서 라세미 화합물의 DN B-유도체와 더 강한 r 국 상호작용을 하며 따라서 더 긴 머무름 시간 및 더 좋은 광학분할 능을보인다. 히단토인 키랄고정상에서의 광학분할 메커니즘은 그립 7-49 에서 보 는 것처럼 키탈고정상과 분석성분 사이의 r-r 상호작용 및 두 자리에 서의 수소결합에 의하여 설명이 가능하다. 죽 히단토인 키랄고정상과
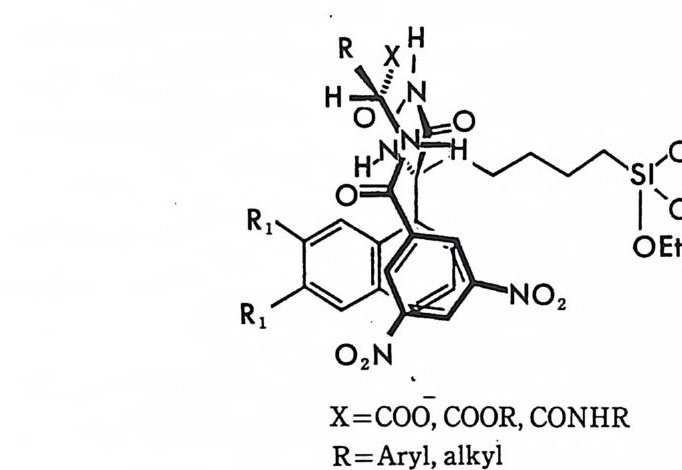 ::江 NOn I\,E,,o? ]
::江 NOn I\,E,,o? ]
그림 7-49 히단토인 키랄고정상에 의한 광학분할 메커니즘
(S) -이성질체는 7C-7C 상호작용 및 두 자리에서의 수소결합(키랄고정상 의 4- 카르보닐 산소와 분석성분의 N-H 수소 사이의 수소결합 및 키랄고정 상의 3 위치 N- H 수소 및 분석성분의 X 기 사이의 수소결합)에 의하여 상 호작용하나 R 구] 및 X- 기가 뒤바뀐 (R) -이성질체와 키랄고정상은 7C -亢 상호작용 및 한 자리에서의 수소결합에 의하여서만 상호작용하므 로 두 개의 광학 이성질체가 구분되는 것이다. 그립 7 - 49 의 키랄성 인지 메커니즘에 의하면 키랄고정상을 이루는 히단토인의 1- 위치 N-H 수소는 키랄성 인지 메커니즘에 전혀 관여하 고 있지 않을 뿐만 아니라 키랄성 인지와는 상관없는 수소결합의 양성 자 주게로 작용할 수 있기 때문에 1- 위치의 N-H 수소를 메틸기로 치 환하였을 때 더 좋은 광학분할능을 보일 것으로 예측되며 이와 같은 예측을 확인하기 위하여 히단토인 키랄고정상 g이 합성되었다. 그러 나 키랄고정상 청은 키랄고정상 l 에 비하여 분석성분의 키랄고정상내 머무름을 크게 감소시키기는 하였으나 예상과는 달리 키랄고정상 g이 키랄고정상 1 보다 더 좋은 광학분할능을 보이지 않았을 뿐만 아니라 어떤 경우에는 용리순서의 역전현상이 관찰되었다. 아마도 l- 위치 N -CH 려 도입으로 인하여 히단토인의 형태 (con fo rma ti on) 가 달라지기 때문에 키랄고정상 g에 의한 키랄성 인지 메커니즘은 그림 7-49 의 키 랄성 인지 메커니즘과는 다른 것으로 예상되고 있다 (4). 페닐글리신 -DNB 키랄고정상에서 광학분할이 잘되는 프탈리드 (ph th a li de ) 1 를 끝에 이중결합이 있는 요오드알켄으로 알킬화한 후 제조용 페닐글리신 -DNB 키랄고정상에서의 광학분할, 히드로실릴화 반응, 실리카 겔에의 고정과정을 거치면 그림 7-50 과 같이 프탈리드 롤 기저로 한 제 때]대의 상호성 키랄고정상 혀} 합성할 수 있다 (5). 키랄고정상 發근 아미노산에스데르, 아미노산아미드, 아미노산인산에 스테르, 아미노알코올, 아민화합물들의 DNB 유도체 광학분할에 이용 되었다. 다른 종류의 제 양 1] 대 상호성 키랄고정상과 비교하여 키랄고 정상 §가 특별히 더 좋은 광학분할능을 보이는 것은 아니나 특히 이차
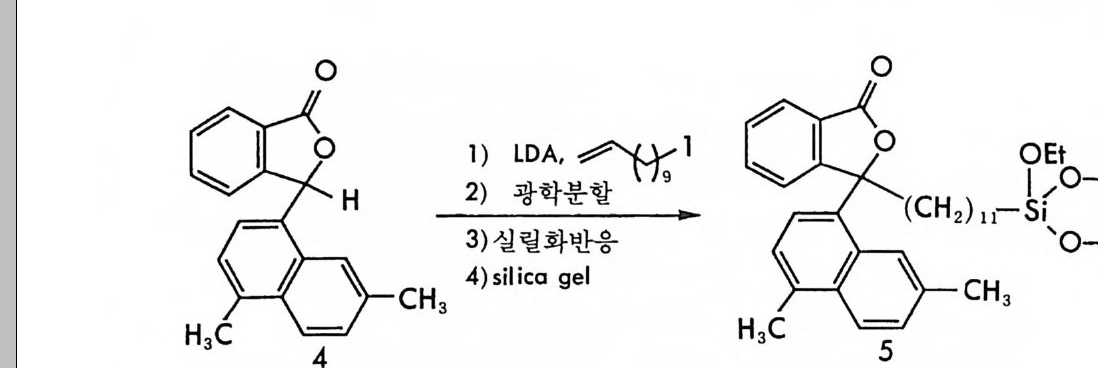 。23l))) 실광L릴D학A화, 분반~할웅 1 `` / 。 \\5Osi E0pIt/ t '1t, t ' f
。23l))) 실광L릴D학A화, 분반~할웅 1 `` / 。 \\5Osi E0pIt/ t '1t, t ' f
그림 7-50 프탈리드 키랄고정상 粹 1 합성과정
알코올의 DNB 유도체를 광학분할할 때 다른 제 3 세대 상호성 키랄고 정상보다 더 좋은 광학분할능울 보이는 것으로 확인되었다 (5). Pir k le 등에 의하여 개발된 제 양1 ] 대 상호성 키 랄고정상들이 여러 종류의 라세미 화합물들의 T- 산성 유도체들을 광학분할하는 데 있어 서 아주 유용하게 사용될 수 있디는 사실이 확인되었으나 이둘 키랄고 정싱들을 제조하기 위하여서는 제조용의 페닐글리신 -DNB 키랄고정 상에서의 광학분할을 통하여 광학적으로 순수한 광학활성 물질을 얻어 야 하는 과정이 포함되기 때문에 아칙 많은 사림들이 이용할 수 있도 록 상품화되어 있지 않다. 이와 같은 점에 있어서, 쉽게 상업적으로 구할 수 있는 광학활성 아미노산과 2- 나프톨 (2-na p h t hol) 을 반응시켜 얻은 N-(2- 나프틸)아미노산을 출발물질로 사용하여 합성되는 N -(2- 나프틸)아미노산 키랄고정상 伊斗 1은 특히 홍미를 끌만한 키랄고 정상들이다. (6) 더구나 라세미 N-(2- 나프틸)아미노산에스테르는 페 닐글리신 -DNB 키랄고정상 혹은 Leu-DNB 키랄고정상에서 광학분할 이 잘 되기 때문에 (7) 키랄성 인지의 상호성 개념에 의하여 N-(2- 나 프틸)아미노산울 기저로 한 키랄고정상 ~ 1 은 아미노산에스데르 혹 은 아미노산아미드 DNB 유도체들을 비롯하여 많은 종류의 T- 산성 라세미 화합물들을 광학분할할 수 있을 것으로 기대된다. 키랄고정상 伊 1 은 광학활성인 N-(2- 나프틸)알라닌 혹은 N-(2- 나프틸)발린과
10 운데센 - 1~ 념t 반응시켜 에스데르알킬기의 끝에 이중결합을 가지 고 있는 에스데르를 얻은 후, 히드로실릴화 반응과 실리카 겔에의 고 정과정을 거쳐 그림 7 - 51 과 같이 합성되었으며 (8,9) 이미 상품화된 바 있다(r제 9o 장 참조~). fC OOH H l)~OH , CH30S02H 2) 히드로실릴화반웅 3) Sil ica gel 二군운 -O-(CH2) n-\\] 67 RR==CCHH3( CH3)2
그림 7- 하 키랄고정상 6i!t 7 의 합성과정
키랄고정상 伊斗 1 은 아민, 알코올, 티울, a - 아미노산, g - 아미노 산, a 쵸댄든 B- 아미노산 에스데르, a 설茂 7 g-아미노산 아미드 등의 DNB 유도체, 3,5- 디니트로페닐우레아 유도체, 3,5- 디니트로페닐카 르밤산 에스테르유도체 등을 광학분할할 뿐만 아니라 카르복실산의 3, 5- 디니트로아닐리드 유도체 동을 광학분할하며 어떤 경우에는 a 값 이 10 이 넘을 정도로 아주 좋은 광학분할능을 보이는 것으로 확인되었 다 (8,9). 예를 들면 키랄고정상 忠o 버 ]서 루이신부릴아미드 DNB 유도 체의 광학분할에 대한 a 값은 무려 15 . 40( 이동상 : 2-프 로판을-핵산= 10/90) 이며 (9) 키랄고정상 1 상에서 루이신부틸아미드 DNB 유도체의 광학분할에 대한 a 값은 17.66 이었다(이동상: 2- 프로판을=핵산, 10/ 90)(8). 이들 의에도 키랄 디올의 3,5- 디니토로페닐카르밤산 에스테
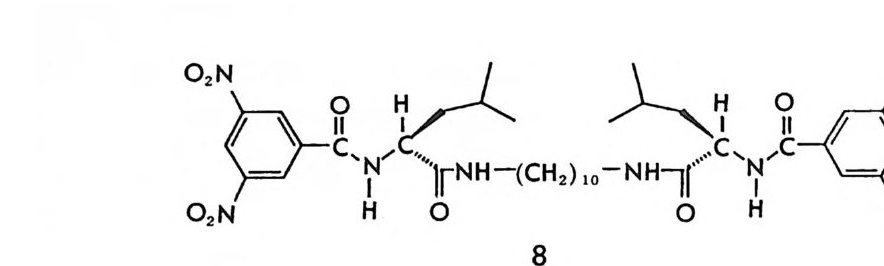 0\\N》 \¢-(CH2)81 0_二 \,,,? :NN0\
0\\N》 \¢-(CH2)81 0_二 \,,,? :NN0\
르유도체 (10 )' 디펩티드 혹은 트리펩티드에스데르의 DNB 유도체 (ll) 등이 키랄고정상 몬} 1 에서 광학분할되는 사실이 보고된 바 있다. 홍미있는 광학분할은 N-(3,5_ 디니트로벤조일)루이신의 이중아미드 인 화합물 針즌 키랄고정상 §에서 a 값이 12~포 료 분리된다는 사실이 다. 이중아미드 유도체를 만듦으로써 두 광학이성질체 (RR 및 SS 이성 질체)와 키랄고정상 사이의 상호작용이 이중으로 일어나서 키랄고정상 과 이중아미드 유도체 8 사이의 상호작용에 대한 66G( 제 2 장 참조) 가 단일아미드 유도체와의 상호작용에 대한 ^^G 에 비교하여 두 배 가 되며 이것은 결국 a 값을 대략 筑]곱으로 하기 때문에 아주 큰 a 값 을 관찰할 수 있게 된다(1 2). 키랄고정상 쑨斗 키랄고정상 1 에 의한 T- 산성 유도체들의 광학분할 메커니즘은 (S)-N-(2- 나프틸)알라닌의 메틸에스테르와 (S)-N _(3,5- 디니트로벤조일)루이신의 n- 프로필아미드사이에형성되는칙물의 1H NMR Overhauser 효과 (Nuclear Overhauser Ef fec t : NOE) 를 연 구하고(1 3, 14) 키 랄고정상 §x.1- 1 에 의한 광학분할의 · 특성을 고찰함으 로써 그립 7-5~ 갇이 제안되었다. 그림 7-52 에서 보는 것처럼 키랄 고정상은 분석성분과의 T- 亢상호작용에 필요한 T- 염기성기 (2- 나프틸 고리), 수소결합의 양성자 주게 (아미노기의 N-H), 수소결합의 양성자 받게 (에스데르의 카르보닐산소)를 가지고 있기 때문에 이에 대응하는 상호작용 부위를 가전 분석성분들의 두 광학 이성질체는 절대 배열에 따라 하나의 광학 이성질체는 키랄고정상과 세 점 상호작용(경우에 따 라 입체장애 상호작용을 포함하여)을 하고 반대의 절대배열을 가전 광학 이성질체는 키랄고정상과 두 점 상호작용을 하게 되어 두 개 광학 이
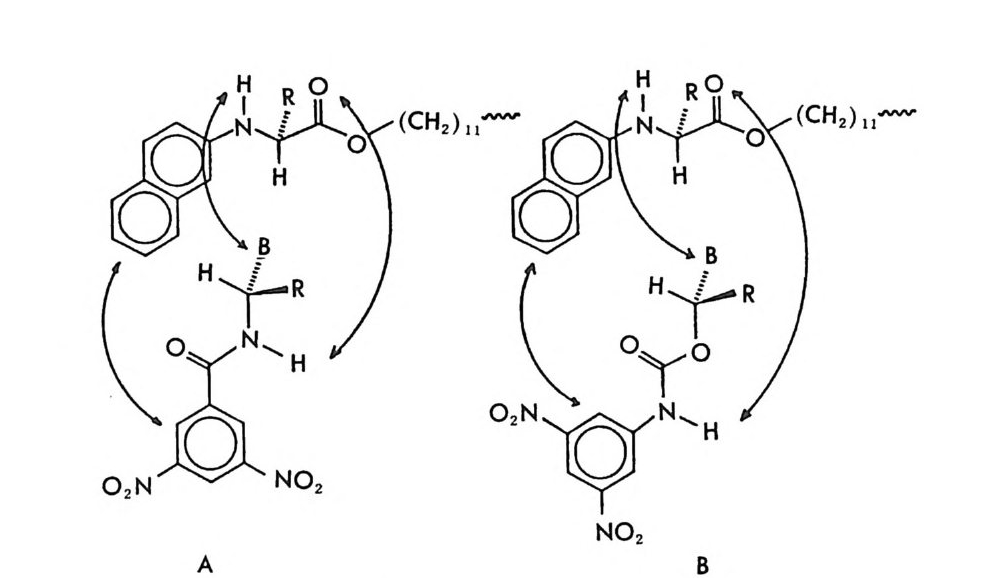 A N02 B
A N02 B
그림 7-52 키랄고정상 6 혹은 7 에 의한 광학분할의 메커니즘. A 는 키랄고정상과 키랄아민 DNB 유도계의 세 점상호작용을 나타내며 因근 키랄고정상과 키랄알코올의 0-(3, 5 - 디니트로페닐) 카르밤산에스데르 유도체의 세 점 상호작용을· 나타낸다. A 및 B 모두 키랄고정상에 더 오래 머무르 는 광학 이성질체를 나타내고 있다.
성질체의 구분이 가능하게 된다. 키랄고정상 § 혹은 1 에서 라세미 화 합물들이 광학분할될 때 두 광학 이성질체의 용리순서는 예의없이 모 두 그림 7-52 의 키랄성 인지 메커니즘으로 예측되는 용리순서와 일치 하는 것으로 확인되었다. 따라서 이와 갇은 규칙적인 용리순서는 절대 배열이 알려져 있지 않은 키랄 화합물의 절대배열을 결정하는 데 유용 하게 응용될 수 있을 것이다. 다른 종류의 홍미있는 제 양1 ] 대 상호성 키랄고정상에는 페닐글리신 -DNB 키랄고정상에서 광학분할이 잘되는 알킬아릴포스핀옥시드 (alky la ryl p h osph in e ox i de) 를 기저로 한 키랄고정상 9 와(1 3) 이소퀴 놀리논(i so qui no li none) 골격을 기저로 한 키랄고정상 lQ이 있다 (14). 1C- 염기성 키랄고정상 堅와 L仮 본 다른 종류의 1C- 염기성 키랄고정상들 과 마찬가지로 아미노산에스데르나 아미노산아미드 DNB 유도체들의
광학분할에 응용될 수 있다. 특히 키랄고정상 !.Q에서 아미노산에스테 르나 아미노산아미드 DNB 유도체를 광학분할할 때 a 값은 이동상으 로 핵산-에탄올 혼합용매를 사용할 때보다 핵산-클로로포름을 사용할 때 훨씬 크며, 아미노산에스데르 DNB 유도체를 광학분할할 때의 두 광학 이성질체의 용리순서와 아미노산아미드 DN B-유도체를 광학분할 할 때의 두 광학 이성질체의 용리순서가 서로 반대라는 사실로부터 Tambute 등은 아미노산아미드 _ DNB 유도체의 아미드 N- H 수소와 키랄고정상의 카르보닐 산소 사이의 수소결합이 키랄성 인지에 중요한 역할을 하고 있는 키랄성 인지 메커니즘을 제안한 바 있다 (14). 5.2 a --0}릴알컬아민을 기저로 하지 않은 1l- 염기성 키랄고정상 의 다른예 a- 아릴알킬아민을 기저로 한 키랄고정상들과 a_ 아릴알킬아민을 기 저로 하지 않은 제 양 1] 대 상호성 키랄고정상들 이의에도 T- 산성 유도
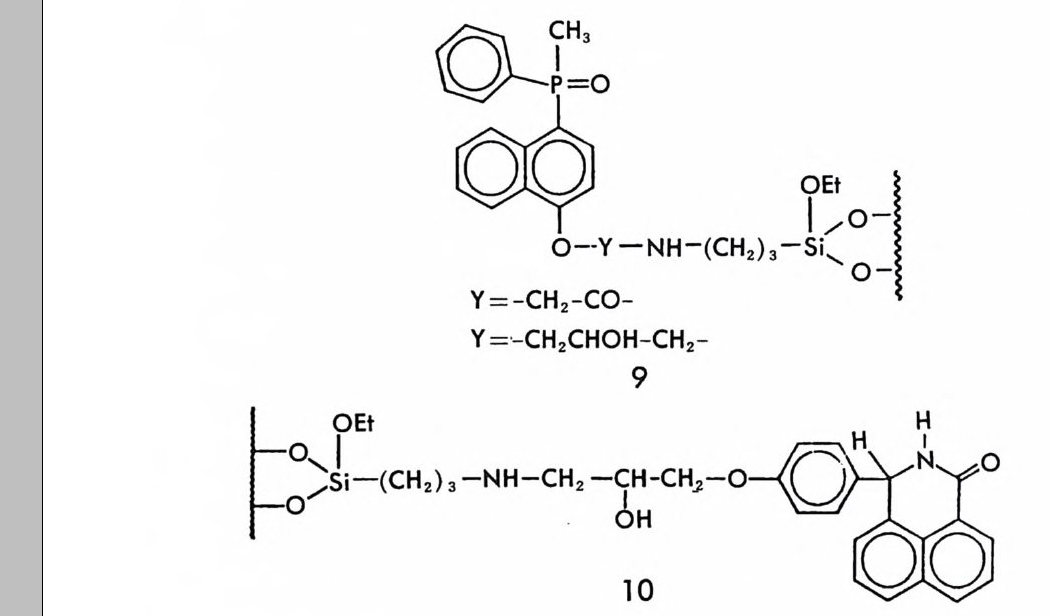 YY ==-·- CC HH22C-CHOO-H-C H2-
YY ==-·- CC HH22C-CHOO-H-C H2-
체로 유도화된 라세미 화합물들의 광학분할에 이용된 7[ _ 염기성 키랄 고정상들이 다수 알려져 있다. Oi 등은 a_(1- 나프틸)에틸아민을 기저로 하고 키랄중심을 둘 가진 키랄고정상 (4 . 3 절의 키랄고정상 겐)이의에도 키랄중심을 두 개 이상 가 지고 있는 키랄고정상으로서 키랄고정상 旦과(1 5) 키랄고정상 旦를 (16 ) 개발하였으며 이들 키랄고정상들을- 이용하여 라세미 화합물둘의 7[ - 산성 유도체를 광학분할하였고 이들 키랄고정상들은 이미 상품화되 어 있다(제 9 장 참조). 키랄고정상 旦과 끄는 특별히 강한 7[_염기성 방향족기를 가지고 있지 않으나(따라서 키랄고정상 브과 브를 7C- 염기성 키랄고정상으로 분류하기에 애매한 점이 있으나, 이들 키랄고정상들이 7C 犬산성 유도체의 라세미 화합철 늘을 광학분할한다는 점을 고려하여 7C- 염기 성 키랄고정상으로 취급하였음) 라세미 알코올 혹은 a- 히드록시카르복 실산에스테르의 3,5- 디니트로페닐카르밤산 에스데르유도체 및 라세미 아민의 DNB 유도체와 같은 라세미 화합물의 7[-산성 유도체의 광학 분할에 성공적으로 응용되었다 (15,16). 그러나 이들 키랄고정상들은 두 개 이상의 키랄중심을 가지고 있기 때문에 이들 키랄고정상들이 어 떤 키랄성 인지 메커니즘에 의하여 두 개의 광학 이성질체를 구별하는-
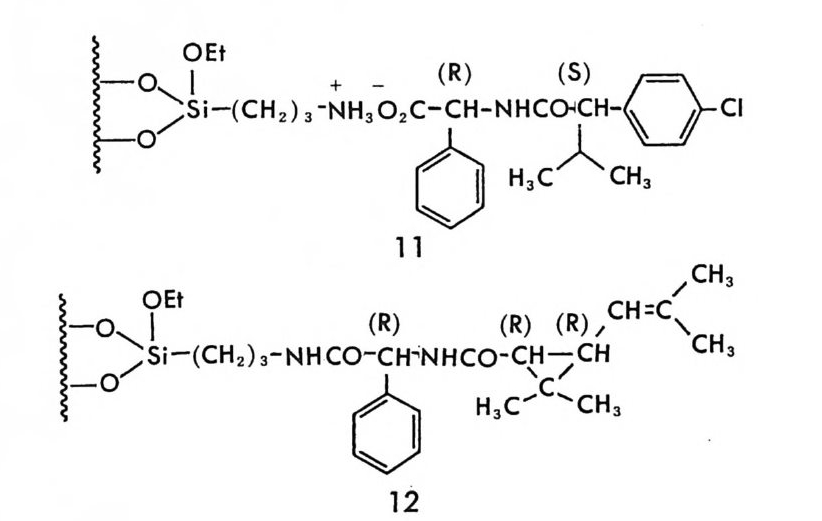 OEtI C
OEtI C
지 설명하기가 매우 어렵다. 득히 키랄고정상 旦 에 서 1 _ 혹은 3 - 0 - 모 노알킬글리세 롤 3, 5- 디니트로페닐카르밤산 에 스 데 르 유도체의 광학분 할(i 7). 긴 알킬기를 가전 알칸 - 1,2 - 디올 3,5 - 디니트로페닐카르밤산 에 스데르 유도체의 광학분할(1 8). 단백질 키나아제 C(p ro t ei n kin a se C) 를 활성화 시키는 것 으로 알려진 디아실 글리 세 몰 (d i ac y l g l y cerol) 의 3, 5- 디니트로페닐카르밤산 에 스데르유도체의 광학분 할(1 9) 등은 흥 미 있는 광학분할의 예이다. 키랄고정상 旦는 라세미 안코운, 라세미 아 민의 r- 산성 유도체 를 광학분할할 뿐만 아니 라 키란고정상 ] _2 의 합상 과정에서 사용된 크리산데미산 ( c hr y sa n t h em i c ac i d) 의 라새미 세 (키안 중심이 둘 이라서 네 개의 입체 이성 질체가 존재함) 의 광학분한 동 다양하 게 응용되고있다. Yamashit a 등 은 비나프 틸 (bin a p h th y l) 형 의 키 안고정 상 旦과 旦를 합성하여 아미노산에스데르의 D NB 유도체, 단일절합주위의 자유회전 의 제한때문에 생기는 키랄화합물안 이 중 아 릴화합물 ( b i ar y l s) , 아 릴알
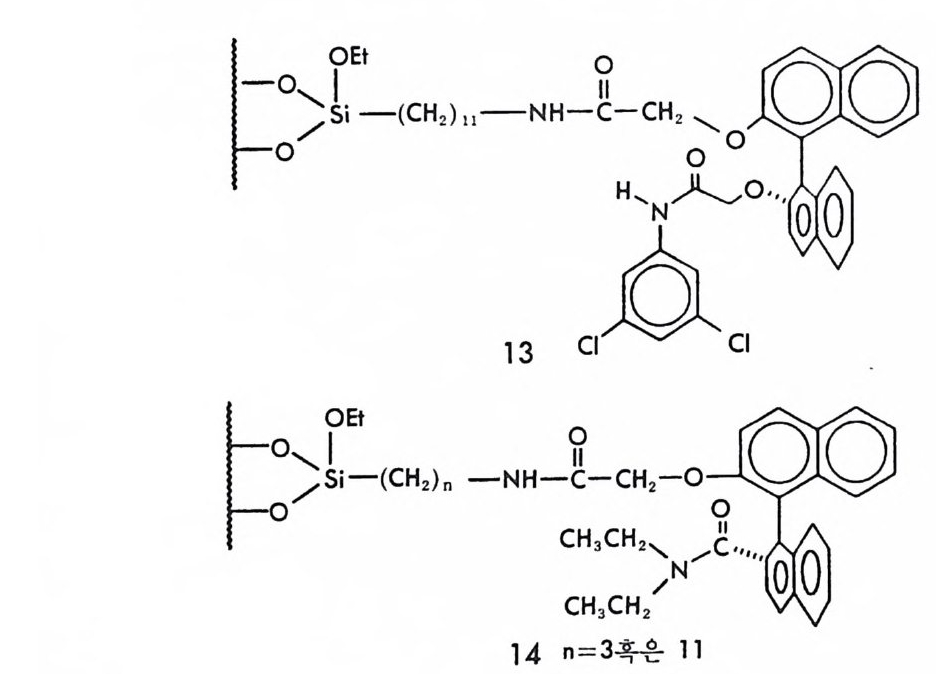 仁〉『 ~(CH ,) ,. -NH- 『 _CH 2 0\ 。
仁〉『 ~(CH ,) ,. -NH- 『 _CH 2 0\ 。
킬카르비놀의 디 클로로페닐 카르밤산 에 스 테 르 등을 광학분할할 수 있 었다 (20) . 최근 에 는 1 , 1' - 비나 프탈렌 ~ 2,2' - 디카르복실산을 출발물질로 사용하여, 두 개의 카르복시기 중 하나 를 광학활성인 페닐에틸아민과 반응시켜 아미 드를 만든 후 나머지 하나의 카르복시기를 이용하여 실 리카 겔 에 고정함으로써 Yamashit a 등은 두 개의 키랄성을 가전 키 란고정상 拉쯔 및 拉 h 를 합성할 수 있었다 ( 2 1). 특히 키랄고정상 巨죤 및 柱b 간 합상하는 과정에 있어서 라세미 1,1' - 바나프탈렌 - 2,2' -디카 로복신산과 (S) 一 ( - ) - 1 _ 페닐에틸아민을 반응시켜 얻은 두 개의 부분 입체 이성 진체 aS~ 라 aRS(a 는 축을 의미하며 비나프틸기의 절대배열을 표현하기 위하여 사용점)는 쉽게 재결정 방법으로 분리되기 때문에 키 랄고정상 拉죤와 巨屈 근 비교적 쉽게 합성된다. 키랄고정상 旦항斗 프 E 는 아미노산에 스데르의 DNB 유도체, 라세미 아민의 DNB 유도체, 라세미 알코 올 의 3, 5- 디니트로페닐카르밤산 에스테르유도체를 광학분 할할 수 있 을 뿐만 아니라 비아릴 (b i ar y l) 계통의 라세미 화합물들도 광학분할한다. 특 히 이 들 라세마 화합물들을 광학분할할 때 키랄고정 상 拉 P 가 키랄고정상 登―따i다 더 좋 은 광학분할 결과를 보이는 것으 로 보아 (aR) _ 비나프탈렌과 (S) -페닐에틸아민이 라세미 화합물의 광 학분한 에 대하여 서로 협동적인 관계에 있음~ 예측할 수 있으며 비아
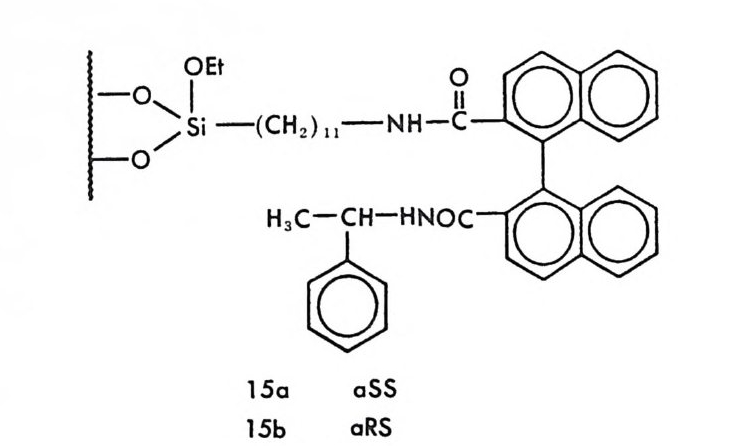 仁〉『\ (CH 2 ) II-NH- 『
仁〉『\ (CH 2 ) II-NH- 『
릴화합물들의 광학분할에서 관찰한 두 광학 이성질체의 용리순서가 키 랄고정상 拉브와 키랄고정상 拉戶에서 서로 반대라는 사실로부터 라세 미 화합물의 키랄성 인지에는 키랄축때문에 키랄성을 가지는 비나프탈 렌이 더 중요한 역할을 하고 있음을 예상할 수 있다. Pett er son 등은 아세틸퀴닌 (ace ty l q u i n i ne) 을 실리카 겔에 고정시 킨 키랄고정상 登을 합성하였다 (22). 키랄고정상 L綴 븐 역상 이동상 을 사용한 페녹시기에 할로겐원자가 치환된 2 - 페녹시프로피온산의 광학 분할, 아미노산의 벤족시카르보닐 (CBZ) 유도체, 단실유도체 및 t- lf 톡시카르보닐 (Boe) 유도체의 광학분할, 만델산과 같은 라세미 카르복 실산의 광학분할, 혈액응고 방지제인 와파린의 광학 분할 등 다양하게 이용되었다. 키랄고정상 L伊 1 기저물질과 비슷한 알칼로이드 를 기저로 한 키랄 고정상 끄은 요도화 N- 메틸퀴니니움 (N - me t h y l qu i n i n i um i od i de) 과 Y- 머캅토프로필 실리카 겔을 반응시켜 간단하게 합성되었으며 정상
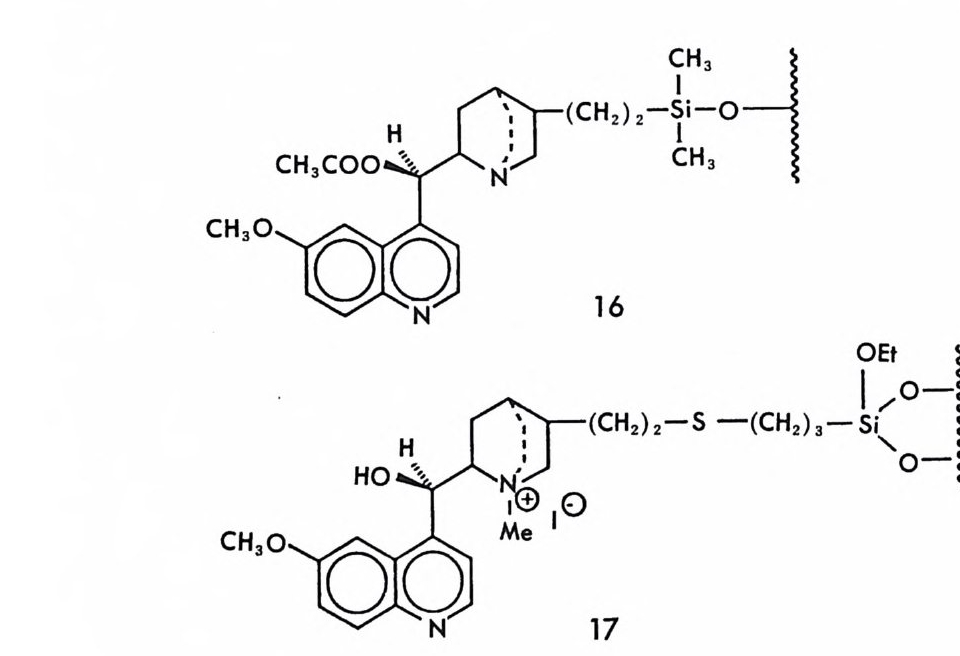 (CH 2 )2_ 『三
(CH 2 )2_ 『三
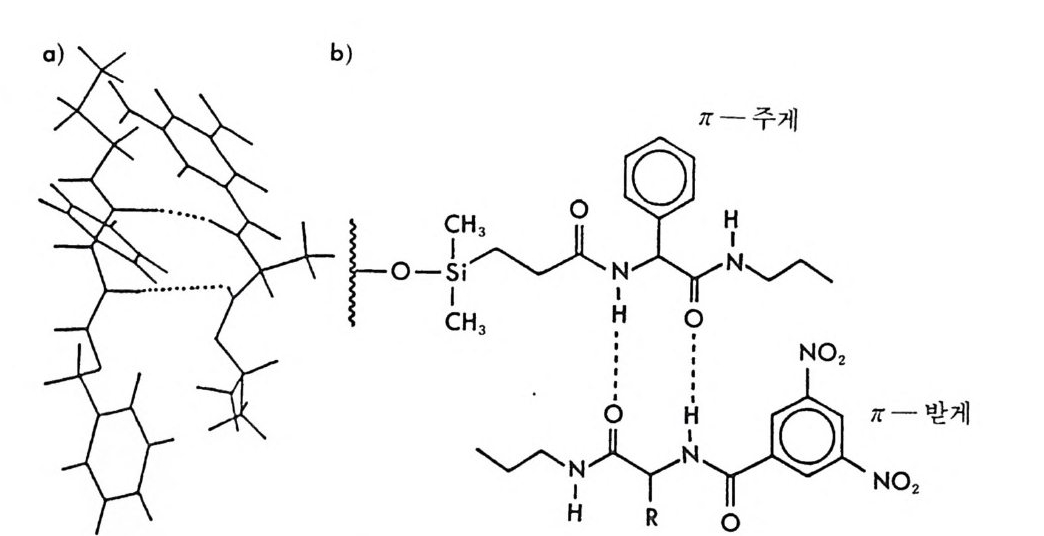 十 0- 〔二\\
十 0- 〔二\\
그림 7-53 (a) 키 안고정 상 1~ :3, 5 - 디니트로벤조일 - L - 알라닌 - 2- 프로필에스테르 의필 아상미호 드작 -용 D 에N &대 시 한상 호컴작퓨용터(그참림고 및문 헌(b ) : 키24랄 및고 정25상). 안과 아미노산 프로
이동상을 사용하여 5 - 아 릴 히단토인, 이미드화합물, 벤조디아제피논 (benz od ia z ep ino ne) , 이뇨제인 에토졸린 (et oz oli ne ) 등을 광학분할할 수 있었다 (23). a - 아 릴 기 를 가진 아미노산의 에스데르 혹은 아미드를 실리카 겔에 공유결합시킨 키 랄 고정상들은 a - 위치의 아릴기가 7C- 염기성기로 작용 하기 때문에 a- 아릴아미노산을 기저로 한 키랄고정상 旦, _12, 챕 및 잎은 아미노산에스데르의 DNB 유도체와 아미노산아미드의 DN B-유도 체 믈 광학분할한다 (24,25). 키랄고정상 登,旦, 쳅 중 7C- 나프틸기를 가전 키랄고정상 씬에서 두 광학 이성질체의 머무릅- 시간이 특별히 긴 것을 관찰하였으며 이것은 키탈고정상과 7C- 산성 라세미 화합물 사이 의 7C-7C 상호작용이 분석성분 분자의 키랄고정상내 머무름에 중요한 역할을 하고 있음을 시사해 주고 있다. 컴퓨터계산에 의하면 키랄고정 상 L料 } 분석성분 (N-3, 5- 디니트로벤조일 -L- 알라닌 -2- 프로필에스테르) 사이의 최소에너지에 해당하는 가장 안정한 착물은 그림 7-53~ 에서
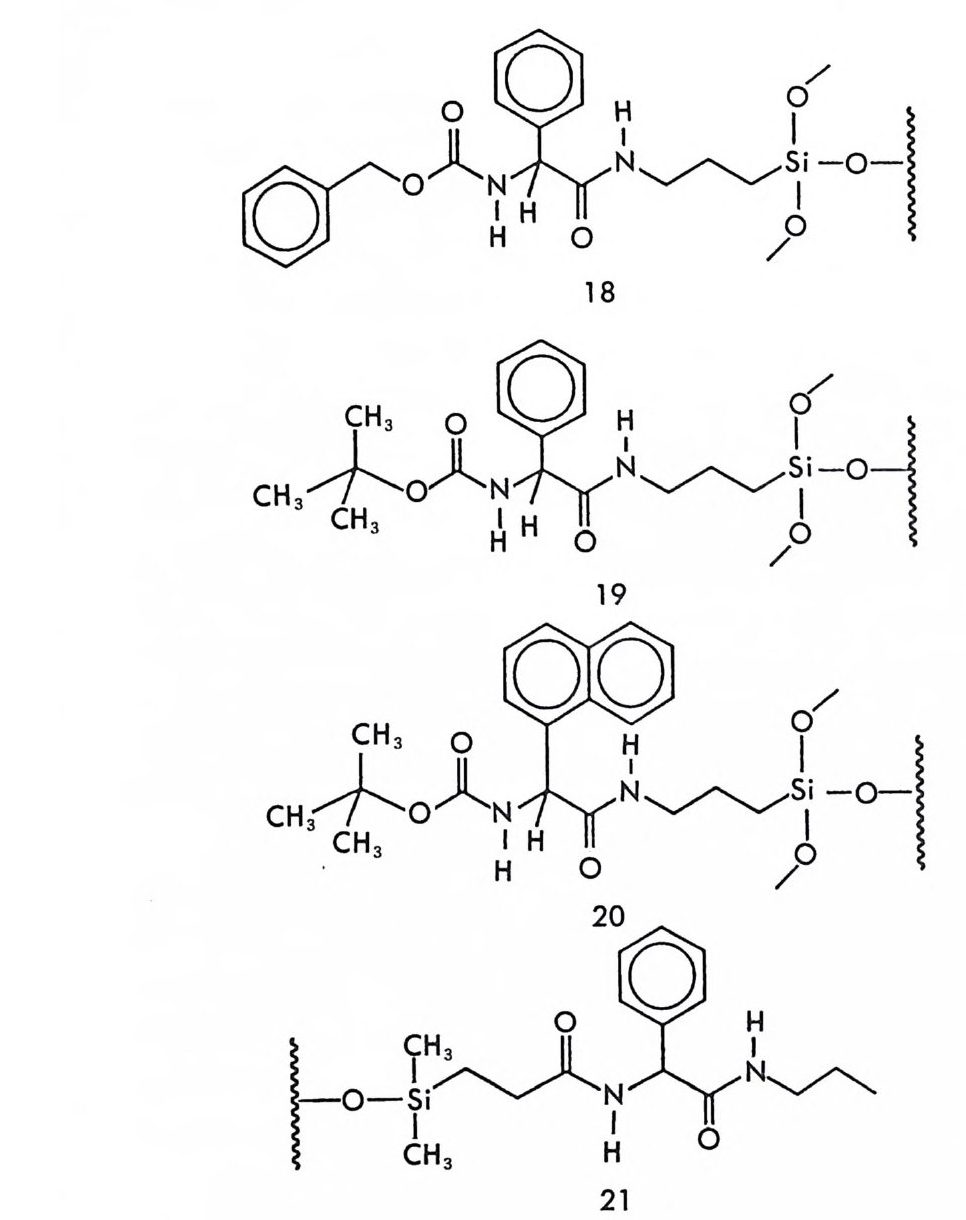 0` 夕 Hl °l /
0` 夕 Hl °l /
보는 바와 같이 키랄고정상의 a - 아릴기와 분석성분의 DNB 사이의 r -7r:상호작용 및 두 자리에서 [N- H … … 0= 이형의 수소결합에 의한 것임이 연구되었다 (24).
키랄고정상 낌에서는 아미노산의 2 - 프로필에스테르 DNB 유도체보 다는 아미노산의 N - 프로필아미드 DNB 유도체를 광학분할할 때 두 광학 이성질체를 분리하는 정도 (a 값)가 2.5 배 이상 증가한다는· 사실 이 확인되었다 (25). 이와 갇은 사실은 아미드기가 에스데르에 비하여 카르보닐산소의 전자밀도를 증가시키기 때문에 그립 7-53~ 에 표시한 키랄고정상과 분석성분 사이의 수소결합 세기를 증가시켜 (수소결합의 에 키안 고 정상의 a - 페닐기 및 분석성분의 DNB 사이에 1C?r 상호작용이 있 유) 키안성 인지의 정도 를 크게 향상시키기 때문인 것으로 설명된다 (25 ). 아미노산의 N, N - 디메틸아미드 DNB 를 키랄고정상 낌상에서 광학분할할 때에는 분석성분의 N, N- 디메틸기가 아미드의 카르보닐 산소의 전자밀도 를 더욱 증가시켜 a 값은 더욱 커지는 것으로 관찰된 바 있다 (25).
참고문헌 (제 7 장, I) 1. R. Foste r , Orga n ic Clwng e- T ransfe r Comp lex es, Academi c Press, New York, 1969. 2. W. Holste i n and H. Hemets b erge r, Chromato g rap h ia , 杓 186 (1982). 3. W. Holste i n and H. Hemets b erge r , Chromato g rph ia , 프, 251 (1982). 4. L. Nondek, J. Chromato g r., 쁘, 61 (19 86). 5. F. Mi ke s, G. Boshart a nd E. Gi l-A v, J. Chromato g r., 쁘, 205 (19 76). 6. M. S. Newman and D. Lednic e r, J. Am. Chem. Soc., 전, 4765 (1956). 7. F. Mi ke s and G. Boshart, J. Chroma t o gr.선언, 455 (19 78). 8. J. M. Fin n , in Chroma gra ph i c Chir a l Sep a rati on s, M. Zie f and L. J. Crane, Eds., Marcel Dekker, New York, 1988, chap 3. 9. P. Macaudie r e, M. Lien ne, A. Tambute and M. Caude, in Chir a l Sep a rati on s by HPLC : Ap pli ca ti on s to Pharmeceuti ca l Comp ou nds, A. M. Krstu l ov ic, Ed., Ell is H orwood, Chic h este r , 1989, chap 14.
10. T. D. Doy le , in Clzh·a/ Liq u id Chromato g 1 p l 1 y , W. J. Louug h , Ed., Blackie , Glasgo w and London, 1989, chap 6. (제 7 장, 2) 1. W. H. Pir k le and D. J. Hoover, i n Top ics in Ste r e o c lzem i st r y,, v ol13, N. L. Al ling e r and E. L. Elie ! , E ds., W ile y Inte r s c i en ce, Now York, 1982, pp. 263-331 . 2. W. H. Pir k le and D. W. House, J. Org, Che m ., 션 , 195 7 (19 79). 3. W. H. Pir k le, D. W. House and J. M. Fin n , J. Chromato g r., 192 , 14:J (1980). (제 7 장, 3) 1. J. Jac q u es, A. Collet and S. H. Wi le n, Enanti om ers, Racemate s and Resoluti on s, Wi ley -l n te r scie n ce, New York, 1981, pp. 306 -3 07 . 2. W. H. Pir k le and J. M. Fin n , J. Org. Chem. , i§_, 2935 (1981). 3. J. M. Fin n , in Clzr0 1n a.fo g r a p liic Chir a l Sep a rati on s, M. Zie f and L. J. Crane, E ds., Marcel Dekker, N ew York, 1988, chap 3. 4. W . H. Pir k le, D. W. House and J. M. Fin n , J. C hromato g r., 192, 143 (19 80). 5. W. H. Pir k le, J. M. Fin n , B. C. Hamp er , J. Schrein e r and J. R. Prib i s h , in As ym metr ic R eacti on s and Proces ses in Chemi stry, E. L. Elie ! , Ed., ACS Serie s 185, ACS, Washin g ton D. C. 1982, chap 18. 6. M. Kasai, C. Froussio s and H. Zif fer , J. Or~. Chem. , 샌 , 459 (19 83). 7. W. H. Pir k le, J. M . Fin n , J. L. Schrein e r and B. C. Hamp er , J. Am. Chem. Soc., 103, 3964 (1981). 8. W. H. Pir k le and J. L. Schrein e r, J. O rg. Chem ., 쁘, 4988 (1981). 9. R. D. Sti pa novic , J. P. McCormi ck , E. 0. Schlemp e r, B. C. Hamp er , T. Shir u ny o zu and W. H. Pir k le, J. Org. Chem., 안, 2500 (1986). 10. S. K. Yang and H. B. Weems, Anal. Chem., 쁘, 2658 (1984). J1 S. K. Yang , M. Mushta q , Z. Bao, H. G. Weems, M. Shou and X. -L.
Lu, J. Chromato g r., 쁘 , 377 (19 89). 12. S. K. Yang , H. B. Weems, M. Mushta q and P. P. Fu, J . chromato g r., 316, 569 (19 84). 13. S. K. Yang , M . Mushta q , H. B. Weems and P. P. Fu, J. Liq . Chromato g r. , 언, 473 (19 86). 14. W. H. Pir k le, A. Ts ip o uras, M. H. Hy u n, D. J. Hart and C. -S . Lee, J. C hromato g r., 쁘~ . 377 (19 86). lS. Z. -Y . Yang , S. Barkan, C. Brunner, J . D. Weber, T. D. Doy le and I. W . Wain e r, J. Chromato g r., 핀, 444 (1985). 16. W. H. Pir k le and A. Tsip o uras, J . Chromato ar. , 쁘, 291 (19 84). 17. W . H. Pir k le and T. ]. Sowi n, J. Chroma t o gr . 쁘7.., 313 (19 87). 18. I. W . Wain e r an d T. D. Do y le , J. Chromato g r., 쯔 117 (1984). 19. D. V. Ni co ll-G rif fith, J. Chromato g r., 쁘 179 (19 87). 20. W. H. Pir k le and J. E. McCune, J. Chromato g r., 쁘 , 67 (1989). 21. I. W. Wain e r, T. D. Doy le . Hami dz adeh and M. Aldrid g e , J. Chromato g r., 268, 107 (19 83). 22. W . H. Pir k le, C. J. Welch and M. H. Hy un , J. Org. Chem., 쁘, 5022 (1983). 23. I. W. Wain e r and T. D. Doy le , J. Chroma t o gr.딴~. 465 (1983). 24. W. H. Pir k le, T. C. Pochap sk y, G. S. Mahler and R. E. Fie l d, J. Chromato g r., 348, 89 (1985). 25. W. H. Pir k le and T. C. Pochap sk y , J. O rg. Chem.' 탄 , 102 (19 86). 26. A. Tambute , P . Gareil , M. Caude and R. Rosset, J. Chromato g r., 쁘 81 (1986). 27. P. Pescher, M. Caude, R. Rosset and A. Tambute , J. Chromato g r., 371, 159 (1986). 28. N. Wad, J. Liq. Chromato g r., 브 , 1107 (19 88). 29. N. Oi, H. Ki tah ara, Y. Mats u moto , H. Nakazim a and Y. Horik a wa, J. Chromato g r., 쁘, 382 (1989). 30. S. L. Abid i , J. Liq. Chromato g r., 핀 , 1085 (1987). 31. P. Cami ller i, C. Dy k e and F. Hossner, J. Chromato g r., 므, 471 (1989). 32. G. R. Cay le y and B. W . Sim p so n, J. Chroma t o gr.관~. 123 (1986).
33. J. K. Baker, A. M. Clark and C. D. Huff or d, J. Liq. Chromato g r., ~. 493 (19 86). 34. W. H. Pir k le and C. J. Welch, J. Org. Chem., 쁘 , 138 (1984). 35. I. W. Wain e r and M. C. Alembik , J. Chroma t o g r . 멘?, 59 (1986). 36. P. Macaudie r e, A. Tambute , M. Caude, R. Rosset, M. A. Alembik and I. W. Wain e r, J. Chromato g r., 판 177 (19 86). 37. T. D. Doy le , W. M. Adams, F. S. Fry. Jr., I. W. Wain e r, J. Liq. Chromato g r., 언, 455 (19 86). 38. W. H. Pir k le, C. J. W elch, G. S. Mahler, A . I. Mey e rs, L. M. Fuente s and M. Boes, J. O rg. Chem., 쁘 , 2504 (19 84). 39. D. F. Smi th and W. H. Pir k le, Psyc hop h armacol. , 쁘, :39 2 (19 86). 40. Q, Yang, Z. -P. Sun and D. -K. Lin g , J. Chromato g r., 뜨, 208 (1988). 41. I. W . Wain e r, T. D. Doy le , Z. Hami dz adeh and M. Aldrid g e , J. Chromato g r., 짝 123 (1983). 42. I. W. Wain e r, T. D. Doy le , F. S. Fry, Jr. and Z. Hami dz adeh, J. Chromato g r., 쁘탄, 149 (19 86). 43. J. Boja r ski, J. Liq . Chromato g r., 브 , 2685 (1989). 44. D. M. McDanie l and B. G. Snid e r, J . Chroma t o gr.씬!, 123 (1987). 45. I. W. Wain e r, T. D. Doy le and C. D. Breder, J. Liq. Chromato g r., 7, 731 (1984). 46. P. Cami ller i, R. E. Dolle, R. Novelli a nd C. J. Salte r , J. C hromato g r., 쁘, 258 (19 89). 47. W. H. Pir k le and J. E. McCune, J. Chroma t o gr.산 1, 419 (19 89). 48. P. Macaudie r e, M. Lien ne, M. Caude, R. Rosset and A. Tambute , J. Chromato g r., 쁘~. 357 (19 89). 49. Unp u bli sh ed Results , M. H. Hy u n and M. S. Ki m . 50. L. E. Weaner at D. C. Hoerr, J. Chroma t o gr.산 z, 109 (1988). 51. M. H. Hy un, Ph. D. Diss erta t i on , Univ e rsit y of Illin o is at Urbana -Champ a ig u, 1984. 52. W . H. Pir k le and J. M. Fin n , J. Org. Chem., 旦, 4037 (1982). 53. W. H. Pir k le and B. C. Hamp e r, in Prepa r ati ve Liq u id Chromato g r a - phy , J. Chromato g r. Libr ary, vol. 38, B. A. Bid l i ng me ye r, Ed., Elsvie r , Amste r dam, 1987, chap 7.
54. W. H. Pir k le, T . C. Pochap s ky , G. S. Mahler, D. E. Corey, D. S. Reno and D. M. Alessi, J. Org. Chem., 탄, 4991 (1986). 55. G. R. Pett it, S. B. Sin g h and G. M. Crag g, J. Org. Chem 견!, 3404 (1985). 56. W. H. Pir k le, A . Tsip o uras and T. J. Sowi n, J. C hroma t o g r. 언안 392 (1985). 57. J. Ki p, P. V. Hap e ren and J. C . Kraak, J. Chroma t o g r. 전쁘 , 423 (1986). 58. L. Sir e t, A. Tambute , M . Caude and R. Rosset, J. Chroma t ogr . 만쁘 67 (19 90). 59. G. Garga ro, F. Gaspa rrin i , D. Mi si t i, G. Palmi er i, M . Pie r in i and C. Vi llan i, Chromato g ra p h ia , 낀, 505 (19 87). 60. W. H. Pir k le and J. E. McCune, J. Chromato g r., 뜨 , 311 (1988). 61. M. H. Hy u n and M. H. Ki m , Bull. Kor. Chem. Soc., 브, 189 (1990). 62. M. H. Hy u n, and M. H. Ki m , J. Liq . Chromato g r., 브 , 3229 (1990). 63. M. Zie f , i n Chr01nato g r ph ic Chir a l Sep a rati on s, M. Zie f and L. J. Crane, Eds., Marcel Dekker, N ew York, 1988, chap 12. 64. M. Zie f , L. J. Crane and J. Horvath , J. Liq . Chromato g r., ?_, 709 (19 84). 65. L. R. Snid e r and J. J. Ki rk land, In tr o d ucti on to Modern Liqu id Clzromato g r aph y , Second Ed., W ile y -foter sci en ce. New York, 1979. 66. P. A. Mourie r , E. Eli ot , M. H. Caude, R. H. Rosset, and A. G. Tambute Anal. Chem. , 57, 2819 (19 85). (제 7 장, 4) 1. W. H. Pir k le and M. H. Hy un, J. Org. Chem., 쁘, 3043 (1984). 2. W. H. Pir k le, M. H. Hy un and B. Bank, J. Chroma t o gr.만§_, 585 (1984). 3. M. H. Hy un , Ph. D. Diss ert at i on , Univ e rsit y of Illin o is at Urbana, 1984. 4. W. H. Pir k le and M. H. Hy un ., J . Chromato g r., 므 1 (1985).
5. W. H. Pir k le. G. S. Mahler and M. H. Hy un , J. Liq . Ch romato g r. , 9 , -1-13 (19 86). 6. W. H. Pir k le, G. S. Mahler, T. C. Pochap sk y and 1\:1. H. Hy un , J. Chromato g r., 388, 307 (198 7). 7. M. H. Hy un and C. -S. Mi n, Bull. Kor. C hem . Soc., 10, 328 (19 89). 8. M. H. Hy un and C. -S . Mi n, Bull. Kor. Chem. Soc., 10, 578 (1989). 9. M. H. Hy un and M. S. Ki m , Bull. Kor. Chem. Soc., 12, 104 (1991) . 10. 0. W. Grif fith, E . B. Camp b ell, W . H. Pir k le, A. Tsip o uras and M. H. Hy un , J. Chromato g r., 쁘혼, 345 (19 86). 11. W. H. Pir k le, A. Tsip o uras, M. H. Hy un , D. J. Hart and C. -S. Lee , J. Chromato g r., 358, 377 (19 8 6). 12. D. J. Hart, C .- S. Lee, W. H. Pir k le, M. H. Hy un and A. Ts ip o uras, J. Am. chem. Soc., 1 08, 6054 (1986). 13. W. H. Pir k le, D. M. Alessi, M. H. Hy un and T. C. Pochap s ky , J. Chromato g r., 398, 203 (1987). 14. M. H. Hy un , 1.- K . Baik and W. H. Pir k le, J. Liq. Chromato g r., 브 1249 (19 88). 15. M. H. Hy un , Y. -W . Park and I. -K. Baik , Tetr a hedron. Lett ., 쌤 4735 (1988). 16. M. H. Hy un and W. H. Pir k le, Bull. Kor. Chem. Soc., ~. 45 (19 87). 17. W. H. Pir k le and M. H. Hy un , J. Chromato g r., 혼, 287 (19 85). 18. W. H. Pir k le and M. H. Hy un , J. Chromato g r., 혼 , 295 (19 85). 19. N. Oi , H . Ki tah ara, T. Doi and Yamamoto , B unseki Kag a ku, 3 2, 345 (19 83). 20. M. H. Huun and W. H. pirk le, J. Chromato g r. 학 , 357 0987 ) . 21. D. W. House, U. S. Pate n t 4,322,310, Mar. 30, 1982. 22. N. Oi and H. Ki tah ara, J . Chromato g r., 쪼 117 (1983). 23. N. Oi, M. Nag a se and T. Doi, J. Chromato g r., 프 111 (1983). 24. N. Oi and H. Ki tah ara, J. Liq. Chromato g r., 왼, 511 (1986). 25. K. Iwaki, S. Yoshid a , N. Ni m ura, T. Ki no shit a, K. Takeda and H. Og ura, J. C hromato g r., 쁘산, 177 (19 87). 2276.. RR.. RD.a pBp er nu,g V ge. rR, . MAe. yR e. r aMnadr Ht i,. AVrm. R, .J . CMheryoe m r atao ng d r. , 쁘H~.. 9A3r (m19, 86J)..
Chromato g r., 쁘, 197 (19 88). 28. R. Dap pe n, H. R. Karfu n kel and F. J. J. Leusen, J. Chroma t o gr.선쁘 101 (19 89). 29. M. J. B. Lloy d , J. Chromato g r.' 판, 219 (1986). (제 7 장, 5) 1. W. H. Pir k le, J. M. Fin n , B. C. Hamp e r, J. Schrein e r and J. R. Prib i s h , in Asym metn · c Re ac ti on s and Processes in Chemi stry, E. L. Elie d , Ed:, ACS Sy m p o siu m Ser. 185, ACS, Washin g ton D. C. 1982, chap 18. 2. M. H. Hy u n, Ph. D. Dis s ert a ti on , Univ e rsit y of Illin o is at Urbana -C hamp a ig n, 198 4. 3. W . H. Pir k le and J. M. Fin n , J. Org. Chem., 무 , 4037 (1982). 4. W. H. Pir k le and M. H. Hy un , J. Chromato g r., 혼 309 (1985). 5. W. H. Pir k le and T. J. Sowi n, J. Chroma t o gr.관座, 83 (1987). 6. W. H. Pir k le and T. C. Pochap sk y , J. Org. Chem. , ~ . 102 (1986). 7. W. H. Pir k le, T. C. Pochap sk y , G. S. Mahler and R. E. Fie l d, J. Chromato g r., 348, 89 (1985). 8. W. H. Pir k le and T. C. Pochap sk y , J. Am. Chem. Soc. 屯뽀, 352 (1986). 9. W. H. Pir k le, T . C. Pochap sk y, G. S. Mahler, D. E. Corey, D. S. Reno and D. M. Alessi, J. Org. Chem., 邑, 4991 (1986). 10. W. H. Pir k le, G. S. Mahler, T. C. Pochaps ky , and M. H. Hy un, J. Chromato g r., 쁘~. 307 (19 87). 11. W. H. Pir k le, D. M. Alessi, M. H. Hy un and T. C. Pocap sk y, J. Chromato g r., 쁘흔, 203 (1987). 12. W. H. Pir k le and T. C. Pochaps ky, J. Chroma t o gr.쁘언, 175 (1986). 13. A. Tambute , A. Beg o s, M. Lie n ne, M. Caude and R. Rosset; J. Chromato g r., 쁘~. 65 (1987). 14. M. Salle, A. Tambute and A. Beg o s, J. Chroma t o gr.산~. 153 (1989). 15. N. Oi and H. Ki tah ara, J. C hromato g r., 쁘던, 117 {1983).
16. N. Oi, M. Nag a se, Y. Inda and T. Doi, J. Chromato g r., 쁘, 487 (1983). 17. T. Takag i and Y. Ita b ashi, J. Chromato g r., 쁘~. 451 (1986). 18. Y. Itab ashi T. Takag i and T. Tsuda, J. C hroma t o g r . 산깐, 271 (19 89). 19. Y. Ita b ashi and T. Takagi , J. Chromato g r., 쁘 단, 257 (1987). 20. J. Yamashit a, T. Numakura, H. Ki ta, T. Suzik i, S. Oi, S. Mi ya no, H. Hashim oto and N. Takai, J. C hromato g r., 쁘던 , 275 (19 87). 21. J. Yamashit a, H .• Sato h , S. Oi, T. Suzuki. S. Mi ya no and N. Takai, J. Chromato g r., 464, 411 (1989). 22. C. Pett er sson and C. Gi oe li, J. Chromato g r., 쁘~. 247 (1987). 23. C. Bertu c ci , C. Rosin i , D. Pin i and P. Salvadori, J. Phann. Biom ed. Anal., ~. 171 (1987). 24. G. Kri.ig e r, J. Grotz i n g e r and H. Berndt, J. Chromato g r. , 쁘~. 223 (19 87). 25. R. Kurop k a, B. Mi llier , H. Hocker and H. Berndt, J. Chromato g r., 481, 380 (19 89).
제 8 장 수소결합키랄고정상 1 서론 독립방식의 키랄고정상에 의한 라세미 화합물의 광학분할에는 대부 분 키 랄고 정상과 분석성분 사이에 형성되는 수소결합이 키랄성 인지에 중요한 작용을 하나 수소결합 이의에도 금속이온을 매개로 한 상호작 용 (LEC 키 랄 고정상), 7'l-7'l 착물형성 상호작용(전하아동 착물 형성 키랄 고정상), 주인분지에 손님분자가 끼어들어서 생기는 착물형성 상호작 용(크라운게테르 키랄고정상) 등이 키랄성 인지의 필수적인 요소로서 작용하고 있다. 한편 몇몇 키랄고정상들에 의한 라세미 화합물의 광학 분할에 있어서는 키랄고정상과 라세미 화합물의 두 광학 이성질체 사 이에 다중의 수소결합에 의하여 두 착물이 형성될 때 두 광학 이성질 체의 절대배열에 따라 형성되는 두 착물의 안정성에 차이가 있기 때문 에 두 광학 이성질체가 구분된다고 생각되고 있다. 이와 같이 키랄고 정상과 라세미 화합물 사이에 수소결합이 중요한 역할을 하여 (입체장 애 상호작용 포함) 라세미 화합물의 광학분할이 이루어질 때 이러한 키 랄고정상들을 특별히 수소결합 키랄고정상이라고 명명하여 제 惡》 ]서
다루고자 한다. 수소 결 합 키 랄고 정상 들은 라세미 화합 물 과 입체선택 적인 다중의 수소결합 을 할 수 있 는 다수의 수 소결 합 양성자 주 게와 수소결합 양성자 받게 를 가지고 있으면 키 랄 고정상으 로 서 충분한 역 할 을 할 수 있다. 경우에 따라서 수소결합 키 랄 고정상 들은 방향 족 기 을 포함하기도 하나 이 들 방향족기는 키랄성 인지의 메커니 즘 에 특 별한 영향을 미치지 않으며 다만 수소결합을 할 수 있 는 작용기 를 분 자내의 적절한 위치에 배열되도록 하는 역할이나 혹 은 입체장애의 역 항을 항 때, 키랄성 인지 메커니즘의 관점에서 이 들을 수 소결 합 키 안고 장성이 라고 할 수 있을 것이다. 따라서 제 7 장 5 . 2 절에서 기 숭한 키 안고 장상 旦-잎둘이 순수한 수소결합 상호작용에 의하여 두 광학 이성 질 체 를 구분할 때 이들 키랄고정상들을 수소결합 키 랄 고정상으 로 - 볼 수 있 을 것 0] 다. 착물형성 키랄고정상이나 수소결합 키 랄 고정상 들 은 모두 전자의 주 고받음에 의하여 광학분할하고자 하는 라세미 화합물과 상호작용을 하 기 때문에 이들 키랄고정상들을 모두 함께 포함하여 주게 - 받게 키랄고 정상 (Donnor-Acce pter CSP:DA-CSP) 으로 취급하기도 하며 (1) 독립 적인 키랄분자 하나하나가 키랄성 인지에 중요한 역할을 하며 독립방 식 키랄고정상들은 독립적인 키랄분자 하나하나가 실리카 겔에 결 합된 모양이 솔 (brush) 과 같다고 하여 솔형 키 랄고정상 (brush type CSP) 이 라고 명명하기도 한다 (2). 그러나 이와 같은 키랄고정상의 명칭 혹은 분류는 모두 키랄고정상을 서술하는 사람의 편의에 따라 붙여진 명칭 임을밝혀두고자한다. 2 아미노산 아미드 키랄고정상 아미노산 아미드를 기저로 한 키랄고정상은 처음 기체 크로마토그래 피에 도입되어 아미노산의 휘발성 유도체들을 광학분할하는 데 사용되
었으며 (3. 4) 현재 아미 노산 아미 드 혹은 아미 노산 디아미드 를 기저로 한 기체 크로마토그괘피용 키랄고정상들 중 몇몇은 Chir a lsil Val 등 의 명칭으로 상품화되어 사용되고 있다 (4). 기체 크로마토그래피용 키랄고정상으로 사용되는 아미노 산 디아미 드 키랄고정상은 Hara 등에 의하여 HPLC 에 응용되기 시작하였다. Hara 등은 DCC 존재하 에서 아미노 프로필 실리카 겔 (APS) 과 N- 아 실 - L - 발린을 결합시킴으로써 (그림 8- 1) 발린 디아미드를 기저로 하는 여러 종 류의 키랄고정상 1 을 합성하였다 (5.6). 이 들 키랄고정상들은-
(Et O )3S i- (CH 2 )3 - N H2 二> 仁〉fi E:(CH 2)3 - NH2 APS APS+ R , C O -L-Val 드노 「。o 〉:i E:(CH 2 )3_\X:/\ II I 0 IH a R1 = H (FVA) d R1 = n- p ro p yl (BVA) b R1 = CH3 ( AVA) e R1 = n- b ut yl (VVA) c R1 = CH2 C H3 (P VA) f R1 = t-b ut yl( Pi V A) 그림 8- 1 디아미드 키랄고정상 1 의 합성
N- 아실아미노산에스데르를 광학분할할 뿐만 아니라 N- 벤질옥시카르 보닐디펩티드에스데르의 네 개 입체 이성질체 를 분리할 수 있으나, 여 섯 종류의 키랄고정상 l 중 R1 이 H 인 FVA(N -F ormy l- L - Va l) 키랄 고정상 브가 가장 좋은 광학분할능을 보이며 R 기의 입체장애가 큰
PiV A (N-Piv a loy l- L-Val) 키 랄고정상 끄는 N - 아실아미노산에 스 데르 에 대하여 광학분할능을 거의 보이지 않는 것으로 확인되었다 (5). 한 편, 디아미드 키랄고정상 특히 FVA 키랄고정상 보상에서 광학분할되 는 N- 아실아미노산에스데르는 N - 아실기가 아세틸기이고 에스테르기 는 0- t부될기일 때 가장 좋은 광학분할능을 보인다 ( 7) . 이 때, 키랄 고정싱에 오래 머무르는 이성질체는 항상 L- 이성질체임이 확인되었으 며 리신, 글루타민 등과 같이 다른 작용기 를 가지고 있 는 아미노산 들 의 N- 아실에스테르보다는 루이신, 알라닌, 발린 등과 같 이 다른 작 용기를 가지고 있지 않은 단순한 아미노산의 N - 아실에 스데르가 더 좋 은 광학분할능을 보이는 것으로 확인되었다. 키랄고정상 l 에서 라세미 화합물의 광학분할에 사용되는 이동상은 보통 2- 프로판을을 소량 포함하는 핵산용액이다. 클로로포름 , 디클로. 로메탄, 디에틸에데르와 같은 비양성자성 극성용매와 핵산의 혼합용 매를 이동상으로 사용하면 이동상옵브 1 성분, 이동상 -키랄 고정상의 수 소결합 상호작용을 작게 하므로 2- 프로판을-핵산의 혼합 용매 를 사용 할 때 보다 더 좋은 광학분할(큰 a 값)을 관찰할 수 있으나 피이크의
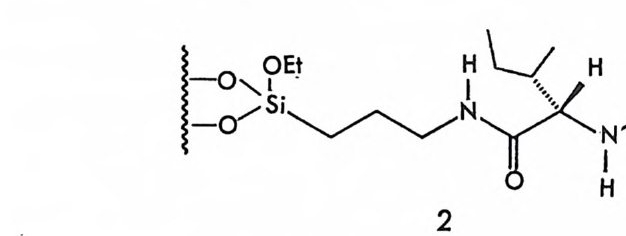 I 二二갑, \[:H?LH
I 二二갑, \[:H?LH
끌립현상을 줄이기 위하여 보통 2 - 프로판울-핵산의 혼합용매를 주로 이동상으로 사용한다. 분석시간을 줄이기 위하여 Hara 등은 초임계 유체 크로마토그래피 (SFC) 를 이용하여 광학분할을 실시한 바 있다 (8). Akany a 등은 N- 포르밀 -L - 이소루이신을 아미노프로필 실리카 겔 과 반응시켜 얻은 N- 포르밀 -L- 이소루이신 키랄고정상 2 에서 (9) 아미 노산에스테르의 광학분할뿐만 아니라 생리활성을 가진 라세미 화합물
들을 포함한 여러 종류의 화합물들(그림 8-2) 을 광학분할할 수 있었다 (10 ). 전형적인 크로마토그램은 그립 8 - 碑} 같다.
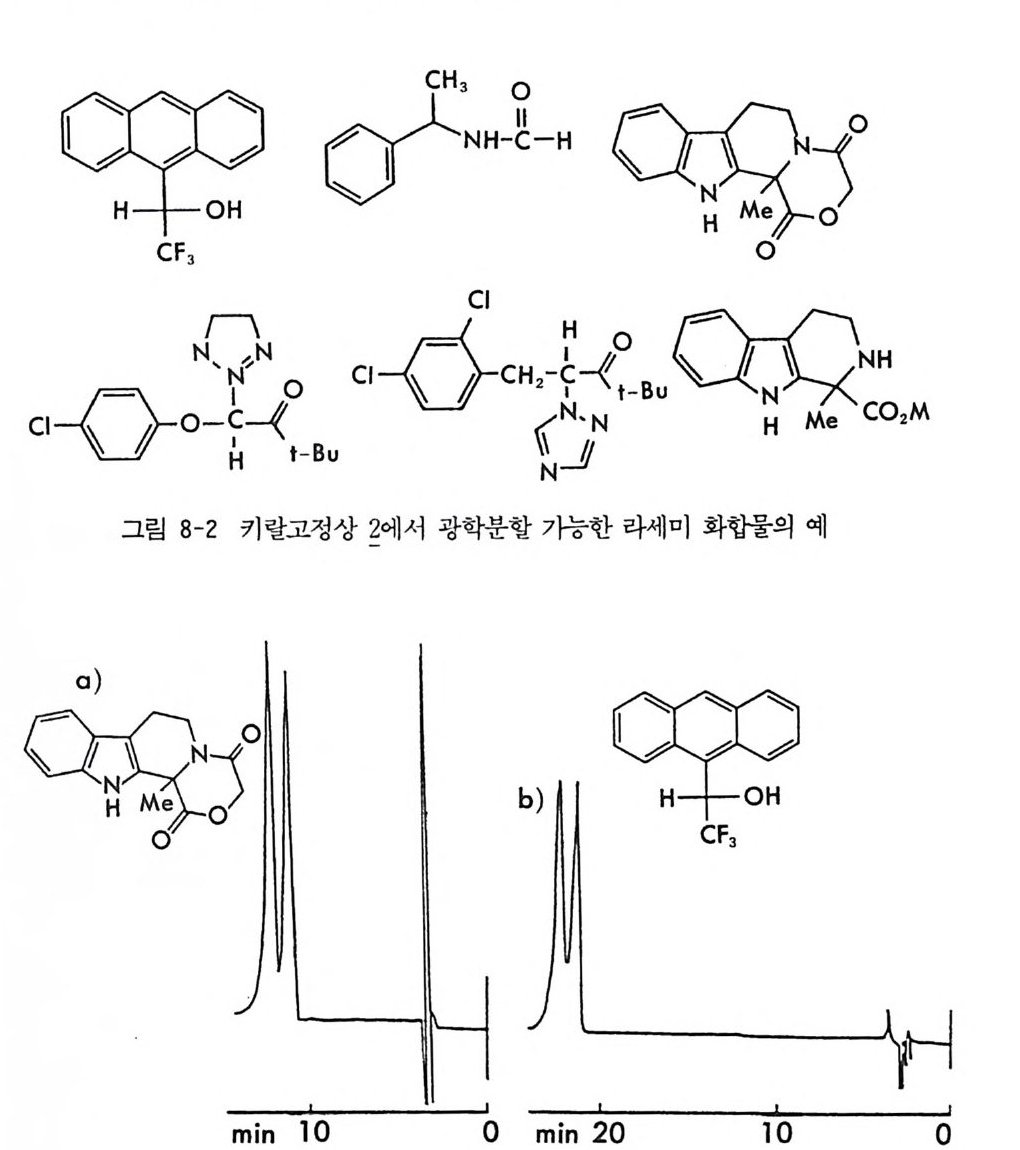 三 H-\-H \::O
三 H-\-H \::O
그림 8-3 키랄고정 2상 에서의 광학분할 예. (a) 이동상 : CH2Cl 2- 핵 산 (60/40), 유속 : 2mL/ mi n, 검 출기 : 254nmUV.(b) 이동상 : 2- 프로판을 : 핵산 (I/ 99) , 유속 : lmL/mi n, 검출기 : 254nm UV (참고문헌 : 10)
아미노산 디아미드 키랄고정상을 합성할 때 키랄고정상 l 혹은 g와 같이 아미노산의 카르복시기를 통하여 실리카 겔에 고정시키는 것과는 달리 아미노산의 아미노기를 통하여 실리카 겔에 결합된 키랄고정상 ? 은 아미노산메틸에스데르의 N- 아세틸유도체나 N - 벤조일유도체를 광 학분할한다 (11). 같은 형태의 키랄고정상으로 최근 Hara 등은 L- V al, L- L eu, L- t -Leu 의 t4--t탈아미드 HCI 염을 염화 10 쉼는데센일 (10 - undecen y l chlo ri de) 과 반응시켜 디아미드를 얻은 후 히드로실릴화 반응 및 실리 카 겔에의 고정과정을 거쳐 긴 연결가지를 가전 키랄고정상 요 를 합성 하였다(1 2). 키랄고정상 1 이나 키랄고정상 g이 N- 아실아미노산에스데르의 광학 분할에 응용되었으나 다른 종류의 라세미 화합물의 광학분할에는 충분 히 좋은 광학분할능을 보이지 않는 반면(키랄고정상 待 l 경우 다른 종류 의 라세미 화합물에 대한 광학분할을 시도해 보았는지 밝혀지지 않았음) 키
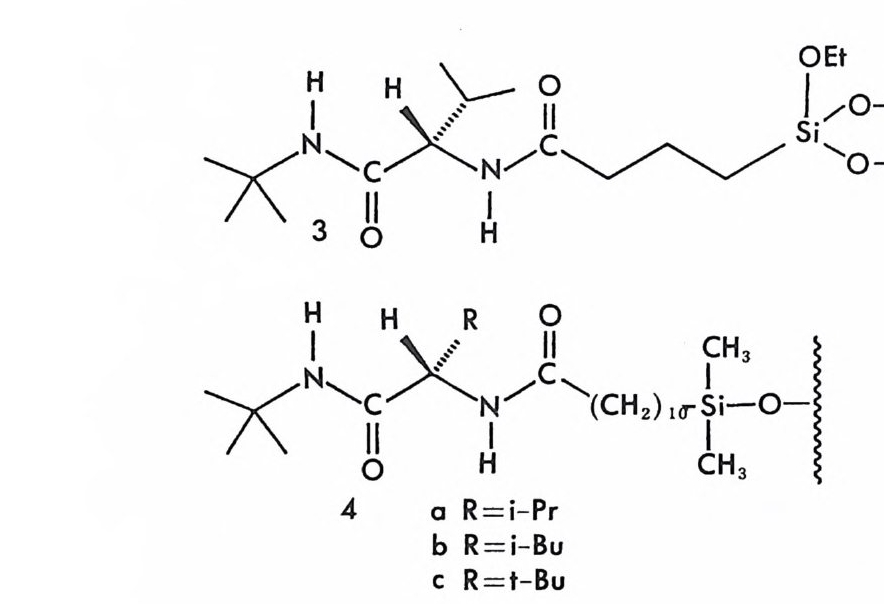 x\\/ 『:J,〈OEt O O 」
x\\/ 『:J,〈OEt O O 」
랄고정상 신는 여러 종류의 라세미 화합물에 대하여 좋은 광학분할능을 보인다. 키랄고정상 4 에서 광학분할되는 라세미 화합물들은 그립 8 -4 에서 보는 바와 같이 a - 히드록시카르복실산 메틸에스데르의 O- 페닐카르밤 산R 에스,데~르 (xPC) 유R도,y체 , a- 히드록 시N카H-르1-보B닐u R화합물의~ O- 페Q닐 -카H 르r밤 OPC OH Q ~HNB X = OCH3 X= R' NHCONHPh NHPC OH\oH R,~R, R1 入 R2 RI R2
그립 8-4 키랄고정상 4 에서 광학분할 가능한 라세미 화합물의 예
산 에스데르 유도체, 아미노산 이소프로필에스테르의 N-4- 벤조일 (NB) 유도체, 아민의 N- 페닐우레아유도체, 라세미 디올 등과 같이 다양하며 모두 2- 프로판올-핵산의 혼합용매를 이동상으로 사용하여 광학분할된다. 키랄고정상 쁘에서 N-(4- 니트로벤조일)루이신 이소프 로필에스데르의 광학분할에 대한 전형적인 크로마토그램은 그림 8-5 와같다. 키랄고정상 쁘에서 초임계 유체 크로마토그래피 (SFC: 이동상은 CO2 -메탄을 혹은 C02-2- 프로판을의 혼합용매)에 의하여 아미노산이소프로
 0 30mi n
0 30mi n
그림 8-5 키랄고정상 산에서 N-( 4- 니트로벤조일)루이신 이소프로필에스데르의 광학분할. 이동상 : 2- 프로판을-핵산(1 / 99), 컬럼크기 : O.lx 50 cm (참 고문헌 : 12)
필에스테르의 N-(4 - 니트로벤조일)유도체를 광학분할하였을 때 광학 분할에 소요된 시간은 앉은 미만으로서 아주 빠르다. 그러나 2 - 프로판 울책산의 혼합용매를 이동상으로 사용한 액체 크로마토그래피에 의 한 광학분할에 비교해 분할능이 떨어진다는 사실이 보고된 바 있다(1 3). Oi 등은 t유 L 틸이소시아네이트와 L- 발린을 반응시켜 얻은 N-( t켜 L 틸아미노카르보닐 )-L- 발린을 아미노프로필 실리카 겔에 결합시켜 디 아미드 키랄고정상 §롤 합성하였으며 (14 )' 이 키랄고정상에서 N - 아실 아미노산메틸에스데르의 광학분할뿐만 아니라 a- 히드록시카르복실산 메틸에스테르의 3, 5- 디니트로페닐카르밤산 에스테르유도체의 광학분 할을 보고하였다(1 5). 키랄고정상 탉근 Sumi ch ir a l OA 3000 이라는 명칭으로 상품화되어 있다(제 9 장 참조) .
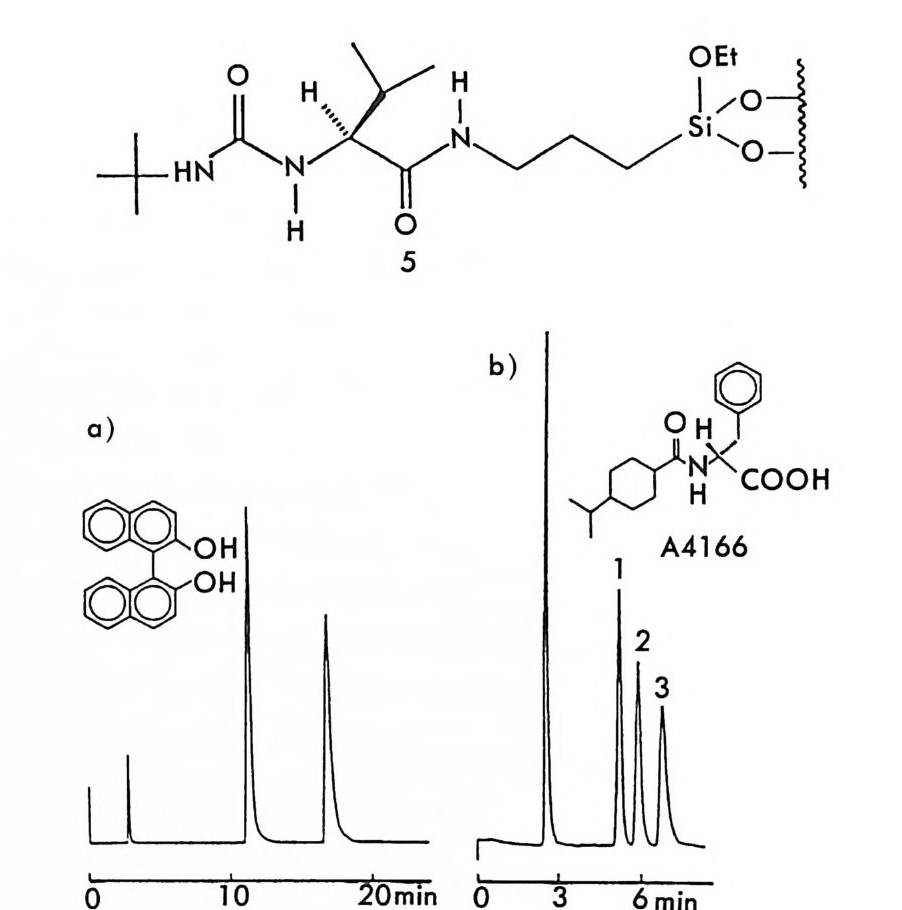 +HNJ l~~\:/『더
+HNJ l~~\:/『더
그림 8-6 키탈고정상 5 에서 (a) 1, 1' -비-나프톨의 광학분할 및 (b) A 4166(2), L - 이성질체 (3)- 및 시스-이성질체 (1)의 분리. (a) 이동상 : 핵산 -1, 2- 디클 로로에탄-에탄을 (90/9/1) , 유속 : l.Om L/mi n, 검출기 : 254nm UV. (b) 이 동상 : n- 핵 산-1, 2- 디 클로로에 탄-에 탄올 (100/20/0. 05) , 유속 : lmL/mi n, 검출기 : 22Orun UV (참고문헌 : 16 및 18)
키랄고정상 §에서의 광학분할 중 홍미있는 것은 1,1' -비-나프톨 (1, l'-b i -na ph t hol) 의 광학분할 (그립 8-6 ~) 이며 (16) 최근에는 상품화된 키랄고정상 혀t 이용하여 새로운 당뇨병 치료제인 N- (트란스 -4- 이소 프로필시클로핵실카르보닐 )-D- 페닐알라닌 (A4166) 과 이것의 광학 이
성질체인 L- 이성질체를 동물의 혈장으로부터 추출하여 직접 광학분할 하였을 뿐만 아니라(1 7) A4166 의 합성과정에서 생기는 부생성물인 L -이성질체 및 D- 이성질체의 c i s - 이성질체를 분리할 수 있었다(그립 8-6 !?_) (18 ) . 이상에서 살펴본 아미노산 디아미드 키랄고정상들은- 라세미 화합물 인 분석성분과 친화적인 상호작용을 할 수 있는 작용기로서 오로지 두 개의 아미드기를 가지고 있기 때문에 Hara 등은 아미노산 디아미 드 키랄고정상에 의한 라세미 화합물들의 광학분할은 키랄고정상과 분석 성분 사이에 형성되는 두 자리에서의 수소결합 상호작용에 의하여 이 루어진다고 생각하였다. 그림 8-7 ~는 N - 아세틸 - L 고갈린 t 유i틸 아미 드를 키랄이동상첨가제 (CMPA) 로 사용하여 N - 아세틸아미노산 t유 L 틸에스데르를 광학분할할 때의 키랄성 인지 메커니즘을 Hara 등이 제안한 것이며 (19 )' 그림 8-7 ~근 키랄고정상 l( 혹은 ? 및 산)에서 N _아세틸아미노산 t냐녁}에스테르의 광학분할에 대하여 제안된 두 자리 에서의 수소결합 키랄성 인지 메커니즘을 나타낸 것이다 (5). 그림 8-7 은 키랄고정상(혹은 키랄이동상 첨가제)과 분석성분 사이에 두 자리에
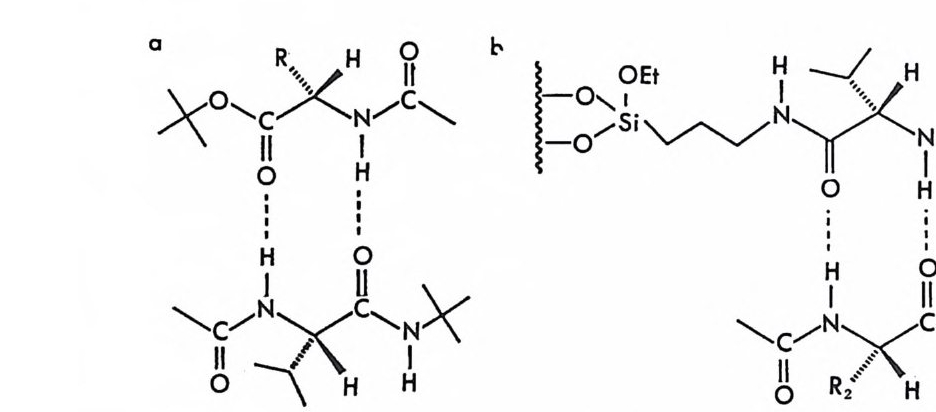 aX \°广。 /」— OCHIN'IIiiI R X \X NCHH0HIU'III / \ ° 방?H \x 」 I二 。。〉\二$o /\0HiIIRI N 2 .. 父xcH H70Ii II I \ Jl 。. X,
aX \°广。 /」— OCHIN'IIiiI R X \X NCHH0HIU'III / \ ° 방?H \x 」 I二 。。〉\二$o /\0HiIIRI N 2 .. 父xcH H70Ii II I \ Jl 。. X,
그림 8-7 아미노산 디아미드 키랄이동상 첨가제 (a) 및 키랄고정상i( b 꽈 N- 아세틸 아미노산 t부될에스테르의 상호작용 모델. 모델은 (L-:_L) -착물형성에 대한것임.
서의 수소결합에 의하여 형성된 수소결합 착물 을 나타낸 것으로써 L - L 착물이 L - D 착물에 비교하여 특별히 더 안정한 이유를 발견하기가 어렵다( 실험결과는 L - 이성 질체 가 키랄고정상에 항상 더 오래 머무름). 따 라서 P i rkl 학본 키랄고정상(혹은 키랄고정상 첨가제)의 두 아미드기와 분석성분의 두 아미드기 사이의 두 자리에서의 수소결합보다는- 두 아 미드기 사이의 그림 8 - 8 와 같은 쌍극자 - 쌍극자 겹침에 의하여 키랄성 인지가 일어난다고 제안하였다 (20). 그림 8- 幹 1 쌍극자 겹침 과정에 의하면 분석성분인 N - 아세틸 - (L) -아마노산 t유 L 릴에스데르의 R 기와 키말고정상(혹은 키 랄 이동상 첨가제)을 이루는 L - 발린의 a- 이소프로필 기가 서로 멀리 떨어져 있어서 입체장애를 덜 받기 때문에 더 안정한 착물 (L - L;착 물 )을 형성하는 반면 N- 아세틸 -D- 아미노산 t숙녀i에스테 르의 R 기는 키랄고정상으로 향하기 때문에 불안정한 착물 (L-D 착물) 을 형성하리라는 것을 쉽게 짐작할 수 있다. Hara 등은 단순한 두 자리 수소결합 메커니즘에 의하여 키랄고정 상 l 에서의 N- 아세틸아미노산 t-Jf l 탈에스테르의 광학분할을 합리적
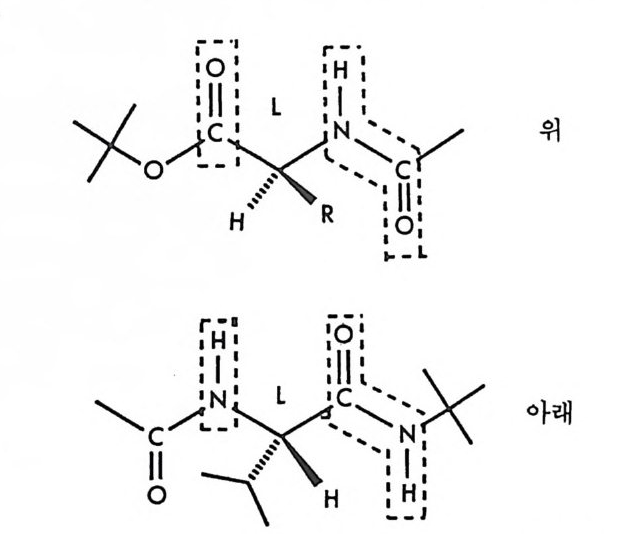 x 。J,:- 0-!·:H, - XLR,r\H- ·.\..: :[►o -:
x 。J,:- 0-!·:H, - XLR,r\H- ·.\..: :[►o -:
그림 8-8 N- 아세틸 - L- 발린 t국 L 틸아미드(키랄이동상 첨가제)와 N- 아세틸 -L -아미노산 t유 L 틸에스데르의 쌍극자뱅, f자 겹침 모델
으로 설명하는 데 무리가 있었기 때문에, 라세미 화합물인 분석성분과 실리카 겔 지지체 사이에 입체장애 상호작용이 존재할 수 있음을 제안 함으로써 수소결합 키랄성 인지 메커니즘을 보완하였다 (6). 키랄고정 상 l 의 경우 실리카 겔에 결합된 아마노산 디아미드는 실리카 겔 표면 에서 뻗어나와 이동상으로 향하고 있는 형태도 가능하며 실리카 겔 표 면의 실라놀기 등과 상호작용에 의하여 실리카 겔 표면에 누워 있는 형태를 취할 수도 있다. 두 형태는 아마도 서로 평형의 관계에 있을 것으로 생각된다 (6). 키랄고정상 l 에서 아미노산의 아미드가 실리카 겔 표면에서 뻗어나온 형태를 취할 경우 키랄고정상과 분석성분은 그 립 8-7 Q와 같이 두 자리에서의 수소결합에 의한 착물을 형성할 것으 로 기대된다(분석성분의 L- 이성질체와 D- 이성질 체사이에 큰 에너지 차이 가 없을 것으로 예상되나 입체칭애때문에 L- 이성질체보다는 D - 이성질체가 더 안정한 착물을 형성할 것으로 생각할 수도 있음). 반면 키랄고정상 l 의 아미노산 아미드가 실리카 겔 표면에 누워있는 형태를 취할 경우 그림 8-9 에서 보는 것처럼 지면을 실리카 겔 표면으로 생각하면, 키
 \\\°H> :
\\\°H> :
그림 8-9 키랄고정상 1 과 N- 아세틸 -L- 아미노산 t유 L 틸에스데르의 작용 모델. 지면은 실리카 겔 표면과 동일함
랄고정상의 이소프로필기와 N - 메틸아니노산 t--¥-f컬에스테르의 R 기가 지면(실리카 겔 표면) 앞으로 향하는 구조를 가진 L - L 착물이 R 기가 지면 (실리카 겔 표면)으로- 향하는- L - D 칙물에 비하여 훨씬 더 안정할 것으로. 기대된다. 따라서 키랄고정상에 의한 분석성분의 키랄고정상 내 머무름을 전체적으로 생각할 때 L - 이성질체가 키랄고정상에 더 오 래 머무르는 것을 그림 8 - 9 의 키랄성 인지 메커니즘으로 설명할 수 있 다 (6). 키랄이동상 첨가제로 사용한 N - 아세틸 -L- 발린 t--¥-틸아미드의 경 우에도, 키 랄 이동상 첨가제가 실리카 겔 표면의 실라놀기와 상호작용 에 의하여 실리카 겔 표면에 강하게 흡착되어 있을 것으로 생각되기 때문에 이것은 일종의 키랄고정상으로 생각할 수 있으며 그림 8-7 ~ 에서 처럼 키랄고정상첨가제의 이소프로필기와 분석성분인 아미노산 의 R 기가 같은 방향으로 향하고 있는 L-L 착물이 더 안정할 것으로 생각할수있다. 3 기타의 수소결합 키랄고정상 Hara 등은 타르타르산의 디이소프로필아미드 §을 키랄이동상 첨가 제로 사용하여 보통의 실리카 겔 컬럼상에서 a 쵸답본 /3-히드록시카르 복실산 아미드, /3-히드록시케돈, a_ 아미노산 에스데르의 N_(4- 니트 로벤조일)유도체, a- 히드록시케톡심. 1, 2- 디올, 비-/3 - 나프톨 등의 두 광학 이성질체를 잘 분리할 수 있었다 (6,21,22). 키랄이동상 첨가 제로서 타르타르산 디아미드의 유용성을 근거로 하여 Hara 등은 타르 타르산아미드를 적절한 방법으로 실리카 겔에 고정시킬 경우 좋은 키 랄고정상으로 이용할 수 있을 것으로 생각하였으며 실제로 (R, R)- 타 르타르산울 출발물질로 사용하여 타르타르산 아미드가 세 개의 메틸렌 기를 통하여 실리카 겔에 고정된 키랄고정상 7_( 23) 및 타르타르산 아
 \° OH H_N \
\° OH H_N \
미드가 11 개의 메틸렌기를 통하여 실리카 겔에 고정된 키말고정상 § 울 합성하였다 (24). (R, R) -타르타르산과 아세트산 무수물을 반응시켜 얻은 (R, R)- 디아세틸타르타르산 무수물을 이소프로필아민과 반응시 켜 반쪽 아미드를 합성하고 이 아미드를 3- 아미노프로필트리에독시실 린과 반응시킨 후 (DCC 를 사용하여 반응함) 실리카 겔에의 고정과정 및 디아세틸기를 제거하는 과정을 거쳐 키랄고정상 1 이 합성되었다(그 림 8-10) (23). 한편 그림 8-10 에 있는 키랄고정상 1 의 합성중간체인
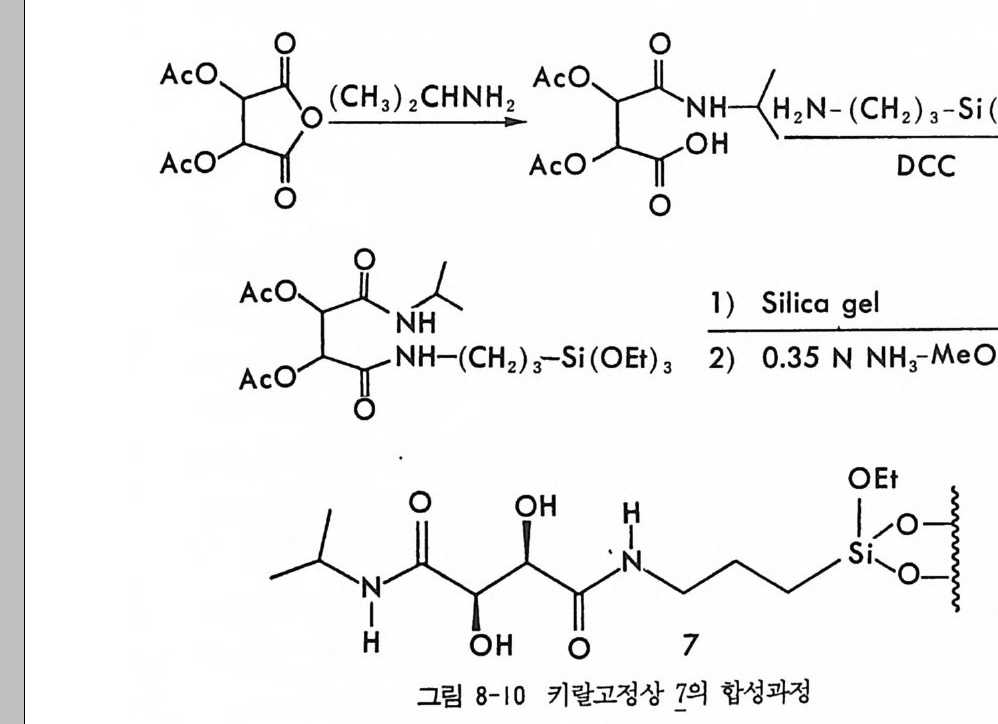 0 0
0 0
반쪽 아미드와 10 권폰데센일아민 (10 - undecen y lam i ne) 을 반옹시켜 끝 에 이중결합을 가전 디아미드를 얻은 후 히드로실릴화 반응, 실리카 겔에의 고정반응, 반웅하지 않은 실라놀기를 트리메틸실릴기로 보호 하는 과정 및 두 아세틸기를 제거하는 과정을 거쳐 키랄고정상 §이 합 성되었다 (24). 키랄고정상 7 의 경우 키랄이동상 첨가제법과 비교하여 라세미 화합물의 광학분할에 있어서 (a 값의 측면에서) 특별히 진보된 점이 없는 반면 (아마도 반응하지 않은 실라놀기의 존재로 인한 분석성분의 비입체 선태성 머무릅- 때문이라고 생각됨) 긴 연결가지들 가지고 있으며 반응하지 않은 실라놀기를 모두 트리메틸실릴기로 보호한 키랄고정상 8 은 광학분할 가능한 라세미 화합물의 범위와 입체 선택성의 크기 (a 값)에 있어서 크게 전보된 것으로 판명되었다 (24). 키랄고정상 g상에 서 광학분할이 가능한 것으로 알려진 라세미 화합물들은 그림 8-11 과 같이 다양하며 그립 8 - 12 는 키랄고정상 §에서 라세미 화합물의 광학 분할에 대한 전형적인 크로마토그램의 예를 제시한 것이다.
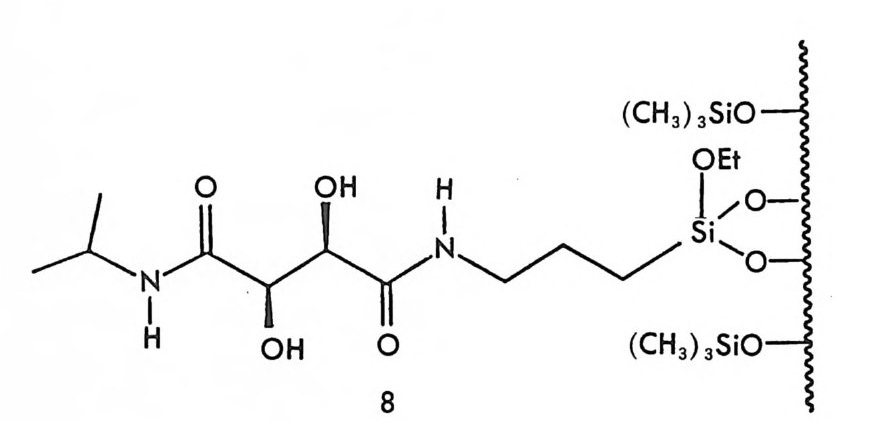 < 。 。 H
< 。 。 H
타르타르산 디아미드를 기저로한 키랄고정상에서 라세미 화합물의 광학분할에 대한 구체적인 키랄성 인지 메커니즘은 아직 논의된 바 없 으나 아미노산 디아미드를 기저로 한 키랄고정상에서의 광학분할에 대
 RRl , ) O人yXXH ZRy R,y ORH I三O Y R ,N)H:\HX< N R2H Y3 RR°I 1\\Y
RRl , ) O人yXXH ZRy R,y ORH I三O Y R ,N)H:\HX< N R2H Y3 RR°I 1\\Y
그림 8-11 키랄고정상 §에서 광학분할가능한 라세미 화합물의 예
한 키랄성 인지 메커니즘과 마찬가지로 키랄고정상과 분석성분 사이의 입체 선택적인 두 자리에서의 수소결합에 의하여 키랄성 인지가 이루 어지고 있다고 생각된다. Feib u sh 등은 DNA 의 염기가 수소결합에 의하여 바아비탈산염 (barbit ur ate ) 계통의 화합물들과 짝을 이룰 수 있음을 모방하여 , 바 아비탈산염 계통의 화합물과 세 자리에서 수소결합을 함으로써 바아비 탈산염 등의 라세미 의약품들을- 광학분할할 수 있는 키랄고정상 읽끌 합성하였다 (25). 켈리담산 (che li dam ic a ci d) 을 출발물질로 사용하여
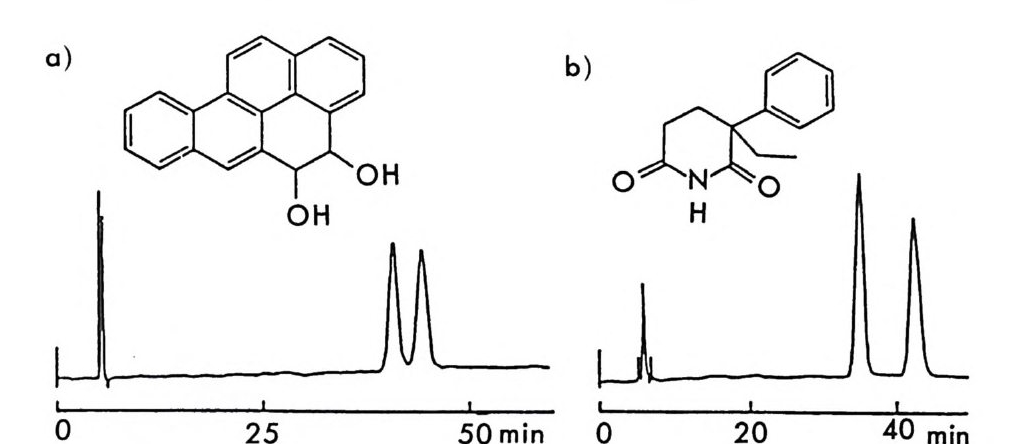 a) b) 。二
a) b) 。二
그림 8-12 키란고정상 § 에서 (a) 디올의 광학분할 및 (비 글루테티미드의 광학분 할 , 컬 럼 크기 : 50 x 0.l cm . 온도 : 2(J' C , 유속 : 60µ L/mi n, 검 출기 : 254nm UV. (a) 이 동상 : THF - 핵 산(1 0 / 90) , (b) 이 동상 : 2 - 프로판 울 : 핵산 (4/96) (참고문헌 : 24)
여러 과정을 거쳐 합성된 2, 6- 디아미노 -4-(10 국는데센일옥시)피리딘 과 염화 (S) - 2- 페닐부티릴 (2- p hen y lbu ty r y l chlor i de) 을 반응시키고 히드로실릴화반응 및 실리카 겔에의 고정과정을 거쳐 키랄고정상 읽춘 합성할 수 있으며 이렇게 합성된 키랄고정상 읽근 그림 8-13 에서 보는 바와 같이 S - 절 대배열을 가진 바아비탈산염과 세 자리에서 수소결합 하여 안정한 착물을 형성한다. R- 절대배열을 가진 바아비탈산염도 키 랄고정상 g와 세 자리에서 수소결합이 가능하나 R- 이성질체의 경우에 는 덩치가 큰 시클로헥센일기가 키랄고정상의 덩치가 큰 페닐기와 같 은 방향으로 향하게 되므로 입체장애에 의하여 덜 안정한 착물을 만들 게 되며 따라서 두 개의 광학 이성질체의 구분이 가능하게 된다. 바아 비탈산염 이의에도 라세미 글루타르이미드, 히단토인 등이 키랄고정 상 9 에서 광학분할되는 것으로 알려져 있으나 광학분할능 (a 값)이 그 렇게 좋은 편은 아니다 (25) . 광학활성인 트란스 -1, 2- 디아미노시클로핵산울 3- 글리시드옥시프로 필 실리카 겔과 반응시켜 합성된 키랄고정상 꼬은 폴리히드록시화합
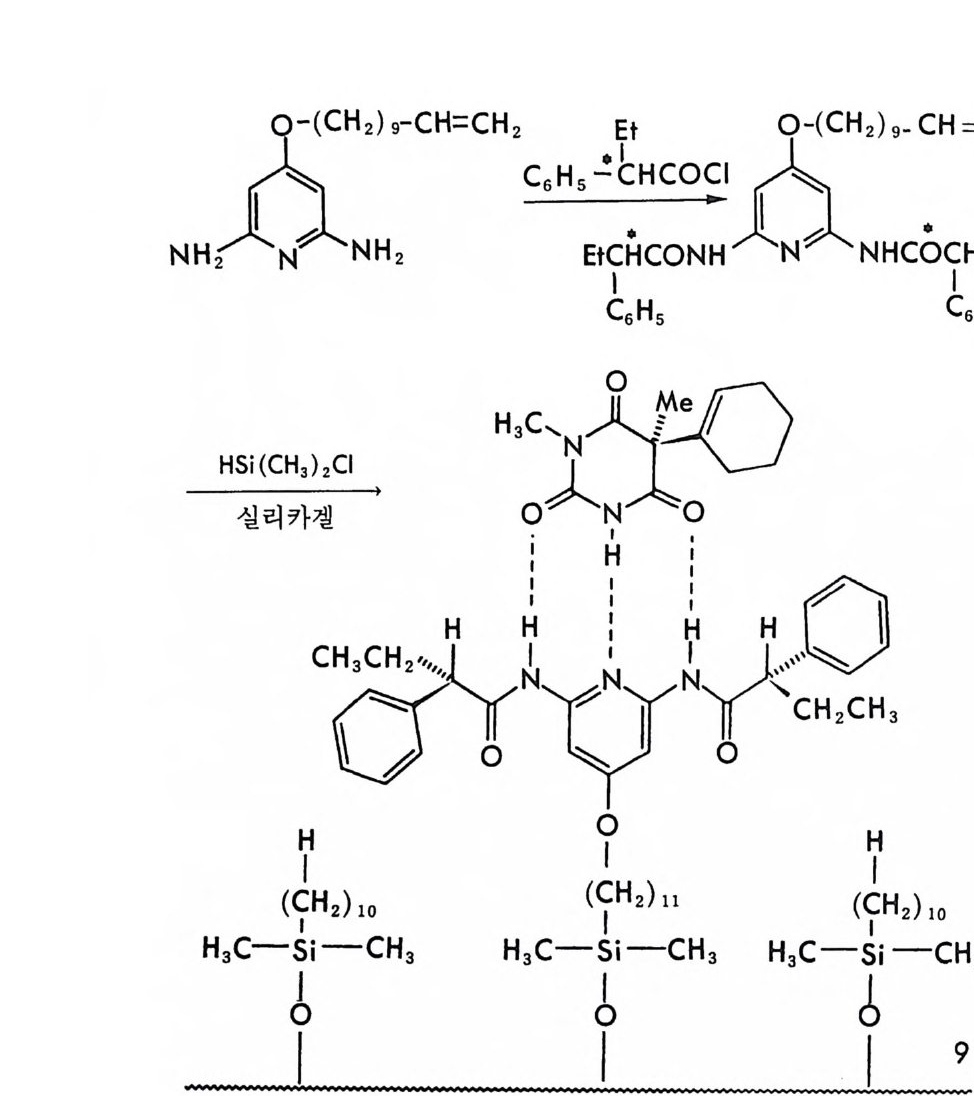 :回 -CH=CH , ~E回[ H= C H2
:回 -CH=CH , ~E回[ H= C H2
그림 8-13 키랄고정상 廻 합성 및 (S) -바아비탈산염과의 세 점 수소작용 모델
물 죽 카테친 (ca t ec hi n) 을 비롯하여 2, 2' -디히드록시 -1, 1' -비나프틸 및 트란스 -1, 2- 시클로핵산디올을 광학분할한다 (26). 키랄고정상 .!Q에 의한 이들 라세미 화합물들의 광학분할은 키랄고정상과 라세미 화합물 入1-o l 의 다중 수소결합에 의하여 가능할 것으로 생각된다. 그림 8-14 는 키랄고정상 1_03!} 2, 2' -디히드록시 -1, 1' -비나프틸과의 두 자리 수
그림 8-14 키랄고정상 L碑 ] 구조 및 키랄고정상 LO:ilJ - 2. 2' -디히드록시 -1, 1'- 비 나프릴의 수소결합 모형도 소결합을 나타내는 모형도이다. 2, 2' -디히드록시 -1, 1' -비나프릴의 두 개 히드록시기 및 키랄고정상의 두 개 아미노기가 입체 화학적으로 특 정한 공간배치를 차지하여 수소결합에 참여하고 있기 때문에 두 개의 광학 이성질체가 구분되는 것으로 생각된다. 참고문헌(제 8 장) 1. W. H. Pir k le and T. C. Pochap s ky, Chem. Rev., 쁘, 347 (19 89). 2. R. Dapp e n, H. Arm and V. R. Mey e r, J. Chroma t o gr.전전, 1 (1986). 3. R. Charles, V. Beit ler , B. Feib u sh and E. Gi l-A v, J. Chromato g r., 112, 121 (1975). 4. R. W. Soute r , Chromato g ra p h ic Se pa ratz o ns of Ste r eois o mers, CRC Press, Florid a , Boca Rato n , 1985, chap 2. 5. S. Hara and A. Dobashi , J. Chroma t o gr.선~' 543 (1979). 6. A. Dobashi , Y. Dobashi and S. Hara, J. Liq. Chromato g r., 언, 243 (1986). 7. A. Dobashi, K. Oka and S. Hara, J. Am. Chem. Soc., 102, 7122 (1980).
8. S. Hara, A. Dobashi, K. Ki no shit a, T. Hondo, M. Sait o and M Senda, J. C hromato g r., 판 153 (1986). 9. J. N . Akany a, S. M. Hitch en, and D. R. Tay lo r, Chromato g r ap h ia , 10. -1J6. ',N 2. 2A4 k(1a9n8y2 a). and D. R. Tay lo r, J. Liq . Chromato g r ., 브 , 805 (1987). 11. Ch. Facklam, H. Praceju s , G. Oehme and H. Much, J. Chromato g r., 쁘 I, 118 (19 83). 12. A. Dobashi, Y. Dobashi, K. Ki no shit a a nd S. Hara, Anal. Chem. , 쁘 1985 (1988). 13. A. Dobashi, Y. Dobashi, T. Ono, S. Hara M . Sait o, S. Higa shid a te and Y. Yamauch i, J. C hromato g r., 쁘 ! , 121 (1989). 14. N. Oi, H. Ki tah ara, T. Doi and S. Yamamoto , Bunseki Kag a ku, 관 345 (1983). 15. N. Oi and H. Ki tah ara, Buneski Kag a ku, 쁘 , 386 (1984). 16. HPLC Ap plica ti on Data ( Sumi ch ir a l OA), Enanti om er Sep a rati on of Vario u s Comp o unds [ I ] , Sumi ka Chemi ca l Analys i s Servic e, Lt d. 17. Y. Sato , M. Ni sh ik a wa, and H Shin k ai, J. L iq. Chromato g r. , 프, 445 (19 89). 18. H. Shin k ai, M. Nashik a wa and Y. Sato , J. L iq. Chromato g r., 프 , 457 (1989). 19. A. Dobash i and S. Hara, Tetr a hedron. Lett ., 싼 1509 (19 83). 20. W. H. Pir k le, Tetr a hedron. Lett ., 낀 , 5707 (1983). 21. Y. Dobashi , A. Dobashi and S. Hara, Tetr a hedron. Lett ., 쁘, 329 (1984). 22. Y. Dobashi and S. Hara, J. Am. Chem. Soc., 쁘~. 3406 (1985). 23. Y. Dobashi and S. Hara, Tetr a hedron. Lett ., 쁘, 4217 (1985). 24. Y. Dobash i and S. Hara, J. Org. Chem., 쁘, 2490 (1987). 25. B. Feib u sh, A. Fig ue roa, R. Charles, K. D. Onan, P. Feib u sh and B. L. Karge r , J. Am. Chem. Soc., 쁘, 3310 (1986). 26. M. Sin iba ldi , V. Carunchio , C, Corradin i and A. M. Gi re ll i, Chromato g rap hia 브, 459 (19 84).
제 9 장 상품화된 키랄고정상 1 서론 지난 10 여 년 동안에 개발되어 라세미 화합물들의 광학분할에 사용 되어온 많은 키랄고정상들 중 현재 상품화되어 있어 쉽게 구입할 수 있는 키랄고정상들은 제한되어 있다. 이러한 제한점은 대부분 키랄고 정상들이 라세미 화합물들의 광학분할에 얼마나 유용하게 또 광범위하 게 사용될 수 있는가 하는 것에 의하여 결정되어진 것이 아니라 키랄 고정상을 얼마나 쉽게 제조할 수 있는가 하는 것에 의하여 결정되어진 것이다. 키랄고정상 제조과정의 쉽고 어려움은 결국 키랄고정상 제조 에 반드시 필요한 99% 이상(혹은 99% 에 가까운)의 광학순도를 가전 광학활성 물질을 얼마나 쉽게 구할 수 있는가 하는 점에 귀결된다. 구 하기 쉬운 광학활성 물질로는 상업적으로 구할 수 있는 것들이거나 혹 은 자연에서 구할 수 있는 천연화합물이다. 따라서 상품화된 대부분의 키랄고정상들은 상업적으로 쉽게 구할 수 있는 광학활성 물질들이나 혹은 자연에서 쉽게 구할 수 있는 광학활성 천연물들을 기본으로 하여 합성된 것들이다. 그러나 키랄고정상에 대한 지속적인 연구의 결과로
이제는 키랄고정싱을 이용하여 한번에 100g 정도의 라세미 화합물을 직접 광학분할할 수 있게 되었기 때문에 (제 7 장 참조) 광학순도가 높은 광학활성 물질을 얻기가 용이하여 앞으로는 라세미 화합물의 두 광학 이성질체에 대하여 좋은 입체 선택성을 보이며 광범위하게 응용할 수 있는 키랄고정상들이 상품화될 것으로 기대된다. 액체 크로마토그래피용 키랄고정상의 중요성은 키랄고정상이 분자 의 키랄성 인지를 연구하는 데 중요한 수단으로 이용될 수 있다는 데 있을 뿐만 아니라 실질적인 면에 있어서는 오히려 입체화학에 관련된 분야 죽 약학, 의학, 유기합성, 분석화학, 농학, 식품과학 등의 연구 에 있어서 키랄컬럼들을 이용하여 라세미 화합물을 어떻게 광학분할하 며 또 광학활성 물질의 광학순도를 어떻게 측정하는가 하는 점에 있 다. 따라서 쉽게 상업적으로 구하여 사용할 수 있는 키랄고정상들은 어떤 것들이 있으며 이들의 특성이 무엇인가를 아는 것은 실제의 키랄 컬럼 사용자들에게는 아주 중요한 문제이다. 따라서 〈 제 9 장-상품화된 키랄고정상 〉 에서는 현재 세계 각지에서 상품으로 구입가능한 HPLC 용 키랄고정상들을 제시하고 이들 상업용 키랄고정상 혹은 키랄컬럼들 의 특성을 간단히 소개함으로써 국내에서 키랄컬럼을 이용하여 입체화 학에 관련된 문제점을 해결하고자 하는 연구자들에게 도움을 주고자 하였다. 현재 세계 각지의 많은 HPLC 관련 품목 제조회사 혹은 공급회사들 이 HPL C-용 키랄컬럼둘을 공급하고 있다. 가장 최근의 정보를 수집 하기 위하여 1989 년 12 월 20 일에 세계 각지의 HPLC 관련 품목제조 회사 혹은 공급회사에 편지를 보내어 이들 회사들이 제조하거나 공급 하고 있는 키랄고정상(혹은 키랄컬럼)에 대한 정보를 수집하였으며 이 들 회사들의 팜플렛과 문헌 등을 참고로 하여 〈제 9 장-상품화된 키랄 고정상났t 기술하였다. 〈제 9 장〉만을 참고하더라도 상품화된 키랄컬럼 사용자들에게 도움이 되도록 어떤 경우에는 제 筑 R] 서부터 제 8 장까지 의 내용을 중복하여 제 9 장에 기술하였다.
상품화된 키랄고정상들의 특성 및 광학분할 범위 등에 대하여 키랄 고정상 제조회사 혹은 키랄컬럼 공급회사에 발간된 몇 권의 지침서가 있다. HPLC 용 키랄컬럼을 이용하여 라세미 화합물의 광학분할을 할 때에 이들 지침서는 좋은 참고가 될 것이며 〈 제 9 장沼t 기술하는 과정 에서도 일부 참조하였다. 2 복합방식 키랄고정상 2.1 셀룰로오스를 기저로 한 키랄고정상 셀룰로오스를 기저로 한 키랄고정상들 중 상품화되어 있는 키랄고정 상은 크게 두 종류로 분류할 수 있다. 즉 미세결정형 셀룰로오스 트리 아세데이트 키랄고정상 및 셀룰로오스 유도체를 실리카 겔에 흡착시켜 얻은 키랄고정상의 두 종류이다. 2.1. 1 미세결정형 셀룰로오스 트리아세테이트 키랄고정상 특 징 : 일정한 입자크기를 가전 미세결정형 셀룰로오스. 트리아세데 이트를 직접 HPLC 컬럼에 충진하여 광학분할에 이용된다. 정확한 키랄성 인지의 메커니즘은 밝혀져 있지 않으며 아마도 셀룰로오스 트리아세데이트의 삼차원적 구조가 키랄성 인지 에 중요한 역할을 하고 있다고 생각되고 있다.
구 조:
 OCOCH3
OCOCH3
싱품명 : Chir a lcel CA-I 공급회사 : Daic e l, Baker Chir a l Tria c el 공급회사 : Macherey -N ag e ! 옹 용 : 미세결정형 셀룰로오스 트리아세테이트 키랄고정싱에서 광학 분할 가능한 라세미 화합물들은 다양하다(제 3 장 2.2 참조). 특히 회전장애 이성질체 (a t ro pi somelr) 가 미세결정형 셀룰로 오스 트리아세데이트 키랄고정상에서 광학분할이 잘되며 라 세미 알코올, 고리형 아미드 및 이미드, 에스데르, 케돈, 락 톤등이 광학분할된다. 미세결정형 셀룰로오스 트리아세데이 트의 나선성 구조가 광학분할에 중요한 역할을 하기 때문에 방향족기나 특별한 작용기를 갖고 있지 않은 라세미 화합물들 도 미세결정형 셀룰로오스 트리아세데이트 키랄고정상에서 광학분할된다. 예를 들면 바아비탈산염, 케타민, 아지리딘, 술폭시드, 만델산 유도체, 피롤리돈 유도체, 아제핀, 이미다 졸 유도체, 이소퀴놀란 히단토인, Tra g er 염기 등이 광학분 할된다. 이동상 : 에탄올/물 혹은 메탄을/물의 혼합용매가 가장 많이 사용된 다. 간혹 핵산/알코올의 혼합용매도 사용된다. 아세톤, THF, 아세토니트랄 클로로포름, CH2Cl2 등은 키랄고정상 울 용해하기 때문에 사용할 수 없다. 2.1. 2 실리카 겔에 흡착된 셀룰로오스 유도체 키랄고정상 특 칭 : 셀룰로오스유도체를 다공성인 실리카 겔에 흡착시킨 키랄고 정상이다. 광학분할하고자 하는 라세미 화합물과 키랄고정상 사이의 T 一亢 상호작용, 수소결합, 쌍극자-쌍극자 겹침 등이 키랄성 인지에 중요한 역할을 하고 있는 것으로 짐작되나 정 확한 키랄성 인지 메커니즘은 아직 알려져 있지 않다.
구조및상품명:
상품명 R= COCH3 Ch ira lcel OA R= COCs H s Chir a lcel O;B R = COCs H 4-p- CH3 Chir a lcel OJ R = CO-CH = CH-C6H5 Ch ira lcel OK R = CONH-CsHs Ch ira lcel OC R = CONH-NH-Cs H 3-(3, 5)-N02 Chir a lcel OD R = CONH-Cs H 4 -p- C l Ch ira lcel OF R = CONH- C s H 4 -p - C H3 Chir a lcel OG 공급회사 : Daic e l, Baker 응 용 : 다양한 라세미 화합물들이 실리카 겔에 흡착된 셀룰로오스유 도체 키랄고정상에서 광학분할된다. 보통 방향족기와 함께 카르보닐기, 니트로기, 술피닐기, 시아노기, 히드록시기 등 울 포함하는 라세미 화합물들이 광학분할된다(제:POl , 2.3 참 조). 방향족기를 가지고 있지 않은 라세미 알코올은 벤조일 에스데르 유도체로서 광학분할이 가능하며 특히 Chir a lcel OA 상계서는 방향족기를 가지고 있지 않은 라세미 /3-락톤, 라세미 a- 히드록시 -Y- 락톤 등이 광학분할된다. 라세미 카르 복실산들도 분자내에 방향족기를 가지고 있을 때 실리카 겔에 흡착된 셀룰로오스 유도체 키랄고정상에서 광학분할이 가능 하다. 이동상 : 2- 프로판올-핵산의 혼합용매가 주로 사용된다. 이 의에 에탄 울-물의 혼합용매, 메탄올품t의 혼합용매가 사용되기도 한다
(2. 3. 2 참조). 실리카 겔에 흡착된 셀룰로오스 유도체를 용해 시킬 수 있는 벤젠, 톨루엔, CH2Cl2, 클로로포름, THF, 디 옥산, 에틸아세데이트, 아세토니트릴 등의 용매는 이동상으 로사용할수 없다. 2.2 시클로덱스트린 키랄고정상 특 칭 : 시클로덱스트린은 라세미 화합물과 입체 선택적인 내포 착화 합물을 형성하여 라세미 화합물을- 광학분할하는· 것으로 알려 져 있다. 시클로덱스트린을 기저로 한 키탈고정상으로서 상 품화되어 있는 것은 디섯 종류이다. 특히 키랄공동의 크기가 각기 다른 a-, /J기 Y- 시클로덱스트린을 실리카 겔에 결합시 켜 키랄고정싱을 합성하였으므로 키랄공동의 크기에 따라 광 학분할되는 라세미 화합물들이 달라전다(제 3 장 3 참조).
상품명 및 구조 (표 3-6 참조) : 구조 I 상품명 공급회사 /3- CD I Cy c lobond I Aste c , Technic o l Chir a l = Dalto s il 100, covalentl y I Serva bound Chir a l = Si 100, covalentl y bound I Serva y-C D I Cy c lobond Il I Aste c , Technic o l Chir a l = Dalto s il 100, covalentl y I Serva bound Ch ira l = Si 100, covalentl y bound I Serva a-CD I Cy c lobond Ill I Aste c , Technic o l Ch ira l = Dalto s il 100, covalentl y I Serva bound
/3- C D I CChy cir l ao lb =on Sdi 1I 00A, cceotvy a la letne dt l y bound \I S Aesrtve ca , Technic o l a-CD I Cy c lobond III Acety la te d I Aste c , Technic o l 응 용 : 바아바탈산염, 고리형 아미드 및 이미드, 아민, ' 아미노알코 울등 방향족기를 가진 라세미 화합물들이 광학분할된다. 아 미노산은 단실 유도체나 혹은 /J-나프틸아미드로 광학분할되 며 a - 아릴프리피온산 중에는 유도체로 만들지 않고 직접 광 학분할된 예가 있다. 제 3 장의 표 3 구 및 표 3-8 , 표 3 - 9 에 CD 키 랄 고정상에서 광학분할된 라세미 화합물들의 예가 제시 되어 있다. 이동상 : 물-알코올, 물 - 아세토니트릴 등의 혼합용매를 이동상(역상 이 동상)으로 사용한다. 혼합용매중 물의 양이 많아질수록 분석 성분의 머무롬 시간이 길어진다. 이온화할 수 있는 작용기를 가전 라세미 화합물을- 광학분할할 때에는 인산염완충용액 - 아 세토니트릴 혹은 인산염완충용액-에탄올울 적절히 혼합하여 p H 가 조정된 이동상을 사용한다. 2.3 단백질 키랄고정상 단백질을 기저로 한 키랄고정상에서는 당 단백질 (a,-aci d gly c o - p ro t e i n) 을 기저로 한 AGP 키 랄고정상 및 소의 혈청 알부민 (bov ine serum album i ne) 을 기저로 한 BSA 키랄고정상이 상업화되어 있다. 2.3.1 AGP 키랄고정상 특 징 : 혈장 단백질인 당 단백질 («1-acid g l y co p ro t e i n) 울 다공성인 구형 실리카 겔에 고정시킨 키랄고정상으로서 키랄고정상과
라세미 화합물 사이의 이온 _ 이온 상호작용 및 소수성 상호작 용이 키랄성 인지에 중요한 역할을 한다. 싱품명 : Chir a l AGP 공급회사 : Chrom Tech, Technic o l, Baker Enanti oP ac 공급회사 : Pharmacia LKB Bio t e c hnolog y 응 용:다양한종류의 양이온성 화합물(아민 계통), 음이온성 화합물 (카르복실산 계통), 중성 화합물들이 광학분할된다(제 3 장의 표 3-11, 표 3~12, 표 3-13, 표 3-1 4 , 표 3-1 5 참조) 광학분할조건 : (제 3 장 4.2.3 참조) 이동상 : 인산염완충용액 (보통 0.02M) 에 알코올, 아세토니트릴 등의 유기변형제를 적당량 첨가한 용액을 이동상으로 사용하며 음 이온성 라세미 화합물(카르복실산 계통)을 광학분할할 때에는 인산염완충용액에 N, N- 디옥틸아민이나 브롬화 테트라부틸 암모늄을 첨가한 용액을 이동상으로 사용한다. p H 범위 : 3.0-7.5 유 속 : 컬럼의 수명을 최대한으로 하기 위하여 최대유속은 0.3 m L/ m i n을 초과하지 않는 것아 좋다. 컬럼의 보관 : 컬럼을 며칠동안 사용하지 않을 때는 인산염완충용액 운기용매의 혼합용매 대신 물을 통과시킨 후 물 -2- 프로판올 (1/1) 의 혼합용매를 통과시켜 물강_프로판을 (1/1) 혼합용매 조건에서 보관하는 것이 좋으며 컬럼이 마르지 않도록 주의하 는 것이 좋다. 보통 4°C 산 C 의 일정한 온도에서 컬럼의 양쪽 끝을 잘 봉합한 상태로 보관하는 것이 좋다. 2.3.2 BSA 키랄고정상 특 칭 : 소의 혈청알부민을 실리카 겔에 고정시킨 키랄고정상이다. 상품명 : Resolvosil BSA-7 공급회사 : Macherey- Nage l.
Chir a l = Si 300 BSA, 공급회사 : Serva cov a len tl y bound 응 용 : 다양한 음이온성 라세미 화합물 및 중성 라세미 화합물들이 광학분할되나 양이온성 라세미 화합물의 광학분할 예는 혼하 지 않다. 보통 아미노산 유도체, 방향족 아미노산, 방향족 술 폭시드, 바아비탈산염 , 벤조디아제피논, 벤조인 및 벤조인 유도체, /3 - 차단제 , 쿠마린 유도체 등이 광학분할된다(제:PJ-, 4.3 참조 ). 광학 분할 의 조 전: 이동상 : 인산영완충용액 혹은 붕산염완충용액에 소량의 1_ 프로판올 (대략 5%) 를 첨가한 용매 를 이동상으로 사용한다. 유기용매 첨 가제로서 아세토니트릴이나 메탄올을 가하면 알부민이 변성 되기 때문에 아세트니트릴이나 메탄올은 유기변형제로 사용 할 수 없다. 분석성분의 머무름 시간과 입체 선택성 (a 값)은 pH , 완충용액의 세기 (0.0l-0.2M), 첨가하는 1- 프로판올의 양을 적절히 바꿈으로써 조절이 가능하다. p H 범위 : 5.0-8.0 분석용량 : 실리카 겔에 고정된 알부민 (BSA) 의 양이 소량이기 때문에 분석하고자 하는 시료의 용량은 시료주입당 1 - 2nmol 을 초 과하면 광학분할능이 저하된다. 컬럼보관 : 사용중려]는 이동성을 낮은 유속으로 계속해서 흘려주어야 좋다. 사용하지 않을 때는 5' C 의 냉장고에 보관하는 것이 좋으며 장시간 사용하지 않을 때는 컬럼을 통하여 0.02% 의 나트륨아지드 (sod i umaz i de) 를 포함하는 중성 인산염완 충용액을 통과시킨 후 보관하는 것이 좋다.
2.4 고분자 키랄고정상 고분자물 기저로 한 키랄고정상 중 상품화된 것으로는 광학활성인 나선성 고분자를 기저로 한 키랄고정성들과 폴리아크릴아미드를 기저 로한 키랄고정상 및 폴리메타크릴아미드를 기저로 한 키랄고정상이 있 다. 2.4 .1 나선성 때문에 광학활성인 고분자를 기저로 한 키랄고정상 특 칭 : 나선성 때문에 광학활성인 고분자 즉 폴리트리페닐메틸메타 크릴레이트 (PTrMA) 혹은 폴리디페닐 -2- 피리딜메틸메타크 릴레이트 (PD2P y MA) 를 다공성인 실리카 겔에 흡착시킨 키랄 고정상으로서 나선성 고분자가 가지고 있는 키랄공동이 광학 분할에 중요한 역할을 하고 있다고 생각될 뿐 정확한 키랄성 인지 메커니즘은 현재 알려져 있지 않다.
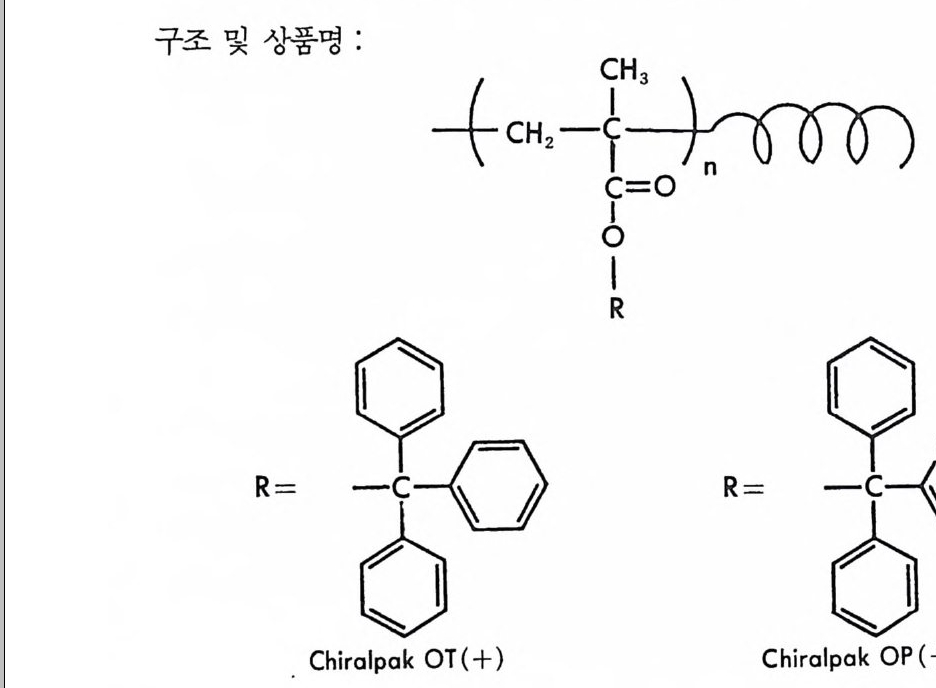 구조및상품명:
구조및상품명:
공급회 사 : Daic e l, Baker 응 용 : 방향족기를 포함하는 라세미 화합물들이 광학분할된다(제 4 장, 2.2 참조) 이동상 .. 주로 메탄올을 이동상으로 사용한다. 헥산 _ 2 군프로판을의 혼합용매, 아세토니트릴, 핵산 등이 이동 상으로 사용되기도 한다. 메탄올 이동상에 물을 첨가하여 (20% 까지) 분석성분의 머무름 시간을 증가시키고 입체 선택 성을 증진시킬 수 있다. 톨루엔, CH2Cl2, 클로로포름, 아세 돈, THF, DMSO 등은 실리카 겔에 흡착된 나선성 고분자를 용해할 수 있으므로 이동상으로서 사용할 수 없다. 2.4 .2 폴리아크릴아미드를 기저로 한 키랄고정상 특 징 : N- 아크릴로일 - (S)- 페닐알라닌에틸에스데르를 실리카 겔에 흡착시켜 라디칼 중합반응을 실시하고 (AIBN, 8(j C ) 실리카 겔의 실라놀기를 제거하여 얻어진 고분자 키랄고정상으로서 실리카 겔의 좋은 기계적 성질로 인하여 HPLC 에 응용하기 에 적합하다(제 4 장, 3 참조).
구 조:
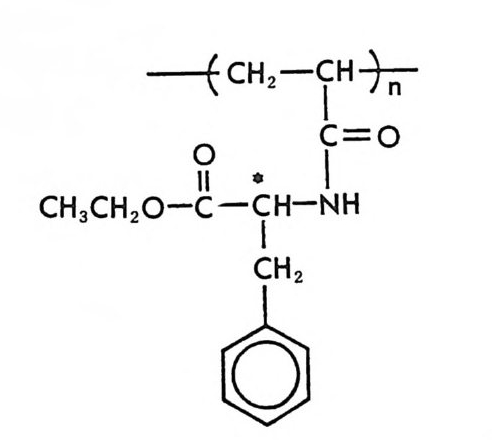 CH3CH2_0--C{운 C— H•<2 ;:-H ~-NCIIH H = 뉴 O
CH3CH2_0--C{운 C— H•<2 ;:-H ~-NCIIH H = 뉴 O
상품명 : Chir a Sp h er 공급회사 : Merck 웅 용 : 다양한 종류의 라세미 의약품들이 Chir a S p her 키랄 HPLC 컬럼상에서 광학분할된다. 죽 바아비탈산, 이뇨제인 벤조티 아디아전 계통의 라세미 의약품, 벤조디아제피논 계통의 라 세미 의약품등이 광학분할된다(제 4 장, 3.2 참조). 이동상 : 비극성 용매에서부터 강한 국성용매까지 다양한 유기용매 를 이동상으로 사용할 수 있으며 Chir a Sp h er 컬럼은 n- 핵산용 매로 채워져 공급되고 있다. 아세토니트릴, 디옥산, THF, 메틸 t유넷에데르` 톨루엔, 2- 프로판을, 에탄올, 메탄올, 물 및 이들의 혼합용매 등 모두 이동상으로 가능하다. 3 독립방식의 키랄고정상 3.1 리간드 교환 키랄고정상 특 징 : 프롤린, 히드록시프롤린, 아미노산 혹은 다른 종류의 두 자 리 리간드로 작용할 수 있는 광학활성 물질을 실리카 겔에 고 정시킨 키랄고정상으로서 키랄고정상 -Cu (Il )-광학이성질체 (두 자리 리간드)의 삼성분 칙물을· 형성할 때 라세미 화합물의 두 광학 이성질체가 서로 안정성이 디른· 삼성분 착물을 형성 하기 때문에 광학분할이 이루어지는 키랄고정상이다. 구조및상품명: 상품명 : Nucleosil C hir a l..:.1 공급회사 : Machery- N ag e l (정확한 구조는 보고되지 않음) 특징 : 실리카 갤에 고정된 L- 히드록시프롤린 -Cu( JI )
상품명 : Chir a lpa k WH 공급회사 : Daic e l, Baker 구 조:
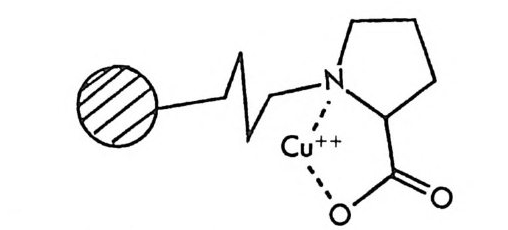
특 징 : 리간드는 프롤린임 상품명 : Chir a lpa k WM 공급회사 : Daic e l, Baker 구 조:
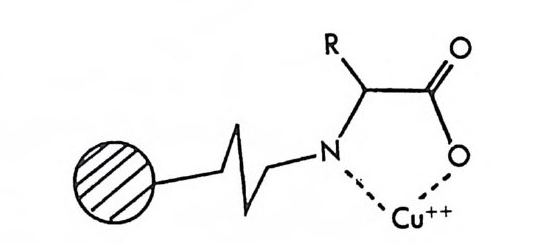 @N曰R 、`、 `Cut ':O °
@N曰R 、`、 `Cut ':O °
특징 : 아미노산을 실리카 겔에 고정시킨 키랄고정상아다. R 은 정 확히 밝혀있지 않음. 상품명 : Chir a lpa k WE (키 공급회사 : Daic e l, Baker
구 조:
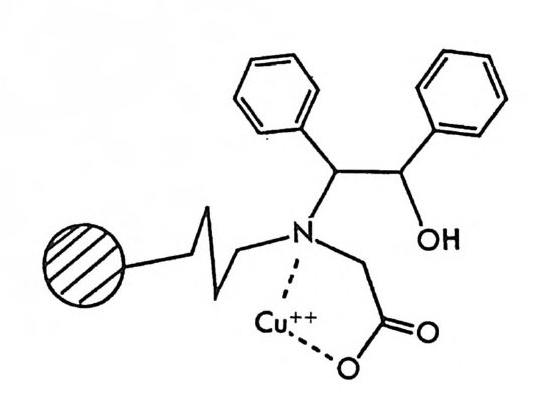 OH
OH
특징 : 아미노알코올의 변형물질을 실리카 겔에 고정시킨 키랄고정 상 O] 다. 이의에 Serva 에서 Chir a l = Si 100 L-Hy pr oCu 및 D-Hy pr oCu Chir a l = Si 100 D-ProCu 및 L-ProCu Chir a l = Si 100 D-ValCu 및 L-ValCu( 모두 공유결합 형태임) 라는 상품명으로 HPLC 키랄컬럼을 공급하고 있을 뿐만 아니 라 리간드 교환 키랄고정상을 컬럼에 충전시키지 않고 직접 5g , 10g , 100g 단위로 제공하고 있다. 응 용 : 리간드 교환 키랄고정상에서는 두 자리 리간드로 작용할 수 있는 라세미 화합물 죽 주로 아미노산이 광학활성되며 a- 히 드록시카르복실산이 광학분할된 예도 있다. 이동상 : 주로 0.02-0.5mM CuS04 수용액 . 머무름 시간을 조정하기 위하여 아세토니트릴과 같은 유기용매를 첨가할 수 있다. 3.2 크리운에데르 키랄고정상 특 칭 : 키랄공동을 가전 광학활성 크리운· 에데르를 기저로 한 키랄고 정상으로서 현재 상품화된 것으로 조사된 것은 한 종류의 크 리운 에테르 키랄고정상이 있다. 이것은 18-Crown-6 화힙물 울 역상 -C18 컬럼에 흡착시킨 것으로써 (제昞} 참조) 분석성분 의 암모니움이온이 크라운 에테르의 키랄공동에 끼어들어가 세 자리에서 수소철합을 형성하고 분석성분의 나머지 부분과 키랄 크라운 에테르와의 입체 선택적인 상호작용(입체장애 등)에 의하여 키랄성 인지가 이루어진다. 구 조:
 广0 ^
广0 ^
상품명 : Crownp a k CR, 공급회 사 : Daic el , Baker 응 용 : 아미노기를 가지고 있는 대부분의 라세미 화합물들이 암모늄 이온형태로 광학분할된다. 죽 아미노산, 아민, 아미노알코올 들이 광학분할된다. 이동상 : 과염소산 수용액 (pH 2.0, pH 1.5 혹은 pH 1.0) , 질산 수용액 , 혹은 삼플르오르아세트산수용액이 이동상으로 사용된다. 유 기용매를 이동상으로- 사용하여서는 아니된다. 3.3 전하이동 착물형성 키랄고정상 키랄고정상과 광학분할하고자 하~즌 라세미 화합물 사이의 다양한 상 호작용 중 7t-7t 상호작용이 키랄성 인지에 중요한 역할을 하는 전하이 동 착물형성 키랄고정상들 중 상품화되어 있는 것들은 아주 다양하다. 크게 T- 산성 키랄고정상및 T- 영기성 키랄고정상으로나~수있다. 3.3.1 1r:-산성 키랄고정상 특 칭 : T- 영기성기를 가지고 있는 라세미 화합물과 7r:-7r:상호작용을 할 수 있는 T- 산성기를 가지고 있는 광학활성 물질을 실리카 겔에 고정시킨 키랄고정상이다.
구조, 상품명 및 공급회사 : 0022口NN 『\ H; x:h:N-(CH2) 』 \0o —— Reg is( R)-혹 은 (S)-공급회사 : Reg is Pheny lg l y c i n e (Ionic ) Bakerbond DNBPG (Ionic ) 공급회사 .• Baker 0022NN〉 :\H; :\°( CH2)3_ 『::二 Reg is(R )- 혹은 (S)- 공급회 사 : Reg is Pheny lg l y c i n e (Covalent) Ba5ke rbon>d DNBNPIGH( C 。ov aN?le' -n (tC) H2공)3급S°長회Ei \ t사 0o :—— Ba ker 5 Reg is( R)-혹 은 (S)-L euc ine 공급회사: Reg is Bakerbond DNBLeu 공급회사: Baker
—o,1O Et (R) /n'-2 Sumi ch ir a l OA- 20 00 공급회사 : Sumi ka , Reg is o.... OIE t + - (R) (S) Sumi ch ir a l OA- 21 00 공급회사 : Sumi ka , Reg is ―:〉『~ (CH , ),- 집 H3 -o 2c亡 」 :k-NH C0:N0N022 -oSuom>i ch Li_r (aC lH O2A)3-- N25H 0C0O i -공N급H회C사 0: S:uNrni0k a 2, Reg is OEt /N02 Sumi ch ir a l OA-3100 공급회사 .: Sumi ka , Reg is
 —0—\0 /oSE i, t_ -cH 2 ,,I 3 -N H eO-H_8aCC -lN_HccH I。 c3 g:g
—0—\0 /oSE i, t_ -cH 2 ,,I 3 -N H eO-H_8aCC -lN_HccH I。 c3 g:g
웅 용:알킬카르비놀, 히단토인, 락탐, 숙신이미드, 프탈리드, 술폭 시드, . 술피드 (sul fi de), 아미드, 고리형 아미드 , 이미드 동이 키랄중심이나 키랄중심에 가까운 위치에 방향족기를 가지고 있을 때 광학분할된다(제 7 장, 3 참조). 아민 등은 나프틸카르 밤산 에스데르유도체로 광학분할되며 카르복실산은 나프틸아 미드 혹은 3,5- 디메틸아닐리드 유도체로 광학분할되고 /3-차 단제는 옥시졸리돈 유도체로 광학분할되며 이의에도 T- 염기 성기를 가지고 있는 많은 종류의 라세미 화합물 및 라세미 화 합물들의 T- 염기성 유도체들이 광학분할될 것으로 기대된다. 이동상 : 이온결합 형태의 키랄고정상에서는 20% 이하의 2- 프로판을
핵산의 혼합용매를 이동상으로 사용한다. 공유결합 키랄고 정상에서는 2 - 프로판올-핵산의 혼합용매를 주로 이동상으로 사용하나 이동상의 극성을 염려할 필요는 없다. 3.3.2 7r- 염기성 키랄고정상 특 징 : 강한 r- 염기성기를 가지고 있어서 광학분할하고자 하는 라세 마 화합물 (7C - 산성기를 가지고 있는 라세미 화합물)과 T?r 상호 작용을 할 수 있는 광학활성 물질을 실리카 겔에 고정시킨 키 랄고정상 0] 다. 구조, 상품명 및 공급회사 :
 二宁; °`(CH2)n- 더
二宁; °`(CH2)n- 더
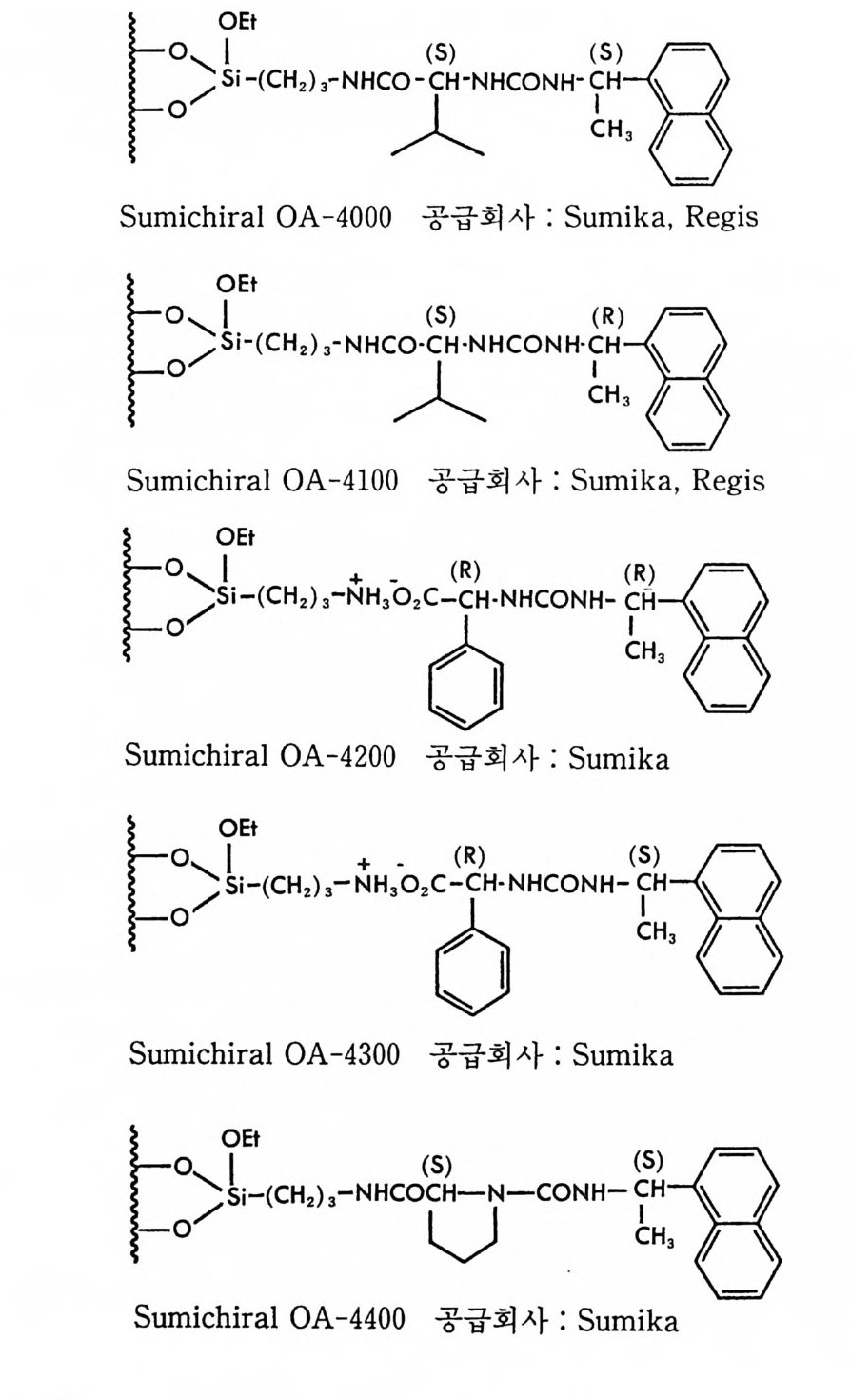 「。o 〉 r(CH , ),-NHCXHCONH-r: i3
「。o 〉 r(CH , ),-NHCXHCONH-r: i3
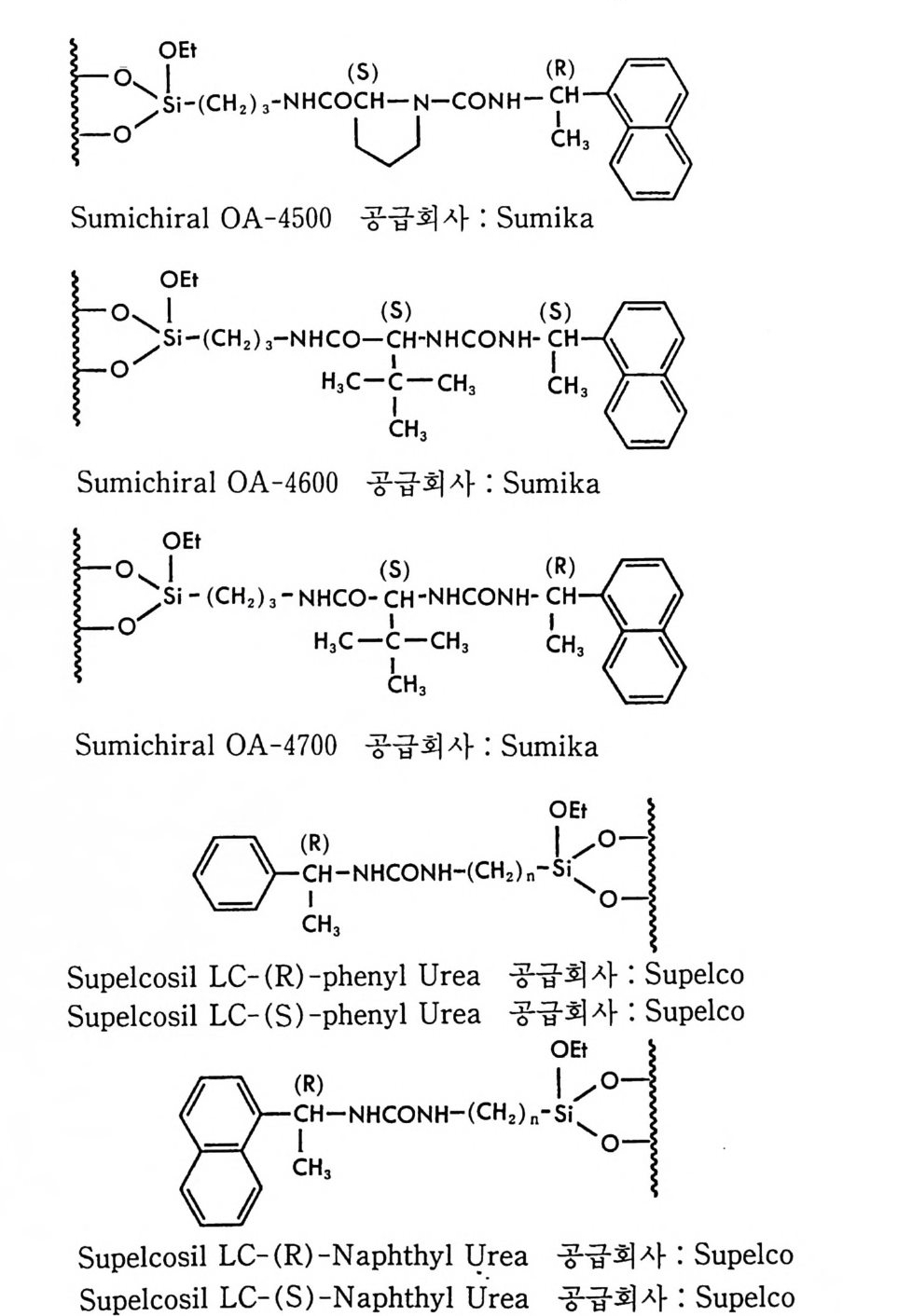 「tt'::',,‘ 'L': l,0,S0'O:', ',:'.,\\ 'tr,'osS/n\So/Ourt.E E- l-ut a tlc(lciCc ChiHHamHh222 ) ) ) i 33 -- il-AO4 NNN A OHlHH _cC-rH 0 35 O?__공 cH 근一o60 :O S 「:〉『_ I(CH,) ,-NHCXNHCONH-\::CH,
「:〉『_ I(CH,) ,-NHCXNHCONH-\::CH,
상품명 : Sumi ch ir a l OA-3000 공급회사 : Sumi k a 옹 용 : 아미노산의 N- 아세틸 -0- t국냐i에스테르의 광학분할이 특히 성공적이다. a- 히드록시카르복실산의 3, 5- 디니트로페닐 카 르방산 에스데르의 광학분할에도 응용된다.
이동상 : 핵산 -2- 프로판을의 혼합용매와 같은 정상 이동상을 사용한 다. 4 상품화된 키랄고정상 제조회사의 요약 상품화된 키랄고정상을 제조하여 공급·하는- 회사들 및 제품의 명칭 간단한 특성을 디음 표에 요약하였다(제조회사순은 알파벳순으로). 제조회 사 키 랄고 정상의 상품명 트-,. 지o 키랄고정상의 키랄단위 Aste c Cy c lobond I 키랄공동 /3 -Al 클로덱스트린 Cy c lobond ll 키랄공동 Y-A] 클로덱스트린 Cy c lobond 冊 키랄공동 c; - Al 클로덱스트린 Cy c lobond I Acety la te d 키랄공동 아세틸화 /3- C D Cy c lobond ill Acety la te d 키랄공동 아세틸화 a-CD Baker IBa kerbond DNBPG 7C - 받게 (R)-DNB 페닐글리신 (Ionic ) Bakerbond DNBPG I n - 받게 l(R)-D NB 페닐글리신 (Covalent) Bakerbond DNBLeu 17C- 받게 l(S)-DNB 루이신 (Covalent) Chrom Chir a l AGP 단백질키랄 당단백질 (a1-Ac id gly c o Tech 고정상 pro te i n ) Daic e l Crownp a k CR (+ ) 키랄공동 18 - 크라운 -6 Chir a lcel CA-I 나선성, 미세결정형 셀룰로오스 트 키랄공동 리 0~ 세데 o] 트 Chir a lcel OA I 나선성, 셀룰로오스트리아세테이트 키랄공동 Chir a lcel OB I 나선성, 1 셀룰로쭈 트리벤조에이트 키랄공동 Chir a lcel OK |中J성, 1 셀룰로오스 트리신남메이트 키릴공동
Chir a lcel OJ | 나선성, 셀 물 로오스 트리 (p - 메 칠 ) 키 랄 공동 벤 조 에이 트 Chir a lcel OC I 나선성, 셀 룰 로오스 트리페닐카바메 키릴공동 이트 Chir a lcel OD I 나선성, 셀 룰 로오 스 트리 (3,5 - 디메 키랄공동 틸 페닐)카바메이트 Chir a lcel OF |니선성, 셀 룰 로오스 트리 (p 을 늄뚝i 키랄공동 페닐)카바메이 트 Chir a lcel OG |나선성, 셀 룰 로오스 트 리 (p - 메 틸 페 키랄공동 닐)카바메이트 Chir a lcel WH 리간드교환 프롤 린 -Cu(Il ) Chir a lcel WM 리간드교환 아미노산 - Cu(ll ) Chir a lcel WE 리간드교환 두자리리간드 - Cu(Il ) Chir a lpa k OT (+ ) 나선성 폴 리 (트 리 페 닐메 틸 메 타크 릴 레 o] 트) Chir a lpa k OP (+ ) I Y'J성 |폴리 (2 - 피리딜디페닐메틸 메타크릴레이트) Macherey -N ucleosil Chir a l-I 리간드교환 L- 히드록시프롤 린 -Cu Nag e l (II ) Nucleosil Chir a l-2 r- 받게 3, 5- 디니트로벤질페닐에 털아민 Resolvosil BSA-7 1 단백질 |소혈청알부민 (BSA) 키랄고정상 Chir a l Tria c el I 나선성 1 미세결정형 셀 룰 로스 트 리아세테이트 Merck IChir a Sp h er |키랄공동 1s 페닐알라닌에틸에스데 르를포함한폴리아미드 Cellulose tri a c eta t e I 키랄공동 1 셀룰로오스 트리아세데이 E Lich rosp h er lOONH2+ l1r- 받게 IDNB 대닐글리신 DNBPG Pharmacia IEnanti oP ac 1 단백질 당단백질 (a1-Ac id gly c o 키랄고정상 pro te i n ) Reg is jRe g is ( R)- Ire- 받게 (R)-DNB- 페닐중신 Pheny lg l y c ine (Ionic )
Reg is ( S)- 1 7[ - 받게 I (S) - DNB 대닐글리신 Pheny lg l y c in e (Covalent) Reg is (R)- 1 7!-받게 I (R)-DNB- 페닐중신 Pheny lg l y c ine (Covalent) Reg is ( S)-Leucin e I7C- 받게 I (S) -D NB Leu (Ionic ) Reg is ( S)- I7C- 받게 I (S) -DNB Leu Leucin e (Covalent) Reg is (R)-N- I7C 주게 I (R) -N- 나프틸알라닌 (2 一 Nap h th y !) alanin e Reg is ( S)- N - I7C 주게 I (S) -N- 나프틸양가닌 (2- N ap h th y l)al anin e Serva !ch ir a l = Dalto sil 100 (구 형입자) D- D N B-Leu(covalent) r- 받게 D-DNB-Leu D- D NB- L eu(io n ic ) r- 받게 D-DNB-Leu L- D NB Leu(covalent) 亢-받게 L-DNB-Leu L-DNB Leu(covalent) 7C- 받게 L-DNB-Leu D- D NB-Phg ly ( c ovalent) 亢-받게 L-DNB-Pheg ly D-DNB-Phg ly ( ion ic ) T- 받게 L-DNB-Phg ly L- D NB-Phg ly ( c ovalent) 7C- 받게 L-DNB-Phg ly L-DNB Phg ly ( ion ic ) r - 받게 L-DNB-Phg ly Chir a l = Si 100 (불 규 칙 입 간 D-DN B-Leu(covalent) TC- 받게 D-DNB-Leu L-DNB-Leu(covalent) 7C- 받게 L-DNB-Leu D-DNB-Phg ly ( c ovalent) r- 받게 D-DNB-Phg ly L-DNB-Phg ly ( c ovalent) 7C- 받게 L-DNB-Phg ly D-DNB-Leu(io n ic ) r- 받게 D-DNB-Leu L-DNB-Leu(io n ic ) 7C- 받게 L-DNB-Leu D-DNB-Phg ly ( ion ic) 1C- 받게 D-DNB-Phg ly L-DNB-Phg ly ( ion ic ) r- 받게 L-DNB-Phg ly Chir a l = Dalto sil 100 (구 형입자) a-cyc lodextr i n ( covalent) |키 랄공동 la- 시클로덱스트린
/3- cy cl odextr i n ( covalent) 키 랄공동 f3 - 디 클 로덱 스트린 Cyh- cir ya c l l o=d Seix t1r 0i 0n ( (co불v규a l칙en 입t) 키 릴공동 y - Al 클로덱스트린 자) a-cyc lodextr i n ( covalent) 키 랄공동 a - Al 클로덱스트린 /3- cy c lodextr i n ( covalent) 키 랄공동 f3-시클로덱스트린 Cy-h ciry a c l l o=d eDxatlr ti on s( cilo v 1a0l0e n(t구) 키 랄공동 y - 디클로덱스트린 형입자) L- H y pr oCu(covalent) 1 리간드 교환 |L - 히드록시프 롤 린 -Cu ( Il ) D-ProCu(covalent) 리간드교환 D- 프롤 린 - Cu( ll ) L-ProCu(covalent) 리간드교환 L - 프롤 린 - Cu ( Il ) D-Va!Cu(covalent) 리간드교환 D- 발린 - Cu(Il ) L-ValCu(covalent) 리간드교환 L- 발린 -Cu(H ) Chir a l = Si 100 (불규 칙 입 잔 L-Hy pro Cu(covalent) 리간드교환 L- 히드록시프롤린 Cu ( Il ) D-ProCu(covalent) 리간드교환 D- 프롤린 - Cu ( Il ) L-ProCu(covalent) 리간드교환 L- 프롤린 -Cu ( ll ) D-ValCu(covalent) 리간드교환 D- 발린 -Cu(Il ) L-ValCu(covalent) 리간드교환 L- 빌린 -Cu(Il ) Ch ira l = Si 300 IB SA(covalent) 단백질 |소혈청알부민 (BSA) Sumi ka Su mich ira l OA-1000 T 주게 a- 나프틸에틸아민 Surnic h ir a l OA-2000 亢-받게 DNB 페닐글리신 Sumi chira lOA-2100 r 주게 클로로페닐 0l 소발레로일페 닐글리신 Surnic h ira l OA-2200 7C 주게 크리산데모일페닐글리신 Sumi ch ira lOA-2500 7C- 받게 N-DNB- 나프릴굴리신 Sum ich ir a l OA-3000 수소결합 • t유틸아미노카르보닐발린 Sumi chira l OA-3100 r- 받게 발린- (3, 5- 디니트로페닐) 우레 °l 드 Su mich ir a l OA-3200 1 7(-받게 I t부될글리신 -(3, 5- 디니 트로페닐우레이드)
Sumi ch ir a l OA- 40 00 I n 주 게 \(S , S) - a 나프릴에틸아미 노카르보닐발린 Sumi ch ir a l OA- 4 1 00 I 7C 주 게 I (R , S )- a - 나프릴에틸아미 노카르보닐발린 Sumi ch ir a l OA- 4 20 0 I 1e - 주게 I (R, R) - a- 나프틸에틸아미 노카르보닐 페닐글리신 Sumi ch ir a l OA- 43 00 I7C 주게 I( S, R)-a- 나프릴에틸아미 노카르보닐 페닐글리신 Sumi ch ir a l OA- 44 00 I 7C 주게 I (S, S)-a - 나프릴에틸아미 노카르보닐프롤린 Sumi ch ir a l OA- 45 00 |T 주게 I(R, S)-a- 나프릴에틸아미 노카르보닐프롤린 Sumi ch ir a l OA- 46 00 I1r 주게 I (S, S)-a- 나프릴에틸아미 노카르보닐 t부될글리신 Sumi ch ir a l OA- 47 00 |7C 주게 I (R, S)-a- 나프틸에틸아미 노카르보닐 t부될굴리신 Sup e lc o lsup e lcosil LC- (R )- 1 7!주게 I (R) -페닐에틸아민 ph eny l Urea Sup e lcosil LC- (S ) - In 주게 I(S) -페닐에틸아민 PSuhpe ne lyc lo Usirl e LaC -(R ) - 1 7!주게 l( R) -나프틸에틸아민 Nap h th yl Urea Sup e lcosil LC-(S ) - 1 7!주게 I (S) -나프틸에틸아민 Nap h th y l Urea 5 키랄 HPLC 컬럼 제조회사 혹은 공급회사의 주소(알파벳순) 세계 각지에 많은 키랄 HPLC 컬럼 제조회사 및 공급회사들이 있으 나 그동안 접촉하여 자료를 얻을 수 있었던 회사의 주소를 수록하였 다. Aste c : Advanced Sep a rati on Technolog ies Inc. 37 Lesli e C ou rt, P. 0. BOX 297, W ippa ny , N J 07981 U.S.A .
Baker : J. T. Baker Inc. 222 Red School Lane, Phil lips burg, New Jer sey 08865, U. S. A Chrom Tech : Chrom Tech AB, BOX 512, S-145 63 Norsborg, Sweden Daic e l : Daic e l Chemi ca l Industr i e s , LTD. 8-1, Kasumi ga seki 3-Chome, Chiy o da-ku, Toky o 100, Jap a n Macherey- N age l : Macherey -N age l GmbH & Co. KG P. 0. BOX 101352, D-5160 Duren, Germany Merck :E. Merck, Frankfu r te r Str a sse 250, D-6100 Darmsta d t 1, Germany Pharmaci a : Pharmacia LKB Bi ot e c hnolog y AB, S-75182 Up ps ala, Sweden Reg is : Reg is Chemi ca l Comp a ny , 8210 Austi n Avenue, Morto n Grove, Illin o is 60053, U.S.A Serva : Serva Fein b io c hemi ca GmbH Co. Carl-Benz-St ra /3 e 7. P. 0. BOX 105260. D-6900 Heid e lberg, Germany Sumi ka : Sumi ka Chemi ca l Analys is S ervi ce , Ltd. S cie n ti cfic Instr u ments Di v. Chromato g rap h y Group , 3-1-135, Kasug a de-naka, Konohana-ku, Osaka 554, Jap a n Sup e lco : Sup el co, Inc. Supe lc o Park, Bellefo n te , PA 16823-0048 U. S. A Tech nico l : Technic o l Ltd. Brook Str e et, High er Hi llga te , Sto c kp o rt, Che-shir e SKI 3HS, U. K.
참고문헌(제 9 장) 다음의 참고문헌들은 상품화된 키랄고정상들에 대한 정보를 얻는데 유 용하게 사용될 수 있을 것이다(무순). 1. R. Dapp e n, H. Arm and V. R. Meye r , J. Chroma t o gr.전전 1 (19 86). 2. S. R. Perrin and W. H. Pir k le, A. Chir a l Hand Book, Reg is C hemi ca l Comp a ny 3. Ap plica ti on Guid e for Chir a l Colum Selecti on , CROWNPAK, CHIRALCEL, CHIRALPAK, Chir a l HPLC Column for Op tica l Resoluti on , Daic e l Chemi ca l Industr i e s , LTD, 1989. 4. I. W. Wain e r, A. Practi ca l Guid e to the Selecti on and Use of HPLC Chir a l Sta t i on ary Phases, J. T. Baker. 5. Cy cl obond Hand book, a Guid e to Usin g Cy cl odextr i n Bonded Phases, Advanced Sep a rati on Technolgi es Inc.(Aste c )
찾아보기
기 가로막기 효과 85 간접 분리방법 65, 68-89 거울상 이성질체 0-16 경사용리 크로마토그래피 250 고유 광회전도 20 공유결합 부분니깁체 이성질체 53 광학분할 19 광학분할 시약 53 광학순도 20 광학 이성질 현상 15-42 광학 이성질체 16 광학활성 18 광학활성 니선상 고분자 234 광학활성 나선성 고분자를 기저로 한 키랄고정상 234-250 광학활성 천연물 120 광학활성 크라운·에데르 322 광학활성 폴리아미드를 기저로 한 키랄고정상 250-266 광학활성 합성고분자 233 광회전도 19 교차결합시약 270 구조 이성질체 15 글루코시드 결합 122, 124 ; 150 기하 이성질체 17 L 나선성 21, 32 내포 크로마토그래피 129
NMR 에 의한 광학분할 60- 6 5 NOE 효과 107, 426 C 다중점 상호작용 105 단백질을 기저로 한 키랄고정상 179-223, 469 단일점 상호작용 105 당단백질 180 대칭면 21 대칭요소 21 대칭중심 21 대칭회전축 21 독립방식의 키랄고정상 444, 474 독립적 방식에 의한 키랄성 인지 109 동반에 의한 광학분할 48 동위원소 희석법 46 D-A 착물 337 E2 (R)-(-)-TAPA 키랄고정상 340 라세미 고용체 19 라세미산 34, 35 라세미체 19, 34 라세미 혼합물 19, 46 라세미화 19 라세미 화합물 19 LUMOol] 너지 준위 337 루이신 -DNB 키랄고정상 348-361
찾아보기
리간드 교환 크로마토그래피 94, 283 리간드 교환 키랄고정상 283- 3 17, 474 □ Marckwald 법칙 56-57 MPL D-]스템 75 머무름시간 66 메조형 26 면과 면의 상호작용 364 (D,L) -명명법 24 (R,S) -명명법 23, 29, 31 미세 결정형 셀룰로오스 트리아세데 이트 키랄고정상 124- 1 32, 465 t:I 반대칭 분자 21 반면상구조 33 반전축 21 배열 광학 이성질체 17 배열 부분 입체 이성질체 17 배열 이성질체 17 보정된 머무름시간 66 복합방식의 키랄고정상 465 복합적 방식에 의한 키랄성 인지 109 부분 입체 이성질체 16, 17 분리인자 65 분배계수 109
분자궤도함수 이론 337 분자의 대칭성 21 분해인자 66 Bi o t의 법칙 19 BSA- 세파로스 키 랄고정상 207 BSA 의 특성 203- 2 05 BSA 키랄고정상 203- 21 7, 470 비대칭 분자 21 비대칭 유발 중합반응 234, 235 비대칭 탄소 22 비대칭 파괴 51 비대칭 합성 41 비등방성 효과 62 비이온성 변형제 195, 196 人 (S)-th i o - DNBTy r- E 키랄고정상 379 (S)-th io -DNBTy r- A 키랄고정상 379 삼성분 착화합물 94, 95, 284, 285, 288 상대배열 26 세 점 상호작용 모델 101- 1 12 셀룰로오스를 기저로 한 키랄고정 상 122-150, 465 셀룰로오스의 구조 121, 123 소의 혈청알부민 180, 203 속도론적 광학분할 50 손님분자 100, 320 솔형 키랄고정상 444
찾아보기
수소결합 과정 396 수소결 합 키 랄고정 상 443-462, 484 순간 부분 입체 이성질체 53, 90 쉬프영기 308 시 클 로덱스트린의 구조 150-153 시클로덱스트린 키랄고정상 150- 1 79, 46 8 씽 국 자쌍극자 상호작용 104, 105 쌍극자겹침 과정 395 쐐기 구조식 22 씨결정 47 。 a - 아 릴 일컬아민 키랄고정상 391- 4 17 AGP 의 특성 808-182 AGP 키랄고정상 180- 2 03, 469 아미노산 아미드 키랄고정상 444 - 45 5 아밀로오스의 구 조 124 액체 크로마토그래피에 의한 광학 분 할 65-112 양이온성 변형제 195, 196 enanti om eric excess 52 에난시오머 16 eryt hr o 26 에피머 36 역상 크로마토그래피 170 열량계 방법 46 오보뮤코이드 217
오보뮤코아드 키랄고정상 217-220 옥심 교환반웅 58 용량인자 66 용매의 국성인자 386 용매의 선택성 파라미터 386 우레아결합 키랄고정상 415 우선성 18 원근법 그림 22 유발광학분할 48 음이온성 변형제 195, 196 이온화 에너지 338 이동상변형제 182, 194, 195 이성분 착물 94, 284 이성질체 15, 16 이성질체의 분류 15- 1 8 이형태체 81 입체 배열 의존 상호작용 104, 105 입체 이성질체 16 입체장애 상호작용 104 입체특이성 103 입체화학 16 大 자물쇠 -열쇠 관계 60, 103 자발광학분할 47 전자 천화도 338 전하이동 상호작용 104, 337 전하이동 착물형성 키랄고정상 3_3 7 -441, 477 전하이동착물 53, 57 절대배열 23
찾아보기
점군 21 정상 크로마토그쾌피 173 정전기적 상호작용 104, 105 제 1 세대 키 랄고정상 348 제 2 세대 키랄고정상 348 제 3 세대 상호성 키랄고정상 392, 393, 424, 428 좌선성 18 주게-받게 키랄고정상 444 주문자방식 의 키 랄고정 상 180, 266, 268 주변수소 368, 392 주인분자 100, 320 준임계 유체 크로마토그래피 141, 175, 247, 390 지침원자 31 직접 분리방법 65, 90-112 天 초임계 유체 크로마토그래피 390, 446, 449 천화적인 상호작용 104 친화크로마토그래피 204 = 카르비놀 키랄고정상 343-347 Cahn-In g old-Prelo g의 순위 결정 규칙 23 Cahn-In g old-Prelo g의 (R.S) -명 명법 24
크라운 에데르 320-323 크라운 에데르 키랄고정상 320-333, 476 키랄고정상법 91, 93, 101-112 키랄공동 58, 99, 108, 266, 267, 275 키랄공동을 가전 고분자 키 랄고정 상 266- 2 79 키랄 란타나이드 이동 시약 61, 63 키랄 리간드 53, 283 키랄면 21, 31 키랄선택자 65 키랄성 18, 21 키랄성 인지의 상호성 개념 347- 3 48 키랄시약 53 키랄 용매화 시약 61, 344 키랄유도체 시약 60, 65, 70 키랄이동상법 93, 284 키랄이동상 첨가제법 91, 93, 284 키랄장벽 321 키랄중심 21 키랄축 21, 28 키랄탄소 22 키랄형판분자 266, 267, 279 키랄 환경 36, 45, 46, 60 a- 키모트립신 220 a- 키모트립신을 기저로 한 ACHT 키랄고정상 220-223 E thr eo 26
찾아보기
Tro g er 염기 27 고 'Tl- 7 C 상호작용 104, 105, 337, 364 'Tl - 산성분자 339 'Tl - 산성 키랄고정상 347- 3 91, 477 'Tl - 염기성 분자 339 'Tl - 영 기 성 키 랄고정 상 391-435, 481 'Tl - 전자받게 337, 339, 342 'Tl - 전자주게 337, 339, 342 퍼센트 광학순도 20 P i rkle - 형 키랄고정상 342 페닐글리신 - DNB 키랄고정상 348 -361 평면편광 18 Frie d el-C raft s 알킬화 반응 393
프탈리드 키랄고정상 424 플래쉬 컬럼 크로마토그래피 376 F i sher 두영 식 2 2 -lo Hammet
현명호 서울대학교 문리과대학 화학과(이학사) 한국과학원 화학과(이학석사) 미국 Univ e rsit y of Illin o is at Urbana-Champ a ig n 대학원 화학과 (Ph . D.) 미국 Univ e rsit y of Illin o is at Urbana-C h amp a ig n 박사후 연구원 미 국 Harvard Univ e rsit y 박사후 연구원 미 국 Univ e rsity of Illin o is at Urbana-Champ a ig n 연구원 현재 부산대학교 자연과학대학 화학과 부교수 논문 A Rati on al Ap pr oach to the Desig n of High ly- E f fec ti ve Chir a l Sta t i on ary Phases 의 다수 LC 에 의한 광학 이성질체의 분리 대우학술총서 자연과학 79 초판 찍음 1992 년 5 월 20 일 초판 펴냄 1992 년 5 월 30 일 지은이 현명호 펴낸이 朴孟浩 펴낸곳 (주)民音社 편집/이갑수·윤석호·이남수·전경심·강일철·임인기 출판등록 1991 . 12. 20 제 16-490 호 • 우편 대체번호 0110041-31-0523282 • 은행 지로번호 3007783 • 135-120 서울 강남구 신사동 506 강남출판문화센터 5F • 515- 2000~2( 영업부) 515-2003~5( 편집부) 55- 2 007, 2101( 팩시밀리) 값 17,500 원 Prin t e d in Seoul, Korea © 현명호, 1992 자연과학 • 화학 KDC / 433 .42 ISBN 89-374-3579-9 94430
대우학술총서 ( 자연과학 ) 1 소립자와 게이지 상호작용 44 한국의 고생물 이하영 김진의 45 질량분석학 김명수 2 동력학특론 이병호 46 급변론 박대현 3 질소고정 송승달 47 생체에너지 주충노 4 상전이와 임계현상 검두칠 48 리이안 기하학 박을룡 5 촉매작용 진종식 49 君表現論 박승안 6 뫼스바우어 分光學 옥항남 50 비선형 편미분 방정식론 7 극미량원소의 영양 승정자 하기식 8 수소화붕소와 유가붕소 51 생체막 김형만 화합물 윤능민 52 수리분류학 고철환 9 항생물질의 全合成 강석구 53 찰스 다윈 정용재 10 국소적 형태의 At iy a h- 54 금속부식 박용수 sin g er 지표이론 지동표 55 양자광학 이상수 11 Mucop o I y saccha r i des 의 56 효소반응 속도론 서정현 생화학 및 생물리학 박준우 111234 프天천로然체스物물타리化학글學라硏 홍딘究승 法수合 우成원 김식성 각 555978 확분화률자성암론 분 성구광자인학홍 론 소 현이수민 성 111675 脂結고 분防晶 자化營에유養 리 김의 숙김한회병 화호학 반응 666120 에백곤터너충신속지경 띠이 론생이 론리양 학재모 현혜부 정경 생 18 과학혁명 검영식 63 수학 기초론 김상문 2109 한정국보이지질론 론한 영장열기 홍 6654 S신BoC경lHo 과부mo학호n 와 부김 호승R업e e이 · d박만- 찬영 옹 21 원자핵반응론 정운혁 66 양자 전기역학 김영덕 22 파괴역학 김상철 67 군환론 박재걸 닳듭쏴궤노i 聞대수,,하학 222645 자1반11 기분응공위속명상도방수른법정贊 경조이훈성현 호구 障7710양 1:1지 청이른 27 플라스마 룰리학파 빼응할 여 최력인 28 천문관측과 분석 이시우 29 석탄에너지변환기술 검상돈 30 빼양미고생를확 뻬탈호 31 편미분 방정식론 김총식 32 대를일 이른 소광실 33 급속전자계의 다체이른 검리주 34 엑정좋합체 진경일 35 복합재료 련속인 36 단백질 생합성 박인원 37 한국의 광를륭 깁수진 38 일반상대른 이철훈 39 레이저 광산란 분꽝학 검증진 40 복소다양체른 갑상룬 41 역학적 연구탈법 김일순 묘빼구조를리화민등팔 43 후라에 해석 綱분작용소김도한